

Abstract
Mesenchymal spindle cell tumors with kinase fusions, often presenting in superficial or deep soft tissue locations, may rarely occur in bone. Herein, we describe the clinicopathologic and molecular data of 8 bone tumors characterized by various kinase fusions from our files and incorporate the findings with the previously reported 7 cases, mainly as single case reports. In the current series all but one of the patients were young children or teenagers, with an age range from newborn to 59 years (mean 19 years). Most tumors (n=5) presented in the head and neck area (skull base, mastoid, maxilla, and mandible), and remaining 3 in the tibia, pelvic bone, and chest wall. The fusions included NTRK1 (n=3), RET (n=2), NTRK3 (n=2), and BRAF (n=1). In the combined series (n=15), most tumors (73%) occurred in children and young adults (< 30 years) and showed a predilection for jaw and skull bones (40%), followed by long and small tubular bones (33%). The fusions spanned a large spectrum of kinase genes, including in descending order NTRK3 (n=6), NTRK1 (n=4), RET (n=2), BRAF (n=2), and RAF1 (n=1). All fusions confirmed by targeted RNA sequencing were in-frame and retained the kinase domain within the fusion oncoprotein. Similar to the soft tissue counterparts, most NTRK3-positive bone tumors in this series showed high-grade morphology (5/6), whereas the majority of NTRK1 tumors were low-grade (3/4). Notably, all 4 tumors presenting in the elderly were high-grade spindle cell sarcomas, with adult fibrosarcoma (FS)-like, malignant peripheral nerve sheath tumor (MPNST)-like and MPNST phenotypes. Overall, 10 tumors had high-grade morphology, ranging from infantile and adult-types FS, MPNST-like, and MPNST, whereas 5 showed benign/low-grade histology (MPNST-like and myxoma-like). Immunohistochemically (IHC), S100 and CD34 positivity was noted in 57% and 50%, respectively, while co-expression of S100 and CD34 in 43% of cases. One-third of tumors (4 high grade and the myxoma-like) were negative for both S100 and CD34. IHC for Pan-TRK was positive in all 8 NTRK-fusion positive tumors tested and negative in 2 tumors with other kinase fusions. Clinical follow-up was too limited to allow general conclusions.
Keywords: NTRK, RET, sarcoma, bone, malignant peripheral nerve sheath tumor, fibrosarcoma, fusion
INTRODUCTION
About twenty years after the initial description in 1998 of the canonical ETV6::NTRK3 fusion in infantile fibrosarcoma (IFS)1, and with the advent of next generation sequencing (NGS), other gain-of-function kinase fusions have been reported in subsets of soft tissue and bone tumors, spanning various clinical presentations, histologic types, and risk of malignancy.2 Apart from the NTRK1-rearranged lipofibromatosis-like neural tumor (LPFNT)3, this emerging tumor family also includes a heterogeneous group of rare but distinct spindle cell tumors with NTRK1–3, RAF1, BRAF, ALK and RET fusions, often displaying a malignant peripheral nerve sheath tumor (MPNST) or fibrosarcoma (FS)-like phenotype and associated with immunohistochemical co-expression of S100 and CD34.4–6 This group of neoplasms encompasses low-grade tumors composed of a monomorphic spindle cell proliferation with haphazard growth, often with distinctive patterns of keloidal stromal and perivascular collagen deposition, as well as intermediate to high-grade tumors composed of cellular solid and fascicular areas of monomorphic spindle cells with hyperchromatic nuclei, scant cytoplasm, and increased mitotic rate. More recently, this family of kinase fusion positive spindle cell tumors has been divided into several subcategories mainly based on morphologic features and immunohistochemical (IHC) profiles, to reflect their similarities with other established soft tissue tumors and allow a more readily pattern-recognition by practicing pathologists. These include: LPFNT reminiscent of a lipofibromatosis, infantile fibrosarcoma (IFS)-like in patients younger than 2 years of age and exclusive of the canonical ETV6::NTRK3 fusion, adult type FS-like, MPNST-like, and myxoma-like (see Table 1).7 Notwithstanding, these tumors also appear to be part of a continuous morphologic spectrum as several cases showed hybrid or overlapping histologic features, e.g., lipofibromatosis-like patterns alternating with solid areas of cellular fascicular growth or tumors displaying both low-grade and high-grade morphology.8 However, these descriptive morphologic designations have been proven effective in pre-selecting tumors that may require further PanNTRK IHC staining or molecular sequencing. Members of this rare family of mesenchymal tumors with kinase fusions may occur in different anatomic sites, including skin, superficial and deep soft tissue, bone, gynecologic tract, and gastrointestinal tract.9–12 Herein, we present 8 primary bone tumors with kinase fusions (BTKF), the largest series to date, and compare our findings to 7 other case reports in the recent literature.
Table 1.
Phenotypic subcategories of kinase gene associated mesenchymal spindle cell tumors (modified from Xu et al. 7)
| IFS | S100-negative primitive (blue) spindle cell tumor, often with fascicular/herringbone arrangement, occurring in patients below age of 2 years old, harboring the canonical ETV6::NTRK3 fusion |
| IFS-like | IFS phenotype as defined above, with alternative kinase fusions other than ETV6::NTRK3 |
| LPFNT | Tumor with monotonous bland spindle cells showing a highly infiltrative pattern within adjacent adipose tissue at the periphery, frequent co-expression of S100 and CD34, prominent bands of stromal collagen, and perivascular hyalinization |
| MPNST-like | S100-positive cellular spindle cell tumor showing exclusive solid architecture, frequent stromal collagen deposits, and perivascular hyalinization |
| Adult FS-like | S100-negative cellular spindle cell tumor showing exclusive solid architecture, often with fascicular/herringbone arrangement, occurring in patients ≥ 2 y |
| Myxoma-like | Hypocellular hypovascular myxoid neoplasm with bland stellate to spindle cells |
MATERIALS AND METHODS
Patient selection and clinicopathologic review
Candidate cases were retrieved from the pathology archives of MSKCC and UMCG and the personal consultation cases of two of the authors (A.J.H.S. and C.R.A.) based on the presence of kinase gene fusion or rearrangement as documented by molecular studies (fluorescence in situ hybridization [FISH] or targeted RNA sequencing). All identified bone tumors with kinase fusions included in this study were reviewed by two pathologists (A.J.H.S. and C.R.A.) to gather relevant histopathologic parameters, demographic, and outcome data. Five of the 8 tumors have been previously reported, as shown in Table 2. IMT cases with typical morphology and ALK or ROS1 fusions were excluded from the study. Each tumor was further subdivided into IFS, IFS-like tumor, LPFNT, MPNST-like, adult FS-like, and myxoma-like based on their histologic features, immunoprofile, and underlying fusion using earlier definitions provided by Xu et al. (Table 1). Immunohistochemistry studies were reviewed for the following primary antibodies: S100 (polyclonal, dilution: 1:8000, DAKO; Agilent), CD34 (clone: QBEnd-10, ready to use [RTU], Ventana; Roche Diagnostics), and pan-TRK (clone EPR17341, dilution 1:100; Abcam). Staining was performed on a Leica-Bond-3 (Leica, Buffalo Grove, IL) or a Ventana Benchmark (Ventana Medical Systems, Tucson, AZ) automated immunostaining platform using a heat-based antigen retrieval method and high pH buffer.
Table 2.
Clinicopathological, molecular and outcome data of 8 bone tumors with kinase fusions
| Case | Age/Sex | Location | Kinase Gene | Partner | Size (cm) | Histology | MI | Atypia | S100 | CD34 | pan-NTRK | FU (months) |
| 1 * | newborn/M | skull base | NTRK3 | ETV6 | 4.7 | IFS | 3 | N | neg | neg | pos | NA |
| 2 ** | 2mo/M | chest wall | RET | CLIP2 | NA | IFS-like | 5 | NA | neg | neg | NA | NED (114) |
| 3 *** | 4y/M | mandible | NTRK1 | LMNA | 4 | LG-MPNST-like | 5 | N | pos | pos | pos | NED (142) |
| 4 *** | 13 y/M | maxilla | NTRK1 | LMNA | 2.5 | LG-MPNST-like | 0 | N | pos | pos | pos | NED (648) |
| 5 | 9y/F | pelvis/ilium | NTRK1 | TPM3 | 8.7 | HG-MPNST-like | >20 | N | pos | neg | pos | recent case |
| 6 | 14y/M | mastoid | BRAF | FNBP1 | NA | HG-adult FS-like | >10 | N - necrotic | neg | neg | NA | recent case |
| 7 | 59y/F | tibia | NTRK3 | UPF2 | 5.6 | HG-MPNST | >10 | Y | neg | neg | pos | NA |
| 8* | 15y/M | skull base | RET | KIF5B | 1.3 | myxoma-like | 0 | N | neg | neg | neg | NA |
Targeted RNA Sequencing and FISH Detection of Kinase Fusions
Targeted RNA Sequencing.
For the Illumina MiSeq platform (used in 3 cases), RNA was extracted from formalin-fixed paraffin embedded (FFPE) tissue using Amsbio’s ExpressArt FFPE Clear RNA Ready kit (Amsbio LLC, Cambridge, MA). All cases had adequate RNA quality and could be further processed and analyzed. RNA-seq libraries were prepared using 20 to 100 ng total RNA with the TruSight RNA Fusion Panel (Illumina, San Diego, CA). Targeted RNA sequencing was performed on an Illumina MiSeq platform, as previously described.4 Reads were independently aligned with STAR (version 2.3) against the human reference genome (hg19) and analyzed by STAR-Fusion. The detailed procedure for the two cases studied by the Archer Anchored Multiplex RNA sequencing assay has also been previously described.13 In short, unidirectional gene-specific primers were designed to target specific exons in 62 genes known to be involved in oncogenic fusions in solid tumors. RNA was extracted from FFPE specimens, followed by cDNA synthesis and library preparation. Anchored Multiplex polymerase chain reaction amplicons were sequenced on Illumina Miseq, and the data was analyzed using the Archer software. In one case the commercially available FoundationOne®Heme comprehensive genomic profiling test was applied. This assay combines DNA and RNA sequencing, amongst others for the sensitive detection of known and novel fusions in sarcomas using FFPE material. In this case the novel finding of UPF2::NTRK3 was confirmed by running a RNA-based NGS panel from a second lab (NeoGenomics sarcoma comprehensive fusion panel).
Fluorescence In Situ Hybridization.
Two cases were tested by FISH for gene abnormalities relevant to that particular case (both detecting LMNA::NTRK1). Custom probes made by bacterial artificial chromosomes (BAC) clones flanking the genes of interest (LMNA, NTRK1) as previously described,3 according to UCSC genome browser (http://genome.ucsc.edu) and obtained from BACPAC sources of Children’s Hospital of Oakland Research Institute (Oakland, CA; bacpac.chori.org). DNA from each BAC was isolated according to the manufacturer’s instructions. The BAC clones were labeled with fluorochromes (fluorescent-labeled dUTPs, Enzo Life Sciences, New York, NY) by nick translation and validated on normal metaphase chromosomes. The 4 μm-thick FFPE slides were deparaffinized, pretreated, and hybridized with denatured probes, as previously described.4 After overnight incubation, the slides were washed, stained with 4’,6-diamidino-2-phenylindole, mounted with an antifade solution, and then examined on a Zeiss1 fluorescence microscope (Zeiss Axioplan, Oberkochen, Germany) controlled by Isis 5 software (Metasystems).
RESULTS
A total of 8 bone tumors with confirmed kinase fusions were selected from our files. The clinicopathological features, IHC profile, underlying fusion events, and clinical outcome are summarized in Table 2.
Clinical features
All patients in the current cohort were young children or teenagers. The patient age ranged from newborn (congenital IFS case) to 59 years (mean 19 yrs., median 11 yrs.). By location, most tumors (n=5) presented in the head and neck area (skull base (n = 2), mastoid, maxilla, and mandible), but few also occurred in tibia, pelvic bone, and chest wall, involving one of the ribs. Data on imaging studies were available in three cases (case #1, case #5, and case #7), while in the remaining the skeletal location was confirmed by radiologic reports and/or gross appearance within the bone. Case #1 concerned a maxillary tumor which by CT scans revealed a well-demarcated and lytic expansile tumor with invasion of surrounding soft tissue (Figure 1A). Case #5 was a pelvic bone tumor originating in the iliac wing, in which the MRI scans showed a 13-month interval size increase, from 4.6 × 2.3 × 4.4 cm to 8.7 × 6.4 × 5.4 cm. The tumor characteristics were that of a well-defined lytic lesion with cortical breakthrough and soft tissue extension with invasion of the iliopsoas muscle. In case #7, plain X-ray and MRI scans showed a 5.6 × 2.8 × 2.5 cm infiltrative destructive lesion in the proximal tibia with cortical breakthrough, extending in surrounding soft tissue, and abutting the neurovascular bundle (Figure 2A).
Figure 1.

BTKF (bone tumor with kinase fusion) with low-grade MPNST-like and myxoma-like morphology. A-E, a 2.5 cm LG-MPNST-like lesion with LMNA::NTRK1 fusion (case #4) involving maxilla showing aggressive clinical features with CT imaging including cortical breakthrough and soft tissue extension (A). This tumor is composed of vague fascicles of bland spindle cells infiltrating bone and cartilage (B). At higher power the tumor shows haphazardly arranged uniform spindle cells, dense keloid-like stromal changes and marked perivascular hyalinization (C). The tumor is positive for Pan-TRK (D). E, mandible tumor representing LG-MPNST with LMNA::NTRK1 fusion (case #3) showing fascicles of uniform eosinophilic myofibroblast-like spindle cells. F, Myxoma-like histology: skull base tumor with KIF5B::RET fusion (case #8). This hypocellular and hypovascular tumor is composed of bland stellate cells in an abundant myxoid background and indistinguishable from myxoma of bone.
Figure 2.
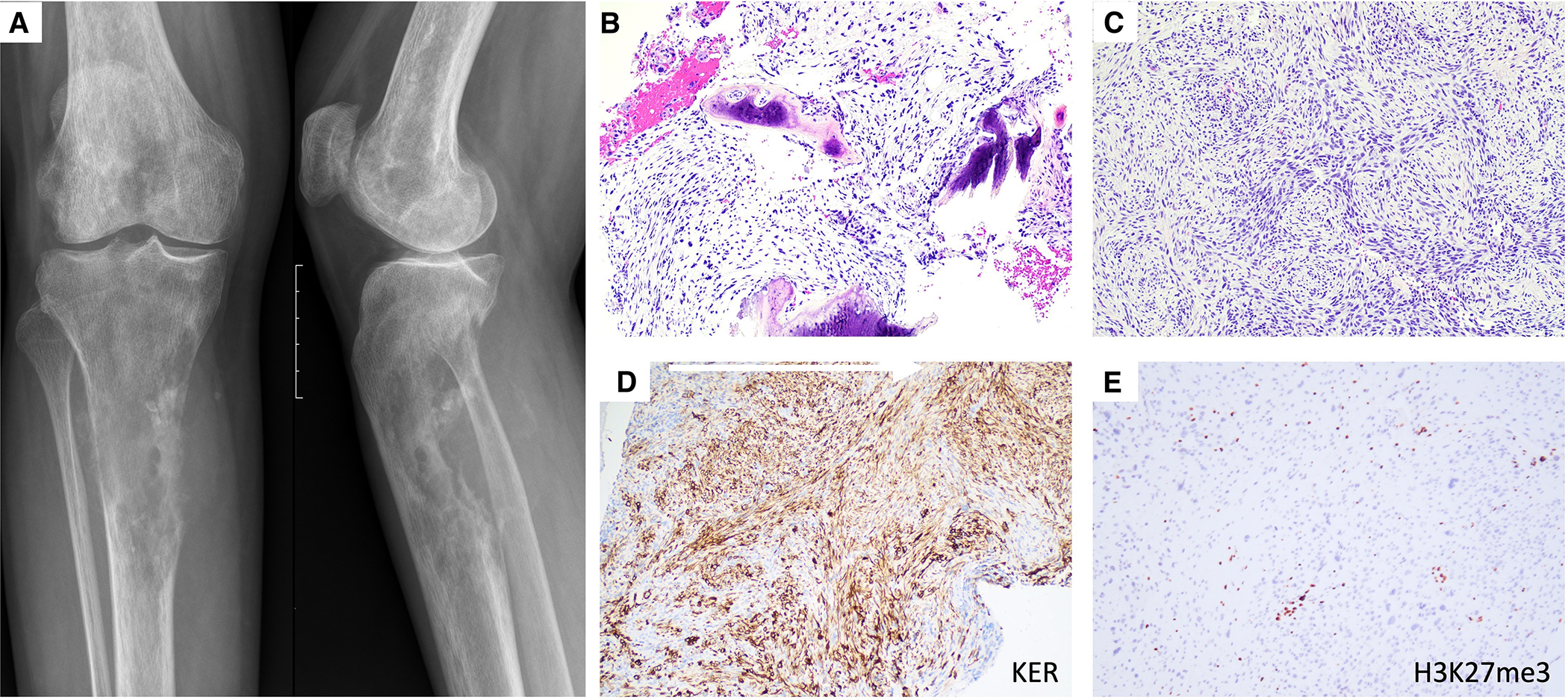
Primary MPNST of bone with UPF2::NTRK3 fusion, occurring in the proximal tibia of a-29-year-old female. Plain radiograph shows an ill-defined destructive bone tumor with cortical breakthrough and soft tissue extension (A). The tumor is composed of perpendicular fascicles of monomorphic hyperchromatic (blue) spindle cells (B-C), which show expression of cytokeratin (D) and loss of trimethylated histone protein H3K27 (E).
Molecular findings
In the current cohort, the kinase genes affected were NTRK1 (n=3), RET (n=2), NTRK3 (n=2), and BRAF (n=1). Kinase fusions were detected by RNA sequencing methods in 6 cases showing in-frame gene products including the kinase domain. The transcript gene fusions were between ETV6 exon 5 and NTRK3 exon 15 (n =1, case #1); between CLIP2 exon 14 and RET exon 12 (case #2); between TPM3 exon 7 and NTRK1 exon 10 (case #5); between FNBP1 exon 10 and BRAF exon 9 (case #6); between KIF5B exon 5 and RET exon 15 (case #8), and between UPF2 exon 3 and NTRK3 exon 4 (case #7). In case #7, representing a primary MPNST of bone, additional molecular findings included: P53 H179L mutation, loss of CDKN2A, and loss of EED. The remaining 2 cases were confirmed to have a LMNA::NTRK1 fusion by FISH (case #3 and case #4).
Histologic Features and IHC profile
All bone lesions were subclassified based on their histologic features and IHC profile into distinctive morphologic phenotypes, as defined in Table 1 and presented in Table 2, including classic IFS (n = 1), IFS-like (n = 1), adult FS-like (n = 1), low-grade and high-grade MPNST-like tumor (n = 3), and myxoma-like tumor (n = 1). In addition, one case represented a primary MPNST arising in bone. The histologic features were diverse. The three MPNST-like tumors appeared to correlate with NTRK1 fusions. In addition to the malignant phenotype displayed by the IFS and IFS-like tumor, there were 3 other tumors that showed features in keeping with a high-grade sarcoma, based on nuclear atypia, elevated mitotic index of ≥ 10 per 10 HPFs, and tumor necrosis, including one MPNST-like tumor with TPM3::NTRK1 (case #5), one adult FS-like tumor with FNBP1::BRAF fusion (case #6), and one MPNST with UPF2::NTRK3 fusion (case #7).
Infantile fibrosarcoma and infantile fibrosarcoma-like
There were 2 bone tumors in this cohort which had a FS phenotype and occurred in infants. One congenital skull base tumor harbored the canonical ETV6::NTRK3 fusion (case #1) was designated as classic IFS albeit with unusual morphologic features, while another chest wall tumor presenting in a 2-months-old newborn male showed the presence of a CLIP2::RET fusion and was considered a molecular variant of IFS (or IFS-like, case #2). Histologically, case #1 revealed a cystic lesion surrounded by a thick fibrous capsule with mural nodules showing cystic hemorrhagic change, and solid growth with an HPC-like vascular pattern, and heavy lymphocytic infiltrate (Figure 3A–C). Case #2 showed hypercellular solid growth with spindle cells arranged in intersecting fascicles with the prototypical herringbone pattern or as sheets of ovoid cells forming ill-formed fascicles (Figure 3E–F). Mitotic index ranged from 3 to 5 mitotic figures (MFs) per 10 HPFs. None of the IFSs showed perivascular hyalinization, dense hyalinized collagen bands, or areas reminiscent of lipofibromatosis. IHC staining for Pan-TRK was performed in IFS with ETV6::NTRK3 fusion (case #1) which showed heterogeneous cytoplasmic immunopositivity (Figure 3D). Both tumors (IFS and IFS-like) were negative for S100 and CD34.
FIGURE 3.

BTKF showing high-grade FS-like patterns. A-D IFS with ETV6::NTRK3 fusion (case #1) composed of a cystic lesion surrounded by a thick fibrous capsule with mural nodules (A) showing cystic hemorrhagic change (B) and solid growth with HPC-like vascular pattern and heavy lymphocytic infiltrate (C), and IHC staining for Pan-TRK (D)plump epithelioid cells (D). E-F IFS-like tumor with CLIP2::RET fusion (case #2) composed of fascicles of spindle cells in herringbone (E) and intersecting patterns (F).
MPNST-like Tumors and MPNST
In three MPNST-like cases (cases #3–5), the tumor had a solid growth and showed features resembling MPNST, composed of primitive uniform blue ovoid to spindle cells with scant cytoplasm arranged haphazardly or in a vague fascicular or angiopericytic pattern (Figures 1B, 1C, and 1E). Perivascular hyalinization and stromal collagen bands were prominent in case #3 (Figure 1C). In this subset, two tumors (case #3 and case #5) had low-grade morphology, whereas an iliac bone tumor harboring TPM3::NTRK1 in a 9-year-old patient (case #4) showed high-grade features with a mitotic index exceeding 20/10 HPFs, but no tumor necrosis. All 3 MPNST-like tumors were positive for S100. CD34 immunopositivity was seen in case #3 and case #5 but was absent in case #4. IHC for Pan-TRK showed diffuse cytoplasmic staining in all three cases, including LMNA::NTRK1 (n=2), and TPM3::NTRK1, respectively (Figure 1D). Case #7 represented a unique case of a primary MPNST of bone with an UPF2::NTRK3 fusion, occurring in the proximal tibia of a 59-year-old female. This high-grade MPNST showed mitotically active monotonous blue spindle cells invading bone and arranged in a fascicular and perpendicular pattern. The diagnosis of MPNST was further confirmed by the NGS report showing in addition to the NTRK3 fusion, the presence of CDKN2A/B loss, TP53 H179L mutation and EED loss. The tumor showed an overall low mutational burden (0 mutations/Mb) and microsatellite stable. Based on this genomic landscape, IHC was performed to investigate alterations of the PRC2 complex, showing complete loss of H3K27me3 in the tumor cells, further support for the diagnosis of MPNST. Additionally, tumor cells were positive for pan-cytokeratin and Pan-TRK (Figure 2B–E).
Adult-type FS
Case #6 was a mastoid bone tumor presenting in a 14-year-old male patient, which harbored a FNBP1::BRAF fusion. Tumor histology was high grade with nuclear atypia, high mitotic index of > 10 mitoses in 10 HPF, and extensive tumor necrosis. CD34 and S100 were negative in this tumor.
Unusual histotype of myxoma-like tumor
Case #8, a skull base tumor arising in a 15-year-old boy, showed an unusual hypocellular, hypovascular myxoid appearance indistinguishable from myxoma of bone (Figure 1F). The tumor was composed entirely of bland stellate and spindle cells in a myxoid background. There was no mitotic activity, cytologic atypia, or tumor necrosis. The initial histologic differential diagnosis of this case was intraosseous myxoma or fibrous dysplasia. IHC was negative for S100, CD34, and Pan-TRK. Targeted RNA sequencing revealed a KIF5B::RET fusion.
Pan-TRK Immunostaining
IHC staining for Pan-TRK, performed in 6/7 cases, was positive with cytoplasmic staining in all 5 cases with NTRK1/3 fusion (Figures 1D and 3D) and negative in the case with RET fusion. No nuclear immunopositivity was seen in this cohort.
Clinical Outcome
Clinical follow-up was limited as most of them represented consultation cases. Clinical outcome was only available in 3 cases with a follow-up period of 114 to 648 months (median: 142 mo.). The 3 cases included the IFS-like tumor with CLIP2::RET presenting in the chest wall of a 2-month-old baby boy and both low-grade MPNST-like tumors with LMNA::NTRK1 occurring in the maxilla and mandible. These three patients showed no evidence of disease; none of them developed recurrences or metastatic disease after major surgery. Two other cases were recent diagnoses.
DISCUSSION
In the past decade, next generation sequencing technology has significantly contributed to refining the classification of soft tissue and bone tumors enabling the detection of genomic alterations in a single step.14 This has resulted, among others, in the discovery of several novel gene fusions that are associated with distinct but until then unrecognized tumor types. A good example of this development are kinase fusions detected across mesenchymal tumors with uniform spindle cell morphology.
In 2016, NTRK1 fusions were first described to define lipofibromatosis-like neural tumors (LPFNT), a monomorphic spindle cell neoplasm with keloidal and perivascular collagen deposition, infiltrating subcutaneous fat in a reticular fashion and often showing IHC co-expression of S100 and CD34.3 Notably, LPFNT lacks SOX10 expression, which distinguish them from other histologic look-alikes, such as lipofibromatosis or nerve sheath tumors. LPFNT has not been associated with history of NF1 and behaves in a benign fashion, with only local recurrences being described if incompletely excised. Anecdotal evidence, based on rare patients with large inoperable tumors with this phenotype showed dramatic clinical responses to kinase inhibitor therapy and became resectable, albeit with positive margins (Antonescu, Suurmeijer personal communication, and Baranov et al.).15 Although initially described as a distinct pathologic entity mostly harboring NTRK1 fusions, it was subsequently recognized that LPFNT may present as a focal component within a tumor with hybrid LPFNT and solid patterns, and can harbor other kinase fusions, including NTRK2/3, RET, ALK and MET. The outcome of patients with tumors with hybrid histology remains mostly benign, with only one isolated case with a low-grade histology in a series of 24 patients developing distant metastatic disease to the lung.8
Another phenotype commonly associated with kinase fusions represents a more heterogeneous group of spindle cell tumors that frequently co-express S100 and CD34 and resemble low-grade MPNSTs.4 These lesions belong to an emerging family of rare mesenchymal spindle cell tumors showing a wide morphologic spectrum and clinical behavior, harboring not only NTRK rearrangements, but also RAF1, BRAF, or RET fusions. Most of these tumors (60%) occur in the superficial or deep soft tissues of the extremities and trunk of children or young adults (<30 years old) of both genders.
Finally, a subgroup of FS-like or MPNST-like tumors represent higher grade lesions, which have a predilection for NTRK3 gene fusions.5 On very rare occasions, these tumors are associated with neurofibromatosis (NF1).16 Clinical follow-up, albeit limited, has emphasized that tumors with a high-grade malignant phenotype have the propensity for distant spread and follow an aggressive clinical course.4,5
Some of the detected NTRK3, RET, and BRAF kinase fusions in this series were novel or uncommon. Notably, in all of these fusion variants, the predicted chimeric amino acid sequence was in-frame and contained the kinase activation domain. Therefore, these kinase fusions likely represent oncogenic drivers. In case #7 representing a prototypical MPNST of bone, a previously unreported UPF2::NTRK3 fusion was detected by two different RNA sequencing platforms (Illumina TruSight and Neogenomics). The notion that this rare occurrence of an NTRK3 fusion in MPNST represents a driver event is supported by a recent case report of a sciatic nerve MPNST with another novel SNRNP70::NTRK3 fusion. In this case, the tumor showed a fast and durable response to the NTRK inhibitor entrectinib for 10 months.17 The KIF5B::RET fusion found in a single myxoma-like tumor (case #8) has also been reported as a molecular event in an adult FS-like tumor arising in the small intestine.18 In addition, this gene fusion represents a common driver alteration in non-small cell lung cancer.19 Although the FNBP1::BRAF fusion detected in an adult FS-like tumor (case #6) has only been reported in a single case of a primary lung melanoma20, several cases of BRAF-rearranged spindle cell neoplasms with many other fusion partners and IFS-like or FS-like morphology have been published in the literature.21,22
To date (including 8 cases from the current study), 15 patients with mesenchymal spindle cell tumors originating from bone and harboring kinase fusions were reported in the literature after excluding prototypical cases of IMT.4,6,7,15,23–26 The clinicopathologic features, fusion events, and IHC profile of these cases are summarized in Table 3. Four tumors presenting in children below the age of 2 years were classified by default as IFS/IFS-like tumors. Most of the other cases (7/11) occurred in children and young adults (< 30 years). This propensity for teenagers and young adults is also reported for mesenchymal neoplasms with kinase fusions from other body sites.7 Notably, all 4 tumors seen in the elderly were high-grade FS-like, MPNST-like tumors or MPNST. By location, bone tumors with kinase fusions (BTKF) had a variable anatomic distribution, often presenting in jaw and skull bones (6/15; 40%) and long and small tubular bones (5/15; 33%), and less commonly in pelvic bones (2/15), vertebrae (1/15), and ribs (1/15). By imaging studies (radiographs, computed tomography, MRI), available in 3 cases, lesions appeared well-demarcated, lytic tumors with aggressive features such as cortical destruction and invasion in surrounding soft tissue. Akin to mesenchymal neoplasms from other body sites the most frequently rearranged kinase genes were NTRK3 (n=6) and NTRK1 (n=4), whereas those involving RET (n=2), BRAF (n=2), and RAF1 (n=1) were less common. BTKF cases displayed a wide morphologic spectrum and were further subdivided in six phenotypic groups, based on age at presentation, histology, and S100 protein expression, as first described by Xu et al. 7 Overall, ten (67%) tumors had high-grade morphology (IFS, IFS-like, adult FS like, HG MPNST-like, and MPNST), whereas five (33%) cases were low-grade (LG MPNST-like and myxoma-like). Similar to soft tissue locations, most NTRK3 bone tumors in this series showed high-grade morphology (5/6), whereas the majority of NTRK1 tumors were low-grade (3/4). By reported IHC findings, S100 and CD34 positivity was noted in 57% and 50%, respectively. The immunoprofiles of KFBT varied according to histologic subcategories. Notably, 5 (33%) tumors (4 high grade and one myxoma-like) were negative for both S100 and CD34. Pan-TRK was seen in all eight NTRK-fusion positive tumors tested but was negative in two tumors with other kinase fusions. Importantly, as was shown in a recent Dutch nationwide analysis of 69 cases including different cancer types, overall, IHC for Pan-TRK has limited sensitivity (around 80%) for NTRK1–3 fusion genes.27 For this reason, although Pan-TRK immunohistochemistry may be used as a screening tool for tumors with possible NTRK fusion genes, molecular tests are required if there is high suspicion based on histologic features (resembling FS or MPNST) and immunoprofiles (CD34 and/or S100). Given the wide variety of gene fusions encountered in these tumors, targeted RNA sequencing is preferred. In this cohort, clinical follow-up was too limited to allow any general conclusions. With respect to the whole group of mesenchymal tumors with kinase gene fusions, studies with larger cohorts would increase our understanding of prognostic factors that determine differences in outcome between low-grade and high-grade morphology, superficial and deep tumor localization, occurrence in children or adult patients, and gene fusion type. To date, the number of such studies is very limited. Historically, classic IFS with ETV6::NTRK3 is considered to be a locally aggressive and rarely metastasizing tumor.28 Notably, in a small series of childhood tumors, no statistical difference was seen in any clinicopathologic feature between the classic IFS with ETV6::NTRK3 and other pediatric NTRK-rearranged mesenchymal tumors29 In contrast, in a meta-analysis of 46 NTRK-rearranged uterine sarcomas, several adverse prognostic features were established, including a mitotic index ≥8 per 10 HPF, lymphovascular invasion, necrosis, and NTRK3 fusion. Patients with uterine tumors lacking any of these 4 features had an excellent prognosis.30 Although patients with BTKF may be eligible for tyrosine kinase inhibitor therapy,31 none of the patients in this series received such treatment, as most of these older diagnoses preceded the availability of tyrosine kinase inhibitors in clinical use.
Table 3.
Fusion events, clinicopathological and outcome data of 15 bone tumors with kinase fusion genes combining our cases with those retrieved from the literature
| Case | Reference | Age/sex | Location | Kinase gene | Fusion partner | Size (cm) | Histology | MI | Atypia | S100 | CD34 | Pan-TRK | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | #16 | newborn/F | lumbosacral | NTRK3 | ETV6 | 5 | IFS | 1 | N | pos | pos | pos | NA |
| 2 | #17 | newborn/M | sphenoid | NTRK3 | ETV6 | NA | IFS | NA | N | NA | NA | NA | NA |
| 3 | #7 | newborn/M | skull base | NTRK3 | ETV6 | 4.7 | IFS | 3 | N | neg | neg | pos | NA |
| 4 | #6 | 2mo/M | chest wall | RET | CLIP2 | NA | IFS-like | 5 | NA | neg | neg | NA | NED (114) |
| 5 | #4 | 4y/M | mandible | NTRK1 | LMNA | 4 | LG-MPNST-like | 5 | N | pos | pos | pos | NED (142) |
| 6 | #16 | 12y/M | pelvis | NTRK1 | TPM3 | 9.9 | LG-MPNST-like | 1 | N | pos | pos | pos | NA |
| 7 | #18 | 21y/F | femur | NTRK3 | HMBOX1 | >5 | LG-MPNST-like | 0 | N | pos | neg | pos | NA |
| 8 | #4 | 13 y/M | maxilla | NTRK1 | LMNA | 2.5 | LG-MPNST-like | 0 | N | pos | pos | pos | NED (648) |
| 9 | #19 | 60y/F | femur | RAF1 | MAP4 | 9.7 | HG-MPNST-like | 12 | N | pos | pos | neg | NA |
| 10 | #20 | 73y/F | metacarpal | BRAF | KIAA1549 | 4.9 | HG-MPNST-like | 4 | focal atypia | pos | pos | NA | NA |
| 11 | #21 | 38y/F | radius | NTRK3 | STRN | 8.7 | adult FS-like | 3 | N | neg | pos | NA | AWD (113) |
| 12 | #7 | 15y/M | skull base | RET | KIF5B | 1.3 | myxoma-like | 0 | N | neg | neg | neg | NA |
| 13 | recent case | 9y/F | pelvis/ilium | NTRK1 | TPM3 | 8.7 | HG-MPNST-like | >20 | N | pos | neg | pos | recent case |
| 14 | recent case | 14y/M | mastoid | BRAF | FNBP1 | NA | HG-adult FS-like | >10 | N - necrotic | neg | neg | NA | recent case |
| 15 | recent case | 59y/F | tibia | NTRK3 | UPF2 | 5.6 | HG-MPNST | >10 | Y | neg | neg | pos | recent case |
AWD, alive with disease; F, female; M, male; N, no; NA, not available; NED, no evidence of disease; neg, negative; pos, positive.
Given their rarity and variable clinicopathologic features in terms of age at presentation, bone location and uniform spindle cell morphology, the differential diagnosis of BTKF is broad and challenging. Primary spindle cell tumors of bone (without kinase gene fusions) entering the differential diagnosis includes both fibrous, osteoblastic, and neural neoplasms. Moreover, as primary bone occurrence of some of these tumor entities all are exceptionally rare, the clinical possibility of metastatic manifestations should be ruled out. In the jaw and skull bones, myxoma-like BTKF must be differentiated from myxoma of jaw bones and (myxoid) fibrous dysplasia.
Solitary fibrous tumor (SFT) of bone may strongly resemble low-grade MPNST-like BTKF as these entities represent monomorphic spindle cell tumors with haphazard growth, stromal collagen, perivascular hyalinization, hemangiopericytoma-like vessels, and expression of CD34.32 However, SFT may be differentiated by STAT6 IHC expression or demonstration of the characteristic NAB2::STAT6 fusion. Fibroblastic osteosarcoma may occasionally display a pure/predominantly spindle cell proliferation, but in a larger sample focal osteoid deposition and multifocal IHC expression of SATB2 can be detected. Other extremely rare skeletal presentations of soft tissue tumors, such as the monophasic variant of synovial sarcoma and classic MPNST, may be confused with high-grade variants of FS-like or MPNST-like BTKF.33–36 Although neurofibromatosis (NF1)-association is common in classic MPNST, rare cases of high-grade MPNST-like kinase fusion tumors have been reported recently to occur in the setting of NF1.16 However, in contrast to BTKF, classic MPNST often shows loss of the H3K27me3 expression 27, as shown in the unique case #7 included in this series, which represents a primary MPNST of bone and harboring UPF2::NTRK3 fusion in addition to TP53 H179L mutation, loss of CDKN2A/B, and loss of EED. The patient did not have clinical history or stigmata of NF1. It is tempting to speculate that the NTRK3 fusion detected in this case represents a late, acquired event in an otherwise phenotypically and genomically typical case of a high grade MPNST. This is further supported by the fact that most if not all NTRK-fusion positive high grade spindle cell neoplasms tested in our experience retain the H3K27me3 expression, which is lost in most high grade MPNST regardless of NF1 status.37
Myxoma of jaw bones and myxoma-like BTKF can only be differentiated by molecular studies, demonstrating the presence of a kinase fusion gene in the latter.7 Myxoid variants of fibrous dysplasia occurring in jaw or skull bones may sometimes lack areas with typical metaplastic ossification (irregular trabeculae of woven bone without rimming of mature osteoblasts) but can be diagnosed by GNAS mutation analysis.38
In conclusion, we reviewed and evaluated 15 BTKF, including 8 cases from our files. These tumors showed highly variable age at presentation and anatomic site distribution. Most tumors were seen in teenagers and young adults. Jaw and skull bones and long tubular bones were most commonly affected. Akin to mesenchymal tumors with kinase fusions in soft tissue locations, NTRK1 and NTRK3 were the most common fusion events, all of which were demonstrable by IHC for Pan-TRK in this series. Other fusion genes included RET, BRAF, and RAF1. Given the wide variety of possible gene rearrangements in BTKF, a targeted RNA sequencing technique is preferred for diagnostic purposes and selecting patients that are eligible for kinase inhibitor therapy.
Disclosures:
Supported in part by: P50 CA217694 (CRA), P30 CA008748, Cycle for Survival (CRA), Kristin Ann Carr Foundation (CRA)
REFERENCES
- 1.Knezevich SR, McFadden DE, Tao W, Lim JF, Sorensen PH. A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat Genet. 1998;18(2):184–187. [DOI] [PubMed] [Google Scholar]
- 2.Antonescu CR. Emerging soft tissue tumors with kinase fusions: An overview of the recent literature with an emphasis on diagnostic criteria. Genes Chromosomes Cancer. 2020;59(8):437–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Agaram NP, Zhang L, Sung YS, Chen CL, Chung CT, Antonescu CR, Fletcher CD. Recurrent NTRK1 Gene Fusions Define a Novel Subset of Locally Aggressive Lipofibromatosis-like Neural Tumors. Am J Surg Pathol. 2016;40(10):1407–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suurmeijer AJH, Dickson BC, Swanson D, et al. A novel group of spindle cell tumors defined by S100 and CD34 co-expression shows recurrent fusions involving RAF1, BRAF, and NTRK1/2 genes. Genes Chromosomes Cancer. 2018;57(12):611–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Suurmeijer AJ, Dickson BC, Swanson D, et al. The histologic spectrum of soft tissue spindle cell tumors with NTRK3 gene rearrangements. Genes Chromosomes Cancer. 2019;58(11):739–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Antonescu CR, Dickson BC, Swanson D, et al. Spindle Cell Tumors With RET Gene Fusions Exhibit a Morphologic Spectrum Akin to Tumors With NTRK Gene Fusions. Am J Surg Pathol. 2019;43(10):1384–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu B, Suurmeijer AJH, Agaram NP, Antonescu CR. Head and Neck Mesenchymal Tumors with Kinase Fusions: A Report of 15 Cases With Emphasis on Wide Anatomic Distribution and Diverse Histologic Appearance. Am J Surg Pathol. 2023;47(2):248–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kao YC, Suurmeijer AJH, Argani P, et al. Soft tissue tumors characterized by a wide spectrum of kinase fusions share a lipofibromatosis-like neural tumor pattern. Genes Chromosomes Cancer. 2020;59(10):575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Llamas-Velasco M, Itlinger-Monshi B, Flucke U, Mentzel T. Dermal lipofibromatosis-like neural tumor. Journal of cutaneous pathology. 2022;49(6):525–531. [DOI] [PubMed] [Google Scholar]
- 10.Surrey LF, Davis JL. NTRK-rearranged soft tissue neoplasms: a review of evolving diagnostic entities and algorithmic detection methods. Cancer Genet. 2022; 260–261:6–13 [DOI] [PubMed] [Google Scholar]
- 11.Chiang S, Cotzia P, Hyman DM, et al. NTRK Fusions Define a Novel Uterine Sarcoma Subtype With Features of Fibrosarcoma. Am J Surg Pathol. 2018;42(6):791–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Atiq MA, Davis JL, Hornick JL, et al. Mesenchymal tumors of the gastrointestinal tract with NTRK rearrangements: a clinicopathological, immunophenotypic, and molecular study of eight cases, emphasizing their distinction from gastrointestinal stromal tumor (GIST). Mod Pathol. 2021;34(1):95–103. [DOI] [PubMed] [Google Scholar]
- 13.Zheng Z, Liebers M, Zhelyazkova B, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nature medicine. 2014;20(12):1479–1484. [DOI] [PubMed] [Google Scholar]
- 14.Nacev BA, Sanchez-Vega F, Smith SA, et al. Clinical sequencing of soft tissue and bone sarcomas delineates diverse genomic landscapes and potential therapeutic targets. Nature communications. 2022;13(1):3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baranov E, Winsnes K, O’Brien M, et al. Histologic characterization of paediatric mesenchymal neoplasms treated with kinase-targeted therapy. Histopathology. 2022;81(2):215–227. [DOI] [PubMed] [Google Scholar]
- 16.Hiemcke-Jiwa LS, Meister MT, Martin E, et al. NTRK rearrangements in a subset of NF1-related malignant peripheral nerve sheath tumors as novel actionable target. Acta neuropathologica. 2023;145(1):149–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobayashi H, Makise N, Shinozaki-Ushiku A, et al. Dramatic response to entrectinib in a patient with malignant peripheral nerve sheath tumor harboring novel SNRNP70-NTRK3 fusion gene. Genes Chromosomes Cancer. 2023;62(1):47–51. [DOI] [PubMed] [Google Scholar]
- 18.Zhao M, Yin X, He H, et al. Recurrent RET fusions in fibrosarcoma-like neoplasms in adult viscera: expanding the clinicopatological and genetic spectrum. Histopathology. 2023;82(3):633–645. [DOI] [PubMed] [Google Scholar]
- 19.Nagasaka M, Brazel D, Baca Y, et al. Pan-tumor survey of RET fusions as detected by next-generation RNA sequencing identified RET fusion positive colorectal carcinoma as a unique molecular subset. Transl Oncol. 2023;36:101744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kervarrec T, Jean-Jacques B, Pissaloux D, Tirode F, de la Fouchardiere A. FNBP1-BRAF fusion in a primary melanoma of the lung. Pathology. 2021;53(6):785–788. [DOI] [PubMed] [Google Scholar]
- 21.Kao YC, Fletcher CDM, Alaggio R, et al. Recurrent BRAF Gene Fusions in a Subset of Pediatric Spindle Cell Sarcomas: Expanding the Genetic Spectrum of Tumors With Overlapping Features With Infantile Fibrosarcoma. Am J Surg Pathol. 2018;42(1):28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Penning AJ, Al-Ibraheemi A, Michal M, et al. Novel BRAF gene fusions and activating point mutations in spindle cell sarcomas with histologic overlap with infantile fibrosarcoma. Mod Pathol. 2021;34(8):1530–1540. [DOI] [PubMed] [Google Scholar]
- 23.Steelman C, Katzenstein H, Parham D, et al. Unusual presentation of congenital infantile fibrosarcoma in seven infants with molecular-genetic analysis. Fetal Pediatr Pathol. 2011;30(5):329–337. [DOI] [PubMed] [Google Scholar]
- 24.Wang Z, Wang J. Primary NTRK-rearranged spindle cell neoplasm of bone harboring an HMBOX1::NTRK3 gene fusion. Genes Chromosomes Cancer. 2023;62(8):477–482. [DOI] [PubMed] [Google Scholar]
- 25.Yeung MCF, Lam AYL, Shek TWH. Novel MAP4::RAF1 Fusion in a Primary Bone Sarcoma: Expanding the spectrum of RAF1 Fusion Sarcoma. International journal of surgical pathology. 2022;30(6):682–688. [DOI] [PubMed] [Google Scholar]
- 26.Yamazaki F, Nakatani F, Asano N, et al. Novel NTRK3 Fusions in Fibrosarcomas of Adults. Am J Surg Pathol. 2019;43(4):523–530. [DOI] [PubMed] [Google Scholar]
- 27.Koopman B, Kuijpers C, Groen HJM, et al. Detection of NTRK Fusions and TRK Expression and Performance of pan-TRK Immunohistochemistry in Routine Diagnostics: Results from a Nationwide Community-Based Cohort. Diagnostics (Basel). 2022;12(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chung EB, Enzinger FM. Infantile fibrosarcoma. Cancer. 1976;38(2):729–739. [DOI] [PubMed] [Google Scholar]
- 29.Davis JL, Lockwood CM, Stohr B, et al. Expanding the Spectrum of Pediatric NTRK-rearranged Mesenchymal Tumors. Am J Surg Pathol. 2019;43(4):435–445. [DOI] [PubMed] [Google Scholar]
- 30.Costigan DC, Nucci MR, Dickson BC, et al. NTRK-Rearranged Uterine Sarcomas: Clinicopathologic Features of 15 Cases, Literature Review, and Risk Stratification. Am J Surg Pathol. 2022;46(10):1415–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laetsch TW, DuBois SG, Mascarenhas L, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. The Lancet Oncology. 2018;19(5):705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Verbeke SL, Fletcher CD, Alberghini M, et al. A reappraisal of hemangiopericytoma of bone; analysis of cases reclassified as synovial sarcoma and solitary fibrous tumor of bone. Am J Surg Pathol. 2010;34(6):777–783. [DOI] [PubMed] [Google Scholar]
- 33.Sbaraglia M, Righi A, Gambarotti M, Vanel D, Picci P, Dei Tos AP. Soft Tissue Tumors Rarely Presenting Primary in Bone; Diagnostic Pitfalls. Surg Pathol Clin. 2017;10(3):705–730. [DOI] [PubMed] [Google Scholar]
- 34.Righi A, Gambarotti M, Benini S, et al. Primary synovial sarcoma of bone: a retrospective analysis of 25 patients. Histopathology. 2022;80(4):686–697. [DOI] [PubMed] [Google Scholar]
- 35.Gambarotti M, Righi A, Sbaraglia M, et al. Primary malignant peripheral nerve sheath tumors of bone: a clinicopathologic reappraisal of 8 cases. Hum Pathol. 2022;122:92–102. [DOI] [PubMed] [Google Scholar]
- 36.Antonescu CR, Erlandson RA, Huvos AG. Primary leiomyosarcoma of bone: a clinicopathologic, immunohistochemical, and ultrastructural study of 33 patients and a literature review. Am J Surg Pathol. 1997;21(11):1281–1294. [DOI] [PubMed] [Google Scholar]
- 37.Prieto-Granada CN, Wiesner T, Messina JL, Jungbluth AA, Chi P, Antonescu CR. Loss of H3K27me3 Expression Is a Highly Sensitive Marker for Sporadic and Radiation-induced MPNST. Am J Surg Pathol. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pereira T, Gomes CC, Brennan PA, Fonseca FP, Gomez RS. `Fibrous dysplasia of the jaws: Integrating molecular pathogenesis with clinical, radiological, and histopathological features. J Oral Pathol Med. 2019;48(1):3–9. [DOI] [PubMed] [Google Scholar]