

Abstract
In multiple sclerosis (MS), abnormally activated immune cells responsive to myelin proteins result in widespread damage throughout the central nervous system (CNS) and ultimately irreversible disability. Immunomodulation by delivering dendritic cells (DCs) utilizes a potent and rapid MS disease progression driver therapeutically. Here, we investigated delivering DCs for disease severity attenuation using an experimental autoimmune encephalomyelitis (EAE) preclinical MS model. DCs treated with interleukin-10 (IL-10) (DC10s) were transplanted using in situ gelling poly(ethylene glycol) (PEG)-based hydrogel for target site localization. DC delivery increased hydrogel longevity and altered the injection site recruited, endogenous immune cell profile within 2 days post-injection. Furthermore, hydrogel-mediated DC transplantation efficacy depended on the injection-site. DCs delivered to the neck local to MS-associated CNS-draining cervical lymph nodes attenuated paralysis, compared to untreated controls, while delivery to the flank did not alter paralysis severity. This study demonstrates that local delivery of DC10s modulates immune cell recruitment and attenuates disease progression in a preclinical model of MS.
Keywords: Dendritic cells, transplantation, multiple sclerosis, localized delivery, in situ gelling hydrogel
Introduction
Multiple sclerosis (MS) is an autoimmune disorder that targets myelin, the insulating layer surrounding axons that controls nerve function and survival, resulting in progressive paralysis of the patient that ultimately leads to their demise [1]. Worldwide, 1.4–2.0% of people are expected to be affected by MS [2] with health care expenses exceeding over 70,000 USD per patient per annum [3–5]. Current treatments aim to slow the rate of paralysis using non-targeting immunosuppressive drugs [3, 6, 7] that hinder the entire immune system. However, this strategy increases patient susceptibility to infections and other medical complications [8]. As a result, alternative strategies to control immune responses are needed to halt or slow the progression of the disorder.
Dendritic cells, as central players of immunity, are a promising strategy to attenuate the immune response against myelin. Dendritic cells are well-recognized as initiators and drivers of many autoimmune disorders including MS [9–14], in part due to their ability to present antigen better than other immune cells [15] and to their rapid (hours to days) changes in phenotype in inflammatory settings [16–18]. At initiation, within 3 to 7 days post model induction, DCs migrate to inflammatory lesions in the CNS to acquire the myelin antigens targeted in MS, and then transit to cervical lymph nodes to augment immune responses against these antigens, progressing this disorder [19, 20]. Delivery of pro-inflammatory DCs to experimental autoimmune encephalomyelitis (EAE) mice, the most widely-used animal model for MS, at this time-frame has been shown to exacerbate the severity of the disorder [11]. While their ability to augment immune responses [21, 22] led to their historic use in vaccination against pathogens [23–25] and cancers [26–28], recent groups have shown that when exposed to tolerogenic cues during differentiation and maturation, DCs can instead inhibit immune responses [29–32].
In this study, we delivered DC10s locally to cervical lymph nodes using an in situ gelling, cell-encapsulating PEG hydrogel [33, 34]. DC10s were delivered prophylactically (day 0 after model induction) in order to evaluate their potential to prevent or attenuate future damage and disability. These cells were distinct from immature and mature DCs via surface marker expression and immunosuppression. Their viability and phenotypic marker expression upon hydrogel encapsulation were characterized in vitro and in vivo. Furthermore, their ability to modulate the immune response in EAE mice based on proximity to disease-progressing lymph was evaluated.
Materials and Methods
Derivation of Dendritic Cells
All studies with animals were according to the Emory University and the Georgia Institute of Technology ACUC guidelines. Bone marrow cells from female C57Bl/6 mice (4–6 month old, Jackson Laboratories) were extracted, incubated in a lysis buffer for 5 min at room temperature to remove red blood cells, and cultured (106 cells/well) in 6-well plates for 8 days with basal media supplemented with granulocyte macrophage-colony stimulating factor (GM-CSF) (20 ng/mL, BD 554586) and IL-4 (4 ng/mL, Peprotech AF-214–14) with half the media exchanged every 2 days. Lipopolysaccharide (LPS) (500 ng/mL, Sigma L4180) or IL-10 (10 ng/mL, eBioscience 14–8101-62) was added to the media on day 6 to generate mature DCs or DC10s, respectively. Immature DCs did not receive additional factors. Basal media consisted of Dulbecco’s Modified Eagle Medium (DMEM) (CellGro 10–017-CV) supplemented with fetal bovine serum (FBS) (10%, Gibco 16000–044), penicillin-streptomycin (1%, CellGro, 30–002-CI), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (1%, CellGro 35–060-CI), sodium pyruvate (1%, CellGro 25–000-CI), nonessential amino acids (1%, CellGro 25–025-CI), and beta-mercaptoethanol (0.1%, Gibco 21985–033). Lysis buffer included sodium chloride (155 mM, Sigma 37653), potassium bicarbonate (10 mM, Sigma P9144), and ethylenediaminetetraacetic acid (EDTA) (0.1 mM, LifeTech 11267–028) in distilled water. On the last day of derivation, supernatants were collected and an ELISA (R&D Systems, M1000B) was performed according to the manufacturer’s instructions to quantify the IL-10 secreted by these DCs.
Flow Cytometry
Cells (2–5 × 106 cells/mL) were blocked for Fc-receptors (1:200, eBioscience 14–0161-86) for 15 min at 4 °C and then incubated with antibodies (Tables 1 and 2) for 30 min at 4 °C (Supplemental Figure 1). For intracellular stains, cells were incubated for 4 hours in media that contained brefeldin-A (1 μg/mL, Sigma B6542) and monensin (2 μM, Sigma M5273) before blocking for Fc-receptors. Additionally, cells were fixed with paraformaldehyde (2% in PBS, Sigma 158127) and permeabilized with Tween-20 (0.5%, Sigma P2287) each for 15 min at room temperature after blocking Fc-receptors. All solutions were in fluorescence-activated cell sorting (FACS) buffer (bovine serum albumin (BSA) (1%, Sigma A4503) and sodium azide (0.01%, Mallinckrodt 195–3-57) in phosphate buffered saline (PBS, Lonza, 17–516F)). Antibodies were diluted 1:100 (fluorescein isothiocyanate, FITC) or 1:200 (others) unless indicated (Tables 1 and 2). Cells in hydrogels were gently digested with collagenase II (2 mg/mL) for 15 min before staining.
Table 1.
Antibodies used for immune cell characterization
| Marker | Manufacturer |
|---|---|
| CD3ε | BD Biosciences Cat# 553064, RRID:AB_394597 BioLegend Cat# 100311, RRID:AB_312676 |
| CD4 | BioLegend Cat# 100512, RRID:AB_312715 BD Biosciences Cat# 553051, RRID:AB_398528 |
| CD8a | BioLegend Cat# 100707, RRID:AB_312746 |
| CD19 | BD Biosciences Cat# 550992, RRID:AB_398483 |
| CD49b | BD Biosciences Cat# 554999, RRID:AB_395633 |
| CD49d | BD Biosciences Cat# 553157, RRID:AB_394670 |
| IL-10 | 1:100, BD Biosciences Cat# 554467, 1:100, RRID:AB_395412 1:100, BioLegend Cat# 505008, RRID:AB_315362 |
| Ly6c | BD Biosciences Cat# 560595, RRID:AB_1727554 |
| Ly6g | BD Biosciences Cat# 551460, RRID:AB_394207 |
Table 2.
Antibodies used for dendritic cell characterization
| Marker | Manufacturer |
|---|---|
| CD11b | BD Biosciences Cat# 562287, RRID:AB_11154216 |
| CD11c | 1:100, BD Biosciences Cat# 558079, RRID:AB_647251 1:100, Thermo Fisher Scientific Cat# 45–0114-82, RRID:AB_925727 |
| CD275 | eBioscience 12–5985-83, RRID:AB_466094 |
| CD45 RA | 1:800, BD Biosciences Cat# 553380, RRID:AB_394822 |
| CD45 RB | BD Biosciences Cat# 562848, RRID:AB_2737835 |
| CD54 | BioLegend Cat# 116120, RRID:AB_10612936 |
| CD86 | Thermo Fisher Scientific Cat# 17–0862-82, RRID:AB_469419 BD Biosciences Cat# 553692, RRID:AB_394994 |
| CD80 | Thermo Fisher Scientific Cat# 11–0801-86, RRID:AB_465135 Biolegend Cat# 104714, RRID:AB_313135 |
| IA B | BD Biosciences Cat# 553551, RRID:AB_394918 1:400, BD Biosciences Cat# 553552, RRID:AB_394919 |
| PirB | R&D, Cat# FAB2754B, RRID:AB_10567003 |
| Qa2 | Thermo Fisher Scientific Cat# 11–5996-85, RRID:AB_465353 |
Lipopolysaccharide (LPS) Stability Assay
Dendritic cells (1 × 106 cells) were cultured in a 12-well plate with basal media (1 mL) with or without LPS (1 μg/mL) for 24 hours and assessed using flow cytometry for changes in the presence of surface markers.
Autologous Immunostimulation Assay
Dendritic cells were cultured for 72 hours with basal media (200 μL) in a U-bottom 96-well plate with autologous splenocytes (5 × 105 cells) at a 1:1 ratio. Prior to co-culture, splenocytes (5 × 106 cells/mL) were treated with ovalbumin (OVA257–264/323–229) (5 μg/mL each, Genscript RP10610/RP10611) antigen for 8 days and then labeled with carboxyfluorescein succinimidyl ester (CFSE) (0.5 μM in PBS, LifeTech C34554) for 15 min at 37 °C. Prior to co-culture, DCs were primed with myelin oligodendrocyte glycoprotein (MOG35–55) (20 μg/mL, fabricated in-house) or OVA (10 μg/mL each), the latter as an antigen-specific control, for 2 hours at 37 °C. Proliferation was evaluated using flow cytometry. Proliferated cells (percentage) were defined as cells with CFSE levels below that of cells (CFSELO) prior to the introduction of proliferation stimulus.
DC Transplantation into EAE mice
Female C57Bl/6 mice (2–3 month old, Jackson Laboratories) were injected subcutaneously on days 0 and 7 with an emulsion containing MOG35–55 (400 μg) in complete Freund’s adjuvant (CFA) supplemented with mycobacterium tuberculosis (750 μg). Pertussis toxin (PT) (250 ng) was injected on days 0 and 2. Animals were monitored daily after induction and blinded scored for clinical disease signs as follows: 0=asymptomatic; 1=atonic tail; 2=partial hind limb paralysis; 3=paraplegia; 4=quadriplegia; and 5=moribund/death due to EAE. At the time of induction (day 0), hydrogel precursor solution (50 μL on each side for a total of 100 μL per mouse) was injected subcutaneously using an insulin (28G) syringe proximal to cervical lymph node (neck) or the flank regions with or without DCs (1–2 × 106 cells). Hydrogel precursor solution included maleimide-functionalized PEG macromer (5% w/v, PEG-4MAL, Laysan Bio,) functionalized with RGD adhesive peptide (2.0 mM, GRGDSPC, Aapptec) to support cell infiltration and a crosslinking, protease-degradable peptide (GCRDVPMSMRGGDRCG, Aapptec) as described previously [33]. Hydrogels polymerize in situ within 1 min. In all studies, mice remained asymptomatic at 10 days post injection.
In order to monitor the retention of DC10s within the hydrogel, female C57Bl/6 (CD45.2+ recipients) mice (2–3 month old, Jackson Laboratories) were injected with EAE induction reagents, as described above. On day 0, mice received hydrogel containing 1 × 106 primed DCs derived from C57Bl/6 (CD45.1+ donors) mice. After 1, 4, 7, and 10 days post injection, hydrogel constructs were extracted and digested, and recovered cells were analyzed by flow cytometry.
Statistics
Single comparisons of two groups were analyzed with a t-test. Multiple comparisons were analyzed using a one-way ANOVA with a Tukey post-hoc. Significance was defined as p < 0.05.
Results
DCs (CD11c+) were derived from bone marrow after 8 days of culture in media containing GM-CSF, IL-4, and IL-10. Traditional markers CD11b, CD11c, IAB (MHC-II), CD80, CD86, and CD54 (percentage of cells gated for CD11c, p < 0.05) distinguished DCs of all three phenotypes from bone marrow cells. High expression levels of CD86 and CD54 (MFI of cells gated for CD11c, p < 0.05) identified mature DCs from the other phenotypes. However, none of these markers were able to distinguish DC10s from immature cells (Fig. 1). The surface molecules CD45RB, CD275, PirB, and Qa2, markers with known anti-inflammatory roles [35–39], were therefore assessed for their ability to further distinguish the three DC phenotypes. The level of CD45RB expression (MFI, p < 0.05) in bone marrow cells was significantly higher than in DCs of all three phenotypes. CD275 was most prevalent (percentage) in immature DCs compared to DC10s (n.s.) or mature DCs (p < 0.05). However, DC10s had the highest expression level (MFI) of CD275 compared to immature (n.s.) and mature (p < 0.05). PirB was most prevalent (percentage, p < 0.05) in DC10s compared to both immature and mature DCs.
Figure 1.
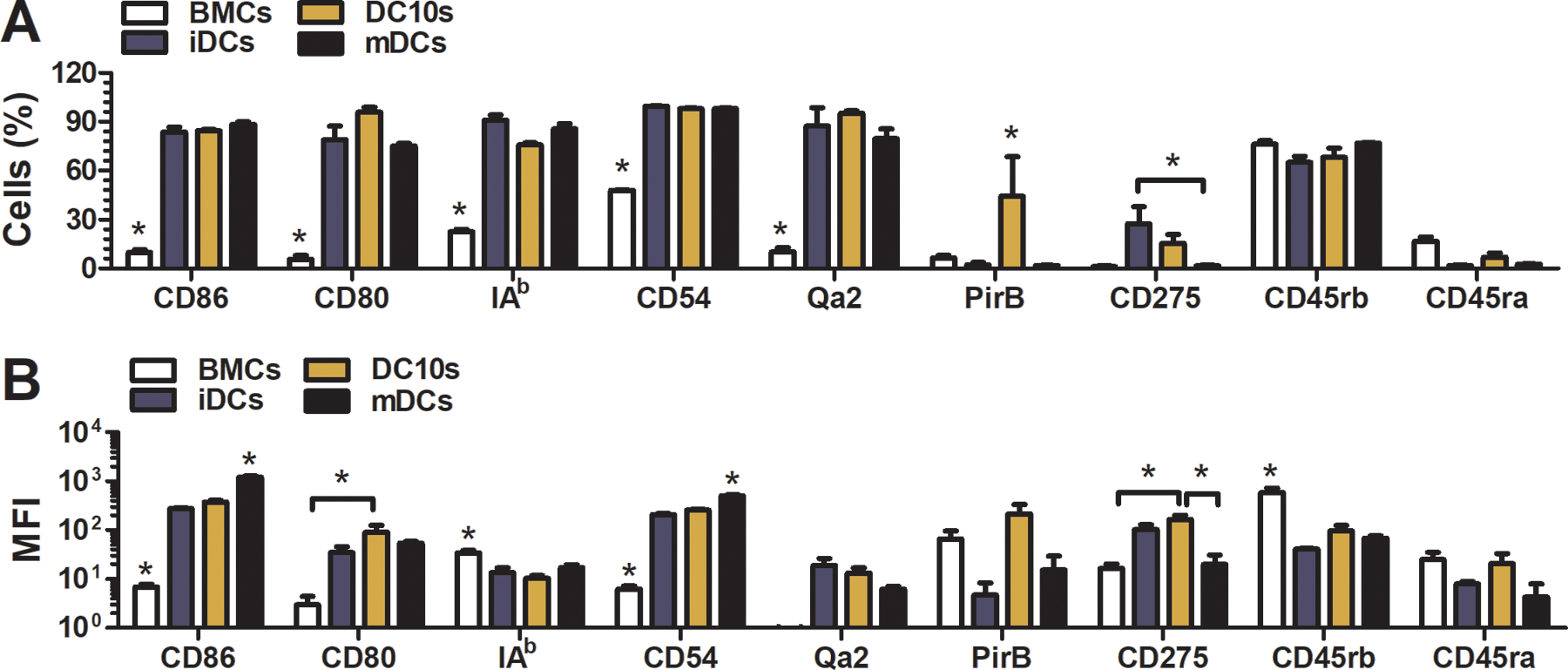
Surface marker profile of DC10s after 8 days of culture. (a) Prevalence (%) and (b) level (MFI) of expression in DC10s compared to bone marrow cells and immature and mature DCs.
Injection-mediated encapsulation within PEG hydrogels did not affect the viability or phenotype of DC10s (Fig. 2). Live/dead staining performed 24 hours after encapsulation revealed DCs encapsulated within PEG hydrogels had similar levels of viability (green) compared to DCs pipetted onto a tissue culture plate. Surface marker analysis revealed that hydrogel encapsulation lowered the expression level of maturation markers (MFI of CD86, CD80, CD54) and enhanced the anti-inflammatory profile (MFI of PirB, CD275) of DCs compared to those pipetted onto tissue culture plates. Furthermore, hydrogel encapsulation maintained the phenotypic stability characteristic of DCs. Hydrogel-encapsulated (+hydrogel, +injection) and non-encapsulated (-hydrogel, -injection) DCs were similarly resistant to LPS-mediated activation (Fig. 2), as indicated by a lack of increase in maturation markers such as CD86 which would be observed for immature DCs treated with LPS.
Figure 2.
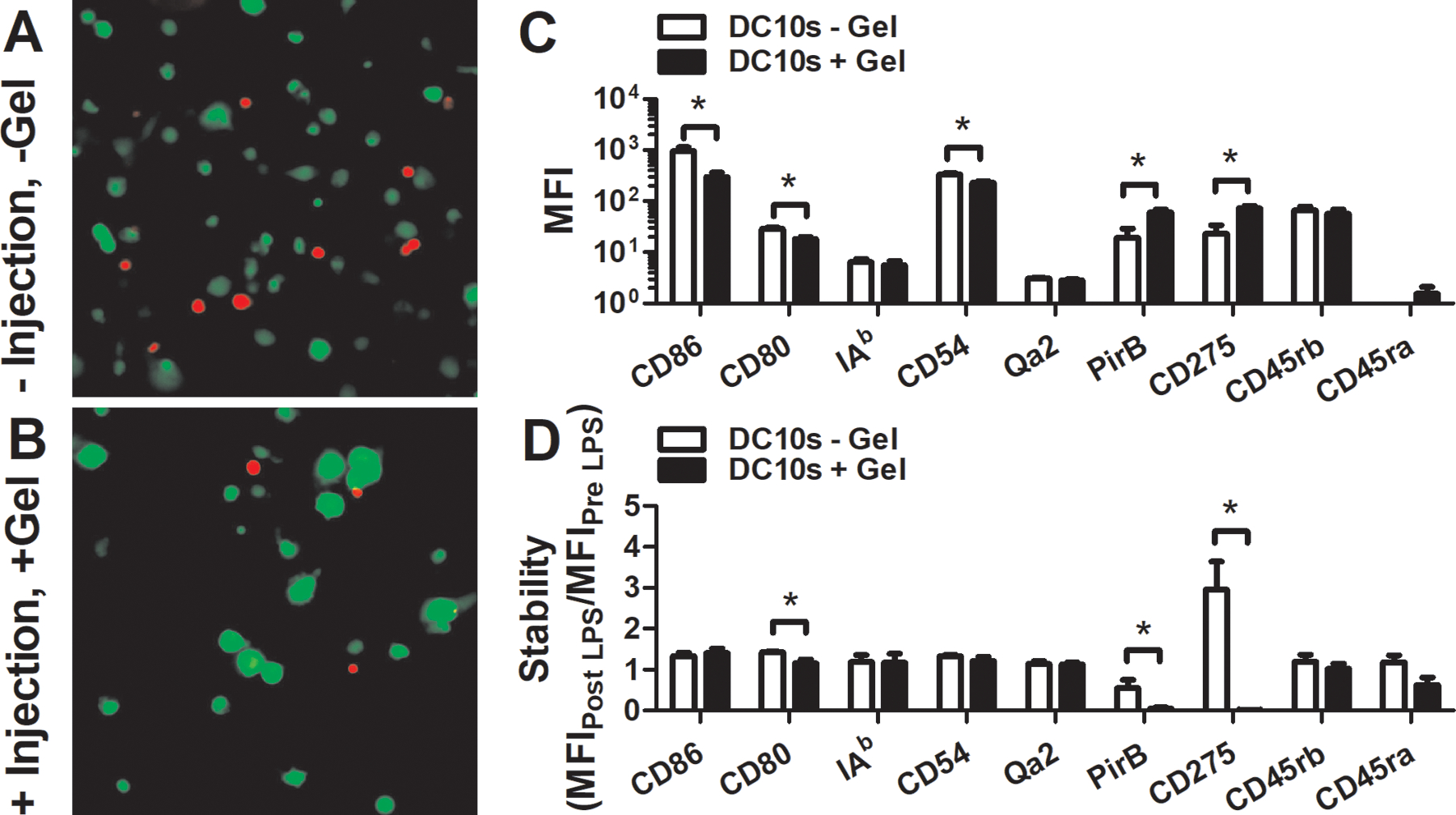
Alterations in viability and phenotype upon encapsulation into and injection with PEG hydrogel. Viability (green) of DCs (a) encapsulated into hydrogel (+ injection + gel) and then injected with a syringe compared to DCs (b) pipetted onto a tissue culture plate (TCP) (- injection – gel) for 24 hours. (c) Expression level (MFI) of DCs encapsulated into and injected with hydrogel gel compared to those cultured on a tissue culture plate for 24 hours. (d) Stability (MFIpost /MFIpre) of DC10s encapsulated into hydrogel and then injected with a syringe compared to those cultured on a TCP for 24 hours in basal media with or without 1 μg/mL LPS.
Subcutaneous injection localized the hydrogel to the neck proximal to where the cervical lymph nodes are located (Fig. 3a). The phenotype of DC10s one day post transplantation via hydrogel was less activated (lower expression levels of CD86 and CD54) than before transplantation, consistent with in vitro observations (Fig. 3c). Two days post injection, hydrogels that did not receive DC10s were not retrievable, unlike those that were co-injected with DC10s (Fig. 3b), which may be attributable to suppression of injection-mediated inflammation by these cells.
Figure 3.
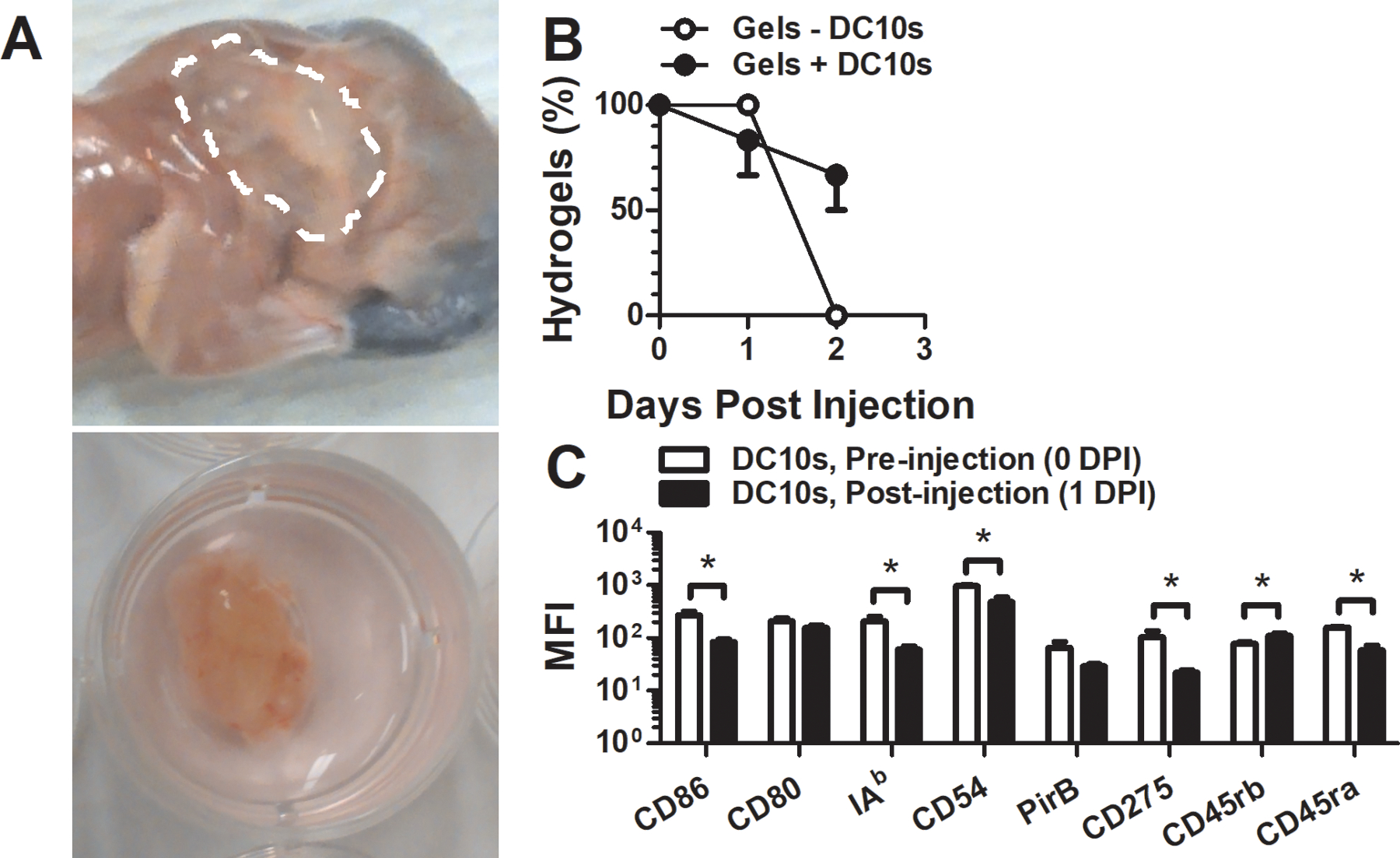
Hydrogel-mediated delivery of DC10s. (a) Visualization of hydrogel at the neck proximal to where 2–3 cervical lymph nodes are located. (b) Retrieval (% of grossly visible) of hydrogels after subcutaneous injection of two 50 μL gels. (c) Stability of DC surface marker expression level (MFI) 1 day after transplantation.
DC10s were labeled with CFSE (0.5 μM in PBS) for 15 min at 37 °C prior to transplantation. At 1 and 2 days post-transplantation, the hydrogels and associated tissues were harvested and digested to evaluate endogenous (CFSE-negative) immune cells (Fig. 4). Modulation of endogenous immune cell behavior and phenotype were observed when DC10s were transplanted. Delivery of DC10s altered endogenous immune cell presence at the implantation site at 1–2 days post injection (Fig. 4a,b). In particular, T-cells (CD3ε), B-cells (CD19), and DCs (CD11c) were found at lower levels in the presence of hydrogel-delivered DCs, while macrophages (CD11b) increased in presence proximal to the hydrogel with DCs, compared to controls. Differences in the phenotypes of endogenous DCs at the hydrogel implantation site upon delivery of DC10s were also observed as early as 1 day post transplantation with novel surface markers, but not with traditional ones (Fig. 4c,d), and were consistent with observations upon hydrogel encapsulation and injection in vitro (Fig. 2c).
Figure 4.
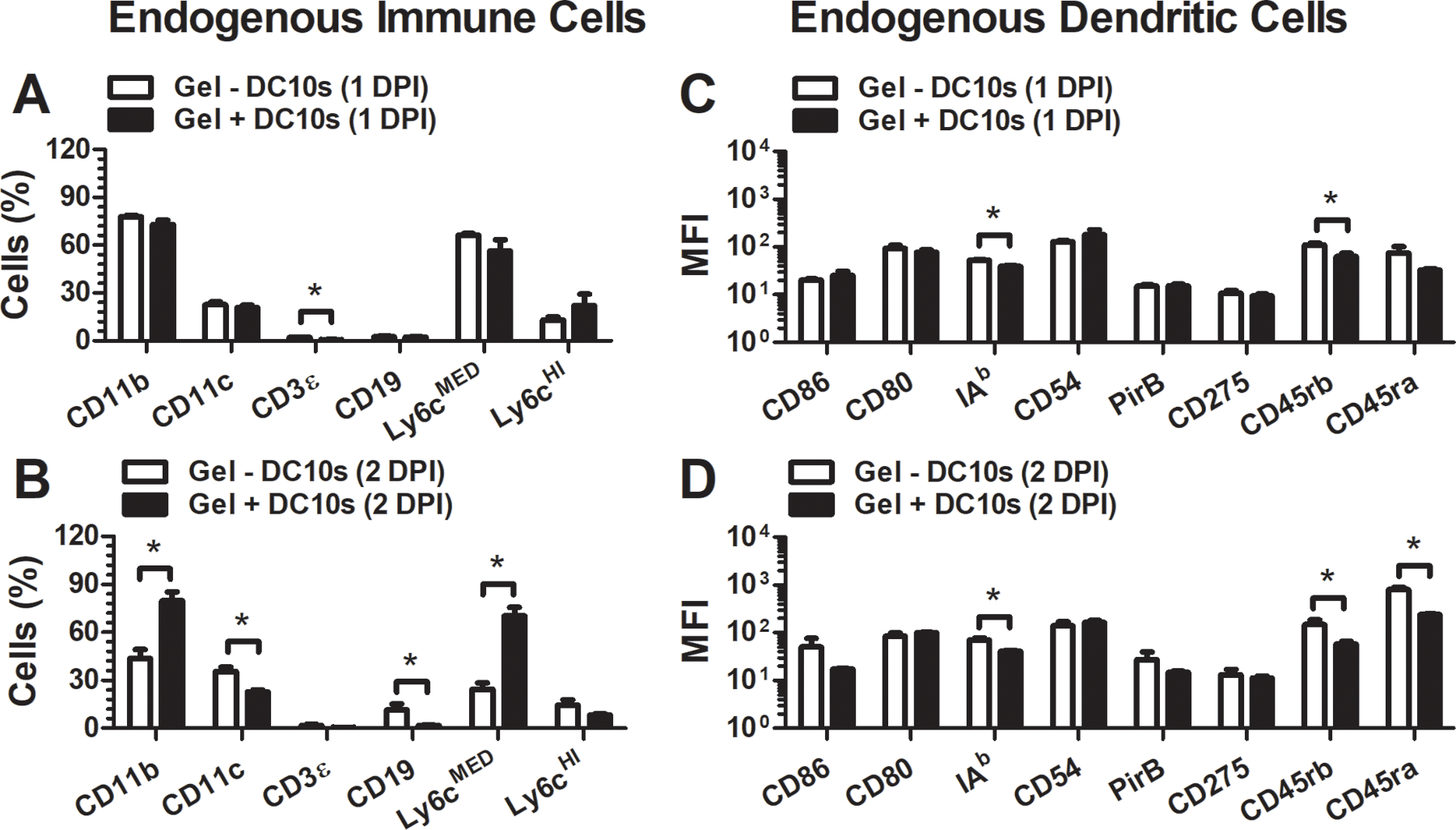
Endogenous immune cell phenotype at the hydrogel injection site after DC10 delivery. Endogenous immune cell presence (a) 1 and (b) 2 days after injection. Endogenous DC surface marker expression level (MFI) (c) 1 and (d) 2 days after injection.
These changes in immune response were not specific to antigen, as indicated by an in vitro immunosuppression assay in which in vitro derived DCs primed with MOG35–55 or OVA257–264/323–229 were co-cultured with autologous splenocytes primed with MOG35–55 (Fig. 5). Both recovered T-cells (CD4) and B-cells (CD19) were less prolific, defined as cells with decreased CSFE levels, when exposed to hydrogel-delivered DCs, regardless of the antigen presented. This observation may partly due to the high prevalence of DCs that produced IL-10, indicated with IL-10-positive staining (Fig. 5e). This observation is consistent with the high levels of IL-10 secretion of these cells compared to the other phenotypes (Supplemental Fig. 2).
Figure 5.
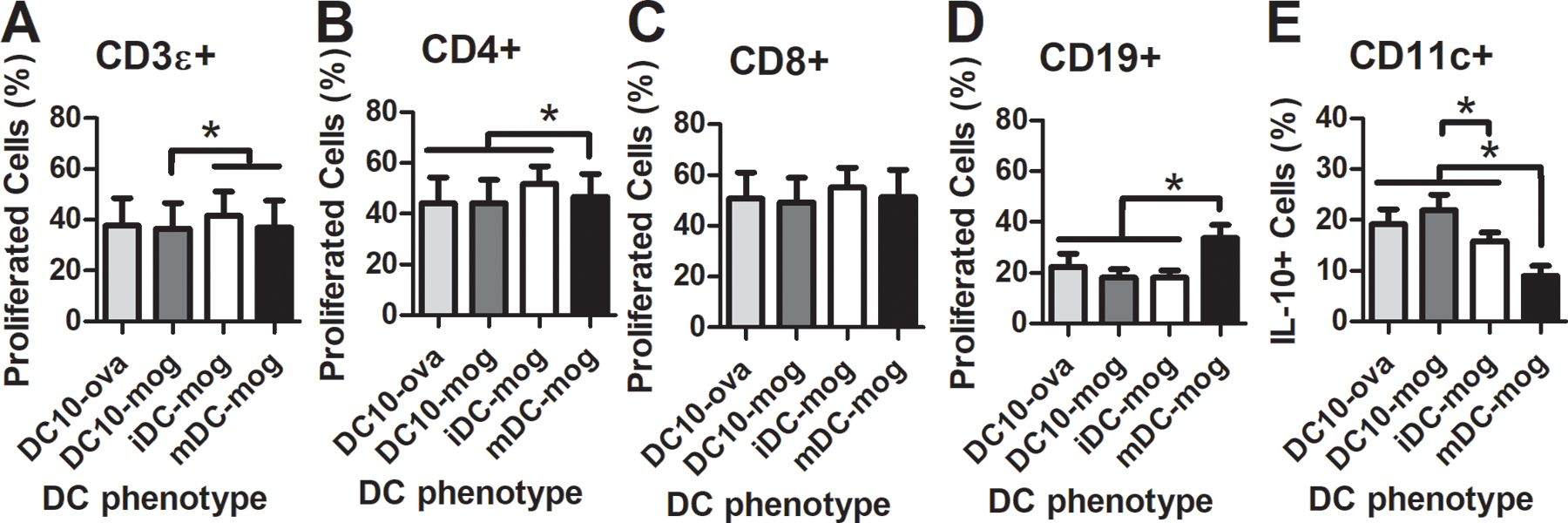
Antigen-dependency of immunosuppression using DC10s. (a-c) T-cell and (d) B-cell proliferation (% cells with decreased CSFE), and (e) IL-10 presence (IL-10+) in dendritic cells when cultured with DC10s compared to immature and mature DC controls.
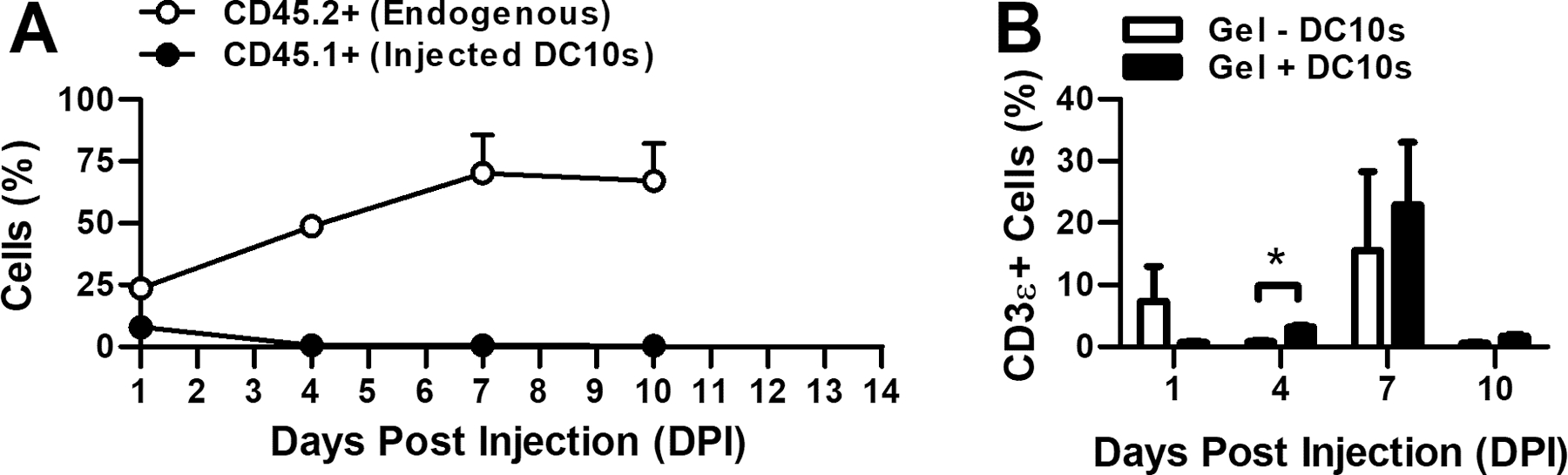
In a separate experiment, we assessed transplanted DC10s persistence and endogenous cell recruitment as a function of time in EAE mice (Fig. 6). Dendritic cells from a congenic (CD45.1+) strain were delivered to the neck of EAE mice (CD45.2+) via subcutaneous injection and hydrogels were explanted 1, 4, 7, or 10 days post injection. Transplanted DC10s in the hydrogels were observed for up to 4 days, while endogenous cell recruitment continued over the course of 10 days. Interestingly, the percentage of T-cells (CD3ε) was initially lower in hydrogels containing DC10s, similar to the study with naïve mice (Fig. 4); however, this phenomenon was transient, and at later time-points T-cell presence was equivalent to the control group.
Figure 6.
Hydrogel-mediated immune cell recruitment. Presence (%) (a) of transplanted (CD45.1+) and recruited (CD45.2+) immune cells and (b) of recruited T-cells in hydrogels after subcutaneous injection of two 50 μL gels on each side of the neck.
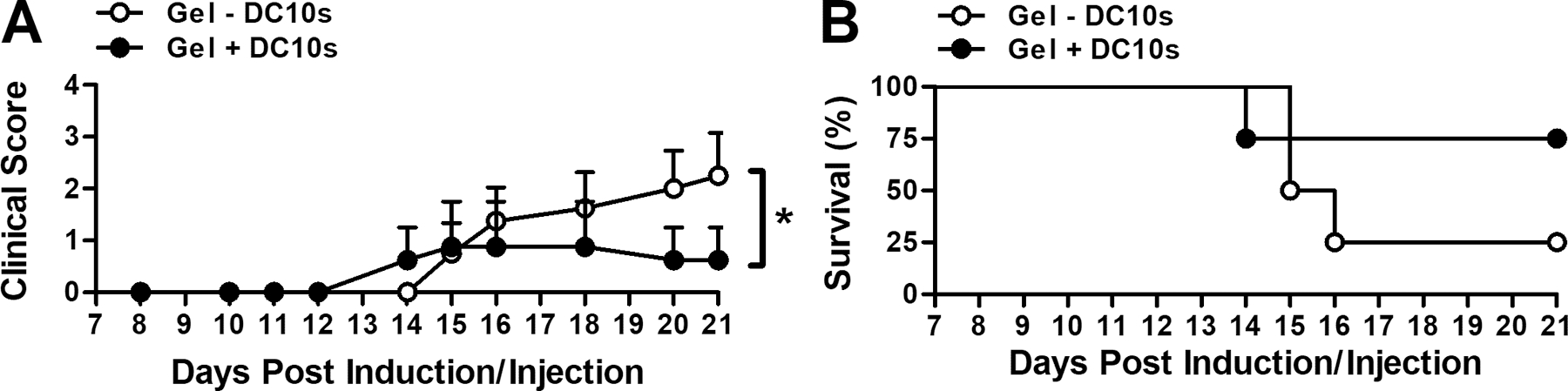
Subcutaneous injection of DC10s using in situ gelling hydrogel ameliorated EAE, the mouse model of MS, when delivered prophylactically at time of EAE induction (Fig. 7a). Mice that received DCs were less paralyzed, indicated by lower clinical scores. Survival, defined as lack of onset of paralysis, was similar between groups (Fig. 7b). In a separate, independent study, we found amelioration of EAE using DC10s to be location-specific when delivered using in situ gelling PEG hydrogel (Fig. 8). Similar to the study in Fig. 7, paralysis was lessened as indicated by a reduction in the clinical score when DC10s were injected subcutaneously into the neck, proximal to the cervical lymph nodes, the latter of which have a known role in the progression of the disorder [40, 41]. However, when these cells were injected within hydrogel subcutaneously into the flank, proximal to the vaccination site, there were no differences between treated and control subjects. Additionally, the control hydrogels without DCs did not significantly differ by location.
Figure 7.
Attenuation of EAE upon delivery of DC10s using in situ gelling PEG hydrogel. (a) EAE mean clinical score of paralysis and (b) survival as defined by longevity of clinical score less than 1.
Figure 8.
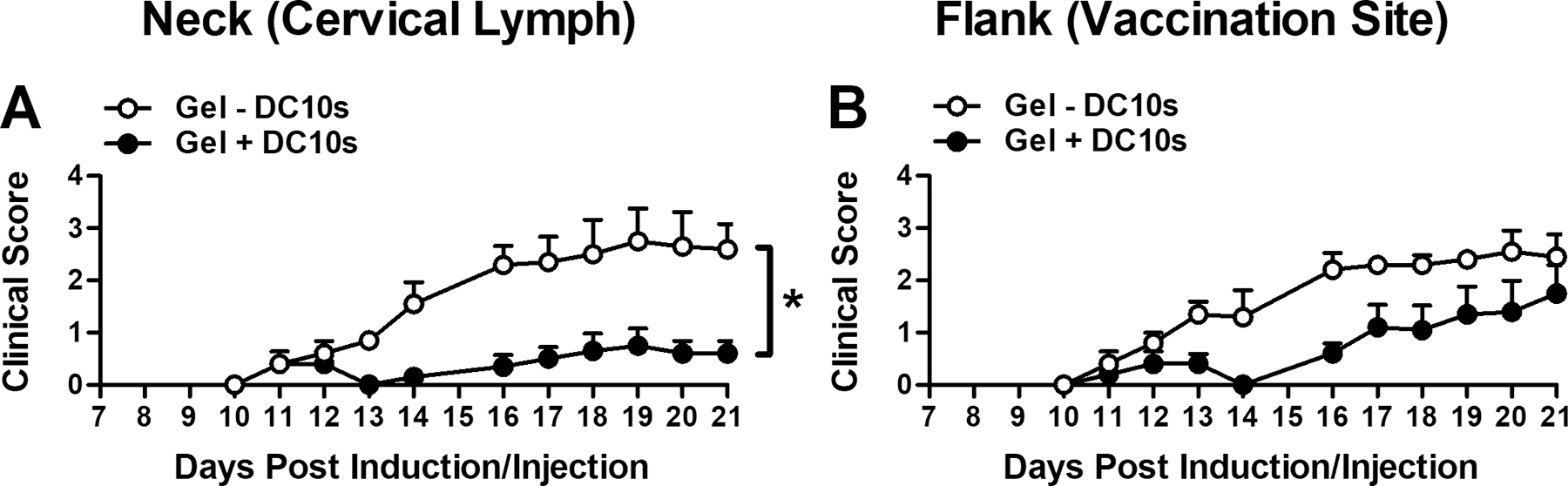
Location-dependency of EAE attenuation upon delivery of DC10s. EAE paralysis (mean clinical score) when tolerogenic DC10-laden hydrogel was delivered (a) to the neck or (b) to the flank.
Discussion
In this report, we performed an initial assessment of the utility of in situ gelling hydrogels to deliver DCs for regenerative medicine applications. DCs were generated from bone marrow cells using IL-10. Exposure of DCs to IL-10 has been shown to inhibit their maturation and activation [42, 43], thereby reducing (auto)immune responses [44, 45]. While characterization using traditional surface markers (CD11b, CD11c, IAB (MHC-II), CD80, CD86, and CD54) did not differentiate these cells from immature DCs, PirB expression distinguished DCs from both immature and mature phenotypes. PirB-mediated signaling (paired Ig-like receptor, a MHC-I receptor) induces immunosuppressive properties in DCs by promoting their secretion of IL-10 [46]. In that study, an immunostimulation assay revealed PirB+ DCs produced IL-10 and suppressed T- and B-cell proliferation.
Dendritic cells are sensitive to the physical and chemical cues of their microenvironment, and therefore, can either enhance or inhibit immunity [47]. To control their environment and their behavior, several groups have explored the use of biomaterials, both alone or in combination with proteins and nucleic acids [48–50]. The most common application of biomaterials in immunity has been to develop more potent vaccines by increasing antigen presentation [49, 51] and by enhancing maturation and activation [48, 51–54]. More recently, micron-sized biomaterials (drug-delivering particles) have been injected into or proximal to lymph nodes to ameliorate systems in the EAE model [55, 56]. Here, we promoted immunosuppression in EAE using a local injection of in situ gelling material, wherein both physical and chemical cues, known to dictate DCs behavior, can be controlled. Encapsulation into a PEG hydrogel lowered the maturation (CD86, CD54) of DC10s after 1 day. Delivery of these hydrogels with DC10s altered endogenous DC phenotype within 2 days of injection.
Hydrogels were used to both define and localize the site of DC transplantation, and thereby, controlling the immunomodulation of the therapy. Dendritic cells were delivered to the cervical lymph nodes, which play a role in the progression of MS, or the flank, proximal to the vaccination site, but irreverent to the disease progression. Delivery proximal to the cervical lymph nodes significantly attenuated paralysis, while delivery to the flank was less effective as an immunotherapy as far as amelioration of EAE symptoms, which we speculate may be due to less interaction with myelin- and CNS-responsive immune cells. While this issue may be mitigated with a higher dose, sites that minimize cell numbers would be more desirable for translation due to limitations in (autologous) cell availability. Traditionally, DCs are delivered systemically, relying on their ability to home to the site of immune activation and immunogenic organs [57, 58]. Localization of DC transplantation and other immunodulatory strategies to target sites, particularly those with antigen nonspecific mechanisms, can enhance the efficacy and improve the safety of these strategies for translation, yet few studies have investigated the effect of local delivery on therapeutic efficacy. For vaccine applications delivering DCs proximal to the lymph nodes or the spleen has been effective strategy for improving efficacy [53, 59]. Localized delivery was shown to reduce the number of doses required to mount an immune response against ovalbumin compared to systemic delivery [59]. For transplantation delivering DCs with the islets prolonged survival compared to off-site and systemic delivery [60].
Conclusion
This report provides initial evidence of the immunomodulatory potential of hydrogel-mediated DC transplantation. Interleukin-10 was used to derive DC10s from bone marrow cells. These DCs were PirB+ and IL-10 secreting, which suppressed the proliferation of activated immune cells in vitro. Using an in situ gelling hydrogel permitted the maintenance of DC10 features in vitro and modulation of endogenous DCs local to the injection site in vivo. Paralysis was ameliorated when delivered to the cervical lymph nodes, but not to the flank. Using materials to deliver DC10s stabilized the phenotype of transplanted cells and by localized these cells to lymph nodes known to control the progression of MS. We provide initial evidence in support of a hydrogel-mediated immunomodulatory strategy that can be applied to both autoimmune disorders and cell transplantation. The translation of this promising strategy will benefit from future investigations that evaluate and optimize the longevity and migration of these DC10s after transplantation and from those that identify and optimize the features of the DCs and hydrogels responsible for immunomodulation. Optimization of these features as well as transplantation parameters can enhance the immunomodulation of this strategy, which can then be combined with therapies that address secondary injury, e.g. Wallerian degeneration, and repair, e.g. remyelination, as a multifunctional platform for treating autoimmune diseases like MS.
Supplementary Material
Supplemental Figure 1. Gating scheme for flow cytometry data. (a) Selection of cells by forward and side scatter. (b) Selection of CD11c+ DCs. (c) Discrimination of CD11b+/CD11c- macrophages from CD11c+/CD11b+ DCs. (d) Negative staining controls for DC and macrophage selection. (e-g) Selection of surface markers in CD11c+ DCs. (h-j) Negative staining controls for selection of these surface markers.
Supplemental Figure 2. IL-10 (pg/mL) secretion by DCs upon derivation, CD11c+.
Acknowledgements
This work was supported by the NIH (ACTSI, UL1TR000454; 1 R21 EB019166-01A1), the Georgia Immunoengineering Consortium’s seed grant, and Georgia Partner’s Regenerative Medicine (REM) seed grant. The first author received support from the ILET2 training grant (1 R90 DK098981, 1 T90 DK097787). The authors would like to thank Devon M. Headen (Georgia Institute of Technology) for technical assistance in hydrogel construction.
Footnotes
Conflict of Interest
No benefit of any kind will be received either directly or indirectly by the author(s).
References
- [1].Maghzi A-H, Borazanci A, McGee J, Steven Alexander J, Gonzalez-Toledo E, Minagar A. 1 - Multiple Sclerosis: Pathophysiology, Clinical Features, Diagnosis, and Management. In: Minagar A, editor. Neuroinflammation. London: Elsevier; 2011. p. 1–23. [Google Scholar]
- [2].Alonso A, Hernan MA. Temporal trends in the incidence of multiple sclerosis: a systematic review. Neurology. 2008;71:129–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bell C, Graham J, Earnshaw S, Oleen-Burkey M, Castelli-Haley J, Johnson K. Cost-effectiveness of Four Immunomodulatory Therapies for Relapsing-Remitting Multiple Sclerosis: A Markov Model Based on Long-term Clinical Data. Journal of Managed Care Pharmacy. 2007;13:245–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kobelt G, Berg J, Lindgren P, Fredrikson S, Jönsson B. Costs and quality of life of patients with multiple sclerosis in Europe. Journal of Neurology, Neurosurgery & Psychiatry. 2006;77:918–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Owens GM, Olvey EL, Shrepnek GH, Pill MW. Perspectives for Managed Care Organizations on the Burden of Multiple Sclerosis and the Cost-Benefits of Disease-Modifying Therapies. Journal of Managed Care Pharmacy. 2013;19:S41–S53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Noyes K, Bajorska A, Chappel A, Schwid SR, Mehta LR, Weinstock-Guttman B, et al. Cost-effectiveness of disease-modifying therapy for multiple sclerosis: a population-based study. Neurology. 2011;77:355–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Rudick RA, Cohen JA, Weinstock-Guttman B, Kinkel RP, Ransohoff RM. Management of multiple sclerosis. N Engl J Med 1997;337:1604–11. [DOI] [PubMed] [Google Scholar]
- [8].Epstein DJ, Dunn J, Deresinski S. Infectious Complications of Multiple Sclerosis Therapies: Implications for Screening, Prophylaxis, and Management. Open Forum Infectious Diseases. 2018;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Allen JS, Pang K, Skowera A, Ellis R, Rackham C, Lozanoska-Ochser B, et al. Plasmacytoid dendritic cells are proportionally expanded at diagnosis of type 1 diabetes and enhance islet autoantigen presentation to T-cells through immune complex capture. Diabetes. 2009;58:138–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Farkas L, Beiske K, Lund-Johansen F, Brandtzaeg P, Jahnsen FL. Plasmacytoid dendritic cells (natural interferon- alpha/beta-producing cells) accumulate in cutaneous lupus erythematosus lesions. The American journal of pathology. 2001;159:237–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T, et al. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nature medicine. 2005;11:328–34. [DOI] [PubMed] [Google Scholar]
- [12].Karni A, Abraham M, Monsonego A, Cai G, Freeman GJ, Hafler D, et al. Innate immunity in multiple sclerosis: myeloid dendritic cells in secondary progressive multiple sclerosis are activated and drive a proinflammatory immune response. Journal of immunology. 2006;177:4196–202. [DOI] [PubMed] [Google Scholar]
- [13].Scheinecker C, Zwolfer B, Koller M, Manner G, Smolen JS. Alterations of dendritic cells in systemic lupus erythematosus: phenotypic and functional deficiencies. Arthritis and rheumatism. 2001;44:856–65. [DOI] [PubMed] [Google Scholar]
- [14].Uno S, Imagawa A, Okita K, Sayama K, Moriwaki M, Iwahashi H, et al. Macrophages and dendritic cells infiltrating islets with or without beta cells produce tumour necrosis factor-alpha in patients with recent-onset type 1 diabetes. Diabetologia. 2007;50:596–601. [DOI] [PubMed] [Google Scholar]
- [15].Bailey SL, Schreiner B, McMahon EJ, Miller SD. CNS myeloid DCs presenting endogenous myelin peptides ‘preferentially’ polarize CD4+ T(H)-17 cells in relapsing EAE. Nature immunology. 2007;8:172–80. [DOI] [PubMed] [Google Scholar]
- [16].Thomas AM, Dong Y, Beskid NM, Garcia AJ, Adams AB, Babensee JE. Brief exposure to hyperglycemia activates dendritic cells in vitro and in vivo. Journal of cellular physiology. 2020;235:5120–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Manickasingham S, Reis e Sousa C. Microbial and T cell-derived stimuli regulate antigen presentation by dendritic cells in vivo. Journal of immunology. 2000;165:5027–34. [DOI] [PubMed] [Google Scholar]
- [18].Itano AA, McSorley SJ, Reinhardt RL, Ehst BD, Ingulli E, Rudensky AY, et al. Distinct dendritic cell populations sequentially present antigen to CD4 T cells and stimulate different aspects of cell-mediated immunity. Immunity. 2003;19:47–57. [DOI] [PubMed] [Google Scholar]
- [19].Hatterer E, Touret M, Belin M-F, Honnorat J, Nataf S. Cerebrospinal Fluid Dendritic Cells Infiltrate the Brain Parenchyma and Target the Cervical Lymph Nodes under Neuroinflammatory Conditions. PLoS ONE. 2008;3:e3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Caravagna C, Jaouen A, Desplat-Jego S, Fenrich KK, Bergot E, Luche H, et al. Diversity of innate immune cell subsets across spatial and temporal scales in an EAE mouse model. Sci Rep 2018;8:5146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Brunner C, Seiderer J, Schlamp A, Bidlingmaier M, Eigler A, Haimerl W, et al. Enhanced dendritic cell maturation by TNF-alpha or cytidine-phosphate-guanosine DNA drives T cell activation in vitro and therapeutic anti-tumor immune responses in vivo. Journal of immunology. 2000;165:6278–86. [DOI] [PubMed] [Google Scholar]
- [22].Mattei F, Schiavoni G, Belardelli F, Tough DF. IL-15 is expressed by dendritic cells in response to type I IFN, double-stranded RNA, or lipopolysaccharide and promotes dendritic cell activation. Journal of immunology. 2001;167:1179–87. [DOI] [PubMed] [Google Scholar]
- [23].Lu W, Arraes LC, Ferreira WT, Andrieu JM. Therapeutic dendritic-cell vaccine for chronic HIV-1 infection. Nature medicine. 2004;10:1359–65. [DOI] [PubMed] [Google Scholar]
- [24].Su H, Messer R, Whitmire W, Fischer E, Portis JC, Caldwell HD. Vaccination against chlamydial genital tract infection after immunization with dendritic cells pulsed ex vivo with nonviable Chlamydiae. The Journal of experimental medicine. 1998;188:809–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].von Stebut E, Belkaid Y, Nguyen BV, Cushing M, Sacks DL, Udey MC. Leishmania major-infected murine langerhans cell-like dendritic cells from susceptible mice release IL-12 after infection and vaccinate against experimental cutaneous Leishmaniasis. European journal of immunology. 2000;30:3498–506. [DOI] [PubMed] [Google Scholar]
- [26].Kugler A, Stuhler G, Walden P, Zoller G, Zobywalski A, Brossart P, et al. Regression of human metastatic renal cell carcinoma after vaccination with tumor cell-dendritic cell hybrids. Nature medicine. 2000;6:332–6. [DOI] [PubMed] [Google Scholar]
- [27].Preynat-Seauve O, Schuler P, Contassot E, Beermann F, Huard B, French LE. Tumor-infiltrating dendritic cells are potent antigen-presenting cells able to activate T cells and mediate tumor rejection. Journal of immunology. 2006;176:61–7. [DOI] [PubMed] [Google Scholar]
- [28].Yu JS, Liu G, Ying H, Yong WH, Black KL, Wheeler CJ. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer research. 2004;64:4973–9. [DOI] [PubMed] [Google Scholar]
- [29].Adorini L, Penna G, Giarratana N, Uskokovic M. Tolerogenic dendritic cells induced by vitamin D receptor ligands enhance regulatory T cells inhibiting allograft rejection and autoimmune diseases. Journal of cellular biochemistry. 2003;88:227–33. [DOI] [PubMed] [Google Scholar]
- [30].Lutz MB, Suri RM, Niimi M, Ogilvie AL, Kukutsch NA, Rossner S, et al. Immature dendritic cells generated with low doses of GM-CSF in the absence of IL-4 are maturation resistant and prolong allograft survival in vivo. European journal of immunology. 2000;30:1813–22. [DOI] [PubMed] [Google Scholar]
- [31].Taner T, Hackstein H, Wang Z, Morelli AE, Thomson AW. Rapamycin-treated, alloantigen-pulsed host dendritic cells induce ag-specific T cell regulation and prolong graft survival. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2005;5:228–36. [DOI] [PubMed] [Google Scholar]
- [32].Terness P, Bauer TM, Rose L, Dufter C, Watzlik A, Simon H, et al. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: mediation of suppression by tryptophan metabolites. The Journal of experimental medicine. 2002;196:447–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Phelps EA, Enemchukwu NO, Fiore VF, Sy JC, Murthy N, Sulchek TA, et al. Maleimide cross-linked bioactive PEG hydrogel exhibits improved reaction kinetics and cross-linking for cell encapsulation and in situ delivery. Advanced materials. 2012;24:64–70, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Phelps EA, Headen DM, Taylor WR, Thule PM, Garcia AJ. Vasculogenic bio-synthetic hydrogel for enhancement of pancreatic islet engraftment and function in type 1 diabetes. Biomaterials. 2013;34:4602–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gahring LC, Ernst DN, Romball CG, Thoman ML, Torbett BE, Hobbs M, et al. The expression of CD45RB on antigen-responsive CD4+ lymphocytes: mouse strain polymorphism and different responses to distinct antigens. Cellular immunology. 1993;148:269–82. [DOI] [PubMed] [Google Scholar]
- [36].Ten Hove T, The Olle F, Berkhout M, Bruggeman JP, Vyth-Dreese FA, Slors JF, et al. Expression of CD45RB functionally distinguishes intestinal T lymphocytes in inflammatory bowel disease. Journal of leukocyte biology. 2004;75:1010–5. [DOI] [PubMed] [Google Scholar]
- [37].Chiang EY, Henson M, Stroynowski I. The nonclassical major histocompatibility complex molecule Qa-2 protects tumor cells from NK cell- and lymphokine-activated killer cell-mediated cytolysis. Journal of immunology. 2002;168:2200–11. [DOI] [PubMed] [Google Scholar]
- [38].Larimore K, Liang L, Bakkour S, Sha WC. B7h-expressing dendritic cells and plasma B cells mediate distinct outcomes of ICOS costimulation in T cell-dependent antibody responses. BMC immunology. 2012;13:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ma G, Pan PY, Eisenstein S, Divino CM, Lowell CA, Takai T, et al. Paired immunoglobin-like receptor-B regulates the suppressive function and fate of myeloid-derived suppressor cells. Immunity. 2011;34:385–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].van Zwam M, Huizinga R, Heijmans N, van Meurs M, Wierenga-Wolf AF, Melief M-J, et al. Surgical excision of CNS-draining lymph nodes reduces relapse severity in chronic-relapsing experimental autoimmune encephalomyelitis. The Journal of Pathology. 2009;217:543–51. [DOI] [PubMed] [Google Scholar]
- [41].van Zwam M, Huizinga R, Melief M-J, Wierenga-Wolf A, van Meurs M, Voerman J, et al. Brain antigens in functionally distinct antigen-presenting cell populations in cervical lymph nodes in MS and EAE. J Mol Med 2009;87:273–86. [DOI] [PubMed] [Google Scholar]
- [42].Allavena P, Piemonti L, Longoni D, Bernasconi S, Stoppacciaro A, Ruco L, et al. IL-10 prevents the differentiation of monocytes to dendritic cells but promotes their maturation to macrophages. European journal of immunology. 1998;28:359–69. [DOI] [PubMed] [Google Scholar]
- [43].Buelens C, Verhasselt V, De Groote D, Thielemans K, Goldman M, Willems F. Interleukin-10 prevents the generation of dendritic cells from human peripheral blood mononuclear cells cultured with interleukin-4 and granulocyte/macrophage-colony-stimulating factor. European journal of immunology. 1997;27:756–62. [DOI] [PubMed] [Google Scholar]
- [44].Moreau A, Vandamme C, Segovia M, Devaux M, Guilbaud M, Tilly G, et al. Generation and in vivo evaluation of IL10-treated dendritic cells in a nonhuman primate model of AAV-based gene transfer. Molecular Therapy — Methods & Clinical Development. 2014;1:14028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Tai N, Yasuda H, Xiang Y, Zhang L, Rodriguez-Pinto D, Yokono K, et al. IL-10-conditioned dendritic cells prevent autoimmune diabetes in NOD and humanized HLA-DQ8/RIP-B7.1 mice. Clinical immunology. 2011;139:336–49. [DOI] [PubMed] [Google Scholar]
- [46].Endo S, Sakamoto Y, Kobayashi E, Nakamura A, Takai T. Regulation of cytotoxic T lymphocyte triggering by PIR-B on dendritic cells. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:14515–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Menges M, Rossner S, Voigtlander C, Schindler H, Kukutsch NA, Bogdan C, et al. Repetitive injections of dendritic cells matured with tumor necrosis factor alpha induce antigen-specific protection of mice from autoimmunity. The Journal of experimental medicine. 2002;195:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Singh A, Suri S, Roy K. In-situ crosslinking hydrogels for combinatorial delivery of chemokines and siRNA–DNA carrying microparticles to dendritic cells. Biomaterials. 2009;30:5187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lewis JS, Zaveri TD, Crooks Ii CP, Keselowsky BG. Microparticle surface modifications targeting dendritic cells for non-activating applications. Biomaterials. 2012;33:7221–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Park J, Babensee JE. Differential functional effects of biomaterials on dendritic cell maturation. Acta Biomaterialia. 2012;8:3606–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Torres MP, Wilson-Welder JH, Lopac SK, Phanse Y, Carrillo-Conde B, Ramer-Tait AE, et al. Polyanhydride microparticles enhance dendritic cell antigen presentation and activation. Acta Biomaterialia. 2011;7:2857–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Bandyopadhyay A, Fine RL, Demento S, Bockenstedt LK, Fahmy TM. The impact of nanoparticle ligand density on dendritic-cell targeted vaccines. Biomaterials. 2011;32:3094–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hori Y, Winans AM, Huang CC, Horrigan EM, Irvine DJ. Injectable dendritic cell-carrying alginate gels for immunization and immunotherapy. Biomaterials. 2008;29:3671–82. [DOI] [PubMed] [Google Scholar]
- [54].Akagi T, Wang X, Uto T, Baba M, Akashi M. Protein direct delivery to dendritic cells using nanoparticles based on amphiphilic poly(amino acid) derivatives. Biomaterials. 2007;28:3427–36. [DOI] [PubMed] [Google Scholar]
- [55].Tostanoski LH, Chiu YC, Gammon JM, Simon T, Andorko JI, Bromberg JS, et al. Reprogramming the Local Lymph Node Microenvironment Promotes Tolerance that Is Systemic and Antigen Specific. Cell Rep 2016;16:2940–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Cho JJ, Stewart JM, Drashansky TT, Brusko MA, Zuniga AN, Lorentsen KJ, et al. An antigen-specific semi-therapeutic treatment with local delivery of tolerogenic factors through a dual-sized microparticle system blocks experimental autoimmune encephalomyelitis. Biomaterials. 2017;143:79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Baas MC, Kuhn C, Valette F, Mangez C, Duarte MS, Hill M, et al. Combining autologous dendritic cell therapy with CD3 antibodies promotes regulatory T cells and permanent islet allograft acceptance. Journal of immunology. 2014;193:4696–703. [DOI] [PubMed] [Google Scholar]
- [58].Peche H, Trinite B, Martinet B, Cuturi MC. Prolongation of heart allograft survival by immature dendritic cells generated from recipient type bone marrow progenitors. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2005;5:255–67. [DOI] [PubMed] [Google Scholar]
- [59].Serody JS, Collins EJ, Tisch RM, Kuhns JJ, Frelinger JA. T Cell Activity After Dendritic Cell Vaccination Is Dependent on Both the Type of Antigen and the Mode of Delivery. The Journal of Immunology. 2000;164:4961–7. [DOI] [PubMed] [Google Scholar]
- [60].Oluwole OO, Depaz HA, Gopinathan R, Ali A, Garrovillo M, Jin M-X, et al. Indirect Allorecognition in Acquired Thymic Tolerance: Induction of Donor-Specific Permanent Acceptance of Rat Islets by Adoptive Transfer of Allopeptide-Pulsed Host Myeloid and Thymic Dendritic Cells. Diabetes. 2001;50:1546–52. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Gating scheme for flow cytometry data. (a) Selection of cells by forward and side scatter. (b) Selection of CD11c+ DCs. (c) Discrimination of CD11b+/CD11c- macrophages from CD11c+/CD11b+ DCs. (d) Negative staining controls for DC and macrophage selection. (e-g) Selection of surface markers in CD11c+ DCs. (h-j) Negative staining controls for selection of these surface markers.
Supplemental Figure 2. IL-10 (pg/mL) secretion by DCs upon derivation, CD11c+.