

Abstract
Optimum genetic delivery for modulating target genes to diseased tissue is a major obstacle for profitable gene therapy. Lipid nanoparticles (LNPs), considered a prospective vehicle for nucleic acid delivery, have demonstrated efficacy in human use during the COVID-19 pandemic. This study introduces a novel biomaterial-based platform, M1-polarized macrophage-derived cellular nanovesicle-coated LNPs (M1-C-LNPs), specifically engineered for a combined gene-immunotherapy approach against solid tumor. The dual-function system of M1-C-LNPs encapsulates Bcl2-targeting siRNA within LNPs and immune-modulating cytokines within M1 macrophage-derived cellular nanovesicles (M1-NVs), effectively facilitating apoptosis in cancer cells without impacting T and NK cells, which activate the intratumoral immune response to promote granule-mediating killing for solid tumor eradication. Enhanced retention within tumor was observed upon intratumoral administration of M1-C-LNPs, owing to the presence of adhesion molecules on M1-NVs, thereby contributing to superior tumor growth inhibition. These findings represent a promising strategy for the development of targeted and effective nanoparticle-based cancer genetic-immunotherapy, with significant implications for advancing biomaterial use in cancer therapeutics.
Key words: Genetic-immunotherapy, M1 macrophage-derived cellular nanovesicles, Lipid nanoparticles (LNPs), Gene therapy, siRNA, Cancer immunotherapy, Solid tumor, Tumor microenvironment (TME)
Graphical abstract
M1-polarized macrophage-derived cellular nanovesicle-coated lipid nanoparticles (M1-C-LNPs) were designed for combined gene and immunotherapy against solid tumors, with superior tumor inhibition efficacy and safety from targeted delivery and immune activation.
1. Introduction
Cancer continues to be a dominant global cause of mortality, with traditional modalities like chemotherapy and radiation therapy frequently associated with a risk of recurrence and substantial side effects1,2. Gene therapy in cancer is gaining prominence due to its capacity for personalization3, 4, 5, modifying the treatment based on the specific genetic alterations found within the patient's cancer cells6,7. In cancer treatment, gene therapy utilizes therapeutic nucleic acids, specifically techniques such as RNA interference (RNAi)8,9, or the clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR associated protein 9 (Cas9)10, with the aim to precisely target and modulate oncogenes within cancer cells. However, for effective cancer treatment through gene therapy, it is crucial to accurately target cancer cells with the delivery of tumor-suppressor genes or the disruption of oncogenes using tools like RNAi or CRISPR, as imprecise targeting may potentially contribute to the risk of recurrence. Recent advancements have marked lipid nanoparticles (LNPs) as robust carriers for siRNA therapeutics11, showcasing potential in the management of genetic diseases, and as delivery vehicles for SARS-CoV-2 mRNA vaccines in combatting infectious diseases12,13.
Cancer immunotherapy centers on mobilizing the body's own immune system to identify and eradicate cancer cells14, 15, 16. Capitalizing on the enduring memory response of the immune system, this approach offers a distinctive advantage, as it facilitates the recognition and elimination of specific cancer cells, thereby potentially preventing recurrence and effectively addressing metastasized cancers17, 18, 19. Cancer immunotherapy strategies, including checkpoint blockade antibodies20, 21, 22 and chimeric antigen receptor (CAR) immune cell therapy23, 24, 25, have transitioned into clinical application and demonstrated encouraging potential in addressing diverse tumor types, such as melanoma, lung, and hematological cancers. Nevertheless, the patient- and cancer-specific immunosuppressive tumor microenvironments (TME) present a hurdle to effective cancer immunotherapy, as they curtail the activity of immune cells and deter them from effectively targeting the cancer cells26,27. Intratumoral delivery of immunotherapeutic agents for TME modulation offers several advantages over systemic administration, including higher local drug concentration, reduced systemic toxicity, and the potential for administration at multiple tumor sites28,29. This approach, increasingly explored in recent clinical trials, is particularly effective for nanomedicines, especially those containing immunostimulants and measuring larger than 100 nm in diameter30,31. Intratumoral injection enhances the retention of these nanomedicines within the tumor, stimulating the TME more effectively and reducing systemic immune-related side effects, thus demonstrating significant promise for future clinical applications in cancer treatment32. To effectively counteract the immunosuppressive TME, a tailored strategy focused on anti-tumoral immunotherapy, primarily involving effector immune cells such as T and natural killer (NK) cells known for their capacity to kill cancer cells, is essential.
Macrophages, a crucial type of white blood cell integral to immune system function, possess the unique ability to alter their roles in response to environmental cues, a phenomenon termed as polarization33,34. These versatile cells can transition between two functional states, classifiable as M1 and M2 macrophages, regulating the immune network by secreting cytokines and expressing ligands or receptors on the surface to interact with other cells35,36. Significantly, M1 macrophages hold the potential to counteract the immunosuppressive TME by secreting pro-inflammatory cytokines such as IL-6 and TNFα37,38. Additionally, their role as antigen-presenting cells enables them to present tumor antigens to T cells, thereby catalyzing a robust adaptive immune response39. This characteristic endows M1 macrophages with the capability to activate NK and T cells40, 41, 42, critical components of the anti-cancer immune response, thereby potentially augmenting the efficacy of immunotherapy.
The broader scientific community has been actively investigating the potential benefits of immune cell-derived nanovesicles (NVs), particularly in terms of enhancing the safety, functionality, and therapeutic effectiveness of immunotherapy, attributes that are inherited from the source cells43,44. Furthermore, the innovative use of immune cell membrane coatings on nanoparticles has emerged as a strategy aimed at amplifying their therapeutic potential45, 46, 47. Recent studies highlight that NVs-derived from M1 macrophages, which are enriched in pro-inflammatory cytokines, can effectively modulate the TME, demonstrating their potential as potent immune therapeutics against cancer48,49.
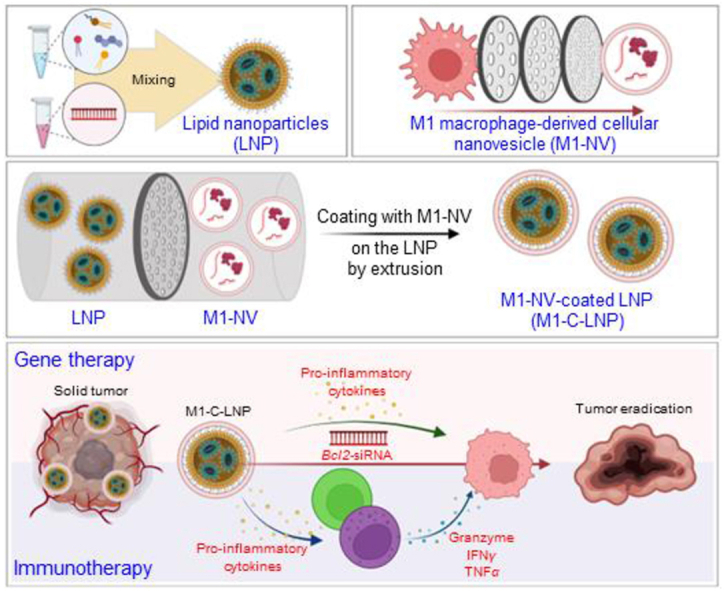
To overcome the limitations of traditional cancer gene therapy, this study advocates for a synergistic approach that combines gene therapy with cancer immunotherapy, using the strategic modulation of the TME as a tool to bolster cancer treatment efficacy. In light of this, our study utilized these LNPs to encapsulate Bcl2-siRNA, recognized for its capacity to induce apoptosis in cancer cells50,51, augmenting the stability of the therapeutic payload and to enhance its delivery rate to cancer cells. As a subsequent step, the surface of these LNPs was hybridized with artificial NVs-derived from M1-polarized macrophages. Keeping these insights in mind, we constructed Bcl2-siRNA-loaded LNPs-coated with NVs-derived from M1-polarized macrophages (termed as M1-NVs-coated LNPs, M1-C-LNPs) (Fig. 1A). Our designed M1-C-LNPs have dual functionality: they induce apoptosis to eradicate cancer cells, and they concurrently modify the TME to stimulate cytotoxic effector immune cells such as NK and T cells within the intratumoral immune system (Fig. 1B).
Figure 1.

Schematic of the dual-functionalized M1 macrophage-derived cellular nanovesicle-coated lipid nanoparticles (M1-C-LNPs) for efficient cancer therapy in the tumor microenvironment. (A) Bcl2-siRNA-loaded lipid nanoparticles (LNPs) consist of phospholipids (DSPC), ionizable lipids (DLin-MC3-DMA), cholesterol, and PEGylated lipids (DMG-PEG-2000). RAW 264.7 cells (mouse macrophage cell line) are polarized to M1 macrophages by lipopolysaccharide (LPS). M1 macrophages are extruded using a mini-extruder, producing M1 macrophage-derived cellular nanovesicles (M1-NVs). M1-NVs and LNPs are estimated by nanoparticle tracking analysis to confirm equal particle numbers. The LNPs and M1-NVs mixture is extruded using a mini-extruder, and passed through a 100 nm pore membrane multiple times, resulting in M1-C-LNPs synthesis. (B) M1-C-LNPs possess both LNPs and M1-NVs functionalities, carrying Bcl2-siRNA and inflammatory cytokines. M1-C-LNPs deliver Bcl2-siRNA and inflammatory cytokines to cancer cells, activating apoptosis by inhibiting Bcl2-mRNA. Additionally, M1-C-LNPs activate NK cells and T cells, increasing perforin and granzyme production for degranulation and cancer cell killing, without delivering siRNA to T and NK cells. M1-C-LNPs exhibit in vivo biocompatibility and enhanced tumor retention due to the properties of M1-NVs, inhibiting tumor growth. Thus, M1-C-LNPs represent a promising nano-platform for improved cancer gene and immunotherapy.
2. Materials and methods
2.1. Materials
DLin-MC3-DMA was acquired from MedChemExpress (Monmouth Junction, NJ, USA). The 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) and 1,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (PEG-DMG) were obtained from Avanti (Alabaster, AL, USA). Cholesterol and lipopolysaccharide (LPS) were purchased from Sigma–Aldrich (St Louis, MO, USA). The Bcl2-siRNA was procured from Origene (Rockville, MD, USA). FAM-siRNA was sourced from Bioneer (Daejeon, Korea). RAW 264.7 murine macrophage cells and CT26 colon cancer cells were acquired from the American Type Culture Collection (ATCC; Rockville, MD, USA). EL4 cells (murine T cell lymphoma) were obtained from the Korea Cell Line Bank (Seoul, South Korea). Mouse natural killer (NK) cells were isolated from mouse spleens using a mouse NK cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany). RAW 264.7 cells and CT26 cells were cultured at 37 °C and 5% CO2 in Dulbecco's modified Eagle's medium (HyClone Laboratories, Logan, UT, USA), supplemented with 10% fetal bovine serum (FBS; Gibco, Carlsbad, CA, USA) and 1% Antibiotic/Antimycotic (A/A; Gibco). The EL4 cells were maintained at 37 °C and 5% CO2 in Roswell Park Memorial Institute medium (HyClone Laboratories), supplemented with 10% FBS and 1% A/A. Mouse NK cells were cultured at 37 °C and 5% CO2 in Roswell Park Memorial Institute medium (HyClone Laboratories), supplemented with 10% FBS, 1% A/A, 2 mmol/L l-glutamine (Lonza, Shanghai, China), 1% nonessential amino acids (Invitrogen), 50 μmol/L β-mercaptoethanol (Sigma–Aldrich), and mouse IL-2 (PeproTech, Rocky Hill, NJ, USA).
2.2. Preparation of LNPs
LNPs were prepared at pH 5.2 to ensure ionization of DLin-MC3-DMA, thereby maximizing siRNA encapsulation. A lipid mixture (DLin-MC3-DMA, DSPC, cholesterol, and PEG-DMG at 50:10:38.5:1.5 Molar ratios) in ethanol was combined with siRNA in sodium acetate buffer (pH 5.2). Lipid and siRNA were mixed at a 1:3 volume ratio (1:20 w/w siRNA to lipid). The resulting LNPs were dialyzed against PBS for 1 h to remove ethanol and restore neutral pH. The diameter, surface zeta-potential, and number of LNPs were assessed through nanoparticle tracking analysis (NTA; Zetaview, Particle Metrix, Germany). The morphology of the LNPs was examined using transmission electron microscopy (TEM; H-7600M, Hitachi, Tokyo, Japan). The stability of LNPs was maintained in DW, PBS, or media for 4 days. Encapsulation of siRNA in LNPs was estimated using a Ribogreen RNA assay kit (ThermoFisher Scientific, MA, USA), following the manufacturer's instructions. Cellular uptake of LNPs in CT26 cells was confirmed with FAM-siRNA-loaded LNPs using a confocal laser scanning microscope (Carl Zeiss, Oberkochen, Germany), and analysed with ZEN software (Carl Zeiss).
2.3. Preparation of M1-NVs
RAW 264.7 cells were treated with LPS (1 μg/mL) to polarize them into M1 macrophages. Twelve h after LPS exposure, RAW 264.7 cells were washed, harvested, and sonicated with a tip sonicator at 20% sonication amplitude (pulsed mode; 3 s ON, 3 s OFF) for 30 s. After centrifugation to remove nuclei, the obtained cell-derived cellular nanovesicles were continuously extruded through a polycarbonate membrane filter (Whatman, UK) with pore sizes of 10, 5, 1, and 0.4 μm, using a mini extruder (Avanti Polar Lipids, AL, USA). The concentration of NVs was determined using a BCA protein assay kit (ThermoFisher Scientific), following the manufacturer's instructions. The diameter, surface zeta-potential, and number of NVs were measured by NTA (Zetaview). TEM (Hitachi) was performed to examine the morphology of the NVs. The expression of RNA and protein for exosome markers and inflammatory cytokines was assessed through real-time PCR and Western blotting. Cellular uptake of NVs in CT26 cells was confirmed with DiR-labelled NVs using a confocal laser scanning microscope (Carl Zeiss), and analysed with ZEN software (Carl Zeiss).
2.4. Preparation of M1-C-LNPs
The number of NVs and LNPs was evaluated by NTA (Zetaview) to ensure the equal mixing of particles. The mixture of NVs and LNPs was extruded using a mini-extruder, and passed through a 100 nm pore membrane several times, synthesizing the M1-C-LNPs.
2.5. Characterization of M1-C-LNPs
NTA was performed to analyse the size, surface zeta-potential, and the number of M1-C-LNPs. The morphology of M1-C-LNPs was estimated by TEM (Hitachi). To assess the maintained NV characteristics in M1-C-LNPs, real-time PCR and Western blotting were performed to confirm the expression of RNA and protein for exosome markers and inflammatory cytokines. The coated ratio of M1-C-LNPs was investigated by flow cytometry (BD Bioscience FacsCanto II Flow Cytometer, San Jose, CA, USA). For flow cytometry, NVs were prepared from GFP-expressed RAW 264.7 cells, and LNPs were labelled with DiR solution. Fluorescent nanoparticles were quantified using FACS Diva software (BD Biosciences).
2.6. Encapsulation efficiency of siRNA in nanoparticles by Ribogreen assay
Encapsulation of siRNA in LNPs or M1-C-LNPs was evaluated using a Ribogreen RNA assay kit (ThermoFisher Scientific, MA, USA), following the manufacturer's protocol. Briefly, LNPs or M1-C-LNPs were either lysed with Triton X-100 or left unlysed, and the total amount of siRNA in the sample was measured using the Quant-It Ribogreen reagent (ThermoFisher Scientific) and a microplate reader (Synergy H1, BioTek, Winooski, USA). After subtracting the blank measurement, the encapsulation efficiency (in percentage) was calculated as in Eq. (1):
| Encapsulation efficiency (%) = [1 – (Non-lysed LNPs or M1-C-LNPs)/Lysed LNPs or M1-C-LNPs] × 100 | (1) |
2.7. In vitro cellular uptake analysis of the nanoparticles by immunocytochemistry
CT26 cells were seeded onto Poly-d-Lysine (Gibco) coated coverslips in 24-well plates to verify the cellular uptake of LNPs, M1-NVs, or M1-C-LNPs. After 12 h, each nanoparticle was treated for 4 h. Cells were then washed with PBS, and fixed with 4% paraformaldehyde for 15 min. Following another PBS wash, cells were permeabilized using 0.2% Triton X-100 in PBS for 15 min, followed by washing with PBS. Cells were blocked with 4% bovine serum albumin (BSA) in PBS for 1 h at room temperature (RT), and subsequently incubated with lysotracker. After washing with PBS, cells mounted with 4′,6-diamidino-2-phenylindole (DAPI) mounting solution were observed using a confocal laser scanning microscope (Carl Zeiss).
2.8. In vitro cytotoxicity of nanoparticles
CT26 cells were seeded and treated with M0-NVs, M1-NVs, LNPs, M0-C-LNPs, or M1-C-LNPs for 4 h. After 24 h, CT26 cell cytotoxicity was confirmed using a CCK-8 assay. The absorbance of each well was measured at 450 nm wavelength using a microplate reader (BioTek). Twelve h after treatment with M1-NVs, LNPs, or M1-C-LNPs, CT26 cells were stained with a Live/Dead kit, according to the manufacturer's protocol. Cell images were observed under inverted fluorescence microscope (ECLIPS Ti2; Nikon, Japan).
2.9. Evaluation of RNA expression in nanoparticle-treated cells by real-time PCR
Total RNA was extracted from cells and nanoparticles using the Trizol reagent (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's protocol. One μg of total RNA was transcribed into cDNA using M-MLV reverse transcriptase (Promega, Madison, WI, USA) with random primers (Promega) and the oligo dT primer (Roche, Mannheim, Germany). To quantify expression levels, real-time PCR was performed using synthesized cDNA and the SYBR Green dye (iQ SYBR Green Supermix, Bio-Rad, Waltham, MA, USA) with specific primers. For comparison of transcript levels between experimental samples, a standard curve was established using cycle thresholds for several serial dilutions of a cDNA sample, which was then used to calculate the relative abundance of each gene. Data were normalized by the level of Rpl7 expression in each individual sample.
2.10. Evaluation of protein expression in nanoparticle-treated cells and tissues by Western blotting
Cells, tissues, or nanoparticles were lysed in lysis buffer including PRO-PREP (iNtRON, Seongnam, Korea) and 1 × phosphatase inhibitor (Roche Applied Science, Indianapolis, IN, USA). Protein concentration was determined using the BCA assay (ThermoFisher Scientific). Proteins from cell lysates, tissues, or nanoparticles were separated using SDS-PAGE, and transferred to nitrocellulose membranes. Blots were blocked with 5% skim-milk for 1 h at RT, incubated overnight at 4 °C with diluted primary antibodies, and then incubated with HRP-conjugated secondary antibody for 1 h at RT. Blots on the membranes were developed using an ECL detection reagent (Bio-Rad, CA, USA). Exosome markers CD9, CD63, and TSG101 were used, as well as apoptosis markers BCL2, Caspase-3, and cleaved Caspase-3. Cyclin B1 was used as a proliferation marker, while COX2 and iNOS were used as inflammation markers. ICAM-1 was used as an adhesion marker, and Collagen I and Collagen IV were used as markers for collagen density in the tumor microenvironment. β-Actin served as a loading control.
2.11. In vitro evaluation of gene delivery of LNPs and M1-C-LNPs by luciferase assay
For quantification of delivered gene expression level, a luciferase assay was performed using luciferase-encoding mRNA. Luciferase-encoding RNA was encapsulated in LNPs and M1-C-LNPs, and treated to CT26, T, and NK cells for 4 h. After washing, treated cells were lysed with Glo lysis buffer, and reacted with Bright-Glo Luciferase assay reagent. Luminescence of each well was measured using a microplate reader (BioTek). Lipofectamine was used as a positive control.
2.12. In vitro analysis of activation of T and NK cells after treatment with M1-C-LNPs
The T and NK cells were seeded and treated with M1-NVs, LNPs, or M1-C-LNPs for 4 h. After 24 h, T and NK cell cytotoxicity was confirmed using a CCK-8 assay. Twelve hours after M1-C-LNP treatment, T and NK cells were fixed with 4% paraformaldehyde, and stained with BioTracker Cytoplasmic membrane dye (Sigma–Aldrich). Cell images were observed under a confocal laser scanning microscope (Carl Zeiss). Protein expression levels in T and NK cells were evaluated by Western blotting. PERFORIN and GRANZYME were used as activation markers for T and NK cells, with β-actin serving as a loading control. Cancer cell-killing capability of T and NK cells was determined by co-culturing with CT26 cells, followed by a CCK-8 assay.
2.13. In vivo experiments
All in vivo experiments were conducted under the guidelines of approved protocols from the Institutional Animal Care and Use Committee of the Catholic University of Korea (Republic of Korea, CUK-IACUC-2021-017-01) and Sungkyunkwan University (Republic of Korea, SKKUIACUC-2022-04-49-1). Eight-week-old female BALB/c and BALB/c nude mice were used for animal experiments after a 1 week acclimatization period. To establish a mouse tumor model, CT26 cells (2 × 106 cells) were subcutaneously inoculated in the right flank of 9-week-old BALB/c and BALB/c nude mice. After 8 days, when the average tumor volume reached 100 mm3, mice were used for the experiments. Tumor size was measured using vernier calipers, with volumes calculated using the following Eq. (2):
| Volume = Width × Width × Length × 0.5 | (2) |
Twenty-five days after tumor inoculation, mice were sacrificed with CO2 gas when the tumor size exceeded 2000 mm3. After complete anesthesia with isoflurane, each nanoparticle was intratumorally injected into CT26 tumor-bearing BALB/c and BALB/c nude mice at 8, 10, and 12 days post-inoculation, using the same dose.
2.14. In vivo retention of M1-C-LNPs in tumor over time
To assess the retention of M1-C-LNPs within tumors, M1-NVs, LNPs, and M1-C-LNPs were labelled with DiR solution. Eight days post-inoculation, when the average tumor volume reached 100 mm³, DiR-labelled M1-NVs, LNPs, and M1-C-LNPs were intratumorally injected into CT26 tumor-bearing BALB/c nude mice. Images were obtained at 2 h, 24 h, 7 days, and 15 days using a fluorescence-labelled organism bio-imaging instrument (FOBI, Neo-Science, Suwon, Korea). The fluorescence images were then normalized against the 2 h M1-NV images for consistency.
2.15. In vivo tumor suppression test by treatment with M1-C-LNPs
Tumor growth inhibition was assessed in the following groups (each consisting of 8 mice): Group 1: PBS only; Group 2: M1-NVs; Group 3: LNPs; Group 4: M1-C-LNPs. Eight days post-inoculation, once the average tumor volume reached 100 mm³, each nanoparticle formulation was intratumorally injected into CT26 tumor-bearing BALB/c mice at 8, 10, and 12 days post-inoculation, using a consistent dose for each injection. Tumor sizes were measured every 2–3 days using vernier calipers.
2.16. In vivo histological and hematological analysis of M1-C-LNPs
Each nanoparticle formulation was administered to BALB/c mice bearing CT26 tumors at 8, 10, and 12 days post-inoculation. The mice were sacrificed at predetermined time points, following which their organs were collected for histocompatibility testing and blood biochemistry analysis. Sera were isolated from whole blood by centrifugation at 3000×g for 30 min. Sera levels of AST, ALT, ALP, CREA, GLU, CPK, CA, and NA/K were quantified using an automated chemical analyzer (Fuji Film DRI-CHEM NX500i, Tokyo, Japan). Major organs were dissected, and fixed with 4% paraformaldehyde for histological analysis. After organs were washed and dehydrated, they were embedded in paraplast (Leica Biosystems, St. Louis LLC, DieMen, Netherlands). Paraffin-embedded tissues were sectioned using a microtome, stained with hematoxylin and eosin (H&E) (Sigma–Aldrich), and observed by ScanScope CS2 (Leica Biosystems).
2.17. In vivo analysis of immune cells in tumor after treatment of M1-C-LNPs
Each nanoparticle formulation was administered to BALB/c mice bearing CT26 tumors at 8, 10, and 12 days post-inoculation. The tumors were harvested on day 19 post-inoculation. Subsequently, the harvested tumor tissues were meticulously dissociated into single cells. The single cells were dispersed in FACS buffer (PBS with 2% FBS and 1 mmol/L EDTA). After that, extracellular materials were removed using a 70 μm cell strainer, and then dispersed in FACS buffer again. The dispersed cells were then incubated for 1 min at 4 °C in RBC lysis buffer (Sigma–Aldrich). RBC lysis buffer was diluted with PBS, and then centrifuged for 3 min at 250×g. After removal of the supernatant, the cells were stained with the following antibody: BV510-conjugated Zombie aqua for live cells; BV785-conjugated anti-CD45, APC-conjugated anti-CD3, and BV650-conjugated anti-CD4 for helper T cells; BV785-conjugated anti-CD45, APC-conjugated anti-CD3, and BV650-conjugated anti-CD8 for cytotoxic T cells; BV785-conjugated anti-CD45, APC-conjugated anti-CD3, and BV650-conjugated anti-NKp46 for natural killer (NK) cells; and FITC-conjugated anti-Granzyme B, APC/Cy7-conjugated anti-TNFα, or BV421-conjugated anti-IFNγ for cytokines. For surface staining, the cells were stained with fluorescence-conjugated antibodies against surface antigens at RT for 20 min. To detect cytokines, the cells were stimulated with the cell stimulation cocktail for 4 h, fixed with IC fixation buffer at RT for 20 min, permeabilized, and stained with fluorescence-conjugated antibodies against cytokines at RT for 30 min. The cells were washed with FACS buffer, and analysed using a flow cytometer (Beckman Coulter, Fullerton, CA, USA) with CytExpert software (Beckman Coulter).
2.18. Statistical analysis
Statistical analysis was performed using GraphPad Prism 8 software (GraphPad Software, San Diego, CA, USA). All data are presented as the mean ± standard deviation (SD) or the mean ± standard error of the mean (SEM). ANOVA was used to evaluate three or more groups, while the t-test was used for two groups. The P-values are indicated in the figure legends.
3. Results and discussion
3.1. Characterization of M1-C-LNPs
Due to their excellent clinical applicability52,53, LNPs have emerged as a leading system for RNAi delivery. They have been utilized in the first FDA-approved siRNA therapy for treating polyneuropathy caused by transthyretin amyloidosis, and have been extensively investigated for various clinical applications that include gene therapies and mRNA vaccines for COVID-1954, 55, 56. In this work, we chose LNPs as the delivery vehicle for siRNA targeting Bcl2, a known anti-apoptotic factor57,58. LNPs were prepared by mixing lipids in ethanol with Bcl2-siRNA in acidic buffer, as shown in Supporting Information Fig. S1A 53,59. We evaluated the prepared LNPs for size, zeta-potential, and morphology (Figs. S1B and S1C). The LNPs had an estimated average diameter of 198 nm and a neutral charge, with a final lipid/siRNA weight ratio of 20:1. They exhibited a spherical morphology. We assessed the stability of the LNPs by measuring their particle size in deionized water (DW), phosphate-buffered saline (PBS), or cell culture media over time (Fig. S1D). The LNPs remained stable for four days in all tested conditions. We used a Ribogreen assay to quantify the encapsulated siRNA within the LNPs (Fig. S1E), revealing an encapsulation efficiency of approximately 65%. Confocal microscopy showed that the FAM-siRNA-loaded LNPs (green) successfully delivered the encapsulated siRNA to cancer cells upon treatment (Fig. S1F). To endow nanoparticles with immune cell activation properties, we employed macrophages to generate NVs. Macrophages interact with other immune cells and function as effector cells in various physiological conditions that include inflammation, tumor microenvironments, and fibrosis35,60,61. They possess a wide range of membrane receptors and cytokines that enable them to recognize and respond to pathogens and other stimuli62. When activated, macrophages can secrete cytokines and signal molecules that activate other immune cells63. As depicted in Supporting Information Fig. S2A, we prepared M1 macrophages by exposing the murine macrophage cell line RAW 264.7 to lipopolysaccharide (LPS), which is known to induce an inflammatory response by binding to toll-like receptor 4 (TLR4) on macrophages64,65. The M0 macrophages not exposed to LPS served as a negative control. We generated macrophage-derived NVs by extruding M0 and M1 macrophages through multiple membrane filters with varying pore sizes, yielding M0-NVs and M1-NVs, respectively. The particle sizes of M0-NVs and M1-NVs were approximately 205 nm and 187 nm, respectively (Fig. S2B). Their zeta-potentials were approximately −24 mV and −24 mV, respectively (Fig. S2C). Next, we identified exosome markers TSG101 and CD63 proteins in both M0-NV and M1-NV using Western blotting (Fig. S2D). The M1-NVs maintained a spherical morphology (Fig. S2E). We estimated the expression of inflammatory cytokines in M1-NVs using real-time PCR and Western blotting, to examine the extruded M1 macrophage's characteristic (Supporting Information Fig. S3A). As demonstrated in Figs. S3B and S3C, the expression of inflammatory cytokines was significantly higher in M1-NVs compared to M0-NVs at both gene and protein levels. To investigate the internalization of M1-NVs into cancer cells, we stained M1-NVs with DiR solution, and observed their fusion with cancer cell membranes (Fig. S3D). Based on these findings, we selected M1-NVs as adjuvants that were capable of activating immune cells for cancer immunotherapy. We designed functional nanoparticles with dual functions by integrating gene therapy and immunotherapy for effective cancer treatment (Fig. 1). The Bcl2-siRNA-loaded LNPs were coated with M1-NVs (Fig. 1A), which serves as an immune adjuvant by delivering inflammatory cytokines. To determine the optimal ratio for coating LNPs with M1-NVs, we performed nanoparticle tracking analysis (NTA) on mixtures with different particle number ratios of M1-NVs and LNPs to prepare core–shell nanoparticles. We extruded the mixtures through a mini-extruder, and passed them through a membrane with 100 nm pores several times, similar to the preparation of cell membrane-coated nanoparticles in previous studies66, 67, 68, 69. This process resulted in M1-C-LNPs. As the number of LNPs in the total number of particles increased, the amount of genes enclosed in M1-C-LNPs gradually increased. Conversely, as the number of M1-NVs in the total number of particles increased, the amount of inflammatory cytokines in M1-C-LNPs gradually increased (Supporting Information Figs. S4A and S4B). We examined the coating efficiency of M1-C-LNPs using flow cytometry. NVs were prepared from a macrophage cell line expressing GFP to detect fluorescent signals, and LNPs were labelled with DiR (Fig. 2A). When mixtures containing equal numbers of GFP-M1-NVs and DiR-LNPs were hybridized in a core–shell format through a mini-extruder, the coating efficiency of M1-C-LNPs at a 1:1 particle number ratio was approximately 66% of the total particles, the highest among the various particle number ratios (Fig. 2B). Based on these results, we chose a 1:1 particle number ratio of M1-NVs to LNPs for the preparation of optimized M1-C-LNPs (Fig. 2C). We used M0 macrophage-derived cellular nanovesicle-coated LNPs (M0-C-LNPs) as a negative control. The particle sizes of M0-C-LNPs and M1-C-LNPs were approximately 193 nm and 203 nm, respectively (Fig. 2D). The zeta-potentials of M0-C-LNPs and M1-C-LNPs were approximately −16 mV and −15 mV, respectively (Fig. 2E). TEM analysis revealed that M1-C-LNPs retained their spherical morphology (Fig. 2F). After coating Bcl2-siRNA-loaded LNPs with macrophage-derived NVs, we found the exosome markers TSG101 and CD63 to be expressed in both M0-C-LNPs and M1-C-LNPs (Fig. 2G). To determine whether FAM-siRNA-loaded on LNPs was retained on M0-C-LNPs and M1-C-LNPs after coating with macrophage-derived NVs, we performed a fluorescence assay (Fig. 2H). We observed that M0-C-LNPs and M1-C-LNPs were loaded with approximately 1.8 μg of siRNA per 1 × 1011 particles, a reduced amount compared to LNPs, likely due to siRNA leakage during the physical extrusion process. To verify the cellular internalization of the nanoparticles, CT26 cancer cells were treated with LNPs, M0-C-LNPs, or M1-C-LNPs, and confocal microscopy was employed to observe the intracellular presence of fluorescently labelled siRNA (FAM-siRNA) within the nanoparticles (Fig. 2I). The analysis revealed that M1-C-LNPs were the most effective in delivering siRNA into the cytoplasm of cancer cells, in comparison to the other groups. To induce their activation, M1 macrophages interact with other immune cells, such as T and NK cells. This process is regulated by mediators like inflammatory cytokines in M1 macrophages63,70,71. In Fig. S3, M1 macrophage-derived NVs used for the preparation of M1-C-LNPs expressed inflammatory cytokines at both the mRNA and protein levels. After coating the LNPs with macrophage-derived NVs, the content of inflammatory cytokines in the particles was examined using real-time PCR and Western blotting (Fig. 2J and K). M1-C-LNPs maintained higher expression levels of inflammatory cytokines, compared to M0-C-LNPs.
Figure 2.

Development and characterization of M1 macrophage-derived cellular nanovesicle-coated lipid nanoparticles (M1-C-LNPs). (A) Illustration and representative gating displaying the synthesis rate of M1-C-LNPs for GFP-expressing M1 macrophage-derived cellular nanovesicle (M1-NVs)-coated DiR-stained LNPs using flow cytometry. (B) Efficiency of lipid nanoparticles (LNPs) with M1-NVs estimated by flow cytometry. (C) Method of synthesizing M1-C-LNPs by coating M1-NVs on siRNA-loaded LNPs through extrusion. (D and E) Average diameter and surface zeta-potential of M0 macrophage-derived NV-coated lipid nanoparticles (M0-C-LNPs) and M1-C-LNPs confirmed by nanoparticle tracking analysis (NTA). (F) Representative transmission electron microscopy (TEM) image of M1-C-LNPs. Scale bar: 100 nm. (G) Protein level expression of exosome markers in M0-C-LNPs and M1-C-LNPs examined by Western blotting. (H) Quantitative evaluation of loaded gene in LNPs, M0-C-LNPs, or M1-C-LNPs using the ribogreen assay. (I) Representative confocal images of CT26 cells 4 h after treatment with LNPs, M0-C-LNPs, or M1-C-LNPs. Green, red, and blue colors in images indicate FAM-siRNA, lysosome, and nucleus, respectively. Scale bar: 10 μm. (J) Expression of mRNA levels of inflammatory cytokines in M0-C-LNPs and M1-C-LNPs, assessed by real-time PCR. (K) Expression of protein levels of inflammatory cytokines in M0-C-LNPs and M1-C-LNPs, analysed by Western blotting. Data are presented as the mean ± SD (n = 3, ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, ∗∗∗∗P < 0.0001).
3.2. In vitro analysis of cancer cell apoptosis induced by Bcl2-siRNA-loaded M1-C-LNPs
Next, we evaluated the induction of apoptosis in CT26 cells by Bcl2-siRNA-encapsulated in M1-C-LNPs compared with other nanoparticles treated groups in vitro (Fig. 3). Bcl2 is an anti-apoptotic gene that plays a key role in regulating programmed cell death (i.e., apoptosis), and is currently being actively researched for the treatment of various cancers that include leukemia, lymphoma, and solid tumor72,73. When the cell viable rate for apoptosis was quantified to compare with each nanoparticle, M1-C-LNPs exhibited a greater cancer-killing effect than LNPs, M1-NVs, and M0-C-LNPs (Fig. 3A). We found that Bcl2 mRNA expression was significantly suppressed in CT26 cells treated with LNPs or M1-C-LNPs encapsulated with Bcl2-siRNA (Fig. 3B). Interestingly, M1-C-LNP treatment in CT26 cells increased apoptosis-related markers at both the gene and protein levels (Fig. 3C and D). Live/dead staining of CT26 cells treated with M1-C-LNPs revealed effective induction of apoptosis (Fig. 3E), consistent with the protein expression results in Fig. 3D. These results indicate that the heightened apoptotic effect of M1-C-LNPs can be attributed to the synergistic combination of the delivered gene within LNPs and the inflammatory cytokines originating from M1-NVs (Figs. S1 and S3).
Figure 3.

In vitro analysis of apoptosis induction in CT26 cells through Bcl2-siRNA delivery by M1 macrophage-derived cellular nanovesicle-coated lipid nanoparticles (M1-C-LNPs). (A) In vitro analysis for inducing apoptosis in CT26 cells through Bcl2-siRNA delivery or/and inflammatory cytokines of each nanoparticle. (B) Expression of Bcl2 mRNA levels in CT26 cells after treatment with PBS, M1-NVs, LNPs, or M1-C-LNPs. (C and D) Representative image and quantitative analysis of expression for protein levels of apoptosis-related markers in CT26 cells after treatment with PBS, M1-NVs, LNPs, or M1-C-LNPs. All values are normalized against the control group. (E) Representative images of CT26 cells stained by Live/Dead staining after treatment with PBS, M1-NVs, LNPs, or M1-C-LNPs. Scale bar: 50 μm. Data are presented as the mean ± SD (n = 3, ∗∗P < 0.01, ∗∗∗P < 0.001, ∗∗∗∗P < 0.0001; ns, not significant).
3.3. Investigating the interaction of M1-C-LNPs with immune cells
Gene delivery of Bcl2 targeting siRNAs is known to inhibit BCL2 expression and induce apoptosis in most cells74,75. Based on the effective delivery of siRNAs-loaded on M1-C-LNPs to cancer cells, as shown in Fig. 3, we examined whether M1-C-LNPs induce apoptosis by delivering the encapsulated Bcl2-siRNA into immune cells such as T and NK cells, which play crucial roles in cancer immunotherapy. Before further quantifying gene transfer via M1-C-LNPs in immune cells, we loaded mRNA encoding luciferase (Luc-mRNA) instead of siRNA into LNPs and M1-C-LNPs, and treated T and NK cells. Both LNPs and M1-C-LNPs effectively delivered Luc-mRNA to CT26 cells, which expressed the protein, but at a low expression rate in both T cells and NK cells (Fig. 4A). To confirm the localization of the loaded siRNA in M1-C-LNPs, we loaded siRNA labelled with the fluorescent molecule FAM (FAM-siRNA) into M1-C-LNPs, stained the cells with a cell membrane tracer, and investigated T and NK cells using confocal microscopy. As illustrated in Fig. 4B, FAM-siRNA loaded into M1-C-LNPs was not delivered into cytoplasm of T and NK cells, but was observed on the cell membrane surface. Next, Western blotting analysis showed that BCL2 expression in T and NK cells was not reduced by either LNPs or M1-C-LNPs loaded with Bcl2-siRNA (Fig. 4C). This finding reconfirms that that the Bcl2-siRNA was not delivered into immune cells via the nanoparticles. Interestingly, the cytotoxicity by apoptosis by each nanoparticle was hardly observed, when T and NK cells were treated with the same concentration of M1-C-LNPs that were administered to cancer cells (Fig. 4D). These results collectively suggest that the delivery of siRNA loaded on M1-C-LNPs to intratumoral immune cells is limited, unlike in cancer cells. This phenomenon may be attributed to immune cells being generally less receptive to foreign substances than other cell types, particularly before activation76,77. Consequently, transfection of immune cells using viral or non-viral vectors is known to be challenging24,78.
Figure 4.

Analysis of gene delivery and activation in T and NK cells after M1 macrophage-derived cellular nanovesicle-coated lipid nanoparticles (M1-C-LNPs) treatment. (A) Evaluation of luciferase activity in CT26, T, and NK cells after treatment with luciferase-RNA-loaded LNPs or M1-C-LNPs. (B) Representative confocal images of T and NK cells 4 h after treatment with PBS, LNPs, or M1-C-LNPs. Pink and green colors in images indicate T cell membrane and FAM-siRNA, respectively. Scale bar: 5 μm. (C) Expression of protein levels of BCL2 in T and NK cells after treatment with PBS, LNPs, or M1-C-LNPs. (D) Cell viability of T and NK cells after treatment with PBS, M1-NVs, LNPs, or M1-C-LNPs. (E) Representative images and quantitative analysis of expression for protein levels of Perforin and Granzyme in PBS, M1-NVs, LNPs, or M1-C-LNPs treated T and NK cells, analysed by Western blotting. (F) Cancer cell-killing capability of T and NK cells treated with PBS, M1-NVs, LNPs, or M1-C-LNPs. CT26 cells were directly co-cultured with T and NK cells after treatment with each nanoparticle. Data are presented as the mean ± SD (n = 3, ∗∗P < 0.01, ∗∗∗P < 0.001, ∗∗∗∗P < 0.0001).
3.4. Cancer killing capability of T and NK cells enhanced by M1-C-LNPs
M1-C-LNPs, the Bcl2-siRNA-loaded LNPs coated with M1-NVs by extrusion, were designed to serve dual-functionality for genetic therapy and immunotherapy against cancer (Figure 1, Figure 2). Especially, M1-NVs were selected as an immune adjuvant by delivering inflammatory cytokines to enhance the efficacy of immunotherapy (Fig. S3). Based on these results, to investigate the activation of T and NK cells by M1-C-LNPs, we investigated the activation of T and NK cells by M1-C-LNPs in aspect of the granule-mediated apoptotic capability (Fig. 4E and F). Granule-mediated apoptosis markers, such as perforin and granzyme, were significantly increased in M1-C-LNP treated T and NK cells (Fig. 4E). To directly evaluate the cancer cell killing ability of these activated effector immune cells, we co-cultured the M1-C-LNP-treated T and NK cells with CT26 cells (Fig. 4F). Control groups (treated only with PBS), along with the M1-NV and LNP-treated groups, demonstrated partial killing of cancer cells. However, T and NK cells treated with M1-C-LNPs were markedly more effective in killing the co-cultured cancer cells. These findings suggest that M1-C-LNPs activate T and NK cells, subsequently inducing granule-mediated cell death in cancer cells. Although M1-C-LNPs have limited entry into T and NK cells, as evidenced in Fig. 4B, the activation of these immune cells is likely attributable to the interaction with membrane proteins from M1-NVs when M1-C-LNPs adhere to the cell membranes71,79, or due to cytokines released from M1-C-LNPs80.
3.5. Activation of intratumoral T and NK cells by long retention of M1-C-LNPs
We next investigated the tumor growth inhibition effect of M1-C-LNPs in a CT26 tumor-bearing mouse model (Fig. 5). A tumor model was established by inoculating CT26 colon cancer cells into BALB/c or BALB/c nude mice to test the in vivo tumor growth inhibition by M1-C-LNP administration. When the average tumor size reached 100 mm³, three intratumoral injections of PBS, M1-NVs, LNPs, or M1-C-LNPs, were given every other day (Fig. 5A). Before considering the antitumor therapeutic effect, we assessed the intratumoral retention of M1-C-LNPs using in vivo fluorescence imaging (Supporting Information Fig. S5 and Fig. 5B). Each nanoparticle was stained with DiR, an NIR fluorescent dye, and the fluorescence signal of the intratumorally injected nanoparticles was monitored over time. Seven days after intratumoral injection, there was no significant difference between the fluorescence intensity of M1-NVs, LNP, and M1-C-LNP (Fig. S5). However, after 15 days, the fluorescence intensity of M1-NVs and M1-C-LNPs persisted in the solid tumor, while the fluorescence signal of LNPs had almost vanished (Fig. 5B). To investigate why M1-NVs and M1-C-LNPs remained in the solid tumor longer, we examined the tumor-interacting proteins expressed on M1-NVs, the outer membrane layer of M1-C-LNPs. ICAM-1 is known to play a role in mediating leukocyte adhesion and the adhesion of inflammatory macrophages to endothelial cells and cancer cells81,82. Western blotting analysis revealed that both M1-NVs and M1-C-LNPs expressed ICAM-1 (Fig. 5C). These results suggest that the longer retention of M1-C-LNPs in the solid tumor is due to adhesion molecules like ICAM-1, which are expressed by M1-NVs. We next investigated the extend for infiltration and activation of effector immune cells by M1-C-LNPs in solid tumor (Fig. 5D and Supporting Information Fig. S6). The populations of intratumoral cytotoxic T, NK, and T helper cells in all groups did not change significantly after treatment, compared to the control group. This result could consistently provide another evidence that the delivery of Bcl2-siRNA loaded on M1-C-LNPs to effector immune cells was limited, and did not result in immune cell death (Fig. 4). However, the expression of granzyme on cytotoxic T and NK cells was significantly increased in tumor treated with M1-NVs or M1-C-LNPs compared to controls, demonstrating that the pro-inflammatory cytokines of M1-NVs explored earlier also induce the expression of granules on intratumoral T and NK cells. The expression of TNFα in cytotoxic T cells and IFNγ in NK cells was significantly higher in solid tumor treated with M1-C-LNPs, compared to other groups, suggesting that M1-C-LNPs could activate cytotoxic T and NK cells to induce granule-mediated cell death against cancer cells both in vitro and in vivo. Furthermore, tumor-associated macrophages (TAMs), which are known for their role in promoting an immunosuppressive environment in tumor, induced cell death with approximately 40%, while the surviving cells were polarized into M1 macrophages, exhibiting enhanced capability to target cancer cells after treatment of M1-C-LNPs in macrophages (Data not shown). These results suggest that the intratumoral injection of M1-C-LNPs can both induce apoptosis in cancer cells, and stimulate the anti-cancer immune activity of T and NK cells.
Figure 5.

In vivo anti-tumor therapeutic effect of M1 macrophage-derived cellular nanovesicle-coated lipid nanoparticles (M1-C-LNPs) for combining gene therapy with immunotherapy on CT26 mouse model. (A) In vivo nanoparticle treatment schedule for PBS, M1-NVs, LNPs, or M1-C-LNPs. CT26 cells were subcutaneously injected into the right flank of BALB/c or BALB/c nude mice. When the tumor size reached a volume of ∼100 mm³, mice began treatment with PBS, M1-NVs, LNPs, or M1-C-LNPs. (B) In vivo quantitative fluorescence analysis of tumor injected with DiR-stained M1-NVs, LNPs, or M1-C-LNPs in BALB/c nude mice. All values are normalized against the M1-NV group. (C) Representative image and quantitative analysis of protein expression levels for ICAM-1 and exosome marker in M1-NVs, LNPs, or M1-C-LNPs analysed by Western blotting. (D) Flow cytometric immunophenotyping of activated cytokine-related markers of T and NK cells in the tumor of CT26 tumor-bearing mice model after intratumoral injection of PBS, M1-NVs, LNPs, or M1-C-LNPs. (E) Representative image and quantitative analysis of protein expression levels for apoptosis and tumor microenvironment-related markers in tumor after the injection of PBS, M1-NVs, LNPs, or M1-C-LNPs estimated by Western blotting. All values are normalized against the control group. (F) Representative images of hematoxylin and eosin (H&E) in tumor after the injection of PBS, M1-NVs, LNPs, or M1-C-LNPs. Scale bar: 300 μm in H&E. (G) Average tumor size growth after the injection of PBS, M1-NVs, LNPs, or M1-C-LNPs. Data are presented as the mean ± SEM (n = 3 or 5, ∗P < 0.05, ∗∗P < 0.01).
3.6. Anti-tumor therapeutic effect of M1-C-LNPs on CT26 mouse model
Next, we evaluated the induction of apoptosis by Bcl2 silencing via M1-C-LNPs in solid tumor. Remarkably, BCL2 expression was completely suppressed only in M1-C-LNPs injected solid tumor, when estimating the expression of apoptosis-related markers. This outcome is likely due to the prolonged persistence of M1-C-LNPs in tumor, leading to efficient gene delivery into tumor cells. Furthermore, in line with the BCL2 expression results, M1-C-LNPs effectively induced apoptosis in solid tumor (Fig. 5E). We also conducted histological analysis to examine tumor density and intratumoral proliferating cells following treatment (Fig. 5F). Compared to the other groups, the M1-C-LNP-treated group had less dense tumor. When monitoring the change in average tumor size over time for each group (Fig. 5G), we observed rapid tumor growth in the PBS, M1-NVs, or LNP-treated groups, while tumor growth was significantly reduced in the M1-C-LNP-treated group. The findings suggest that due to their long retention in the tumor, and their excellent gene delivery efficiency, M1-C-LNPs can effectively inhibit tumor growth. Then, we assessed the toxicity of M1-C-LNPs by verifying blood biochemistry, histocompatibility, and body weight after administration of the nanoparticles (Supporting Information Fig. S7). Blood biochemical and histological analyses revealed that mice treated with M1-NVs exhibited significantly increased ALP levels and alveolar hemorrhage, while M1-C-LNPs demonstrated biocompatibility, and did not cause any adverse effects (Figs. S7A and S7B). No differences in body weight changes were observed among all experimental groups (Fig. S7C). Interestingly, administering a simple mixture of M1-NVs and LNPs prior to extrusion for coating led to increased levels of ALP, TG, and CPK in blood biochemistry tests, similar to the results seen in the group treated with M1-NVs alone (Supporting Information Fig. S8A). The residence time of LNP in solid tumors was relatively short, resulting in a less effective tumor growth inhibition rate (Fig. S8B). These findings suggest that M1-NVs alone may induce in vivo toxicity, potentially due to the presence of inflammatory cytokines such as IL-6, iNOS, TNFα, and IL-1β. However, M1-C-LNPs exhibited relatively reduced toxicity, which could be attributed to the controlled cytokine release, owing to the dense lipid structure in M1-C-LNPs (Fig. 2F).
4. Conclusions
In this study, we developed bifunctionalized M1-C-LNPs containing Bcl2-siRNA and immunomodulatory cytokines to combine gene therapy and immunotherapy in cancer treatment. M1-C-LNPs successfully integrated the properties of LNPs encapsulated with siRNA through an extrusion process and M1-NVs enriched with inflammatory cytokines. The siRNA-loaded on M1-C-LNPs was specifically delivered to cancer cells, inducing cell death only in cancer cells, without affecting immune cells, such as effector immune cells in tumor microenvironment. However, the inflammatory cytokines in M1-C-LNPs stimulated T cells and NK cells, activating granule-mediated cancer cell killing function. Intratumoral injection of M1-C-LNPs demonstrated excellent intratumoral retention due to adhesion molecules on the surface of M1-NVs, contributing to the therapeutic efficacy and safety of the nanoparticles. Ultimately, M1-C-LNPs exhibited superior efficacy in inhibiting tumor growth, compared to other groups. These findings indicate that M1-C-LNPs could be utilized as an optimal nanoplatform for effective genetic-immunotherapy against solid tumor.
Author contributions
Ha Eun Shin, Chun Gwon Park, and Wooram Park conceived the conceptualization of the project, devising the experimental design and overseeing manuscript revisions. Ha Eun Shin, Jun-Hyeok Han, Seungyong Shin, and Ga-Hyun Bae participated part of the experiments. Ha Eun Shin wrote the manuscript. Boram Son, Tae-Hyung Kim, Hee Ho Park, Chun Gwon Park, and Wooram Park reviewed and edited the manuscript. Chun Gwon Park and Wooram Park were responsible for acquiring the funding for this project. All of the authors have read and approved the final manuscript.
Conflicts of interest
The authors have no conflicts of interest to declare.
Acknowledgments
The illustrations were created with BioRender.com. This work was supported by a Basic Science Research Program grant through the National Research Foundation of Korea (NRF) grants (Nos. 2021R1A2C4001776, RS-2023-00218648, RS-2023-00242443, and 2023–00208913) of the Republic of Korea, funded by the Ministry of Science and ICT (MSIT) of the Republic of Korea, a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (No. RS-2023-00266015), and the KIST Institutional Program (No. 2E32351-23-130) of the Republic of Korea.
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting information to this article can be found online at https://doi.org/10.1016/j.apsb.2024.03.004.
Contributor Information
Chun Gwon Park, Email: chunpark@skku.edu.
Wooram Park, Email: parkwr@skku.edu.
Appendix A. Supporting information
The following is the Supporting information to this article:
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Soerjomataram I., Bray F. Planning for tomorrow: global cancer incidence and the role of prevention 2020–2070. Nat Rev Clin Oncol. 2021;18:663–672. doi: 10.1038/s41571-021-00514-z. [DOI] [PubMed] [Google Scholar]
- 3.Mousazadeh H., Pilehvar-Soltanahmadi Y., Dadashpour M., Zarghami N. Cyclodextrin based natural nanostructured carbohydrate polymers as effective non-viral siRNA delivery systems for cancer gene therapy. J Control Release. 2021;330:1046–1070. doi: 10.1016/j.jconrel.2020.11.011. [DOI] [PubMed] [Google Scholar]
- 4.Lu Y., Zhong L., Jiang Z., Pan H., Zhang Y., Zhu G., et al. Cationic micelle-based siRNA delivery for efficient colon cancer gene therapy. Nanoscale Res Lett. 2019;14:1–9. doi: 10.1186/s11671-019-2985-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen D., Yang X., Liu M., Zhang Z., Xing E. Roles of miRNA dysregulation in the pathogenesis of multiple myeloma. Cancer Gene Ther. 2021;28:1256–1268. doi: 10.1038/s41417-020-00291-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mirza Z., Karim S. Nanoparticles-based drug delivery and gene therapy for breast cancer: recent advancements and future challenges. Semin Cancer Biol. 2021;69:226–237. doi: 10.1016/j.semcancer.2019.10.020. [DOI] [PubMed] [Google Scholar]
- 7.Yahya E.B., Alqadhi A.M. Recent trends in cancer therapy: a review on the current state of gene delivery. Life Sci. 2021;269 doi: 10.1016/j.lfs.2021.119087. [DOI] [PubMed] [Google Scholar]
- 8.Kanasty R., Dorkin J.R., Vegas A., Anderson D. Delivery materials for siRNA therapeutics. Nat Mater. 2013;12:967–977. doi: 10.1038/nmat3765. [DOI] [PubMed] [Google Scholar]
- 9.Setten R.L., Rossi J.J., Han S.-P. The current state and future directions of RNAi-based therapeutics. Nat Rev Drug Discov. 2019;18:421–446. doi: 10.1038/s41573-019-0017-4. [DOI] [PubMed] [Google Scholar]
- 10.Yin H., Kauffman K.J., Anderson D.G. Delivery technologies for genome editing. Nat Rev Drug Discov. 2017;16:387–399. doi: 10.1038/nrd.2016.280. [DOI] [PubMed] [Google Scholar]
- 11.Akinc A., Maier M.A., Manoharan M., Fitzgerald K., Jayaraman M., Barros S., et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat Nanotechnol. 2019;14:1084–1087. doi: 10.1038/s41565-019-0591-y. [DOI] [PubMed] [Google Scholar]
- 12.Baden L.R., El Sahly H.M., Essink B., Kotloff K., Frey S., Novak R., et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N Engl J Med. 2021;384:403–416. doi: 10.1056/NEJMoa2035389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Polack F.P., Thomas S.J., Kitchin N., Absalon J., Gurtman A., Lockhart S., et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N Engl J Med. 2020;383:2603–2615. doi: 10.1056/NEJMoa2034577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen D.S., Mellman I. Elements of cancer immunity and the cancer–immune set point. Nature. 2017;541:321–330. doi: 10.1038/nature21349. [DOI] [PubMed] [Google Scholar]
- 15.Abbott M., Ustoyev Y. Cancer and the immune system: the history and background of immunotherapy. Semin Oncol Nurs. 2019;35 doi: 10.1016/j.soncn.2019.08.002. [DOI] [PubMed] [Google Scholar]
- 16.Park W., Heo Y.J., Han D.K. New opportunities for nanoparticles in cancer immunotherapy. Biomater Res. 2018;22:1–10. doi: 10.1186/s40824-018-0133-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu J., Liew S.S., Wang J., Pu K. Bioinspired and biomimetic delivery platforms for cancer vaccines. Adv Mater. 2022;34 doi: 10.1002/adma.202103790. [DOI] [PubMed] [Google Scholar]
- 18.Huang A.C., Zappasodi R. A decade of checkpoint blockade immunotherapy in melanoma: understanding the molecular basis for immune sensitivity and resistance. Nat Immunol. 2022;23:660–670. doi: 10.1038/s41590-022-01141-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sellars M.C., Wu C.J., Fritsch E.F. Cancer vaccines: building a bridge over troubled waters. Cell. 2022;185:2770–2788. doi: 10.1016/j.cell.2022.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Topalian S.L., Taube J.M., Pardoll D.M. Neoadjuvant checkpoint blockade for cancer immunotherapy. Science. 2020;367 doi: 10.1126/science.aax0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lima-Sousa R., Melo B.L., Alves C.G., Moreira A.F., Mendonça A.G., Correia I.J., et al. Combining photothermal-photodynamic therapy mediated by nanomaterials with immune checkpoint blockade for metastatic cancer treatment and creation of immune memory. Adv Funct Mater. 2021;31 [Google Scholar]
- 22.He C., Yu L., Yao H., Chen Y., Hao Y. Combinatorial photothermal 3D-printing scaffold and checkpoint blockade inhibits growth/metastasis of breast cancer to bone and accelerates osteogenesis. Adv Funct Mater. 2021;31 [Google Scholar]
- 23.June C.H., O'Connor R.S., Kawalekar O.U., Ghassemi S., Milone M.C. CAR T cell immunotherapy for human cancer. Science. 2018;359:1361–1365. doi: 10.1126/science.aar6711. [DOI] [PubMed] [Google Scholar]
- 24.Shin S., Lee P., Han J., Kim S.N., Lim J., Park D.H., et al. Nanoparticle-based chimeric antigen receptor therapy for cancer immunotherapy. J Tissue Eng Regen Med. 2023;20:371–387. doi: 10.1007/s13770-022-00515-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shao M., Teng X., Guo X., Zhang H., Huang Y., Cui J., et al. Inhibition of calcium signaling prevents exhaustion and enhances anti-leukemia efficacy of CAR-T cells via SOCE-Calcineurin-NFAT and glycolysis pathways. Adv Sci. 2022;9 doi: 10.1002/advs.202103508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wculek S.K., Cueto F.J., Mujal A.M., Melero I., Krummel M.F., Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20:7–24. doi: 10.1038/s41577-019-0210-z. [DOI] [PubMed] [Google Scholar]
- 27.Hou J., Karin M., Sun B. Targeting cancer-promoting inflammation—have anti-inflammatory therapies come of age?. Nat Rev Clin Oncol. 2021;18:261–279. doi: 10.1038/s41571-020-00459-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Momin N., Palmeri J.R., Lutz E.A., Jailkhani N., Mak H., Tabet A., et al. Maximizing response to intratumoral immunotherapy in mice by tuning local retention. Nat Commun. 2022;13:109. doi: 10.1038/s41467-021-27390-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muñoz N.M., Williams M., Dixon K., Dupuis C., McWatters A., Avritscher R., et al. Influence of injection technique, drug formulation and tumor microenvironment on intratumoral immunotherapy delivery and efficacy. J Immunother Cancer. 2021;9 doi: 10.1136/jitc-2020-001800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Irvine D.J., Dane E.L. Enhancing cancer immunotherapy with nanomedicine. Nat Rev Immunol. 2020;20:321–334. doi: 10.1038/s41577-019-0269-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han J.-H., Lee Y.Y., Shin H.E., Han J., Kang J.M., Wang C.-P.J., et al. Image-guided in situ cancer vaccination with combination of multi-functional nano-adjuvant and an irreversible electroporation technique. Biomaterials. 2022;289 doi: 10.1016/j.biomaterials.2022.121762. [DOI] [PubMed] [Google Scholar]
- 32.Melero I., Castanon E., Alvarez M., Champiat S., Marabelle A. Intratumoural administration and tumour tissue targeting of cancer immunotherapies. Nat Rev Clin Oncol. 2021;18:558–576. doi: 10.1038/s41571-021-00507-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen S., Saeed A.F., Liu Q., Jiang Q., Xu H., Xiao G.G., et al. Macrophages in immunoregulation and therapeutics. Signal Transduct Targeted Ther. 2023;8:207. doi: 10.1038/s41392-023-01452-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meli V.S., Veerasubramanian P.K., Atcha H., Reitz Z., Downing T.L., Liu W.F. Biophysical regulation of macrophages in health and disease. J Leukoc Biol. 2019;106:283–299. doi: 10.1002/JLB.MR0318-126R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang P.M.K., Nikolic-Paterson D.J., Lan H.Y. Macrophages: versatile players in renal inflammation and fibrosis. Nat Rev Nephrol. 2019;15:144–158. doi: 10.1038/s41581-019-0110-2. [DOI] [PubMed] [Google Scholar]
- 36.Schlundt C., Fischer H., Bucher C.H., Rendenbach C., Duda G.N., Schmidt-Bleek K. The multifaceted roles of macrophages in bone regeneration: a story of polarization, activation and time. Acta Biomater. 2021;133:46–57. doi: 10.1016/j.actbio.2021.04.052. [DOI] [PubMed] [Google Scholar]
- 37.Shapouri-Moghaddam A., Mohammadian S., Vazini H., Taghadosi M., Esmaeili S.A., Mardani F., et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. 2018;233:6425–6440. doi: 10.1002/jcp.26429. [DOI] [PubMed] [Google Scholar]
- 38.Sun Y.Y., Li X.F., Meng X.M., Huang C., Zhang L., Li J. Macrophage phenotype in liver injury and repair. Scand J Immunol. 2017;85:166–174. doi: 10.1111/sji.12468. [DOI] [PubMed] [Google Scholar]
- 39.Guerriero J.L. Macrophages: their untold story in T cell activation and function. Int Rev Cell Mol Biol. 2019;342:73–93. doi: 10.1016/bs.ircmb.2018.07.001. [DOI] [PubMed] [Google Scholar]
- 40.Murray P.J., Allen J.E., Biswas S.K., Fisher E.A., Gilroy D.W., Goerdt S., et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou J., Zhang S., Guo C. Crosstalk between macrophages and natural killer cells in the tumor microenvironment. Int Immunopharm. 2021;101 doi: 10.1016/j.intimp.2021.108374. [DOI] [PubMed] [Google Scholar]
- 42.Cess C.G., Finley S.D. Multi-scale modeling of macrophage—T cell interactions within the tumor microenvironment. PLoS Comput Biol. 2020;16 doi: 10.1371/journal.pcbi.1008519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moller A., Lobb R.J. The evolving translational potential of small extracellular vesicles in cancer. Nat Rev Cancer. 2020;20:697–709. doi: 10.1038/s41568-020-00299-w. [DOI] [PubMed] [Google Scholar]
- 44.Simeone P., Bologna G., Lanuti P., Pierdomenico L., Guagnano M.T., Pieragostino D., et al. Extracellular vesicles as signaling mediators and disease biomarkers across biological barriers. Int J Mol Sci. 2020;21:2514. doi: 10.3390/ijms21072514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pan H., Zheng M., Ma A., Liu L., Cai L. Cell/bacteria-based bioactive materials for cancer immune modulation and precision therapy. Adv Mater. 2021;33 doi: 10.1002/adma.202100241. [DOI] [PubMed] [Google Scholar]
- 46.Dash P., Piras A.M., Dash M. Cell membrane coated nanocarriers―an efficient biomimetic platform for targeted therapy. J Control Release. 2020;327:546–570. doi: 10.1016/j.jconrel.2020.09.012. [DOI] [PubMed] [Google Scholar]
- 47.Wang Y., Zhang P., Wei Y., Shen K., Xiao L., Miron R.J., et al. Cell-membrane-display nanotechnology. Adv Healthcare Mater. 2021;10 doi: 10.1002/adhm.202001014. [DOI] [PubMed] [Google Scholar]
- 48.Rao L., Wu L., Liu Z., Tian R., Yu G., Zhou Z., et al. Hybrid cellular membrane nanovesicles amplify macrophage immune responses against cancer recurrence and metastasis. Nat Commun. 2020;11:4909. doi: 10.1038/s41467-020-18626-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang E., Phan P., Zhao Z. Cellular nanovesicles for therapeutic immunomodulation: a perspective on engineering strategies and new advances. Acta Pharm Sin B. 2023;13:1789–1827. doi: 10.1016/j.apsb.2022.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Milane L., Ganesh S., Shah S., Duan Z.F., Amiji M. Multi-modal strategies for overcoming tumor drug resistance: hypoxia, the Warburg effect, stem cells, and multifunctional nanotechnology. J Control Release. 2011;155:237–247. doi: 10.1016/j.jconrel.2011.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Butowska K., Han X., Gong N., El-Mayta R., Haley R.M., Xue L., et al. Doxorubicin-conjugated siRNA lipid nanoparticles for combination cancer therapy. Acta Pharm Sin B. 2023;13:1429–1437. doi: 10.1016/j.apsb.2022.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haussecker D. Current issues of RNAi therapeutics delivery and development. J Control Release. 2014;195:49–54. doi: 10.1016/j.jconrel.2014.07.056. [DOI] [PubMed] [Google Scholar]
- 53.Kulkarni J.A., Witzigmann D., Chen S., Cullis P.R., van der Meel R. Lipid nanoparticle technology for clinical translation of siRNA therapeutics. Acc Chem Res. 2019;52:2435–2444. doi: 10.1021/acs.accounts.9b00368. [DOI] [PubMed] [Google Scholar]
- 54.Brown D.G., Wobst H.J. A decade of FDA-approved drugs (2010–2019): trends and future directions. J Med Chem. 2021;64:2312–2338. doi: 10.1021/acs.jmedchem.0c01516. [DOI] [PubMed] [Google Scholar]
- 55.Teijaro J.R., Farber D.L. COVID-19 vaccines: modes of immune activation and future challenges. Nat Rev Immunol. 2021;21:195–197. doi: 10.1038/s41577-021-00526-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guerrini G., Magrì D., Gioria S., Medaglini D., Calzolai L. Characterization of nanoparticles-based vaccines for COVID-19. Nat Nanotechnol. 2022;17:570–576. doi: 10.1038/s41565-022-01129-w. [DOI] [PubMed] [Google Scholar]
- 57.Kehr S., Haydn T., Bierbrauer A., Irmer B., Vogler M., Fulda S. Targeting BCL-2 proteins in pediatric cancer: dual inhibition of BCL-XL and MCL-1 leads to rapid induction of intrinsic apoptosis. Cancer Lett. 2020;482:19–32. doi: 10.1016/j.canlet.2020.02.041. [DOI] [PubMed] [Google Scholar]
- 58.Cimmino A., Calin G.A., Fabbri M., Iorio M.V., Ferracin M., Shimizu M., et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kulkarni J.A., Darjuan M.M., Mercer J.E., Chen S., Van Der Meel R., Thewalt J.L., et al. On the formation and morphology of lipid nanoparticles containing ionizable cationic lipids and siRNA. ACS Nano. 2018;12:4787–4795. doi: 10.1021/acsnano.8b01516. [DOI] [PubMed] [Google Scholar]
- 60.Siveen K., Kuttan G. Role of macrophages in tumour progression. Immunol Lett. 2009;123:97–102. doi: 10.1016/j.imlet.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 61.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 62.Mosser D.M., Edwards J.P. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Arango Duque G., Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol. 2014;5:491. doi: 10.3389/fimmu.2014.00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu T.-T., Chen T.-L., Chen R.-M. Lipopolysaccharide triggers macrophage activation of inflammatory cytokine expression, chemotaxis, phagocytosis, and oxidative ability via a toll-like receptor 4-dependent pathway: validated by RNA interference. Toxicol Lett. 2009;191:195–202. doi: 10.1016/j.toxlet.2009.08.025. [DOI] [PubMed] [Google Scholar]
- 65.Xu Y., Jagannath C., Liu X.-D., Sharafkhaneh A., Kolodziejska K.E., Eissa N.T. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27:135–144. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shin H.E., Oh S.W., Park W. Hybrid nanovesicle of chimeric antigen receptor (CAR)-engineered cell-derived vesicle and drug-encapsulated liposome for effective cancer treatment. J Ind Eng Chem. 2023;122:127–137. [Google Scholar]
- 67.Lau H.C., Han D.W., Park J., Lehner E., Kals C., Arzt C., et al. GMP-compliant manufacturing of biologically active cell-derived vesicles produced by extrusion technology. J Extracell Vesicles. 2022;1:e70. doi: 10.1002/jex2.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hong J., Kang M., Jung M., Lee Y.Y., Cho Y., Kim C., et al. T-cell-derived nanovesicles for cancer immunotherapy. Adv Mater. 2021;33 doi: 10.1002/adma.202101110. [DOI] [PubMed] [Google Scholar]
- 69.Kang M., Lee S.H., Kwon M., Byun J., Kim D., Kim C., et al. Nanocomplex-mediated in vivo programming to chimeric antigen receptor-M1 macrophages for cancer therapy. Adv Mater. 2021;33 doi: 10.1002/adma.202103258. [DOI] [PubMed] [Google Scholar]
- 70.Najafi M., Hashemi Goradel N., Farhood B., Salehi E., Nashtaei M.S., Khanlarkhani N., et al. Macrophage polarity in cancer: a review. J Cell Biochem. 2019;120:2756–2765. doi: 10.1002/jcb.27646. [DOI] [PubMed] [Google Scholar]
- 71.Bellora F., Castriconi R., Dondero A., Reggiardo G., Moretta L., Mantovani A., et al. The interaction of human natural killer cells with either unpolarized or polarized macrophages results in different functional outcomes. Proc Natl Acad Sci U S A. 2010;107:21659–21664. doi: 10.1073/pnas.1007654108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kang M.H., Reynolds C.P. Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res. 2009;15:1126–1132. doi: 10.1158/1078-0432.CCR-08-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Carneiro B.A., El-Deiry W.S. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol. 2020;17:395–417. doi: 10.1038/s41571-020-0341-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Carrington E.M., Zhan Y., Brady J.L., Zhang J.G., Sutherland R.M., Anstee N.S., et al. Anti-apoptotic proteins BCL-2, MCL-1 and A1 summate collectively to maintain survival of immune cell populations both in vitro and in vivo. Cell Death Differ. 2017;24:878–888. doi: 10.1038/cdd.2017.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chipuk J.E., Fisher J.C., Dillon C.P., Kriwacki R.W., Kuwana T., Green D.R. Mechanism of apoptosis induction by inhibition of the anti-apoptotic BCL-2 proteins. Proc Natl Acad Sci U S A. 2008;105:20327–20332. doi: 10.1073/pnas.0808036105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Medzhitov R., Janeway C.A., Jr. Decoding the patterns of self and nonself by the innate immune system. Science. 2002;296:298–300. doi: 10.1126/science.1068883. [DOI] [PubMed] [Google Scholar]
- 77.Riaz M.K., Riaz M.A., Zhang X., Lin C., Wong K.H., Chen X., et al. Surface functionalization and targeting strategies of liposomes in solid tumor therapy: a review. Int J Mol Sci. 2018;19:195. doi: 10.3390/ijms19010195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kim K.S., Han J.H., Park J.H., Kim H.K., Choi S.H., Kim G.R., et al. Multifunctional nanoparticles for genetic engineering and bioimaging of natural killer (NK) cell therapeutics. Biomaterials. 2019;221 doi: 10.1016/j.biomaterials.2019.119418. [DOI] [PubMed] [Google Scholar]
- 79.Mattiola I., Pesant M., Tentorio P.F., Molgora M., Marcenaro E., Lugli E., et al. Priming of human resting NK cells by autologous M1 macrophages via the engagement of IL-1β, IFN-β, and IL-15 pathways. J Immunol. 2015;195:2818–2828. doi: 10.4049/jimmunol.1500325. [DOI] [PubMed] [Google Scholar]
- 80.Tang L., Zheng Y., Melo M.B., Mabardi L., Castaño A.P., Xie Y.Q., et al. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat Biotechnol. 2018;36:707–716. doi: 10.1038/nbt.4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bui T.M., Wiesolek H.L., Sumagin R. ICAM-1: a master regulator of cellular responses in inflammation, injury resolution, and tumorigenesis. J Leukoc Biol. 2020;108:787–799. doi: 10.1002/JLB.2MR0220-549R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lawson C., Wolf S. ICAM-1 signaling in endothelial cells. Pharmacol Rep. 2009;61:22–32. doi: 10.1016/s1734-1140(09)70004-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.