

Key Points
-
•
Treatment with the oral CXCR4 antagonist mavorixafor resulted in increased levels of absolute neutrophil and lymphocyte counts vs placebo.
-
•
Infection frequency, severity, and duration were decreased with mavorixafor treatment vs placebo. Mavorixafor was well tolerated.
Visual Abstract
Abstract
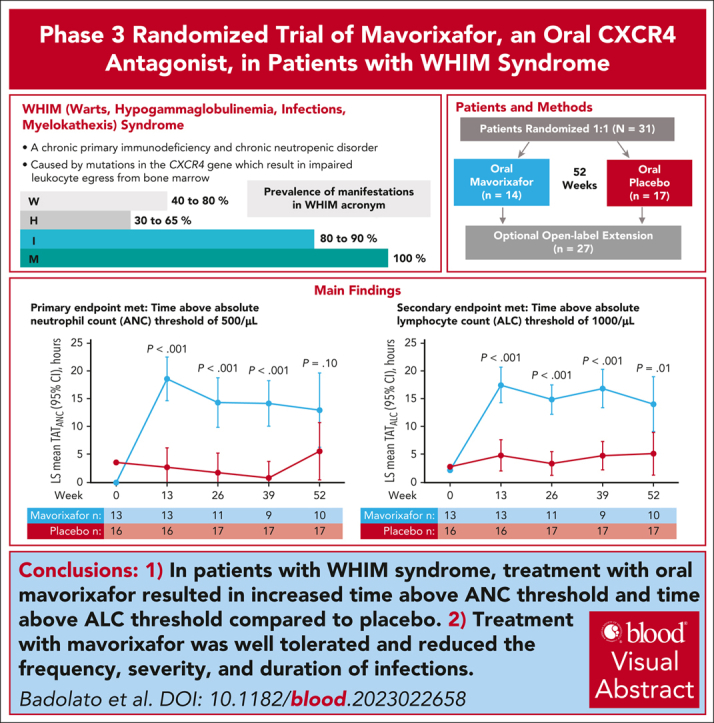
We investigated efficacy and safety of mavorixafor, an oral CXCR4 antagonist, in participants with warts, hypogammaglobulinemia, infections, and myelokathexis (WHIM) syndrome, a rare immunodeficiency caused by CXCR4 gain-of-function variants. This randomized (1:1), double-blind, placebo-controlled, phase 3 trial enrolled participants aged ≥12 years with WHIM syndrome and absolute neutrophil count (ANC) ≤0.4 × 103/μL. Participants received once-daily mavorixafor or placebo for 52 weeks. The primary end point was time (hours) above ANC threshold ≥0.5 × 103/μL (TATANC; over 24 hours). Secondary end points included TAT absolute lymphocyte count ≥1.0 × 103/μL (TATALC; over 24 hours); absolute changes in white blood cell (WBC), ANC, and absolute lymphocyte count (ALC) from baseline; annualized infection rate; infection duration; and total infection score (combined infection number/severity). In 31 participants (mavorixafor, n = 14; placebo, n = 17), mavorixafor least squares (LS) mean TATANC was 15.0 hours and 2.8 hours for placebo (P < .001). Mavorixafor LS mean TATALC was 15.8 hours and 4.6 hours for placebo (P < .001). Annualized infection rates were 60% lower with mavorixafor vs placebo (LS mean 1.7 vs 4.2; nominal P = .007), and total infection scores were 40% lower (7.4 [95% confidence interval [CI], 1.6-13.2] vs 12.3 [95% CI, 7.2-17.3]). Treatment with mavorixafor reduced infection frequency, severity, duration, and antibiotic use. No discontinuations occurred due to treatment-emergent adverse events (TEAEs); no related serious TEAEs were observed. Overall, mavorixafor treatment demonstrated significant increases in LS mean TATANC and TATALC, reduced infection frequency, severity/duration, and was well tolerated. The trial was registered at www.clinicaltrials.gov as #NCT03995108.
Warts, hypogammaglobulinemia, infections, and myelokathexis (WHIM) syndrome is a rare, autosomal-dominant immunodeficiency usually caused by gain-of-function variants in the CXCR4 gene that leads to impaired neutrophil and lymphocyte trafficking. Plerixafor, an injectable CXCR4 inhibitor, improves cytopenias and infections in patients with WHIM syndrome. Badolato et al present a phase 3 trial of mavorixafor, an oral CXCR4 inhibitor, in 31 patients, reporting increases in peripheral counts and decreased frequency, severity, and duration of infections.
Introduction
Warts, hypogammaglobulinemia, infections, and myelokathexis (WHIM) syndrome is a rare, autosomal-dominant immunodeficiency predominantly caused by gain-of-function variants in the CXCR4 gene that typically results in truncation of the carboxyl terminus of C-X-C chemokine receptor type 4 (CXCR4), leading to impaired leukocyte trafficking between bone marrow and blood.1, 2, 3, 4 Key clinical features include chronic and severe neutropenia, lymphopenia, monocytopenia, hypogammaglobulinemia, and myelokathexis.3, 4, 5, 6, 7, 8, 9 Infections are the most common clinical manifestation, with long-term recurrent complications such as bronchiectasis.3,8,9 Wart susceptibility, impaired humoral immunity, and malignancy are also often observed.3,7, 8, 9 There are no targeted approved treatments; current therapeutic options are primarily supportive, consisting of granulocyte colony-stimulating factor (G-CSF), immunoglobulin replacement therapy (IgRT), and antimicrobial prophylaxis, none of which target the full range of disease manifestations.3,7,10 Hematopoietic stem cell transplantation, although corrective, is associated with transplant-related complications and mortality risk.11
Improvement of clinical symptoms of WHIM syndrome as well as absolute neutrophil count (ANC) and absolute lymphocyte count (ALC) have been observed with plerixafor, an injectable CXCR4 inhibitor.12,13 In a proof-of-concept, open-label trial (n = 3), clinical symptoms and hematologic biomarkers of WHIM syndrome improved with plerixafor. In a phase 3 crossover trial, plerixafor was nonsuperior to G-CSF for total infection severity score (P = .54), noninferior to G-CSF in maintaining ANC above 0.5 × 103/μL (P = .023), and superior to G-CSF in maintaining ALC above 1.0 × 103/μL (P < .0001).12 Mavorixafor, an oral, small-molecule, selective CXCR4-antagonist, increased ANCs and ALCs and reduced annualized infection rates and the number of cutaneous warts in a phase 2 study (ClinicalTrials.gov NCT03005327).14 Here, we report the results of the first randomized, placebo-controlled phase 3 trial of a selective CXCR4 antagonist, mavorixafor, in participants with WHIM syndrome.
Methods and patients
Trial design and oversight
The trial included an initial 12-month (52-week) randomized, double-blind, placebo-controlled period followed by an open-label extension (OLE; ongoing) and was conducted at 20 sites in 12 countries in Australia, North America, Europe, and Asia. A list of investigators is provided in the supplemental Appendix, available on the Blood website. The trial complied with Good Clinical Practice, the Declaration of Helsinki, and other applicable regulatory requirements. An institutional review board or independent ethics committee at each site approved the protocol, protocol amendments, and informed consent documents. Participants and/or their parent(s)/guardian(s) gave written consent before trial initiation.
The trial sponsor, X4 Pharmaceuticals, Inc, along with collaborating physicians, designed the trial and were involved in the writing of this manuscript and the decision to submit it for publication. The trial (clinicaltrials.gov NCT03995108, Efficacy and safety study of mavorixafor in participants with warts, hypogammaglobulinemia, infections, and myelokathexis [WHIM] syndrome) was conducted and reported in accordance with the protocol and statistical analysis plan. Data were interpreted jointly with the authors. An independent data monitoring committee reviewed unblinded safety data throughout the trial. LLX Solutions performed all statistical analyses.
Participants
Key inclusion criteria included age ≥12 years, clinical diagnosis of WHIM syndrome with a genotype-confirmed CXCR4 variant, and ANC ≤0.4 × 103/μL (or total white blood cell [WBC] count ≤0.4 × 103/μL if ANC below the lower limit of detection) at screening. Exclusion criteria included clinically diagnosed active infection (excluding warts) and any of the following treatments within the specified time frame preceding trial start: plerixafor (≤6 months), chronic or prophylactic antibiotics (determined at the discretion of the investigator; ≤4 weeks), chronic or prophylactic G-CSF, granulocyte-macrophage colony-stimulating factor, or systemic glucocorticoid (≤2 weeks), or any investigational therapy (≤5 half-lives or 2 weeks, whichever was longer).
Randomization and intervention
A centralized randomization procedure assigned participants in a 1:1 ratio to receive oral mavorixafor 400 mg once daily for adults and adolescents >50 kg at screening, 200 mg once daily for adolescents ≤50 kg at screening, or matched placebo. Dosing was increased to 400 mg once daily in participants who reached age 18 years or >50 kg during the trial. Randomization was stratified according to whether participants received IgRT (starting ≤5 months before screening). The sponsor, study teams, investigators, and participants were blinded. Participants who completed the randomized period or were granted early release because of infections (supplemental Methods) were eligible to enroll in the OLE and receive mavorixafor per the dosing criteria until commercial availability or trial termination by the sponsor.
Participants in the prior-IgRT stratum continued their same IgRT regimen, avoiding IgRT administration ≤4 days before each visit. Participants in the no-prior-IgRT stratum could not receive IgRT during the trial. Permissible use of other symptom-treating therapies is outlined in the supplemental Methods. Participants were administered vaccines according to a predetermined schedule starting at week 13 (supplemental Methods).
End points and assessments
The primary end point in the randomized period was time above threshold ANC (TATANC), defined as time (hours) above ANC threshold ≥500 cells per μL over a 24-hour period, assessed every 3 months for 52 weeks. Key secondary efficacy end points in hierarchical order were time above threshold ALC (TATALC), defined similarly to TATANC but with ALC threshold ≥1.0 × 103/μL, a composite efficacy score calculated by summing the ranks of total infection score and total wart change score at 52 weeks, total wart change score at 52 weeks, and total infection score (further details provided in the protocol and supplemental Methods).15 Other selected secondary end points included annualized infection rate; tetanus vaccine titers; and mavorixafor pharmacokinetics, safety, and tolerability (supplemental Methods). Adverse events were graded using the Common Terminology Criteria for Adverse Events v5.0. Results from the OLE will be described in a separate publication.
Statistical analysis
A sample size of 18 participants (9 in each group) was based on estimates of the mean TATANC for mavorixafor (11.4 hours), and placebo (1.4 hours), and a pooled SD of 4.7 hours from the phase 2 trial.14 It was predicted to provide >90% power using a 2-sample t test on the mean TATANC over a 24-hour period, given the null hypothesis that the difference between the 2 treatment groups is equal to 0 with a 2-sided significance level (alpha) of 0.05. The univesafety population, comprising all participants randomly assigned to treatment who received ≥1 dose of study treatment (analyzed according to the treatment they actually received), was used for safety analyses. The intent-to-treat population, comprising all participants randomly assigned to treatment, was used for efficacy analyses. The pharmacokinetic population, comprising all participants who received ≥1 dose of study treatment and had ≥1 blood sample providing pharmacokinetic data, was used for pharmacokinetic analyses.
The primary end point (TATANC) was analyzed using mixed-model repeated measures, using TAT as a dependent variable, covariates (per protocol), and participant as the repeated random effect, and was reported as least squares (LS) mean. To control for multiplicity, a single primary end point and hierarchical approach to the analysis of key secondary end points were prespecified. Each end point was tested at the 2-sided alpha level of 0.05. Further information regarding statistical analyses is included in the supplemental Methods.
Institutional review board (IRB) approvals were obtained from the following: UnitingCare Health Human Research Ethics Committee, Queensland, Australia; Children's Health Queensland Hospital and Health Service Human Research Ethics Committee, Queensland, Australia; Ethikkommission der Medizinischen Universität Wien, Vienna, Austria; De Videnskabsetiske Komiteer for Region Midtjylland, Viborg, Denmark; Comité de Protection des Personnes Sud-Est II, Bron, France; Ethics Committee of HaEmek Medical Center, Afula, Israel; Comitato Etico di Brescia-ASST Spedali Civili, Brescia, Italy; Amsterdam UMC, Location AMC, Amsterdam, The Netherlands; Local EC at the Federal State Budgetary Educational Institution of Higher Education I. P. Pavlov First Saint Petersburg State Medical University of the Ministry of Health of the Russian Federation, St Petersburg, Russia; Seoul National University Hospital Institutional Review Board, Seoul, South Korea; CEIm del Hospital 12 de Octubre, Madrid, Spain; Central Research Ethics Committee, Manchester, United Kingdom; Johns Hopkins Medicine Office of Human Subjects Research Institutional Review Boards, Baltimore, MD; WCG IRB (Central IRB), Puyallup, WA; Duke University Health System Institutional Review Board, Durham, NC; University of Texas Southwestern Medical Center Institutional Review Board, Dallas, TX.
Results
Between 24 October 2019 and 9 September 2021, 35 participants were assessed for eligibility. In the intent-to-treat population, 31 participants meeting eligibility criteria were randomly assigned to receive either mavorixafor (n = 14) or placebo (n = 17) (supplemental Figure 1). Baseline characteristics between the 2 groups were balanced (Table 1). At the data cutoff on 11 November 2022, median follow-up times were 359 days (32-372 days) and 364 (336-427 days) for the mavorixafor and placebo groups, respectively. Relative dose intensity ([total actual dose/total planned dose]∗100) for adolescent participants ≤50 kg and adults/adolescents >50 kg was 96.5% (range, 96.5%-96.5%) and 99.6% (range, 82.7%-104.3%), respectively, with mavorixafor and 98.5% (range, 75.6%-100.0%) and 99.6% (range, 83.0%-104.8%) with placebo. Dose modifications (interruptions, reductions, delays, and holds) occurred in 3 (21.4%) and 5 (29.4%) participants receiving mavorixafor and placebo, respectively, with 75% of mavorixafor modifications occurring before week 26. Eleven (78.6%) and 17 (100%) participants in the mavorixafor and placebo groups, respectively, completed treatment in the randomized placebo-controlled period, with 1 (7.1%) participant receiving mavorixafor eligible for early release.
Table 1.
Demographics and clinical characteristics of participants at baseline (safety population)
| Characteristic | Mavorixafor (n = 14)∗ | Placebo (n = 17)† |
|---|---|---|
| Age, median (range), y | 17.5 (12-58) | 23 (13-72) |
| 12 to <18 y, no. (%) | 7 (50.0) | 8 (47.1) |
| ≥18 y, no. (%) | 7 (50.0) | 9 (52.9) |
| Sex, no. (%) | ||
| Female | 9 (64.3) | 9 (52.9) |
| Male | 5 (35.7) | 8 (47.1) |
| Race, no. (%) | ||
| Asian | 0 | 1 (5.9) |
| White | 13 (92.9) | 16 (94.1) |
| Other | 1 (7.1) | 0 |
| Body mass index, median (range), kg/m2 | 21.6 (17.9-30.5) | 22.0 (16.7-33.0) |
| IgRT use, no. (%) | 6 (42.9) | 8 (47.1) |
| Screening blood count, median (range), ×103/μL | ||
| ANC | 0.15 (0.04-0.39) | 0.2 (0-0.4) |
| ALC | 0.42 (0.26-1.07) | 0.52 (0.1-8.56) |
| AMC | 0.07 (0.03-0.39) | 0.1 (0-0.42) |
| WBC | 0.6 (0.30-1.80) | 0.8 (0.2-9.3) |
| Platelets | 182 (75-341) | 188 (18.0-260) |
| CXCR4 variant, no. (%) | ||
| C terminus‡ | 14 (100) | 17 (100) |
| Any infection in the 12 mo before trial,§ no. (%) | 6 (42.9) | 11 (64.7) |
| Time since WHIM syndrome diagnosis, median (range), y | 8.3 (1.0-24.6) | 8.5 (1.9-22.9) |
| Time since WHIM syndrome symptom,‖ median (range), y | 13.5 (8.6-58.6) | 16.6 (4.7-65.8) |
| Region, no. (%) | ||
| United States | 2 (14.3) | 4 (23.5) |
| Non-United States | 12 (85.7) | 13 (76.5) |
Percentages may not equal 100 because of rounding.
In the mavorixafor group, 1 participant received G-CSF on 2 separate occasions for a total of 10 days for neutropenia. One participant received G-CSF for 3 days for prophylaxis for a vaginal procedure, dexamethasone 4 mg IV once for the vaginal procedure, and oral hydrocortisone 15 mg/d as ongoing replacement therapy after surgical removal of pituitary adenoma. One participant received triamcinolone hexacetonide 20 mg infiltration twice for bilateral popliteal cyst and oral prednisone 50 mg for 16 days with an oral prednisone taper over 37 days for arthritis.
In the placebo group, 1 participant received G-CSF for 10 days for cellulitis. One participant had methylprednisolone 60 mg IV listed as a concomitant medication for rituximab prophylaxis for Evans syndrome with rituximab administered once on day 119.
All CXCR4 variants identified had been previously described as pathogenic.16
Infection history included any infections for the 12 months before dosing (including the screening period before the first dose) from all possible sources of medical records, regardless of the severity of the infection.
Time since WHIM syndrome symptom was defined as first dose date minus date of WHIM syndrome symptom divided by 365.25. Date of WHIM syndrome symptom was recorded per patient reported medical history.
Efficacy
Overall LS mean TATANC in the mavorixafor and placebo groups was 15.0 hours (95% confidence interval [CI], 11.2-18.9) and 2.8 hours (95% CI, 0.0-5.9) (P < .001; 5.3-fold increase), respectively (Table 2; Figure 1A). This increase was sustained and significantly higher with mavorixafor than placebo at weeks 13, 26, and 39. Overall LS mean TATALC in the mavorixafor and placebo groups was 15.8 hours (95% CI, 13.0-18.7) and 4.6 hours (95% CI, 2.2-6.9; P < .001; a 3.5-fold increase), respectively (Table 2; Figure 1B).
Table 2.
Primary, key secondary, and select secondary end points (ITT population)
| End point | Mavorixafor (n = 14) | Placebo (n = 17) | LS mean difference from placebo (SE), LS mean difference 95% CI | P value |
|---|---|---|---|---|
| Primary end point | ||||
| Overall TATANC,∗ LS mean (SE), h, 95% CI | 15.0 (1.9) 11.2-18.9 |
2.8 (1.5) 0-5.9 |
12.3 (2.5) 7.2-17.4 |
P < .001 |
| Key secondary end point | ||||
| Overall TATALC,† LS mean (SE), h, 95% CI | 15.8 (1.4) 13.0-18.7 |
4.6 (1.1) 2.2-6.9 |
11.3 (1.8) 7.5-15.0 |
P < .001 |
| Composite efficacy score at 52 wk, LS mean (SE), 95% CI | n = 11 26.7 (3.5) 19.9-33.5 |
n = 17 33.4 (2.8) 27.9-38.8 |
−6.6 (4.5) −15.5 to 2.2 |
P = .14‡ |
| Total wart change score at 52 wk (based on CGI-C), LS mean (SE), 95% CI | n = 11 −1.1 (0.7) −2.5 to 0.3 |
n = 17 −1.2 (0.5) −2.2 to −0.1 |
0.1 (0.9) −1.7 to 1.8 |
P = .94‡ |
| Total infection score, LS mean (SE), 95% CI | 7.4 (2.8) 1.6-13.2 |
12.3 (2.4) 7.2-17.3 |
−4.9 (3.7) −12.6 to 2.9 |
P = .21‡ |
| Select secondary end point | ||||
| Annualized infection rate, LS mean (SE) | 1.7 (0.5) | 4.2 (0.7) | 0.4 (0.2-0.8)§ | P = .007‡ |
| Infection severity based on CTCAE grade, n (%) | — | — | ||
| Grade 1 | 8 (57.1) | 8 (47.1) | — | — |
| Grade 2 | 2 (14.3) | 3 (17.6) | — | — |
| Grade 3 | 1 (7.1) | 4 (23.5) | — | — |
| Grade 4 | 0 | 1 (5.9) | — | — |
| Grade 5 | 0 | 0 | — | — |
| Proportion of participants with infections, n (%) | ||||
| ≥5 infections | 1 (7.1) | 5 (29.4) | — | P = .13‡ |
| <5 infections | 13 (92.9) | 12 (70.6) | — | |
| Infection duration, median (range), d | 8.5 (0-43) | 32.0 (8-134) | — | — |
| Overall vaccine titer (tetanus‖), LS mean (95% CI) | 1.3 (0.8-2.0) | 0.8 (0.5-1.2) | 1.6 (1.0-2.6)¶ | P = .039‡ |
CGI-C, Clinical Global Impression of Change; CTCAE, Common Terminology Criteria for Adverse Events; ITT, intent-to-treat.
TATANC of ≥0.5 × 103/μL over a 24-hour period, assessed every 3 months for 12 months.
TATALC of ≥1.0 × 103/μL over a 24-hour period assessed every 3 months for 12 months.
If the analysis for a specific end point was not statistically significant, results for subsequent end points were considered nominal.
Data reported as ratio (95% CI).
Eight participants receiving placebo and 10 participants receiving mavorixafor were vaccinated with tetanus vaccines at week 13. All participants in the ITT population were followed through week 52.
Data reported as LS mean ratio (95% CI).
Figure 1.

LS mean TATANC and TATALC. TATANC (A) and TATALC (B) vs time on treatment with mavorixafor vs placebo over 52 weeks (intent-to-treat [ITT] population). TATANC LS mean (95% CI) for mavorixafor vs placebo at weeks 13, 26, 39, and 52 were 18.6 hours (14.5-22.7) vs 2.8 (0-6.4), P < .001; 14.4 (9.7-19.0) vs 1.8 (0.0-5.5), P < .001; 14.2 (9.9-18.5) vs 0.8 (0.0-4.0), P < .001; and 13.0 (6.0-20.0) vs 5.6 (0.2-11.1), P = .10, respectively. TATALC LS mean (95% CI) for mavorixafor vs placebo at weeks 13, 26, 39, and 52 were 17.5 (14.1-20.8) vs 4.9 (1.9-7.8), P < .001; 14.9 (12.1-17.6) vs 3.4 (1.2-5.7), P < .001; 16.8 (13.2-20.4) vs 4.8 (2.1-7.5), P < .001; and 14.0 (8.9-19.2) vs 5.1 (1.1-9.2), P = .01, respectively. At week 52, 3 of 17 participants receiving placebo were given mavorixafor in advance of their week 52 assessments. One participant receiving mavorixafor did not take mavorixafor at week 52. Some samples collected were not measurable; 3 participants in the mavorixafor group discontinued mavorixafor treatment.
Mean ANC and ALC were increased above 0.5 × 103/μL and 1.0 × 103/μL thresholds, respectively, by week 13 in the mavorixafor group and remained consistently higher than the placebo group at time points assessed (Figure 2). Mean absolute monocyte count (AMC) and WBC counts were also increased at week 13 in the mavorixafor group and remained higher than the placebo group at time points assessed (Figure 2). Overall absolute and fold change from baseline for total ANC, ALC, AMC, and WBC counts were increased 2.8- to 3.3-fold with mavorixafor (supplemental Table 1).
Figure 2.

LS Mean ANC, ALC, AMC, and WBC counts from baseline (week 0) to 52 weeks (ITT population). (A) LS Mean ANC over post–dose dense sampling period. (B) LS Mean ALC over post–dose dense sampling period. (C) LS Mean AMC over post–dose dense sampling period. (D) LS Mean WBC counts over post–dose dense sampling period. All P values are nominal. At week 52, 3 of 17 participants receiving placebo were given mavorixafor in advance of their week 52 assessments as they entered the open-label portion of the trial. One participant receiving mavorixafor did not take mavorixafor at week 52. Some samples collected were not measurable; 3 participants in the mavorixafor group discontinued mavorixafor treatment.
LS mean composite efficacy score (total wart change and total infection score) in the mavorixafor and placebo groups was 26.7 (95% CI, 19.9-33.5) and 33.4 (95% CI, 27.9-38.8), respectively, an LS mean difference of −6.6 (95% CI, 15.5-2.2; P = .14; Table 2). When the 2 components of the composite efficacy score were evaluated separately, nonsignificant differences in total wart change score at 52 weeks were observed (Table 2; supplemental Table 2). Fewer participants receiving mavorixafor vs placebo developed warts at a new location at week 52 (21.4% vs 35.3%).
Reductions in total infection score in the mavorixafor vs placebo group (−4.9; 95% CI, −12.6 to 2.9; nominal P = .21) of ∼40% were observed but were not significant (Table 2). Reductions were evident after 3 months of mavorixafor and decreased by ≥80% vs placebo after 6 months (Figure 3A). Median infection rates in the 12 months before the trial in the mavorixafor and placebo groups were 0 (range, 0-4) and 1 (range, 0-3), respectively. The annualized infection rate was reduced by ∼60% in the mavorixafor vs placebo group (1.7 vs 4.2, respectively; 95% CI, 0.2-0.8; nominal P = .007; Table 2). After 6 months of treatment, mean annualized infection rates were reduced by 79% with mavorixafor vs placebo (Figure 3B) with penicillin use in 3 participants receiving mavorixafor vs 10 receiving placebo. In the mavorixafor and placebo groups, participants experienced infections including upper respiratory (7 [50.0%] and 13 [76.5%]), skin (2 [14.3%] and 7 [41.2%]), lower respiratory (0 and 3 [17.6%]), or digestive (2 [14.3%] and 2 (11.8%]), respectively (supplemental Table 3). Five participants in the placebo group and 1 in the mavorixafor group had grade ≥3 infections. The mean annualized infection rate for grade ≥3 infections in the mavorixafor group was 0.4 (95% CI, −0.5 to 1.3) for months ≤3 and then remained at 0 for other time points assessed. In the placebo group, the mean annualized infection rate for grade ≥3 infections was 0.7 (95% CI, −0.80 to 2.2) and 0.2 (95% CI, −0.3 to 0.7) for months ≤3 and 3 to ≤6, respectively, and was consistent at 0.5 (95% CI, −0.2 to 1.2) and 0.5 (95% CI, −0.5 to 1.5) for months 6 to ≤9 and 9 to ≤12, respectively (Figure 3C). The median total duration of infection in the mavorixafor and placebo groups was 8.5 days (range, 0-43 days) and 32 days (8-134 days), respectively, exceeding 10 days in 6 (42.9%) and 16 (94.1%) participants receiving mavorixafor and placebo, respectively. Nine participants (64.3%) receiving mavorixafor experienced ≤1 infection, with only 1 participant (7.1%) experiencing ≥5 infections (Figure 3D). In comparison, 2 (11.8%) participants receiving placebo experienced ≤1 infection, and 5 (29.4%) experienced ≥5 infections.
Figure 3.

Infection parameters (ITT population). (A) Total infection score (mean, 95% CI) by 3-month interval with mavorixafor vs placebo. (B) Annualized infection rate (mean, 95% CI) by 3-month interval with mavorixafor vs placebo. (C) Annualized infection rate (mean, 95% CI) with CTCAE grade ≥3 by 3-month interval with mavorixafor vs placebo. (D) Proportion of participants experiencing infection events. CTCAE, Common Terminology Criteria for Adverse Events.
Overall, tetanus titers were numerically higher in participants receiving mavorixafor than placebo. Analyses of vaccine titers are ongoing and will be presented in a separate manuscript.
Overall, quality of life was not different between participants receiving mavorixafor or placebo, as assessed by Patient Global Impression of Change, Patient Global Impression of Severity, the Short-Form 36-Item Survey, the EuroQol 5-Dimension, 5-Level Visual Analogue score, and the Dermatology Life Quality Index (supplemental Table 4). Small numbers of adolescent participants (mavorixafor, n = 7; placebo, n = 8) included in the study and who completed the Pediatric Quality of Life Inventory Teens surveys or parent survey precluded conclusions based on these survey results (supplemental Table 4).
Other secondary end points results are provided in supplemental Results, supplemental Figure 2, and supplemental Tables 1-6.
Safety
No treatment-emergent adverse events (TEAEs) led to treatment discontinuations or deaths; there were no treatment-limiting toxicities. Treatment-emergent serious adverse events occurred in 5 (35.7%) and 2 (11.8%) participants receiving mavorixafor and placebo, respectively (Table 3), of which none were deemed related to treatment. Seven (50%) participants receiving mavorixafor experienced treatment-related TEAEs compared with 3 (17.6%) receiving placebo. Treatment-related TEAEs were reported in ≤1 participant each in both treatment groups with gastrointestinal (eg, dyspepsia, nausea, and vomiting) and skin disorders (eg, dry skin, rash, dermatitis psoriasiform, and pruritus) occurring with mavorixafor vs infections (eg, cellulitis, conjunctivitis, localized infection, skin infection, subcutaneous abscess, and lower and upper respiratory infections) occurring with placebo (Table 3).
Table 3.
Summary of AEs (safety population)
| Mavorixafor (n = 14) | Placebo (n = 17) | |||
|---|---|---|---|---|
| Participants with any TEAE, no. (%) | 14 (100) | 17 (100) | ||
| Participants with treatment-related TEAEs, no. (%) | 7 (50) | 3 (17.6) | ||
| Participants with any TESAE∗,†,‡, no. (%) | 5 (35.7) | 2 (11.8) | ||
| Participants with treatment-related TESAE, no. (%) | 0 | 0 | ||
| TESAEs†,‡, no. (%) | ||||
| COVID-19 | 1 (7.1) | 0 | ||
| Campylobacter gastroenteritis | 1 (7.1) | 0 | ||
| Cellulitis | 0 | 1 (5.9) | ||
| Endocarditis | 1 (7.1) | 0 | ||
| Sepsis | 1 (7.1) | 0 | ||
| Thrombocytopenia | 2 (14.3) | 0 | ||
| Febrile neutropenia | 1 (7.1) | 0 | ||
| Lipase increased | 1 (7.1) | 0 | ||
| Platelet count decreased | 1 (7.1) | 0 | ||
| Malignant glioma | 1 (7.1) | 0 | ||
| Pneumonitis | 0 | 1 (5.9) | ||
| TEAE or treatment-related TEAE leading to discontinuation, no. (%) | 0 | 0 | ||
| TEAE or treatment-related TEAE leading to death, no. (%) | 0 | 0 | ||
| Treatment-limiting toxicity, no. (%) | 0 | 0 | ||
| TEAEs reported in ≥2 participants in either group,∗,†no. (%) | Any grade | Grade ≥3 | Any grade | Grade ≥3 |
| COVID-19 | 4 (28.6) | — | 5 (29.4) | — |
| Upper respiratory tract infection | 3 (21.4) | 1 (7.1) | 6 (35.3) | 0 |
| Thrombocytopenia | 3 (21.4) | 1 (7.1) | 0 | 0 |
| Dizziness | 2 (14.3) | — | 1 (5.9) | — |
| Epistaxis | 2 (14.3) | — | 1 (5.9) | — |
| Pityriasis | 2 (14.3) | — | 0 | — |
| Rhinitis | 2 (14.3) | — | 0 | — |
| Rash | 2 (14.3) | — | 0 | — |
| Vomiting | 2 (14.3) | — | 0 | — |
| Bronchitis | 1 (7.1) | — | 4 (23.5) | — |
| Cellulitis | 1 (7.1) | 0 | 3 (17.6) | 2 (11.8) |
| Headache | 1 (7.1) | — | 2 (11.8) | — |
| Nasopharyngitis | 1 (7.1) | 0 | 7 (41.2) | 1 (5.9) |
| Urinary tract infection | 1 (7.1) | — | 2 (11.8) | — |
| Conjunctivitis | 0 | — | 3 (17.6) | — |
| Acarodermatitis | 0 | — | 2 (11.8) | — |
| Ear infection | 0 | 0 | 2 (11.8) | 1 (5.9) |
| Ear pain | 0 | — | 2 (11.8) | — |
| Lower respiratory tract infection | 0 | 0 | 3 (17.6) | 1 (5.9) |
| Sinusitis | 0 | 0 | 2 (11.8) | 1 (5.9) |
| Skin infection | 0 | 0 | 2 (11.8) | 1 (5.9) |
| Skin laceration | 0 | — | 2 (11.8) | — |
| Tinea versicolor | 0 | — | 2 (11.8) | — |
| Treatment-related TEAEs,∗,†no. (%) | ||||
| Acute kidney injury | 1 (7.1) | — | 0 | — |
| Dermatitis psoriasiform | 1 (7.1) | — | 0 | — |
| Dizziness | 1 (7.1) | — | 0 | — |
| Dry eye | 1 (7.1) | — | 0 | — |
| Dry skin | 1 (7.1) | — | 0 | — |
| Dysgeusia | 1 (7.1) | — | 0 | — |
| Dyspepsia | 1 (7.1) | — | 0 | — |
| Nausea | 1 (7.1) | — | 0 | — |
| Product aftertaste | 1 (7.1) | — | 0 | — |
| Pruritus | 1 (7.1) | — | 0 | — |
| Rash | 1 (7.1) | — | 0 | — |
| Syncope | 1 (7.1) | 1 (7.1) | 0 | 0 |
| Vomiting | 1 (7.1) | — | 0 | — |
| Cellulitis | 0 | — | 1 (5.9) | — |
| Conjunctivitis | 0 | — | 1 (5.9) | — |
| Headache | 0 | — | 1 (5.9) | — |
| Localized infection | 0 | — | 1 (5.9) | — |
| Lower respiratory tract infection | 0 | — | 1 (5.9) | — |
| Skin infection | 0 | — | 1 (5.9) | — |
| Subcutaneous abscess | 0 | — | 1 (5.9) | — |
| Tonsillitis | 0 | — | 1 (5.9) | — |
| Upper respiratory tract infection | 0 | — | 1 (5.9) | — |
AE, adverse event; COVID-19, coronavirus disease 2019; TESAE, treatment-emergent serious adverse event.
Participants may have experienced ≥2 different AEs.
Preferred term.
TESAEs were reported in 5 participants receiving mavorixafor: 1 participant (thrombocytopenia, grade 2; febrile neutropenia, grade 3); 1 participant (Campylobacter gastroenteritis, grade 2; thrombocytopenia, grade 4; sepsis, grade 4; endocarditis, grade 3); 1 participant (platelet count decrease, grade 3); 1 participant (lipase increased, grade 4; COVID-19, grade 1); and 1 participant (malignant glioma, grade 4). TESAEs were reported in 2 participants receiving placebo: 1 participant (cellulitis, grade 3) and 1 participant (pneumonitis, grade 4).
Neoplasms were reported in 2 participants in the mavorixafor group (1 treatment-emergent serious adverse event of glioma; 1 nonserious TEAE of stage 0 vaginal cancer), both of which were deemed unrelated to mavorixafor. Thrombocytopenia was reported in 3 participants receiving mavorixafor and was deemed unrelated to mavorixafor. There was no evidence that mavorixafor lowered platelets over the course of the study (supplemental Table 6). In fact, in all participants, platelet counts increased from baseline to week 52 by a mean of 34 × 103/μL (standard deviation, 75.7) in the mavorixafor group and decreased by a mean of 1.1 × 103/μL (standard deviation, 63.0) in the placebo group. One participant receiving mavorixafor was transfused with platelets for grade 4 thrombocytopenia (platelet count 17 × 103/μL), unrelated to mavorixafor but concurrent with sepsis from a gastrointestinal infection. Grade 4 thrombocytopenia and sepsis resolved in 4 days, and the participant continued on mavorixafor and subsequently entered the OLE.
Both local and central ophthalmologic examinations showed no shift from baseline values to abnormal/clinically significant values in both groups. Eye disorders were reported in 2 (14.3%) participants receiving mavorixafor and 3 (17.6%) receiving placebo. Dry eye in 1 participant was considered related to mavorixafor; no ocular TEAEs in the placebo group were deemed treatment-related.
Discussion
To our knowledge, this is the first randomized, placebo-controlled, multinational trial in participants with WHIM syndrome. The trial met its primary end point with a significantly increased (5.5-fold) LS mean TATANC and its first key secondary end point with a significantly increased (3.4-fold) LS mean TATALC in the mavorixafor vs placebo group. Mavorixafor was generally well tolerated with no discontinuations due to safety reasons, and pharmacokinetic parameters were stable over the 52 weeks.
Decreases in total infection score and annualized infection rate in the mavorixafor group were seen as early as 3 months and were greater at 6 to 12 months. No participant experienced infections ≥ grade 3 after receiving mavorixafor for 3 months. These results suggest early benefits regarding infection prevention and control that have continued to improve over time. This is important, as people with WHIM syndrome experience recurrent severe infections, which can affect quality of life and increase morbidity; thus, agents offering long-term protection are needed.3,7,10
Despite no difference in wart change score, minor improvements in wart change scores were observed in both the mavorixafor and placebo groups. A previous phase 2 trial of mavorixafor showed an average 75% reduction in the number of warts observed up to 18 months on trial.14 In our trial, wart change up to 52 weeks was assessed by a central review using photographs of targeted regions, which may not have permitted enough time for significant reduction in wart change score or to monitor the effect of treatment on improvement of newly emerged warts. Therefore, longer observation times and an alternative form of wart assessment may be needed. There was no assessment by this committee of anogenital warts. Data on wart reduction and new wart formation over longer treatment periods are being collected in the OLE.
Treatment-related vomiting, dyspepsia, and nausea (1 participant each) occurred in participants receiving mavorixafor. No treatment-related ocular issues or retinal abnormalities were reported with mavorixafor except dry eye in 1 participant.14 WHIM syndrome is a cancer-prone disease, with ∼30% of people experiencing malignancy by age 40 years.7 The glioma in 1 participant was deemed by study investigator unrelated to mavorixafor. Mavorixafor has not been studied in any glioma models or in people with glioma.
Limitations of this study include a small participant number given the rarity of the genetic disorder. Despite this, the primary and first key secondary end points showed significant improvement. Five participants (placebo, n = 2; mavorixafor, n = 3) received concomitant medications that may have influenced outcomes. However, the inclusion of these participants provides real-world results and highlights the complexity of patients with WHIM syndrome. In addition, >90% of participants in this trial self-identified as White, and results should be extrapolated with caution to different races. Because the first assessment was not made until 3 months, it is unknown whether clinically important changes in WBC, ANC, ALC, and AMC occurred earlier. Furthermore, because no acute assessments were made at the time of infections, it is unclear how levels of these indices (eg, ANC, ALC, AMC, and WBC) may have changed at the onset of or during an infection. In this trial, no differences in quality of life were seen between the 2 groups using the Patient Global Impression of Change, Patient Global Impression of Severity, the Short-Form 36-Item Survey, the EuroQol 5-Dimension, 5-Level Visual Analogue score, and the Dermatology Life Quality Index. However, these instruments have not been validated in patients with WHIM syndrome. In addition, it is unclear how soon after immune reconstitution the effects of WHIM syndrome are expected to improve, which may require prolonged evaluation of patients with continued treatment.
Individuals with WHIM syndrome have an estimated median life expectancy of ∼55 years, and therapies that may be protective should be evaluated not only for their short-term effect but also from a long-term perspective.3,7 There is an unmet need in the treatment of WHIM syndrome to prevent immunodeficiency-associated morbidity and mortality. Current therapies like G-CSF and IgRT do not address the underlying molecular defect in WHIM syndrome and cannot be administered orally. Data from this trial suggest daily, oral administration of mavorixafor is effective, safe, and well tolerated, with improvements in cytopenias and reductions in serious infections.
Conflict-of-interest disclosure: R.B. is a consultant for X4 Pharmaceuticals, Inc, Angelini Pharma, and Janssen. L.A. has received research funding (to Institu de Recerca Sant Joan de Déu) from CSL Behring, Pharming, and Grifols and is a speaker for Novartis, Sanofi, Roche, and UCB Pharmaceuticals. A.A. has received research funding/grants from X4 Pharmaceuticals, Inc, Grifols, and Argenx; and is a consultant for Grifols, Argenx, Takeda Pharmaceuticals, Adma Biologics, Inc, and Octapharma. A.A.B. has received research funding from X4 Pharmaceuticals, Inc, and the National Institutes of Health. D.D. has consulted, received research funding from, and received honoraria from X4 Pharmaceuticals, Inc. K.E.D. is on the advisory board of Agios. H.J.K. receives research funding from Amgen and is a member on the board of directors or advisory committees for Amgen, Novartis, GPCR Therapeutics, and Cartexell. A.K. has received research funding (to Pavlov University) from X4 Pharmaceuticals, Inc, Alexion, and Apellis and is a speaker for Novartis, Generium, Sobi, AstraZeneca, and Johnson & Johnson. D.L. is a board member for RCPA. C.L. receives research grants from Emek Center Pediatric Hematology University Hospital. J.P. is on the advisory board of Allergy & Anaphylaxis Australia, Food and Allergy Standards Australia and New Zealand, and National Blood Authority and is the director of the Australasian Society of Clinical Immunology and Allergy (QPIAS). A.S. is a speaker for Sobi, Novartis, and Octapharma. T.K.T. is a consultant for X4 Pharmaceuticals, Inc and also receives research funding from X4 Pharmaceuticals, Inc, AbbVie, Inc, Viela Bio, Horizon, and Chiesi. M.G.V. has received research funding outside the current work from Austrian National Bank, a grant from Pfizer, and honoraria from Gilead, Astro Pharma, and Menarini. J.D. is a consultant for X4 Pharmaceuticals, Inc. A.B., K.C., S.D., Y.H., H.J., S.L., R.M., T.Y., and A.G.T. are current employees and/or have equity ownership in X4 Pharmaceuticals, Inc. M.S. was employed by X4 Pharmaceuticals, Inc, at the time of this work, has equity ownership in X4 Pharmaceuticals, Inc, and is a member of the board of directors of X4 Pharmaceuticals, Inc. G.J.B. is a member of the board of directors of X4 Pharmaceuticals, Inc, and has stock options in the company. The remaining authors declare no competing financial interests.
Acknowledgments
The authors thank the trial participants and their families and caregivers, investigators, and investigational site staff. The authors also acknowledge Atil Bisgin, Diego Cadavid, Eloisa Chapa, Sarah Cohen, Elisa Cordero, Candida Fratazzi, Ken Gorelick, Joanna Haas, Hal Hoffman, Lori Neri, Paula Ragan, Anjali Sharathkumar, Felipe Suarez, Weihua Tang, Valerie Tjon-a-Koy; Istvan Varkonyi, the LLX Solutions Team; and Syneos Health. Members of the data monitoring committee included Charles Davis, Eric Gershwin, and John Levine. Members of the independent adjudication committee included Esther de Vries, Kathryn Edwards, and Craig Platt.
The trial was supported by X4 Pharmaceuticals, Inc. Editorial and writing assistance was provided by PRECISIONscientia in Yardley, PA, with financial support from X4 Pharmaceuticals, Inc, and in compliance with international good publication practice guidelines.
Authorship
Contribution: R.B., D.D., T.K.T., G.J.B., Y.H., H.J., R.M., and J.D. contributed to the concept and design of the study; R.B., L.A., A.A., Y.B., A.A.B., D.D., À.D.-M., K.E.D., N.E., H.H., H.J.K., S.K.-A., T.W.K., A.K., D.L., C.L., O.N., P.O., Y.R., C.E.R., A.S., M.G.V., S.D., Y.H., H.J., S.L., T.Y., and J.D. provided data acquisition; N.E., T.W.K., D.L., T.K.T., K.C., S.D., Y.H., H.J., S.L., R.M., M.S., A.G.T., and T.Y. performed data analysis; R.B., L.A., D.D., À.D.-M., K.E.D., N.E., H.H., H.J.K., S.K.-A., T.W.K., A.K., D.L., C.L., O.N., P.O., J.P., Y.R., A.S., T.K.T., C.A.W., K.C., S.D., Y.H., H.J., S.L., R.M., M.S., A.G.T., T.Y., and J.D. provided data interpretation; and R.B., L.A., A.A., Y.B., A.A.B., D.D., À.D.-M., K.E.D., N.E., H.H., H.J.K., S.K.-A., T.W.K., A.K., D.L., C.L., O.N., P.O., J.P., Y.R., C.E.R., A.S., T.K.T., M.G.V., C.A.W., A.B., G.J.B., K.C., S.D., Y.H., H.J., R.M., M.S., A.G.T., and J.D. were involved in drafting and revising the manuscript.
Footnotes
X4 Pharmaceuticals, Inc, will share the redacted trial protocol in the publication supplement. Qualified scientific and medical researchers may make requests for individual participant data that underlie the results (text, tables, figures, and appendixes) reported in this article, after deidentification, at medicalinfo@x4pharma.com. Methodologically sound proposals for such data will be evaluated and approved by X4 Pharmaceuticals, Inc, in its sole discretion. All approved researchers must sign a data access agreement before accessing the data. Data will be available as soon as possible but no later than within 1 year of the acceptance of the article for publication, and for 3 years after article publication. X4 Pharmaceuticals, Inc, will not share identified participant data or a data dictionary.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Supplementary Material
References
- 1.Balabanian K, Lagane B, Pablos JL, et al. WHIM syndromes with different genetic anomalies are accounted for by impaired CXCR4 desensitization to CXCL12. Blood. 2005;105(6):2449–2457. doi: 10.1182/blood-2004-06-2289. [DOI] [PubMed] [Google Scholar]
- 2.Hernandez PA, Gorlin RJ, Lukens JN, et al. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat Genet. 2003;34(1):70–74. doi: 10.1038/ng1149. [DOI] [PubMed] [Google Scholar]
- 3.Heusinkveld LE, Majumdar S, Gao JL, McDermott DH, Murphy PM. WHIM syndrome: from pathogenesis towards personalized medicine and cure. J Clin Immunol. 2019;39(6):532–556. doi: 10.1007/s10875-019-00665-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roselli G, Martini E, Lougaris V, Badolato R, Viola A, Kallikourdis M. CXCL12 mediates aberrant costimulation of B lymphocytes in warts, hypogammaglobulinemia, infections, myelokathexis immunodeficiency. Front Immunol. 2017;8:1068. doi: 10.3389/fimmu.2017.01068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wetzler M, Talpaz M, Kleinerman ES, et al. A new familial immunodeficiency disorder characterized by severe neutropenia, a defective marrow release mechanism, and hypogammaglobulinemia. Am J Med. 1990;89(5):663–672. doi: 10.1016/0002-9343(90)90187-i. [DOI] [PubMed] [Google Scholar]
- 6.Gorlin RJ, Gelb B, Diaz GA, Lofsness KG, Pittelkow MR, Fenyk Jr., JR. WHIM syndrome, an autosomal dominant disorder: clinical, hematological, and molecular studies. Am J Med Genet. 2000;91(5):368–376. [PubMed] [Google Scholar]
- 7.Beaussant Cohen S, Fenneteau O, Plouvier E, et al. Description and outcome of a cohort of 8 patients with WHIM syndrome from the French Severe Chronic Neutropenia Registry. Orphanet J Rare Dis. 2012;7:71. doi: 10.1186/1750-1172-7-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dotta L, Notarangelo LD, Moratto D, et al. Long-term outcome of WHIM syndrome in 18 patients: high risk of lung disease and HPV-related malignancies. J Allergy Clin Immunol Pract. 2019;7(5):1568–1577. doi: 10.1016/j.jaip.2019.01.045. [DOI] [PubMed] [Google Scholar]
- 9.Geier CB, Ellison M, Cruz R, et al. Disease progression of WHIM syndrome in an international cohort of 66 pediatric and adult patients. J Clin Immunol. 2022;42(8):1748–1765. doi: 10.1007/s10875-022-01312-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Badolato R, Donadieu J, WHIM Research Group How I treat warts, hypogammaglobulinemia, infections, and myelokathexis syndrome. Blood. 2017;130(23):2491–2498. doi: 10.1182/blood-2017-02-708552. [DOI] [PubMed] [Google Scholar]
- 11.Laberko A, Deordieva E, Krivan G, et al. Multicenter experience of hematopoietic stem cell transplantation in WHIM syndrome. J Clin Immunol. 2022;42(1):171–182. doi: 10.1007/s10875-021-01155-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McDermott DH, Velez D, Cho E, et al. A phase III randomized crossover trial of plerixafor versus G-CSF for treatment of WHIM syndrome. J Clin Invest. 2023;133(19) doi: 10.1172/JCI164918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McDermott DH, Pastrana DV, Calvo KR, et al. Plerixafor for the treatment of WHIM syndrome. N Engl J Med. 2019;380(2):163–170. doi: 10.1056/NEJMoa1808575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dale DC, Firkin F, Bolyard AA, et al. Results of a phase 2 trial of an oral CXCR4 antagonist, mavorixafor, for treatment of WHIM syndrome. Blood. 2020;136(26):2994–3003. doi: 10.1182/blood.2020007197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O'Brien PC. Procedures for comparing samples with multiple endpoints. Biometrics. 1984;40(4):1079–1087. [PubMed] [Google Scholar]
- 16.Zmajkovicova K, Nykamp K, Badarau A, et al. 8-10 November 2023. Expanding C-X-C chemokine receptor 4 variant landscape in warts, hypogammaglobulinemia, infections, myelokathexis (WHIM) syndrome: integrating clinical and functional data for variant interpretation. Paper presented at: International Primary Immunodeficiencies Congress (IPIC 2023) Rotterdam, The Netherlands. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.