

Abstract
Background
Pulmonary arterial hypertension (PAH) ultimately leads to right ventricular failure and premature death. The identification of circulating biomarkers with prognostic utility is considered a priority. As chronic inflammation is recognized as key pathogenic driver, we sought to identify inflammation‐related circulating proteins that add incremental value to current risk stratification models for long‐term survival in patients with PAH.
Methods and Results
Plasma levels of 384 inflammatory proteins were measured with the proximity extension assay technology in patients with PAH (n=60) and controls with normal hemodynamics (n=28). Among these, 51 analytes were significantly overexpressed in the plasma of patients with PAH compared with controls. Cox proportional hazard analyses and C‐statistics were performed to assess the prognostic value and the incremental prognostic value of differentially expressed proteins. A panel of 6 proteins (CRIM1 [cysteine rich transmembrane bone morphogenetic protein regulator 1], HGF [hepatocyte growth factor], FSTL3 [follistatin‐like 3], PLAUR [plasminogen activator, urokinase receptor], CLSTN2 [calsyntenin 2], SPON1 [spondin 1]) were independently associated with death/lung transplantation at the time of PAH diagnosis after adjustment for the 2015 European Society of Cardiology/European Respiratory Society guidelines, the REVEAL (Registry to Evaluate Early and Long‐Term PAH Disease Management) 2.0 risk scores, and the refined 4‐strata risk assessment. CRIM1, PLAUR, FSTL3, and SPON1 showed incremental prognostic value on top of the predictive models. As determined by Western blot, FSTL3 and SPON1 were significantly upregulated in the right ventricle of patients with PAH and animal models (monocrotaline‐injected and pulmonary artery banding‐subjected rats).
Conclusions
In addition to revealing new actors likely involved in cardiopulmonary remodeling in PAH, our screening identified promising circulating biomarkers to improve risk prediction in PAH, which should be externally confirmed.
Keywords: biomarkers, inflammation, prognosis, proteomics, pulmonary hypertension
Subject Categories: Vascular Disease, Biomarkers
Nonstandard Abbreviations and Acronyms
- CRIM1
cysteine rich transmembrane bone morphogenetic protein regulator 1
- ESC/ERS
European Society of Cardiology/European Respiratory Society
- FSTL3
follistatin‐like 3
- PAB
pulmonary artery banding
- PAH
pulmonary arterial hypertension
- PLAUR
plasminogen activator, urokinase receptor
- REVEAL
Registry to Evaluate Early and Long‐Term PAH Disease Management
- SPON1
spondin 1
Research Perspective.
What Is New?
Proteomic analysis identified several inflammatory‐related proteins independently associated with death or lung transplantation at the time of pulmonary arterial hypertension diagnosis, with 4 (CRIM1 [cysteine rich transmembrane bone morphogenetic protein regulator 1], PLAUR [plasminogen activator, urokinase receptor], FSTL3 [follistatin‐like 3], and SPON1 [spondin 1]) showing incremental prognostic value on top of the 2015 European Society of Cardiology/European Respiratory Society guidelines, the REVEAL (Registry to Evaluate Early and Long‐Term PAH Disease Management) 2.0 risk scores, and the refined 4‐strata risk assessment.
FSTL3 and SPON1 were significantly upregulated in the right ventricle of patients with pulmonary arterial hypertension and pulmonary arterial hypertension animal models, suggesting that the stressed right ventricle is a source of these circulating proteins.
What Question Should Be Addressed Next?
Further studies are needed to confirm the predictive value of these proteins and to clarify their function in the pathophysiology of the disease.
Pulmonary arterial hypertension (PAH) is a complex and multifactorial cardiopulmonary disorder characterized by sustained vasoconstriction and obliterative remodeling of small pulmonary arteries (PA), leading to progressive elevation of pulmonary vascular resistance, right ventricular failure and premature death. 1 Despite advances in drug therapy that target the pulmonary vasoconstrictive component of the disease, long‐term prognosis for patients with PAH is still poor with a survival rate around 40% to 75% at 3 years 1 , 2 due, at least in part, to nonspecific symptoms making diagnosis and severity assessment challenging, thus delaying initiation or uptitration of PAH‐specific supportive therapies.
The development of clinically useful risk stratification models is an essential step to assess disease severity and progression, as well as to modulate appropriate proactive interventions. To date, the US REVEAL (Registry to Evaluate Early and Long‐Term PAH Disease Management) risk score 3 and the European Society of Cardiology/European Respiratory Society (ESC/ERS) guidelines risk assessment table 4 are most often used to guide treatment decisions for patients with PAH with various causes. These models integrate patient demographics, exercise capacity, hemodynamics, and natriuretic peptides to establish risk scores predicting prognosis. Although these existing tools are of recognized value, they suffer from the invasiveness of right heart catheterization. Moreover, most patients are categorized at intermediate risk, limiting their discriminative properties for a significant proportion of patients. To provide a more nuanced assessment of patients' prognosis, a refined noninvasive 4‐strata risk stratification model was recently proposed 5 and externally validated 6 to prognosticate patients with PAH, modestly increasing the discrimination of the model. Thus, refinements in risk stratification are mandatory and incorporation of new predictors of disease severity is needed. Because circulating biomarkers allow longitudinal monitoring, the identification of new circulating biomarker that add incremental benefit to already existing clinical predictors is considered a key priority. 7 , 8
Although the exact pathophysiology underlying lung and right ventricular adverse remodeling remains unclear, chronic inflammation and altered immune processes are recognized as key pathogenic drivers of PAH. 9 Indeed, perivascular inflammatory infiltrates and associated release of inflammatory mediators are prominent features of PAH lungs promoting adventitial fibroblast activation, PA smooth muscle cell proliferation, and endothelial‐mesenchymal transition, 9 which together establish a self‐perpetuating inflammatory loop and contribute to the progressive narrowing of the PA lumen. In addition, persistent inflammation is also intimately linked to cardiomyocyte death and fibrosis that adversely affect right ventricular function. 10
In view of the importance of inflammation in PAH initiation and progression, we hypothesized that changes in circulating inflammatory factors levels may provide insights into the prognosis of well‐characterized patients with idiopathic or connective tissue disease‐associated PAH. Using proximity extension assay, we examined the plasma levels and discriminative properties of 384 immune‐related proteins as biomarkers for the assessment of the severity and prognosis of the disease.
METHODS
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Human Lung and Right Ventricle Tissue Acquisition
All experimental procedures using human tissues or plasma were performed in accordance with the principles of the Declaration of Helsinki, as well as the approval of the Institut Universitaire de Cardiologie et Pneumologie de Québec‐University of Laval Ethics Committee (CER‐20735 and CER‐20773). Written informed consent was obtained from all study subjects.
Tissues were obtained from patients who had previously given signed informed consent. PAH and control tissues were obtained from Respiratory Health Network tissue bank (www.rsr‐qc.ca). Healthy lung tissues (controls) were obtained during lung resection for tumors. Lung samples were taken at distance from the tumor and demonstrated normal lung parenchyma. PAH tissues were from lung explants or warm autopsies. PAH diagnosis was previously confirmed by right heart catheterization. The patients' characteristics are described in Tables S1 and S2.
Proteomic Analysis in Human Plasma Samples
Study Population
Sixty patients with idiopathic PAH or PAH associated with connective tissue disease undergoing right heart catheterization at the Institut Universitaire de Cardiologie et Pneumologie de Québec in Canada were enrolled from June 2012 to March 2019. Diagnostic criteria for PAH included mean PA pressure ≥25mm Hg with PA wedge pressure ≤15mm Hg and pulmonary vascular resistance >3 Wood units, together with the exclusion of known associated diseases, according to contemporary guidelines. 11 More specifically, exclusion criteria included patients with left ventricular ejection fraction <40%, restrictive (lung fibrosis on computed tomography or total lung capacity <80% predicted) or obstructive (forced expiratory volume in 1 s/forced vital capacity <70%) lung disease, perfusion defects on lung scintigraphy, or active or recent (<5 years) cancer. Patients were diagnosed and treated according to the guidelines. 4 Patients with PAH were categorized as compensated right ventricle (RV, defined as cardiac index >2.2 L/min per m2) versus decompensated RV (defined as cardiac index ≤2.2 L/min per m2). Similarly, 28 subjects referred for right heart catheterization resulting in normal pulmonary hemodynamics and the absence of cardiac or respiratory diseases were enrolled as a control group.
Blood samples were obtained from each subject in EDTA tubes during right heart catheterization using the central venous line. The blood samples were immediately centrifuged to obtain plasma samples and kept frozen at −80 °C until proteomics assay. Baseline clinical information was obtained from the clinical database (age, sex, PAH type, World Health Organization functional class, exercise capacity, hemodynamics, NT‐proBNP [N‐terminal fragment of brain natriuretic peptide], estimated glomerular filtration rate, and treatment). Estimated glomerular filtration rate was calculated by Modification of Diet in Renal Disease formula. 12
Plasma of the cohort was screened using proximity extension, a high‐throughput multiplex proteomic immunoassay, for changes in the expression of 384 immune‐related proteins, primarily cytokines and chemokines (Olink Explore Inflammation Panel, Olink Proteomics, Uppsala, Sweden). The assay uses epitope‐specific binding and hybridization of a set of paired oligonucleotide antibody probes, which is subsequently amplified and detected by next generation sequencing, resulting in log base‐2 normalized protein expression values. Technicians from Olink were blinded to patient status. This study complied with Strengthening the Reporting of Observational Studies in Epidemiology guidelines. 13
Animal Models of PAH and RV Remodeling
All animal protocols were approved by the Institut Universitaire de Cardiologie et Pneumologie de Québec‐University of Laval Biosafety and Ethics Committees (#2022‐1049 and 2023‐1317). RV tissues from monocrotaline‐injected and pulmonary artery banding (PAB)‐operated rats used in our previous publications were de novo examined. 14 , 15 Proteins were extracted from human and rodent RVs and PAs using a 2% Chaps lysis buffer supplemented with a protease inhibitor cocktail (Roche). Protein concentrations were determined by the Bradford method. Equal amounts of proteins (15 μg) were separated by electrophoresis in SDS‐polyacrylamide gels and transferred to polyvinylidene fluoride membranes. Membranes were subsequently blocked in 5% nonfat dry milk in TBS‐T buffer for 1 hour. The membranes were incubated with a rabbit polyclonal anti‐SPON1 (spondin 1) antibody (#PA5‐116176, ThermoFisher Scientific) or a rabbit polyclonal anti‐FSTL3 (follistatin‐like 3) antibody (#PA1915, Boster Biological Technology) overnight at 4 °C. After being rinsed 3 times with TBS‐T buffer, membranes were incubated with goat‐anti‐rabbit antibody conjugated to horseradish peroxidase for 2 hours at room temperature in 5% nonfat dry milk in TBS‐T buffer. Immunolabeling was detected using ECL reagents (Perkin–Elmer) with the imaging Chemidoc MP system (Bio‐Rad Laboratories). Protein expression was quantified using the Image lab software (Bio‐Rad Laboratories) and normalized to Amido black as previously described. 14 , 15
Statistical Analysis
Normally distributed variables, nonnormally distributed variables, and categorical variables were shown as mean±SD, median (interquartile range), and counts and percentages, respectively. Normally distributed variables were compared by the Student's t test, and nonnormally distributed variables were compared by the Mann–Whitney U test. For more than 2 groups, the 1‐way ANOVA for normally distributed variables or the Kruskal–Wallis test for nonnormally distributed variables was performed for comparison. Categorical variables were compared by the chi‐square test or the Fisher's exact test. The relationship between circulating protein levels and hemodynamics (pulmonary vascular resistance, cardiac index, and stroke volume), NT‐proBNP, and estimated glomerular filtration rate were assessed using Pearson's correlation analysis.
We first compared the expression of the immune‐related proteins between controls and patients with PAH and between controls and patients with PAH categorized as decompensated RV (cardiac index ≤2.2 L/min per m2). Proteins identified by Olink proteomic extension assay with an adjusted P value ≤0.01 (using Benjamini–Hochberg correction) and a fold change ≥1.4 were considered to be differentially expressed in the plasma of patients with PAH. We then performed univariate and multivariate Cox proportional hazard analyses for death or lung transplantation using differentially expressed proteins. Event‐free survival was analyzed from sampling to death or lung transplantation. The cutoff date was September 30, 2021. No patients were lost to follow‐up.
The multivariate models were adjusted for age, sex, and the 2015 ESC/ERS risk score (model 1), the REVEAL 2.0 risk score (model 2), and the refined 4‐strata noninvasive risk assessment (model 3). The multivariate logistic regression analyses for the discrimination of patients with death or lung transplantation were used for calculation of the combined models. The C‐statistics between models were compared by DeLong's test. 16 Calibration was tested using the Hosmer–Lemeshow goodness‐of‐fit test for the logistic regression model.
As a complementary approach, receiver operating characteristics curves and time‐dependent receiver operating characteristics curves of 1 and 3 years with area under the curve were used to assess the performance of the proteins found to be independently associated with long‐term outcomes alone or in combination to validated risk stratification models to predict PAH transplant‐free survival. The Kaplan–Meier analysis was used to estimate transplant‐free survival rate and a log‐rank test was used for comparisons between groups using the optimal cutoff point determined by the curve point nearest to the 100% of axis of the ordinates on receiver operating characteristics curves for the survival curve. 17
A significance level inferior to 5% (P<0.05) was considered statistically significant, unless specifically stated. All statistical analyses were made with Graph Pad Prim version 9.0.0 (GraphPad Software, Inc., San Diego, CA), SPSS version 27 (IBM, Armonk, NY), or R version 4.0.5 (R Foundation for Statistical Computing, Vienna, Austria).
RESULTS
Study Subjects
Baseline clinical characteristics of the cohort are shown in Table 1. Patients with PAH were well matched to controls. As expected, patients with PAH had severe hemodynamic impairment, elevated NT‐proBNP, and poor exercise capacity, whereas patients with connective tissue disease‐PAH were older than patients with idiopathic PAH (Table S3).
Table 1.
Baseline Clinical Characteristics of Control Patients and Patients With PAH
| Control (n=28) | PAH (n=60) | P value | |
|---|---|---|---|
| Female sex, n (%) | 18 (64) | 39 (65) | 0.948 |
| Age, y | 61±16 | 65±14 | 0.297 |
| World Health Organization functional class, n (%) | |||
| I | 0 (0) | ||
| II | 10 (17) | ||
| III | 41 (68) | ||
| IV | 9 (15) | ||
| Hemodynamics | |||
| Mean pulmonary arterial pressure, mm Hg | 17±3 | 47±9 | <0.001 |
| Mean right atrium pressure, mm Hg | 5±4 | 8±4 | 0.005 |
| Cardiac index, L/min per m2 | 3.0±0.6 | 2.4±0.8 | <0.001 |
| Pulmonary vascular resistance, dynes·s·cm−5 | 123±58 | 832±379 | <0.001 |
| Heart rate, bpm | 71±13 | 81±11 | 0.002 |
| Mixed venous oxygen blood saturation, % | 72±8 | 63±9 | <0.001 |
| N‐terminal pro‐B‐type natriuretic peptide, pg/mL* | 155 (79–244) | 1375 (369–4082) | < 0.001 |
| Estimated glomerular filtration rate (Modification of Diet in Renal Disease), mL/min per 1.73 m2 | 83±28 | 69±22 | 0.009 |
| 6‐minute walking distance, m | 306±121 | ||
| PAH medication categories, n (%) | |||
| None | 28 (100) | 37 (62) | |
| Mono therapy | 0 (0) | 15 (25) | |
| Double therapy | 0 (0) | 7 (12) | |
| Triple therapy | 0 (0) | 1 (2) | |
| Specific PAH therapy at enrollment, n (%) | |||
| Endothelin receptor antagonist | 0 (0) | 10 (17) | |
| Phosphodiesterase type 5inhibitor | 0 (0) | 19 (32) | |
| Epoprostenol | 0 (0) | 3 (5) | |
Data are expressed by mean±SEM, median (interquartile range)*, or n (%). PAH indicates pulmonary arterial hypertension.
Identification of Inflammatory Biomarkers for RV Function and Long‐Term Outcomes
Out of the 384 proteins analyzed, 26 immune‐related analytes were significantly overexpressed in plasma of patients with PAH compared with controls (adjusted P≤0.01 and fold change ≥1.4). Using the same thresholds, 49 proteins were significantly upregulated in patients with PAH with decompensated RV compared with controls, of which 24 were found to overlap (Figure 1A and 1B). From the correlation and cluster analysis summarized in Figure 1C, it emerges that, among the measured proteins, some of them strongly correlated with each other. The major cluster consisted of MGMT (O‐6‐methylguanine‐DNA methyltransferase), IRAK1 (interleukin 1 receptor associated kinase 1), ATP5IF1 (ATP synthase inhibitory factor subunit 1), HEXIM1 (HEXIM P‐TEFb complex subunit 1), LAP3 (leucine aminopeptidase 3), DFFA (DNA fragmentation factor subunit alpha), and GMPR (guanosine monophosphate reductase). Another independent secondary cluster of highly correlated proteins consisted of TGF‐α (transforming growth factor alpha), FSTL3, PLAUR [plasminogen activator, urokinase receptor], ESM1 (endothelial cell specific molecule 1), CRIM1 (cysteine rich transmembrane bone morphogenetic protein regulator 1), SPON1, and CXCL17 (C‐X‐C motif chemokine ligand 17). The correlation between these proteins suggests potential coregulation and connection between them into pathologic processes.
Figure 1. Increased plasma levels of inflammation‐related proteins in patients with PAH.
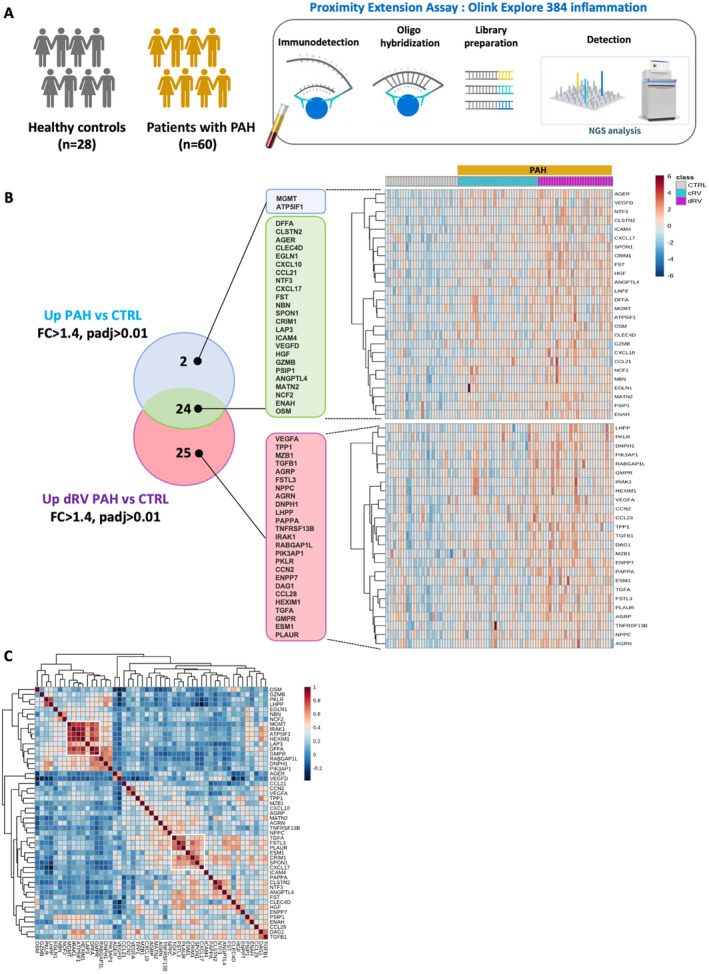
A, Schematic overview of the study design. B, Heatmap of the 51 plasma proteins found differentially expressed (fold change ≥1.4 and adjusted P value ≤0.01) between patients with PAH and controls. On top of the heatmap are control samples in gray, cRV PAH sample in blue, and patients with dRV PAH in purple. Color key indicates expression value: dark blue: lowest; dark red: highest. C, Heatmap of correlation matrix (Pearson) for the 51 circulating proteins significantly increased in patients with PAH (PAH vs CTRL and dRV PAH vs CTRL). Two major clusters of correlating proteins were identified. cRV indicates compensated right ventricle; CTRL, controls; dRV, decompensated right ventricle; FC, fold change; NGS, next‐generation sequencing; and PAH, pulmonary arterial hypertension.
During a median follow‐up of 2.5 years (interquartile range, 0.9–4.3 years) after blood sample was drawn, 31 patients with PAH died and 2 underwent lung transplantation. Among the 51 analytes found to differ significantly between PAH and controls, 17 were significantly associated with death or lung transplantation in patients with PAH using univariate Cox proportional hazards analyses (Table S4). Only 6 proteins, namely CRIM1 (also called cysteine‐rich motor neuron 1), HGF (hepatocyte growth factor), CLSTN2 (calsyntenin 2), SPON1 (also called F‐spondin), PLAUR, and FSTL3 remained independent prognostic factors after adjusting for the 2015 ESC/ERS, the REVEAL 2.0 risk scores, and the refined 4‐strata risk assessment (Figure 2 and Table 2). Of interest, CRIM1, HGF, CLTSN2, and FSTL3 were also upregulated in patients with decompensated RV compared with compensated RV and were similarly overexpressed in idiopathic PAH and connective tissue disease‐PAH (Figure 2). Moreover, all proteins correlated with mean pulmonary artery pressure, pulmonary vascular resistance, cardiac index, stroke volume, and NT‐proBNP at baseline (Figure S1).
Figure 2. Expression levels (NPX) of CLSTN2, HGF, SPON1, CRIM1, PLAUR, and FSTL3 in plasma from CTRL and patients with PAH.
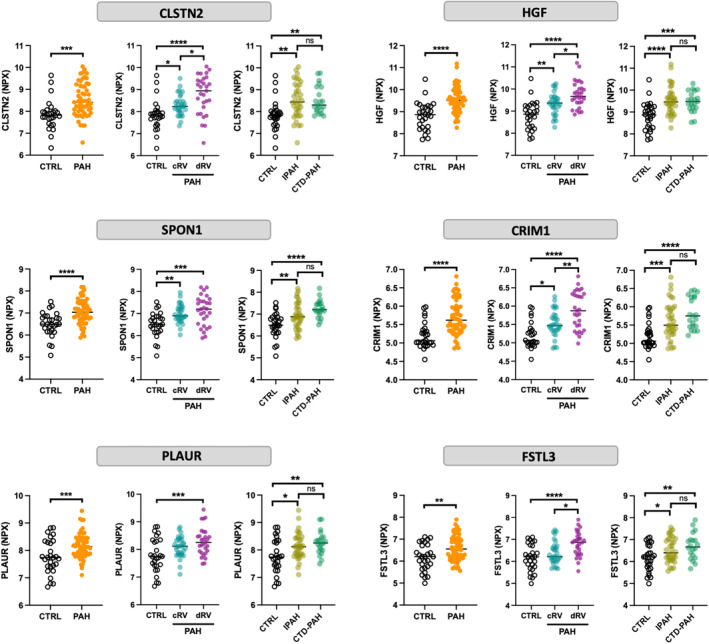
According to contemporary cardiac index measured by right heart catheterization at the time blood sample was drawn, patients with PAH were categorized as cRV (cardiac index >2.2 L/[min·m2]) and dRV (cardiac index ≤2.2 L/[min·m2]). Statistical analyses were performed using the Student's t test or 1‐way ANOVA followed by Tukey post hoc test. *P< .05, **P<0.01, ***P<0.001, and ****P<0.0001. CLSTN2 indicates calsyntenin 2; cRV, compensated right ventricle; CRIM1, cysteine rich transmembrane bone morphogenetic protein regulator 1; CTD‐PAH, connective tissue disease‐associated pulmonary arterial hypertension; CTRL, controls; dRV, decompensated right ventricle; FSTL3, follistatin‐like 3; HGF, hepatocyte growth factor; IPAH, idiopathic pulmonary hypertension; NPX, normalized protein expression; PAH, pulmonary arterial hypertension; PLAUR, plasminogen activator, urokinase receptor; and SPON1, spondin 1.
Table 2.
Multivariate Cox Proportional Hazard Analyses for Death or Lung Transplantation in Patients With PAH
| Protein | Adjusted for age, sex, and 2015 ESC/ERS risk score | Adjusted for REVEAL 2.0 risk score | Adjusted for age, sex, the 4‐strata noninvasive method | ||||||
|---|---|---|---|---|---|---|---|---|---|
| HR | 95% CI | P value | HR | 95% CI | P value | HR | 95% CI | P value | |
| CRIM1 * | 3.085 | 1.020–9.334 | 0.046 | 3.494 | 1.497–8.156 | 0.004 | 2.956 | 1.224–7.139 | 0.016 |
| HGF * | 2.368 | 1.033–5.428 | 0.042 | 2.137 | 1.028–4.444 | 0.042 | 2.370 | 1.092–5.147 | 0.029 |
| MATN2 | 1.356 | 0.424–4.333 | 0.607 | 1.558 | 0.542–4.480 | 0.411 | 1.496 | 0.512–4.371 | 0.462 |
| EGLN1 | 1.192 | 0.849–1.675 | 0.310 | 1.037 | 0.743–1.447 | 0.830 | 1.130 | 0.805–1.587 | 0.480 |
| ANGPTL4 | 1.322 | 0.602–2.902 | 0.486 | 1.796 | 0.884–3.648 | 0.105 | 1.501 | 0.727–3.102 | 0.272 |
| FST | 1.242 | 0.488–3.160 | 0.649 | 1.547 | 0.662–3.616 | 0.313 | 1.211 | 0.482–3.047 | 0.684 |
| DNPH1 | 1.130 | 0.523–2.444 | 0.756 | 0.981 | 0.488–1.971 | 0.957 | 1.021 | 0.474–2.201 | 0.958 |
| AGER | 0.989 | 0.583–1.678 | 0.968 | 1.382 | 0.881–2.166 | 0.159 | 1.055 | 0.629–1.769 | 0.839 |
| DFFA | 1.462 | 0.733–2.765 | 0.243 | 1.208 | 0.650–2.243 | 0.550 | 1.410 | 0.758–2.623 | 0.278 |
| RABGAP1L | 0.786 | 0.386–1.604 | 0.509 | 0.646 | 0.324–1.288 | 0.215 | 0.813 | 0.396–1.669 | 0.573 |
| TGF‐α | 2.201 | 0.857–5.653 | 0.101 | 2.238 | 0.805–6.226 | 0.123 | 1.680 | 0.610–4.626 | 0.315 |
| CLEC4D | 1.122 | 0.655–1.922 | 0.674 | 1.056 | 0.628–1.775 | 0.837 | 1.150 | 0.684–1.935 | 0.598 |
| CLSTN2 * | 2.056 | 1.022–4.135 | 0.043 | 2.059 | 1.142–3.711 | 0.016 | 2.033 | 1.126–3.670 | 0.019 |
| PKLR | 0.520 | 0.233–1.159 | 0.110 | 0.380 | 0.173–0.835 | 0.016 | 0.507 | 0.233–1.102 | 0.087 |
| NBN | 0.949 | 0.395–2.279 | 0.907 | 1.327 | 0.629–2.799 | 0.458 | 0.932 | 0.386–2.248 | 0.875 |
| NTF3 | 1.342 | 0.865–2.081 | 0.189 | 1.220 | 0.833–1.785 | 0.307 | 1.219 | 0.799–1.858 | 0.358 |
| ENPP7 | 1.260 | 0.914–1.736 | 0.158 | 1.228 | 0.881–1.711 | 0.225 | 1.266 | 0.918–1.746 | 0.150 |
| DAG1 | 3.070 | 0.944–9.984 | 0.062 | 2.611 | 1.036–6.582 | 0.042 | 2.957 | 0.933–9.370 | 0.065 |
| FSTL3 * | 2.845 | 1.352–5.988 | 0.006 | 2.918 | 1.419–6.002 | 0.004 | 2.213 | 1.099–4.458 | 0.026 |
| TGF‐ β1 | 1.236 | 0.424–3.601 | 0.698 | 1.562 | 0.587–4.156 | 0.372 | 1.500 | 0.553–4.067 | 0.425 |
| TPP1 | 2.373 | 0.899–6.267 | 0.081 | 1.692 | 0.726–3.944 | 0.223 | 2.293 | 0.884–5.948 | 0.088 |
| CCL28 | 1.167 | 0.543–2.511 | 0.692 | 1.215 | 0.561–2.632 | 0.622 | 1.283 | 0.598–2.752 | 0.522 |
| OSM | 1.089 | 0.748–1.587 | 0.656 | 0.850 | 0.578–1.250 | 0.409 | 0.982 | 0.648–1.488 | 0.933 |
| AGRN | 0.807 | 0.399–1.630 | 0.549 | 1.296 | 0.766–2.193 | 0.334 | 0.994 | 0.527–1.876 | 0.985 |
| PSIP1 | 1.505 | 0.738–3.070 | 0.261 | 1.396 | 0.722–2.701 | 0.322 | 1.512 | 0.755–3.027 | 0.243 |
| TNFRSF13B | 1.530 | 0.809–2.896 | 0.191 | 1.729 | 0.909–3.288 | 0.095 | 1.302 | 0.643–2.635 | 0.463 |
| PAPPA | 1.436 | 0.695–2.967 | 0.328 | 1.532 | 0.767–3.060 | 0.227 | 1.323 | 0.654–2.680 | 0.436 |
| CXCL10 | 0.873 | 0.554–1.378 | 0.561 | 0.976 | 0.665–1.432 | 0.901 | 0.960 | 0.627–1.471 | 0.851 |
| SPON1 * | 3.129 | 1.268–7.722 | 0.013 | 3.675 | 1.716–7.869 | 0.001 | 2.882 | 1.172–7.086 | 0.021 |
| VEGFD | 1.156 | 0.595–2.246 | 0.668 | 1.750 | 0.936–3.274 | 0.080 | 1.326 | 0.670–2.627 | 0.418 |
| CXCL17 | 1.458 | 0.637–3.338 | 0.373 | 2.041 | 1.096–3.798 | 0.024 | 1.583 | 0.701–3.577 | 0.269 |
| NCF2 | 1.381 | 0.819–2.328 | 0.226 | 1.361 | 0.833–2.223 | 0.219 | 1.362 | 0.792–2.344 | 0.264 |
| CCN2 | 0.512 | 0.225–1.165 | 0.110 | 0.651 | 0.325–1.304 | 0.226 | 0.518 | 0.235–1.142 | 0.103 |
| HEXIM1 | 1.354 | 0.896–2.044 | 0.150 | 1.242 | 0.822–1.877 | 0.303 | 1.370 | 0.909–2.066 | 0.133 |
| IRAK1 | 1.118 | 0.626–1.998 | 0.706 | 1.095 | 0.649–1.848 | 0.733 | 1.113 | 0.623–1.990 | 0.718 |
| ESM1 | 1.617 | 0.866–3.021 | 0.132 | 1.978 | 1.096–3.572 | 0.024 | 1.501 | 0.800–2.816 | 0.206 |
| GZMB | 0.829 | 0.496–1.387 | 0.475 | 0.782 | 0.498–1.227 | 0.284 | 0.718 | 0.418–1.234 | 0.230 |
| CCL21 | 0.933 | 0.455–1.912 | 0.850 | 1.180 | 0.695–2.003 | 0.541 | 0.997 | 0.484–2.057 | 0.995 |
| PIK3AP1 | 0.994 | 0.534–1.852 | 0.986 | 0.846 | 0.475–1.506 | 0.570 | 0.980 | 0.541–1.776 | 0.947 |
| ENAH | 1.561 | 0.662–3.677 | 0.309 | 1.667 | 0.854–3.253 | 0.134 | 1.335 | 0.658–2.712 | 0.424 |
| LAP3 | 0.885 | 0.473–1.656 | 0.702 | 0.806 | 0.436–1.490 | 0.491 | 0.883 | 0.468–1.666 | 0.701 |
| GMPR | 1.325 | 0.746–2.355 | 0.337 | 1.031 | 0.587–1.810 | 0.916 | 1.321 | 0.747–2.335 | 0.338 |
| PLAUR * | 3.780 | 1.310–10.903 | 0.014 | 4.301 | 1.496–12.362 | 0.007 | 2.780 | 1.022–7.565 | 0.045 |
| AGRP | 1.350 | 0.716–2.548 | 0.354 | 1.300 | 0.722–2.340 | 0.382 | 1.419 | 0.778–2.589 | 0.254 |
| NPPC | 1.151 | 0.609–2.177 | 0.665 | 1.251 | 0.712–2.199 | 0.435 | 0.988 | 0.533–1.831 | 0.970 |
| LHPP | 0.769 | 0.402–1.469 | 0.426 | 0.531 | 0.275–1.025 | 0.059 | 0.605 | 0.290–1.261 | 0.180 |
| ICAM4 | 1.128 | 0.688–1.850 | 0.633 | 1.148 | 0.714–1.845 | 0.569 | 0.992 | 0.594–1.657 | 0.977 |
| VEGFA | 0.699 | 0.360–1.358 | 0.291 | 0.684 | 0.376–1.245 | 0.214 | 0.633 | 0.320–1.249 | 0.187 |
| MZB1 | 1.507 | 0.861–2.638 | 0.151 | 1.504 | 0.904–2.503 | 0.116 | 1.452 | 0.840–2.509 | 0.181 |
| MGMT | 1.100 | 0.850–1.425 | 0.469 | 1.072 | 0.827–1.390 | 0.600 | 1.087 | 0.837–1.413 | 0.530 |
| ATP5IF1 | 1.141 | 0.831–1.566 | 0.416 | 1.073 | 0.809–1.423 | 0.623 | 1.162 | 0.852–1.586 | 0.343 |
AGER indicates advanced glycosylation end‐product specific receptor; AGRN, agrin; AGRP, agouti related neuropeptide; ANGPTL4, angiopoietin like 4; ATP5IF1, ATP synthase inhibitory factor subunit 1; CCL21, C‐C motif chemokine ligand 21; CCL28, C‐C motif chemokine ligand 28; CCN2, cellular communication network factor 2; CLEC4D, C‐type lectin domain family 4 member D; CLSTN2, calsyntenin 2; CRIM1, cysteine rich transmembrane bone morphogenetic protein regulator 1; CXCL10, C‐X‐C motif chemokine ligand 10; CXCL17, C‐X‐C motif chemokine ligand 17; DAG1, dystroglycan 1; DFFA, DNA fragmentation factor subunit alpha; DNPH1, 2′‐deoxynucleoside 5′‐phosphate N‐hydrolase 1; EGLN1, Egl‐9 family hypoxia inducible factor 1; ENAH, ENAH actin regulator; ENPP7, ectonucleotide pyrophosphatase/phosphodiesterase 7; ERS European Respiratory Society; ESC, European Society of Cardiology; ESM1, endothelial cell specific molecule 1; FST, follistatin; FSTL3, follistatin‐like 3; GMPR, guanosine monophosphate reductase; GZMB, granzyme B; HEXIM1, HEXIM P‐TEFb complex subunit 1; HGF, hepatocyte growth factor; HR, hazard ratio; ICAM4, intercellular adhesion molecule 4; IRAK1, interleukin 1 receptor associated kinase 1; LAP3, leucine aminopeptidase 3; LHPP, phospholysine phosphohistidine inorganic pyrophosphate phosphatase; MATN2, matrilin‐2; MGMT, O‐6‐methylguanine‐DNA methyltransferase; MZB1, marginal zone B and B1 cell specific protein; NBN, nibrin; NCF2, neutrophil cytosolic factor 2; NPPC, natriuretic peptide C; NTF3, neurotrophin 3; OSM, oncostatin M; PAH, pulmonary arterial hypertension PAPPA, pappalysin 1; PIK3AP1, phosphoinositide‐3‐kinase adaptor protein 1; PKLR, pyruvate kinase L/R; PLAUR, plasminogen activator, urokinase receptor; PSIP1, PC4 and SRSF1 interacting protein 1; RABGAP1L, RAB GTPase activating protein 1 like; SPON1, spondin 1; TGF‐α, transforming growth factor alpha; TGF‐β1, transforming growth factor beta 1; TNFRSF13B, tumor necrosis factor receptor superfamily member 13B; TPP1, tripeptidyl peptidase 1; VEGFA, vascular endothelial growth factor A; and VEGFD, vascular endothelial growth factor D.
Rows identify biomarkers that remain independent prognostic factors after adjusting for the 2015 ESC/ERS, the REVEAL 2.0 risk scores, and the refined 4‐strata risk assessment.
Incremental Predicted Performance of Identified Biomarkers and Stratification of Prognosis
In agreement with regression analyses, CRIM1, HGF, SPON1, PLAUR, and FSTL3 significantly discriminated patients with PAH with poorer transplant‐free survival, with area under the curve >0.667 (Figure 3). Of note, these proteins had numerically better discriminative properties than NT‐proBNP (area under the curve 0.630) (Figure S2). In addition, time‐dependent receiver operating characteristics analyses of 1 and 3 years also showed that CRIM1, CLSTN2, FSTL3, SPON1, and PLAUR had significant discriminated capacity for transplant‐free survival in patients with PAH (Table S5). We then evaluated the performance of adding these identified biomarkers to the REVEAL 2.0 and the 2015 ESC/ERS risk scores, as well as the refined 4‐strata risk assessment for prognosis. Although all predicted transplant‐free survival, CRIM1 and SPON1 consistently exhibited significant additional prognostic value on top of all 3 predictive models, as confirmed by improved C‐statistics. (Figure 4 and Table S6). FSTL3 and PLAUR also displayed incremental prognostic value on top of the 2015 ESC/ERS and refined 4‐strata predictive models (Figure 4 and Table S6).
Figure 3. Survival ROC curves and Kaplan–Meier curves showing progression free survival in patients with PAH with baseline biomarker levels above or below the identified threshold.
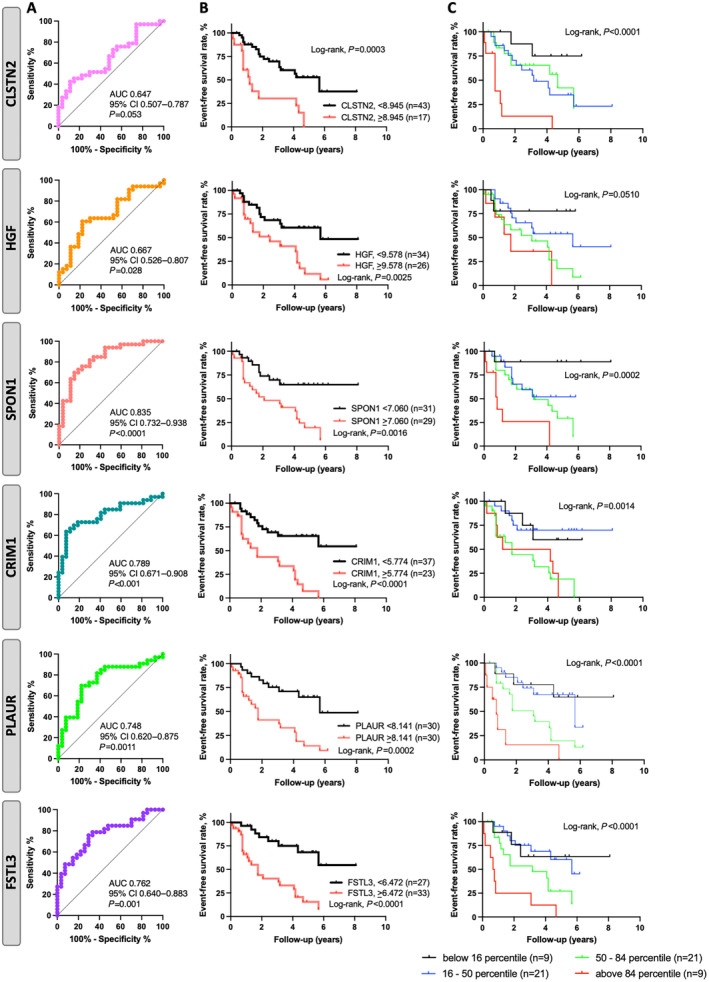
A, ROC assessing the prognostic accuracy of CLSTN2, HGF, SPON1, CRIM1, PLAUR, and FSTL3. B, Kaplan–Meier survival curves according to ROC cutoff values for CLSTN2, HGF, SPON1, CRIM1, PLAUR, and FSTL3. C, Higher percentiles of the biomarker levels were also associated with lower event‐free survival, suggesting their ability to identify patients at high event risk. AUC indicates area under the curve. CLSTN2, calsyntenin 2; CRIM1, cysteine rich transmembrane bone morphogenetic protein regulator 1; FSTL3, follistatin‐like 3; HGF, hepatocyte growth factor; PAH, pulmonary arterial hypertension; PLAUR, plasminogen activator, urokinase receptor; ROC, receiver operating characteristics; and SPON1, spondin 1.
Figure 4. C‐statistics of biomarkers with 2015 ERS/ESC risk score, REVEAL 2.0 risk score and the refined four‐strata risk assessment method in patients with PAH for death and lung transplantation.
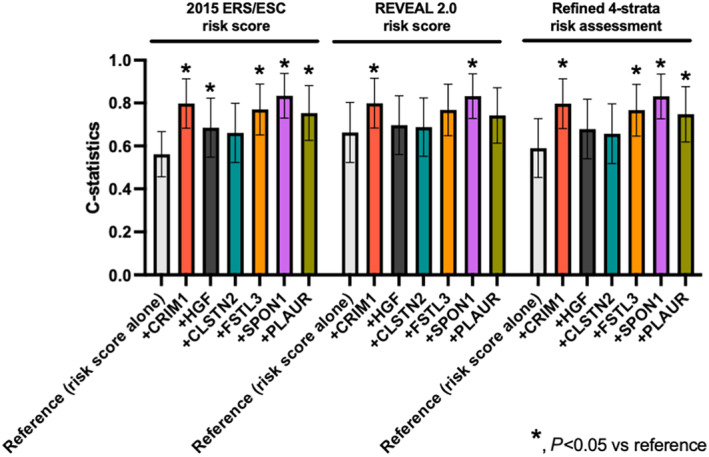
Graph of C‐statistic with 95% CI for each prognostic biomarker. C‐statistics were compared using the DeLong test with each reference. *P<0.05 vs reference. CLSTN2 indicates calsyntenin 2; CRIM1, cysteine rich transmembrane bone morphogenetic protein regulator 1; ESC/ERS, European Society of Cardiology/European Respiratory Society; FSTL3, follistatin‐like 3; HGF, hepatocyte growth factor; PAH, pulmonary arterial hypertension; PLAUR, plasminogen activator, urokinase receptor; REVEAL, Registry to Evaluate Early and Long‐Term PAH Disease Management; and SPON1, spondin 1.
Expression of SPON1 and FSTL3 in Lung and Right Ventricle From Patients With PAH and Animal Models
Our data showing that PLAUR FSTL3, SPON1, and CRIM1 improve risk stratification when combined to different models led us to measure their protein expression levels in lungs and RVs from patients with PAH and animal models. Examination of comparative transcriptomic analysis of RVs in human and rat models (PAB and monocrotaline) revealed that PLAUR, FSTL3, and SPON1 but not CRIM1 were increased in decompensated RV. 15 , 18 Unfortunately, no suitable anti‐CRIM1 antibody was available and the immunoblot for PLAUR yielded inconclusive results due to the presence of multiple bands. Western blot analysis revealed significantly higher levels of SPON1 in the RVs of patients with PAH and in the RVs of monocrotaline‐and PAB‐challenged rats (Figure 5). No significant difference was observed in dissected PAs from control and patients with PAH (Figure 5). Similarly, protein levels of SPON1 within dissected PAs were unchanged between control and monocrotaline rats (Figure 5). For FSTL3, the protein levels were significantly increased in the RV of patients with PAH, whereas no change was observed in the pulmonary vasculature (Figure 6). FSTL3 protein levels were significantly increased in remodeled RVs from monocrotaline and PAB rats (Figure 6). Taken together, these results suggest that the stressed RV is a source of circulating SPON1 and FSTL3 in patients with PAH.
Figure 5. Upregulation of SPON1 in remodeled RVs from patients with PAH and animal models.
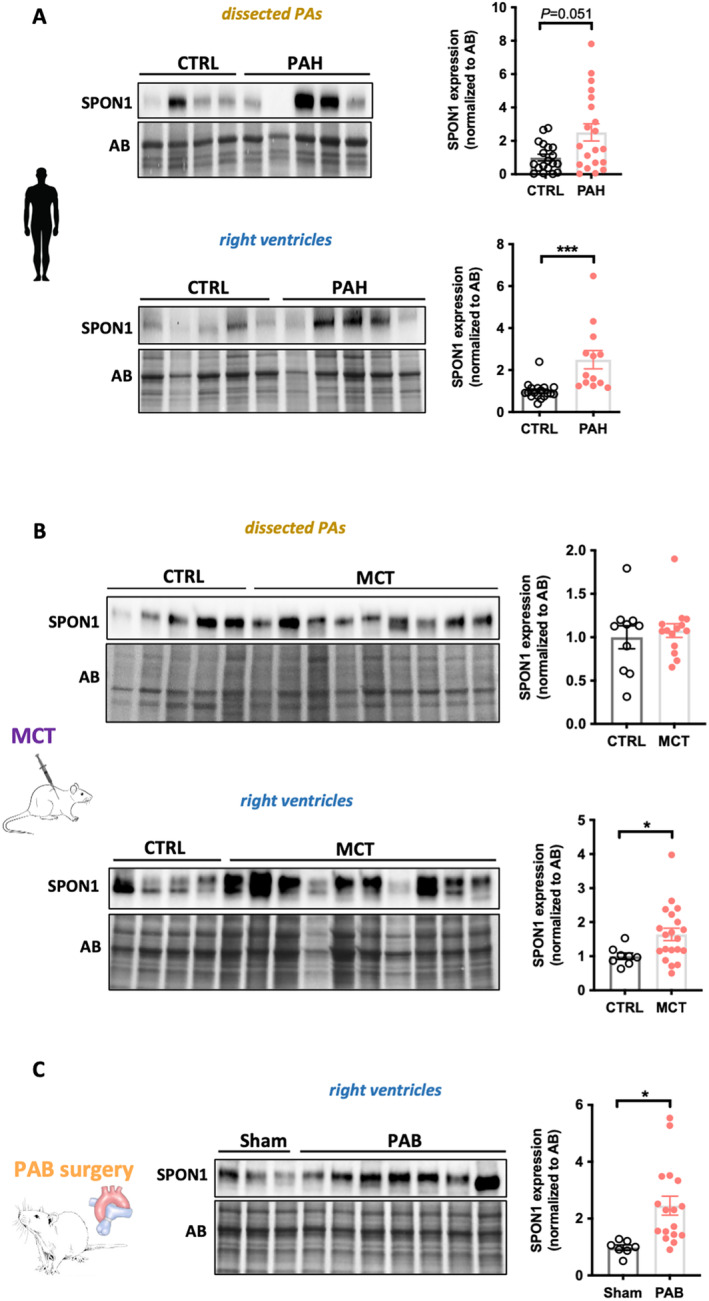
A, Representative Western blots and corresponding quantifications of SPON1 in dissected PAs and RVs from CTRL (n=18–19) and patients with PAH (13–20). B, Representative Western blots and corresponding quantifications of SPON1 in dissected PAs and RVs from CTRL (n=8–10) and monocrotaline‐injected (n=14–20) rats. C, Representative Western blot and corresponding quantification of SPON1 in RVs from sham (n=7) and PAB‐subjected (n=17) rats. Protein expression was normalized to amido black. Scatter dot plots show individual values and mean±SEM. Statistical analyses were performed using Student's t test. *P<0.05 and ***P<0.001. AB indicates amido black; CTRL, controls; MCT, monocrotaline; PA, pulmonary arteries; PAB, pulmonary artery banding; PAH, pulmonary arterial hypertension; RV, right ventricle; and SPON1, spondin 1.
Figure 6. Upregulation of FSTL3 in remodeled RVs from patients with PAH and animal models.
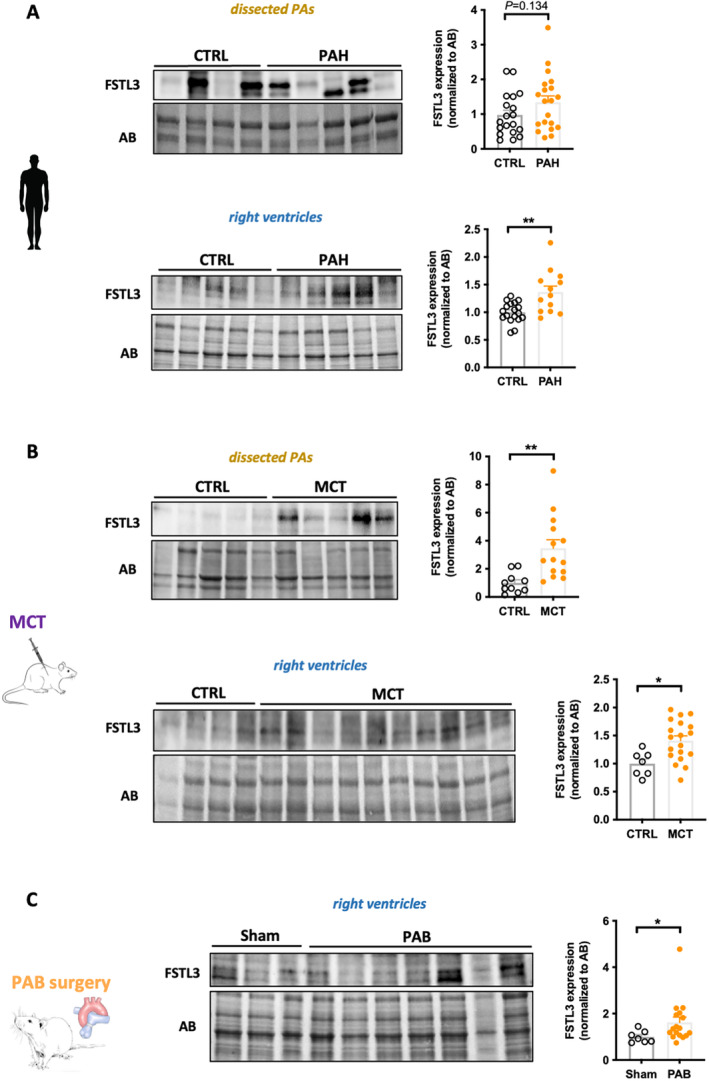
A, Representative Western blots and corresponding quantifications of FSTL3 in dissected pulmonary arteries and RVs from CTRL (n=18–19) and patients with PAH (n=13–20). B, Representative Western blots and corresponding quantifications of FSTL3 in dissected PAs and RVs from CTRL (n=7–10) and monocrotaline‐injected (n=14–19) rats. C, Representative Western blot and corresponding quantification of FSTL3 in RVs from sham (n=7) and PAB‐subjected (n=17) rats. Protein expression was normalized to amido black. Scatter dot plots show individual values and mean±SEM. Statistical analyses were performed using Student's t test. *P<0.05 and **P<0.01. AB indicates amido black; CTRL, controls; FSTL3, follistatin‐like 3; MCT, monocrotaline; PA, pulmonary ateries; PAB, pulmonary artery banding; PAH, pulmonary arterial hypertension; and RV, right ventricle.
DISCUSSION
Over the past few years, the efforts to identify reliable noninvasive biomarkers to predict outcomes and aid clinical decision‐making in PAH were intensified because of the development of high‐throughput proteomic technologies. 15 , 19 , 20 The present study is a part of this initiative focusing on inflammation‐related circulating factors due to their well‐established implication in PAH progression. 9 Although the nature of the analytes tested (ie, inflammation/immune‐related factors) makes them unlikely to be specific for PAH, we revealed using proximity extension assay that HGF, SPON1, CRIM1, PLAUR, CLSTN2, and FSTL3 are predictors of death or transplantation. We also demonstrated that SPON1, CRIM1, PLAUR, and FSTL3 add performance to current risk stratification tools.
Among the 384 inflammation‐related proteins tested, we identified CRIM1, SPON1, and PLAUR as new plasma proteins closely related to the severity of the disease. CRIM1 is a type I transmembrane glycoprotein bearing an N‐terminal IGFBP (insulin‐like growth factor‐binding protein motif) and 6 cysteine‐rich von Willebrand factor C repeats located in the extracellular domain and known to be subjected to a juxtamembrane cleavage that generates a secreted ectodomain. 21 Although its biological function remains largely unknown, CRIM1 was shown to serve an important role in the development of multiple organs, including the heart, likely via regulating the bioavailability of growth factors and integrin signaling. 22 , 23 , 24 Interestingly, CRIM1 was documented to be expressed in coronary vascular smooth muscle cells and coronary endothelial cells during cardiac development 24 and serum levels of CRIM1 were reported to positively correlate with the severity of left ventricular failure in human. 25 SPON1 is a secreted and extracellular matrix‐attached protein of the thrombospondin family with obscure functions. Early work on this protein indicated that it stimulates vascular smooth muscle cell proliferation 26 and inhibits angiogenesis, 27 suggesting that increased plasma levels in patients with PAH may contribute to RV ischemia. uPAR (urokinase plasminogen activator receptor), encoded by the PLAUR gene, is a component of the plasminogen activation system implicated in a variety of biological processes such as extracellular matrix remodeling, inflammation, and cell behavior. 28 Levels of soluble uPAR were repeatedly shown to exhibit predictive and prognostic value in patients with cardiovascular diseases 29 and kidney dysfunction. 30 Because uPAR regulates many biological processes involved in PAH progression, further studies are required to determine its precise function during maladaptive events and confirm its prognostic significance.
A major challenge to clinical implementation of new biomarkers to complement or improve current approaches for prognosis of PAH is the lack of replicability. In direct connection with this, we validated recent published data showing that HGF and FSTL3 are augmented in blood of patients with PAH and could serve as promising prognostic biomarkers. 20 , 31 , 32 , 33 The ability to obtain similar results from different cohorts and using distinctive quantifications of protein concentration is a fundamental step for research credibility. Although FSTL3 was documented to be upregulated in remodeled pulmonary vessels of patients with PAH, we provided additional evidence that FSTL3 protein levels are increased in human PAH RVs. This finding coupled to the fact that FSTL3 is also upregulated in RVs from PAB rats suggests a combined cardiac and pulmonary origin of circulating FSTL3. FSTL3 is known to be induced by activin receptor type II activation but also functions as a negative regulator of the pathway activity by antagonizing the action of activin A 34 , 35 making it difficult to determine with certainty the impact of its high circulating expression. Although the role of activin A signaling in cardiac remodeling has been the subject of controversy, 36 recent findings indicate a detrimental function. Indeed, increasing circulating activin A in mice was shown to induce cardiac activin receptor type II signaling and FSTL3 expression resulting in compromised cardiac function. 36 Conversely, genetic or pharmacological inhibition of activin receptor type II signaling preserved cardiac function in several animal models of heart failure. 35 In support of this, engagement of activin A signaling was reported to directly impair cardiomyocyte contractile function, 37 whereas secretion of FSTL3 by cardiomyocytes promotes proliferation and activation of cardiac fibroblasts. 38 Of note, FST (follistatin), an another activin antagonist, was found increased in patients with PAH (Figure 1 and Table S4), stressing the importance of the signaling pathway in PAH pathogenesis. Given that addition of sotatercept, an activin signaling inhibitor, to existing therapy showed promise for the treatment of PAH, 39 the clinical significance of circulating FST/FSTL3 in assessing response to therapy will be of great interest.
In addition to validating or identifying promising circulating factors that may serve as prognostic biomarkers in PAH, our work also pinpointed alterations of several plasma proteins that may be involved in PAH pathogenesis. For instance, OSM (oncostatin‐MOSM), a member of the interleukin‐6 family cytokines, was found significantly increased in PAH. Although not explored in the setting of PAH, compelling data documenting that OSM stimulates vascular smooth muscle cell proliferation, 40 induces pulmonary inflammation and fibrosis in mice, 41 and promotes progression of heart failure when continuous activated 42 , 43 suggest that increased circulating levels could simultaneously contribute to adverse pulmonary and RV remodeling in PAH. In addition, elevated circulating levels of the serine protease GZMB (granzyme B) were detected in patients with PAH. Because GZMB is essentially stored by immune cells and initiates multiple cell death pathways, it can be envisaged that increased expression of GZMB triggers adverse ventricular remodeling by promoting cardiomyocyte apoptosis and associated interstitial fibrosis. 44
Several limitations should be acknowledged. First, although our work includes a substantial number of patients, the single‐center nature is an inherent limitation of our study. However, the fact that some circulating proteins identified by our study are already documented to provide a prognostic utility in the context of PAH increases the credibility of our data. Another limitation lies in the fact that uncertainties remain regarding the tissue origin of these circulating biomarkers (pulmonary, cardiac, or both). In addition, the absence of multiple testing adjustments for Cox proportional hazard analyses may have resulted in type I error. The findings of this study should be interpreted as exploratory. Finally, the evolution of these biomarkers in response to therapies is lacking, a point that needs to be addressed in a future study.
CONCLUSIONS
In conclusion, the present study using high‐throughput proteomic technologies enabled us to identify potential novel immune‐related plasma biomarkers, which may help improve outcome prediction in PAH if externally confirmed in future studies.
Sources of Funding
This work was supported by the Canadian Institutes of Health Research grant to Steeve Provencher (CIHR, #IC138603), as well as an unrestricted grant from the Fondation Institut Universitaire de Cardiologie et Pneumologie de Québec. Olivier Boucherat holds a junior scholar award and Sébastien Bonnetholds a distinguished research scholar award from the Fonds de Recherche du Québec: Santé.
Disclosures
None.
Supporting information
Tables S1–S6
Figures S1–S2
Acknowledgments
We thank the Institut Universitaire de Cardiologie et Pneumologie de Québec Biobank of the Quebec Respiratory Health Research Network as well as the department of cytology and pathology from the institute for providing access to tissue and clinical data.
This article was sent to Yen‐Hung Lin, MD, PhD, Associate Editor, for review by expert referees, editorial decision, and final disposition.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.123.032888
For Sources of Funding and Disclosures, see page 13.
References
- 1. Hoeper MM, Humbert M, Souza R, Idrees M, Kawut SM, Sliwa‐Hahnle K, Jing ZC, Gibbs JS. A global view of pulmonary hypertension. Lancet Respir Med. 2016;4:306–322. doi: 10.1016/S2213-2600(15)00543-3 [DOI] [PubMed] [Google Scholar]
- 2. Lajoie AC, Lauziere G, Lega JC, Lacasse Y, Martin S, Simard S, Bonnet S, Provencher S. Combination therapy versus monotherapy for pulmonary arterial hypertension: a meta‐analysis. Lancet Respir Med. 2016;4:291–305. doi: 10.1016/s2213-2600(16)00027-8 [DOI] [PubMed] [Google Scholar]
- 3. Benza RL, Gomberg‐Maitland M, Miller DP, Frost A, Frantz RP, Foreman AJ, Badesch DB, McGoon MD. The REVEAL registry risk score calculator in patients newly diagnosed with pulmonary arterial hypertension. Chest. 2012;141:354–362. doi: 10.1378/chest.11-0676 [DOI] [PubMed] [Google Scholar]
- 4. Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J. 2015;46:903–975. doi: 10.1183/13993003.01032-2015 [DOI] [PubMed] [Google Scholar]
- 5. Hoeper MM, Pausch C, Olsson KM, Huscher D, Pittrow D, Grunig E, Staehler G, Vizza CD, Gall H, Distler O, et al. COMPERA 2.0: a refined four‐stratum risk assessment model for pulmonary arterial hypertension. Eur Respir J. 2022;60:2102311. doi: 10.1183/13993003.02311-2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boucly A, Weatherald J, Savale L, de Groote P, Cottin V, Prevot G, Chaouat A, Picard F, Horeau‐Langlard D, Bourdin A, et al. External validation of a refined four‐stratum risk assessment score from the French pulmonary hypertension registry. Eur Respir J. 2022;59:2102419. doi: 10.1183/13993003.02419-2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Leopold JA, Kawut SM, Aldred MA, Archer SL, Benza RL, Bristow MR, Brittain EL, Chesler N, DeMan FS, Erzurum SC, et al. Diagnosis and treatment of right heart failure in pulmonary vascular diseases: a National Heart, Lung, and Blood Institute workshop. Circ Heart Fail. 2021;14:e007975. doi: 10.1161/CIRCHEARTFAILURE.120.007975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lahm T, Douglas IS, Archer SL, Bogaard HJ, Chesler NC, Haddad F, Hemnes AR, Kawut SM, Kline JA, Kolb TM, et al. Assessment of right ventricular function in the research setting: knowledge gaps and pathways forward. An official American Thoracic Society research statement. Am J Respir Crit Care Med. 2018;198:e15–e43. doi: 10.1164/rccm.201806-1160ST [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Perros F, Humbert M, Dorfmuller P. Smouldering fire or conflagration? An illustrated update on the concept of inflammation in pulmonary arterial hypertension. Eur Respir Rev. 2021;30:30. doi: 10.1183/16000617.0161-2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sydykov A, Mamazhakypov A, Petrovic A, Kosanovic D, Sarybaev AS, Weissmann N, Ghofrani HA, Schermuly RT. Inflammatory mediators drive adverse right ventricular remodeling and dysfunction and serve as potential biomarkers. Front Physiol. 2018;9:609. doi: 10.3389/fphys.2018.00609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Frost A, Badesch D, Gibbs JSR, Gopalan D, Khanna D, Manes A, Oudiz R, Satoh T, Torres F, Torbicki A. Diagnosis of pulmonary hypertension. Eur Respir J. 2019;53:53. doi: 10.1183/13993003.01904-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Levey AS, Coresh J, Greene T, Stevens LA, Zhang YL, Hendriksen S, Kusek JW, Van Lente F; Chronic Kidney Disease Epidemiology C . Using standardized serum creatinine values in the modification of diet in renal disease study equation for estimating glomerular filtration rate. Ann Intern Med. 2006;145:247–254. doi: 10.7326/0003-4819-145-4-200608150-00004 [DOI] [PubMed] [Google Scholar]
- 13. von Elm E, Altman DG, Egger M, Pocock SJ, Gotzsche PC, Vandenbroucke JP, Initiative S. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Int J Surg. 2014;12:1495–1499. doi: 10.1016/j.ijsu.2014.07.013 [DOI] [PubMed] [Google Scholar]
- 14. Shimauchi T, Boucherat O, Yokokawa T, Grobs Y, Wu W, Orcholski M, Martineau S, Omura J, Tremblay E, Shimauchi K, et al. PARP1‐PKM2 Axis mediates right ventricular failure associated with pulmonary arterial hypertension. JACC Basic Transl Sci. 2022;7:384–403. doi: 10.1016/j.jacbts.2022.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boucherat O, Yokokawa T, Krishna V, Kalyana‐Sundaram S, Martineau S, Breuils‐Bonnet S, Azhar N, Bonilla F, Gutstein D, Potus F, et al. Identification of LTBP‐2 as a plasma biomarker for right ventricular dysfunction in human pulmonary arterial hypertension. Nature Cardiovascular Research. 2022;1:748–760. doi: 10.1038/s44161-022-00113-w [DOI] [Google Scholar]
- 16. DeLong ER, DeLong DM, Clarke‐Pearson DL. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988;44:837–845. doi: 10.2307/2531595 [DOI] [PubMed] [Google Scholar]
- 17. Schisterman EF, Perkins NJ, Liu A, Bondell H. Optimal cut‐point and its corresponding Youden index to discriminate individuals using pooled blood samples. Epidemiology. 2005;16:73–81. doi: 10.1097/01.ede.0000147512.81966.ba [DOI] [PubMed] [Google Scholar]
- 18. Khassafi F, Chelladurai P, Valasarajan C, Nayakanti SR, Martineau S, Sommer N, Yokokawa T, Boucherat O, Kamal A, Kiely DG, et al. Transcriptional profiling unveils molecular subgroups of adaptive and maladaptive right ventricular remodeling in pulmonary hypertension. Nat Cardiovasc Res. 2023;2:917–936. doi: 10.1038/s44161-023-00338-3 [DOI] [Google Scholar]
- 19. Rhodes CJ, Wharton J, Ghataorhe P, Watson G, Girerd B, Howard LS, Gibbs JSR, Condliffe R, Elliot CA, Kiely DG, et al. Plasma proteome analysis in patients with pulmonary arterial hypertension: an observational cohort study. Lancet Respir Med. 2017;5:717–726. doi: 10.1016/S2213-2600(17)30161-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Amsallem M, Sweatt AJ, Arthur Ataam J, Guihaire J, Lecerf F, Lambert M, Ghigna MR, Ali MK, Mao Y, Fadel E, et al. Targeted proteomics of right heart adaptation to pulmonary arterial hypertension. Eur Respir J. 2021;57:57. doi: 10.1183/13993003.02428-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wilkinson L, Kolle G, Wen D, Piper M, Scott J, Little M. CRIM1 regulates the rate of processing and delivery of bone morphogenetic proteins to the cell surface. J Biol Chem. 2003;278:34181–34188. doi: 10.1074/jbc.M301247200 [DOI] [PubMed] [Google Scholar]
- 22. Pennisi DJ, Wilkinson L, Kolle G, Sohaskey ML, Gillinder K, Piper MJ, McAvoy JW, Lovicu FJ, Little MH. Crim1KST264/KST264 mice display a disruption of the Crim1 gene resulting in perinatal lethality with defects in multiple organ systems. Dev Dyn. 2007;236:502–511. doi: 10.1002/dvdy.21015 [DOI] [PubMed] [Google Scholar]
- 23. Zhang Y, Fan J, Ho JW, Hu T, Kneeland SC, Fan X, Xi Q, Sellarole MA, de Vries WN, Lu W, et al. Crim1 regulates integrin signaling in murine lens development. Development. 2016;143:356–366. doi: 10.1242/dev.125591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Iyer S, Chou FY, Wang R, Chiu HS, Raju VK, Little MH, Thomas WG, Piper M, Pennisi DJ. Crim1 has cell‐autonomous and paracrine roles during embryonic heart development. Sci Rep. 2016;6:19832. doi: 10.1038/srep19832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eleuteri E, Di Stefano A, Vallese D, Gnemmi I, Pitruzzella A, Tarro Genta F, Delle Donne L, Cappello F, Ricciardolo FL, Giannuzzi P. Fibrosis markers and CRIM1 increase in chronic heart failure of increasing severity. Biomarkers. 2014;19:214–221. doi: 10.3109/1354750X.2014.896946 [DOI] [PubMed] [Google Scholar]
- 26. Miyamoto K, Morishita Y, Yamazaki M, Minamino N, Kangawa K, Matsuo H, Mizutani T, Yamada K, Minegishi T. Isolation and characterization of vascular smooth muscle cell growth promoting factor from bovine ovarian follicular fluid and its cDNA cloning from bovine and human ovary. Arch Biochem Biophys. 2001;390:93–100. doi: 10.1006/abbi.2001.2367 [DOI] [PubMed] [Google Scholar]
- 27. Terai Y, Abe M, Miyamoto K, Koike M, Yamasaki M, Ueda M, Ueki M, Sato Y. Vascular smooth muscle cell growth‐promoting factor/F‐spondin inhibits angiogenesis via the blockade of integrin alphavbeta3 on vascular endothelial cells. J Cell Physiol. 2001;188:394–402. doi: 10.1002/jcp.1122 [DOI] [PubMed] [Google Scholar]
- 28. Mahmood N, Mihalcioiu C, Rabbani SA. Multifaceted role of the Urokinase‐type plasminogen activator (uPA) and its receptor (uPAR): diagnostic, prognostic, and therapeutic applications. Front Oncol. 2018;8:24. doi: 10.3389/fonc.2018.00024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Koller L, Stojkovic S, Richter B, Sulzgruber P, Potolidis C, Liebhart F, Mortl D, Berger R, Goliasch G, Wojta J, et al. Soluble Urokinase‐type plasminogen activator receptor improves risk prediction in patients with chronic heart failure. JACC Heart Fail. 2017;5:268–277. doi: 10.1016/j.jchf.2016.12.008 [DOI] [PubMed] [Google Scholar]
- 30. Meijers B, Poesen R, Claes K, Dietrich R, Bammens B, Sprangers B, Naesens M, Storr M, Kuypers D, Evenepoel P. Soluble urokinase receptor is a biomarker of cardiovascular disease in chronic kidney disease. Kidney Int. 2015;87:210–216. doi: 10.1038/ki.2014.197 [DOI] [PubMed] [Google Scholar]
- 31. Guignabert C, Savale L, Boucly A, Thuillet R, Tu L, Ottaviani M, Rhodes CJ, De Groote P, Prevot G, Bergot E, et al. Serum and pulmonary expression profiles of the Activin signaling system in pulmonary arterial hypertension. Circulation. 2023;147:1809–1822. doi: 10.1161/CIRCULATIONAHA.122.061501 [DOI] [PubMed] [Google Scholar]
- 32. Mickael C, Kheyfets VO, Langouët‐Astrié C, Lee MH, Sanders LA, Trentin CO, Sweatt AJ, Zamanian RT, Bull TM, Stenmark K, et al. Peripheral blood inflammation profile of patients with pulmonary arterial hypertension using the high‐throughput Olink proteomics platform. Am J Respir Cell Mol Biol. 2022;66:581–583. doi: 10.1165/rcmb.2021-0369LE [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sweatt AJ, Hedlin HK, Balasubramanian V, Hsi A, Blum LK, Robinson WH, Haddad F, Hickey PM, Condliffe R, Lawrie A, et al. Discovery of distinct immune phenotypes using machine learning in pulmonary arterial hypertension. Circ Res. 2019;124:904–919. doi: 10.1161/Circresaha.118.313911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bartholin L, Maguer‐Satta V, Hayette S, Martel S, Gadoux M, Corbo L, Magaud JP, Rimokh R. Transcription activation of FLRG and follistatin by activin a, through Smad proteins, participates in a negative feedback loop to modulate activin a function. Oncogene. 2002;21:2227–2235. doi: 10.1038/sj.onc.1205294 [DOI] [PubMed] [Google Scholar]
- 35. Roh JD, Hobson R, Chaudhari V, Quintero P, Yeri A, Benson M, Xiao C, Zlotoff D, Bezzerides V, Houstis N, et al. Activin type II receptor signaling in cardiac aging and heart failure. Sci Transl Med. 2019;11:11. doi: 10.1126/scitranslmed.aau8680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Oshima Y, Ouchi N, Shimano M, Pimentel DR, Papanicolaou KN, Panse KD, Tsuchida K, Lara‐Pezzi E, Lee SJ, Walsh K. Activin a and follistatin‐like 3 determine the susceptibility of heart to ischemic injury. Circulation. 2009;120:1606–1615. doi: 10.1161/CIRCULATIONAHA.109.872200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. MacDonnell S, Megna J, Ruan Q, Zhu O, Halasz G, Jasewicz D, Powers K, Del Pilar Molina‐Portela M, Jin X, Zhang D. Activin a directly impairs human cardiomyocyte contractile function indicating a potential role in heart failure development. Front Cardiovasc Med. 2022;9:1038114. doi: 10.3389/fcvm.2022.1038114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Panse KD, Felkin LE, Lopez‐Olaneta MM, Gomez‐Salinero J, Villalba M, Munoz L, Nakamura K, Shimano M, Walsh K, Barton PJ, et al. Follistatin‐like 3 mediates paracrine fibroblast activation by cardiomyocytes. J Cardiovasc Transl Res. 2012;5:814–826. doi: 10.1007/s12265-012-9400-9 [DOI] [PubMed] [Google Scholar]
- 39. Hoeper MM, Badesch DB, Ghofrani HA, Gibbs JSR, Gomberg‐Maitland M, McLaughlin VV, Preston IR, Souza R, Waxman AB, Grunig E, et al. Phase 3 trial of Sotatercept for treatment of pulmonary arterial hypertension. N Engl J Med. 2023;388:1478–1490. doi: 10.1056/NEJMoa2213558 [DOI] [PubMed] [Google Scholar]
- 40. Grove RI, Eberhardt C, Abid S, Mazzucco C, Liu J, Kiener P, Todaro G, Shoyab M. Oncostatin M is a mitogen for rabbit vascular smooth muscle cells. Proc Natl Acad Sci USA. 1993;90:823–827. doi: 10.1073/pnas.90.3.823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mozaffarian A, Brewer AW, Trueblood ES, Luzina IG, Todd NW, Atamas SP, Arnett HA. Mechanisms of oncostatin M‐induced pulmonary inflammation and fibrosis. J Immunol. 2008;181:7243–7253. doi: 10.4049/jimmunol.181.10.7243 [DOI] [PubMed] [Google Scholar]
- 42. Poling J, Gajawada P, Richter M, Lorchner H, Polyakova V, Kostin S, Shin J, Boettger T, Walther T, Rees W, et al. Therapeutic targeting of the oncostatin M receptor‐beta prevents inflammatory heart failure. Basic Res Cardiol. 2014;109:396. doi: 10.1007/s00395-013-0396-3 [DOI] [PubMed] [Google Scholar]
- 43. Kubin T, Poling J, Kostin S, Gajawada P, Hein S, Rees W, Wietelmann A, Tanaka M, Lorchner H, Schimanski S, et al. Oncostatin M is a major mediator of cardiomyocyte dedifferentiation and remodeling. Cell Stem Cell. 2011;9:420–432. doi: 10.1016/j.stem.2011.08.013 [DOI] [PubMed] [Google Scholar]
- 44. Santos‐Zas I, Lemarie J, Zlatanova I, Cachanado M, Seghezzi JC, Benamer H, Goube P, Vandestienne M, Cohen R, Ezzo M, et al. Cytotoxic CD8(+) T cells promote granzyme B‐dependent adverse post‐ischemic cardiac remodeling. Nat Commun. 2021;12:1483. doi: 10.1038/s41467-021-21737-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S6
Figures S1–S2