

Abstract
At one time considered a possible form of neuromyelitis optica (NMO) spectrum disorder (NMOSD), it is now accepted that myelin oligodendrocyte glycoprotein (MOG) antibody (Ab)–associated disorder (MOGAD) is a distinct entity from either NMO or multiple sclerosis (MS) and represents a broad spectrum of clinical phenotypes. Whereas Abs targeting aquaporin-4 (AQP4) in NMO are pathogenic, the extent that anti-MOG Abs contribute to CNS damage in MOGAD is unclear. Both AQP4-specific Abs in NMO and MOG-specific Abs in MOGAD are predominantly IgG1, a T cell-dependent immunoglobulin (Ig) subclass. Key insights in neuroimmunology and MOGAD pathogenesis have been learned from MOG experimental autoimmune encephalomyelitis (EAE), described 2 decades before the term MOGAD was introduced. MOG-specific T cells are required in MOG EAE, and while anti-MOG Abs can exacerbate EAE and CNS demyelination, those Abs are neither necessary nor sufficient to cause EAE. Knowledge regarding the spectrum of MOGAD clinical and radiologic presentations is advancing rapidly, yet our grasp of MOGAD pathogenesis is incomplete. Understanding both the humoral and cellular immunology of MOGAD has implications for diagnosis, treatment, and prognosis.
Introduction
Historically, 2 opposite paths led to the identification of myelin oligodendrocyte glycoprotein (MOG) and aquaporin-4 (AQP4) as autoantigens in human CNS neuroinflammatory syndromes. The phenotype of neuromyelitis optica (NMO) was well recognized before the discovery of its target autoantigen, AQP4.1 By contrast, MOG was a target in search of a human disease.2 Initial studies of MOG focused on its role as an autoantigen in experimental autoimmune encephalomyelitis (EAE) and possibly in MS.3,4 Antibodies to MOG found in patients with clinically isolated syndrome were once thought to predict the development of clinically definite MS.5 However, MOG antibody assays were unreliable at that time. Subsequent improvement in MOG IgG testing by cell-based assay (CBA) along with the insight to search for these antibodies in neuroinflammatory syndromes besides MS led to the identification of MOG-specific antibodies in a range of CNS inflammatory conditions, now consolidated by the term MOG antibody–associated disorder (MOGAD).6,7
MOGAD is a difficult disease to tackle. First, it encompasses an expanding spectrum of phenotypes that now includes acute disseminated encephalomyelitis (ADEM), optic neuritis, transverse myelitis, cortical encephalitis, cerebral lesions, and brainstem or cerebellar syndromes.7-9 Additional heterogeneity exists with respect to age at disease onset and course, with a proportion of patients exhibiting a relapsing course.10 Such pronounced heterogeneity in MOGAD affects our ability to test and advance disease-modifying therapies.
What Is the Role of MOG-Specific Antibodies in MOGAD?
By definition, the diagnosis of MOGAD requires detection of IgG Abs that bind exposed conformational epitopes of MOG, commonly measured by CBA.6,11,12 Antigen-specific IgG can facilitate complement deposition, antibody-dependent cell-mediated cytotoxicity, or antibody-dependent cellular phagocytosis. Antibodies from patients with MOGAD can induce these effector functions when exposed in vitro to cells expressing full-length MOG.13 MOG is exposed on the outer oligodendrocyte surface, possibly permitting antibody exposure to this target antigen.14 However, current data supporting the pathogenicity of anti-MOG antibodies in vivo is indirect: IgG staining is present in some MOGAD lesions, MOG IgG can be detected in the CSF of many patients with MOGAD, and MOG antibody titers are often elevated around the time of an attack.15-17 It is possible that MOG IgG contributes secondarily to disease, or in some cases merely serves as a marker of ongoing inflammatory pathology.18 For example, low titers of MOG IgG have been detected in individuals with other neurologic syndromes, questioning their relevance in those conditions.19 Further, MOG seropositivity is not always stable, and relapses may occur after seroreversion.20 Some patients with MOGAD experience rapid improvement after corticosteroid treatment for acute flares, an effect unlikely mediated by potential changes in MOG-specific antibody levels. Mechanistic investigations evaluating pathogenicity of MOG-specific IgG in MOGAD using animal models are limited due to species specificity of most human MOG-reactive antibodies.21 Notably, MOG-specific IgGs that cross-react with rodent or marmoset MOG enhance disease in recipient animals if cell-mediated CNS inflammation is established beforehand, but those MOG-specific Abs do not cause disease when administered alone.21,22
In contrast to MOG antibodies, AQP4 IgG clearly has a direct role in NMO pathogenesis (Figure 1).25 AQP4 is expressed in multiple tissues, yet AQP4 orthogonal arrays of particles (OAPs), the target of pathogenic AQP4-specific Abs in NMO, are expressed primarily on astrocyte end-foot processes, and NMO lesions occur where the density of OAPs is highest.23,24 AQP4 OAPs provide a lattice-like platform for IgG clustering and subsequent complement deposition.24 Thus, NMO may be considered a “humoral OAP autoimmune disease.”26 Although individual MOG isoforms expressed in vitro influence the binding of MOG-specific IgG, it is not clear how the structure, number, or density of MOG molecules on oligodendrocytes in vivo promotes MOG IgG binding and complement deposition in MOGAD.12 Complement staining is not consistently identified in MOGAD lesions, whereas complement deposition is a hallmark feature of NMO lesions.15,27 Complement inhibition is beneficial in NMO, yet the extent that pathology in MOGAD is complement-dependent is unresolved.
Figure 1. Models Illustrating How Antibodies and Complement May Participate in NMO and MOGAD.

AQP4, a transmembrane (TM) protein, is expressed in the astrocytes, kidneys, muscle, and lung, yet the AQP4-specific IgG1 in NMO damages astrocytes primarily.18,23 AQP4 is expressed abundantly in astrocyte end-foot processes at the blood-brain barrier (BBB) (A), where tetramers of AQP4 (M23:M23 and M23:M21 isoforms) assemble into orthogonal arrays of particles (OAPs)23 (B), which provides a lattice-like platform that permits monovalent binding of AQP4-specific IgG1 in a distribution that is optimal for recruitment of C1q (C) and initiation of the classical complement cascade that results in cytotoxicity to astrocytes from formation of the membrane attack complex (MAC).24 (D–G) MOG, also a TM protein, is exposed on the outer oligodendrocyte surface. Unlike AQP4-specific IgG1 in NMO, the role of anti-MOG antibodies in CNS damage and the potential mechanisms (e.g., complement-dependent cytotoxicity [CDC], antibody-dependent cellular cytotoxicity [ADCC], or antibody-dependent cellular phagocytosis [ADCP]) employed by MOG-specific IgG1 in MOGAD are less clear. Complement deposition is identified inconsistently in MOGAD lesions.15 Unlike AQP4-specific IgG1 from patients with NMO, MOG-specific IgG1 from patients with MOGAD may preferentially bind MOG in a bivalent manner that is less efficient for recruitment of C1q and activation of CDC12 but may promote ADCC or ADCP.13 Natural killer cell (NK); myeloid APC (M). Copyright Xavier Studio, reprinted with permission.
Animal models can provide invaluable insight into pathophysiologic mechanisms of human diseases. For instance, the discovery that myelin-specific T-cell clones can cause relapsing paralysis and CNS demyelination in mice represented the first demonstration that individual self-antigen–specific T-cell clones can cause autoimmune disease.28 These findings underscored the importance of clonal expansion and the requirement for self-antigen recognition in autoimmunity.28 Some approved MS disease-modifying therapies were successfully developed through testing in EAE.29,30 Yet, testing candidate medications in animal models of disease is not an obligate requirement. Indeed, the 3 currently approved medications for NMO were advanced largely without leveraging an established NMO animal model.31-33 MOG is different. MOG EAE shares key clinical and pathologic features with MOGAD. MOG EAE is characterized by myelitis, optic neuritis, and encephalitis.34,35 MOG EAE has informed how T and B cells can contribute to MOG CNS autoimmunity and may serve as an ideal model for many immunologic features of MOGAD.
MOG-Specific T Cells, a Critical Link in MOG Autoimmunity
There are distinct immunopathogenic variants of MOG EAE (Figure 2). One model, which is induced by immunization with MOG peptide, requires MOG-specific CD4+ T cells and is B-cell-independent.37 We refer to this model as T-dependent MOG EAE. CD4+ T cells, which are the predominant T-cell subset in MOGAD lesions,27 recognize 9–14 amino acid peptide fragments of antigen in association with self-MHC II molecules expressed on antigen-presenting cells (APCs).41 Mature MOG protein contains 218 amino acids,14 so how is MOG recognized by T cells? APCs are capable of processing protein antigens into peptide fragments that can be presented on MHC II molecules and recognized by antigen-specific T cells (Figure 2). Because many peripheral APCs express MHC II constitutively, immunization with encephalitogenic MOG peptides bypasses the need for peripheral antigen processing.34 However, MOG-specific T cells that enter the CNS encounter endogenous oligodendrocyte-derived MOG protein that must be endocytosed, processed, and presented by APCs in situ for recognition by MOG-specific T cells.34,38
Figure 2. MHC II Endocytic Antigen Processing and Presentation to T Cells in MOG CNS Autoimmunity.

This figure illustrates types of APCs, including myeloid cells (A), B cells (B), and microglia (C) that may participate in MOG CNS autoimmunity. MOG EAE is typically induced by subcutaneous immunization with either MOG protein or MOG peptide (pMOG), which leads to peripheral (outside CNS) activation of pathogenic MOG-specific T cells that traffic into the CNS and initiate CNS inflammation.36 Human MOG protein, which contains proline-42, causes T-B-dependent EAE.36,37 MOG-specific T-cell activation requires recognition of pMOG in association with MHC II molecules expressed on APCs.34 Antigen processing of MOG protein through the MHC II endocytic pathway is required in at least at 2 stages in MOG protein-induced EAE, for initial recognition by MOG-specific T cells in the periphery and for reactivation within the CNS (in situ).34,38 Several molecules, including invariant chain (Ii, CD74), HLA-DM (H-2M) (DM), and proteases participate in MHC II maturation and in orchestrating steps within the endocytic pathway.39 MHC II (α/β) molecules associate with Ii in the endoplasmic reticulum (ER) forming a trimer (Ii-MHC II) and travel through the Golgi to the endosomal compartment. Ii is enzymatically degraded yielding a fragment of Ii, class II Ii peptide (CLIP), which remains bound within the MHC II peptide-binding groove. The MHC II chaperone DM facilitates removal of CLIP, permitting exchange for antigenic peptide (e.g., pMOG). Peripheral myeloid cells capture native antigenic proteins (MOG) (top of A) via phagocytosis or pinocytosis and deliver them to the endolysosomal compartment where they are degraded by proteases into 9–14 amino acid fragments that can bind the MHC II peptide-binding groove.39 Endosomes containing peptide-MHC II complexes fuse with the plasma membrane permitting presentation of peptides (e.g., pMOG-MHC II) to encephalitogenic MOG-specific T cells. In contrast to MOG protein, pMOG immunization (lower left in A) supplies peptide that can bind cell surface MHC II molecules directly, bypassing the need for endocytic processing.34 B cells are exceptionally efficient APCs when they bind antigens (e.g., MOG protein) (top of B) with their B-cell antigen-specific receptors (anti-MOG-BCR) and deliver them to the endolysosomal compartment for processing and association with MHC II molecules.39,40 Independent of whether MOG EAE is induced by immunization with MOG protein, pMOG, or by adoptive transfer of encephalitogenic MOG-specific T cells (not shown), endocytic Ag processing by APCs (e.g., resident microglia) (shown in C) in situ is required for recognition of cognate MOG peptide by MOG-specific T cells that initiate CNS inflammation.34,38 Copyright Xavier Studio, reprinted with permission.
A second EAE model, induced by immunization with human MOG protein, requires both CD4+ T cells and B cells.36,37 We refer to this model as T-B-dependent MOG EAE. Besides serving as the source of MOG antibody–secreting plasma cells, B cells can function as APCs. Like other professional APCs, B cells express MHC II constitutively.36,40 They also express their B-cell antigen-specific receptor (BCR). In 1985, Lanzavecchia taught us that when engaging specific antigen via their BCR, these B cells can process and present antigen to T cells that recognize the same antigen >10,000 fold more efficiently than other B-cell APCs.40 Similarly, MOG-specific B cells are more efficient than other B cells when presenting MOG to MOG-specific T cells.35,42 B cells can also produce cytokines that either promote or protect against CNS autoimmunity. For example, B-cell IL-6 production promotes expansion of Th17 cells and exacerbates MOG protein EAE.36 It is difficult to distinguish the relative contribution of each of these B-cell functions in MOGAD, a condition defined by the detection of MOG-specific antibody. Indeed, one may question whether MOG-specific IgG represents the tip of the iceberg in MOGAD pathogenesis (Figure 3). MOG protein EAE has provided the key insights. B-cell MHC II expression is a requirement for activation of pathogenic MOG-specific T cells and development of EAE when induced by MOG protein.36 Further, mice containing B cells that express membrane MOG-specific BCR, but are incapable of secreting MOG-specific antibodies, are susceptible to MOG protein EAE.36 Collectively, these results underscore the contribution of B-cell APC function in MOG protein EAE.
Figure 3. Cellular Immunity May Have a Prominent Role in MOGAD Pathogenesis.
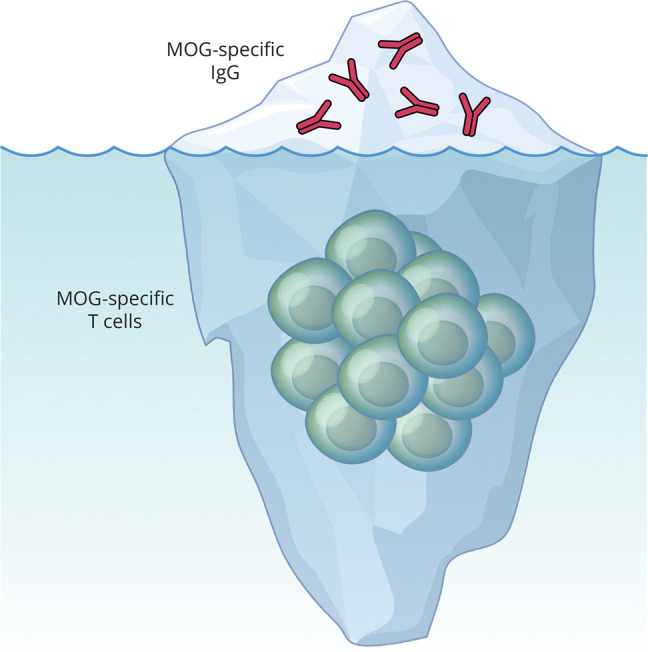
MOG IgG (above water surface) is necessary for diagnosis of MOGAD, yet cellular immunity (beneath water surface) is required in models of MOG CNS autoimmunity. MOG-specific antibodies in MOGAD and MOG EAE are IgG, a T-cell-dependent Ig subclass. CD4+ T cells predominate in MOGAD lesions27 and in EAE lesions. Other cells (e.g., resident microglia, infiltrating myeloid cells, fewer CD8+ T cells, and B cells) also contribute to CNS inflammation (not depicted). In general, MOG-specific antibodies do not induce disease in the absence of inflammation.43 MOG-specific T cells can induce EAE in the absence of B cells or antibodies.36,37 Collectively, such findings suggest that seronegative MOG-targeted autoimmune disease (“sMOG”) may exist in some individuals who manifest clinical symptoms associated with MOGAD in the absence of detectable MOG-specific antibodies. Graphics created using BioRender.com. Copyright Xavier Studio, reprinted with permission.
The pivotal role of MOG-specific T cells is also highlighted in spontaneous MOG EAE models.35,36 MOG-specific T-cell receptor (TCR) transgenic mice develop spontaneous optic neuritis.44 Although MOG-specific BCR knock-in mice secrete high levels of MOG-specific antibodies, they do not develop EAE spontaneously.43 Further, mice that contain both MOG-specific T cells and MOG-specific B cells, but do not secrete any antibodies, develop spontaneous optic neuritis and myelitis.36 Although anti-MOG Abs are not required, they can potentiate severity of this “opticospinal” EAE.
What determinants are targeted by pathogenic MOG-specific T cells in EAE and MOGAD? Most studies of MOG in mice and in humans have focused upon recognition of its N-terminal extracellular 1-117 Ig-like domain. MOG 35-55, which causes EAE in C57BL/6, PL/J, and NOD mice, is recognized in association with MHC II I-Ab, I-Au, and I-Ag7, respectively, whereas MOG 92-106 causes EAE in SJL/J mice and is recognized in association with I-As molecules.14,45 Thus, MOG T-cell epitopes can be recognized in association with multiple MHC II molecules. It is logical to focus on the extracellular domain for recognition by Abs because it contains contact residues of conformational MOG exposed on the outer surface of myelin.46 However, the extracellular Ig domain only accounts for roughly one-half of the MOG protein, and T cells, unlike most Abs, can recognize processed epitopes from the extracellular, transmembrane, or intracellular domains. Although MOG 35-55 contains the immunodominant determinant in the extracellular domain in C57BL/6 mice, the transmembrane region 119–132 contains the immunodominant pathogenic epitope of full-length MOG, highlighting the need to study T recognition of full-length MOG in MOGAD.47 While certain MOG-specific T-cell determinants have been identified in healthy controls and persons with MS, MOG-specific T-cell epitope(s) in MOGAD have not yet been identified. Organ-specific autoimmune diseases are often associated with HLA risk alleles. For example, NMO is associated with HLA DRB1*03:01.18 No consistent HLA association has yet been defined in MOGAD, although currently published studies have been relatively small.48,49 It is conceivable that phenotypic heterogeneity or the presence of multiple HLA allelic associations could contribute to the lack of a specific HLA association in early MOGAD studies. It may be important to stratify the MOGAD phenotypes in larger studies that evaluate potential HLA associations. Characterization of MOG-specific T-cell epitopes in patients may also help identify candidate HLA associations in MOGAD.
Does MOGAD represent the true prevalence of MOG-targeted CNS inflammatory disease? This important question is not answered easily. While it is known that pathogenic Abs recognize conformational determinants of MOG, not all MOG Abs that recognize conformational MOG determinants are pathogenic.50 Further, low-titer MOG-specific Abs that may be detected in other CNS conditions are not necessarily pathogenic.19 Collectively, those findings raise concern that the diagnosis of MOGAD may overestimate the true prevalence of pathogenic MOG-targeted CNS inflammatory disease. Conversely, MOGAD could underestimate the prevalence of MOG-targeted autoimmunity. Anti-MOG Abs do not persist in some individuals. Thus, due to such seroreversion, one may miss detection of anti-MOG Abs.20 As described, T-dependent MOG EAE occurs independently of pathogenic conformational MOG-specific Abs, although it may be associated with nonpathogenic Abs. Is there a human equivalent of T-dependent MOG EAE? Specifically, does seronegative MOG (“sMOG”)–targeted CNS autoimmunity exist? If so, our current diagnosis of MOGAD could conceivably underestimate true MOG-targeted CNS autoimmune disease, a possibility that may be relevant to some patients diagnosed with AQP4 and MOG seronegative (i.e. “double negative”) NMOSD.
What stimuli initiate MOGAD? It is well recognized that a febrile prodrome, often associated with a viral infection or vaccination (e.g., Tdap), can precede ADEM.51 It is estimated that 40% of MOGAD attacks follow infection by one of a variety of pathogens, including SARS-CoV-2, Epstein-Barr virus, and influenza.52 MOGAD onset has also been described after PD-1 checkpoint inhibition, which is known to worsen EAE.53 Rarely, cases of malignancy exist concurrently suggesting that a paraneoplastic etiology could also contribute to development of MOGAD.54 Whether these candidate triggers act by molecular mimicry, by causing CNS damage leading to immunogenic MOG exposure, or by bystander proinflammatory cytokine–mediated activation of MOG-specific lymphocytes is unknown (Figure 4). The normal immune repertoire contains T cells that recognize self-antigen, possibly at frequencies as high in healthy persons as those with a given autoimmune disease.56 A better understanding of the immunologic triggers should yield further insights into the pathogenesis of MOGAD and may identify predictors of monophasic or relapsing courses.
Figure 4. Potential Triggers of MOG Autoimmunity.

Several possible mechanisms may promote MOG CNS autoimmune disease. MOG autoimmunity could be triggered by molecular mimicry (left panel), a process that can occur when antigenic determinants of pathogens cross-react with self-antigens. Immunogenic MOG exposure (middle panel), either from CNS damage secondary from another disease,55 or ectopic MOG expression54 could promote activation of MOG-specific immune cells. The normal immune repertoire contains MOG-reactive T cells. Thus, bystander activation (right panel) of preexisting MOG-specific T cells by proinflammatory cytokines from an unrelated stimulus (e.g., systemic infection) could theoretically lead to proinflammatory polarization and expansion of MOG-specific T cells. Copyright Xavier Studio, reprinted with permission.
Currently, there are no approved therapies for MOGAD. Acute exacerbations are commonly treated with high-dose steroids along with additional plasma exchange or IV immunoglobulin (IVIg) when refractory to initial treatment. Different classes of potential disease-modifying therapeutics are being tested. Evidence suggests anti-CD20 B-cell depletion likely reduces relapses in a subset of patients with MOGAD but may be less efficacious than in MS or NMO.57 Given the heterogeneity in MOGAD, one questions whether B-cell depletion may prove effective only in a subset of patients, similar to the benefit of anti-CD20 B-cell depletion in T-B-dependent MOG EAE but not in T-dependent MOG EAE.42 Therapeutics that directly alter the level of circulating anti-MOG Ab are being evaluated in MOGAD. The neonatal Fc receptor (FcRn) prevents Ab degradation and can extend the half-life of pathogenic auto-Abs. Rozanolixizumab, a monoclonal Ab that blocks the interaction of Abs and FcRn, is currently being tested in MOGAD and may provide insight into the pathogenicity of anti-MOG Ab.58 IL-6, a pleiotropic cytokine that promotes Th17 differentiation, may be markedly elevated in the CSF of patients with MOGAD,59 and treatment with tocilizumab, an anti-IL-6R monoclonal Ab, has been associated with benefit in some severe MOGAD cases.60,61 Currently, the anti-IL-6R monoclonal Ab satralizumab, which has been approved for treatment of AQP4-seropositive NMO,33 is being tested in a phase III placebo-controlled trial in MOGAD.62 Although effective in NMO, it is thought that complement inhibitors may not be as beneficial in MOGAD, given the lack of clear complement-mediated pathology. The spectrum of MOGAD presentations and demographic differences in age, sex, and ethnicity that may influence risk of relapse underscore the challenges when designing prospective treatment trials and highlight the need to stratify patients appropriately.
Conclusion
Knowledge regarding clinical, pathologic, and imaging features of MOGAD is growing quickly. Clinical experience and improvements in MOG IgG assays have permitted creation of initial MOGAD diagnostic criteria. However, gaps exist in our understanding of the basic immunopathology of MOGAD.
Although animal models alone cannot replicate human disease, the investigators, who discovered MOG and advanced the understanding of MOG EAE, can be commended for having successfully introduced an invaluable animal model of MOGAD, paradoxically before the human disease was recognized.2,14 Studying MOGAD in parallel with MOG EAE will accelerate our understanding of MOGAD pathogenesis and possibly facilitate development of selective immunotherapy.
Acknowledgment
The authors thank Collin Spencer, Sharon Sagan, Patsi Nelson, and Kevin O'Connor for helpful discussions.
Glossary
- Ab
antibody
- ADEM
acute disseminated encephalomyelitis
- APCs
antigen-presenting cells
- AQP4
aquaporin-4
- CBA
cell-based assay
- EAE
experimental autoimmune encephalomyelitis
- FcRn
neonatal Fc receptor
- MOG
myelin oligodendrocyte glycoprotein
- MOGAD
MOG antibody–associated disorder
- NMO
neuromyelitis optica
- NMOSD
NMO spectrum disorder
- OAP
orthogonal arrays of particles
Appendix. Authors
| Name | Location | Contribution |
| Carson E. Moseley, MD, PhD | Department of Neurology, Weill Institute for Neurosciences, University of California, San Francisco | Drafting/revision of the manuscript for content, including medical writing for content; study concept or design |
| Akash Virupakshaiah, MD | Department of Neurology, Weill Institute for Neurosciences, University of California, San Francisco | Drafting/revision of the manuscript for content, including medical writing for content; study concept or design |
| Thomas G. Forsthuber, MD, PhD | Department of Molecular Microbiology and Immunology, University of Texas at San Antonio | Drafting/revision of the manuscript for content, including medical writing for content; study concept or design |
| Lawrence Steinman, MD | Department of Neurology and Neurological Science, Stanford University, CA | Drafting/revision of the manuscript for content, including medical writing for content; study concept or design |
| Emmanuelle Waubant, MD, PhD | Department of Neurology, Weill Institute for Neurosciences, University of California, San Francisco | Drafting/revision of the manuscript for content, including medical writing for content; study concept or design |
| Scott S. Zamvil, MD, PhD | Department of Neurology, Weill Institute for Neurosciences; Program in Immunology, University of California, San Francisco | Drafting/revision of the manuscript for content, including medical writing for content; study concept or design |
Study Funding
This investigation was supported by a Clinician Scientist Development Award Grant FAN-2107-38301 from the National Multiple Sclerosis Society (NMSS) (CEM), the C.D. Spangler Foundation (CEM), a Clinician-Scientist Investigator Award from the Weill Neurohub (CEM), NMSS Sylvia Lawry Fellowship Award FP-2307-41848 (AV), and by NIH grants 1 R01 AI131624-01A1 (SSZ) and 1 RO1 AI170863-01A1 (SSZ), and the Sumaira Foundation (SSZ).
Disclosure
The authors report no relevant disclosures. Go to Neurology.org/NN for full disclosures.
References
- 1.Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202(4):473-477. doi: 10.1084/jem.20050304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Linnington C, Webb M, Woodhams PL. A novel myelin-associated glycoprotein defined by a mouse monoclonal antibody. J Neuroimmunol. 1984;6:387-396. doi: 10.1016/0165-5728(84)90064-x [DOI] [PubMed] [Google Scholar]
- 3.Kerlero de Rosbo N, Mendel I, Ben-Nun A. Chronic relapsing experimental autoimmune encephalomyelitis with a delayed onset and an atypical clinical course, induced in PL/J mice by myelin oligodendrocyte glycoprotein (MOG)-derived peptide: preliminary analysis of MOG T cell epitopes. Eur J Immunol. 1995;25(4):985-993. doi: 10.1002/eji.1830250419 [DOI] [PubMed] [Google Scholar]
- 4.Kerlero de Rosbo N, Milo R, Lees MB, Burger D, Bernard CC, Ben-Nun A. Reactivity to myelin antigens in multiple sclerosis. Peripheral blood lymphocytes respond predominantly to myelin oligodendrocyte glycoprotein. J Clin Invest. 1993;92(6):2602-2608. doi: 10.1172/jci116875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berger T, Rubner P, Schautzer F, et al. Antimyelin antibodies as a predictor of clinically definite multiple sclerosis after a first demyelinating event. N Engl J Med. 2003;349(2):139-145. doi: 10.1056/nejmoa022328 [DOI] [PubMed] [Google Scholar]
- 6.Waters P, Woodhall M, O'Connor KC, et al. MOG cell-based assay detects non-MS patients with inflammatory neurologic disease. Neurol Neuroimmunol Neuroinflamm. 2015;2(3):e89. doi: 10.1212/nxi.0000000000000089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O'Connor KC, McLaughlin KA, De Jager PL, et al. Self-antigen tetramers discriminate between myelin autoantibodies to native or denatured protein. Nat Med. 2007;13(2):211-217. doi: 10.1038/nm1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sato DK, Callegaro D, Lana-Peixoto MA, et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology. 2014;82(6):474-481. doi: 10.1212/wnl.0000000000000101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Armangue T, Olive-Cirera G, Martinez-Hernandez E, et al. Associations of paediatric demyelinating and encephalitic syndromes with myelin oligodendrocyte glycoprotein antibodies: a multicentre observational study. Lancet Neurol. 2020;19(3):234-246. doi: 10.1016/s1474-4422(19)30488-0 [DOI] [PubMed] [Google Scholar]
- 10.Jurynczyk M, Messina S, Woodhall MR, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain. 2017;140(12):3128-3138. doi: 10.1093/brain/awx276 [DOI] [PubMed] [Google Scholar]
- 11.Banwell B, Bennett JL, Marignier R, et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: international MOGAD Panel proposed criteria. Lancet Neurol. 2023;22(3):268-282. doi: 10.1016/s1474-4422(22)00431-8 [DOI] [PubMed] [Google Scholar]
- 12.Macrini C, Gerhards R, Winklmeier S, et al. Features of MOG required for recognition by patients with MOG antibody-associated disorders. Brain. 2021;144(8):2375-2389. doi: 10.1093/brain/awab105 [DOI] [PubMed] [Google Scholar]
- 13.Yandamuri SS, Filipek B, Obaid AH, et al. MOGAD patient autoantibodies induce complement, phagocytosis, and cellular cytotoxicity. JCI Insight. 2023;8(11):e165373. doi: 10.1172/jci.insight.165373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bernard CC, Johns TG, Slavin A, et al. Myelin oligodendrocyte glycoprotein: a novel candidate autoantigen in multiple sclerosis. J Mol Med. 1997;75(2):77-88. doi: 10.1007/s001090050092 [DOI] [PubMed] [Google Scholar]
- 15.Takai Y, Misu T, Kaneko K, et al. Myelin oligodendrocyte glycoprotein antibody-associated disease: an immunopathological study. Brain. 2020;143(5):1431-1446. doi: 10.1093/brain/awaa102 [DOI] [PubMed] [Google Scholar]
- 16.Carta S, Cobo Calvo A, Armangue T, et al. Significance of myelin oligodendrocyte glycoprotein antibodies in CSF: a retrospective multicenter study. Neurology. 2023;100(11):e1095-e1108. doi: 10.1212/wnl.0000000000201662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lui A, Chong J, Flanagan E, et al. High titers of myelin oligodendrocyte glycoprotein antibody are only observed close to clinical events in pediatrics. Mult Scler Relat Disord. 2021;56:103253. doi: 10.1016/j.msard.2021.103253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zamvil SS, Slavin AJ. Does MOG Ig-positive AQP4-seronegative opticospinal inflammatory disease justify a diagnosis of NMO spectrum disorder?. Neurol Neuroimmunol Neuroinflamm. 2015;2(1):e62. doi: 10.1212/nxi.0000000000000062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sechi E, Buciuc M, Pittock SJ, et al. Positive predictive value of myelin oligodendrocyte glycoprotein autoantibody testing. JAMA Neurol. 2021;78(6):741-746. doi: 10.1001/jamaneurol.2021.0912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huda S, Whittam D, Jackson R, et al. Predictors of relapse in MOG antibody associated disease: a cohort study. BMJ Open. 2021;11:e055392. doi: 10.1136/bmjopen-2021-055392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spadaro M, Winklmeier S, Beltran E, et al. Pathogenicity of human antibodies against myelin oligodendrocyte glycoprotein. Ann Neurol. 2018;84(2):315-328. doi: 10.1002/ana.25291 [DOI] [PubMed] [Google Scholar]
- 22.Genain CP, Nguyen MH, Letvin NL, et al. Antibody facilitation of multiple sclerosis-like lesions in a nonhuman primate. J Clin Invest. 1995;96(6):2966-2974. doi: 10.1172/jci118368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verkman AS, Ratelade J, Rossi A, Zhang H, Tradtrantip L. Aquaporin-4: orthogonal array assembly, CNS functions, and role in neuromyelitis optica. Acta Pharmacol Sin. 2011;32(6):702-710. doi: 10.1038/aps.2011.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soltys J, Liu Y, Ritchie A, et al. Membrane assembly of aquaporin-4 autoantibodies regulates classical complement activation in neuromyelitis optica. J Clin Invest. 2019;129(5):2000-2013. doi: 10.1172/JCI122942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hillebrand S, Schanda K, Nigritinou M, et al. Circulating AQP4-specific auto-antibodies alone can induce neuromyelitis optica spectrum disorder in the rat. Acta Neuropathol. 2019;137(3):467-485. doi: 10.1007/s00401-018-1950-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sagan SA, Moinfar Z, Moseley CE, et al. T cell deletional tolerance restricts AQP4 but not MOG CNS autoimmunity. Proc Natl Acad Sci U S A. 2023;120(30):e2306572120. doi: 10.1073/pnas.2306572120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoftberger R, Guo Y, Flanagan EP, et al. The pathology of central nervous system inflammatory demyelinating disease accompanying myelin oligodendrocyte glycoprotein autoantibody. Acta Neuropathol. 2020;139(5):875-892. doi: 10.1007/s00401-020-02132-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zamvil S, Nelson P, Trotter J, et al. T-cell clones specific for myelin basic protein induce chronic relapsing paralysis and demyelination. Nature 1985;317(6035):355-358. doi: 10.1038/317355a0 [DOI] [PubMed] [Google Scholar]
- 29.Teitelbaum D, Meshorer A, Hirshfeld T, Arnon R, Sela M. Suppression of experimental allergic encephalomyelitis by a synthetic polypeptide. Eur J Immunol. 1971;1(4):242-248. doi: 10.1002/eji.1830010406 [DOI] [PubMed] [Google Scholar]
- 30.Yednock TA, Cannon C, Fritz LC, Sanchez-Madrid F, Steinman L, Karin N. Prevention of experimental autoimmune encephalomyelitis by antibodies against α4βl integrin. Nature. 1992;356(6364):63-66. doi: 10.1038/356063a0 [DOI] [PubMed] [Google Scholar]
- 31.Pittock SJ, Berthele A, Fujihara K, et al. Eculizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. N Engl J Med. 2019;381(7):614-625. doi: 10.1056/nejmoa1900866 [DOI] [PubMed] [Google Scholar]
- 32.Cree BAC, Bennett JL, Kim HJ, et al. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): a double-blind, randomised placebo-controlled phase 2/3 trial. Lancet. 2019;394(10206):1352-1363. doi: 10.1016/s0140-6736(19)31817-3 [DOI] [PubMed] [Google Scholar]
- 33.Yamamura T, Kleiter I, Fujihara K, et al. Trial of satralizumab in neuromyelitis optica spectrum disorder. N Engl J Med. 2019;381(22):2114-2124. doi: 10.1056/nejmoa1901747 [DOI] [PubMed] [Google Scholar]
- 34.Slavin AJ, Soos JM, Stuve O, et al. Requirement for endocytic antigen processing and influence of invariant chain and H-2M deficiencies in CNS autoimmunity. J Clin Invest. 2001;108(8):1133-1139. doi: 10.1172/jci200113360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bettelli E, Baeten D, Jager A, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T and B cells cooperate to induce a Devic-like disease in mice. J Clin Invest. 2006;116(9):2393-2402. doi: 10.1172/jci28334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Molnarfi N, Schulze-Topphoff U, Weber MS, et al. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J Exp Med. 2013;210(13):2921–2937. 10.1084/jem.20130699. 10.1084/jem.20130699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lyons JA, San M, Happ MP, Cross AH. B cells are critical to induction of experimental allergic encephalomyelitis by protein but not by a short encephalitogenic peptide. Eur J Immunol. 1999;29(11):3432-3439. doi: [DOI] [PubMed] [Google Scholar]
- 38.Tompkins SM, Padilla J, Dal Canto MC, Ting JP, Van Kaer L, Miller SD. De novo central nervous system processing of myelin antigen is required for the initiation of experimental autoimmune encephalomyelitis. J Immunol. 2002;168(8):4173-4183. doi: 10.4049/jimmunol.168.8.4173 [DOI] [PubMed] [Google Scholar]
- 39.Pishesha N, Harmand TJ, Ploegh HL. A guide to antigen processing and presentation. Nat Rev Immunol. 2022;22(12):751-764. doi: 10.1038/s41577-022-00707-2 [DOI] [PubMed] [Google Scholar]
- 40.Lanzavecchia A. Antigen-specific interaction between T and B cells. Nature. 1985;314(6011):537-539. doi: 10.1038/314537a0 [DOI] [PubMed] [Google Scholar]
- 41.Zamvil SS, Mitchell DJ, Moore AC, Kitamura K, Steinman L, Rothbard JB. T-cell epitope of the autoantigen myelin basic protein that induces encephalomyelitis. Nature. 1986;324(6094):258-260. doi: 10.1038/324258a0 [DOI] [PubMed] [Google Scholar]
- 42.Weber MS, Prod'homme T, Patarroyo JC, et al. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Ann Neurol. 2010;68(3):369-383. doi: 10.1002/ana.22081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Litzenburger T, Fassler R, Bauer J, et al. B lymphocytes producing demyelinating autoantibodies: development and function in gene-targeted transgenic mice. J Exp Med. 1998;188(1):169-180. doi: 10.1084/jem.188.1.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med. 2003;197(9):1073-1081. doi: 10.1084/jem.20021603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Slavin A, Ewing C, Liu J, Ichikawa M, Slavin J, Bernard CC. Induction of a multiple sclerosis-like disease in mice with an immunodominant epitope of myelin oligodendrocyte glycoprotein. Autoimmunity. 1998;28(2):109-120. doi: 10.3109/08916939809003872 [DOI] [PubMed] [Google Scholar]
- 46.Breithaupt C, Schubart A, Zander H, et al. Structural insights into the antigenicity of myelin oligodendrocyte glycoprotein. Proc Natl Acad Sci U S A. 2003;100(16):9446-9451. doi: 10.1073/pnas.1133443100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shetty A, Gupta SG, Varrin-Doyer M, et al. Immunodominant T-cell epitopes of MOG reside in its transmembrane and cytoplasmic domains in EAE. Neurol Neuroimmunol Neuroinflamm. 2014;1(2):e22. doi: 10.1212/nxi.0000000000000022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bruijstens AL, Wong YYM, van Pelt DE, et al. HLA association in MOG-IgG- and AQP4-IgG-related disorders of the CNS in the Dutch population. Neurol Neuroimmunol Neuroinflamm. 2020;7(3). doi: 10.1212/nxi.0000000000000702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grant-Peters M, Passos GRD, Yeung HY, et al. No strong HLA association with MOG antibody disease in the UK population. Ann Clin Transl Neurol. 2021;8(7):1502-1507. doi: 10.1002/acn3.51378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Piddlesden SJ, Lassmann H, Zimprich F, Morgan BP, Linington C. The demyelinating potential of antibodies to myelin oligodendrocyte glycoprotein is related to their ability to fix complement. Am J Pathol. 1993;143:555-564. [PMC free article] [PubMed] [Google Scholar]
- 51.Baxter R, Lewis E, Goddard K, et al. Acute demyelinating events following vaccines: a case-centered analysis. Clin Infect Dis. 2016;63(11):1456-1462. doi: 10.1093/cid/ciw607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jarius S, Ruprecht K, Kleiter I, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflammation. 2016;13(1):280. doi: 10.1186/s12974-016-0718-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu Q, Wang B, Zhao W. MOG-IgG-associated demyelination induced by pembrolizumab treatment in a patient with malignant melanoma. Neurology. 2022;98(12):501-502. doi: 10.1212/wnl.0000000000200055 [DOI] [PubMed] [Google Scholar]
- 54.Wildemann B, Jarius S, Franz J, Ruprecht K, Reindl M, Stadelmann C. MOG-expressing teratoma followed by MOG-IgG-positive optic neuritis. Acta Neuropathol. 2021;141(1):127-131. doi: 10.1007/s00401-020-02236-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 2013;12(2):157-165. doi: 10.1016/s1474-4422(12)70310-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cao Y, Goods BA, Raddassi K, et al. Functional inflammatory profiles distinguish myelin-reactive T cells from patients with multiple sclerosis. Sci Transl Med. 2015;7(287):287ra274. doi: 10.1126/scitranslmed.aaa8038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barreras P, Vasileiou ES, Filippatou AG, et al. Long-term effectiveness and safety of rituximab in neuromyelitis optica spectrum disorder and MOG antibody disease. Neurology. 2022;99(22):e2504-e2516. doi: 10.1212/wnl.0000000000201260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.clinicaltrials.gov/study/NCT05063162 Accessed April 9, 2024.
- 59.Kaneko K, Sato DK, Nakashima I, et al. CSF cytokine profile in MOG-IgG+ neurological disease is similar to AQP4-IgG+ NMOSD but distinct from MS: a cross-sectional study and potential therapeutic implications. J Neurol Neurosurg Psychiatry. 2018;89(9):927-936. doi: 10.1136/jnnp-2018-317969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ringelstein M, Ayzenberg I, Lindenblatt G, et al. Interleukin-6 receptor blockade in treatment-refractory MOG-IgG-associated disease and neuromyelitis optica spectrum disorders. Neurol Neuroimmunol Neuroinflamm. 2022;9(1):e100. doi: 10.1212/nxi.0000000000001100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Virupakshaiah A, Moseley CE, Elicegui S, et al. Life-threatening MOG antibody-associated hemorrhagic ADEM with elevated CSF IL-6. Neurol Neuroimmunol Neuroinflamm. 2024;11(4):e200243. doi: 10.1212/nxi.0000000000200243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Accessed April 9, 2024. clinicaltrials.gov/study/NCT05271409