

Abstract
The DNA polymerase δ complex (PolD), comprising catalytic subunit POLD1 and accessory subunits POLD2, POLD3, and POLD4, is essential for DNA synthesis and is central to genome integrity. We identified, by whole exome sequencing, a homozygous missense mutation (c.1118A > C; p.K373T) in POLD3 in a patient with Omenn syndrome. The patient exhibited severely decreased numbers of naïve T cells associated with a restricted T-cell receptor repertoire and a defect in the early stages of TCR recombination. The patient received hematopoietic stem cell transplantation at age 6 months. He manifested progressive neurological regression and ultimately died at age 4 years. We performed molecular and functional analysis of the mutant POLD3 and assessed cell cycle progression as well as replication-associated DNA damage. Patient fibroblasts showed a marked defect in S-phase entry and an enhanced number of double-stranded DNA break-associated foci despite normal expression levels of PolD components. The cell cycle defect was rescued by transduction with WT POLD3. This study validates autosomal recessive POLD3 deficiency as a novel cause of profound T-cell deficiency and Omenn syndrome.
Keywords: POLD3, Omenn syndrome, Replication-induced stress, Cell-cycle defect
Introduction
The B-family DNA polymerases are responsible for eukaryotic DNA replication [1]. The DNA polymerase delta complex (PolD), a holoenzyme comprising the catalytic subunit POLD1 and the accessory subunits POLD2, POLD3, and POLD4, synthesizes both the leading and lagging strand during genome duplication [2, 3]. In addition, PolD is essential for the maintenance of genome integrity, as it is the polymerase predominantly responsible for homologous recombination, promotes alternative non-homologous end joining (NHEJ) at sites of double strand breaks, controls break-induced replication, and has a translesion synthesis (TLS) activity [4–7]. POLD3, with POLD2 and POLD4, regulate the activity of POLD1, the activity and stability of PolD, and establish protein–protein interactions [3]. In particular, POLD3 mediates interaction with proliferating cell nuclear antigen (PCNA), which is essential for PolD activity and processivity, and promotes TLS activity within PolD and the polymerase ζ complex (Pol ζ) [8, 9].
Mutations which affect PolD components have been identified. Heterozygous mutations affecting the proofreading domain of POLD1 have been found in patients with a predisposition to colorectal and endometrial cancer. These mutations impair the endonuclease activity of POLD1, leading to genomic instability and a hypermutator phenotype [10, 11]. Meanwhile, heterozygous mutations in the polymerase and CysB domains of POLD1, abrogating POLD1 polymerase but not exonuclease activity, have been associated with autosomal-dominant mandibular hypoplasia, deafness, progeroid features, and lipodystrophy (MDPL) syndrome [12–14]. Recently, biallelic loss-of-function mutations in POLD1 and POLD2, leading to reduced functionality of PolD, have been reported in 6 subjects from 4 unrelated families [15–17]. All identified variants in POLD1 and POLD2 have been missense variants with the exception of one family with an in-frame deletion resulting in the loss of one amino acid [14]. The phenotype of patients with deficient polymerase δ includes immunodeficiency, developmental abnormalities, and replicative stress. This immunodeficiency presents as upper and lower respiratory tract infections [12, 15–17], otitis media [16, 17], and lymphopenia [15–17] with patients requiring immunoglobulin replacement [15–17]. Developmental abnormalities include growth retardation [12, 15, 17], teeth abnormalities [12, 13, 17], and dysmorphic facial features [12–14]. Replicative stress has been shown to increase cancer predisposition in one patient so far [18]. These discoveries revealed a role for Polδ in T and B-cell development and confirmed the emergence of a new subgroup of primary immunodeficiency disorders caused by diminished function of DNA polymerases.
Here, we describe a patient with a novel homozygous mutation in POLD3 associated with a defect in S-phase entry, increased replication-associated DNA damage, and defective T-cell development. The patient presented with profound T-cell deficiency, Omenn syndrome, and developmental defects, consistent with previously described polymerase δ deficient patients.
Methods
Study Oversight
The Ethics Committee of the University Hospitals Leuven (UZLeuven) approved this study (S66910, S63807). The study was performed in compliance with the Declaration of Helsinki for research involving human subjects. Written informed consent was obtained from the parents prior to DNA, PBMCs, and fibroblast isolation.
Sequencing
WES was performed on the index patient and his parents. Genomic DNA samples were prepared from heparinized peripheral blood using QIAamp DNA Blood Midi Kit (QIA-GEN) or Chemagen DNA extraction kit (PerkimElmer). WES libraries were prepared using SureSelect Human V4/V5 enrichment kit (Agilent Technologies). 150 bp paired-end sequencing was performed on the Illumina NovaSeq (Macrogen Europe). BWA software was used for alignment to the Human Reference Genome Build hg19 (http://bio-bwa.sourceforge.net). GATK Unified Genotyper (https://software.broadinstitute.org/gaks.bestpractices) was used to identify SNVs and insertions/ deletions. ANNOVAR (http://anovar.openbioinformatics.org) was used for annotation. For Sanger sequencing confirmation in the patient, parents, and sibling, genomic DNA samples were prepared from heparinized peripheral blood using QIAamp DNA Blood Mini kit (QIAGEN). The region of interest around the end of exon 10 of P0LD3 was sequenced using the primers 5′-TGTAAAACGACGGCCAGTAAGGCGGCATGGGATAGTTG-3′ and 5′-GGTCATAGCTGTTTCCTGAACGCCCAT TCTGTACAACC-3′. Sanger sequencing was performed on an ABI 3730 XL Genetic Analyzer (Applied Biosystems) at the LGC Genomics Facility in Berlin, Germany. Sequencing data were analyzed using Chromas 2.6.5 (http://www.technelysium.com.au).
Cell Culture
Primary dermal fibroblasts were established from skin biopsies following standard procedures and maintained in DMEM/F-12 (1:1) containing L-glutamine and HEPES with 10% FBS and 1% penicillin/streptomycin. HEK 239 T cells were cultured in DMEM + 10% FBS and 1% penicillin/streptomycin. To assess fibroblast proliferation, cells were seeded in 6-well plates at ~ 30% density and imaged with an EVOS M7000 microscope (Thermo Fisher Scientific). Images were analyzed with ImageJ software (NIH).
Plasmids
Myc-DDK tagged POLD3 expression plasmid was purchased from Origene (RC223072). The Q5 Site-Directed Mutagenesis Kit (New England Biolabs) was used to generate the POLD3 mutation in the plasmid. The following specific primers were used for mutagenesis: F: 5′-CCTTCTGTCAcGAGCTCAAGTG-3′, R: 5′-AGGTTCAGGCTCAGTCTT-3′. The cDNA sequence was confirmed by Sanger sequencing.
Lentiviral Transductions
p3D lentiviral vectors with or without wild-type POLD3 were transfected together with p3A, p3B, and p3C packing vectors (kind gifts from Bertrand Boisson) into HEK293T cells. Lentiviral particles were collected, and primary dermal fibroblasts were transduced. Transduced cells were selected with puromycin treatment.
Immunoblot Analysis
Cells were lysed in NP-40 lysis buffer with Halt™ protease inhibitor cocktail (Thermo Fisher Scientific). Cell lysates were separated alongside SeeBlue Plus2 Prestained standard protein ladder (Invitrogen) with a 4–12% Bis–Tris Gel (Invitrogen) and transferred onto PVDF membranes. Membranes were probed overnight with primary antibodies including anti-POLD1 (1:500, sc-17776, Santa Cruz Biotechnology); anti-POLD2 ( 1:1000, HPA026745, MilliporeSigma); anti-POLD3 (1:500, A301-244A-M, Bethyl Laboratories); and anti-βactin (1:9000, A2228, Sigma-Aldrich). Membranes were then incubated with HRP-conjugated anti-mouse IgG (1:5000, 71,045, EMD Millipore) and HRP-conjugated anti-rabbit IgG (1:5000, sc-2357, Santa Cruz Biotechnology) antibodies. Antibody binding was detected by chemiluminescence using ECL, PicoPlus, or Femto substrates (Thermo Fisher Scientific), on a Chemidoc Imager (BioRad).
Real-Time Quantitative PCR (qPCR)
RNA was extracted from primary fibroblasts lysed in TRI-zol reagent (Life Technologies) using the RNeasy Mini Kit (QIAGEN). Reverse transcription was performed using random hexamers and Superscript III reverse strand synthesis kit according to the manufacturer’s instructions (Thermo Fisher Scientific). qPCR for POLD1, POLD2, and POLD3 was performed using SYBR™ Select Master Mix (Applied Biosystems) with a QuantStudio Real-Time PCR System (Thermo Fisher Scientific). Intron-spanning primers were used for POLD1 (5′- CCTACGAGGCCAACGTCG ACT-3′ and 5′- CCACAGCACGTCCGCCTCC −3′), POLD2 (5′-ACTGACCCGTTCATCTTC CCAG-3′ and 5′-CAACAGCACTGTCTGGTCCTCA-3′), and POLD3 (5′-TGAGCCTGAACC TCCTTCTGTC-3′ and 5′- CTGTGCAGGATTCACTCTCGTAG-3′). GAPDH was used for normalization, and relative expression was calculated using the ΔΔCt method (Applied Biosystems).
Vα7.2 Staining
Whole blood was incubated with CD3 (47–0036-42, eBioscience) and Vα7.2 (351,705, BioLegend) antibodies for 30 min on ice. Data were collected with a Canto II cytometer (BD Biosciences) and analyzed with FlowJo software (TreeStar).
Cell-Cycle Analysis
Assays for EdU incorporation into patient fibroblasts were performed using the Click-iT™ EdU Cell Proliferation Kit for Imaging (C10337, Thermo Fisher Scientific), following the manufacturer’s instructions. Fibroblasts were synchronized with a 72-h starvation in media with 0.5% FBS. Cells were then released in media with 20% FBS for the required time and incubated with 10 μM EdU for 1.25 h prior to collection. For the S-phase progression assay, cells were released in media with 20 μM EdU and 100 ng/mL nocodazole (Abcam) for the indicated time. Cells were trypsinized, fixed, permeabilized, and stained for total DNA with FxCycle™ PI/RNase Staining Solution (Thermo Fisher Scientific). Data were collected with a Canto II cytometer (BD Biosciences) and analyzed with FlowJo software (TreeStar).
Imaging Flow Cytometry
Fibroblasts were cultured for 24 h. Cells were trypsinized, washed with PBS, fixed, and permeabilized. Fibroblasts were stained with anti-γH2AX antibody (1:400; 05–636, Millipore) for 30 min on ice followed by staining with Goat Anti-Mouse IgG H&L FITC (ab6785, Abcam) and DRAQ5 (Biostatus). Data were collected with an Amnis ImageStream MKII cytometer (EMD Millipore) and analyzed with IDEAS software (EMD Millipore). Quantification of foci was performed using CellProfiler software (https://cellprofiler.org/) [19].
High-Throughput Sequencing of the TRB Repertoire and IgH Repertoire
Blood-derived DNA from the index patient (3 months), a patient with Omenn syndrome due to RAG1 deficiency (RAG1: c.1210C > T; c.1677G > T) (3 months), a patient with ataxia telangiectasia (ATM: c.1564_1565delGA; c.1402_1403delAA) (13y), and a healthy donor (3 months) were used for determining the TRB and IgH repertoire. TRB and IgH VDJ rearrangement analysis was performed by NGS according to published protocols [20, 21]. Data were analyzed with the Vidjil Platform [22], and the chord plots were generated with the circlize R package (R version 4.2.2) [23].
Results
Case Presentation
The patient is a 3-year-old boy with consanguineous parents of Moroccan descent. He presented at 3 months with recurrent upper and lower respiratory tract infections, leading to acute respiratory distress syndrome requiring hospitalization. He had alopecia, generalized lymphadenopathy, hepatosplenomegaly, and erythroderma, with preceding recurrent bacterial superinfection (Fig. 1A, B). Chest X-ray and ultrasound failed to show a thymus. The patient was diagnosed with Omenn syndrome based on the clinical presentation and as he fulfilled 3 out of 4 PIDTC criteria [24] and for which he received steroids, cyclosporine, and alemtuzumab. He underwent hematopoietic stem cell transplantation (HSCT) at 6 months with a 10/10 matched unrelated donor, following conditioning with alemtuzumab, treosulfan, and fiudarabine, with the need for a stem cell boost ~ 8 months later because of chimerism < 20%. The boost HSCT was complicated by acute graft versus host disease, which was treated according to EBMT guidelines. At 3 years, he had severe growth retardation, developmental delay, and facial dysmorphy. Immunological analysis prior to HSCT showed severely reduced naiïve CD4+ and CD8+ cells, low memory B cells, and high NK cells (Table 1). The patient had elevated serum IgE, eosinophilia, and an oligoclonal T-cell receptor variable β-chain (TCR Vβ) repertoire as measured by flow cytometry (Table 1). There was no maternal engraftment, as verified by karyotype and molecular chimerism assessment (case report in supplement).
Fig. 1.
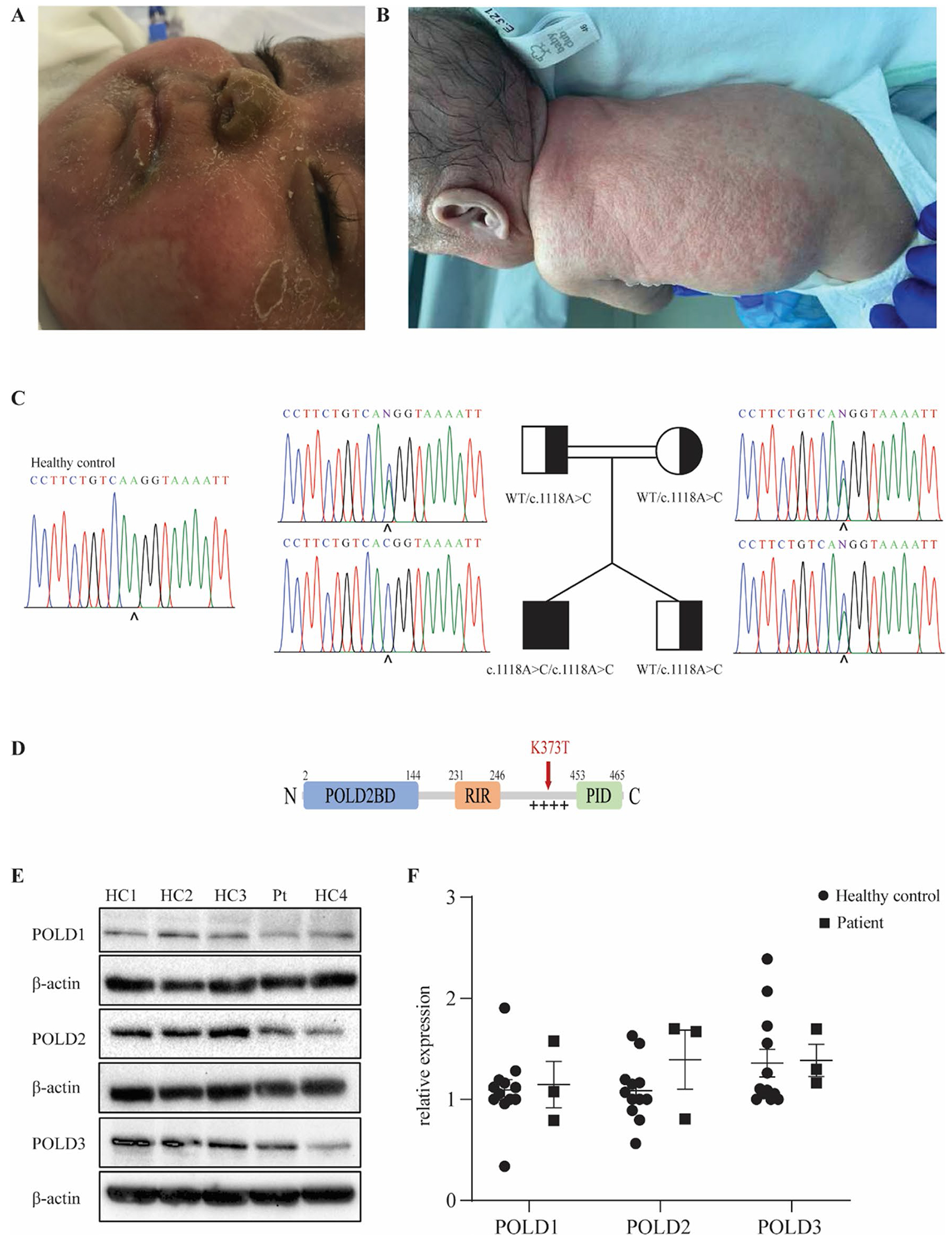
Identification of a homozygous mutation in POLD3 in a patient presenting with severe combined immunodeficiency and developmental defects. A Skin eczema due to neutrophil infiltration in the patient. B Skin infection with pustulosis and clinical alopecia in the patient. C Patient pedigree and familial segregation of the identified POLD3 mutation by Sanger sequencing compared to a healthy control. D Schematic representation of POLD3 domain structure. POLD2BD, POLD2 binding domain; RIR, RIV-1 interacting region; + + + + , disordered positively charged region; PID, PCNA interacting domain. The identified K373T mutation site is indicated in red. E Protein expression levels of POLD1, POLD2, and POLD3 in primary fibroblasts from four healthy controls (HC) and the patient (PT). β-actin was used as a loading control. The blot is representative of 3 independent experiments. F Quantification of POLD1, POLD2, and POLD3 protein expression in primary fibroblasts of four healthy controls and the patient normalized with β-actin expression (n = 3). Results represent mean ± SEM
Table 1.
Hematological and Immunological parameters In the patient prior to transplant at age 3 months
| Patient | Normal range | |
|---|---|---|
| Peripheral blood | ||
| WBC (cells/μL) | 18.73 | 5.00–19.50 |
| Neutrophils (cells/μL) | 3.8 | 1.0–8.5 |
| Lymphocytes (cells/μL) | 8.5 | 3.7–9.6 |
| Eosinophils (cells/μL) | 5.7 | 0.3 |
| Basophils (cells/μL) | 0.1 | ≤ 0.1 |
| Monocytes (cells/μL) | 0.6 | 0.7 |
| Lymphocyte subset analysis | ||
| CD3 + (%) | 58 | 53–84 |
| CD4 + (%) | 52 | 35–64 |
| Naïve CD4 (%) (CD27 + CD45RA +) | < 1 | 73–100 |
| CD8 + (%) | 3 | 12–28 |
| Naïve CD8 (%) (CD27 + CD45RA +) | 1 | 47–100 |
| CD19 + (%) | 8 | 6–32 |
| Non-switched memory B (%) (CD27 + IgD +) | 1 | 3–9 |
| NK (%) | 32 | 4–18 |
| TCR Vβ repertoire | ||
| Vβ 1 (%) | 2.40 | 1.89–11.70 |
| Vβ 2 (%) | 0.24 | 4.03–23.48 |
| Vβ 3 (%) | 1.99 | 0.52–15.71 |
| Vβ 4 (%) | 10.32 | 0.79–3.26 |
| Vβ 5.3 (%) | 0.28 | 0.37–2.98 |
| Vβ 7.1 (%) | 0.30 | 0.64–20.01 |
| Vβ 7.2 (%) | 0.07 | 0.05–5.45 |
| Vβ 8 (%) | 5.83 | 2.26–29.47 |
| Vβ 9 (%) | 4.38 | 1.10–9.30 |
| Vβ 11 (%) | 1.28 | 0.25–5.11 |
| Vβ 12 (%) | 0.38 | 1.00–4.76 |
| Vβ 13.1 (%) | 1.88 | 1.62–8.16 |
| Vβ 13.2 (%) | 0.29 | 0.80–5.28 |
| Vβ 13.6 (%) | 0.16 | 0.84–8.80 |
| Vβ 14 (%) | 2.67 | 1.33–8.03 |
| Vβ 16 (%) | 0.06 | 0.42–1.90 |
| Vβ 17 (%) | 0.27 | 2.28–12.61 |
| Vβ 18 (%) | 0.05 | 0.58–5.23 |
| Vβ 20 (%) | 33.82 | ≤ 9.73 |
| Vβ 21.3 (%) | 0.03 | 1.08–5.97 |
| Vβ 22 (%) | 0.14 | 1.99–9.89 |
| Serum immunoglobulin levels | ||
| IgG (g/L) | 1.07 | 2.32–14.11 g/L |
| IgA (g/L) | < 0.04 | 0.0–0.83 g/L |
| IgM (g/L) | 0.16 | 0.0–1.45 g/L |
| IgE (U/ml) | 1471 | < 4.1 U/ml |
Values outside the normal range are in bold.
Identification of a Biallelic Missense Variant in POLD3
WES analysis of the patient revealed a total of 52,459 variants, none of which were within known IEI-causing genes [25]. Variants present in 1000 Genomes, ExAC, or GnomAD databases with a minor allele frequency > 1% were filtered out leaving 7439 variants of which 1823 were present in coding or essential splicing regions. Twenty-seven of these variants were homozygous, and 3 were found to be within the connectome of known IEI-causing genes (Supp Fig. 1A). Given that the variants in MAP3K11 and HERC2 were classed as benign by PolyPhen while the variant in POLD3 was predicted damaging and also more promising in terms of deleteriousness score, the POLD3 variant was prioritized in the investigation. Moreover, the phenotype of the patient shared features with other reported PolD-deficient patients [12–17]. The identified variant in the POLD3 gene (NM_006591) was a homozygous variant in exon 10 (c.1118A > C) with a CADD score of 25 that segregated with autosomal recessive inheritance, as confirmed by Sanger sequencing (Fig. 1C). As the POLD3c.1118A>C variant is situated in the last codon of exon 10 which could lead to either a missense or splicing variant, gel migration and Sanger sequencing analysis of cDNA from patient fibroblasts was performed which distinguished the variant as a missense variant resulting in the K373T protein change (Supp Fig. 1B, 1C). The lysine residue affected by the variant shows a high degree of conservation across mammalian species (Supp Fig. 1D). This variant is located in a disordered interdomain region, enriched in positively charged residues, thought to be involved in enhancing polymerase activity and processivity [9] (Fig. 1D and Supp Fig. 1E). Taken together, the high CADD score of this variant and the similarities between this patient and previously reported patients with mutations in POLD1 and POLD2 led us to consider POLD3K373T as the underlying cause of disease in this patient.
POLD3K373T Results in Normal mRNA and Protein Expression in Patient Fibroblasts
We first examined the protein expression of POLD3 and other components of PolD in primary dermal fibroblasts from the patient. Expression of not only POLD3 but also POLD1 and POLD2 were comparable to healthy control fibroblasts (Fig. 1E, F). Interestingly, this finding differs from that of patients with mutations in POLD1 and POLD2 as they showed reduced expression of all 3 PolD components. Quantitative PCR analysis also showed normal mRNA levels of POLD1, POLD2, and POLD3 in the patient’s fibroblasts (Supp Fig. 1F).
POLD3 Deficiency Leads to Defective Thymocyte Development but Only a Marginally Affected B Cell IgH Repertoire
Prior to HSCT, the patient displayed severely reduced levels of naïve T cells and a restricted TCR Vβ repertoire (Table 1). To further explore T-cell development, we investigated early recombination events of the TCRα chain in patient T cells. Staining of CD3+ T cells with the monoclonal Vα7.2 antibody which recognizes the highly upstream TRAV1-2 gene revealed a drastic reduction in the proportion of T cells with a rearranged TRAV1-2 in the patient as compared to 4 healthy controls (Fig. 2A, B). This reduction was greater than that seen in an ataxia telangiectasia patient (AT) but not as severe as that observed in a RAG1-deficient patient with Omenn syndrome (RAG1) (Fig. 2A, B). As patients with ataxia telangiectasia and RAG1 deficiency have demonstrated skewed T cell receptor repertoires [26], T cell receptor β (TRB) diversity was assessed in the patient via high throughput sequencing. Visualization of this data in chord diagrams shows TRB diversity is greatly impacted in the patient (Fig. 2C). The impact seen in the patient was of a greater degree than in the AT patient but of a similar level to the RAG1-deficient patient (Fig. 2C). B cell IgH diversity, however, appears to be much less altered in the patient than TRB diversity which is also the case for the AT patient (Fig. 2D). Furthermore, Shannon diversity index analysis also revealed that patient TRB diversity was only 31% of that of a healthy control while IgH diversity was 85% of what was observed in a healthy control (Fig. 2E). These data point to an integral role of POLD3 in maintaining normal T-cell development.
Fig. 2.
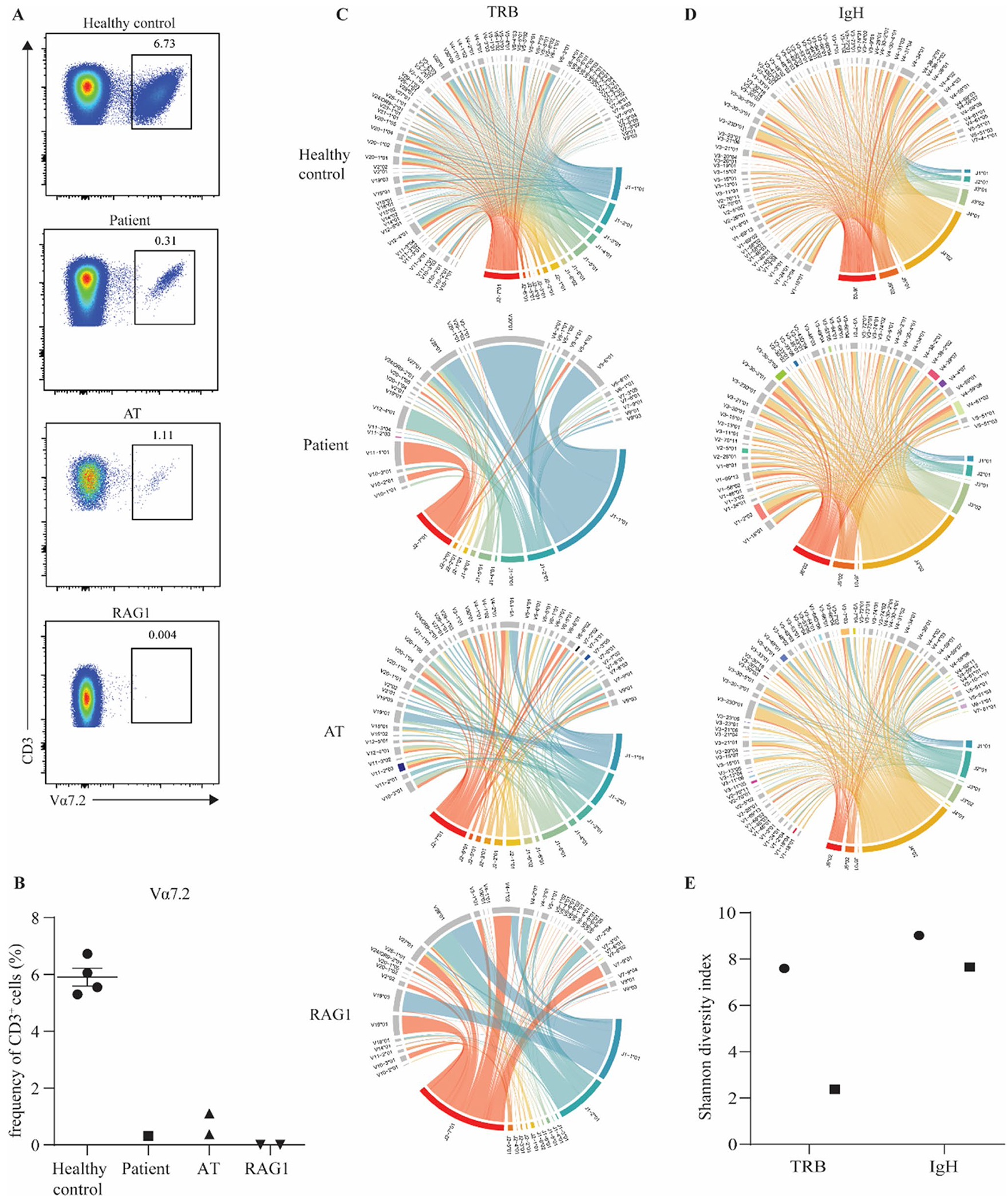
Impaired rearrangement of the TRAV1-2 locus and reduced T and B cell receptor diversity in patient cells. A FACS plots of a healthy control, index patient, a patient with AT, and a patient with RAG1 deficiency showing the frequency of CD3 + Vα7.2 + cells. B Quantification of the frequency of Va7.2 cells within the CD3+ compartment. Chord plot of C T cell receptor β (TRB) repertoire and D IgH repertoire in a healthy control, index patient, a patient with AT, and a patient with RAG1 deficiency. E Shannon diversity index of TRB and IgH in the index patient and a healthy control
POLD3K373T Results in Reduced Cell Proliferation due to Defective S-Phase Entry
Given the role of PolD in DNA replication, we analyzed the proliferative activity and cell cycle progression of primary fibroblasts from the patient in comparison to controls. We imaged and counted fibroblasts after 1, 4, and 7 days in culture. The patient’s fibroblasts grew aberrantly, lacking the consistent shape seen in healthy control fibroblasts (Fig. 3A). The proliferation of the patient’s fibroblasts was markedly reduced compared to a healthy control over a 7-day period (Fig. 3B). To determine whether this reduced proliferation seen in the patient was due to cell cycle perturbations, we explored cell cycle dynamics in primary fibroblasts. Following synchronization at the G1 phase, cells were released for 24, 26, or 28 h. Fibroblasts were then labeled for newly synthetized DNA in S-phase cells and stained for DNA content. Results showed a decreased proportion of patient cells in the S and G2 phases as compared to healthy controls at 24 h (Fig. 3C, D). The same pattern was also observed at 26 and 28 h (data not shown). To gain more insight into the entry and progression of patient fibroblasts in the S-phase, we performed a similar assay in which cells were synchronized and released for 24 or 48 h in the presence of a microtubule polymerization inhibitor which arrests cells at mitosis and blocks entry into a new cycle so that we could observe a single cell cycle. We saw that patient cells did progress into the S and G2 phases but at a much slower rate than healthy control fibroblasts. At 24 h, a small number of patient cells were in the S phase and even fewer in G2; by 48 h, 1/4 of cells were in G2 but almost 3/4 remained in G1 (Fig. 3E, F). Hence, it appears that the reduced proliferation in patient fibroblasts is caused by cellular arrest at the entry to the S phase, which may be due to a defect in DNA replication.
Fig. 3.
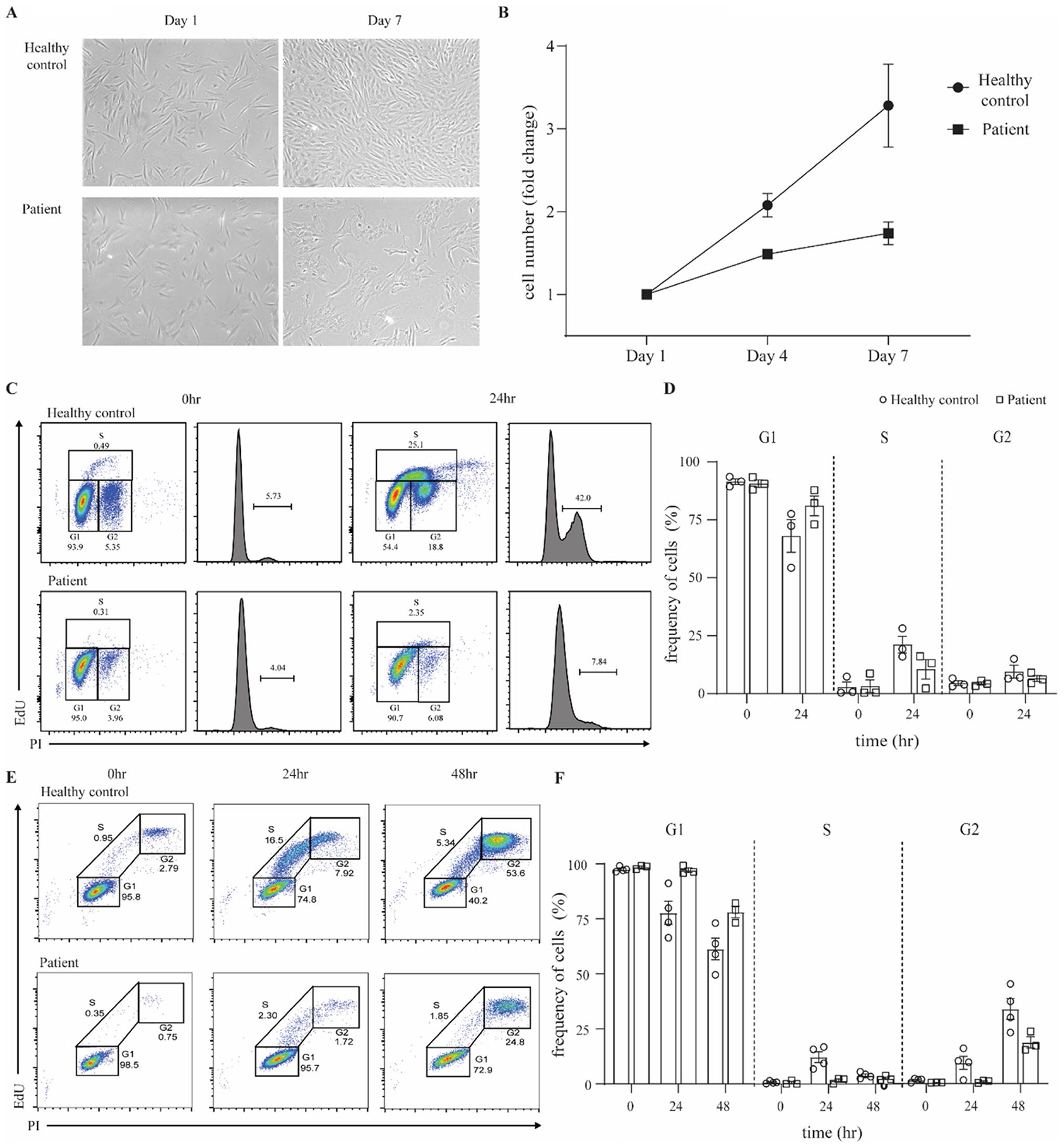
Patient fibroblasts show reduced proliferation and impaired S-phase entry. A Representative images of skin-derived primary fibroblasts from a healthy control and the patient, 1 day and 7 days after seeding. Images were acquired with an EVOS7000 microscope, original magnification × 4. B Fibroblast proliferation in healthy controls and patient. Cell counts were performed with ImageJ. Data are representative of 2 independent experiments. C Representative FACS plots and histograms of cell-cycle analysis of healthy control and patient primary fibroblasts upon synchronization at G1 and release at 0 and 24 h. Data are representative of 3 independent experiments. D Percentage of cells in G1, S, and G2 phase in healthy controls compared to the patient, upon synchronization at G1 and release at 0 and 24 h. E Representative FACS plots from S phase entry and progression analysis of healthy control and patient primary fibroblasts upon synchronization at G1 and release. Nocodazole was used in the release medium to inhibit mitosis and block cells in G2 phase. Data are representative of 3 independent experiments. F Percentage of cells in G1, S, and G2 phase in healthy controls and the patient, upon synchronization at G1 and release for 0, 24, 48 h. Results represent means ± SEM
POLD3K373T Causes Replication-Associated DNA Damage
Given that defective DNA replication could underlie the S phase entry arrest observed in patient fibroblasts, we next investigated replication-associated DNA damage. As replicative stress, if not resolved, commonly leads to double-strand DNA breaks (DSB), we stained primary fibroblasts for γ-H2AX, a marker of DSBs. We found that the mean number of γ-H2AX foci per cell was significantly higher in the patient compared to 2 healthy controls (Fig. 4A, B). The increased numbers of γ-H2AX foci further support the idea that the delayed entry into the S phase of patient cells is due to problems with DNA replication.
Fig. 4.
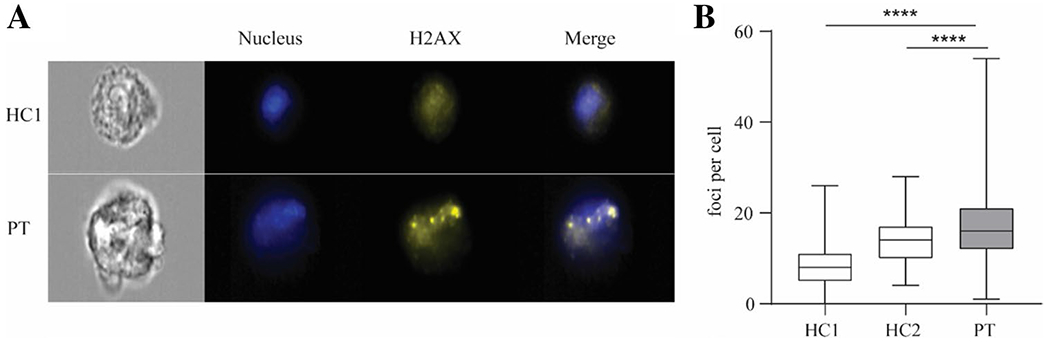
Patient fibroblasts display replication-associated DNA damage. A Representative images from imaging flow cytometry analysis of γH2AX staining in healthy control and patient fibroblasts. DRAQ5 dye was used to stain the nucleus. Original magnification × 40. B Quantification of γH2AX foci per cell. 252, 244, 287 cells were counted respectively for healthy control 1, healthy control 2, and the patient. Image analyses were performed using CellProfiler, v2.0. Results represent median + min to max.****p < 0.0001, Mann–Whitney test
The Patient Fibroblast Cell Cycle Defect Is Rescued by Stable Transduction with WT POLD3
To further assess whether POLD3K373T was responsible for the cell cycle delay seen in the patient, we stably overexpressed POLD3 in patient and control fibroblasts. Restoration of WT POLD3 in the patient cells rescued the cell cycle delay (Fig. 5A, B). This was seen at 2 different lentiviral MOI for which there was comparable POLD3 expression between the healthy control and patient cells (Fig. 5C). These results suggest that the p.K373T variant impacts POLD3 function which causes an impairment in cell cycle progression.
Fig. 5.
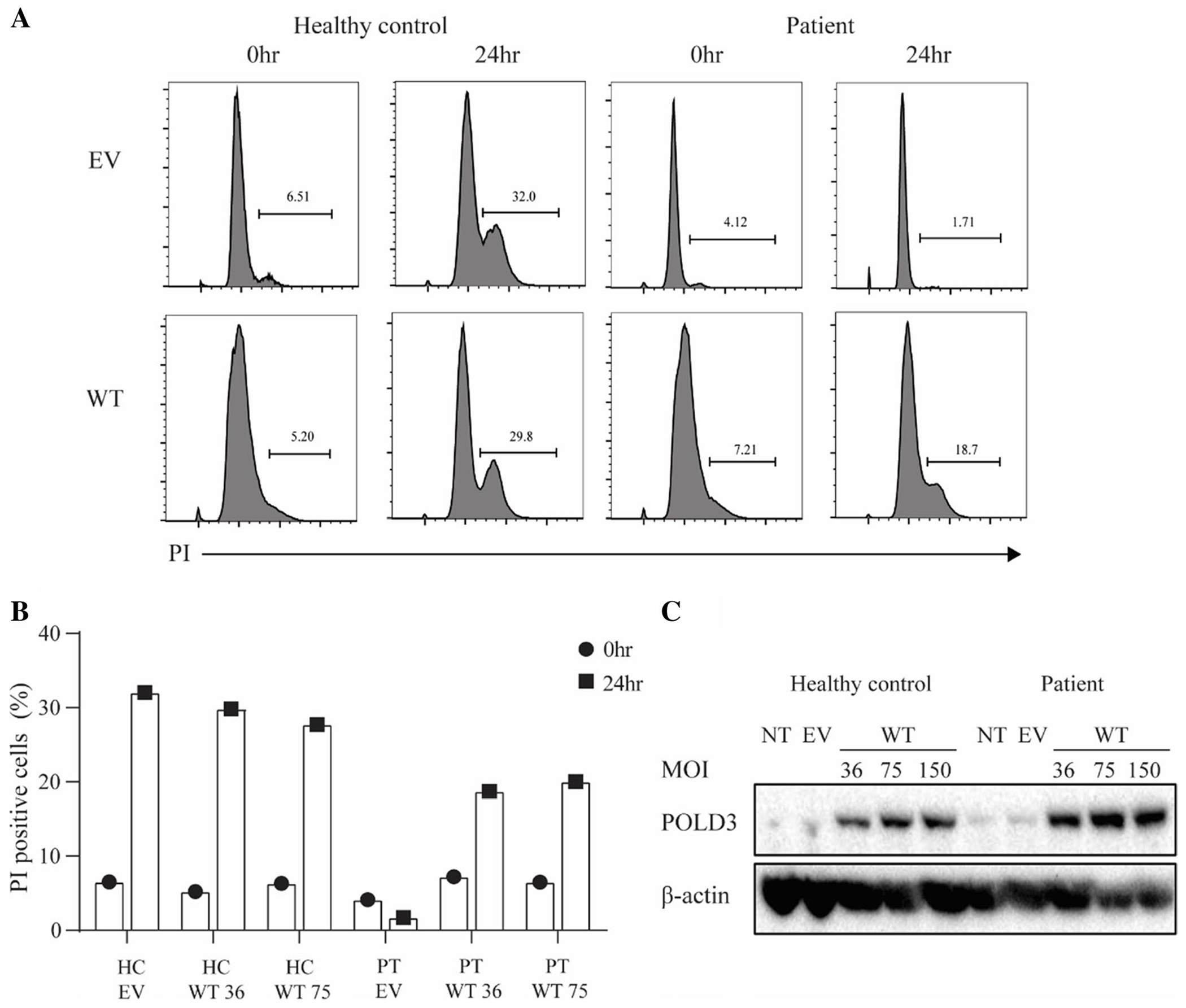
Delayed cell cycle progression of patient fibroblasts is rescued by WT POLD3 transduction. A Representative FACS histograms of cell cycle analysis of healthy control and patient fibroblasts transduced with EV or WT POLD3 (MOI 36) synchronized at G1 upon release for 24 h, measured by propidium iodide (PI) DNA staining. B Frequency of cycling cells In healthy control and patient fibroblasts transduced with EV or WT POLD3. C Western blot analysis of POLD3 and β-actin expression in healthy control and patient fibroblasts. NT, non-transduced; EV, transduced with empty vector; WT, transduced with WT POLD3
Discussion
In recent years, a new subgroup of inborn errors of immunity has emerged, categorized as those that cause disruption to DNA polymerases. This subgroup includes, to date, 18 patients with immunodeficiency found to have biallelic mutations in POLE1 and POLE2, which encode subunits of the polymerase ε complex (Polε), POLD1 and POLD2 [15–17, 27, 28]. This report adds to this subgroup by identifying a patient with a biallelic missense mutation in POLD3 in a patient with Omenn syndrome and developmental defects.
Staining of TCR VB and Va7.2 + cells showed that the patient had a reduced capacity for recombination which was confirmed by high throughput sequencing which emphasized the restricted repertoire of patient T cells which was as severe as in a patient with Omenn syndrome caused by a mutation in RAG1. Reports of patients with POLD1 mutations have also shown decreased Va7.2 + cells [17] or skewed T cell repertoires, specifically in CD8 + T cells [16], highlighting the importance of PolD to T-cell development. This is consistent with the knowledge that antigen receptor recombination requires heightened levels of proliferation that necessitate accurate DNA replication and repair and that POLD3 is required to repair stalled replication forks [29, 30]. The IgH repertoire of patient B cells, however, was affected to a much lesser extent despite B cell levels at the low end of the normal range. This lower B-cell frequency but intact B-cell maturation, based on Ig repertoire, was also observed in the reported POLD2 deficient patient [15]. Interestingly, the translesion synthesis activity of PolD and Polζ, both of which contain POLD3, has been shown to be required for efficient hypermutation of immunoglobulin variable genes in chicken DT40 cells [6]. It is possible that while the mutation present in the reported patient affects the polymerase activity of PolD, it does not affect the translesion synthesis activity of PolD and Polζ, resulting in a relatively unaffected B cell repertoire.
Analysis of patient fibroblasts showed reduced proliferation associated with a striking defect in S-phase entry which was rescued with WT POLD3. Unfortunately, due to the lack of patient samples because of transplantation, it was not possible to analyze the expansion of patient lymphocytes. While the replication dynamics in lymphocytes differ from that in fibroblasts, it is likely that the decreased cell cycle progression seen in primary fibroblasts would also be seen in lymphocytes, as it was for patients with mutations in POLD1 and POLD2 [15].
Given that normal levels of POLD1, POLD2, and POLD3 were found in the patient, it is likely that PolD complex formation is unaffected in contrast to reported patients with mutations in POLD1 and POLD2. Rather, it is probable that the compromised function of the PolD complex underlies the defects in the patient presented here. This may explain why previously reported POLD1 and POLD2 deficient patients displayed a milder phenotype, as although they had reduced levels of complex formation, residual activity was still present [15]. The mutation in our patient is within a positively charged area of POLD3 that may be important for DNA binding. As the mutation causes a change from a positively charged lysine to an uncharged threonine, it may impact the DNA binding capacity of the PolD complex and consequently its function. Additionally, ADP-ribosylation of POLD3 has recently been shown to be critical in break-induced replication and replication fork recovery [31]. This ADP-ribosylation was found to occur at residue 422 which is within the vicinity of the patient’s mutation and hence could be disrupted by the POLD3K373T mutation.
While this manuscript was in preparation, a case report on another patient with a homozygous mutation in POLD3 was published [32]. The index case presented at 4y with a history of recurrent infections from 7 months. His siblings had died at ages 3, 5, and 5 years. He had low naïve T cells but normal B cell counts, and he had normal IgG and IgM but partially deficient IgA. Furthermore, the patient had sensorineural hearing loss and developmental delay. WES identified a homozygous variant (c.29 T > C; p.I10T) in the POLD2 binding region of POLD3, and patient cells showed no expression of POLD3, POLD1, or POLD2. Strikingly, while the patient presented here had normal expression of POLD3 as well as POLD1 and POLD2, he presented in infancy with a more severe phenotype. However, cell replication, cell cycle dynamics, and DNA damage were not examined in the other reported patient, so a formal comparison to investigate the difference in phenotypic severity cannot be made.
Overall, this work identifies POLD3K372T as a cause of severe combined immunodeficiency with developmental defects featuring disrupted cell cycle progression and increased DNA damage. This work also highlights that while POLD3 is considered an accessory subunit of PolD, its role is crucial for the functioning of the complex. Indeed, we were able to assess the effects of a deleterious mutation in POLD3 that does not disrupt the entire complex. Identification of additional patients with mutations in POLD3 will hopefully enable a better understanding of the contributions of POLD3 and PolD to immune function and neurodevelopment.
Supplementary Material
Acknowledgements
We acknowledge the Centre for Multimodal Oncology Research (C-MORE) at KU Leuven for the use of the ImageStream. We thank Dr Rebeca Perez-De Diego for fruitful discussion. We thank the parents of the patient for their trust.
Funding
This work was supported by UZ Leuven Klinische Onderzoeksraad Funding – KOOR. MRR was supported by an ESID fellowship. LE is supported by a PhD Fellowship from the Research Foundation – Flanders (FWO) (grant 11E0123N). SD is supported by a PhD Fellowship from the Research Foundation – Flanders (FWO) (grant 11F4421N). LDN is supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health (grant AI001222). IM is supported by the Jeffrey Modell Foundation.
Competing Interests
IM receives funding from CSL Behring and consultancy fees from Boehringer-Ingelheim, unrelated to this submission, paid to KU Leuven.
Abbreviations
- CADD
Combined annotation dependent depletion
- DSB
Double strand breaks
- HSCT
Hematopoietic stem cell transplant
- IEI
Inborn error of immunity
- IgH
Immunoglobulin heavy chain
- NHEJ
Non-homologous end joining
- PCNA
Proliferating cell nuclear antigen
- PolD
Polymerase delta complex
- Polζ
Polymerase zeta complex
- TLS
Translesion synthesis
- TRB
T cell receptor B locus
- WES
Whole exome sequencing
Footnotes
Ethics Approval The Ethics Committee of the University Hospitals Leuven (UZLeuven) approved this study (S66910, S63807). The study was performed in compliance with the Declaration of Helsinki for research involving human subjects.
Consent to Participate Written informed consent was obtained from the parents prior to DNA, PBMCs, and fibroblast isolation.
Consent for Publication Informed consent was provided by the parents for the publication of the images in Fig. 1a and b.
Supplementary Information The online version contains supplementary material available at https://doi.org/10.1007/s10875-023-01627-z.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- 1.Loeb LA, Monnat RJ. DNA polymerases and human disease. Nat Rev Genet. 2008;9(8):594–604. [DOI] [PubMed] [Google Scholar]
- 2.Johnson Robert E, Klassen R, Prakash L, Prakash S. A major role of DNA polymerase δ in replication of both the leading and lagging DNA strands. Mol Cell. 2015;59(2):163–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xie B, Mazloum N, Liu L, Rahmeh A, Li H, Lee MYWT. Reconstitution and characterization of the human DNA polymerase delta four-subunit holoenzyme. Biochemistry. 2002;41(44):13133–42. [DOI] [PubMed] [Google Scholar]
- 4.Donnianni RA, Zhou Z-X, Lujan SA, Al-Zain A, Garcia V, Glancy E, et al. DNA polymerase delta synthesizes both strands during break-induced replication. Mol Cell. 2019;76(3):371–81.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hirota K, Yoshikiyo K, Guilbaud G, Tsurimoto T, Murai J, Tsuda M, et al. The POLD3 subunit of DNA polymerase δ can promote translesion synthesis independently of DNA polymerase ζ. Nucleic Acids Res. 2015;43(3):1671–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Layer JV, Debaize L, Van Scoyk A, House NC, Brown AJ, Liu Y, et al. Polymerase δ promotes chromosomal rearrangements and imprecise double-strand break repair. Proc Natl Acad Sci. 2020;117(44):27566–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maloisel L, Fabre F, Gangloff S. DNA polymerase δ is preferentially recruited during homologous recombination to promote heteroduplex DNA extension. Mol Cell Biol. 2008;28(4):1373–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ducoux M, Urbach S, Baldacci G, Hübscher U, Koundrioukoff S, Christensen J, et al. Mediation of proliferating cell nuclear antigen (PCNA)-dependent DNA replication through a conserved p21Cip1-like PCNA-binding motif present in the third subunit of human DNA polymerase δ*. J Biol Chem. 2001;276(52):49258–66. [DOI] [PubMed] [Google Scholar]
- 9.Lee Y-S, Gregory MT, Yang W. Human Pol ζ purified with accessory subunits is active in translesion DNA synthesis and complements Pol η in cisplatin bypass. Proc Natl Acad Sci. 2014;111(8):2954–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bellido F, Pineda M, Aiza G, Valdés-Mas R, Navarro M, Puente DA, et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: review of reported cases and recommendations for genetic testing and surveillance. Genet Med. 2016;18(4):325–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palles C, Cazier J-B, Howarth KM, Domingo E, Jones AM, Broderick P, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013;45(2):136–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elouej S, Beleza-Meireles A, Caswell R, Colclough K, Ellard S, Desvignes JP, et al. Exome sequencing reveals a de novo POLD1 mutation causing phenotypic variability in mandibular hypoplasia, deafness, progeroid features, and lipodystrophy syndrome (MDPL). Metabolism. 2017;71:213–25. [DOI] [PubMed] [Google Scholar]
- 13.Pelosini C, Martinelli S, Ceccarini G, Magno S, Barone I, Basolo A, et al. Identification of a novel mutation in the polymerase delta 1 (POLD1) gene in a lipodystrophic patient affected by mandibular hypoplasia, deafness, progeroid features (MDPL) syndrome. Metabolism. 2014;63(11):1385–9. [DOI] [PubMed] [Google Scholar]
- 14.Weedon MN, Ellard S, Prindle MJ, Caswell R, Allen HL, Oram R, et al. An in-frame deletion at the polymerase active site of POLD1 causes a multisystem disorder with lipodystrophy. Nat Genet. 2013;45(8):947–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Conde CD, Petronczki ÖY, Baris S, Willmann KL, Girardi E, Salzer E, et al. Polymerase δ deficiency causes syndromic immunodeficiency with replicative stress. J Clin Investig. 2019;129(10):4194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cui Y, Keles S, Charbonnier L-M, Julé AM, Henderson L, Celik SC, et al. Combined immunodeficiency caused by a loss-of-function mutation in DNA polymerase delta 1. J Allergy Clin Immunol. 2020;145(1):391–401.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nichols-Vinueza DX, Delmonte OM, Bundy V, Bosticardo M, Zimmermann MT, Dsouza NR, et al. POLD1 deficiency reveals a role for POLD1 in DNA repair and T and B cell development. J Clin Immunol. 2021;41(1):270–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strobl J, Huber B, Heredia RJ, Kirnbauer R, Boztug K, Stary G. Polymerase-δ-deficiency as a novel cause of inborn cancer pre-disposition associated with human papillomavirus infection. Br J Dermatol. 2023;188(5):684–5. [DOI] [PubMed] [Google Scholar]
- 19.Stirling DR, Swain-Bowden MJ, Lucas AM, Carpenter AE, Cimini BA, Goodman A. Cell Profiler 4: improvements in speed, utility and usability. BMC Bioinformatics. 2021;22(1):433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Villarese P, Abdo C, Bertrand M, Thonier F, Giraud M, Salson M, Macintyre E. One-Step next-generation sequencing of immunoglobulin and t-cell receptor gene recombinations for mrd marker identification in acute lymphoblastic leukemia. Methods Mol Biol. 2022;2453:43–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brüggemann M, Kotrová M, Knecht H, Bartram J, Boudjogrha M, Bystry V, et al. Standardized next-generation sequencing of immunoglobulin and T-cell receptor gene recombinations for MRD marker identification in acute lymphoblastic leukaemia; a Euro-Clonality-NGS validation study. Leukemia. 2019;33(9):2241–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giraud M, Salson M, Duez M, Villenet C, Quief S, Caillault A, et al. Fast multiclonal clusterization of V(D)J recombinations from high-throughput sequencing. BMC Genomics. 2014;15(1):409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gu Z, Gu L, Eils R, Schlesner M, Brors B. circlize implements and enhances circular visualization in R. Bioinformatics. 2014;30(19):2811–2. [DOI] [PubMed] [Google Scholar]
- 24.Dvorak CC, Haddad E, Heimall J, Dunn E, Buckley RH, Kohn DB, et al. The diagnosis of severe combined immunodeficiency (SCID): the primary immune deficiency treatment consortium (PIDTC) 2022 definitions. J Allergy Clin Immunol. 2023;151(2):539–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human inborn errors of immunity: 2022 update on the classification from the international union of immunological societies expert committee. J Clin Immunol. 2022;42(7):1473–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berland A, Rosain J, Kaltenbach S, Allain V, Mahlaoui N, Melki I, et al. PROMIDISα: a T-cell receptor α signature associated with immunodeficiencies caused by V(D)J recombination defects. J Allergy Clin Immunol. 2019;143(1):325–34.e2. [DOI] [PubMed] [Google Scholar]
- 27.Frugoni F, Dobbs K, Felgentreff K, Aldhekri H, Al Saud BK, Arnaout R, et al. A novel mutation in the POLE2 gene causing combined immunodeficiency. J Allergy Clin Immunol. 2016;137(2):635–8.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pachlopnik Schmid J, Lemoine R, Nehme N, Cormier-Daire V, Revy P, Debeurme F, et al. Polymerase ε1 mutation in a human syndrome with facial dysmorphism, immunodeficiency, livedo, and short stature (“FILS syndrome”). J Exp Med. 2012;209(13):2323–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murga M, Lecona E, Kamileri I, Díaz M, Lugli N, Sotiriou SK, et al. POLD3 is haploinsufficient for DNA replication in mice. Mol Cell. 2016;63(5):877–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ogi T, Limsirichaikul S, Overmeer RM, Volker M, Takenaka K, Cloney R, et al. Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol Cell. 2010;37(5):714–27. [DOI] [PubMed] [Google Scholar]
- 31.Richards F, Llorca-Cardenosa MJ, Langton J, Buch-Larsen SC, Shamkhi NF, Sharma AB, et al. Regulation of Rad52-dependent replication fork recovery through serine ADP-ribosylation of PolD3. Nat Commun. 2023;14(1):4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mehawej C, Chouery E, Azar-Atallah S, Shebaby W, Delague V, Mansour I, et al. POLD3 deficiency is associated with severe combined immunodeficiency, neurodevelopmental delay, and hearing impairment. Clin Immunol. 2023;251:109326. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.