

Abstract
Crosslinking mass spectrometry (XL-MS) supports structure analysis of individual proteins and highly complex whole-cell interactomes. The identification of crosslinked peptides from enzymatic digests remains challenging, especially at the cell level. Empirical methods that use gas-phase cleavable crosslinkers can simplify the identification process by enabling an MS3-based strategy that turns crosslink identification into a simpler problem of detecting two separable peptides. However, the method is limited to select instrument platforms and is challenged by duty cycle constraints. Here we revisit a pseudo-MS3 concept that incorporates in-source fragmentation, where a fast switch between gentle high-transmission source conditions and harsher in-source fragmentation settings liberates peptides for standard MS2-based peptide identification. We present an all-in-one method where retention time matches between the crosslink precursor and the liberated peptides establish linkage and MS2 sequencing identifies the source-liberated peptides. We demonstrate that DC4, a very labile cleavable crosslinker, generates high-intensity peptides in-source. Crosslinks can be identified from these liberated peptides as they are chromatographically well-resolved from mono-links. Using bovine serum albumin (BSA) as a crosslinking test case, we detect 27% more crosslinks with pseudo-MS3 over a best-in class MS3 method. While performance is slightly lower for whole cell lysates (generating two-thirds of the identifications of a standard method), we find that 60% of these hits are unique, highlighting the complementarity of the method.
Graphical Abstract
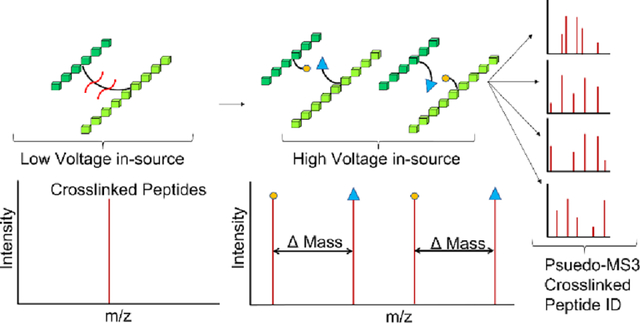
Image shows that in-source fragmentation toggle can be dynamically incorporated with MS2 measurements to achieve and on-the-fly pseudoMS3 measurement for crosslink detection.
INTRODUCTION
Crosslinking mass spectrometry (XL-MS) can generate distance measurements that are useful in the structure analysis of proteins. By crosslinking protein lysates and even whole cells, it is possible to determine protein structures in their native state, avoiding the challenges of protein production associated with classical methods in structural biology1–3. Even high-resolution mapping of protein networks should be possible with XL-MS when supported by AI-based fold prediction methods4. Crosslinks are difficult to detect. Workflows involve the digestion of the crosslinked proteins followed by the MS detection of crosslinked peptides. These are invariably low-abundance reaction products. Chromatographic methods5 and affinity tags6 help enrich for crosslinks but even when enriched, identifying the sequences of the linked peptides is not straightforward. Any given MS2 spectrum of a putative crosslink has two peptides contributing to the fragmentation pattern. The pairwise assessment of peptides greatly increases the search space even when it is tightly constrained by high accuracy mass measurements7–9. Some computational solutions have made good progress in addressing this problem, but biased fragmentation makes it challenging to detect both peptides with equal sensitivity10.
Alternative methodologies offer solutions to the problem of crosslink detection. For example, gas-phase cleavable crosslinking reagents have been developed that return some simplicity to peptide identification11–15. Here, crosslinking agents are designed with a labile functional group that can be readily cleaved at reduced energies in the CID or HCD collision cell of a tandem mass spectrometer. This step yields individual peptides with crosslinker-derived modifications. After cleavage, the resulting characteristic signature fragments can then be selected and further fragmented in an MS3 experiment, effectively reducing the search complexity to simple linear peptides once again. A variety of cleavable moieties have been installed in crosslinkers and made commercially available in recent years. Most amine-targeting crosslinkers are cleavable to some degree, as fragmentable amide bonds are formed upon crosslinking16, but intentionally-designed cleavable crosslinkers usually incorporate a more labile sulfoxide, like disuccinimidyl sulfoxide (DSSO)14, a cleavable urea, like disuccinimidyl dibutyric urea (DSBU)15, or a RINK group as in protein interaction reporters (PIR)17. Quaternary amines such as disuccinimidyl 1,4-diazabicyclo[2.2.2]octane (DC4) have also been used13.
Cleavable reagents that are processed in collision cells are not a perfect solution. Efficient gas-phase cleavage is required to generate clean fragmentation products for subsequent selection in MS3, but mixed fragmentation occurs in many cases, leading to signal splitting and potentially lower sensitivity than might otherwise be the case. Additionally, an MS3 routine limits the duty cycle of the acquisition, and many instruments (e.g. TOFs) are not able to perform the method. It has been established recently that peptides coupled with gas-phase cleavable linkers can be processed at normal energies in regular MS2 scans (e.g., stepped HCD)18. The approach can even increase the degree of backbone fragmentation, leading to more confident assignments. This adaptation has become popular for larger scale crosslinking experiments, but it returns some of the complexity of database searching that the reagents were designed to overcome.
We were interested to see if alternative fragmentation modes could recover some of the advantages of the MS3 approach. One possible way to improve duty cycle yet still simplify the search space could be to invoke in-source fragmentation19. Here, crosslinked peptides would be cleaved in an elevated electric field during the ion desolvation and transmission process. In-source fragmentation was often used when MS3 modes of operation were less well-developed. While generally regarded as a nuisance to be avoided, in-source fragmentation has been embraced recently to improve performance in metabolite detection20. It has also been shown to generate rich and intense fragmentation spectra for peptides21, creating opportunities for improved S/N measurements on simpler instrument platforms. A method was introduced by the Bruce lab using the PIR crosslinker22, where one LC-MS run used alternating high and low source voltages to generate new peptides in-source, followed by a separate LC-MS/MS run at the higher energy condition to identify liberated peptides. Here, we describe an all-in-one approach where source conditions are toggled rapidly in a single method for triggering data dependent MS2 acquisitions of the liberated peptides. We explore additional cleavable crosslinker chemistries and develop a new software tool to exploit the method for whole proteome crosslinking experiments.
EXPERIMENTAL SECTION
Materials.
Bovine serum albumin (BSA) was purchased from Sigma-Aldrich (P/N A7906). DSSO was purchased from ThermoFisher Scientific (P/N A33545). DC4 was purchased from the Cayman Chemical Company (P/N 14734).
BSA crosslinking.
Bovine Serum Albumin (BSA) at 1 mg/mL was crosslinked using 0.5 mM and 1 mM DSSO or DC4, from stocks of 50 mM and 100 mM crosslinker, respectively. Crosslinking buffer contained 50 mM HEPES (pH 7.4) and 100 mM NaCl. Crosslinking was conducted for 20 minutes at room temperature. Samples were then quenched by the introduction of 500 mM ammonium bicarbonate to a final concentration of 50 mM. After 20 minutes of quench, the protein was denatured by the addition of sodium deoxycholate to a final concentration of 1%. The protein was then reduced and alkylated by the addition of DTT to 10 mM final concentration for 20 minutes, followed by the addition of 2-chloroacetamide to 40 mM final concentration, for 40 minutes. The sample was then trypsin digested at a 1:40 enzyme:protein ratio overnight. Samples were then acidified with trifluoracetic acid and centrifuged at 14000 x g for 10 minutes to remove the precipitated deoxycholate. Digests were zip-tipped and peptides eluted with 50% acetonitrile and 0.1% formic acid (FA), partially dried under vacuum, and made up to 20 μL in 0.1% formic acid for LC-MS analysis.
Lysate crosslinking.
A 500 mL overnight culture of E. coli BL21 was pelleted and re-suspended in 40 mL of cold lysis buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 10 mM MgCl2, 1 X Roche protease inhibitor, 1 mM DTT), lysed by sonication, and cleared by centrifugation at 20000 x g for 30 minutes. Lysate concentration was measured using a Pierce BCA assay kit, and the lysate was then diluted in lysate crosslinking buffer (20 mM HEPES pH 7.4, 150 mM NaCl) to 1 mg/mL. 200 μg of protein was crosslinked at room temperature for 40 minutes with 0.75 mM of DSSO or DC4, from 100- or 50-mM stocks, respectively. Crosslinking was quenched by the addition of 500 mM ammonium bicarbonate to a 50 mM final concentration and incubated for 20 minutes at room temperature. Sodium deoxycholate was added to a final concentration of 1% to assist with denaturation of the protein, supplemented with 3% acetonitrile. The protein was then reduced and alkylated by the addition of DTT to 10 mM final concentration for 20 minutes, followed by the addition of 2-chloroacetamide to 40 mM final concentration for 40 minutes. The sample was trypsin digested at a 1:40 enzyme:protein ratio overnight. Samples were acidified with trifluoracetic acid and centrifuged at 14000 x g for 10 minutes to remove the precipitated deoxycholate. Peptides were then desalted using a Pierce C18 Spin Column, lyophilized to dryness, and stored at −80 °C. For analysis, peptides were resuspended with 30% acetonitrile and 0.1% FA, partially dried under vacuum and made up to 20 μL in 0.1 % FA for LC-MS analysis.
LC-MS/MS analysis of BSA on the Orbitrap Velos.
BSA samples on the Orbitrap Velos were analyzed using a nano-HPLC (Easy-nLC 1000, ThermoFisher Scientific) coupled to the nano-ESI source of an Orbitrap Velos (ThermoFisher Scientific). Samples were injected onto a 75 μm x 2 cm PepMap 100 trap column (C18, 3 μm, 100 Å, ThermoFisher Scientific) and desalted via a short wash with solvent A (3 % acetonitrile: 97% water, 0.1% FA). Peptides were separated on a 75 μm x 15 cm self packed column of Kinetix 2.6 μm XB-C18 beads (Phenomenex) in a PF360–75-8-N Picofrit (New Objective) format using a multistep gradient from 5–30 % solvent B (97% acetonitrile: 3% water, 0.1% FA) for 60 minutes, 30–40 % solvent B for 10 minutes, 40–100 % solvent B for 1 minute, and a 10-minute wash at 100% solvent B.
Several different Velos MS methods were used in these experiments. To create a ground-truth dataset, a conventional crosslink detection experiment was used for DSSO. Here, stepped HCD and a typical data-dependent analysis (DDA) was employed, selecting charge states of 4+ and higher to be fragmented and scanned in the orbitrap. Resolutions for the MS1 and MS2 scans were 60,000 and 15,000, respectively. HCD MS2 scans had a normalized collision energy of 30 with a step of +/− 3. Dynamic exclusion was set for 60 seconds with a repeat count of 1. This method would not work for DC4 given the lability of the reagent, vide infra. We used an MS3 method to create the ground-truth data set as described in the next section. For in-source voltage ramp experiments, an initial MS1 scan was acquired, followed by 4 successive MS1 scans with increasing source-driven dissociation voltages applied: 40V, 50V, 60V and 70V. Experiments with alternating voltages had a similar setup, alternating between 0 and 45V SDD voltage MS1 scans. In the experiments where in-source fragmentation was used for crosslink identification, two different ion trap DDA methods were used. The first was an untargeted DDA method in which all ions of charge 2+ and higher were selected for DDA in a top 6 method, using CID with a normalized collision energy of 30, before scanning the ion trap in the rapid scan mode. In the second DDA method, a mass tag rule was used. If a signature mass difference corresponding to the crosslinker cleavage was detected (112.10 Da in the case of DC4, 31.97 in the case of DSSO), the ions were selected for fragmentation. In the case of DC4, only the low mass fragment of the pair was instructed to be selected for fragmentation.
LC-MS/MS analysis of E. coli on the Orbitrap Eclipse.
E. coli samples were analyzed on the Orbitrap Eclipse using a nano-HPLC (Easy-nLC 1200, ThermoFisher Scientific) coupled to the nano-ESI source of an Orbitrap Eclipse (ThermoFisher Scientific). Samples were injected onto a 75 μm x 2 cm PepMap 100 trap column (C18, 3 μm, 100 Å, ThermoFisher Scientific) and desalted via a short wash with solvent A (0.1 % FA). Peptides were separated on a 75 μm x 15 cm EASY-Spray HPLC column (3 μm beads, ThermoFisher Scientific, P/N ES900) using a multistep gradient from 5–30 % solvent B (80% acetonitrile : 20% water, 0.1% FA) for 190 minutes, 30–45% solvent B for 45 minutes, 1 minute to 100% solvent B, and a 10-minute wash with 100% solvent B.
Several acquisition methods were also used on the Eclipse. For conventional crosslink detection of DSSO, stepped HCD runs with charge states of 4+ and higher were selected for MS2. The Orbitrap MS1 resolution was set to 120,000, and the Orbitrap MS2 resolution was set to 30,000. HCD collision energy was set to 30 +/− 3. Dynamic exclusion was set for 30 seconds, with a repeat count of 1. For DC4 MS3 runs (including the DC4-crosslinked BSA mentioned above), a single normalized CID MS2 energy 25 of was used. From these scans, mass tags were used to select a pair of ions with a mass difference of 112.1001 with an error tolerance of 10 ppm, and the lighter ion of each pair was further fragmented in MS3 with a normalized CID MS3 energy of 35, scanning the ion trap in rapid scan mode.
In-source runs on the Eclipse used 120,000 resolution for the MS1, followed by a 120,000 resolution orbitrap scan with an in-source acceleration voltage of 45 V applied. Ions for MS2 fragmentation were selected from the in-source voltage scans based on a targeted mass difference (31.9721 for DSSO, 112.1001 for DC4), and subjected to CID with a normalized collision energy of 30, before scanning the ion trap in the rapid scan mode.
Data analysis.
In-source BSA and E. coli data (both DSSO and DC4) were analyzed using a new app within the Mass Spec Studio. In this app, we bundled MS-GF+ with Hardklör and developed a new scoring method. Briefly, all peptides identified by MS-GF+ from the high energy source condition are mass paired with MS1 features (putative crosslink precursors) detected by Hardklör in the low voltage condition, bounded by the retention time of a candidate MS1 feature. The MS-GF+ searches used default parameters, including a mass tolerance of 10 ppm, methionine oxidation as a variable modification, and cysteine carbamidomethylation as a fixed modification. MS2 mass tolerance was set to 0.5 Th. Hardklör parameters were similarly left to the default settings for either the 60,000 resolution orbitrap scan (BSA data from the Velos) or the 120,000 resolution orbitrap scan (E. coli data from the Eclipse).
When determining pairings, the MS1 tolerance for the crosslink precursor was set to 5 ppm. Precursors were validated by assigning charge states and monoisotopic ions and scoring for local complexity and potential overlap (boosted for redundancies). Only high-scoring and high-quality signals were accepted for pairing. The final score for each pair meeting these criteria was the sum of the MS-GF+ identification scores for the two cleaved products, using the best scoring identification in case of redundancies (determined at 5% FDR). Scores were scaled from 0–100 using the CLAM utility in the standard CRIMP search22. Q values were calculated from decoy hits arising from a decoy database search, where decoys are defined as matches that contain at least one false assignment in the MS-GF+ search. A false discovery rate of 5% was set for the export. Finally, the results were then manually evaluated to ensure high accuracy. A positive crosslink identification was one that included a positive identification of both peptides from the MS2 data. The extracted ion chromatograms (XICs) for the crosslink precursor and at least two liberated peptides (one of either heavy or light for each of the two peptides) were evaluated to ensure no spurious correlations were present.
For the conventional MS2-based crosslink detection methods, data were searched using CRIMP 2.0 with the default parameters, with oxidized methionine as a variable modification and cysteine carbamidomethylation as a fixed modification. For MS3-based crosslink detection, data were searched in Proteome Discoverer 2.5 using the XlinkX node with default parameters, including methionine oxidation as a dynamic modification and cysteine carbamidomethylation as a static modification. A precursor mass tolerance of 5 ppm was used, and a 0.5 Da tolerance was used for linear ion trap fragments. Results were filtered to include only those hits that positively identified both the peptides in the crosslink, using the same level of stringency as was applied to the in-source results. E. coli DSSO stepped HCD data were searched in CRIMP 2.0 using a slight modification of the default parameters: the Eα percent threshold was set to 20, the Eβ percent threshold was set to10, with a top N of 10. The FDR was set to 5%, corresponding to a score of 28. The MS1 mass accuracy was set to 5 ppm, and the MS2 mass accuracy was set to 10 ppm.
RESULTS AND DISCUSSION
We developed a modified instrument duty cycle that mimics a traditional data-dependent acquisition (DDA) method, with one exception (Figure 1). The usual MS1-based precursor ion acquisition scan is replaced with a combination that uses a low source accelerating voltage acquisition and a high source accelerating voltage acquisition. Applying a low source voltage allows us to measure the mass of the intact crosslinked peptide at high resolution, whereas the high source voltage induces fragmentation of the crosslinked peptide and supports mass measurements of the liberated peptides. This high voltage acquisition is immediately followed by a series of data-dependent ion trap MS2 experiments at the high voltage setting, so that liberated peptides can be sampled for sequence identification. The linkage between the liberated peptides and the putative crosslinked peptide is established both by mass and chromatographic retention time. That is, the liberated peptides (when accounting for the cleaved crosslinker masses) must match the mass of a detected higher mass ion and generate overlapping chromatographic features. This concept has similarities to a data-independent acquisition (DIA) method23.
Figure 1.
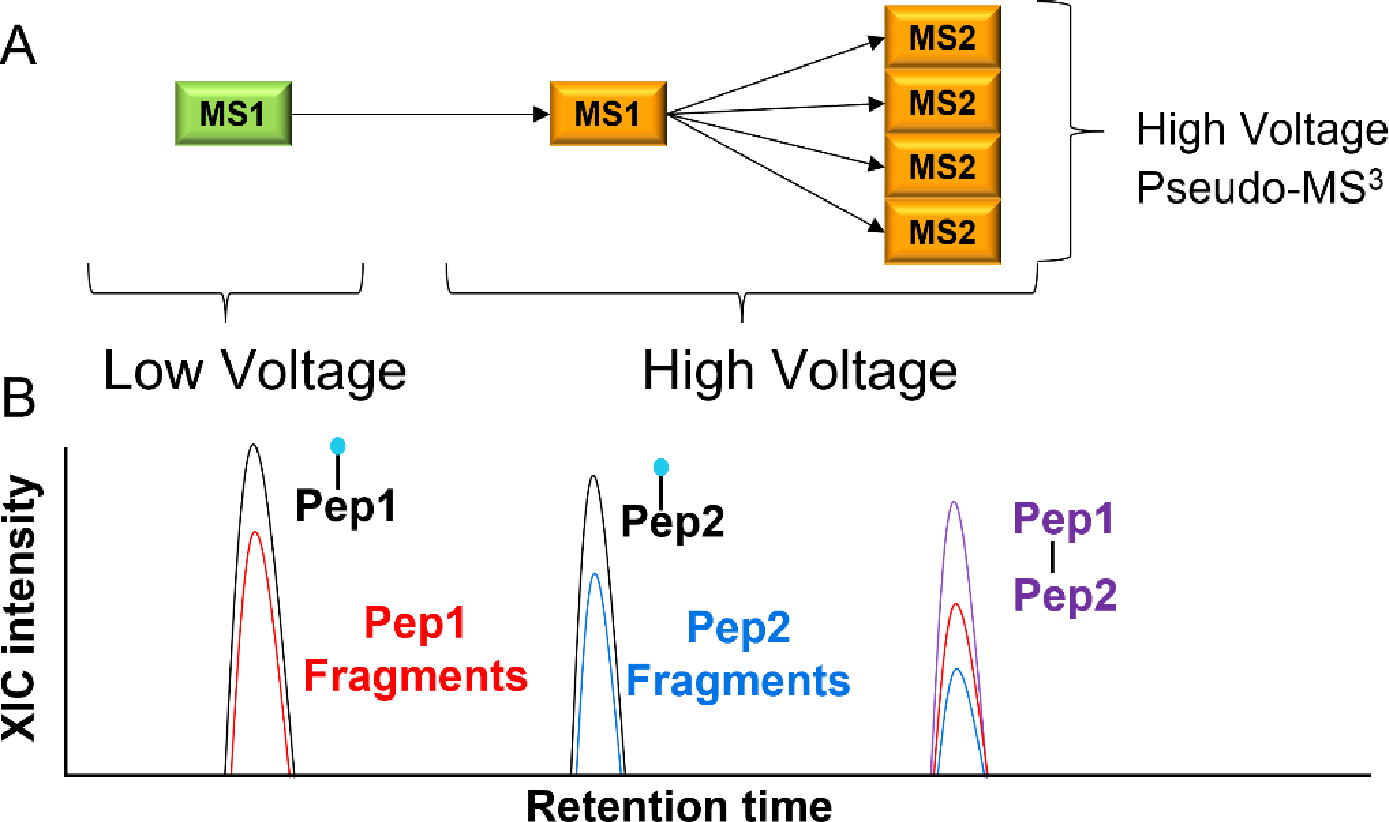
In-source fragmentation data acquisition cycle and search strategy. (A) In an in-source experiment for crosslink detection, the declustering and/or desolvation source voltage setting is initially set at a typical value for the first high-resolution scan, to support the detection of the intact crosslinked peptide. In a second high-resolution scan, this source voltage is quickly increased to fragment the crosslinker for mass measurement of the component peptides. Subsequent MS2 scans are performed at this elevated voltage to sequence the resulting fragmented peptides. (B) The peptides identified at the high source voltage setting are linked back to a potential crosslinked peptide using accurate masses from both MS1 scans. The figure shows that chromatographic separation is required to resolve potential interferents.
To build upon the potential of in-source fragmentation first demonstrated by the Bruce lab with PIR22, we selected two gas-phase cleavable crosslinkers: DSSO and DC4. DSSO fragments with an onset voltage (at low pressure) of around 25 eV24,25, depending on charge and mass. DC4 may require a similar voltage, although an in-depth study has not yet been performed to our knowledge. Both generate pairs of ions upon fragmentation and thus will create a doublet of doublets for each crosslinked peptide (Figure 2).
Figure 2.
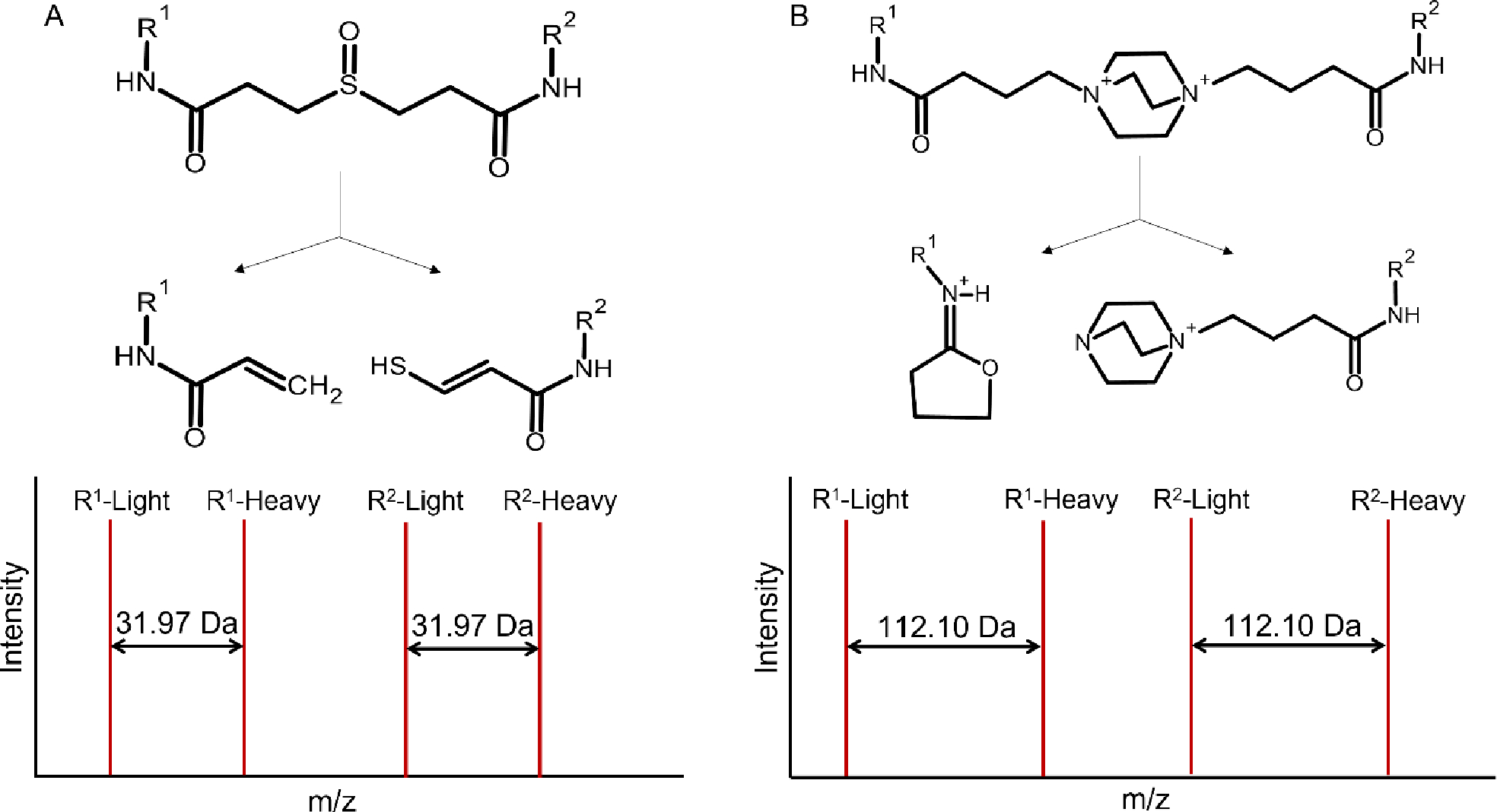
Cleavable crosslinkers and their fragmentation products. (A) DSSO, with short and long reaction products having a Δmass = 31.97. (B) DC4, with short and long reaction products having a Δmass = 112.10.
Establishing a ground truth data set
The fragmentation energies measured from beam-style or resonance-based collision energies do not transfer directly to in-source fragmentation conditions. In the source region, ions are accelerated through variable pressure zones and scattering losses can occur in a m/z-dependent manner, depending on the nature of the source26–28. Indeed, most modern sources are designed to minimize in-source fragmentation, thus we first needed to determine what acceleration voltage was necessary to fragment cleavable crosslinkers, and if acceptable ion transmission could be retained. To this end, we created a dataset of known crosslinked peptides. We crosslinked BSA with both DSSO and DC4 and extensively sampled crosslinked peptide spectra in the conventional fashion for gas-phase cleavable crosslinkers, attempting maximize the number of hits. We detected 81 crosslinks in the DSSO dataset, and 37 crosslinks in the DC4 dataset (Table S1). The number of crosslinks detected for DC4 with stepped HCD is low, because these peptides generated intense crosslinker cleavage ions at the expense of sequence ions (Figure S1)13. This lowers the score of a spectrum match. Therefore, these DC4 samples were also acquired on an Orbitrap Eclipse using an MS3 based approach. We identified 85 crosslinks using this method (Table S1), which agrees well with number of crosslinks reported in a community-wide study of conventional methods29.
In-source crosslinker fragmentation – an inspection
Armed with this list of identified crosslinks, we then performed a series of runs in which the accelerating voltage in the source of the Velos instrument was increased from 40–70 V in increments of 10 V. Five crosslinked peptides for each crosslinker were selected across the retention time range, and the expected linear peptides were monitored in the MS1 spectra from the high-voltage scan. While there was some minor dependence on the specific crosslinked peptide examined, the optimal voltage for fragmentation ranged from 40 – 50 V, and the range was similar for both reagents (Figure 3). We selected an acceleration voltage of 45 V as a balance between good fragmentation and high transmission efficiency. Select MS1 spectra obtained in both low (0V) and high (45V) modes are shown in Figure S2, highlighting the liberation and detection of two peptide doublets.
Figure 3. Dependence of crosslinker fragmentation on in-source acceleration voltage.
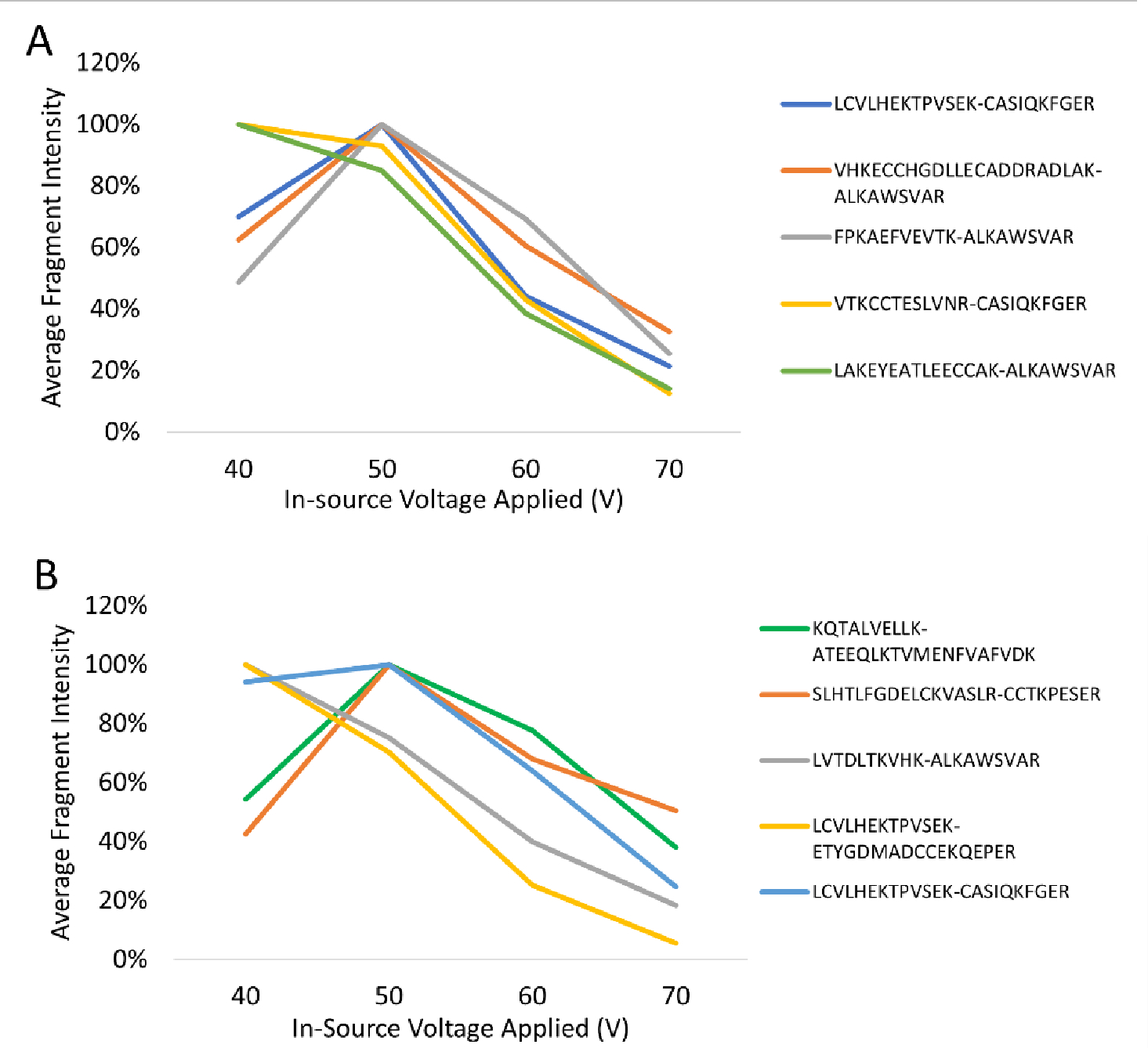
(A) DSSO-crosslinked peptides and (B) DC4-crosslinked peptides. Fragment intensities determined in FreeStyle and normalized to the intensity of the most intense voltage for each fragment.
Occasionally additional fragmentation products were formed. For DSSO, the predominant reaction upon crosslinker cleavage was loss of water from the sulfoxide to generate a thiol, but in some cases a significant amount of the sulfoxide remains. This increases the complexity of the fragmentation and causes some degree of signal splitting, lowering the yield of the desired product. We also observed some cleavage products of the peptide backbone. In contrast, DC4 generated very few fragments beyond the expected crosslinker cleavage. Indeed, the cleavage of the crosslinker was the predominant reaction even in MS2 (Figure S1).
A fast switching of the source voltage showed a strong inverse correlation between the disappearance of the crosslinked peptide signal and the appearance of the liberated peptides(Figure 4A). To explore this further, the intensities of the liberated peptides were determined and referenced to the intensity of the precursor for all crosslinked peptides we detected from BSA (Figure 4B). For DSSO, the combined fragment intensity was usually less than the intensity of the crosslinked peptides. However, the more labile DC4 generated peptide intensities that were much higher than the crosslinked peptide in all its forms. This phenomenon was not due to signal coalescence. That is, multiple charge states of the crosslinked peptide will fragment simultaneously and collapse down to the liberated peptides, which typically have only one major charge state, but at best this would only generate equivalent combined intensities. There are three possible reasons for this intriguing discrepancy. Other charge states outside of our m/z scan window may contribute, but this seems unlikely to contribute a large fraction of new ion density. Alternatively, the gas dynamics and electric field in the source may not be as well tuned for larger species like the crosslinked peptides, but such biases have a m/z dependency, rather than a mass dependency. More likely the high in-source accelerating voltage leads to transmission losses for the lower m/z range due to enhanced scattering27, allowing for a comparatively richer population of peptide ions for a given trap fill-time. We note that a few DC4-linked peptides generated peptide intensities more than 15-fold higher than the crosslinked peptide precursor. For most of these, the crosslinked peptide intensity was suppressed because of crosslinker cleavage even at the low source voltage setting.
Figure 4. Impact of toggling the accelerating voltage during chromatographic separation of crosslinked BSA peptides.
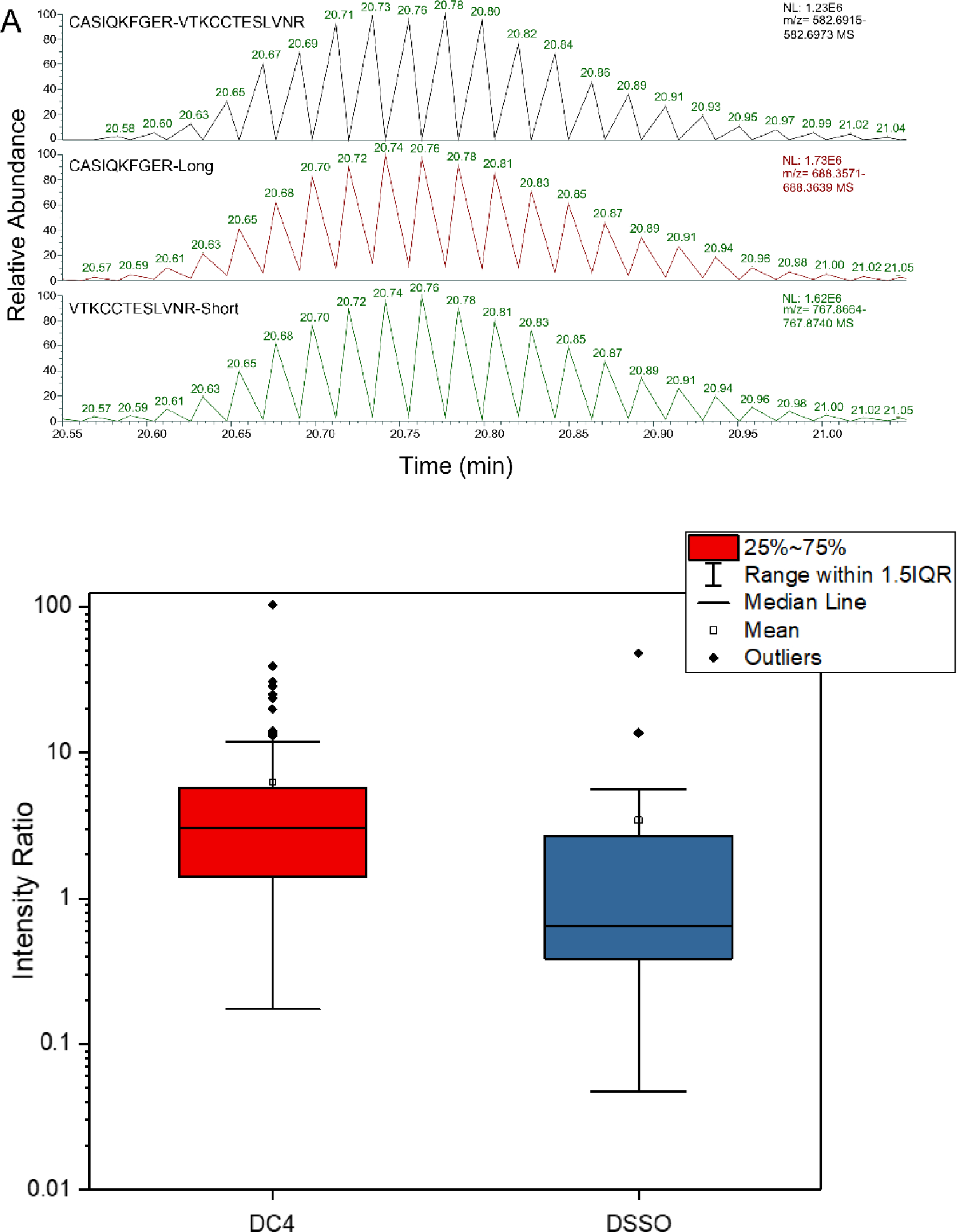
(A) Rapid switching between low and high voltages for DC4 over the chromatographic feature for the indicated crosslinked BSA peptides. (B) Intensity ratios of product ions produced by the voltage toggle to precursor ions over a chromatographic feature, for all BSA crosslinks detected by the pseudo-MS3 method.
We next determined if the generation of peptides from in-source fragmentation would conflict with other peptide signals in the analysis. XICs were generated using MS1 data from both the low and the high accelerating voltage settings. New signals for peptides were observed that correlated with the crosslinked peptide feature, but we also generated signals from fragmented mono-linked products of the crosslinking reaction. The latter can be more intense than the crosslinked products (e.g., Figure S3), at least when no attempt at crosslinked peptide enrichment is made. Because these are indistinguishable from the fragments of the crosslinked peptide, a chromatographic separation is absolutely required to resolve one from the other. A sampling of crosslinks shows that this can be readily achieved in practice, although occasionally the elution times can be close (Figure 5). Taken together, fast switching between low and high voltage acceleration can generate strong signals for the liberated peptides (at least for labile crosslinkers like DC4) that can be sufficiently well-resolved from potential interferents. These liberated peptides can be sampled by MS2 for standard proteomics-grade peptide identification.
Figure 5. Chromatographic separation of monolinks from crosslinks.
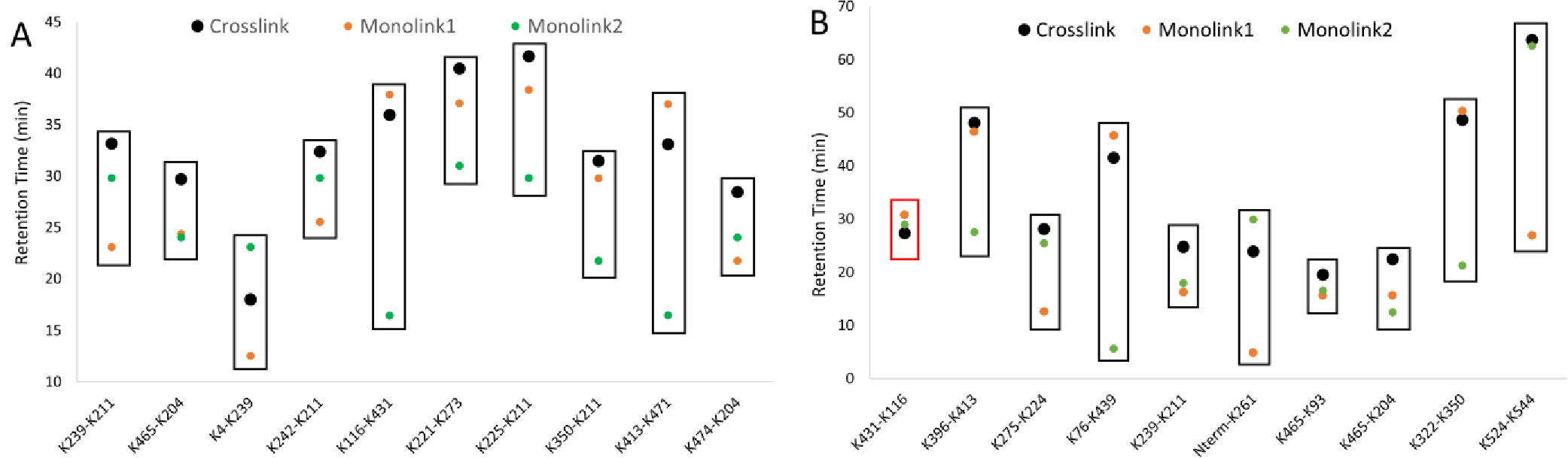
(A) DSSO reaction products and (B) DC4 reaction products, for a random selection of 10 different crosslinked peptides. Data denotes peak apices, and sufficiently resolved features are boxed in black. Crosslinks were considered resolved from monolinks if full baseline separation was achieved.
Automated detection of crosslinks from in-source fragmented data
To evaluate the pseudo-MS3 concept for more complex datasets, we developed automated crosslink analysis software that uses the identified liberated peptides and their chromatographic alignment with the crosslinked peptide precursor, as in a DIA experiment. We generated a plugin for the Mass Spec Studio30,31 that only requires raw data and a FASTA sequence database as inputs (Figure 6).
Figure 6. Flow chart of the data analysis pipeline.
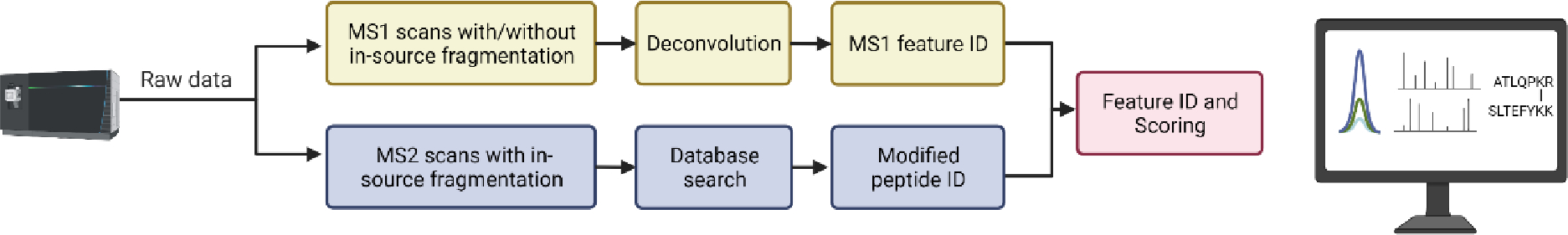
Chromatographic features are identified in the low and high in-source acceleration MS1 data sets. In parallel, peptides are identified from MS2 scans and then paired with their corresponding features. Features are aligned with putative crosslink precursors and comprehensively scored.
In this plugin, peptides are identified from ion trap MS2 scans using the MS-GF+ search utility32. During acquisition, these scans can be collected for all peptides or restricted with real-time mass filters to peptides with the requisite mass difference between long and short version of the fragmented crosslinker (Figure 2). Peptide spectrum matches (PSMs) that contain crosslink-derived modifications can then be paired to match the precursor ion(s) of the crosslinked peptide, but chromatographic alignment of the corresponding MS1 features is required. To achieve this goal, XICs are generated, and the aligned feature sets are identified. Scoring evaluates the quality of the PSMs and the alignment, and additional filters are applied to reject hits that have low quality isotopic envelopes in either the high or low accelerating voltage scans. The result is a strongly discriminating score. Indeed, a conventional target-decoy search does not return enough decoy hits for an accurate estimation of error (not shown). False negatives are more acceptable than false positives in establishing a proof-of-concept, thus a restrictive scoring strategy is appropriate in this instance. The hits can be manually evaluated with the support of a spectrum viewer (Figure S4). Searches are fast compared to conventional crosslinker database searches, as the list of peptides to be paired is modest, especially when using the real-time mass filters.
Crosslinking of BSA
Using the new pseudo-MS3 plugin, we analysed the full set of data collected from the BSA digests, for both the DSSO and DC4 crosslinking experiments, and compared the results to the established ground-truth data collected using conventional approaches (Figure 7).
Figure 7. Comparison of crosslinking methods.
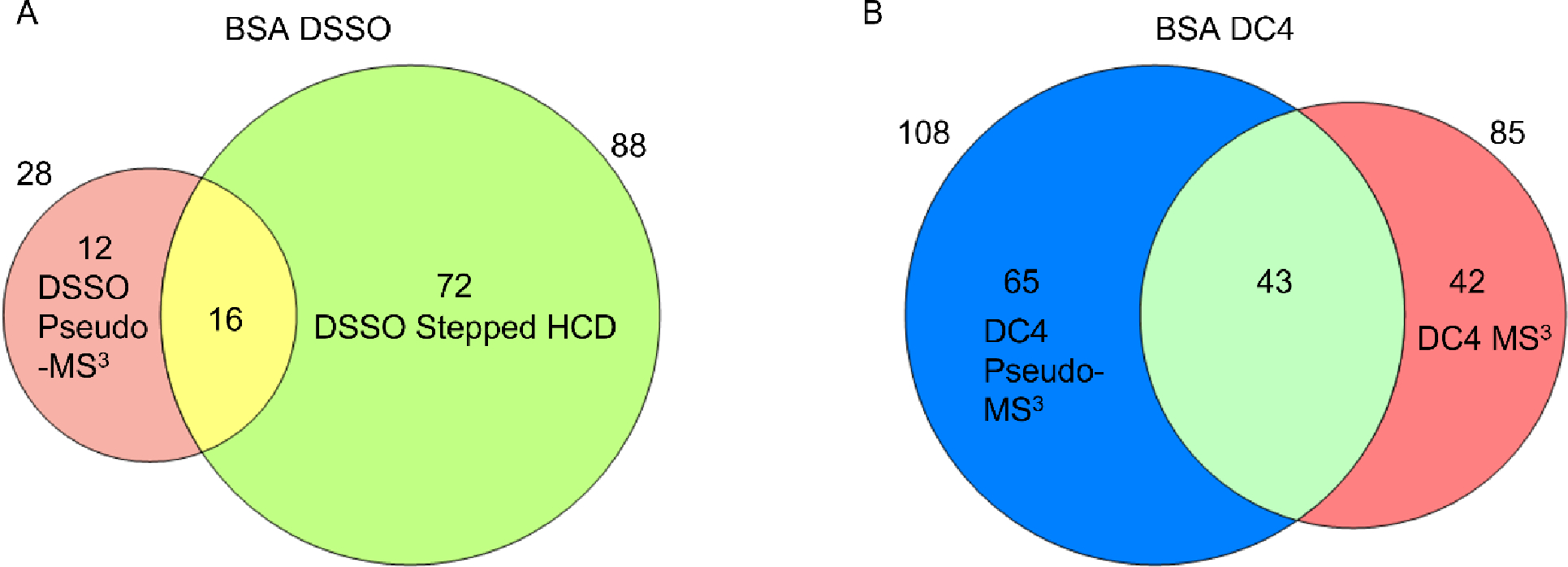
(A) DSSO crosslinking of BSA, comparing stepped HCD and MS2 with the pseudo-MS3 approach, (B) DC4 crosslinking of BSA, comparing MS3 with the pseudo-MS3 approach.
We detected only 28 crosslinks for DSSO, significantly lower than the 88 identified by fragmenting the crosslinked peptides in the collision cell. This is not surprising given the low fragmentation efficiency of DSSO under our chosen in-source conditions. However, 12 of the 28 were uniquely determined by the pseudo-MS3 method. The results generated for the labile DC4 are more compelling. Approximately 27% more crosslinks were identified over the MS3-based approach, and 65 crosslinks were unique to the in-source approach. The number of unique hits is surprising. The ground-truth dataset reflects a saturation of detectable crosslinks, at least using conventional MS3 methods. More than 38,000 MS3 events were triggered over two runs and a highly redundant hit list was obtained. A large unused capacity for crosslink identifications was obviously available. A community-led assessment of yield also indicates that 73 to 88 crosslinks can be reproducibly detected for this protein29.
The reasons for the unique hits are not immediately obvious. The fragmentation mode for identifying the liberated peptides (in-source or in-cell) was otherwise identical (CID). The in-source method samples the peptide size distribution differently, with a slight preference for longer peptides (Figure S5). The peptides liberated in the source may have better transmission compared to the precursor crosslinked peptide, and perhaps also different charge states. It is also possible that lower charge-state liberated peptides are easier to identify and select “on the fly” by the instrument than higher charge-state crosslinked peptides. Whatever the reason, false positive hits from the pseudo-MS3 method do not inflate these numbers. The scoring approach is highly conservative. To confirm this, we conducted a crosslinking analysis of E.coli lysate using DSS, a noncleavable reagent. A standard MS2 method detected 230 crosslinks, whereas the pseudo-MS3 method detected only 7, with most of them discarded after our manual validation routine.
The unique crosslinks found by the pseudo-MS3 method sample BSA structure with a distance distribution that is similar to the set determined by the MS3-based approach. In the DC4 MS3 data, 10% of crosslinks were overlength. In the in-source approach, 18% of crosslinks were overlength. The slight increase is explained by some ambiguity in the detection of a specific crosslinked site. That is, in the Orbitrap Velos, the noisier ion trap scans increase the uncertainty in the specific location of the linkage point.
Crosslinking of E. coli lysate
A more complex sample allowed us to test the effect of higher spectral complexity on crosslink peptide assignment. Again, to establish a reference set, lysates of E. coli were prepared and crosslinked with both DSSO and DC4. Digests were analyzed on an Orbitrap Eclipse using a 4-hour gradient, with no additional enrichment step. DSSO samples were processed using stepped HCD and DC4 was acquired with MS3, as with BSA. We identified 451 unique peptide pairs for DSSO, and 467 for DC4. Many of these crosslinks were found within and between ribosomal proteins, which are highly abundant in the lysate.
We then analysed the same digests with a matched gradient and the pseudo-MS3 approach on the same instrument. Although the source geometry is different than the Velos, testing with BSA showed that a voltage toggle of +45V was also optimal for in-source fragmentation (data not shown). We found only 89 crosslinks in the DSSO sample but 366 in the DC4 sample, again demonstrating the greater effectiveness of a more labile crosslinker for this application (Figure 8). The comparative advantage of the pseudo-MS3 method that we observed for BSA is somewhat eroded, probably due to greater complexity in the MS1 spectra during in-source fragmentation, but the coverage is comparable between the MS3 and pseudo-MS3 methods. We also note that 227 crosslinked peptides were not found with the conventional MS3 method, even though their MS features were of sufficient quality for sampling.
Figure 8.
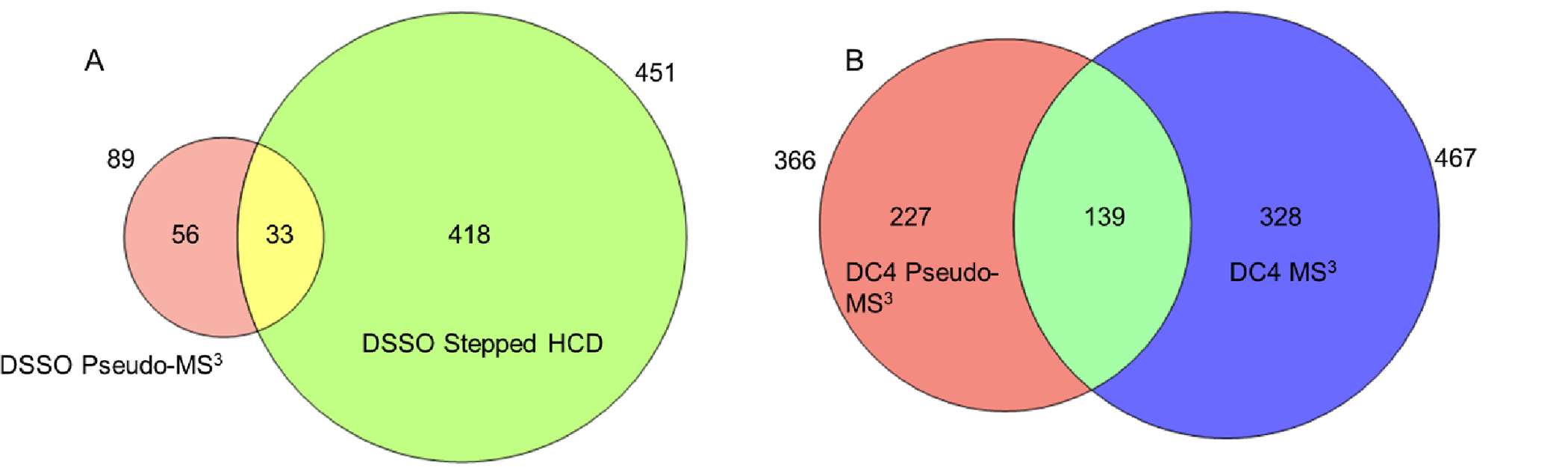
(A) Comparison between the pseudo-MS3 and the stepped HCD analysis methods for DSSO-crosslinked E. coli lysates.(B) Comparison between the pseudo-MS3 and the MS3 analysis methods for DC4-crosslinked E. coli lysates.
Taken together, the pseudo-MS3 approach appears able to tolerate a significant background signal and can detect crosslinks missed by standard methods. The complementarity of the data is encouraging and suggests that improved algorithms could return additional sensitivity. For example, our analysis does not include the detection of the offset “sawtooth” pattern (Figure 5), which could also help identify pairings between liberated peptides and their crosslinked precursor. The manual inspection of MS1 features can also be replaced with an automated strategy that is less subjective, relaxing our current restriction on highest quality MS1 isotope profiles.
CONCLUSIONS
Gas-phase cleavable crosslinkers have improved performance in XL-MS applications, particularly for cell-based reactions and complex protein states in general. However, MS3 methods are not available on every instrument platform, and duty cycle remains an issue even for systems that are MS3-capable. The alternative MS2-based data acquisition method is effective, but database searching remains very time-intensive. A pseudo-MS3 method, first introduced by the Bruce lab, provides an alternative that is strongly complementary and shows promise in boosting coverage of crosslinked peptides when used together with conventional methods. Spectral acquisitions are of similar quality and in many cases better. Improvements in data analysis should lead to additional gains in sensitivity. Using CID in a linear ion trap to sequence the liberated peptides is an advantage of the method, as it is both fast and sensitive. It is clear, however, that conventional gas phase cleavable reagents such as DSSO are not effective and more labile reagents are needed. DC4 may not be the ideal reagent for in situ applications as cell permeability would be low, but other functional groups are possible. Overall, a pseudo-MS3 method is a useful XL-MS routine for collecting structural data on complex systems and worth developing further.
Supplementary Material
Figure S1. Typical HCD MS2 spectrum of a DC4 crosslink.
Figure S2. Typical MS1 spectra of DSSO and DC4 crosslinks, in low and high in-source voltage conditions.
Figure S3. Extracted ion chromatogram of the fragment ion for crosslinker-labeled YICDNQDTISSKLK.
Figure S4. Example output and validation view of the CRIMP In-source plug-in for Mass Spec Studio.
Figure S5. Comparison of shared and unique crosslink peptides for BSA
Table S1: Unique Peptide pair results from all analyses.
ACKNOWLEDGMENTS
This work was supported by CANARIE (project RS-326 to DCS) and NSERC (RGPIN-2017-04879 to DCS).
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting information
The supporting information is available free of charge on the ACS Publications website.
REFERENCES
- (1).Piersimoni L; Kastritis PL; Arlt C; Sinz A Cross-Linking Mass Spectrometry for Investigating Protein Conformations and Protein-Protein Interactions—A Method for All Seasons. Chem. Rev. 2022, 122 (8), 7500–7531. 10.1021/ACS.CHEMREV.1C00786. [DOI] [PubMed] [Google Scholar]
- (2).Slavin M; Zamel J; Zohar K; Eliyahu T; Braitbard M; Brielle E; Baraz L; Stolovich-Rain M; Friedman A; Wolf DG; Rouvinski A; Linial M; Schneidman-Duhovny D; Kalisman N Targeted in Situ Cross-Linking Mass Spectrometry and Integrative Modeling Reveal the Architectures of Three Proteins from SARS-CoV-2. Proc. Natl. Acad. Sci. U. S. A. 2021, 118 (34), e2103554118. 10.1073/PNAS.2103554118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wang Y; Hu Y; Höti N; Huang L; Zhang H Characterization of In Vivo Protein Complexes via Chemical Cross-Linking and Mass Spectrometry. Anal. Chem. 2022, 94 (3), 1537–1542. 10.1021/ACS.ANALCHEM.1C02410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Jumper J; Evans R; Pritzel A; Green T; Figurnov M; Ronneberger O; Tunyasuvunakool K; Bates R; Žídek A; Potapenko A; Bridgland A; Meyer C; Kohl SAA; Ballard AJ; Cowie A; Romera-Paredes B; Nikolov S; Jain R; Adler J; Back T; Petersen S; Reiman D; Clancy E; Zielinski M; Steinegger M; Pacholska M; Berghammer T; Bodenstein S; Silver D; Vinyals O; Senior AW; Kavukcuoglu K; Kohli P; Hassabis D Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596 (7873), 583–589. 10.1038/S41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Shi Y; Fernandez-Martinez J; Tjioe E; Pellarin R; Kim SJ; Williams R; Schneidman-Duhovny D; Sali A; Rout MP; Chait BT Structural Characterization by Cross-Linking Reveals the Detailed Architecture of a Coatomer-Related Heptameric Module from the Nuclear Pore Complex. Mol. Cell. Proteomics 2014, 13 (11), 2927–2943. 10.1074/mcp.M114.041673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Wheat A; Yu C; Wang X; Burke AM; Chemmama IE; Kaake RM; Baker P; Rychnovsky SD; Yang J; Huang L Protein Interaction Landscapes Revealed by Advanced in Vivo Cross-Linking-Mass Spectrometry. Proc. Natl. Acad. Sci. U. S. A. 2021, 118 (32), e2023360118. 10.1073/PNAS.2023360118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Yu C; Huang L Cross-Linking Mass Spectrometry: An Emerging Technology for Interactomics and Structural Biology. Anal. Chem. 2018, 90 (1), 144–165. 10.1021/ACS.ANALCHEM.7B04431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Iacobucci C; Götze M; Ihling CH; Piotrowski C; Arlt C; Schäfer M; Hage C; Schmidt R; Sinz A A Cross-Linking/Mass Spectrometry Workflow Based on MS-Cleavable Cross-Linkers and the MeroX Software for Studying Protein Structures and Protein-Protein Interactions. Nat. Protoc. 2018, 13 (12), 2864–2889. 10.1038/S41596-018-0068-8. [DOI] [PubMed] [Google Scholar]
- (9).Netz E; Dijkstra TMH; Sachsenberg T; Zimmermann L; Walzer M; Monecke T; Ficner R; Dybkov O; Urlaub H; Kohlbacher O OpenPepXL: An Open-Source Tool for Sensitive Identification of Cross-Linked Peptides in XL-MS. Mol. Cell. Proteomics 2020, 19 (12), 2157. 10.1074/MCP.TIR120.002186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Giese SH; Fischer L; Rappsilber J A Study into the Collision-Induced Dissociation (CID) Behavior of Cross-Linked Peptides. Mol. Cell. Proteomics 2016, 15 (3), 1094. 10.1074/MCP.M115.049296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Tang X; Bruce JE A New Cross-Linking Strategy: Protein Interaction Reporter (PIR) Technology for Protein-Protein Interaction Studies. Mol. Biosyst. 2010, 6 (6), 939–947. 10.1039/B920876C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Petrotchenko EV; Serpa JJ; Borchers CH An Isotopically Coded CID-Cleavable Biotinylated Cross-Linker for Structural Proteomics. Mol. Cell. Proteomics 2011, 10 (2), S1–S8. 10.1074/mcp.M110.001420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Clifford-Nunn B; Showalter HDH; Andrews PC Quaternary Diamines as Mass Spectrometry Cleavable Crosslinkers for Protein Interactions. J. Am. Soc. Mass Spectrom. 2012, 23 (2), 201–212. 10.1007/s13361-011-0288-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Liu F; Lössl P; Scheltema R; Viner R; Heck AJR Optimized Fragmentation Schemes and Data Analysis Strategies for Proteome-Wide Cross-Link Identification. Nat. Commun. 2017, 8, 15473. 10.1038/NCOMMS15473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Müller MQ; Dreiocker F; Ihling CH; Schäfer M; Sinz A Cleavable Cross-Linker for Protein Structure Analysis: Reliable Identification of Cross-Linking Products by Tandem MS. Anal. Chem. 2010, 82 (16), 6958–6968. 10.1021/ac101241t. [DOI] [PubMed] [Google Scholar]
- (16).Trnka MJ; Baker PR; Robinson PJJ; Burlingame AL; Chalkley RJ Matching Cross-Linked Peptide Spectra: Only as Good as the Worse Identification. Mol. Cell. Proteomics 2014, 13 (2), 420–434. 10.1074/MCP.M113.034009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Tang X; Munske GR; Siems WF; Bruce JE Mass Spectrometry Identifiable Cross-Linking Strategy for Studying Protein-Protein Interactions. Anal. Chem. 2005, 77 (1), 311–318. 10.1021/AC0488762. [DOI] [PubMed] [Google Scholar]
- (18).Kolbowski L; Lenz S; Fischer L; Sinn LR; O’Reilly FJ; Rappsilber J Improved Peptide Backbone Fragmentation Is the Primary Advantage of MS-Cleavable Crosslinkers. Anal. Chem. 2022, 94 (22), 7779–7786. 10.1021/acs.analchem.1c05266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Sleno L; Volmer DA Ion Activation Methods for Tandem Mass Spectrometry. J. Mass Spectrom. 2004, 39 (10), 1091–1112. 10.1002/JMS.703. [DOI] [PubMed] [Google Scholar]
- (20).Xue J; Domingo-Almenara X; Guijas C; Palermo A; Rinschen MM; Isbell J; Benton HP; Siuzdak G Enhanced In-Source Fragmentation Annotation Enables Novel Data Independent Acquisition and Autonomous METLIN Molecular Identification. Anal. Chem. 2020, 92 (8), 6051–6059. 10.1021/ACS.ANALCHEM.0C00409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Xue J; Derks RJE; Hoang L; Giera M; Siuzdak G Proteomics with Enhanced In-Source Fragmentation/Annotation: Applying XCMS-EISA Informatics and Q-MRM High-Sensitivity Quantification. J. Am. Soc. Mass Spectrom. 2021, 32 (11), 2644–2654. 10.1021/JASMS.1C00188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Hoopmann MR; Weisbrod CR; Bruce JE Improved Strategies for Rapid Identification of Chemically Cross-Linked Peptides Using Protein Interaction Reporter Technology. J. Proteome Res. 2010, 9 (12), 6323–6333. 10.1021/PR100572U. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ludwig C; Gillet L; Rosenberger G; Amon S; Collins BC; Aebersold R Data-Independent Acquisition-Based SWATH-MS for Quantitative Proteomics: A Tutorial. Mol. Syst. Biol. 2018, 14 (8), e8126. 10.15252/MSB.20178126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Winter SV; Meier F; Wichmann C; Cox J; Mann M; Meissner F EASI-Tag Enables Accurate Multiplexed and Interference-Free MS2-Based Proteome Quantification. Nat. Methods 2018, 15 (7), 527–530. 10.1038/s41592-018-0037-8. [DOI] [PubMed] [Google Scholar]
- (25).Szabó D; Schlosser G; Vékey K; Drahos L; Révész Á Collision Energies on QTof and Orbitrap Instruments: How to Make Proteomics Measurements Comparable? J. Mass Spectrom. 2021, 56 (1), e4693. 10.1002/jms.4693. [DOI] [PubMed] [Google Scholar]
- (26).Prasad S; Wouters ER; Dunyach JJ Advancement of Atmospheric-Vacuum Interfaces for Mass Spectrometers with a Focus on Increasing Gas Throughput for Improving Sensitivity. Anal. Chem. 2015, 87 (16), 8234–8241. 10.1021/ACS.ANALCHEM.5B01140. [DOI] [PubMed] [Google Scholar]
- (27).Bruins AP Mass Spectrometry with Ion Sources Operating at Atmospheric Pressure. Mass Spectrom. Rev. 1991, 10 (1), 53–77. 10.1002/MAS.1280100104. [DOI] [Google Scholar]
- (28).Javaheri H; Schneider BB Ion Guide for Improved Atmosphere to Mass Spectrometer Vacuum Ion Transfer. J. Am. Soc. Mass Spectrom. 2021, 32 (8), 1945–1951. 10.1021/JASMS.0C00394. [DOI] [PubMed] [Google Scholar]
- (29).Iacobucci C; Piotrowski C; Aebersold R; Amaral BC; Andrews P; Bernfur K; Borchers C; Brodie NI; Bruce JE; Cao Y; Chaignepain S; Chavez JD; Claverol S; Cox J; Davis T; Degliesposti G; Dong MQ; Edinger N; Emanuelsson C; Gay M; Götze M; Gomes-Neto F; Gozzo FC; Gutierrez C; Haupt C; Heck AJR; Herzog F; Huang L; Hoopmann MR; Kalisman N; Klykov O; Kukačka Z; Liu F; Maccoss MJ; Mechtler K; Mesika R; Moritz RL; Nagaraj N; Nesati V; Neves-Ferreira AGC; Ninnis R; Novák P; O’Reilly FJ; Pelzing M; Petrotchenko E; Piersimoni L; Plasencia M; Pukala T; Rand KD; Rappsilber J; Reichmann D; Sailer C; Sarnowski CP; Scheltema RA; Schmidt C; Schriemer DC; Shi Y; Skehel JM; Slavin M; Sobott F; Solis-Mezarino V; Stephanowitz H; Stengel F; Stieger CE; Trabjerg E; Trnka M; Vilaseca M; Viner R; Xiang Y; Yilmaz S; Zelter A; Ziemianowicz D; Leitner A; Sinz A First Community-Wide, Comparative Cross-Linking Mass Spectrometry Study. Anal. Chem. 2019, 91 (11), 6953–6961. 10.1021/ACS.ANALCHEM.9B00658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ziemianowicz DS; Sarpe V; Crowder DA; Pells TJ; Raval S; Hepburn M; Rafiei A; Schriemer DC Harmonizing Structural Mass Spectrometry Analyses in the Mass Spec Studio. J. Proteomics 2020, 225. 10.1016/J.JPROT.2020.103844. [DOI] [PubMed] [Google Scholar]
- (31).Rey M; Sarpe V; Burns KM; Buse J; Baker CAH; Van Dijk M; Wordeman L; Bonvin AMJJ; Schriemer DC Mass Spec Studio for Integrative Structural Biology. Structure 2014, 22 (10), 1538. 10.1016/J.STR.2014.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Kim S; Pevzner PA MS-GF+ Makes Progress towards a Universal Database Search Tool for Proteomics. Nat. Commun. 2014. 10.1038/ncomms6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Typical HCD MS2 spectrum of a DC4 crosslink.
Figure S2. Typical MS1 spectra of DSSO and DC4 crosslinks, in low and high in-source voltage conditions.
Figure S3. Extracted ion chromatogram of the fragment ion for crosslinker-labeled YICDNQDTISSKLK.
Figure S4. Example output and validation view of the CRIMP In-source plug-in for Mass Spec Studio.
Figure S5. Comparison of shared and unique crosslink peptides for BSA
Table S1: Unique Peptide pair results from all analyses.