

Abstract
Background:
The etiology of lung cancer among individuals who never smoked remains elusive, despite 15% of lung cancer cases in men and 53% in women worldwide being unrelated to smoking. Epigenetic alterations, particularly DNA methylation (DNAm) changes, have emerged as potential drivers. Yet, few prospective epigenome-wide association studies (EWAS), primarily focusing on peripheral blood DNAm with limited representation of never-smokers, have been conducted.
Methods:
We conducted a nested case-control study of 80 never-smoking incident lung cancer cases and 83 never-smoking controls within the Shanghai Women’s Health Study and Shanghai Men’s Health Study. DNAm was measured in pre-diagnostic oral rinse samples using Illumina MethylationEPIC array. Initially, we conducted an EWAS to identify differentially methylated positions (DMPs) associated with lung cancer in the discovery sample of 101 subjects. The top-50 DMPs were further evaluated in a replication sample of 62 subjects, and results were pooled using fixed-effect meta-analysis.
Results:
Our study identified three DMPs significantly associated with lung cancer at the genome-wide significance level of p<8.22×10−8. These DMPs were identified as cg09198866 (MYH9;TXN2), cg01411366 (SLC9A10), and cg12787323. Furthermore, examination of the top 1000 DMPs indicated significant enrichment in epithelial regulatory regions and their involvement in small GTPase-mediated signal transduction pathways. Additionally, GrimAge acceleration was identified as a risk factor for lung cancer (OR=1.19 per year; 95%CI: 1.06–1.34).
Conclusions:
While replication in a larger sample size is necessary, our findings suggest that DNAm patterns in pre-diagnostic oral rinse samples could provide novel insights into the underlying mechanisms of lung cancer in never-smokers.
Keywords: Lung cancer, Never-smokers, DNA methylation, Oral rinse samples, Epigenetic clock, GrimAge
INTRODUCTION
Lung cancer is the second most common cancer and the leading cause of cancer deaths globally, accounting for an estimated 2.21 million new cases and 1.80 million deaths in 2020 [1]. Although smoking is a well established risk factor, a substantial proportion of cases (15% in men and 53% in women worldwide) occurs in non-smokers [2]. Notably, the rate of lung cancer among never-smokers is the highest among Asian women [3], a trend mirrored in ethnically Chinese women in the United States [4]. For instance, 57% of never-smoking women with lung cancer in the United States are of Asian or Pacific Islander descent [5]. The etiology of lung cancer in never-smokers is complex, involving environmental, occupational, lifestyle, and genetic factors. Despite ongoing research, a complete understanding of the precise mechanisms behind lung cancer in never smokers remains elusive.
Factors unrelated to smoking, notably exposures to fine particulate matter (PM2.5) [6, 7] and secondhand tobacco smoke, share common carcinogenic constituents with tobacco smoke [7] and are associated with lung cancer among never-smokers [8]. Epigenetic modifications, particularly changes in DNA methylation (DNAm), have emerged as potential markers to understand the cumulative effect of non-smoking environmental exposures in lung cancer development in never-smokers. For example, AHRR gene hypomethylation, typically associated with smoking, is observed in never-smokers exposed to PM2.5 and secondhand smoke [9, 10]. Additionally, exposure to benzo(a)pyrene (B(a)P), a potent carcinogenic polycyclic aromatic hydrocarbon (PAH) derived from smoking and household coal and biomass combustion, is associated with altered blood DNAm patterns and lung cancer [11].
A few prospective studies have linked DNAm in pre-diagnostic samples, identifying CpG sites, most notably within the AHRR gene, that were associated with lung cancer [12, 13]. However, these studies included a limited number of never smokers, and none of the CpG sites achieved genome-wide significance within this subgroup [12, 14, 15]. Additionally, these studies exclusively relied on DNAm measurements in peripheral blood leukocytes, which are histologically different from the cells that give rise to lung cancer. Given that most lung cancers develop from epithelial cells lining the airways, DNAm in peripheral blood connective tissue cells may not adequately capture the underlying molecular changes directly related to lung cancer development.
The current study examines DNAm in oral rinse samples, encompassing a mixture of epithelial and other upper airway cell types directly exposed to air pollution. This non-invasive and easily collectable sample provides a valuable medium for identifying potential etiologic markers of diseases originating from epithelial tissues, such as lung cancer. We conducted this investigation within a never-smoking population from two prospective cohorts in Shanghai, making it the largest prospective study on DNAm and lung cancer to date among never-smokers. This approach minimizes the possibility of residual confounding from smoking and aids in identifying DNAm signatures that might be unique to this population. The aim of this study was to identify DNAm biomarkers associated with lung cancer using pre-diagnostic oral rinse samples from individuals who never smoked.
METHODS
Study participants and design
The study, depicted in Figure 1, is nested case-control study within the Shanghai Women’s Health Study (SWHS) and the Shanghai Men’s Health Study (SMHS), with respective recruitments between 1996–2000 (n=74,941) and 2002–2006 (n= 61,480) [16, 17]. These population-based prospective cohort studies in Shanghai, China, involved multiple in-person interviews to obtain information on demographics, occupational and environmental exposures, lifestyle, dietary, and other factors. The participation rates in both cohorts were high (SWHS=92.7%; SMHS=74.0%) [16, 17]. Cohort members were followed for cancer diagnoses through in-person surveys administered every 2–3 years and annual record linkage with the Shanghai Cancer Registry and Vital Statistics Unit.
Figure 1.
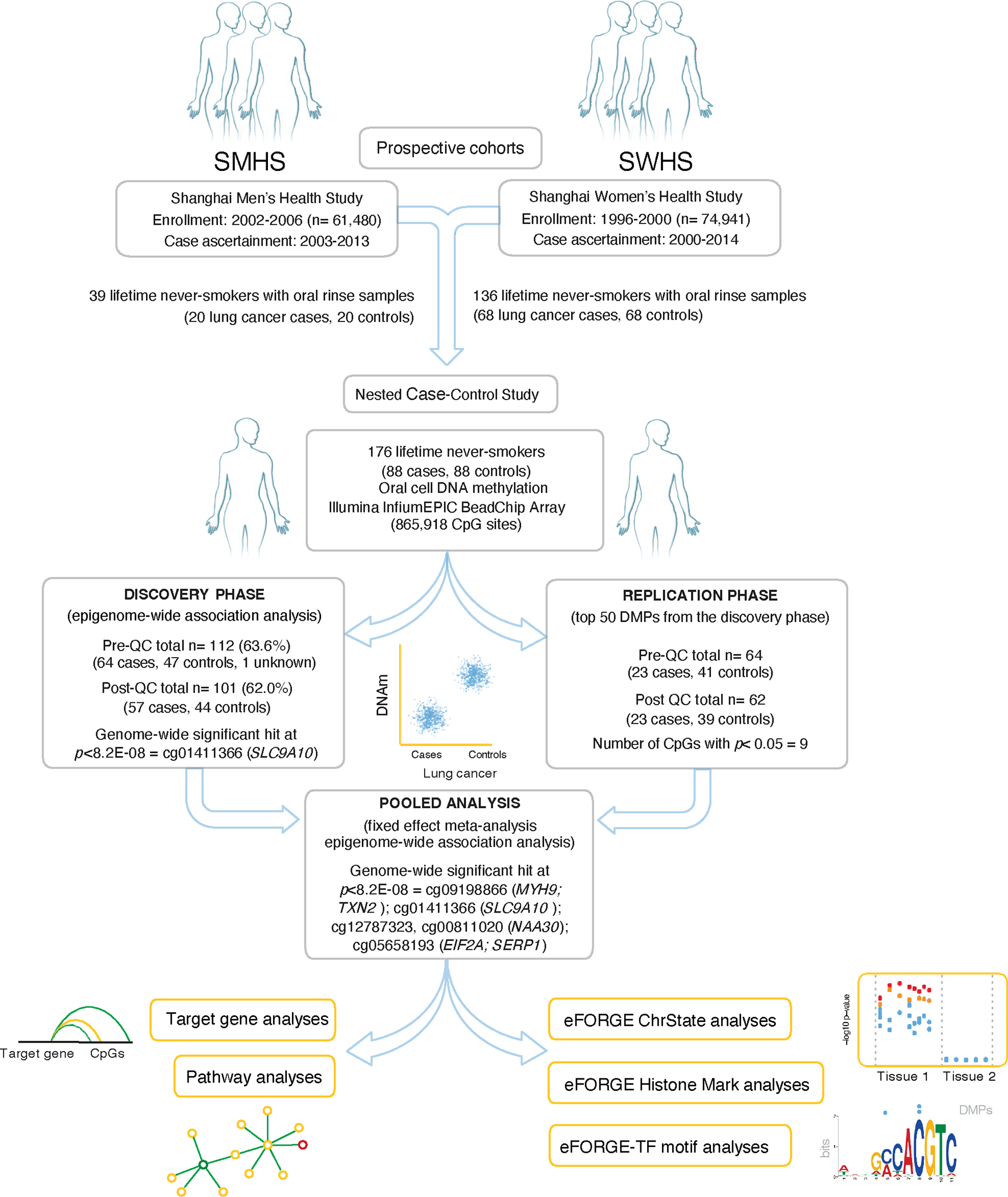
Overview of oral cell DNA methylation and future risk of lung cancer in never-smokers from SMHS and SWHS: Study design, study information, sample size, and number of significant DMPs for discovery, replication, and pooled analyses.
Eligible cases included incident lung cancer in individuals who never smoked (<100 cigarettes in lifetime) with available oral rinse samples. Using the incidence-density method, we randomly selected one never-smoking control for each index case matched on age at baseline (within ± 2 years), sex, sampling time (morning/afternoon), recent use of antibiotics, and menopausal status. The diagnosis of lung cancer was determined using the International Classification of Diseases-Ninth revision (ICD-9) code 162. Lung cancer diagnoses spanned 2000–2014 in SWHS (average follow-up: 7.0 years, range: 0–13 years) and 2003–2013 in SMHS (average follow-up: 5.2 years, range: 1–10 years).
All study participants provided written informed consent before being interviewed, and the study protocols were approved by the institutional review boards of all participating institutions.
DNA extraction and DNA methylation measurements
Oral rinse samples were collected during study enrollment, using a mouth rinse technique. DNA was isolated using the DNeasy PowerSoil Kit (Qiagen). Bisulfite-converted DNA was employed to assess genome-wide DNAm using Infinium MethylationEPIC BeadChip Array (Illumina, Inc., CA, USA). Samples were grouped into three projects based on the shipment date, with the largest project (project 2) including 101 (62.0%) samples.
Calculation of epigenetic aging biomarkers
Epigenetic age was calculated using the online Horvath calculator (http://dnamage.genetics.ucla.edu/) with advance analysis option [18]. Epigenetic Age Acceleration (EAA) measures were obtained, indicating biological aging relative to chronological age. Extrinsic EAA (EEAA) and Intrinsic EAA (IEAA) for Horvath’s clock were also assessed, considering the age-related changes in blood cell counts due to immune system aging.
Statistical Analysis
We processed raw DNA methylation image files using R statistical software (www.r-project.org/) and several Bioconductor packages, including the ChAMP pipeline [19], with default parameters. Samples exhibiting poor performance (>10% failed probes), were excluded, removing 12 samples. Further exclusions comprised 177,239 probes with detection p-value <0.01, 332 probes with bead count <3 in at least 5% of samples, 1,511 no-CpG probes, 78,915 single-nucleotide polymorphism (SNP) probes, and 11 multi-hit probes. A total of 607,910 probes meeting quality control criteria were used in subsequent statistical analyses. Missing values were imputed using the K-nearest neighbor method, and data were normalized using Beta Mixture Quantile normalization (BMIQ) [20]. Correction for potential batch effects related to sample plate, array, and slide was done using ComBat [21]. We visualized density distributions for samples at all processing steps (Supplemental Figure 1). DNAm at each CpG site is reported as β-values, indicating the fraction of methylated DNA molecules at the target CpG (ranging from 0 to 1). Epigenetic Dissection of Intra-Sample Heterogeneity (EpiDISH), a reference-based method, estimated cell-type proportions [22].
Differentially methylated positions (DMPs) and lung cancer risk
In a two-stage analysis, comprising a the discovery phase (n = 101) and a replication phase (n=62), we aimed to identify DMPs associated with lung cancer. Robust linear regression models were employed on an M-value scale with adjustments for age, sex, sample plate, and estimated cell type proportions. Nonetheless, we reported coefficients and standard errors on the beta value scale for easy interpretation. Unmatched analysis was performed to preserve the maximum sample size. EWAS model fit was evaluated using quantile-quantile plots and the genomic inflation factor (λ). Manhattan and volcano plots illustrated EWAS findings, with a genome-wide significance threshold set at p<8.22×10−08, applying a Bonferroni correction. The top 50 DMPs identified based on p-values from the discovery phase were tested for replication in the second set using similar models. Findings from both phases were combined using a fixed-effect meta-analysis. DMPs achieving a p-value threshold of <8.22×10−08 were selected for subsequent analysis.
Unconditional logistic regression models were then applied to these DMPs, using DNAm M-values as both continuous and categorical variables (divided into tertiles based on distributions in controls). The Firth bias correction method addressed potential separation issues in categorical model, using the “logistf” function in R [23]. The assessment of trends across tertiles of methylation M-values was conducted using a linear trend test or Cochran-Armitage trend test, as appropriate. In the sensitivity analysis, we conducted an EWAS in the combined sample (n= 163). Additionally, we conducted a stratified analysis by sex for selected DMPs surpassing the genome-wide significance threshold, along with testing for an interaction between sex and the respective DMP in the combined sample.
In silico annotation of DMPs using eFORGE, and pathway analyses
We performed a functional overlap analysis using eFORGE (https://eforge.altiusinstitute.org/) with default settings, examining the top 1000 DMPs from the discovery and pooled EWAS for enrichment across 1) Hidden Markov Model (HMM) Chromatin States and 2) Histone mark broadPeaks from the Roadmap Epigenomics Consortium [24]. Additionally, we conducted pathway enrichment analysis, utilizing gene information from the top 1000 DMPs in GOrilla (http://cbl-gorilla.cs.technion.ac.il/), applying false discovery rate (FDR) to identify significantly enriched pathways.
Epigenetic aging and lung cancer risk
Estimated epigenetic age was evaluated through scatterplots and examining empirical correlations with chronological age. Unconditional logistic regression models, with adjustments for age and sex, were employed to evaluate the association between DNAm againg and lung cxancer.
RESULTS
Out of the 163 participants (80 lung cancer cases and 83 controls), 38 individuals were enlisted in the SMHS (19 cases and 19 controls), while 125 individuals were enlisted in the SWHS (64 cases and 61 controls) (Table 1). The discovery analysis included 101 participants, representing 62% of the total sample, which included 44 lung cancer cases and 57 controls. Among the lung cancer cases with available tumor histology data, 31 (38.8%) were classified as adenocarcinoma, and 41 (51.3%) remained unclassified.
Table 1.
Distribution of selected characteristics by lung cancer status
| Discovery phase | Replication phase | Pooled sample | ||||
|---|---|---|---|---|---|---|
| Characteristics | Controls (n = 44) | Cases (n = 57) | Controls (n = 39) | Cases (n = 23) | Controls (n = 83) | Cases (n = 80) |
| Age, mean (SD) | 57.70 (8.49) | 59.02 (9.11) | 58.54 (9.59) | 62.09 (7.48) | 58.10 (8.97) | 59.90 (8.74) |
| BMI, mean (SD) | 24.57 (3.39) | 24.89 (3.84) | 25.47 (5.10) | 23.78 (2.14) | 24.99 (4.27) | 24.57 (3.46) |
| Sex, n(%) | ||||||
| Male (SMHS) | 5 (11.4) | 6 (10.5) | 14 (35.9) | 13 (56.5) | 19 (22.9) | 19 (23.8) |
| Female (SWHS) | 39 (88.6) | 51 (89.5) | 25 (64.1) | 10 (43.5) | 64 (77.1) | 61 (76.2) |
| Secondhand Smoking, n(%) | ||||||
| Never | 12 ( 30.8) | 15 ( 29.4) | 4 ( 10.3) | 2 ( 8.7) | 16 ( 19.3) | 17 (21.2) |
| Ever | 27 ( 69.2) | 36 ( 70.6) | 21 ( 53.8) | 8 (34.8) | 48 ( 57.8) | 44 (55.0) |
| missing | 5 (11.4) | 6 (10.5) | 14 ( 35.9) | 13 (56.5) | 19 ( 22.9) | 19 (23.8) |
| Histological Subtypes, n(%) | ||||||
| Adenocarcinoma | 21 (36.8) | 10 (43.5) | 31 (38.8) | |||
| Non-adenocarcinoma | 7 (12.3) | 0 (0) | 7 (8.8) | |||
| Unclassified | 29 (50.9) | 12 (52.2) | 41 (51.2) | |||
| Unknown | 0 (0.0) | 1 (4.3) | 1 (1.2) | |||
Abbreviations: SMHS= Shanghai Men’s Health Study; SWHS= Shanghai Women’s Health Study; SD= standard deviation
Differentially methylated positions and lung cancer risk
Findings from the discovery analysis, follow-up replication, and subsequent pooled analysis using meta-analysis are illustrated in Figure 2 and summarized in Table 2 (with complete findings detailed in Supplemental Table 1). In the discovery analysis, a single DMP was identified, showing a significant association with lung cancer at the genome-wide level of p <8.22E-08 (cg01411366: coefficient= −0.095, p = 7.23E-08; SLC9A10). This association was confirmed in the replication analysis (β= −0.079, p = 0.047). In the meta-analysis, two additional DMPs were identified: cg09198866 (p = 5.39E-09, MYH9; TXN2) and cg12787323 (p = 2.72E-08), while cg01411366 (p = 1.02E-08) remained statistically significant with lung cancer at p <8.22E-08. Specifically, the meta-analysis reveals that hypomethylation of cg09198866 and cg01411366, alongside hypermethylation of cg12787323, corresponding to alterations of 1.7%, 9.3% and 3.1% on the beta value scale, respectively, were associated with a higher risk of developing lung cancer. It is noteworthy that the magnitude and direction of these associations remained consistent across the different stages of analysis. The findings broadly aligned with the combined epigenome-wide analysis, pooling together discovery and replication samples, with the exception that cg05658193 (EIF2A; SERPA) demonstrated a significant association with lung cancer at p < 8.22E-08 (Supplemental Table 2).
Figure 2: Overview of epigenome-wide DNAm analyses for lung cancer in the Discovery phase.
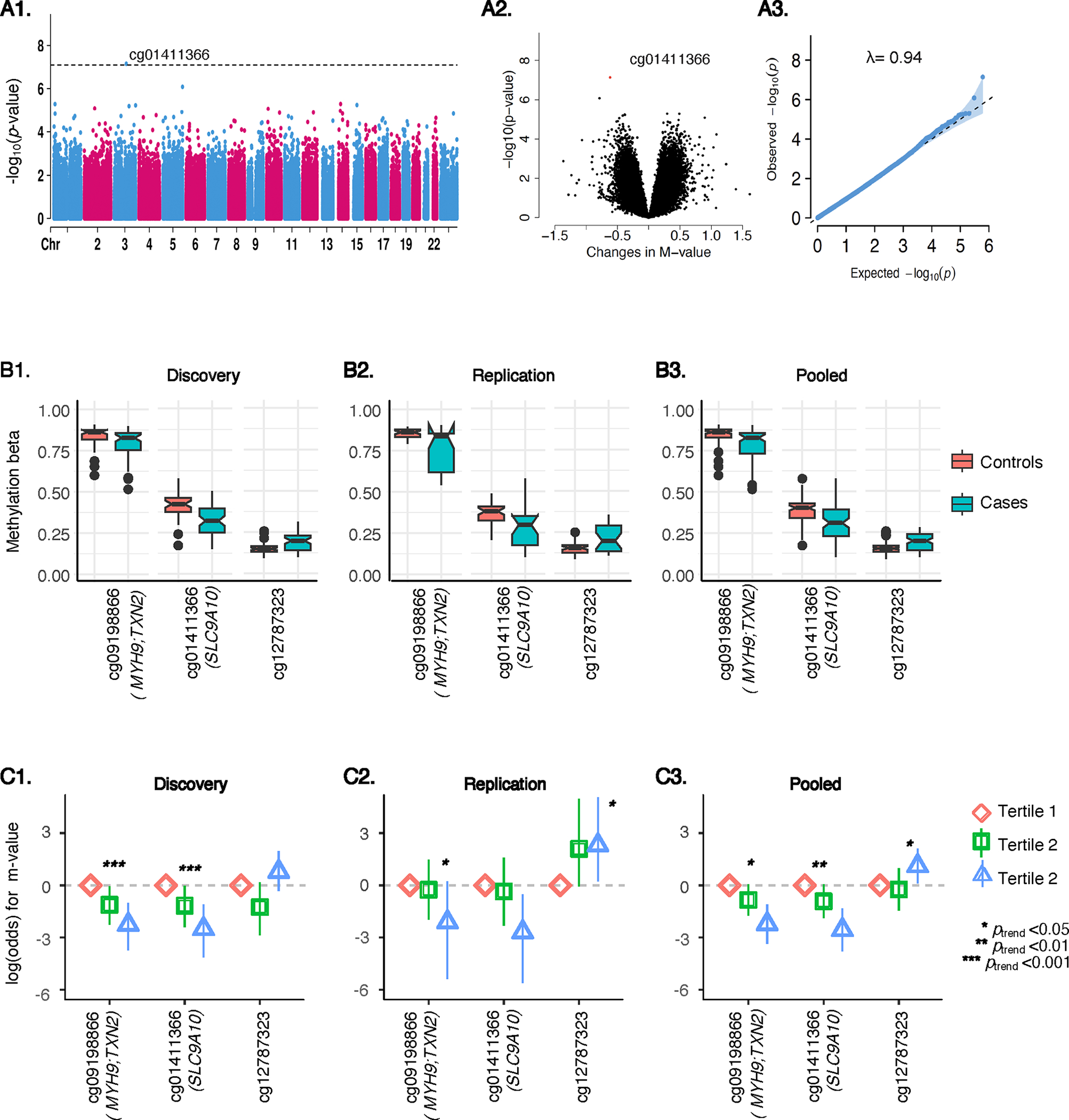
A1, A2, A3 illustrates the Manhattan, volcano, and quantile-quantile plots. The Manhattan plot (A1) presents a graphical representation of the association between DNAm sites (CpGs) across the genome and their corresponding p-values. Each dot represents a CpG site; its position along the x-axis indicating genomic location, and the y-axis shows the negative logarithm of the p-value. Significant associations are indicated by dots that rise above the dotted horizontal line, representing the genome-wide significance level of p<8.22E-08. The volcano plot (A2) displays statistical significance (p-values) against the effect size (changes in DNAm M-value) for each CpG site. The quantile-quantile plot (A3) compares the distribution of observed p-values to the expected distribution under the null hypothesis, along with the genomic inflation parameter, lamda (λ) value. B1, B2, B3 illustrates box plots comparing top-hit DMPs in methylation beta values between lung cancer cases and controls in Discovery (B1), Replication (B2), and pooled (B3) samples. C1, C2, and C3 illustrates odds ratios (ORs) and 95% confidence intervals (CIs) for lung cancer across tertiles of DNAm M-values for top-hit DMPs in Discovery (C1), Replication (C2), and Pooled (C3) samples.
Table 2.
Top DMPs with lung cancer risk in the in the discovery, replication, and pooled analyses, sorted by the smallest p-value in the pooled analysis.
| Discovery phase (57 cases, 44 controls) | Replication phase (23 cases, 39 controls) | Pooled analysis (80 cases, 83 controls) | Annotation | |||||
|---|---|---|---|---|---|---|---|---|
| DMPs | Adjusted % DNAm differencea | p-value | Adjusted % DNAm differencea | p-value | Adjusted % DNAm differenceb | p-value | CHR | Gene symbol |
| cg09198866 | −1.5 | 3.82E-05 | −2.1 | 3.05E-05 | −1.7 | 9.68E-09 | 22 | MYH9; TXN2 |
| cg01411366 | −9.5 | 7.23E-08 | −7.9 | 4.76E-02 | −9.3 | 1.02E-08 | 3 | SLC9A10 |
| cg12787323 | 3.0 | 1.32E-05 | 3.4 | 4.69E-04 | 3.1 | 2.72E-08 | 10 | |
DMPs that achieved an epigenome-wide significance level of p-value <8.22E-08 (bold faced) in meta-analysis are presented. Complete results are presented in Supplemental Table 1. Estimates are presented in beta value scale.
Robust linear regression models, adjusted for age, sex, sample plate (except for replication phase), study project (except for the discovery phase), and estimated cell type proportions were used. All the discovery samples originated from a single project, while all replication samples were processed on a single plate. Estimates are presented in beta value scale for easy interpretation.
Findings from discovery and replication sets were combined using a fixed effect meta-analysis.
The logistic regression models consistently supported these findings, demonstrating significant associations between these three DMPs and lung cancer across discovery, replication, and meta-analyses (Figure 2, Supplemental Table 3). Moreover, the categorical analysis revealed a significant and largely consistent monotonic trend across tertiles of these DMPs. In analysis stratified by sex, the effect of these three DMPs on lung cancer appeared to be more pronounced in males, although none of the DMPs exhibited a significant interaction with sex in the pooled sample (Supplemental Table 4).
Overlap of lung cancer-associated DMPs with genes and regulatory elements
To understand the regulatory context of DMPs, we mapped them to the nearest gene, epigenomic peaks, tissue-specific gene expression via RNA-seq, and chromatin interaction annotations (Figure 3). Notably, cg01411366 is located within a regulatory element linked to the SLC9A10 gene. Conversely, cg09198866 resides in an intergenic region distal to TXN2 and MYH9 genes. Transcription factor binding site (TFBS) analysis and other annotations suggest a regulatory element overlap, yet the specific target gene remains unclear. For cg12787323, located intergenically around 200kb from the nearest gene, it did not coincide with known enhancer elements or enhancer-promoter interactions according to GeneHancer, presenting interpretational challenges.
Figure 3. Lung cancer-associated DMPs.
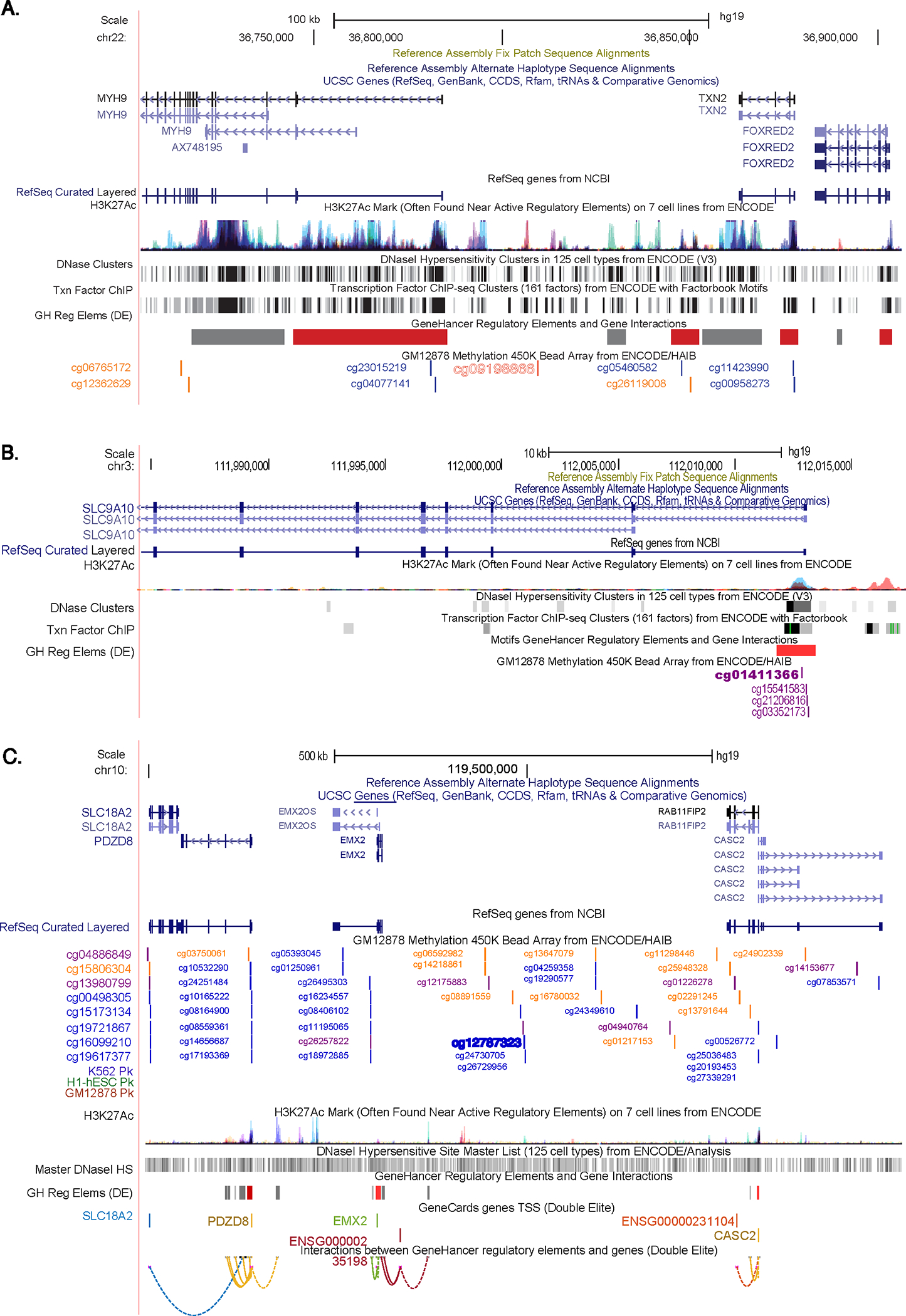
DMP browser shot displays (in order from the top): genomic coordinates, gene locations, H3K27ac peaks for 7 ENCODE cell lines, and ENCODE chromatin state segmentations and chromatin accessibility data of A) cg09198866 (MYH9; TXN2); B) cg01411366 (SLC9A10); and C) cg12787323. These data indicate that cg09198866 is located in an intergenic region distal to the TXN2 and MYH9 genes. TFBS analysis and other annotations suggested a regulatory element overlap, but the specific target gene is unclear. Conversely, cg01411366 is located on the regulatory element of the SLC9A10 gene. For cg12787323, located intergenically around 200kb from the nearest gene, it does not overlap with known enhancer elements or enhancer-promoter interactions according to GeneHancer, presenting interpretational challenges. This browser shot was generated using the UCSC Genome Browser (https://genome.ucsc.edu/) on human genome build hg19.
Integrative epigenomics analysis of DMPs using eFORGE, and pathway analyses
Integrative epigenomics analysis using eFORGE [24] showed enrichment for HMM chromatin states and histone mark broadPeaks related to epithelial, muscle, and lung tissues (Figure 4, complete findings are presented in Supplemental Figure 2. Consistent results were observed for top CpGs from the discovery EWAS across diverse study subsets (Supplemental Figure 3). Pathway analysis indicated enrichment for several pathways (Supplemental Figure 4), with the “regulation of small GTPase mediated signal transduction” being the sole significant pathway after multiple testing correction using FDR<0.05. Taken together, GO and FORGE2 results suggest a potential involvement of small GTPases and epithelial regulatory elements in the identified top DMPs in our study.
Figure 4. Tissue-specific integrative analysis indicates a potential effect on lung regulatory elements.
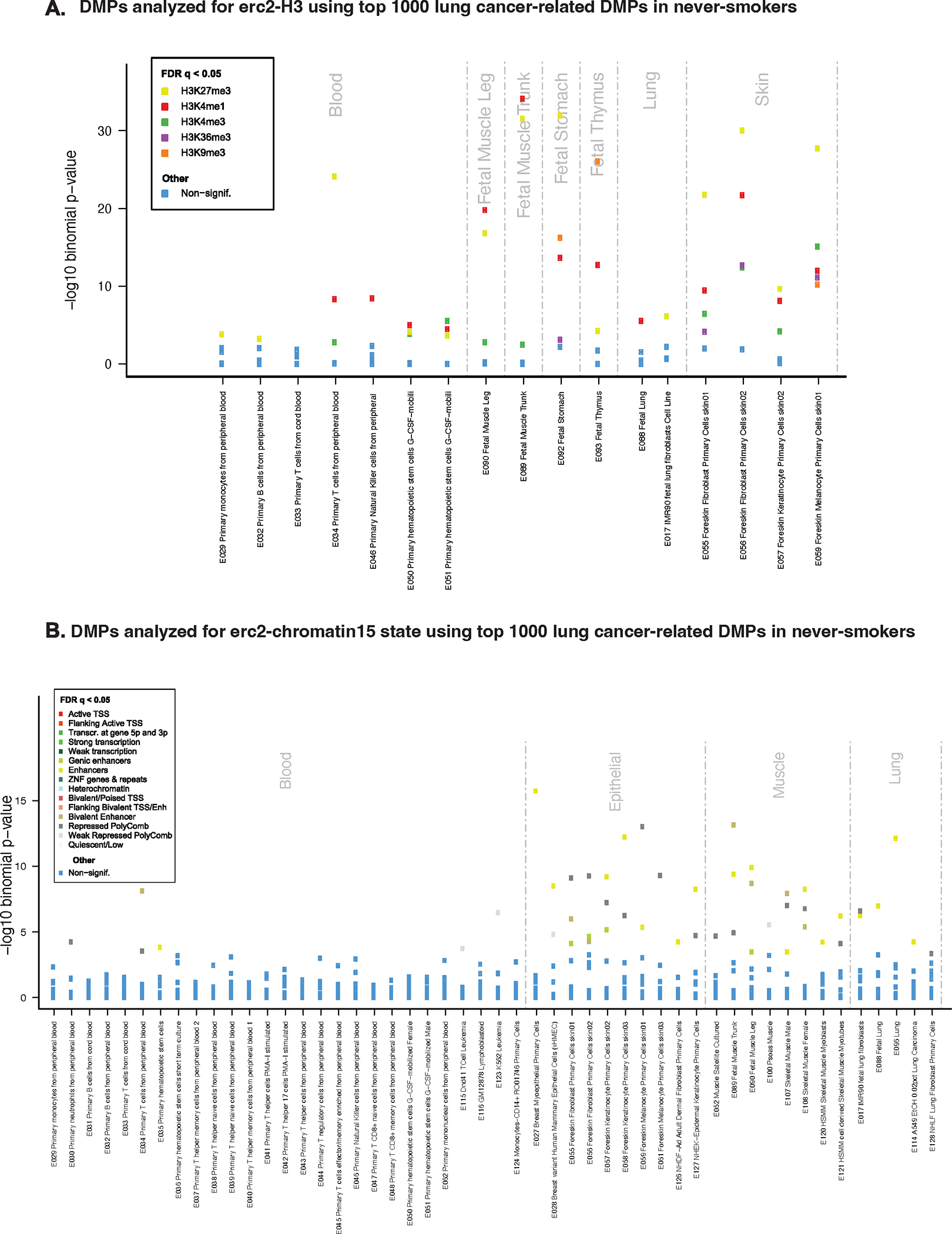
A) eFORGE analysis for top 1000 lung cancer-related DMPs in the pooled EWAS in never-smokers: the x-axis indicates tissues/cell type samples used in the analysis; the y-axis shows eFORGE enrichment (−log10 p-value) with Roadmap Epigenomics histone mark broadpeaks. The highest ranked sample set (indicated by the highest points) shows significant enrichment for lung, muscle, thymus, and skin, including for H3K4me1, an enhancer-associated histone mark. B) eFORGE analysis for the top 1000 lung cancer-related DMPs in the pooled EWAS: the x-axis indicates tissues/cell type samples used in the analysis; the y-axis shows eFORGE enrichment (−log10 p-value) with Roadmap Epigenomics chromatin states for a range of tissue samples. The highest-ranked sample set (indicated by the highest points) shows the most significant enrichment in epithelial cells with notable enrichment also seen in muscle and lung tissues, including for the “Enhancer” chromatin state category.
Epigenetic age acceleration and the risk of lung cancer
Our analysis demonstrated robust correlations (Pearson’s correlation coefficient, r = 0.61 to 0.87) between chronological age and epigenetic clocks, which are DNAm-based markers originally developed from various tissues and organ types (Supplemental Figure 5). This indicates that the epigenetic age markers calculated based on oral cell DNAm in our study performed well, despite potential differences in tissue specificity among the clocks. In logistic regression models, epigenetic age acceleration, as measured by GrimAge clock, was associated with a higher risk of lung cancer (odds ratio, OR= 1.19 per year of acceleration; 95% CI: 1.06, 1.34) (Figure 5). These associations remained consistent in sensitivity analyses, even with further adjustments for secondhand smoke exposure (data not included). No associations with lung cancer were observed for other epigenetic clocks assessed in this study.
Figure 5. Associations between epigenetic age acceleration and incident lung cancer in never-smokers.
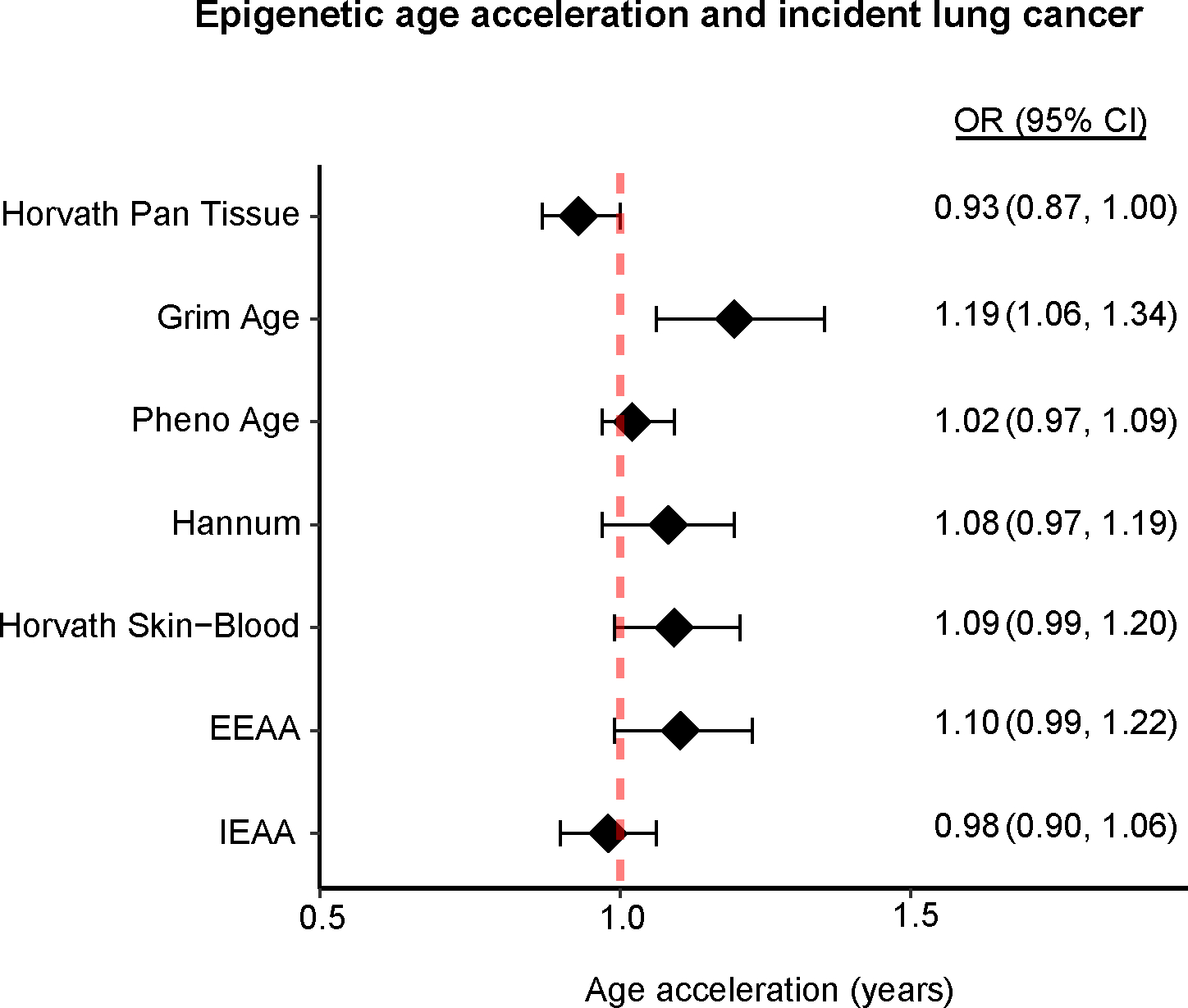
Diamonds represent odds ratios, error bars indicate 95% confidence intervals per year of epigenetic age acceleration, and the red dotted line signifies the null hypothesis.
DISCUSSION
In this study, involving lung cancer cases and controls nested within two large prospective cohorts in Shanghai, China, we assessed DNAm in oral rinse samples and identified three DMPs associated with the development of lung cancer using an agnostic epigenome-wide approach. Specifically, hypomethylation of cg09198866 (MYH9; TXN2) and cg01411366 (SLC9A10), as well as hypermethylation of cg12787323, were associated with an increased risk of lung cancer. Lung cancer-related DMPs exhibited significant enrichment in regions that regulate epithelial functions and were linked to the regulation of small GTPase-mediated signal transduction pathways, which are implicated in lung carcinogenesis. While our findings require confirmation through studies with larger sample sizes, they suggest that DNAm patterns in pre-diagnostic oral rinse samples could potentially serve as novel etiologic markers of lung cancer.
The precise biological mechanisms underlying the associations between the notably identified DMPs and lung cancer are not fully understood. The DMP cg09198866 is located in an intergenic region, distant from two genes – TXN2 and MYH9. This DMP overlaps with a regulatory element; however, the specific gene it affects remains undetermined. TXN2 encodes a mitochondrial thioredoxin family member, essential for modulating mitochondrial membrane potential and defending against oxidative stress. It significantly impacts ferroptosis, a novel iron-dependent lipid peroxidation-induced cell death, distinct from apoptosis. Cancer cells require robust antioxidative and anti-ferroptosis mechanisms for survival in oxidizing conditions. The upregulation of the Na+-independent cystine/glutamate antiporter (system Xc-), comprised of SLC3A2 and SLC7A11, augments antioxidative responses and inhibits ferroptosis, promoting tumor growth, survival, and cancer stem cell maintenance [25]. Overexpression of TXN2 has been linked to resistance against Erastin/RSL3-induced ferroptosis in lung cancer cells in Xuanwei, China [26], a region known for its high lung cancer incidence due to household smoky (bituminous) coal combustion [27], which is a significant source of redox-active iron particles in this population. Conversely, MYH9 encodes the heavy chain of nonmuscle myosin IIA, a novel cancer stem cell marker implicated in tumorigenesis via the PI3K/AKT/mTOR pathway. MYH9 is involved with a bidirectional promoter it shares with FOXE1 and PTCSC2, suppressing promoter activity. FOXE1 is a key regulator in autophagy and matrix metalloproteinases pathways in lung cancer development [28], while PTCSC2 is a ferroptosis-associated long noncoding RNA linked to head and neck squamous cell carcinoma [29].
The DMP cg01411366 resides within a regulatory element proximate to SLC9A10, a member of the sodium-hydrogen exchanger family implicated in transport of various substances. SLC9A10 was noted in a study on lung adenocarcinoma in never smokers, indicating a possible connection to the disease [30]. In contrast, cg12787323, located approximately 200kb from the nearest gene, lacks enhancer-promoter interactions, complicating its interpretation. Meanwhile, cg00811020 near the Nα-acetyltransferase 30 (NAA30) gene promoter is noteworthy. NAA30 encodes the catalytic subunit of N-terminal acetyltransferase (NAT) complex C, essential for peptide acetylation and cellular functions such as proliferation, apoptosis, and protein trafficking. Hypomethylation in the NAA30 promoter is linked to increased lung cancer risk and exposure to carcinogenic PAHs in tobacco smoke and coal combustion byproducts [31]. Pathway analysis of the top DMPs suggested small GTPase-mediated signal transduction pathways play a role in lung cancer development. These GTPases, particularly K-Ras from the KRAS oncogene, are downstream mediators in the epidermal growth factor receptor (EGFR) signaling pathway, affecting cell proliferation. In East Asia, lung cancer oncogenic mutations have been identified in both EGFR (60–78% cases) and KRAS (1% cases) genes in never-smokerers [32]. Intriguingly, KRAS mutations correlate with PAH-rich coal combustion exposures in Xuanwei, China [32], indicating a link between environmental factors, genetic mutations, and DNAm alterations in lung cancer development.
Our findings align with previous prospective studies that used DNAm in peripheral blood cells, showing higher age-adjusted GrimAge acceleration associated with an increased lung cancer risk [33], while other DNAm-based aging markers showed no association [34]. GrimAge acceleration, defined as the residual from epigenetic age regressed on chronological age, includes data from 1,030 CpG sites related to smoking pack-years and seven plasma proteins [35]. Remarkably, GrimAge predicts lifespan, including in never-smokers, and correlates with various comorbidities [36]. Previous studies have linked GrimAge with exposure to environmental contaminants such as smoking [37], PM2.5 [6], and household air pollution [38] and PAHs [39], all are established lung cancer risk factors [40]. Participants from this study were from homogenous urban areas in Shanghai, minimizing potential environmental exposure variation. Available data also suggested no significant difference in the distribution of chronic diseases, including chronic respiratory conditions at enrollment between participants who developed lung cancer and those who did not (data not presented). Future investigations with detailed exposure data are essential to clarify the environmental factors potentially influencing the GrimAge-lung cancer risk relationship observed in this study.
Our study has several strengths, including a comprehensive analysis of over 850,000 DNAm biomarkers using the Illumina InfiumEPIC BeadChip array. This array boasts nearly twice the coverage of its predecessor, the 450k array, used in nearly all previous prospective studies [12–14]. A novel finding of our research is that oral rinse samples, rich in epithelial cells from the upper respiratory tract directly exposed to air pollutants, could serve as non-invasive and easily obtainable specimens for DNAm analysis, particularly for diseases like lung cancer that originate from epithelial tissues. Notably, our most significant DMPs associated with lung cancer showed significant enrichment within epithelial regulatory regions. Moreover, our research is the largest prospective EWAS of lung cancer among never-smokers conducted to date. We have identified DMPs with genome-wide significance, which denotes their association with future lung cancer risk in this subgroup. Nonetheless, it is vital to recognize the limitation posed by our modest sample size, which heightens the risk of false positive findings. External validation with larger cohorts is necessary to affirm the robustness and broader applicability of our findings.
Diverse epigenetic clocks have been developed, each tailored based on calibration methods, tissues, sample size, and statistical approaches. We calculated epigenetic clocks using DNAm data from oral rinse samples, comprising various cell types such as buccal cells and saliva. While Horvath’s Pan Tissue and Skin-Blood clocks were developed using a variety of tissues, such as buccal cells and saliva [41], it is important to note that none of the epigenetic clocks evaluated in our study were specifically designed for oral cell. Additionally, not all clocks demonstrated consistent accuracy across various tissue types. Despite this, we observed robust correlations between DNAm age and chronological age (Pearson’s r = 0.61 to 0.87) in this study, suggesting the applicability of these markers despite potential tissue specificity variations.
Additionally, the study followed the U.S. CDC definition of “never smokers” but did not distinguish between individuals who had never smoked and those who smoked <100 cigarettes in their lifetime. Evidence suggests minimal exposure to smoking (serum cotinine concentrations <5 nmol/L) among never smokers in a large Asian lung cancer cohort, predominantly comprised of male participants [42]. Given the low smoking prevalence among Chinese women (around 2%) [43], and the high percentage of women (more than 76%) in our study, any substantive confounding due to unaccounted smoking appears unlikely. It is also notable that DNAm patterns are influenced by cumulative smoking dose and time since cessation [44, 45]. While certain smoking-related DNAm signatures may persist, quitting can revert methylation levels to those resembling non-smokers [44, 45]. Thus, confounding from unaccounted smoking in our cohort is expected to be negligible. Additional limitations of this study include the lack of detailed data on lung cancer histological subtypes. Since DNAm patterns vary across different histologic subtypes of lung cancer [46], future research with comprehensive histological information is warranted to identify both common and distinct DNAm patterns associated with various lung cancer subtypes.
CONCLUSIONS
In summary, our genome-wide analysis revealed multiple distinct DMPs in pre-diagnostic oral rinse samples from individuals who never smoked, including both lung cancer cases and controls. In addition, we found that an accelerated GrimAge clock was associated with an increased risk of developing lung cancer in this population. While our findings require confirmation in larger cohorts, they imply that DNAm assessment in pre-diagnostic oral rinse samples could offer novel insights into the risk factors and pathogenesis of lung cancer among never-smokers.
Supplementary Material
What is already known on this topic
Few prospective studies on genome-wide DNA methylation (DNAm), DNAm-based aging, and lung cancer have ever been conducted; all were focused on DNAm patterns in peripheral blood and had limited inclusion of never-smokers. These studies identified specific CpG sites associated with lung cancer, influenced by smoking, with the AHRR gene standing out. Despite some shared CpG findings between ever- and never-smokes, none of these CpG sites demonstrated a broad genome-wide significance in the relatively small subset of never-smokers studied.
What this study adds
To our knowledge, this study marks the first prospective EWAS of lung cancer among never-smokers, using oral rinse samples. Multiple differentially methylated positions (DMPs) associated with lung cancer in never-smokers were identified. Top DMPs exhibited significant enrichment in epithelial regulatory regions and linked to small GTPase-mediated signal transduction pathways, implicated in lung carcinogenesis. Additionally, GrimAge acceleration was associated with an increased lung cancer risk among never-smokers.
How this study might affect research, practice or policy
Subject to validation in a larger sample size, our findings suggest that changes in DNAm patterns in pre-diagnostic oral rinse samples may provide novel insights into the pathogenesis of lung cancer and its risk factors, particularly in individuals who have never smoked. These non-invasive samples, which consist of a mixture of epithelial and other upper airway cell types directly exposed to various air pollutants, offer a valuable medium to identify etiologic markers of diseases originating from epithelial tissues, such as lung cancer.
Acknowledgements:
The authors acknowledge the research contributions of the Cancer Genomics Research Laboratory of the Intramural Research Program, National Cancer Institute, National Institutes of Health for their expertise, execution, and support of this research in the areas of project planning, wet laboratory processing of specimens, and generating the data. This research was accepted for a presentation at the 2023 annual American Association for Cancer Research (AACR) meeting, and an associated abstract is available in publication.
Funding:
This research was supported in part by the Intramural Research Program of the National Cancer Institute, National Institutes of Health (grant number: NA) and the National Institute of Environmental Health Sciences of National Institutes of Health (research grants R01 CA70867, CA082729, UM1 CA173640).
Footnotes
Declaration of interests: The authors declare that they have no competing interests
Ethics approval and consent to participate: All study participants provided written informed consent before being interviewed, and the study protocols were approved by the institutional review boards of all participating institutions (number OH98CN006).
Copyright statement: Journal acknowledges that Author retains the right to provide a copy of the final peer-reviewed manuscript to the NIH upon acceptance for Journal publication, for public archiving in PubMed Central as soon as possible but no later than 12 months after publication by Journal.
Data sharing:
All data generated or analyzed during this study are included in this published article and its supplementary information files. For original data, please contact the corresponding author, Mohammad L. Rahman at mohammad.rahman2@nih.gov
References
- 1.Sung H, et al. , Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: A Cancer Journal for Clinicians, 2021. 71(3): p. 209–249. [DOI] [PubMed] [Google Scholar]
- 2.Sun S, Schiller JH, and Gazdar AF, Lung cancer in never smokers — a different disease. Nature Reviews Cancer, 2007. 7(10): p. 778–790. [DOI] [PubMed] [Google Scholar]
- 3.Chen W, et al. , Disparities by province, age, and sex in site-specific cancer burden attributable to 23 potentially modifiable risk factors in China: a comparative risk assessment. The Lancet Global Health, 2019. 7(2): p. e257–e269. [DOI] [PubMed] [Google Scholar]
- 4.Siegel DA, et al. , Proportion of Never Smokers Among Men and Women With Lung Cancer in 7 US States. JAMA Oncology, 2021. 7(2): p. 302–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeRouen MC, et al. , Incidence of Lung Cancer Among Never-Smoking Asian American, Native Hawaiian, and Pacific Islander Females. JNCI: Journal of the National Cancer Institute, 2021. 114(1): p. 78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nwanaji-Enwerem JC, et al. , Associations between long-term exposure to PM(2.5) component species and blood DNA methylation age in the elderly: The VA normative aging study. Environ Int, 2017. 102: p. 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bukowska B, Mokra K, and Michałowicz J, Benzo[a]pyrene-Environmental Occurrence, Human Exposure, and Mechanisms of Toxicity. Int J Mol Sci, 2022. 23(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hosgood HD 3rd, et al. , Household coal use and lung cancer: systematic review and meta-analysis of case-control studies, with an emphasis on geographic variation. Int J Epidemiol, 2011. 40(3): p. 719–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tantoh DM, et al. , Methylation at cg05575921 of a smoking-related gene (AHRR) in non-smoking Taiwanese adults residing in areas with different PM2.5 concentrations. Clin Epigenetics, 2019. 11(1): p. 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reynolds LM, et al. , Secondhand Tobacco Smoke Exposure Associations With DNA Methylation of the Aryl Hydrocarbon Receptor Repressor. Nicotine Tob Res, 2017. 19(4): p. 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meng H, et al. , Epigenome-wide DNA methylation signature of benzo[a]pyrene exposure and their mediation roles in benzo[a]pyrene-associated lung cancer development. J Hazard Mater, 2021. 416: p. 125839. [DOI] [PubMed] [Google Scholar]
- 12.Fasanelli F, et al. , Hypomethylation of smoking-related genes is associated with future lung cancer in four prospective cohorts. Nat Commun, 2015. 6: p. 10192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baglietto L, et al. , DNA methylation changes measured in pre-diagnostic peripheral blood samples are associated with smoking and lung cancer risk. Int J Cancer, 2017. 140(1): p. 50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sandanger TM, et al. , DNA methylation and associated gene expression in blood prior to lung cancer diagnosis in the Norwegian Women and Cancer cohort. Sci Rep, 2018. 8(1): p. 16714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao N, et al. , Epigenome-wide scan identifies differentially methylated regions for lung cancer using pre-diagnostic peripheral blood. Epigenetics, 2022. 17(4): p. 460–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shu XO, et al. , Cohort Profile: The Shanghai Men’s Health Study. Int J Epidemiol, 2015. 44(3): p. 810–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng W, et al. , The Shanghai Women’s Health Study: rationale, study design, and baseline characteristics. Am J Epidemiol, 2005. 162(11): p. 1123–31. [DOI] [PubMed] [Google Scholar]
- 18.Horvath S, DNA methylation age of human tissues and cell types. Genome Biol, 2013. 14(10): p. R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tian Y, et al. , ChAMP: updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics, 2017. 33(24): p. 3982–3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teschendorff AE, et al. , A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics, 2013. 29(2): p. 189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Price EM and Robinson WP, Adjusting for Batch Effects in DNA Methylation Microarray Data, a Lesson Learned. Front Genet, 2018. 9: p. 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teschendorff AE, Breeze CE, Zheng SC, and Beck S, A comparison of reference-based algorithms for correcting cell-type heterogeneity in Epigenome-Wide Association Studies. BMC Bioinformatics, 2017. 18(1): p. 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heinze G and Schemper M, A solution to the problem of separation in logistic regression. Statistics in Medicine, 2002. 21(16): p. 2409–2419. [DOI] [PubMed] [Google Scholar]
- 24.Breeze CE, Cell Type-Specific Signal Analysis in Epigenome-Wide Association Studies. Methods Mol Biol, 2022. 2432: p. 57–71. [DOI] [PubMed] [Google Scholar]
- 25.Ishimoto T, et al. , CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(−) and thereby promotes tumor growth. Cancer Cell, 2011. 19(3): p. 387–400. [DOI] [PubMed] [Google Scholar]
- 26.Li G, et al. , Dysregulation of ferroptosis may involve in the development of non-small-cell lung cancer in Xuanwei area. J Cell Mol Med, 2021. 25(6): p. 2872–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lan Q, et al. , Variation in lung cancer risk by smoky coal subtype in Xuanwei, China. Int J Cancer, 2008. 123(9): p. 2164–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ji GH, Cui Y, Yu H, and Cui XB, Profiling analysis of FOX gene family members identified FOXE1 as potential regulator of NSCLC development. Cell Mol Biol (Noisy-le-grand), 2016. 62(11): p. 57–62. [PubMed] [Google Scholar]
- 29.Lu R, Li Z, and Yin S, Constructing a Ferroptosis-related Long Non-coding RNA Signature to Predict the Prognostic of Head and Neck Squamous Cell Carcinoma Patients by Bioinformatic Analysis. Biochem Genet, 2022. 60(5): p. 1825–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Job B, et al. , Genomic aberrations in lung adenocarcinoma in never smokers. PLoS One, 2010. 5(12): p. e15145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Armstrong B, Hutchinson E, Unwin J, and Fletcher T, Lung cancer risk after exposure to polycyclic aromatic hydrocarbons: a review and meta-analysis. Environ Health Perspect, 2004. 112(9): p. 970–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hosgood HD 3rd, et al. , Driver mutations among never smoking female lung cancer tissues in China identify unique EGFR and KRAS mutation pattern associated with household coal burning. Respir Med, 2013. 107(11): p. 1755–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dugué PA, et al. , Biological Aging Measures Based on Blood DNA Methylation and Risk of Cancer: A Prospective Study. JNCI Cancer Spectr, 2021. 5(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Michaud DS, et al. , Epigenetic age and lung cancer risk in the CLUE II prospective cohort study. Aging (Albany NY), 2023. 15(3): p. 617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu AT, et al. , DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging, 2019. 11(2): p. 303–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu AT, et al. , DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging (Albany NY), 2019. 11(2): p. 303–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cardenas A, et al. , Epigenome-wide association study and epigenetic age acceleration associated with cigarette smoking among Costa Rican adults. Sci Rep, 2022. 12(1): p. 4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blechter B, et al. , Household air pollution and epigenetic aging in Xuanwei, China. Environment International, 2023. 178: p. 108041. [DOI] [PubMed] [Google Scholar]
- 39.Li J, et al. , Exposure to Polycyclic Aromatic Hydrocarbons and Accelerated DNA Methylation Aging. Environ Health Perspect, 2018. 126(6): p. 067005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Myers R, et al. , High-Ambient Air Pollution Exposure Among Never Smokers Versus Ever Smokers With Lung Cancer. J Thorac Oncol, 2021. 16(11): p. 1850–1858. [DOI] [PubMed] [Google Scholar]
- 41.Horvath S, et al. , Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging (Albany NY), 2018. 10(7): p. 1758–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Larose TL, et al. , Circulating cotinine concentrations and lung cancer risk in the Lung Cancer Cohort Consortium (LC3). Int J Epidemiol, 2018. 47(6): p. 1760–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang M, et al. , Trends in smoking prevalence in urban and rural China, 2007 to 2018: Findings from 5 consecutive nationally representative cross-sectional surveys. PLoS Med, 2022. 19(8): p. e1004064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shenker NS, et al. , DNA Methylation as a Long-term Biomarker of Exposure to Tobacco Smoke. Epidemiology, 2013. 24(5): p. 712–716. [DOI] [PubMed] [Google Scholar]
- 45.Guida F, et al. , Dynamics of smoking-induced genome-wide methylation changes with time since smoking cessation. Human Molecular Genetics, 2015. 24(8): p. 2349–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun S, Schiller JH, and Gazdar AF, Lung cancer in never smokers--a different disease. Nat Rev Cancer, 2007. 7(10): p. 778–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files. For original data, please contact the corresponding author, Mohammad L. Rahman at mohammad.rahman2@nih.gov