

Abstract
The pathophysiology of immunoglobulin E (IgE) mediated food allergy has been understood on a superficial level for several decades. Surveillance by dendritic cells for exogenous antigens leads to a high-affinity IgE response that arms effector cells (sensitization), such that subsequent exposures can trigger a type 1 hypersensitivity recall response. However, merely scratching the surface, whether confronting unmet needs in a clinical setting or probing the basic immunology of allergic immunity, quickly reveals the many unmet fundamental questions that lie there. This review article focused on the following such questions. Why are common allergens common? How does sensitization most often occur? How is IgE maintained over long time periods, even in the apparent absence of exposure? What distinguishes sensitization from clinical allergy? Can we stratify risk (i.e., sensitivity and severity)? What distinguishes the pathophysiology of non–IgE-mediated allergy when so much of it seems to overlap?
LEARNING OBJECTIVES
To discuss the current paradigms of how and why food allergy develops in terms of its pathogenesis.
To discuss recent data that address the physiology that underlies areas of current unmet needs (e.g., prognosis).
To compare and contrast the pathophysiology of different presentations of food allergy (e.g., immunoglobulin E [IgE], non-IgE).
PATHOPHYSIOLOGY 101
Immediate hypersensitivity reactions by definition are provoked by immunoglobulin E (IgE), which has been appreciated for decades as something of a paradox in food allergy, given that most clinical reactions occur on the first known ingestion of the culprit food. This paradox is not lost on many parents of young patients who often ask their allergist how it is that their child had such a reaction when they have never knowingly consumed the food before the reaction.
Studies, beginning more than a hundred years ago and culminating in the identification of IgE >50 years ago,1,2 have led to the current paradigm that sensitization to an antigen consists of initial priming of specificCD4+ T-helper type 2 (Th2) cells by dendritic cells, which results in interleukin (IL) 4 receptor (4R) dependent E IgH class switching in B cells and their differentiation to IgE-secreting plasma cells. This secreted IgE then binds to the high-affinity IgE receptor, Fc« RI, constitutively expressed on the surface of mast cells and basophils (among others). With this sensitization in place (a processthat requiresdaysto weeks), secondary exposure to the same antigen (now referred to as an allergen) induces cross-linking of the IgE-Fc« RI with consequent release of histamine and other mediators of anaphylaxis (please see the first sections of Larché et al.3 for an excellent review of the basics of sensitization as well as late-phase IgE-dependent inflammation and the potential role of IgE on amplifying the recall response by binding to Fc« RI on dendritic cells and other innate immune cells).
PATHOPHYSIOLOGY: SENSITIZATION TO AMPLIFICATION AND PERSISTENCE
What any clinician familiar with testing for food allergy will know, however, is that sensitization defined simply as a nominally positive test result for IgE by a skin-prick test or serum assay is not synonymous with clinical sensitivity; it is generally overly sensitive for a diagnosis.4 Even worse, however, from a practical diagnostic standpoint, although there is a clear correlation between the strength of sensitization (i.e., size of skin test reactivity, concentration of allergen specific IgE), the variance is large, such that some individuals with strong sensitization are not clinically allergic and that some with low sensitization are allergic. Although there are likely to be a number of factors, such as the allergen specificnon- IgE antibody repertoire (e.g., IgG, IgA), intestinal permeability, effector cell features (e.g., the special case of increased sensitivity among those with concomitant food allergy and mastocytosis), and end organ sensitivity, variations in the “quality” of the IgE response, not routinely evaluated, contribute to that variance.
The features of the specificIgE quality that are associated with its efficiency for cross-linking, and correlated with clinical utility as well, are the following: affinity, specific activity (the ratio of specificto totalIgE), and clonaldiversity.5 In the typically low allergen dose exposure range that provokes reactions, high-affinity IgE is generally necessary. The generation of such high-affinity IgE, anaphylactic IgE, likely requires very specific T-cell help to generate. Recently, a distinct phenotype of T cell, characterized by both B-Cell Lymphoma 6 (BCL6) and GATA Binding Factor 3 (GATA3) dependent transcriptional programs to induce a type 2 T follicular helper (Tfh), was found to be necessary to induce anaphylaxis in mice and is associated with disease in humans.6 The investigators named this cell population “Tfh13” because IL-13 expression in the Tfh lineage is necessary for inducing anaphylaxis.
The identification of this T-cell population, which has also been observed in patients with peanut allergy (Tu et al., unpublished data), is consistent with what is understood more generally about the generation of a high-affinity antibody response of other isotypes as well. It is an iterative process of T- and B-cell interactions, amplifying the number of participating clones, selecting those with the highest affinity, and, therefore, diversifying the response in the process. This process can progress with subsequent rounds of antigen exposure as well, each time potentially producing IgE-secreting plasma cells and expanding the antigen-specific memory B-cell pool. A unique feature of IgE, however, is that, although IgE-producing plasma cells can be long lived, membrane IgE expressingmemoryB cells do not survive; and rounds of recall stimulation, expansion, and selection of IgE are from a pool of IgG1+ B cells.7 Although IgE class switching can and does occur from naive IgD+ IgM+ B cells directly and without this amplification process, the “quality” of the IgE response (the overall population of affinity and clonality, which determines its avidity) is poor and less likely to drive strong, clinically relevant, hypersensitivity reactions.
PATHOPHYSIOLOGY: SENSITIZATION VERSUS HOMEOSTASIS
An implication of this IgE sensitization and amplification paradigm is that there may be separate “checkpoint” determinants of food allergy that influence the propensity toward a Th2 immune response (i.e., what we term atopy) and the propensity toward diversification and amplification of an antigen-specific response (sometimes referred to in the autoimmune literature as immune progression). Those determinants may be either intrinsic or extrinsic. Consistent with the role of oral exposure as an extrinsic factor, Du Toit et al.8 had an enormous impact on practice with their demonstration that the early introduction of a common allergen, peanut, to the diet of infants at high risk to develop peanut allergy was profoundly protective. The dual allergen exposure hypothesis advanced by Du Toit et al.8 posit that cutaneous exposure, particularly in the setting of allergic inflammation and barrier breach, is sensitizing, whereas oral exposure is tolerogenic.
Less widely appreciated, has been the striking observation of an impressive intrinsic factor. In a Genome wide association study (GWAS) of the Learning Early About Peanut (LEAP) cohort, Winters et al.9 identified a strong association between peanut allergy and the MALT1 locus for LEAP participants in the peanut avoidance group, with 58.6% of carriers developing peanut allergy at 60 months compared with just 12.7% of noncarriers. The odds ratio for peanut allergy associated with the peak single nucleotide variation was an astounding 10.99 (p = 6.49 108). There was no associated risk in the protected peanut-consumption group, a remarkable example of gene by environment interaction. The MALT1 paracaspase is a central player in the activation of the transcription factor Nuclear Factor Kappa B Subunit 1 (NF-κ B) from both innate and adaptive (i.e., B cell receptor (BCR) and T cell receptor (TCR)) signaling pathways and has been proposed to be key in balancing immunity versus tolerance.10
IMMUNE PROGRESSION AS AN ENDOTYPE
An hypothesis to link these phenotypes of persistence, sensitivity, and severity is the immune progression hypothesis (Fig. 1). Immune progression itself is not a mechanism; it is a candidate endotype in search of its underlying mechanisms (e.g., genetic polymorphisms, key exposures, dysbiosis). When drawing on the LEAP findings as an example, oral exposure is a protective determinant, atopic dermatitis severity and duration is a risk determinant (personal communication) and MALT1 SNV, rs57265082, is a risk determinant. The hypothesis, consistent with the data so far, is that these factors drive the diversification of either protective regulatory or pathogenic type 2 inflammatory responses.11 This hypothesis also fits with the observations over many years that markers of immune diversification correlate with disease severity and/or persistence. Recent examples include the association between the likelihood of having persistent egg allergy and the diversification of the specific IgE response to multiple egg allergens12 and the greater sensitivity on peanut challenge with the size of the antigen-specific T-cell repertoire.13
Figure 1.
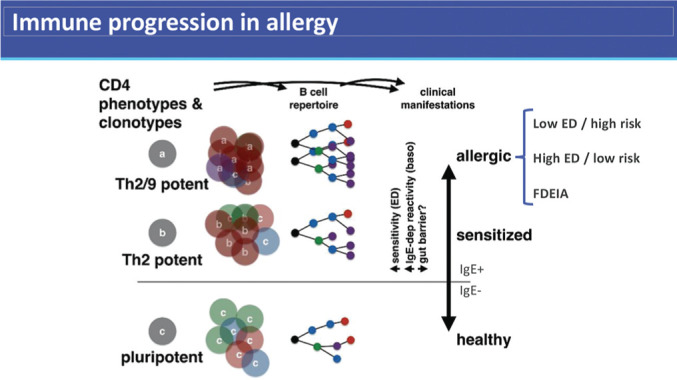
The progression to a polarized allergic state from clinically insignificant sensitization to increasing clinical sensitivity and lifelong persistence is associated with an expansion of the antigen-specific adaptive immune response and determined by intrinsic and extrinsic risk factors. This may be reversible as in individuals who lose clinical allergy spontaneously. T-helper type 2 (Th2) cells are characterized by production of interleukin (IL) 4, often with IL-13 and IL-5. Th2/9 refers to IL-9 + Th2 cells, as described in Ref. 18.
NON–IgE-MEDIATED FOOD ALLERGY IN CONTRAST
As the pathophysiology of IgE-mediated food allergy is better understood, a striking feature that has emerged is the significant degree of overlap, with at least one non-IgE manifestation of food allergy, eosinophilic esophagitis (EoE). As Rothenberg et al.14 have shown, numerous genetic loci that affect barrier function, Th2 responses, and regulatory responses overlap between IgE-mediated food allergy and EoE, and it is now well documented that oral immunotherapy–induced suppression of IgE-mediated food allergy can lead to EoE. From both clinical trials that target it and from animal models, IgE does not seem to be important in the pathogenesis of EoE, and yet highly differentiated and Th2 polarized effector T cells (Th2a;15) seem to be present in both diseases. Trials of biologics that target allergic pathways common to both (e.g., IL-4Rα, IL-33) are arguably the most well founded, given the current understanding of disease pathogenesis.
ALLERGENICITY
Approximately 20 protein motifs, <0.2% of those known, account for ~80% of recognized food allergens. Contrary to the view that allergens are innocuous environmental substances, many of them cause damage to host cells (e.g., proteases, defensins) and some are directly toxic (e.g., phospholipases A2, hyaluronidases). Resistance to pathogens is a prerequisite for the survival of any species, and, in plants, the seeds require the highest protection. Many proteins of peanut seeds that likely contribute to the plant's own defense against pathogens are also allergenic. Although clearly more complex than the relatively constrained pathogen-associated molecular patterns that drive Th1 or Th17 responses, these observations strongly imply that innate immune recognition is a feature that plays a role in allergenicity, and this remains a fundamentally important area of investigation, reviewed elsewhere.16,17
CLINICAL PEARLS
Sensitization and progression to clinical food allergy often occurs before any known oral ingestion, likely, in part, from exposure to allergens via other routes, especially inflamed skin.
Sensitization alone is not sufficient for clinical allergy.
Introduction of allergens to the diet early in the life of at risk infants can prevent both sensitization and progression to clinical allergy.
Measures of immune progression are beginning to be for prognosis of food allergy.
Footnotes
The author has no conflicts of interest to declare pertaining to this article
Funded by Food Allergy Research & Education (FARE)
REFERENCES
- 1.Ishizaka K, Ishizaka T.. Physicochemical properties of reaginic antibody. 1. Association of reaginic activity with an immunoglobulin other than gammaA- or gammaG-globulin. J Allergy. 1966; 37:169–185. [DOI] [PubMed] [Google Scholar]
- 2.Johansson SG, Bennich H, Wide L.. A new class of immunoglobulin in human serum. Immunology. 1968; 14:265–272. [PMC free article] [PubMed] [Google Scholar]
- 3.Larché M, Akdis CA, Valenta R.. Immunological mechanisms of allergen-specific immunotherapy. Nat Rev Immunol. 2006; 6:761–771. [DOI] [PubMed] [Google Scholar]
- 4.Renz H, Allen KJ, Sicherer SHet al. Food allergy. Nat Rev Dis Primers. 2018; 4:17098. [DOI] [PubMed] [Google Scholar]
- 5.Christensen LH, Holm J, Lund Get al. Several distinct properties of the IgE repertoire determine effector cell degranulation in response to allergen challenge. J Allergy Clin Immunol. 2008; 122:298–304. [DOI] [PubMed] [Google Scholar]
- 6.Gowthaman U, Chen JS, Zhang Bet al. Identification of a T follicular helper cell subset that drives anaphylactic IgE. Science. 2019; 365:eaaw6433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asrat S,Kaur N,Liu Xet al. Chronic allergen exposure drives accumulation of long-lived IgE plasma cells in the bone marrow, giving rise to serological memory. Sci Immunol. 2020; 5:eaav8402. [DOI] [PubMed] [Google Scholar]
- 8.Du Toit G, Roberts G, Sayre PHet al. Randomized trial of peanut consumption in infants at risk for peanut allergy. N Engl J Med 2015; 372:803–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Winters A, Bahnson HT, Ruczinski Iet al. The MALT1 locus and peanut avoidance in the risk for peanut allergy. J Allergy Clin Immunol. 2019; 143:2326–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jaworski M, Marsland BJ, Gehrig Jet al. Malt1 protease inactivation efficiently dampens immune responses but causes spontaneous autoimmunity. EMBO J. 2014; 33:2765–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tordesillas L, Berin MC, Sampson HA.. Immunology of food allergy. Immunity. 2017; 47:32–50. [DOI] [PubMed] [Google Scholar]
- 12.Dang TD, Peters RL, Koplin JJet al. Egg allergen specific IgE diversity predicts resolution of egg allergy in the population cohort HealthNuts. Allergy. 2019; 74:318–326. [DOI] [PubMed] [Google Scholar]
- 13.Ruiter B, Smith NP, Monian Bet al. Expansion of the CD4+ effector T-cell repertoire characterizes peanut-allergic patients with heightened clinical sensitivity. J Allergy Clin Immunol. 2020; 145:270–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Azouz NP, Rothenberg ME.. Mechanisms of gastrointestinal allergic disorders. J Clin Invest. 2019; 129:1419–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wambre E, Bajzik V, DeLong JHet al. A phenotypically and functionally distinct human TH2 cell subpopulation is associated with allergic disorders. Sci Transl Med. 2017; 9: eaam9171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ozias-Akins P, Breiteneder H.. The functional biology of peanut allergens and possible links to their allergenicity. Allergy. 2019; 74:888–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palm NW, Rosenstein RK, Medzhitov R.. Allergic host defences. Nature. 2012; 484:465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Micossé C, von Meyenn L, Steck Oet al. Human “TH9” cells are a subpopulation of PPAR-g + TH2 cells. Sci Immunol. 2019; 4:eaat5943. [DOI] [PubMed] [Google Scholar]