

Abstract
In the realm of high-throughput screening (HTS), macrocyclic peptide libraries traditionally necessitate decoding tags, essential for both library synthesis and identifying hit peptide sequences post-screening. Our innovation introduces a tag-free technology platform for synthesizing cyclic peptide libraries in solution and facilitates screening against biological targets to identify peptide binders through unconventional intramolecular CyClick and DeClick Chemistries (CCDC) discovered within our research. This combination allows for the synthesis of diverse cyclic peptide libraries, the incorporation of various amino acids, and facile linearization and decoding of cyclic peptide binder sequences. Our sensitivity-enhancing derivatization method, utilized in tandem with nano LC-MS/MS, enables the sequencing of peptides even at exceedingly low picomolar concentrations. Employing our technology platform, we’ve successfully unearthed novel cyclic peptide binders against a monoclonal antibody and the first cyclic peptide binder of HIV CAPSID protein responsible for viral infections as validated by microscale thermalshift assays (TSA), biolayer Interferometry (BLI) and functional assays.
Keywords: Tag-Free Approach, Peptide Macrocyclization, CyClick Chemistry, Cyclic Peptide Library, Affinity Selection
Graphical Abstract
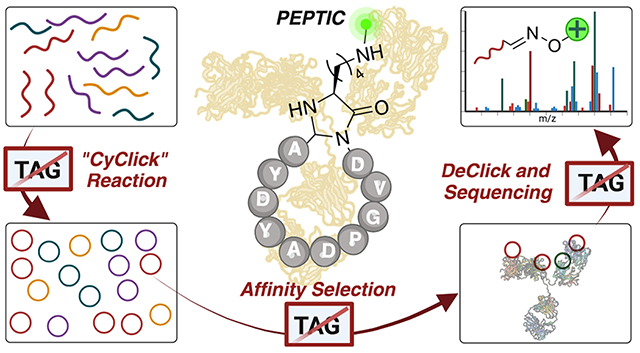
PEPTIC represents a pivotal step forward in the realm of cyclic peptide-based drug discovery, offering a tag-free, efficient, and reliable platform for the synthesis, sequencing, and identification of cyclic peptide binders. This innovative approach is expected to expedite the discovery of novel cyclic peptide binders against biological targets.
Introduction
The generation of cyclic peptide libraries and their high throughput screening against various biological targets has revolutionized the discovery of novel peptide therapeutics.1,2 These macrocycles exhibit higher stability against proteases than their linear counterparts3,4 and occupy a unique conformational space that allows for high affinity binding to the long and shallow interfaces of protein-protein interactions.5,6
Current strategies for the synthesis and screening of cyclic peptide libraries rely on the use of encoding tags like mRNA and phage which utilizes cellular machinery to synthesize peptide library members and physically link them to the RNA/phage that encode each amino acid,7,8 thus avoiding oligomerization. Post-screening PCR amplification allows for straightforward sequence deconvolution with well-established DNA sequencing technologies. Although highly powerful, encoding tags method have challenges associated with the incorporation of non-canonical amino acids9–11 and the size of the peptide relative to the “encoding tag”. This results in reduced library diversity and less control over the introduction of drug-like characteristics, such as N-methylation for cell permeability, and unnatural amino acids for proteolytic stability. Additionally, the interference of large and densely charged tags is suspected to play a role in the prevalence of false positive hit identification. Another encoding tag approach that is fully synthetic is one-bead-two-compound (OBTC). OBTC displays cyclized peptides on the outer layer of solid-phase resin with linear peptide decoding tags featured on the interior. While this method benefits from the full range of solid-phase peptide synthetic techniques, imperfect cyclization efficiency can generate libraries with inseparable mixtures of cyclized vs. linear peptides and the presence of multiple copies of library members can enable multidentate interactions in affinity selections that lead to false positives.12,13 Lastly, DNA-encoded libraries are a promising strategy that powerfully combines the advantages of fully synthetic chemistry with the ease of nucleic acid-based sequence deconvolution. While promising, this method is currently hampered by the uncertainty of cyclization yield and inability to purify the cyclic from linear products before high-throughput screening.14 Moreover, the continued need for a nucleic-acid encoding tag results in a vulnerability to pre-sequencing tag degradation as well as the potential to influence peptide binding during the selection process.
However, the synthesis and screening of library of cyclic peptides without any encoding tags are conspicuously absent, mainly because the chemistry of cyclization in solution is also chemistry of oligomerization thus generate numerous crosslinked and oligomeric byproducts when employed above pseudo-dilute concentrations.15–17 Additionally, the complex fragmentation spectra of macrocyclic peptides significantly complicate the unambiguous post-selection sequencing of hit compounds by traditional tandem mass spectrometry techniques. To address these hurdles, we pursued the exclusively intramolecular CyClick chemistry (CC)18 to generate libraries of cyclic peptides from linear peptide aldehydes in solution without the generation of crosslinked products irrespective of the concentrations.
This technological advancement directly introduces reactive 4-imidazolidinone ring onto cyclic peptides, enabling ring opening via De-Click Chemistry (DC) to regenerate linear peptide aldehydes providing the bioorthogonal handle for tagging with mass enhancers thus enable efficient peptide deconvolution post screening. The integration of these methodologies, known as the “PEPTIC” (Peptide Exploration Platform with Tag-Free Intramolecular Chemistry), facilitates swift synthesis of library of diverse cyclic peptides with a wide breadth of non-canonical residues and backbone modifications to introduce drug-like properties, marking a substantial leap forward in discovery of cyclic peptide therapeutics. The entire workflow for PEPTIC is outlined in Figure 1.
Figure 1.
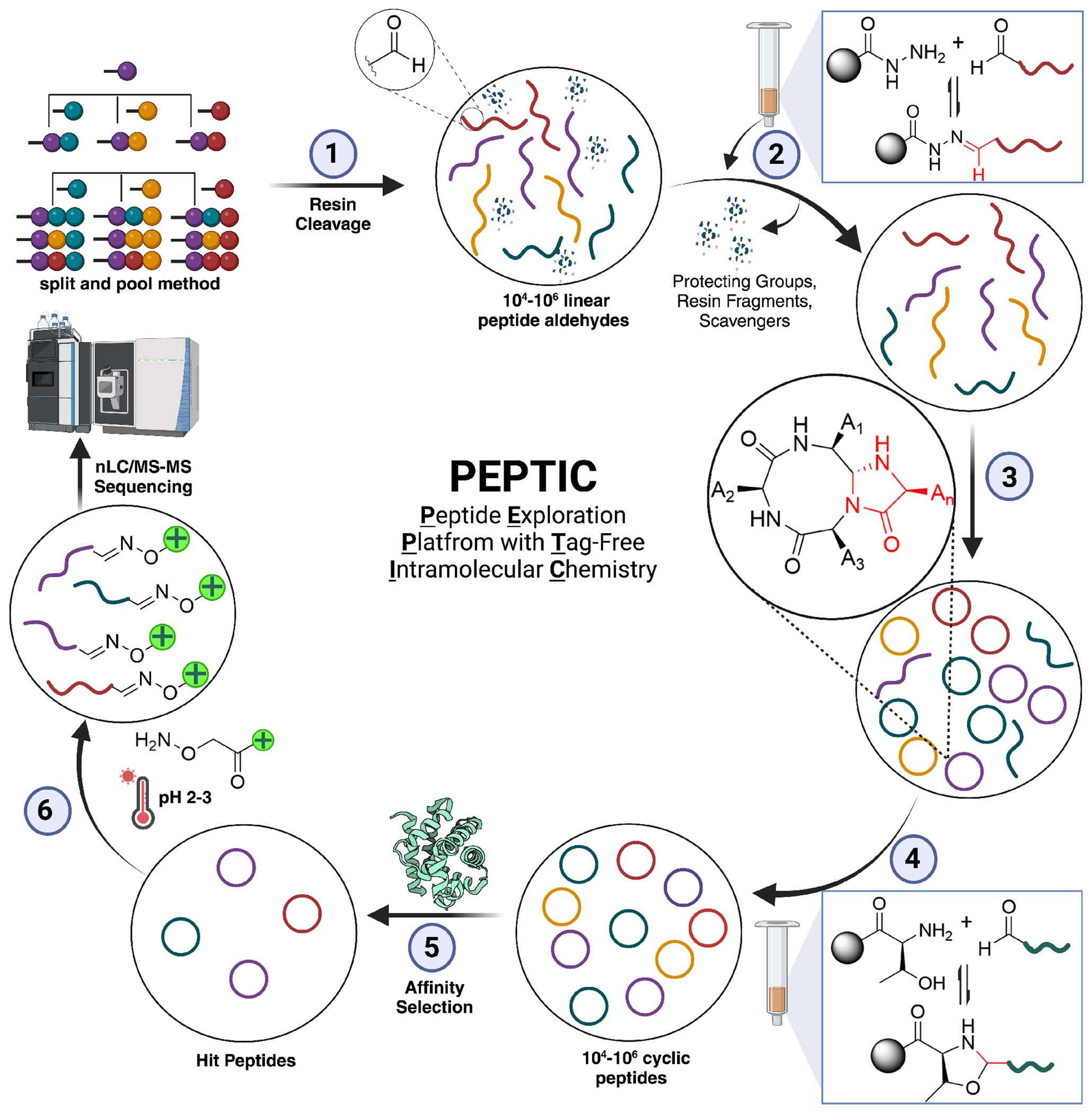
PEPTIC work flow for the synthesis, screening and sequencing of tag-free cyclic peptide libraries. (1) Synthesis of library of linear peptide aldehydes by split and pool method, (2) purification of library of linear peptide aldehydes in one step using reversible capture-release strategy, (3) synthesis of library of tag-free cyclic peptide library in one pot using our exclusively intramolecular CyClick chemistry in solution in one pot, (4) purification of uncyclized linear peptide aldehydes from mixture of cyclic peptide library by capturing strategy, (5) Screening of library of cyclic peptide library with a target protein, (6) Opening of the cyclic peptide binders to linear peptide aldehyde by our De-Click chemistry and the functionalization with mass tag followed by the analysis of sequence of cyclic peptide binders by nano LC-MS/MS.
Results and Discussion
Synthesis and purification of linear peptide aldehydes
To synthesize a diverse library of cyclic peptides in solution, we initiated our investigation by accessing a library of C-terminal linear peptide aldehydes for use in CyClick chemistry (CC).18 Our approach involved a strategic process: first, loading resin with threonine followed by coupling with C-terminal aldehyde amino acid to generate an oxazolidine handle. This handle facilitated the incorporation of diverse amino acids on solid support through a split and pool method, resulting in the creation of a library of linear peptides (Supplementary Information). Subsequent detachment from the solid support under acidic conditions yielded a library of C-terminal linear peptide aldehydes. To purify the library of linear peptide aldehydes in solution, we leveraged the orthogonal aldehyde handle. This involved incubating the peptide libraries with scavenger resins capable of reversibly binding to the aldehyde. Post-incubation, resin washing was employed to eliminate impurities generated during peptide synthesis and cleavage, subsequently releasing pure linear peptide aldehydes back into the solution.
Several aldehyde-scavenger resins were screened, including hydrazine-,19,20 sulfonyl hydrazide-,21 and threonyl- (TG)/cysteinyl- (CG)22,23 functionalized resins on a model peptide aldehdye. These resins formed stable hydrazone, sulfonyl hydrazone, and oxazolidine/thiazolidine structures, respectively, upon condensation with an aldehyde of a model peptide, H2N-AVGPFEYA-CHO (Supplementary Table 1). Among the screened scavengers, the hydrazide resin yielded the most promising outcomes. Optimized conditions for scavenging peptide aldehyde from a crude mixture included 2% acetic acid in acetonitrile at 40-60°C. Consequently, the pure peptide aldehyde was released back into solution via hydrolysis of the hydrazone to reform the aldehyde, accomplished by altering the reaction solvent to 50:50 ACN:H2O with 2% acetic acid (Supplementary Table 1). With these established conditions, we successfully demonstrated the efficient scavenging, washing, and hydrolysis of the individual model peptide aldehyde 1a, H2N-AVGPFEYA-CHO directly from a crude mixture, resulting in a solution of pure linear peptide aldehyde 1a (Figure 2A and 2B, Supplementary Table 1). Consistent recoveries between 75-90% were achieved with known quantities of pure starting peptide. Moreover, a “mini” split-and-pool library comprising 16 peptide aldehydes underwent the purification protocol, showcasing efficient recovery and enrichment of all 16 peptide aldehydes by LC-MS post-hydrazide-resin purification (Figure 2C).
Figure 2.
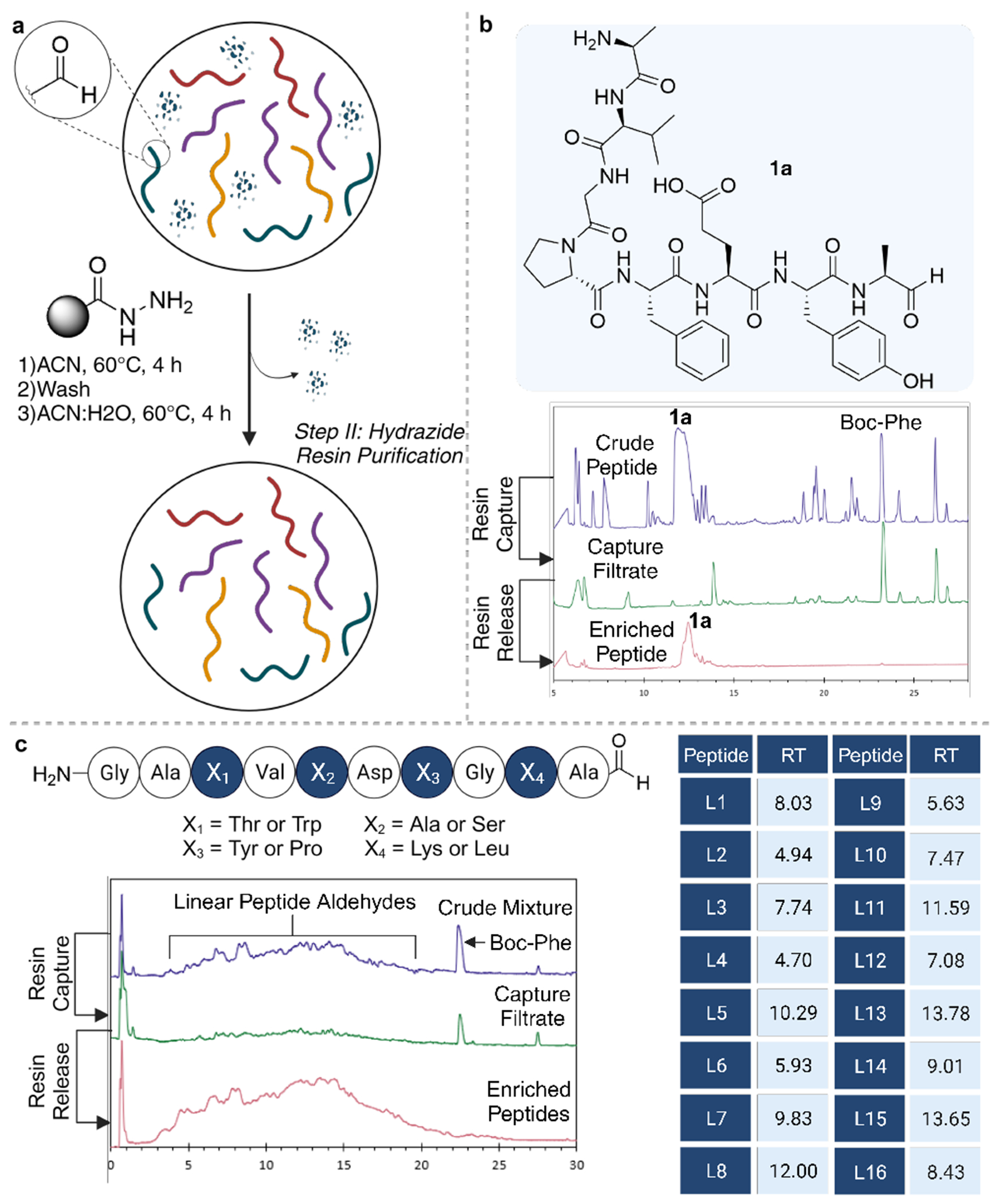
Hydrazide resin approach for purification of linear peptide aldehydes from a complex mixture. (a) General scheme for the capture of linear peptide aldehydes from a mixture using hydrazide resin and the release of pure linear peptide aldehydes in solution. (b) Structure of model peptide 1a with stacked chromatograms (DAD) of the crude model peptide 1a, the filtrate collected after hydrazide resin scavenging, and the filtrate after peptide hydrolysis from hydrazide resin to generate pure peptide aldehyde 1a. (c) Purification of peptide aldehyde “mini” library containing 16 sequences, stacked chromatograms (DAD) from hydrazide purification steps, and table displaying the retention time of each linear peptide aldehydes (L1-L16) from the purified “mini” library.
Tag-free approach for synthesis of cyclic peptides library from linear peptide aldehydes
Armed with pure linear peptide aldehydes, our approach harnesses the capabilities of CyClick chemistry (CC) to efficiently generate a library of cyclic peptides in a single pot, devoid of any crosslinked products. A distinctive aspect of our CyClick chemistry lies in its exclusive intramolecularity, allowing the formation of cyclic peptides solely between the N-terminus and the aldehyde, irrespective of concentrations, size, or sequence of amino acid residues. This distinctive feature sets our methodology apart, as other existing tag-free cyclization methods often lead to the formation of both cyclic and oligomerized products because the chemistry of cyclization is also the chemistry of oligomerization. The intrinsic intramolecularity of the CyClick reaction was further corroborated by DFT calculations, elucidating the role of transannular intramolecular hydrogen bonding in stabilizing the transition state, favoring the formation of cyclic products featuring 4-imidazolidinone moieties at the cyclization site over competing intermolecular reactions.24 In our initial investigation, we started with a mini library of 16 unprotected linear peptide aldehydes and subjected it to CyClick reaction conditions using 1% DIEA in DMF at 60 °C (Supplementary Information). The resulting reaction mixture was analyzed by LC-HRMS before and after the cyclization to investigate the formation of any crosslinked byproducts, for which we found no evidence (Figure 3A). Notably, we observed the formation of all the 16 cyclic peptides under the reaction conditions further confirming the potential of CC to generate tag-free cyclic peptide libraries in solution without crosslinked by products.
Figure 3.
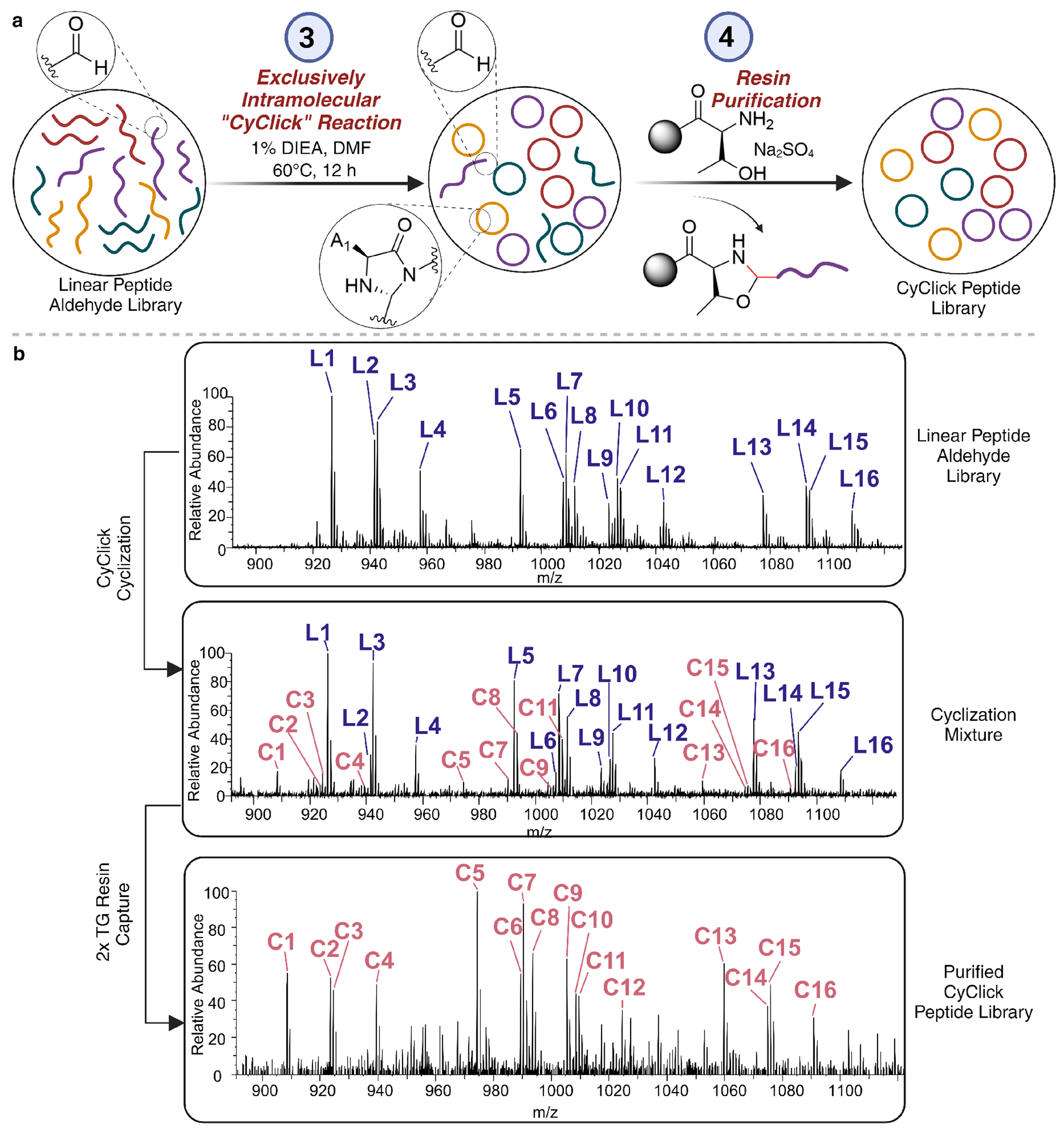
Cyclization and purification of a mixture of linear peptide aldehydes. (a) Scheme for the cyclization of mixture of linear peptide aldehydes and purification of cyclized peptides from uncyclized linear peptide aldehydes. (b) Spectra of direct infusion analysis of 16 peptide ”mini” library, including the linear mixture (top), cyclization mixture (middle) and pure cyclic peptide mixture after two successive incubations with TG functionalized scavenger resin under optimized conditions (bottom).
Subsequent endeavors focused on purifying cyclic peptides from uncyclized linear peptide aldehydes utilizing a hydrazide scavenger resin. However, while the hydrazide resin effectively reduced the absolute quantity of linear peptide aldehydes, it concurrently decreased cyclic product yield. This reduction was attributed to the acidic conditions fostering both hydrazone formation and linearization of our cyclic peptides. To circumvent the undesired linearization associated with acidic scavenging conditions, we employed TG-functionalized resin operating under basic conditions, trapping aldehydes through oxazolidine formation (Supplementary Table 2). Strikingly, conditions for capturing linear peptide aldehydes mirrored those utilized for synthesizing peptide macrocycles. By incubating the cyclization mixture from a model peptide 1a with 5 equivalents of TG functionalized resin, 5 equivalents of sodium sulfate, in 1% DIEA in DMF at 50-60 °C, we efficiently removed linear peptide aldehydes without concurrent linearization of cyclic product 2a (Supplementary Table 2, Supplementary Figure 1). Notably, sodium sulfate addition was critical for achieving oxazolidine crosslinking and complete removal of linear peptide aldehydes from the cyclization mixture.
Subsequently, we successfully applied TG-resin for purifying a mini library of 16 cyclic peptides from the one-pot cyclization mixture following two rounds of TG resin purification. (Figure 3B, Supplementary Figure 2). These data showed the ability of our approach in synthesizing cyclic peptide libraries in solution without the requirement of any encoding tag.
Optimization of macrocycle linearization and analysis
Another major role of encoding tags on cyclic peptide libraries is to simplify the sequencing of cyclic peptide binders enriched in affinity selections because sequencing of macrocyclic peptides by tandem mass spectrometry is significantly challenging due to the complexity of the resulting spectra. Methods that utilize MS3 level fragmentation and corresponding data analysis workflows for deconvolution of cyclic peptide sequences are still early in their development.25,26 To nullify the requirement of encoding tags for sequencing cyclic peptide binders, we focused on developing de-clicking chemistry (DC) as a viable approach. This method selectively opens cyclic peptides at 4-imidazolidinone moieties, converting them into linear peptides possessing bioorthogonal aldehyde handles facilitating direct sequencing of binders through established shotgun proteomics workflows, alleviating the complexity associated with sequencing macrocyclic peptides via tandem mass spectrometry (MS/MS). The strategy revolved around hydrolyzing the 4-imidazolidinone moiety under acidic conditions using high temperature, initiating protonation followed by hydrolysis to generate a linear peptide aldehyde.27–29 Initial attempts involved incubating peptide macrocycle 2a in water under acidic conditions, resulting in limited ring opening. However, elevating the temperature to 80°C prompted full conversion of peptide macrocycle 2a to linear peptide aldehyde 1a within 8 hours, as confirmed by HPLC and MS analysis (Supplementary Figure 3). Notably, no significant ring opening of cyclic peptide was observed under physiological conditions. Low pH (3.0) and high temperature is required for the opening of cyclic peptides. The reversible nature of CyClick peptides not only aids in predicting peptide fragmentation but also unveils an orthogonal aldehyde handle, facilitating ionization-boosting derivatization for efficient sequencing of peptide binders at low concentrations.30 To achieve this, we synthesized a simple arginine-containing tetrapeptide linked with N-terminal hydroxylamine, H2N-O-GGRG for the derivatization of peptide aldehyde 1a to generate oxime 3a (Supplementary Figure 4). This derivatization of peptide aldehyde 3a significantly enhanced signal intensity of both the precursor and fragment ions by an order of magnitude (Supplementary Figure 5).
The MS2 spectrum of the derivatized model peptide 3a also featured a prominent y-ion ladder from y1 to yn-2, where n is the number of residues in the peptide (Supplementary Figure 5). Despite some limitations in unambiguously sequencing the N-terminal dipeptide sequence, adjustments in library designs such as fixing the identity of the amino acid at either of the two positions or ensuring no isobaric sequences are included in the library design can address this concern. To determine the limit-of-detection for sequencing, serial dilutions of peptide 3a were performed followed by analysis using nanoLC/MS-MS. A clear y-ion ladder with signal intensity above 1x104 was achieved down to 0.169 femtomoles of peptide 3a (Supplementary Figure 6). We noted the significant overlap in the reaction conditions required for the linearization of cyclic peptides that includes low pH (3.0) and high temperature (60 °C) and derivatization and attempted to perform both steps simultaneously in one-pot on macrocyclic peptide 2a. We were delighted to observe the quantitative conversion of the macrocycle 2a to the linear oxime product 3a in the one-pot reaction with no unwanted byproducts (Figure 4A and 4B). We further tested the stability of our cyclic peptides in varying nucleophiles under physiological conditions including Lys, Cys, glutathione and hydroxylamine. No ring opening of cyclic peptide was observed even in the presence of the excess of these nucleophiles under the conditions required for affinity selection and screening and biological assays for determining the binding affinity of cyclic peptides (Supplementary Figure 7).
Figure 4.
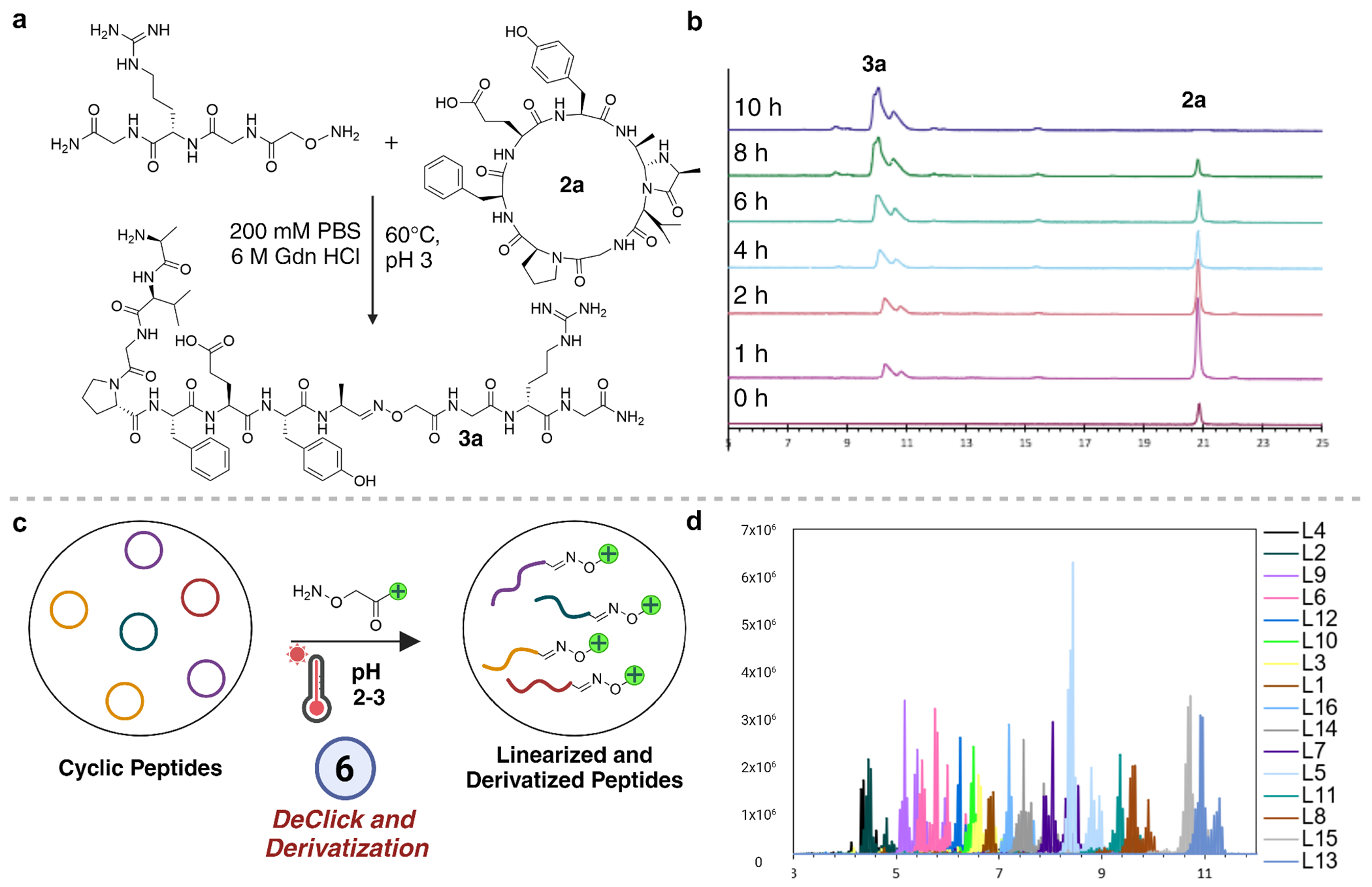
Linearization and derivatization of cyclic peptides. (a) Reaction scheme of one-pot linearization and derivatization of cyclic model peptide 2a to derivatized linear peptide 3a. (b) Stacked chromatograms displaying time-point analysis of one-pot linearization and derivatization of cyclic model peptide 2a to derivatized linear peptide 3a. (c) Scheme for the linearization of library of cyclic peptides by DeClick chemistry and derivatization of resulting linear peptide aldehyde library with mass enhancers. (d) Extracted ion chromatograms of derivatized a mini cyclic peptide library
We sought to demonstrate the strength of this one-pot linearization/derivatization by applying the optimized conditions to a mini library of 16 cyclic peptides. We observed successful derivatization of all 16 peptides in a mixture and achieved high sequencing confidence afforded by the near complete y-ion ladder for all identified peptides (Figure 4C). During LC-MS/MS analysis of the mixture, we successfully detected and sequenced peptides with overlapping retention times by enabling dynamic exclusion in our data-dependent acquisition settings (Figure 4D, Supplementary Figure 8). This is an important capability as identified hits from affinity selections are likely to have similar polarity and chemical characteristics.
PEPTIC for affinity selection and hit identification
Moving forward, we employed the optimized PEPTIC workflow for the synthesis of 1x104 cyclic peptide library incorporating DXXDY sequence and incubated a pure library of cyclic peptides in PBS at an estimated ~450 nM per member concentration with anti-HA mAb clone 12ca5 that is known to recognize the sequence DXXDY antigen, derived from influenza virus hemagglutinin (HA)31 followed by affinity selection using high pressure size exclusion chromatography (HPSEC) (Figure 5A).32,33 Due to the inherent separation capability of size exclusion, the breakthrough fraction containing protein and bound peptide was free of significant contamination. The fraction was acidified to separate peptide binders from 12ca5 and subjected to our one-pot linearization/derivatization conditions, desalted, and submitted for nLC/MS-MS analysis. We were excited to see clear MS2 spectra with the characteristic y-ion ladder associated with our derivatized peptides. From the database search we narrowed down 25 sequences for which we had high confidence in the sequencing results (Supplementary Table 3).
Figure 5.
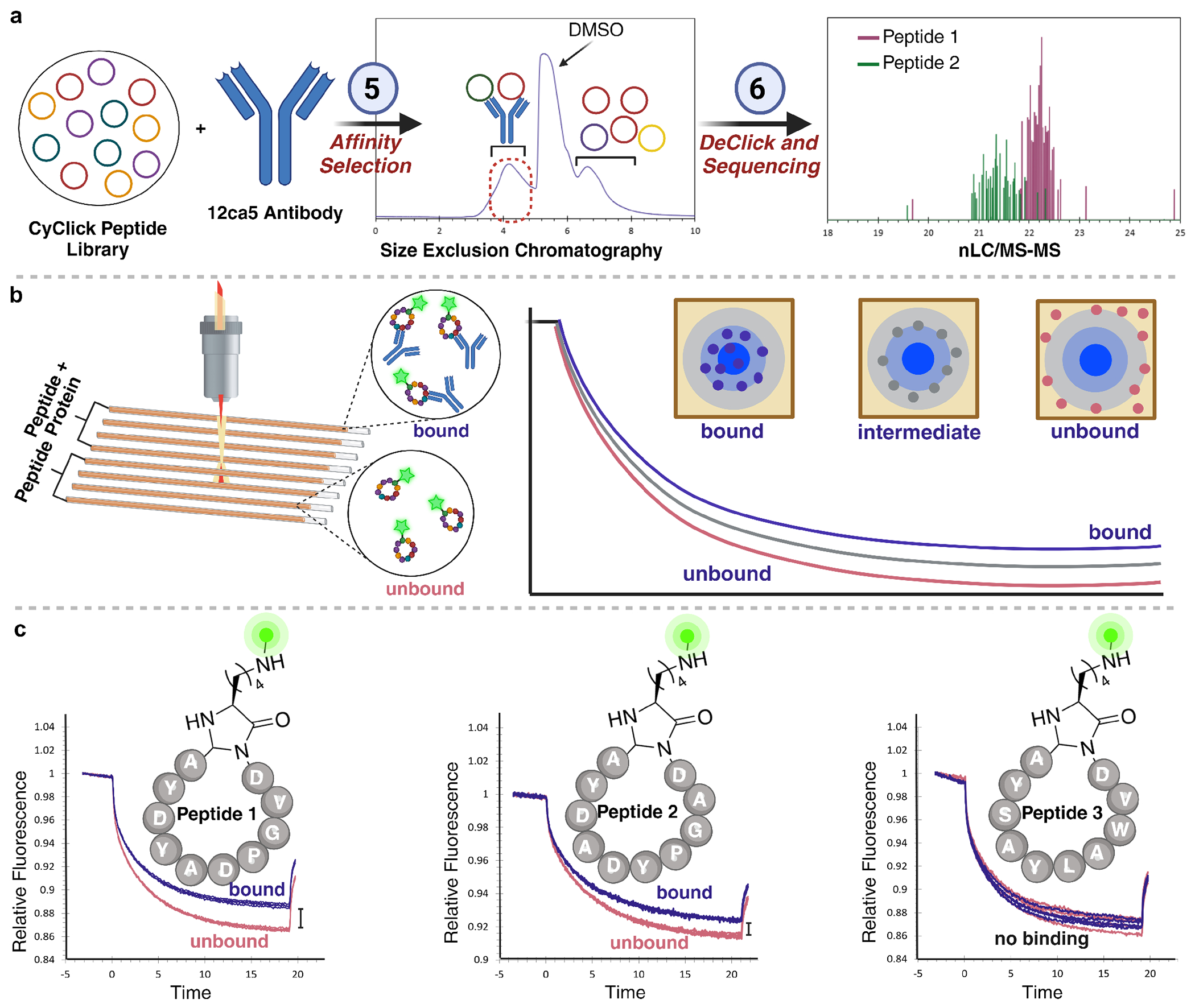
Affinity selection experiments with mixture of cyclic peptides. (a) General workflow for HPSEC affinity selection and identification of cyclic peptide binders of 12ca5 antibody from a cyclic peptide library by PEPTIC workflow using linearization of cyclic peptide binders, derivatization of linear peptide aldehydes followed by analysis using nano LC-MS/MS. (b) Representation of microscale thermophoretic assay and analysis of bound from unbound. (c) Individual cyclic peptides based on enriched peptide sequences using HPSEC affinity selection against 12ca5 antibody showed qualitative binding to 12ca5 antibody as analyzed by microscale thermophoretic assay, pink: peptide only; blue: peptide + protein. Microscale thermophoretic assay with random sequence not enriched by PEPTIC workflow did not show any binding with 12ca5 antibody
Out of 25 cyclic peptide binders, two cyclic peptide binders (GDVGPDAYDYA and GDAGPYDADYA) were chosen based on their MS2 spectra (Supplementary Figure 9) for resynthesis and binding assessment via microscale thermophoresis (MST) (Figure 5B). For each cyclic peptide binder, the N-terminal Gly of the cyclic peptide binder was replaced with Lys and the side chain of Lys was labeled with the fluorophore, fluorescein isothiocyanate (FITC) (Figure 5C, Supplementary Figure 10).
To qualitatively assess binding, 40 nM solutions of each peptide were incubated with 3 μM of 12ca5 antibody. Thermophoretic time traces were collected in quadruplicate with peptide only and peptide-antibody solutions (Figure 5C). For both enriched peptide sequences (GDVGPDAYDYA and GDAGPYDADYA) there was a significant shift in the thermophoretic time traces when incubated with the antibody. Qualitative assessment confirmed binding to 12ca5 antibody for the enriched peptide sequences, validating their preferential selection based on binding interaction. Conversely, a random sequence (GDVWALYASYA) from the library, not enriched in the selection, served as a negative control and exhibited no observed binding to the antibody.
We further applied our PEPTIC method to identify a cyclic peptide that interacts with the HIV-1 capsid protein (CA). The binding site in the HIV-1 capsid core is a frequently targeted cavity in CA hexamers and natively interacts with multiple host proteins during viral infection.34,35 The protein•protein interactions in this binding pocket are driven by a common phenylalanine-glycine (FG) dipeptide motif found in several host proteins, which is an essential factor for HIV-1 nuclear entry and viral replication. Based on the binding site of HIV-1 capsid core (PDB: 6AYA),36 we synthesized FG containing cyclic peptide library with FG-motif (“FG-SIDE”) at the C-terminus or several resides downstream (“FG-END”) in a library of >50 peptides (Figure 6A and 6B). Using a thermal shift assay (TSA), we found that the C-terminal position increased the thermal stability of disulfide-stabilized CA hexamers (CAHEX)37 in vitro, and thus was the best to work from this scaffold (Figure 6D). Notably, similar results were observed with “FG-SIDE” cyclic peptide library for point mutated HIV capsid protein (CAN74D and CAM66I) (Supplemental Figure S11), The observed change in melting temperatures is relatively small for FG-SIDE, but is statistically significant (p = 0.038). We then selected a representative peptide from the library, “cyFG-3”, to investigate binding affinity using TSA and observed significant high binding as compared to the library of cyclic peptides (Figure 6C and 6D). Biolayer Interferometry (BLI) was used to determine the binding constants of cyFG-3 to a modified CAHEX construct that includes a C-terminal 6xHis Tag (CAHEX:6HIS). After immobilizing CA121HEX:6HIS to the HIS-affinity probes, we report the biochemical constants for cyFG-3 are KD = 0.963 ± 0.06 μM, kon = 8.90 ± 0.53 x103 M−1s−1, koff = 8.58 ± 0.136x10−3 s−1 (Figure 6E). We confirmed our result with a cyclic peptide by carrying out a BLI assay with a positive control, a small molecule antiviral Lenacapvir (LEN) that was recently approved for use in highly treatment-experienced individuals who are HIV-1–positive and on a failing antiretroviral therapy (ART) regimen.34,38–42 The biochemical constants for LEN to CAHEX:6HIS are KD = 28.6 ± 0.2 nM, kon = 1.040 ± 0.007 x104 M−1s−1, koff = 2.978 ± 0.015 x10−4 s−1 (Supplemental Figure S12). It is reported that LEN increases the assembly rate of CA in vitro, as determined using a plate-based assay measuring absorbance at 350 nm (A350) over time as a proxy of CA protein association.34,43–44 Using this functional A350 assay, we found that cyFG-3 also binds to the same site as the cyclic peptide and increases the rate of CA assembly, similar to but less drastic than LEN (Figure 6F and 6G, Supplemental Figure S12). This is important to note, as compounds and mutations that increase or decrease the stability of CA often impair viral fitness.36,39,45 Future investigation is needed into the specific mechanisms of CA•cyFG-3 interactions and for increasing the binding affinity by iteration of the lead cyFG-3 cyclic peptide, but this data shows the utility of our platform to identify cyclic peptides binders as lead compounds for a clinically relevant site.
Figure 6.
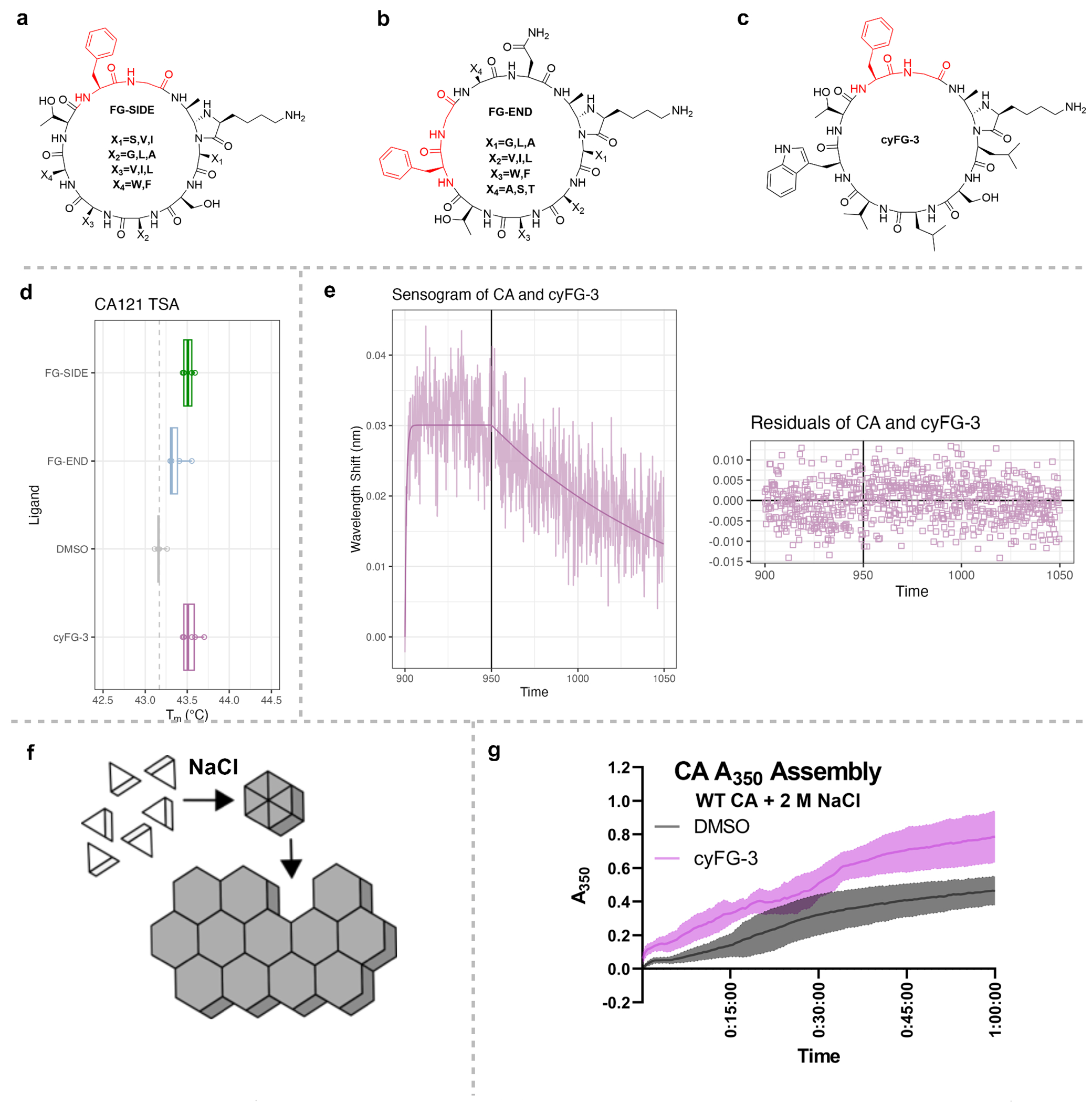
Example of library screening for cyclic peptides with phenylalanine-glycine (FG)-motifs that bind the HIV-1 Capsid protein (CA). Structures of (a) FG-SIDE (b) FG-END, (c) cyFG-3. (d) Average melting temperature (Tm) values as determined by Thermal Shift Assay (TSA) of CA hexamers incubated with the libraries of peptides containing the FG motif on the side (FG-SIDE) or end (FG-END), and the purified cyFG-3 from the FG-SIDE library. (e) Representative Biolayer Interferometry (BLI) sensograms of double background subtracted data and modeled fit for CA Hexamers with cyFG-3 at 75 μM (blue). Global KD = 0.963 ± 0.06 μM, kon = 8.90 ± 0.53 x103 M−1s−1, koff = 8.58 ± 0.136x10−3 s−1. Residuals of the model are shown below the plot as a difference between the modeled line and observed data. (f) Cartoon schematic of in vitro CA assembly. Purified CA monomers are clear in solution, but when a high concentration of NaCl increases the ionic strength of the solution, CA multimerizes into hexamers and large assemblies that turn the solution cloudy, which can be tracked with A350nm as a proxy of assembly. (g) Kinetics of in vitro CA assembly increase with pre-treatment with cyFG-3 (pink), as CA assembles faster than the control DMSO (grey). Average of 6 technical replicates (N = 3) with standard deviation shown.
Conclusion
In summary, this study introduces a pioneering approach, termed PEPTIC (Peptide Exploration Platform with Tag-Free Intramolecular Chemistry), for the synthesis, sequencing, and identification of cyclic peptide binders without the reliance on encoding tags. The innovation of employing CyClick chemistry (CC) for the synthesis of diverse cyclic peptide libraries from linear peptide aldehydes without the formation of crosslinked byproducts is a pivotal stride forward. This tag-free approach, paired with the DeClick Chemistry (DC) methodology, enables the selective linearization of cyclic peptides at 4- imidazolidinone moieties, transforming them into linear peptides with bioorthogonal aldehyde handles. This innovation facilitated the efficient sequencing of cyclic peptide binders even at low concentrations through mass-enhanced derivatization, overcoming the limitations posed by complexities encountered in tandem mass spectrometry-based sequencing of tag-free cyclic peptides, a longstanding challenge in the field of peptide-based drug discovery.
Our findings highlight the successful generation of diverse cyclic peptide libraries, showcasing the adaptability to incorporate various non-canonical amino acids and backbone modifications to introduce drug-like properties. The seamless transition from library synthesis to screening against biological targets demonstrates the potential of PEPTIC in identifying novel cyclic peptide binders. The affinity selection experiments, validated by microscale thermophoresis and BLI assays, reaffirm the efficacy of this platform in identifying cyclic peptides that specifically bind to target proteins. By using this platform we have discovered the cyclic peptide binder of the antibody and the first cyclic peptide binder of the therapeutically relevant HIV Capsid protein. Further the functional assay with the cyclic peptide showed that it helps in the association of the HIV Capsid protein, thus impair viral fitness. Additionally, the expansion of this technology’s application to diverse biological targets and disease models are pivotal for harnessing the full potential of this innovative technology.
Supplementary Material
Acknowledgements
This research was supported by NIH (No. 1R35GM133719-01) and NSF (Grant No. CHE-2108774) to M.R. Monika Raj, Ph.D. was supported by a Research Scholar Grant, RSG-22-025-01-CDP, from the American Cancer Society. A.B. graduate studies is supported by NSF-GRFP. This research was supported in part by the National Institutes of Health (R01 AI120860, U54 AI170855, P30 AI050409) to SGS; WMM was supported in part by T32 GM135060 and F31 AI174951). SGS acknowledges funding from the Nahmias-Schinazi Distinguished Chair in Research. All the images are created with biorender.com.
Footnotes
Supporting Information
All data supporting the findings of this study are available within the Supplementary Information. These include reaction procedures for the synthesis of library of peptide aldehydes, procedure for purification of library of linear peptide aldehydes and cyclic peptides using varying capturing reagents, the procedure of optimized reaction for opening cyclic peptides to linear peptide aldehydes, the procedure for tagging linear peptide aldehydes with mass tag and the procedure for screening to library of cyclic peptides by High pressure size exclusion chromatography followed by the analysis of the sequence of the cyclic peptide binders by LC-MS/MS and the procedure for carrying out affinity assay with pure cyclic peptide binders using microscale thermophoresis. The Supporting Information is available free of charge on the website.
References
- [1].Vinogradov AA, Yin Y, Suga H, J. Am. Chem. Soc 2019, 141, 4167–4181. [DOI] [PubMed] [Google Scholar]
- [2].Zhang H, Chen S, RSC Chem. Biol 2022, 3, 18–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bhat A, Roberts LR, Dwyer JJ, Eur. J. Med. Chem 2015, 94, 471–479. [DOI] [PubMed] [Google Scholar]
- [4].Gentilucci L, De Marco R, Cerisoli L, CPD 2010, 16, 3185–3203. [DOI] [PubMed] [Google Scholar]
- [5].Bruzzoni-Giovanelli H, Alezra V, Wolff N, Dong C-Z, Tuffery P, Rebollo A, Drug Discov. Today 2018, 23, 272–285. [DOI] [PubMed] [Google Scholar]
- [6].Zorzi A; Deyle K; Heinis C Cyclic peptide therapeutics: past, present and future. Curr. Opin. Chem. Bio 2017, 38, 24–29. [DOI] [PubMed] [Google Scholar]
- [7].Josephson K, Ricardo A, Szostak JW, Drug Discov. Today, 2014, 19, 388–399. [DOI] [PubMed] [Google Scholar]
- [8].Owens AE, Iannuzzelli JA, Gu Y, Fasan R, ACS Cent. Sci 2020, 6, 368–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tian F, Tsao M-L, Schultz PG, J. Am. Chem. Soc 2004, 126, 15962–15963. [DOI] [PubMed] [Google Scholar]
- [10].Katoh T; Tajima K; Suga H Consecutive elongation of D-amino acids in translation. Cell Chem.l Bio 2017, 24, 46–54. [DOI] [PubMed] [Google Scholar]
- [11].Iskandar SE, Haberman VA, Bowers AA, ACS Comb. Sci 2020, 22, 712–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Thakkar A, Trinh TB, Pei D, ACS Comb. Sci 2013, 15, 120–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lian W, Upadhyaya P, Rhodes CA, Liu Y, Pei D, J. Am. Chem. Soc 2013, 135, 11990–11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Plais L, Scheuermann J, RSC Chem. Biol 2022, 3, 7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ehrlich A, Heyne H-U, Winter R, Beyermann M, Haber H, Carpino LA, Bienert M, J. Org. Chem 1996, 61, 8831–8838. [DOI] [PubMed] [Google Scholar]
- [16].Skropeta D, Jolliffe KA, Turner P, J. Org. Chem 2004, 69, 8804–8809. [DOI] [PubMed] [Google Scholar]
- [17].Marsault E, Peterson ML, J. Med. Chem 2011, 54, 1961–2004. [DOI] [PubMed] [Google Scholar]
- [18].Adebomi V, Cohen RD, Wills R, Chavers HAH, Martin GE, Raj M, Angew. Chem., Int. Ed 2019, 58, 19073–19080. [DOI] [PubMed] [Google Scholar]
- [19].Furukawa J, Shinohara Y, Kuramoto H, Miura Y, Shimaoka H, Kurogochi M, Nakano M, Nishimura S-I, Anal. Chem 2008, 80, 1094–1101. [DOI] [PubMed] [Google Scholar]
- [20].Abe M, Shimaoka H, Fukushima M, Nishimura S-I, Polym. J 2012, 44, 269–277. [Google Scholar]
- [21].Wang C, Yuan J, Li X, Wang Z, Huang L, Analyst 2013, 138, 5344–5356. [DOI] [PubMed] [Google Scholar]
- [22].Ede NJ, Bray AM, Tetrahedron Lett. 1997, 38, 7119–7122. [Google Scholar]
- [23].Malins LR, deGruyter JN, Robbins KJ, Scola PM, Eastgate MD, Ghadiri MR, Baran PS, J. Am. Chem. Soc 2017, 139, 5233–5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Shao H, Adebomi V, Bruce A, Raj M, Houk KN, Angew Chem Int Ed. 2023, 62, e202307210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Townsend C, Furukawa A, Schwochert J, Pye CR, Edmondson Q, Lokey RS, Bioorg. Med. Chem 2018, 26, 1232–1238. [DOI] [PubMed] [Google Scholar]
- [26].Behsaz B, Mohimani H, Gurevich A, Prjibelski A, Fisher M, Vargas F, Smarr L, Dorrestein PC, Mylne JS, Pevzner PA, Cell Syst. 2020, 10, 99–108. [DOI] [PubMed] [Google Scholar]
- [27].Zehavi U, Ben-Ishai D, J. Org. Chem 1961, 26, 1097–1101. [Google Scholar]
- [28].Larsen SW, Sidenius M, Ankersen M, Larsen C, Eur. J. Pharm. Sci 2003, 20, 233–240. [DOI] [PubMed] [Google Scholar]
- [29].Wu C, Zhang H, Kong N, Wu B, Lin X, Wang H, Angew. Chem. Int. Ed 2023, 62, e202303455. [DOI] [PubMed] [Google Scholar]
- [30].Singudas R, Reddy NC, Rai V, Chem. Commun 2019, 55, 9979–9982. [DOI] [PubMed] [Google Scholar]
- [31].Quartararo AJ, Gates ZP, Somsen BA, Hartrampf N, Ye X, Shimada A, Kajihara Y, Ottmann C, Pentelute BL, Nat. Commun 2020, 11, 3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhang G, Li C, Quartararo AJ, Loas A, Pentelute BL, Chem. Sci 2021, 12, 10817–10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Touti F, Gates ZP, Bandyopadhyay A, Lautrette G, Pentelute BL, Nat. Chem. Biol 2019, 15, 410–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Link JJO, Rhee MS, Tse WC, Zheng J, Somoza JR, et al. Nature, 2020, 584, 614–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gres AT, Kewal KA, Ramani VN, Tanner JJ, Pornillos O, Sarafianos SG, Science 2015, 349, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gres AT, Kirby KA, McFadden WM, Du H, Liu D, Xu C, Bryer AJ, Perilla JR, Shi J, Aiken C, Fu X, Zhang P, Francis AC, Melikyan GB, Sarafianos SG, Nat. Commun 2023, 14, 5614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Pornillos O, Ganser-Pornillos BK, Banumathi S, Hua Y, Yeager M, J. Mol. Biol 2010, 401, 985–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].McFadden WM, Sarafianos SG, Viruses, 2023, 15, 896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].McFadden WM, Snyder AA, Kirby KA, Tedbury PR, Raj M, Wang Z, Sarafianos SG, Retrovirology, 2021, 18, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Selyutina A, Hu P, Miller S, Simons LM, Yu HJ, Hultquist JF, Lee K, KewalRamani VN, Diaz-Griffero F, iScience, 2022, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Matreyek KA, Yücel SS, Li X, Engelman A, PLoS Pathog, 2013, 9, e1003693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Dubrow A, Zuniga B, Topo E, Cho J-H, ACS Omega, 2022, 7, 9206–9211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Miyazaki Y, Doi N, Koma T, Adachi A, Nomaguchi M, Front Microbiol, 2017, 8, 1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lanman J, Sexton J, Sakalian M, Prevelige PE Jr, J. Virol 2002, 76, 6900–6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rihn SJ, Wilson SJ, Loman NJ, Alim M, Bakker SE, Bhella D, Gifford RJ, Rixon FJ, Bieniasz PD, PLOS Pathog. 2013, 9, e1003461. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.