

Abstract
Tuberculosis is the leading cause of death for people living with HIV (PLWH). We hypothesized that altered functions of innate immune components in the human alveolar lining fluid of PLWH (HIV-ALF) drive susceptibility to Mycobacterium tuberculosis (M.tb) infection. Our results indicate a significant increase in oxidation of innate proteins and chemokine levels and significantly lower levels and function of complement components and Th1/Th2/Th17 cytokines in HIV-ALF versus control-ALF (non-HIV-infected people). We further found a deficiency of surfactant protein D (SP-D) and reduced binding of SP-D to M.tb that had been exposed to HIV-ALF. Primary human macrophages infected with M.tb exposed to HIV-ALF were significantly less capable of controlling the infection, which was reversed by SP-D replenishment in HIV-ALF. Thus, based on the limited number of participants in this study, our data suggest that PLWH without antiretroviral therapy (ART) have declining host innate defense function in their lung mucosa, thereby favoring M.tb and potentially other pulmonary infections.
INTRODUCTION
Worldwide, 38.4 million people are living with HIV/AIDS, including approximately 1.5 million people becoming newly infected with HIV annually, with a mortality rate of 650,000 people per year1. Currently, in the US, 50% of the people living with HIV (PLWH) are 50 years old or older2; this number is predicted to be 70% by 20303, indicating an increasingly high burden of HIV-infected individuals as people live longer with chronic HIV infection4–9. Aging-related concerns in PLWH >50 years of age include accelerated CD4+ T cell loss, decreased immune recovery, and increased risk of serious non-AIDS illnesses, further complicating antiretroviral therapy (ART) management.
The spectrum of lung diseases associated with HIV includes both infectious and non-infectious etiologies, but the mechanisms behind this are not well understood10,11. HIV infection results in immune dysregulation, dysfunction, and deficiency12–14. These immunologic abnormalities are most marked in PLWH not receiving ART. Even though immune functions can be partially restored in many PLWH receiving ART; persistent systemic inflammation driving their immunodeficiency may occur, particularly in PLWH who have lower CD4+ T cell counts at the time of ART initiation15–18. Indeed, the chronic phase of HIV infection is associated with chronic immune activation and cell exhaustion, where recurrent episodes of respiratory infections (bacterial, viral, and fungal pneumonia) further increase chronic pulmonary inflammation and predispose PLWH to lung cancer19.
Our understanding of the role of the lung alveolar environment in the development of these lung diseases in the current ART era is still limited. Oxidative stress levels in the healthy lung alveolar space are highly controlled throughout most of the individual’s life, typically increasing only with old age20,21 and progression of disease. At the initiation of the current study, we hypothesized that basal lung alveolar oxidation levels are increased in PLWH, driving immune dysfunction within the alveolar space. To test our hypothesis, we assessed lung alveolar space immune responses that are linked to oxidation and inflammation in HIV-negative control subjects and PLWH without ART. Our results indicate that PLWH have immune dysfunction in their alveolar lining fluid (ALF). This dysfunction is characterized by an increase in lung protein oxidation and chemokine levels, reduction of Th1/Th2/Th17 cytokines, and growth factors in ALF. Furthermore, there was a reduction in complement and surfactant proteins A and D (SP-A and SP-D) levels and activity. The latter innate immune components play important roles in host responses to respiratory pathogens such as Mycobacterium tuberculosis (M.tb)22–25. This innate immunodeficiency in the lungs of PLWH without ART was further validated by the observation that macrophages from healthy human donors were infected with M.tb that had been exposed to ALF from PLWH (HIV-ALF); there was an increase in both M.tb association with macrophages and in intracellular growth compared to M.tb exposed to control-ALF. This reduction in efficiency to control M.tb growth in macrophages was due to a decrease in phagosome maturation below baseline levels. This was reversed by addition of SP-D to HIV-ALF. Thus, our results indicate that innate dysfunction in the lung mucosa of PLWH could drive susceptibility to respiratory infections such as one caused by M.tb.
RESULTS
Levels of oxidized proteins are high in the alveolar environment of PLWH compared to control donors
First, we normalized the ALF samples based on their physiological phospholipid content within the lung, as we described previously20,21,26–33. To analyze the effects of oxidative stress in the lung environment of PLWH, we measured carbonyl and 3-nitrotyrosine protein modification in control- and HIV-ALFs, indicators of irreversible protein oxidation by reactive oxygen species (ROS) and reactive nitrogen species (RNS), respectively (Figs. 1A, B)20,21,30,34. We also measured myeloperoxidase (MPO) levels, indicative of an oxidative environment partially driven by a neutrophil influx (Fig. 1C). Our results indicate that HIV-ALF soluble proteins contained significantly higher levels of carbonyl and nitrotyrosine modifications relative to control-ALF. MPO levels trended downward in HIV-ALF but did not reach statistical significance.
Fig. 1. Oxidized protein levels are high in the alveolar environment of PLWH compared to control donors.
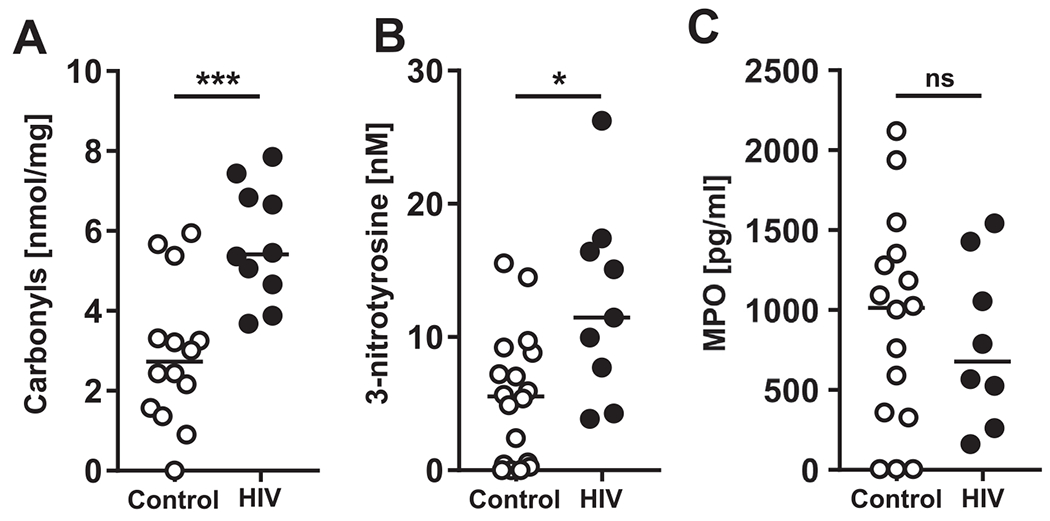
Each dot represents ALF from an individual subject. ALFs from control (n = 14-17) and PLWH (HIV+, n = 8-10) subjects. (A) Protein carbonyl, (B) 3-nitrotyrosine residues were detected by ELISA, and (C) myeloperoxidase (MPO) was detected by human multiplex Luminex assay. Unpaired Student’s t test, *p < 0.05; ***p < 0.0005. Each sample corresponds to ALF obtained from different human donors.
The ALF proteome is altered in PLWH
To assess if this high protein oxidative status observed in HIV-ALF negatively impact also their levels, we next performed a proteomic assessment of both HIV-ALF and control-ALF by data-independent acquisition mass spectrometry. To identify differentially abundant proteins (DAPs) of interest, we calculated their log2 fold-changes in the HIV-ALF relative to those in control-ALF. In general, HIV-ALF contained higher levels of inflammation-associated proteins (Fig. 2A, Table S1) and lower levels of anti-oxidation, complement component, and surfactant proteins (Fig. 2B, D and Table S1, S2). However, the relative levels of antimicrobial proteins (Fig. 2C, Table S2), immunoglobulins (IG) and IG receptor proteins (Fig. 2E, Table S3), and hydrolytic proteins (Fig. 2F, Table S3) in HIV-ALF did not follow this pattern; indeed, they showed distinct differential abundance. These results indicate that HIV infection seems to alter global protein abundance in the alveolar airspace. Of note, a major antibody of the lung mucosa, IgA (composed of IgA heavy chain alpha IgHA1 and its analog IgHA2), was detected at a lower abundance in the HIV-ALF (Fig. 2E). IgJ, which is required for IgA and IgM secretion into the lung mucosa35, was also detected at lower levels in HIV-ALF (Fig. 2E).
Fig. 2. Proteomic analyses and measurement of innate soluble mediators in ALF of both PLWH and control individuals.
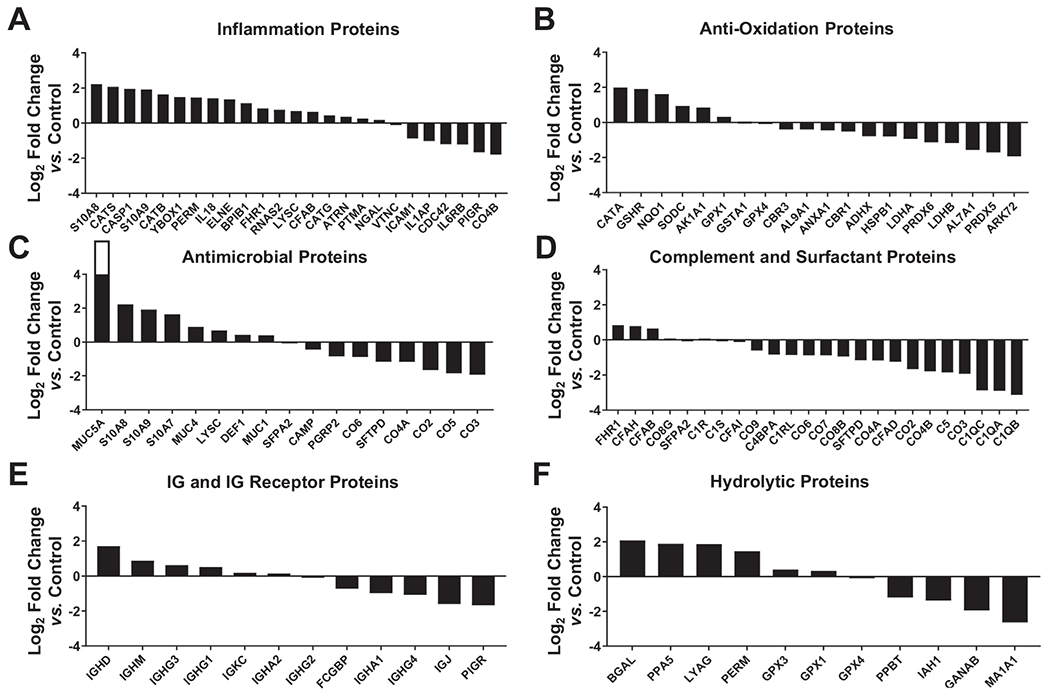
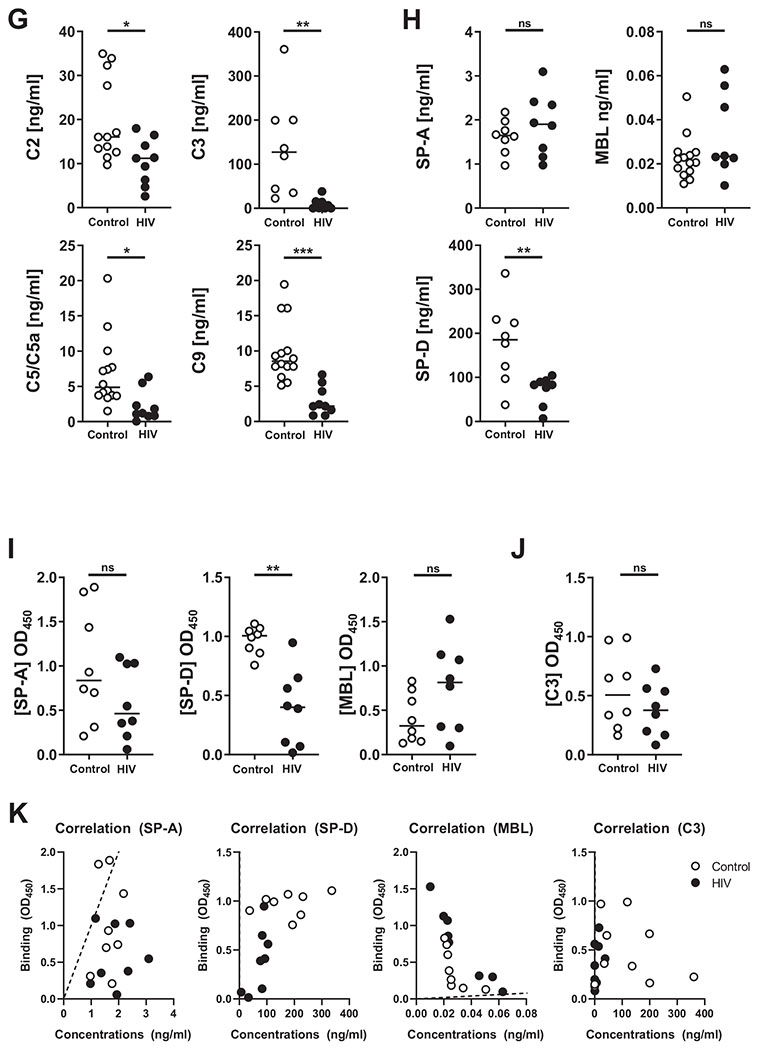
Six major categories of ALF-derived proteins were identified: (A) inflammation proteins, (B) antioxidation proteins, (C) antimicrobial proteins, (D) complement and surfactant proteins, (E) IgG & IgG receptor proteins, and (F) hydrolytic enzymes or hydrolases. Differentially abundant proteins (DAPs) were identified by calculating their log2 fold-changes in the HIV-ALF relative to those in control-ALF. Each sample corresponds to ALF obtained from different human donors. For names of the proteins depicted and statistics, see supplemental material and Table S1 to S3. (G) Complement components: C2, C5/C5a, and C9; and (H) collectins SP-D and MBL were detected by Luminex. Complement component C3 and collectin SP-A were detected by ELISA. M.tb Erdman strain single-cell suspensions were exposed to control-ALF or HIV-ALF. Exposed-M.tb bacteria were washed, suspended in isotonic buffer, and plated onto 96-well plates. Monoclonal antibodies directed against (I) collectins SP-D, SP-A, MBL, and (J) complement component C3 were used to determine their amounts bound to M.tb. For assay controls, purified SP-D, SP-A, MBL, and C3 were used. Relative quantities of bound protein were quantified by standard ELISA by measuring the absorbance at OD450. (K) Correlations between the concentration levels and binding of SP-A, SP-D, MBL, and C3. Notice that regression line for SP-D and C3 overlaps with the y-axis. In G-K, each dot represents ALF from an individual subject. ALFs from control donors (n = 8-17) and PLWH subjects (HIV+, n = 7-10). Unpaired Student’s t test, *p < 0.05; **p < 0.005, ***p < 0.0005. Each sample corresponds to ALF obtained from different human donors.
Innate opsonins are altered in ALF from PLWH
Several ALF innate immune soluble proteins function as opsonic factors for microbes by directing them to specific phagocytic pathways in phagocytes. Based on our results in Fig. 2D, we opted to focus on the levels and functions of innate opsonins including components of the complement cascade (C2, C3, C5, and C9) and collectins, such as SP-A, SP–D, and mannose binding lectin (MBL), given their prominent innate role in the lung alveolar compartment and M.tb infection36–38. Here, we confirmed our proteomics results and found that HIV-ALF samples had significantly lower levels of complement components, specifically C2, C3, and those involved in the formation of the membrane attack complex, C5 and C9 (Fig. 2G). We also observed significantly lower SP-D levels in HIV-ALF, whereas SP-A and MBL levels in HIV-ALF were not significantly different from control-ALF (Fig. 2H).
Next, we used an M.tb-binding assay to further assess the functions of several of these innate immune proteins present in ALF, which play important roles as opsonins during M.tb infection37,38. SP-D binding to M.tb was significantly decreased in HIV-ALF, while SP-A, MBL, and C3 binding remained unaltered in HIV-ALF compared to control-ALF (Figs. 2I, J). It is worth noting that although C3 levels were lower in HIV-ALF, C3 binding to M.tb was similar in HIV-ALF and control-ALF (Fig. 2G, J). In contrast, SP-D had both lower levels and lower binding in HIV-ALF when compared to control-ALF (Figs. 2H, I). In both control-ALF and HIV-ALF, the amount of SP-A, SP-D, MBL, and C3 that bound to M.tb. did not directly correlate with their concentrations (Fig. 2K), indicating that the observed lower binding of SP-D is not solely due to its lower level.
Exposure of M.tb to HIV-ALF provides a bacterial association and growth advantage in primary human macrophages
To test our hypothesis that HIV-induced oxidative stress in the lung alveolar environment results in dysfunctional soluble innate responses predisposing PLWH to a higher risk of respiratory infections, including M.tb. we determined if exposure of M.tb to HIV-ALF influences the capacity of human macrophages (MDMs) to control M.tb infection. First, we assessed whether HIV-ALF had an impact on the capacity of macrophages to recognize M.tb. Control- and HIV-ALF-exposed M.tb were used to infect MDM monolayers, and bacterial-cell association was determined. Our results indicate that HIV-ALF-exposed M.tb has a significantly higher association with MDMs when compared to control-ALF-exposed M.tb (Fig 3A, supplementary Fig S1A).
Fig. 3. Association, intracellular bacterial growth, replication, and P-L fusion events in human macrophages infected with M.tb pre-exposed to control-ALF or HIV-ALF.
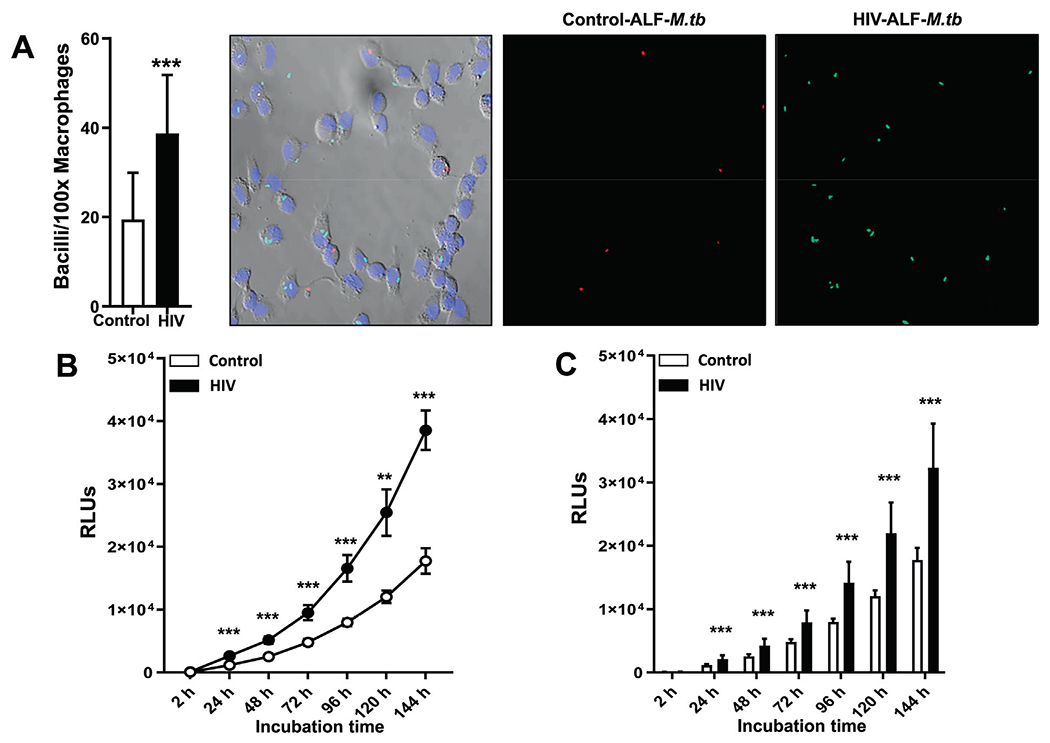
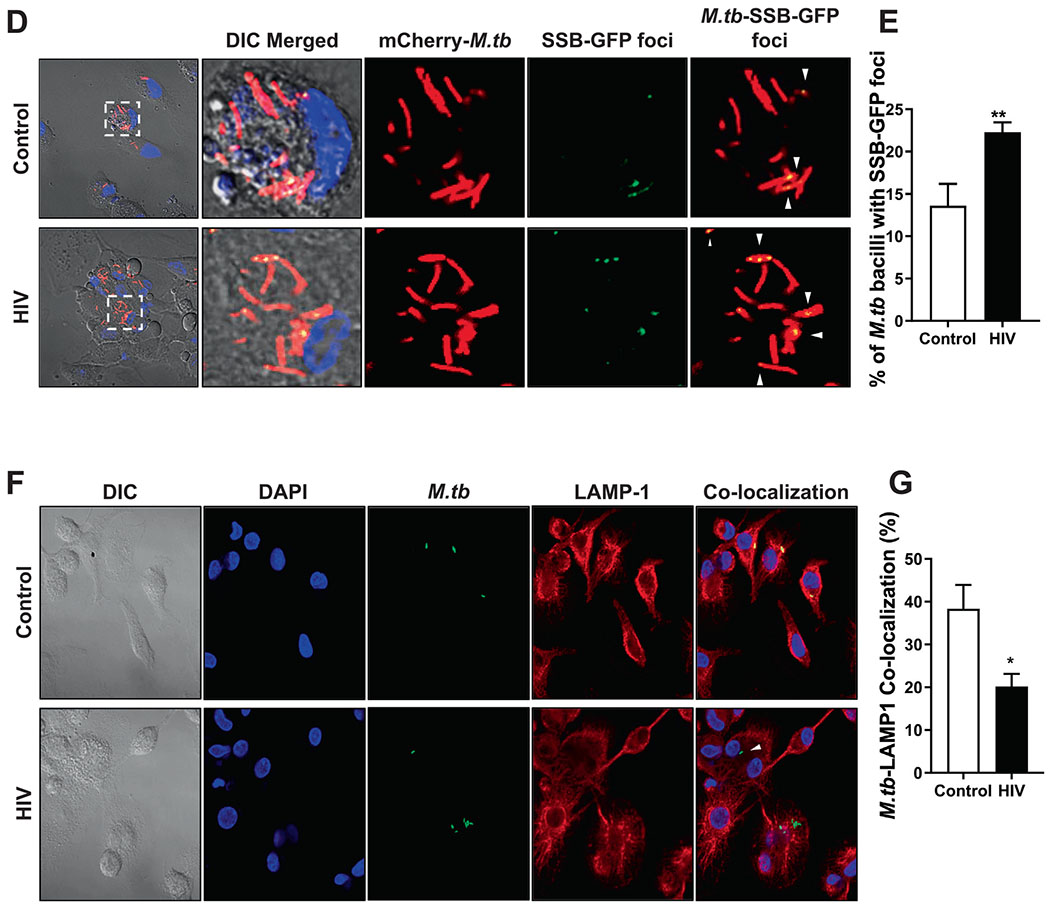
(A) GFP-M.tb Erdman and Red-Cherry tomato M.tb Erdman bacteria were exposed to HIV-ALF or control-ALF, respectively. After exposure, these bacteria were mixed at 1:1 ratio, and macrophages on coverslips were infected with an MOI 10:1 of the bacterial mixture (n = 3/group). Coverslips were processed for and analyzed by confocal microscopy. Shown are GFP-M.tb Erdman exposed to HIV-ALF (Green) and Red-Cherry tomato M.tb Erdman exposed to control-ALF (red). Graph shows percentage of bacterial association with macrophages infected with M.tb exposed to healthy-ALF or HIV-ALF. Counted >150 macrophages per coverslips. Unpaired Student’s t test; Healthy versus HIV+; ***p < 0.0005. (B) M.tb H37Rv-Lux was exposed to HIV-ALF or control-ALF. Macrophage monolayers were infected with ALF-exposed Mtb-Lux (MOI 1:1), and M.tb intracellular growth was assessed by measuring bacterial bioluminescence in terms of RLUs at different time points, representative experiment, and (C) overall data from n = 4. ANOVA Tukey-Posttest; Healthy versus HIV+; **p < 0.005; ***p < 0.0005. Each sample corresponds to ALF obtained from different human donors. (D) Reporter fluorescent strain SSB-GFP, smyc’::mCherry M.tb was exposed to HIV-ALF or control-ALF prior to infection of human macrophages on coverslips for 72 h and M.tb replication rate was determined by confocal microscopy. Shown are representative confocal images of M.tb-infected macrophages on left panels (top and bottom), where region indicated by white dashed-line is shown in large magnification on right with control-ALF exposed samples on top panels and HIV-ALF exposed samples on bottom panels. SSB-GFP+ve M.tb are indicated by white arrowheads, showing merged (yellow) foci. (E) Percentage of SSB-GFP-positive M.tb exposed to control-ALF or HIV-ALF quantified by counting >150 events per coverslip (n = 3). Unpaired Student’s t test; HlV-ALF versus Control-ALF; **p < 0.005. (F) GFP-M.tb Erdman was exposed to HIV-ALF or control-ALF prior to infection of human macrophages on coverslips for 2 h (n = 3). P-L fusion events were visualized with confocal microscopy and enumerated by counting at least >150 independent events per coverslip. Shown are DIC, macrophage nucleus (DAPI, blue), phagosome containing GFP-M.tb (green), lysosomes (LAMP-1+, red), and P-L fusion events (co-localization, yellow). (G) Graph shows the percentage of co-localization of LAMP-1 (lysosomal marker) with phagosomes containing M.tb. Unpaired Student’s t test; Healthy versus HIV; n = 3 *p < 0.05; **p < 0.005. Each sample corresponds to ALF obtained from different human donors.
For evaluate intracellular growth of HIV-ALF-exposed M.tb versus control-ALF-exposed M.tb. we employed both bacterial relative luminescence unit (RLU) (Fig. 3B, C) and traditional colony-forming unit (CFU) (supplementary Fig S1B) assays in MDMs by using a luciferase-expressing strain M.tb-Lux and GFP-expressing M.tb Erdman, respectively20,26,31,39. Although we observed a significant difference in association (Fig. 3A), the uptake (quantity of intracellular bacilli at 2 h) was similar (Figs. 3B, C, and supplementary Fig. S1B). Despite this similar uptake, M.tb exposed to HIV-ALF showed significantly increased growth in MDMs compared to M.tb exposed to control-ALF, which became more apparent over time (although we note that the luciferase and CFU assays are less sensitive than the association assay by microscopy) (Figs. 3B, C).
Exposure of M.tb to HIV-ALF increases bacterial replication rate and reduces its P-L fusion in primary human macrophages
To investigate the characteristics of the growth of HIV-ALF-exposed M.tb in MDMs, we evaluated M.tb intracellular replication and intracellular trafficking. Using a fluorescent M.tb replication reporter strain (SSB-GFP, smyc′::mCherry M.tb)20,30,40, the number of M.tb bacilli with SSB-GFP foci were counted, and then the percentages were calculated to determine the rate of active replication. Our results indicate that HIV-ALF-exposed M.tb had a small but significantly increased replication rate compared to control-ALF-exposed M.tb (control: 13.62 ± 1.48% versus HIV: 22.28 ± 0.68%) (Figs. 3D, E).
To measure intracellular trafficking, we quantified the number of M.tb phagosomes fused with lysosomes (LAMP-1+) and expressed the result as a percentage of M.tb-LAMP-1 co-localization. It is known that M.tb phagosomes have limited fusion with lysosomes41. Our results show that there was less co-localization with lysosomes by phagosomes from HIV-ALF-exposed M.tb than control-ALF-exposed M.tb (HIV: 20.2 ± 1.7% vs.control: 38.37 ± 37%) (Figs.3F,G). Collectively, our data indicate that a higher replication rate and significantly reduced P-L fusion contribute to the increased growth of HIV-ALF-exposed M.tb in MDMs.
Supplementation of HIV-ALF with SP-D restores the macrophage capacity to control HIV-ALF-exposed M.tb infection
SP-D binds avidly to the surface of M.tb via lipoarabinomannan23, driving an increase in P-L fusion of SP-D-coated M.tb, resulting in a better control of M.tb growth in MDMs24. We supplemented HIV-ALF with SP-D to test the hypothesis that the increased M.tb growth observed in MDMs infected with HIV-ALF-exposed M.tb (Figs. 3B, C) is due mainly to the low levels and low binding of SP-D in HIV-ALF compared to control-ALF (Fig. 2H, I). Subsequently, M.tb bacilli (M.tb-Lux for growth assay and GFP-M.tb for P-L fusion assay) were exposed to SP-D-supplemented HIV-ALF and control-ALF prior to infection of human MDMs20. Results showed that the addition of SP-D to the HIV-ALF allowed macrophages to regain control of the infection at the same levels that were observed for control-ALF-exposed M.tb (Figs. 4A, B) and that this effect is via increasing P-L fusion events to reach the same levels as those observed in MDMs infected with control-ALF-exposed M.tb (a 17.6% increase in P-L fusion) (Figs. 4C, D). These data support the concept that the binding of SP-D to M.tb is impaired in HIV-ALF and that its supplementation restores the capacity of human macrophages to control HIV-ALF-exposed M.tb growth.
Fig. 4. Addition of SP-D to HIV-ALF restores macrophage control of M.tb growth.
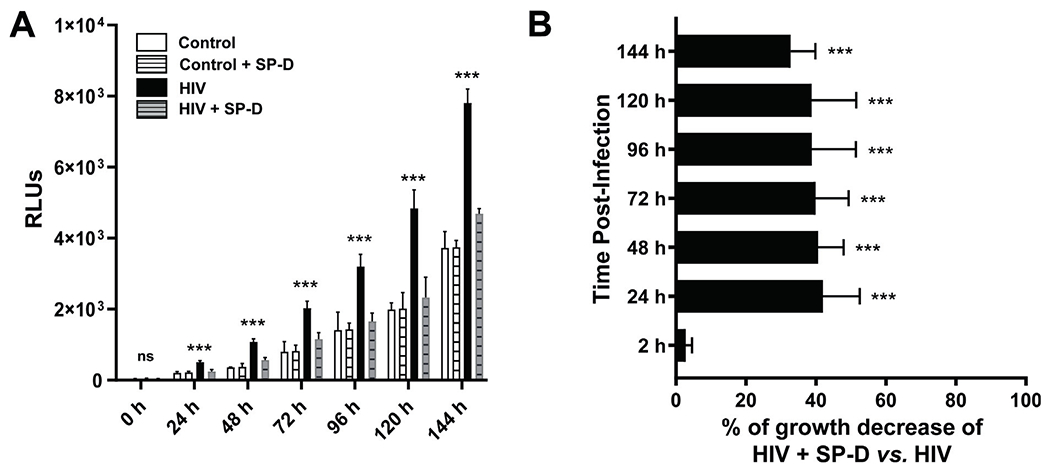
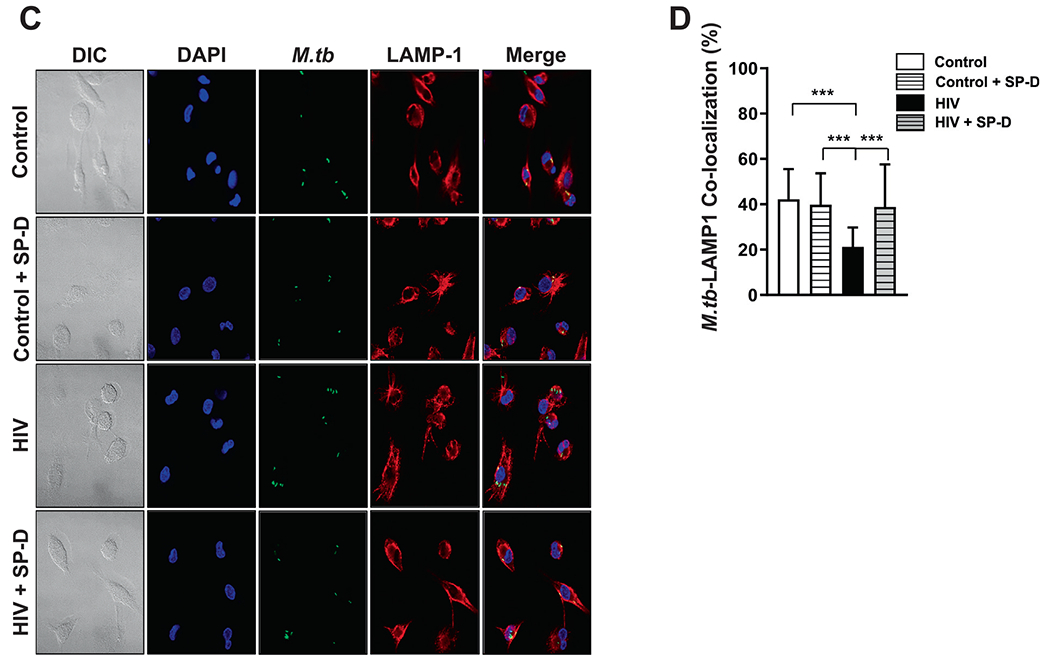
(A) M.tb H37Rv-Lux was exposed to HIV-ALF, control-ALF, or to HIV-ALF or control-ALF each being supplemented with SP-D at its physiological concentration. Human macrophages were infected with these differently-exposed M.tb, and bacterial intracellular growth was measured in RLUs at different time points through 144 h. Representative experiment of n = 3, using ALF and macrophages in each case from different human donors. ANOVA Tukey-Posttest; HIV versus Control; ***p < 0.0005. (B) Overall data (n = 3) showing % of growth decrease for HIV-ALF + SP-D exposed M.tb versus HIV-ALF exposed M.tb. ***p < 0.0005. (C) GFP-M.tb Erdman was exposed to HIV-ALF, control-ALF, HIV-ALF+SP-D, or control-ALF+SP-D as described above prior to infection of macrophages on coverslips for 2 h (n = 3). P-L fusion events were visualized with confocal microscopy and enumerated by counting at least >150 independent events per coverslip. Shown are DIC, macrophage nucleus (DAPI, blue), phagosome containing GFP-M.tb (green), lysosomes (LAMP-1+, red), and P-L fusion events (co-localization, yellow). (D) Graph shows percentage of P-L fusion in terms of M.tb-LAMP-1 co-localization. Unpaired Student’s t test; Healthy versus HIV+; n = 3 ***p < 0.0005. Each sample corresponds to ALF obtained from different human donors.
DISCUSSION
PLWH have a 25-fold increased risk of respiratory tract infections42–44; even modest insufficiency of the immune system in the lung makes PLWH more vulnerable to microbial infections42,43, as well as, to other chronic diseases. In fact, M.tb is a frequent respiratory tract infection among PLWH, and TB is the major cause of death in PLWH.
In this study, we demonstrated that PLWH have an alveolar environment marked by a high degree of protein oxidation and decreased levels of anti-oxidation proteins. This is accompanied by a decrease in Th1/Th2/Th17 responses (supplementary Fig. S2), as well as lower levels of complement proteins and SP-D. In the case of SP-D, its lower levels did not correlate with its lower binding to M.tb. In this regard, SP-D binding to M.tb leads to bacterial aggregation and reduced bacterial growth in macrophages by increasing P-L fusion24.
In this context, a role has been reported for some ALF components during HIV infection42. SP-A and SP-D can modulate HIV infection by inhibiting infectivity of CD4 T cells or by stimulating HIV transfer from dendritic cells (DCs) to CD4 T cells42. SP-D binds to HIV envelope glycoprotein 120 (gp120), blocking HIV binding and entry into macrophages45. Our results indicate that HIV-ALF contains low levels of SP-D with decreased binding to microbes. These SP-D alterations may be a contributing factor for the increased susceptibility of PLWH to respiratory infections. Our in vitro studies with M.tb support this concept since supplementation of HIV-ALF with SP-D restored the ability of macrophage to control M.tb growth. These in vitro studies suggest that the low levels of SP-D and its decreased binding are major contributors to the altered innate immune control of M.tb during HIV infection, although its role in vivo remains to be determined. Reduced levels and reduced binding of SP-D in the lungs may correlate with negative pulmonary outcomes in PLWH without ART42. This is the case for other lung diseases such as pulmonary fibrosis, where lower levels of SP-D correlate with increased mortality46. In contrast to SP-D, SP-A levels and binding to M.tb in the PLWH ALF did not significantly differ from those in control individuals. SP-A can regulate the macrophage response to infection in that SP-A facilitates the transfer of HIV from dendritic cells (DCs) to CD4 T cells47,48. Decreased binding of SP-D and unaltered SP-A may favor establishment of M.tb infection in lung macrophages. Conversely, SP-A promotes attachment of M.tb to macrophages by increasing surface exposure of the mannose receptor (CD206) on macrophages49, which is increased during HIV infection driving IL-6 production. In this regard, we observed an overall decrease in Th1/Th2/Th17 cytokine levels in the lungs of PLWH while IL-6 remained unaltered. This decrease in cytokine levels in HIV-ALF is also observed in in vitro experiments showing that HIV infection impairs the production of TNF by macrophages50,51. Thus, HIV infection may weaken the lung immune response against subsequent microbial infections because of dysregulation of inflammatory responses in the lung, generating an environment that favors pulmonary microbial infections42. Our model is illustrated in Fig. 5.
Fig. 5. Model of the host responses to M.tb in the lung of PLWH compared to control.
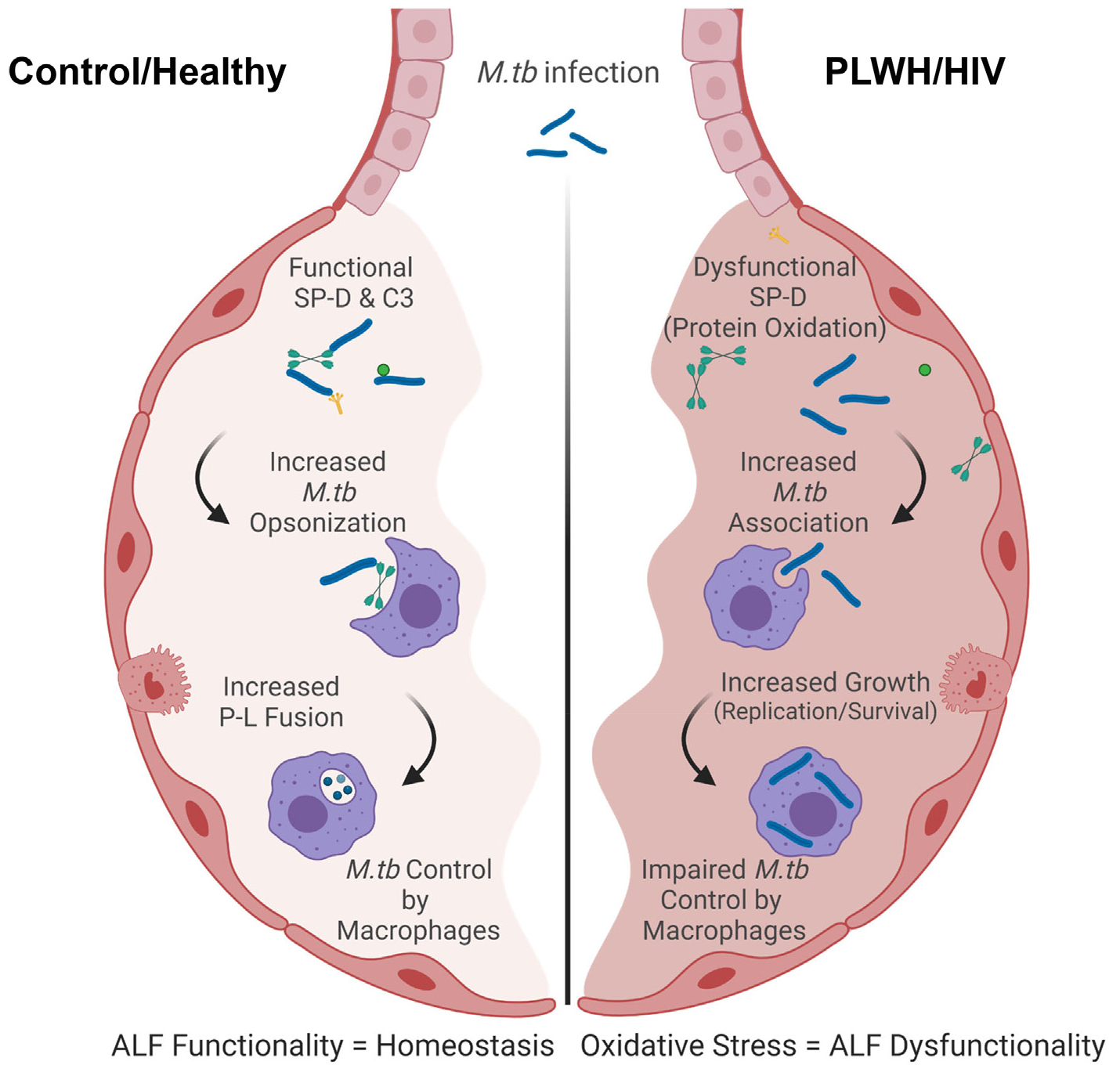
Local oxidative stress is detected in the lung of PLWH. This oxidative stress may drive the dysfunction of soluble immune components and inflammation in the lungs marked by decreased levels of Th1/Th2/Th17 cytokines. Lower levels of complement and SP-D are also detected in PLWH leading to their lower capacity to opsonize and/or modify the M.tb bacterial cell surface prior contacting host cells. This drives better recognition of HIV-ALF exposed M.tb by macrophages. Phagocytosed HIV-ALF exposed M.tb bacilli grow faster within macrophages by further impairing P-L fusion and replicating faster within phagosomes. In vitro, this is related to SP-D function since addition of excess of SP-D to HIV-ALF restores the capacity of macrophages to control HIV-ALF-exposed M.tb; thus, SP-D plays an important role in maintaining lung homeostasis and innate immune activity against lung respiratory infections. Created in BioRender.com.
Previous studies have shown that alveolar macrophages in PLWH are functionally impaired42,52–55, with a reduced capacity for phagocytosis56,57. However, components in HIV-ALF may not be responsible for this observed effect, as exposure of M.tb to HIV-ALF resulted in an increase of M.tb association with macrophages obtained from control individuals.
Although the complement system is systemically activated by HIV infection58, within the HIV-ALF of PLWH, we observed low levels of the classical complement proteolytic cascade, including both early complement components (e.g. C1qa and C1qc) and terminal factors (e.g. C5b-9) involved in the formation of the membrane attack complex. C1q also binds to several receptors on myeloid cells, regulating their functions, including phagocytosis59. C1q deficiency is associated with increased infections and autoimmunity, and C5 and C7 deficiencies are linked to M.tb pathogenesis59–62. The role of complement in M.tb pathogenesis remains unclear, although C3 binding to M.tb cell envelope components leads to M.tb recognition by the complement receptor 3 (CR3) on human macrophages22,63. The outcome of this interaction in terms of M.tb control is unknown. Our results indicate that PLWH have significantly less C3 than HIV negative individuals, which has implications for the establishment of M.tb infection in PLWH. Regarding the complement lectin pathway, MBL binds the gp120 on HIV and M.tb58,64–66; however, MBL levels and binding remained unaltered in the HIV-ALF of the PLWH studied.
We also found lower levels of IgJ and pIgR in HIV-ALF. IgJ is involved in the secretion of IgA and IgM into the lung mucosa. It is essential for the binding of polymeric immunoglobulin to pIgR, which forms a secretory component, facilitating the transcytosis of soluble polymeric isoforms of IgA and immune complexes from basal to the apical sides of alveolar epithelial cells67. Deficiencies of IgJ and pIgR in HIV-ALF could contribute to the decrease in IgA levels observed in HIV-ALF, detected as lower levels of IgA heavy chain alpha, IgHA1, and its analog IgHA2. IgA deficiency in the lung could contribute to the susceptibility to respiratory infections observed in PLWH. Of relevance, IgJ plays a role in the activation of complement68.
Our proteomics results confirm that HIV-ALF contains an array of inflammatory, antimicrobial, and oxidant-scavenging proteins that are upregulated [e.g. calprotectin (S10A8/S10A9) protein and mucins (MUC1, 4, and 5A), among others]. However, these could have altered functional consequences, as we observed for SP-D. Further studies are required to determine the impact of HIV infection on the overall physiological and immunological status of the lung and how this influences the establishment of acute (e.g. SARS-CoV-2) and chronic (e.g. M.tb) respiratory infections. Our study does not address the impact of ART on the lung environment. Systemically, ART fails to restore full recovery of the innate and adaptive systems and does not reduce systemic inflammation69. ART is known to improve the adaptive immune response in the lung; however, inflammation persists despite the recovery of CD4 T cells and HIV viral suppression42. ART also induces the immune reconstitution inflammatory system (IRIS) in the lungs, particularly influencing latent M.tb infection or failed TB treatment42. Thus, the effects of ART on soluble innate immune components and their functions in the lung need to be evaluated further. We anticipate that HIV infection disturbs the oxidative status of the lung. This perturbation of the oxidative status of the lung may be partially restored by ART70,71, but irreversible levels of protein oxidation (e.g. irreversible modification of tyrosine to 3⊠nitrotyrosine by RNS)34 will persist; thus, explaining why PLWH treated with ART still have a higher susceptibility to respiratory infections. We also observed that MPO levels trended downward in HIV-ALF but carbonyl and 3-nitrotyrosine protein modification increase. A plausible explanation is that there is a poor recruitment or activation of neutrophils in the lungs of PLWH. This can be supported by the overall decreased inflammatory response observed in HIV-ALF. MPO in ALF is mainly secreted by neutrophils reaching the alveolar environment. Thus, MPO cannot be the major contributor to the oxidative status observed in the HIV-ALF proteome. As indicated, carbonylation and nitrotrosylation of proteins are a result of the action of ROS and NOS, respectively. We can speculate that both ROS and NOS can result from local cellular oxidation due to potential mitochondrial dysfunction. Several studies have demonstrated mitochondrial impairment in PLWH. We observed this phenomenon in the elderly population21,72.
In our experimental model, the addition of SP-D to HIV-ALF restored the capacity of macrophages to control HIV-ALF exposed M.tb by doubling P-L fusion events (from 20% to 40). Interestingly, after replenishment of SP-D in HIV-ALF, of all the innate immunomodulators measured, only IL-6 (Th2), IL-1β/IL-18 (inflammasome), and GM-CSF (growth factor) were reduced by the presence of functional SP-D (supplementary Fig. S3), indicating a potential negative impact of the high levels of these immunomodulators on the macrophage control of M.tb growth in the initial stages of the infection.
A potential limitation of this study is the number of participants in this study per group (n = 10 to 17), and that PLWH participants declared that they were never or former smokers at the time of providing HIV-ALF samples. This was clinically recorded, trusting the participants’ responses. Further, there is a bias of 90% male (PLWH) versus 60% male (control) study participants potentially skewing the data. We normalized our ALF samples to the physiological phospholipid concentrations in the human lung. The actual status related to former smoking is not expected to influence our results, as published studies have shown that never and former smokers have similar ALF phospholipid content73. It could also be rationalized that HIV infection drives lower levels of SP-D and thus its observed decrease in binding to M.tb. However, our data show that there is no direct correlation between SP-D levels and binding. Our results are in contrast with other studies conducted with a cohort in Malawi, where SP-D levels were higher in PLWH. However, the subjects in that study had AIDS with CD4 T cells <200, and SP-D binding was not assessed74. A plausible explanation for our results is that the decrease in SP-D binding to M.tb could be explained by the presence of the soluble HIV Env gp120 subunit in HIV-ALF since HIV gp120 binds SP-D and could, thereby, block SP-D binding to M.tb. Alternatively, gp120 could also directly bind to M.tb. In these two scenarios, the presence of gp120 in HIV-ALF could redirect M.tb to a pathway favoring its intracellular replication and survival. However, neither explanation appears to be justified. First of all, we did not detect the presence of gp120 in HIV-ALF by MS analysis, and second, gp120 also binds to MBL75, and no reduction in MBL binding to M.tb was observed in this study.
Overall, our data highlight the importance of lower levels and reduced binding of SP-D in PLWH, which we posit plays a role in respiratory infection outcomes. Indeed, SP-D decreased binding could be owing to the plausible increased proteases affecting SP-D and other soluble immunomodulators as indicated by the observed increased inflammation related proteins in the HIV-ALFs. This low SP-D binding could also be because SP-D is produced less or because SP-D became dysfunctional (oxidized) due to an oxidative stress environment. This needs to be further explored in subsequent future studies.
An interesting observation is that all the cytokines and chemokines (except IL-8 and CCL-2) were significantly reduced in HIV-ALF from asymptomatic PLWH without ART. We can speculate that it could be due to an overexpression of negative regulators of inflammation lowering the expression of pro-inflammatory cytokines as an intent to keep the lungs healthy in PLWH, but at the same time, providing a favorable environment for opportunistic infections. Another possibility is that HIV infection could prevent the active cellular secretion of some cytokines into the alveolar environment. At the same time, IL-8 and CCL-2 levels did not get affected, thus, we can also speculate that as a result, the capacity of recruiting cells to the lungs (e.g. neutrophils, monocytes, memory T cells, and dendritic cells) may remain unaltered. However, if recruited, these cells may do not get easily activated due to low inflammatory alveolar environment observed. Future studies will be needed to address these speculations76.
This low inflammatory lung environment observed in PLWH differs from the high inflammatory lung environment that we observed in the elderly leading to their susceptibility to respiratory infections, including M.tb20,21,77. In both cases, however, PLWH and the elderly, there is a high degree of oxidative stress in their lungs, leading to higher levels of oxidation of proteins in the innate immune system, which significantly alters the host response and is associated with susceptibility to respiratory infections. Identifying the outcomes of HIV-associated lung oxidative stress on alveolar innate immune responses will be valuable for the development of host-directed therapies for PLWH and the elderly, reducing their risk of respiratory infections.
MATERIALS AND METHODS
Human subjects and ethics statement -
All experimental procedures with human subjects were carried out in strict accordance with the US Code of Federal and Local Regulations. Healthy (control) participants came from the Ohio State University IRB 2007H0263 [Influence of the human lung hydrolases on Mycobacterium tuberculosis infection (bronchoalveolar lavage)]. HIV (PLWH) participants came from the Ohio State University IRB 2005H0197 (HIV surveillance affects macrophage function). Recombinant DNA and Biohazards Safe Practice was under The Ohio State University 2003R0057. In the case of healthy (controls), Bronchoalveolar lavages (BALs) in these participants were performed to study the impact of hydrolases on the M.tb infection (line of research in Dr. Torrelles’ laboratory). In the case of PLWH, BALs were performed to isolated alveolar macrophage and study their function (line of research in Dr. Wewers’ laboratory). Bronchoalveolar lavage fluid (BALF) to obtain ALF was collected from control and PLWH subjects. All subjects for this study provided informed written consent. We obtained BALF from healthy donors (median age 33.5, without comorbidities, non-smokers, non-drug users, 60% male, and a median of 92% alveolar macrophages), and PLWH who were not receiving ART (undisclosed smoking, never or former smoking status, or drug use but currently declared non-smokers), with median CD4+ T cell count of 378.5 cells/μl (93–661 cells/μl), a median viral load of 54,812 RNA copies/ml (<100-210,504 RNA copies/ml), a median age of 23 (23.2-50.9), a median BMI of 18.82 (16.6-21.5), 90% male, and a median of 87.5% alveolar macrophages20,21,26,27,29,30,32. (Table 1 and Table 2)
Table 1.
Participant demographic parameters.
| Control | |||||||
|---|---|---|---|---|---|---|---|
| BAL ID | Weight** (Pounds) | BMI | Sex | Age***(years) | Viral copies | CD4 T cell counts | ART |
| D4 | 180 | U* | F | 26 | N/A | N/A | N/A |
| D5 | 156 | U* | F | 26 | N/A | N/A | N/A |
| D7 | 195 | U* | M | 24 | N/A | N/A | N/A |
| D8 | 145 | U* | F | 22 | N/A | N/A | N/A |
| D9 | 199 | U* | M | 23 | N/A | N/A | N/A |
| D11 | 148 | U* | F | 46 | N/A | N/A | N/A |
| D12 | 178 | U* | F | 22 | N/A | N/A | N/A |
| D13 | 132 | U* | M | 27 | N/A | N/A | N/A |
| D15 | 134 | U* | M | 18 | N/A | N/A | N/A |
| D16 | 175 | U* | F | 25 | N/A | N/A | N/A |
| D19 | 166 | U* | M | 19 | N/A | N/A | N/A |
| D22 | 146 | U* | F | 19 | N/A | N/A | N/A |
| D23 | 110 | U* | F | 19 | N/A | N/A | N/A |
| D24 | 124 | U* | M | 23 | N/A | N/A | N/A |
| D26 | 182 | U* | M | 23 | N/A | N/A | N/A |
| D27 | 64 | U* | F | 20 | N/A | N/A | N/A |
| D28 | 122 | U* | F | 19 | N/A | N/A | N/A |
| PLWH | |||||||
| BAL ID | Weight** (Pounds) | BMI | Sex | Age*** (years) | Viral copies | CD4 T cell counts | ART |
| 154 | 209 | 28.34 | M | 54 | 48 | 227 | No |
| 155 | U* | U* | F | 32 | 48 | 661 | No |
| 161 | U* | U* | M | 45 | 48 | 605 | No |
| 179 | 189 | 25.63 | M | 44 | 883 | 532 | No |
| 182 | 135 | 21.14 | M | 35 | 65,083 | 251 | No |
| 186 | 157 | 24.59 | M | 29 | 58,836 | 286 | No |
| 190 | 150 | 25.74 | F | 20 | 48 | 710 | No |
| 204 | 174 | 27.25 | M | 23 | 150,187 | 301 | No |
| 210 | 181 | 26.73 | M | 43 | 9,413 | 429 | No |
| 219 | 191 | 23.25 | M | 30 | 210,504 | 125 | No |
Unknown/no collected; M: Male, F: Female; Age (years), Viral copies (per μl), CD4 T cell counts (per μl), ART: antiretroviral treatment.
There is not a statistical difference in the weight between control versus PLWH participants.
There is a statistical difference in the age between control versus PLWH participants (student t test, p = 0.0013).
Table 2.
Cell types in BALs from participants in this study
| BAL ID | Total Cell Count (x106) | Mφ Count (x106) | % Mφ | % L | % N | % E | % Ep |
|---|---|---|---|---|---|---|---|
| 154 | 16.81 | 13.95 | 83 | 9 | 4 | 1 | 3 |
| 155 | U* | U* | U* | U* | U* | U* | U* |
| 161 | U* | U* | U* | U* | U* | U* | U* |
| 179 | 33.65 | 31.3 | 93 | 7 | 0 | 0 | 0 |
| 182 | 24.89 | 22.9 | 92 | 5 | 3 | 0 | 0 |
| 186 | 31.18 | 29 | 93 | 4 | 0 | 0 | 3 |
| 190 | 3.13 | 2.6 | 83 | 8 | 8 | 0 | 1 |
| 204 | 16.20 | 8.1 | 50 | 50 | 0 | 0 | 0 |
| 210 | 23.49 | 19.5 | 83 | 17 | 0 | 0 | 0 |
| 219 | 27.87 | 22.3 | 80 | 20 | 0 | 0 | 0 |
Unknown; Mφ: Macrophage; L: Lymphocytes; N: Neutrophils; E: Eosinophils; Ep: Epithelial cells.
Collection of human BALF and ALF -
BALF was collected, and ALF obtained at its physiological concentration present within the lung (at 1–1.5 mg of phospholipid/mL) as we previously described20,21,26,27,29–32. Specific protein amounts were used for each assay, as specified below. Status of being either a never or a former smoker does not affect the phospholipid content in the donor’s lung mucosa73. Concentrated ALF samples were stored in 50- to 100-μL aliquots at −80°C. Total protein content in ALF samples was determined by using a BCA protein assay kit (Thermo Fisher).
Isolation and culture of human monocyte-derived macrophages -
Peripheral blood mononuclear cells (PBMCs) were isolated from healthy donor blood, and monocyte-derived macrophages (MDMs) were prepared from PBMCs as described26,31. Briefly, heparinized blood was layered on a Ficoll-Paque cushion (GE Healthcare, Uppsala, Sweden) to allow for the collection of PBMCs. PBMCs were then cultured in RPMI (Life Technologies, Carlsbad, CA) with 20% autologous serum in Teflon wells (Savillex, Eden Prairie, MN) for 5 days at 37°C/5% CO2 to allow for monocyte maturation into macrophages. PBMCs were harvested and MDMs purified by their adhesion to tissue culture dishes for 2 h in RPMI with 10% autologous serum, followed by washing away the lymphocytes. MDMs were then incubated overnight in RPMI with 10% autologous serum before their use in the downstream experiments.
M.tb strains and culture -
M.tb H37Rv (ATCC# 27294, Manassas, VA), GFP-M.tb Erdman (kindly provided by Dr. Marcus Horwitz, University of California, Los Angeles, CA) and M.tb Erdman-Red-Cherry tomato (kindly provided by Dr. William Jacobs Jr., Albert Einstein Institute, New York, NY) were grown as we previously described31. M.tb H37Rv-Lux was kindly provided by Dr. Larry S. Schlesinger (Texas Biomed, San Antonio, TX)39. SSB-GFP, smyc’::mCherry M.tb Erdman (kindly provided by Dr. David Russell, Cornell University, Ithaca, NY) was grown as described30,40. M.tb strains were cultured on 7H11 agar media with and without applicable antibiotics for 12-14 days at 37°C, 5% CO2.
Determination of protein oxidation markers in human ALF -
Levels of protein carbonyls [a stable marker of ROS-induced oxidation] in ALF (using 8 μg of protein) were determined using OxiSelect Protein Carbonyl ELISA Kit (Cell Biolabs, Inc.). Levels of 3-nitrotyrosine-containing proteins [a stable marker of RNS-induced oxidation] in ALF (using 10 μg of protein) were determined using OxiSelect Nitrotyrosine ELISA Kit (Cell Biolabs, Inc., San Diego, CA). Myeloperoxidase (MPO, indicative of tissue oxidation) levels in ALF (using 10 μg of protein) were determined by using LUMINEX (R&D Systems, Minneapolis, MN).
Proteomic analyses of human ALF -
ALF sample aliquots corresponding to 10 μg protein (EZQ™ Protein Quantitation Kit; Thermo Fisher) were mixed with 5% SDS/50 mM triethylammonium bicarbonate in the presence of protease and phosphatase inhibitors (Halt; Thermo Scientific). Then, these ALF samples were reduced with tris(2-carboxyethyl)phosphine hydrochloride, alkylated in the dark with iodoacetamide, and applied to S-Traps (micro; Protifi) for tryptic digestion (sequencing grade; Promega, Madison, WI) in 50 mM TEAB. Peptides were eluted from the S-Traps with 0.2% formic acid in 50% aqueous acetonitrile and quantified using Pierce™ Quantitative Fluorometric Peptide Assay (Thermo Scientific).
Data-independent acquisition mass spectrometry was conducted on an Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific). On-line HPLC separation was accomplished with an RSLC NANO HPLC system (Thermo Scientific/Dionex): column, PicoFrit™ (New Objective; 75 μm i.d.) packed to 15 cm with C18 adsorbent (Vydac; 218MS 5 μm 300 Å); mobile phase A, 0.5% acetic acid (HAc)/0.005% trifluoroacetic acid (TFA) in water; mobile phase B, 90% acetonitrile/0.5% HAc/0.005% TFA/9.5% water; gradient 3 to 42% B in 120 min; flow rate, 0.4 μL/min. A pool was made of all of the samples, and 1-μg peptide aliquots were analyzed using gas-phase fractionation and 4-m/z windows (30k resolution for precursor and product ion scans, all in the orbitrap) to create a DIA chromatogram library by searching against a panhuman spectral library78,79. Experimental samples were randomized for sample preparation and analysis. Injections of 1 μg of peptides were employed. MS data for experimental samples were acquired in the orbitrap using 12-m/z windows (staggered; 30k resolution for precursor and product ion scans) and searched against the chromatogram library. Scaffold DIA (v3.3.1 Proteome Software, Portland, OR) was used for all DIA data processing.
Determination of immune mediators in human ALF -
Protein levels in ALFs were determined using custom Human Magnetic Luminex Assays (R&D Systems) for IFNγ, TNF, IL-12/23p40, IL-2, IL-4, IL-10, IL-6, IL-13, IL-17, IL-21, IL-22, IL-27, GM-CSF, TREM-1, IL-8, CCL2, complement component (C) 2, C5/C5a, C9, SP-D, and mannose binding lectin (MBL). SP-A levels were measured by Human SP-A ELISA kit (LifeSpan Biosciences, Inc., Seattle, WA) and C3 levels by Human Complement 3 ELISA kit (Abcam, Cambridge, MA) as per manufacturer instructions.
Preparation of human ALF-exposed M.tb -
M.tb exposure to human ALF was performed as described20,26,27,29,31,32. Briefly, M.tb single cell suspensions were prepared and counted as previously described, and 1 × 108 bacteria were exposed to physiological concentrations of ALF (50 μL as described above) for 12 h at 37°C, 5% CO2. After exposure, ALF-exposed M.tb bacilli were washed twice with sterile endotoxin-free saline and immediately used for analysis and in vitro infections (see below)31,80,81.
Determination of functionality of ALF innate proteins by M.tb-protein binding assays -
Human ALF-exposed M.tb was washed with TBS + 2 mM CaCl2 (TBS-C) to remove residual ALF and subsequently suspended in 500 μL of TBS-C. Human ALF-exposed M.tb (1x107) was added to triplicate wells of 96-well plates and dried overnight. Binding of SP-A, SP-D, C3, and MBL contained in human ALF samples to M.tb was determined by indirect ELISA using primary monoclonal antibodies (mouse anti-human SP-A, mouse anti-human SP-D, mouse anti-human C3/C3b, and mouse anti-human MBL; all from Abcam) and secondary antibodies (donkey anti-mouse HRP and goat-anti rabbit HRP; Santa Cruz). Amounts of protein binding to M.tb were quantified by colorimetric measurements by absorbance at 450 nm using a GloMax plate reader (Version 3.0; Promega Corporation) and plotted based on absorbance values relative to the baseline (negative control, No ALF).
ALF-exposed M.tb association with, and trafficking, replication, and survival within human macrophages
To determine M.tb-macrophage association, GFP-M.tb Erdman and Red-Cherry tomato M.tb Erdman bacteria were exposed to HIV-ALF or control-ALF, respectively, and washed to remove all traces of ALF before both bacterial strains were mixed at 1:1 ratio. MDM monolayers on coverslips were then infected with the bacterial mixture at an MOI of 10:1 for 2 h. Coverslips were fixed with 4% paraformaldehyde, followed by mounting with ProLong diamond antifade mount with DAPI (Invitrogen, Waltham, MA)20,31. Coverslips were imaged by using a Zeiss LSM 800 Confocal Microscope. At least 300 bacteria were quantified per condition. This study was repeated by switching the ALF exposure: GFP-M.tb Erdman was exposed to control-ALF and Red-Cherry tomato M.tb Erdman to HIV-ALF. In both cases, we observed the same phenotype.
To determine intracellular trafficking, MDM monolayers on coverslips were infected with GFP-M.tb Erdman exposed to HIV-ALF or healthy-ALF with an MOI of 10:1 for 2 h. Coverslips were fixed with 4% paraformaldehyde and permeabilized with cold methanol as described26,31. To assess M.tb intracellular trafficking, macrophage lysosomal compartments were stained for LAMP1 marker using a mouse anti-human LAMP1 antibody (H4AB; Developmental Hybridoma Bank, Iowa City, IA) overnight at 4°C, followed by staining with an APC-fluorescent anti-mouse secondary antibody (Molecular Probes, Eugene, OR) or with matched isotype controls for 60 min at 37°C. Coverslips were mounted as described above for confocal microscopy. Phagosome-containing M.tb was seen in green (GFP), late endosomal-lysosomal compartments in red (LAMP-1), and co-localization indicative of phagosome-lysosome (P-L) fusion in yellow. Co-localization between phagosomes containing GFP-M.tb and lysosomes containing LAMP1 marker was quantified by counting >150 events per coverslip.
To determine M.tb replication within macrophages, the reporter M.tb strain (SSB-GFP, smyc’::mCherry M.tb Erdman) was grown on 7H11 agar containing 50 μg/mL hygromycin B30,40. Macrophage monolayers on glass coverslips were infected with this M.tb strain pre-exposed to control- or HIV-ALF at an MOI of 1:1 for 72 h. Fixed coverslips were mounted as above and imaged by confocal microscopy. At least 300 bacteria were quantified per condition. Quantification of the SSB-GFP reporter was accomplished by counting the number of bacteria with and without SSB-GFP replication foci. All microscopy data were analyzed by using the Zeiss ZEN software.
For intracellular survival, MDM monolayers were infected with ALF-exposed M.tb (GFP, Red-Cherry tomato, or Lux) with an MOI of 1:1 and cells were incubated for 2 h at 37°C, 5% CO2, as described20,31. Subsequently, infected MDM monolayers were washed with warm RPMI (at 37°C) and further incubated at 37°C, 5%CO2 in RPMI with 2% autologous human serum for the indicated times post-infection (2 h for association and 24 h to 144 h for survival and bacterial replication). M.tb intracellular survival was assessed by two approaches. For the luciferase assay, MDMs were infected with M.tb-Lux, and bacterial bioluminescence was measured in RLUs every 24 h for up to seven days using a GloMax Multi Detection System (Promega)39. For CFU assays, infected MDMs at the indicated time period postinfection were lysed, diluted, and plated on 7H11 agar plates. The numbers of CFU were enumerated after bacterial growth for 3 to 4 weeks at 37°C26,31,82.
M.tb growth and intracellular trafficking after human SP-D replenishment in ALF -
M.tb-Lux was exposed to control- or HIV-ALF with or without (as control) supplemented human recombinant SP-D (5 μg/mL, R&D Systems). After exposure, ALF-exposed M.tb was washed to remove all traces of ALF and re-suspended in single bacterial suspensions using sterile 0.9% NaCl. MDMs were infected with ALF-exposed M.tb at an MOI of 1:1 for RLU growth assay and at an MOI of 10:1 for intracellular trafficking studies as described above.
Statistical analysis -
Statistical significance was determined using GraphPad 9.3.1 Prism software. In these studies, “n” values represent the number of times that an experiment was performed using different human donors for cells and different human donors for ALFs (for both control- and HIV-ALFs). Statistical analyses were performed using an unpaired, 2-tailed Student’s t test to compare the difference between two groups, and one-way analysis of variance and two-way analysis of variance to compare differences among multiple groups, followed by Tukey’s post-hoc analysis.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank all the PLWH that participated in providing samples to this study. We would like to thank Drs. Joanne Turner and Mark A. Endsley for their careful review of this manuscript and/or guidance in the discussion, and Dr. Anna Allué-Guardia for her assistance with the HIV-1 protein database. Mass spectrometry analyses were conducted at the University of Texas Health Science Center at San Antonio (UTHSCSA) Institutional Mass Spectrometry Laboratory, with expert technical assistance of Sammy Pardo and Dana Molleur.
FUNDING
This study was supported by the Robert J. Kleberg, Jr and Helen C. Kleberg Foundation to JBT and F99 AG-079802 to AOF. AA, AOF, and JBT are part of the Interdisciplinary NextGen Tuberculosis Research Advancement Center (IN-TRAC) at Texas Biomed, which is supported by the NIAID/NIH under the award number P30 AI-168439. The UTHSCSA Institutional Mass Spectrometry Laboratory is supported in part by UTHSCSA and by the University of Texas System Proteomics Core Network for the purchase of the Orbitrap Fusion Lumos mass spectrometer. The content in this publication is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
DECLARATIONS OF COMPETING INTEREST
The authors have no competing interests to declare.
APPENDIX A. SUPPLEMENTARY DATA
Supplementary data to this article can be found online at https://doi.org/10.1016/j.mucimm.2023.12.003.
REFERENCES
- 1.Team – global HIV HaSTIP. Global health sector strategies on, respectively, HIV, viral hepatitis and sexually transmitted infections for the period. Available at: www.who.int/publications/i/item/9789240053779 (2022)[Date accessed: 29 September 2023].
- 2.Prevention CDC. (Centers for Disease Control and Prevention). Available at: www.cdc.gov/hiv/basics/statistics.html Altlanta: (2023) [Date accessed; 25 August 2023 ]. [Google Scholar]
- 3.Wing EJ The aging population with HIV infection. Trans. Am. Clin. Climatol. Assoc 128, 131–144 (2017). [Google Scholar]
- 4.US D.H.H.S. & Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in adults and adolescents living with HIV. Available at: https://aidsinfo.nih.gov/contentfiles/adultandadolescentgl.pdf (2018) [Date accessed: 23 August 2023].
- 5.Samji H. et al. Closing the gap: increases in life expectancy among treated HIVpositive individuals in the United States and Canada. PLOS ONE. 8, e81355 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prevention CPCfDCa. HIV surveillance reports. Available at: https://www.cdc.gov/hiv/library/reports/hiv-surveillance.html (2017) [Accessed: ].
- 7.Shah M. et al. Effect of the US National HIV/AIDS Strategy targets for improved HIV care engagement: a modelling study. Lancet HIV 3, e140–e146 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schouten J. et al. Cross-sectional comparison of the prevalence of age-associated comorbidities and their risk factors between HIV-infected and uninfected individuals: the AGEhIV cohort study. Clin. Infect. Dis 59,1787–1797 (2014). [DOI] [PubMed] [Google Scholar]
- 9.HIV.gov. Aging and HIV. (Centers for Disease Control and Prevention (U.S.), US)&P; Available at: https://www.hiv.gov/hiv-basics/living-well-with-hiv/taking-care-of-yourself/aging-with-hiv Washington DC: (2019) [Accessed: ]. [Google Scholar]
- 10.Crothers K. et al. HIV infection and risk for incident pulmonary diseases in the combination antiretroviral therapy era. Am. J. Respir. Crit. Care Med 183, 388–395 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crothers K. et al. HIV-associated lung infections and complications in the era of combination antiretroviral therapy. Proc. Am. Thorac. Soc 8, 275–281 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mildvan D. et al. Opportunistic infections and immune deficiency in homosexual men. Ann. Intern. Med 96, 700–704 (1982). [DOI] [PubMed] [Google Scholar]
- 13.Edelman AS & Zolla-Pazner S AIDS: a syndrome of immune dysregulation, dysfunction, and deficiency. FASEB J. 3, 22–30 (1989). [DOI] [PubMed] [Google Scholar]
- 14.Jambo KC et al. Small alveolar macrophages are infected preferentially by HIV and exhibit impaired phagocytic function. Mucosal Immunol. 7, 1116–1126 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grossman Z, Meier-Schellersheim M, Paul WE & Picker LJ Pathogenesis of HIV infection: what the virus spares is as important as what it destroys. Nat. Med 12, 289–295 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Derdeyn CA & Silvestri G Viral and host factors in the pathogenesis of HIV infection. Curr. Opin. Immunol 17, 366–373 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Deeks SG HIV infection, inflammation, immunosenescence, and aging. Annu. Rev. Med 62, 141–155 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deeks SG & Phillips AN HIV infection, antiretroviral treatment, ageing, and non-AIDS related morbidity. BMJ. 338:a3172. [DOI] [PubMed] [Google Scholar]
- 19.Cribbs S Pulmonary complications of HIV. Feldman CPER JA, editor. (European Respiratory Society, Lausanne, Switzerland: ), (2014). [Google Scholar]
- 20.Moliva JI et al. The lung mucosa environment in the elderly increases host susceptibility to Mycobacterium tuberculosis infection. J. Infect. Dis 220, 514–523 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moliva JI et al. Molecular composition of the alveolar lining fluid in the aging lung. Age (Dordr) 36, 9633 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schlesinger LS Mycobacterium tuberculosis and the complement system. Trends Microbiol. 6, 47–49 (1998). discussion 49. [DOI] [PubMed] [Google Scholar]
- 23.Ferguson JS, Voelker DR, McCormack FX & Schlesinger LS Surfactant protein D binds to Mycobacterium tuberculosis bacili and lipoarrabinomannan via carbohydrate-lectin interactions resulting in reduced phagocytosis of the bacteria by macrophages. J. Immunol 163, 312–321 (1999). [PubMed] [Google Scholar]
- 24.Ferguson JS et al. Surfactant protein D increases fusion of Mycobacterium tuberculosis-containing phagosomes with lysosomes in human macrophages. Infect. Immun 74, 7005–7009 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferguson JS, Voelker DR, Ufnar JA, Dawson AJ & Schlesinger LS Surfactant protein D inhibition of human macrophage uptake of Mycobacterium tuberculosis is indepenndent of bacterial agglutination. J. Immunol 168, 1309–1314 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Arcos J. et al. Mycobacterium tuberculosis Cell Wall released Fragments by the Action of the Human Lung Mucosa modulate Macrophages to Control Infection in an IL-10-Dependent Manner. Mucosal Immunol. 10, 1248–1258 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scordo JM et al. Mycobacterium tuberculosis cell Wall fragments released upon bacterial contact with the human lung mucosa alter the neutrophil response to infection. Front. Immunol 8, 307 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hill PJ et al. Modifications of Pseudomonas aeruginosa cell envelope in the cystic fibrosis airway alters interactions with immune cells. Sci. Rep 7, 4761 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arcos J. et al. Lung mucosa lining fluid modifies Mycobacterium tuberculosis to reprogram human neutrophil killing mechanisms. J. Infect. Dis 212, 948–958 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scordo JM et al. The human lung mucosa drives differential Mycobacterium tuberculosis infection outcome in the alveolar epithelium. Mucosal Immunol. 12, 795–804 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arcos J. et al. Human lung hydrolases delineate Mycobacterium tuberculosis-macrophage interactions and the capacity to control infection. J. Immunol 187, 372–381 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moliva JI et al. Exposure to human alveolar lining fluid enhances Mycobacterium bovis BCG vaccine efficacy against Mycobacterium tuberculosis infection in a CD8(+) T-cell-dependent manner. Mucosal Immunol. 11, 968–978 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garcia-Vilanova A. et al. The aging human lung mucosa: A proteomics study. J. Gerontol. S. A 2022, glac091 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cai Z & Yan LJ Protein oxidative modifications: beneficial roles in disease and health. J. Biochem. Pharmacol. Res 1, 15–26 (2013). [PMC free article] [PubMed] [Google Scholar]
- 35.Schroeder HWD & Greenspan N Chapter 4. Immunoglubulins: structure and function. In Fundamental immunology (Paul WE, ed 125–151 (Lippincott WIlliams & Wilkins, Philadelphia, PA, 2008). [Google Scholar]
- 36.Sasindran SJ & Torrelles JB Mycobacterium tuberculosis infection and inflammation: what is beneficial for the host and for the bacterium? Front. Microbiol 2, 2 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Torrelles JB, Azad AK, Henning LN, Carlson TK & Schlesinger LS Role of C-type lectins in mycobacterial infections. Curr. Drug Targets 9, 102–112 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Carlson TK et al. Pulmonary innnate immunity: soluble and cellular host defenses of the lung. In Regulation of innate immune function (Marsh C, Tridandapani S & Piper M, eds) 165–211 (Transworld Research Network, Kerala, 2010). [Google Scholar]
- 39.Salunke SB et al. Design and synthesis of novel anti-tuberculosis agents from the celecoxib pharmacophore. Bioorg. Med. Chem 23, 1935–1943 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Sukumar N, Tan S, Aldridge BB & Russell DG Exploitation of Mycobacterium tuberculosis reporter strains to probe the impact of vaccination at sites of infection. PLOS Pathog. 10, e1004394 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clemens DL, Lee BY & Horwitz MA Purification, characterization, and genetic analysis of Mycobacterium tuberculosis urease, a potentially critical determinant of host-pathogen interaction. J. Bacteriol 177, 5644–5652 (1995). 10.1128/jb.177.19.5644-5652.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cribbs SK, Crothers K & Morris A Pathogenesis of HIV-related lung disease: immunity, infection, and inflammation. Physiol. Rev 100, 603–632 (2020). 10.1152/physrev.00039.2018. [DOI] [PubMed] [Google Scholar]
- 43.Benito N, Moreno A, Miro JM & Torres A Pulmonary infections in HIV-infected patients: an update in the 21st century. Eur. Respir. J 39, 730–745 (2012). 10.1183/09031936.00200210. [DOI] [PubMed] [Google Scholar]
- 44.Feikin DR, Feldman C, Schuchat A & Janoff EN Global strategies to prevent bacterial pneumonia in adults with HIV disease. Lancet Infect. Dis 4, 445–455 (2004). 10.1016/S1473-3099(04)01060-6. [DOI] [PubMed] [Google Scholar]
- 45.Kanmogne GD, Primeaux C & Grammas P Induction of apoptosis and endothelin-1 secretion in primary human lung endothelial cells by HIV-1 gp120 proteins. Biochem. Biophys. Res. Commun 333, 1107–1115 (2005). 10.1016/j.bbrc.2005.05.198. [DOI] [PubMed] [Google Scholar]
- 46.Barlo NP et al. Surfactant protein-D predicts survival in patients with idiopathic pulmonary fibrosis. Sarcoidosis Vasc. Diffuse Lung Dis 26, 155–161 (2009). [PubMed] [Google Scholar]
- 47.Gaiha GD et al. Surfactant protein A binds to HIV and inhibits direct infection of CD4+ cells, but enhances dendritic cell-mediated viral transfer. J. Immunol 181, 601–609 (2008). 10.4049/jimmunol.181.1.601. [DOI] [PubMed] [Google Scholar]
- 48.Madsen J. et al. Surfactant protein D modulates HIV infection of both T-cells and dendritic cells. PLOS ONE 8, e59047 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beharka AA et al. Pulmonary surfactant protein A activates a phosphatidylinositol 3-kinase/calcium signal transduction pathway in human macrophages: participation in the up-regulation of mannose receptor activity. J. Immunol 175, 2227–2236 (2005). 10.4049/jimmunol.175.4.2227. [DOI] [PubMed] [Google Scholar]
- 50.Patel NR et al. HIV impairs TNF-alpha mediated macrophage apoptotic response to Mycobacterium tuberculosis. J. Immunol 179, 6973–6980 (2007). 10.4049/jimmunol.179.10.6973. [DOI] [PubMed] [Google Scholar]
- 51.Tachado SD, Zhang J, Zhu J, Patel N & Koziel H HIV impairs TNF-alpha release in response to toll-like receptor 4 stimulation in human macrophages in vitro. Am. J. Respir. Cell Mol. Biol 33, 610–621 (2005). 10.1165/rcmb.2004-0341OC. [DOI] [PubMed] [Google Scholar]
- 52.Agostini C, Trentin L, Zambello R & Semenzato G HIV-1 and the lung. Infectivity, pathogenic mechanisms, and cellular immune responses taking place in the lower respiratory tract. Am. Rev. Respir. Dis 147, 1038–1049 (1993). 10.1164/ajrccm/147.4.1038. [DOI] [PubMed] [Google Scholar]
- 53.Agostini C. et al. Alveolar macrophages in HIV-1 infection express accessory molecules, activation markers, and release increased biological response modifiers. Chest 103(Suppl.), 108S–S111 (1993). 10.1378/chest.103.2_supplement.108s. [DOI] [PubMed] [Google Scholar]
- 54.Cribbs SK, Lennox J, Caliendo AM, Brown LA & Guidot DM Healthy HIV-1-infected individuals on highly active antiretroviral therapy harbor HIV-1 in their alveolar macrophages. AIDS Res. Hum. Retrovir 31, 64–70 (2015). 10.1089/AID.2014.0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lipman MC, Johnson MA & Poulter LW Functionally relevant changes occur in HIV-infected individuals’ alveolar macrophages prior to the onset of respiratory disease. AIDS 11, 765–772 (1997). 10.1097/00002030-199706000-00010. [DOI] [PubMed] [Google Scholar]
- 56.Musher DM et al. The effect of HIV infection on phagocytosis and killing of Staphylococcus aureus by human pulmonary alveolar macrophages. Am. J. Med. Sci 299, 158–163 (1990). 10.1097/00000441-199003000-00003. [DOI] [PubMed] [Google Scholar]
- 57.Bandres JC, Trial J, Musher DM & Rossen RD Increased phagocytosis and generation of reactive oxygen products by neutrophils and monocytes of men with stage 1 human immunodeficiency virus infection. J. Infect. Dis 168, 75–83 (1993). 10.1093/infdis/168.1.75. [DOI] [PubMed] [Google Scholar]
- 58.Yu Q, Yu R & Qin X The good and evil of complement activation in HIV-1 infection. Cell. Mol. Immunol 7, 334–340 (2010). 10.1038/cmi.2010.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lu J & Kishore U C1 complex: an adaptable proteolytic module for complement and non-complement functions. Front. Immunol 8, 592 (2017). 10.3389/fimmu.2017.00592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Welsh KJ, Lewis CT, Boyd S, Braun MC & Actor JK Complement factor C7 contributes to lung immunopathology caused by Mycobacterium tuberculosis. Clin. Dev. Immunol 2012:. 10.1155/2012/429675 429675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jagannath C. et al. Hypersusceptibility of A/J mice to tuberculosis is in part due to a deficiency of the fifth complement component (C5). Scand. J. Immunol 52, 369–379 (2000). 10.1046/j.1365-3083.2000.00770.x. [DOI] [PubMed] [Google Scholar]
- 62.Actor JK et al. A role for complement C5 in organism containment and granulomatous response during murine tuberculosis. Scand. J. Immunol 53, 464–474 (2001). 10.1046/j.1365-3083.2001.00902.x. [DOI] [PubMed] [Google Scholar]
- 63.De La Fuente J, Gortázar C & Juste R Complement component 3: a new paradigm in tuberculosis vaccine. Expert Rev. Vaccines 15, 275–277 (2016). 10.1586/14760584.2016.1125294. [DOI] [PubMed] [Google Scholar]
- 64.Eisen S, Dzwonek A & Klein NJ Mannose-binding lectin in HIV infection. Future Virol. 3, 225–233 (2008). 10.2217/17460794.3.3.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bonar A, Chmiela M, Rudnicka W & Rózalska B Mannose-binding lectin enhances the attachment and phagocytosis of mycobacteria in vitro. Arch. Immunol. Ther. Exp. (Warsz) 53, 437–441 (2005). [PubMed] [Google Scholar]
- 66.Eisen DP Mannose-binding lectin deficiency and respiratory tract infection. J. Innate Immun 2, 114–122 (2010). 10.1159/000228159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kaetzel CS The polymeric immunoglobulin receptor: bridging innate and adaptive immune responses at mucosal surfaces. Immunol. Rev 206, 83–99 (2005). [DOI] [PubMed] [Google Scholar]
- 68.Johansen FE, Braathen R & Brandtzaeg P Role of J chain in secretory immunoglobulin formation. Scand. J. Immunol. 52, 240–248 (2000). [DOI] [PubMed] [Google Scholar]
- 69.Bjerk SM et al. Biomarkers and bacterial pneumonia risk in patients with treated HIV infection: a case-control study. PLOS ONE 8, e56249 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Saison J. et al. Association between discordant immunological response to highly active anti-retroviral therapy, regulatory T cell percentage, immune cell activation and very low-level viraemia in HIV-infected patients. Clin. Exp. Immunol 176, 401–409 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang X. et al. Incomplete immune reconstitution in HIV/AIDS patients on antiretroviral therapy: challenges of immunological non-responders. J. Leukoc. Biol 107, 597–612 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Headley CA et al. Extracellular delivery of functional mitochondria rescues the dysfunction of CD4(+) T cells in aging. Adv. Sci. (Weinh) 2023, e2303664 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Moré JM et al. Smoking reduces surfactant protein D and phospholipids in patients with and without chronic obstructive pulmonary disease. BMC Pulm. Med 10, 53 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jambo KC, French N, Zylstra E & Gordon SB AIDS patients have increased surfactant protein D but normal mannose binding lectin levels in lung fluid. Respir. Res 8, 42 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Saifuddin M, Hart ML, Gewurz H, Zhang Y & Spear GT Interaction of mannose-binding lectin with primary isolates of human immunodeficiency virus type 1. J. Gen. Virol 81, 949–955 (2000). [DOI] [PubMed] [Google Scholar]
- 76.Alexandrova Y, Costiniuk CT & Jenabian MA Pulmonary immune dysregulation and viral persistence during HIV infection. Front. Immunol 12:808722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lafuse WPR et al. Identification of an increased alveolar macrophage subpopulation in old mice that displays unique inflammatory characteristics and is permissive to Mycobacterium tuberculosis infection. J. Immunol 203, 2252–2264 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Searle BC et al. Chromatogram libraries improve peptide detection and quantification by data independent acquisition mass spectrometry. Nat. Commun 9, 5128 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rosenberger G. et al. A repository of assays to quantify 10,000 human proteins by SWATH-MS. Sci. Data 1:140031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schlesinger LS, Bellinger-Kawahara CG, Payne NR & Horwitz MA Phagocytosis of Mycobacterium tuberculosis is mediated by human monocyte complement receptors and complement component C3. J. Immunol 144, 2771–2780 (1990). [PubMed] [Google Scholar]
- 81.Schlesinger LS & Horwitz MA, Phagocytosis of Mycobacterium leprae by human monocyte-derived macrophages is mediated by complement receptors CR1(CD35), CR3(CD11b/CD18), and CR4(CD11c/CD18) and interferon gamma activation inhibits complement receptor function and phagocytosis of this bacterium. J. Immunol 147, 1983–1994 (1991). [PubMed] [Google Scholar]
- 82.Olakanmi O, Britigan BE & Schlesinger LS Gallium disrupts iron metabolism of mycobacteria residing within human macrophages. Infect. Immun 68, 5619–5627 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.