

Key Points
-
•
JAK2V617F mutation contributes to hemorrhagic lesions and to mortality in an experimental model of cerebral venous thrombosis.
-
•
JAK2V617F mutation exacerbates thromboinflammatory response during cerebral venous thrombosis.
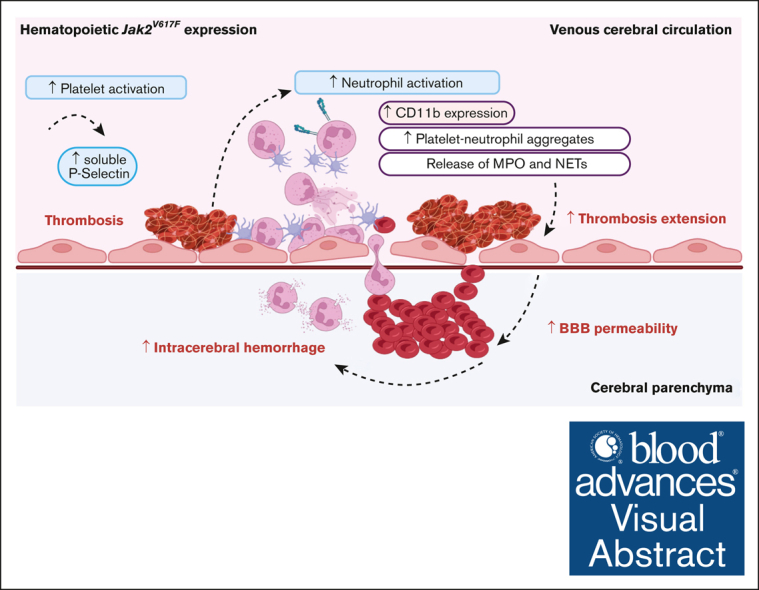
Visual Abstract
Abstract
Cerebral venous sinus thrombosis (CVST) is an uncommon venous thromboembolic event accounting for <1% of strokes resulting in brain parenchymal injuries. JAK2V617F mutation, the most frequent driving mutation of myeloproliferative neoplasms, has been reported to be associated with worse clinical outcomes in patients with CVST. We investigated whether hematopoietic JAK2V617F expression predisposes to specific pathophysiological processes and/or worse prognosis after CVST. Using an in vivo mouse model of CVST, we analyzed clinical, biological, and imaging outcomes in mice with hematopoietic-restricted Jak2V617F expression, compared with wild-type Jak2 mice. In parallel, we studied a human cohort of JAK2V617F-positive or -negative CVST. Early after CVST, mice with hematopoietic Jak2V617F expression had increased adhesion of platelets and neutrophils in cerebral veins located in the vicinity of CVST. On day 1, Jak2V617F mice had a worse outcome characterized by significantly more frequent and severe intracranial hemorrhages (ICHs) and higher mortality rates. Peripheral neutrophil activation was enhanced, as indicated by higher circulating platelet–neutrophil aggregates, upregulated CD11b expression, and higher myeloperoxydase plasma level. Concurrently, immunohistological and brain homogenate analysis showed higher neutrophil infiltration and increased blood-brain barrier disruption. Similarly, patients with JAK2V617F-positive CVST tended to present higher thrombotic burden and had significantly higher systemic immune-inflammation index, a systemic thromboinflammatory marker, than patients who were JAK2V617F-negative. In mice with CVST, our study corroborates that Jak2V617F mutation leads to a specific pattern including increased thrombotic burden, ICH, and mortality. The exacerbated thromboinflammatory response, observed both in mice and patients positive for JAK2V617F, could contribute to hemorrhagic complications.
Introduction
Cerebral venous sinus thrombosis (CVST) is an atypical site of venous thromboembolism. It represents an uncommon form of stroke with highly variable clinical course that mainly affects young adults.1 CVST leads to impaired venous drainage, intracranial hypertension, and parenchymal lesions including cerebral edema, ischemia, and intracerebral hemorrhage (ICH) reported in up to 40%.2, 3, 4, 5, 6 In CVST, because the thrombosis initiation depends largely on the presence of prothrombotic conditions (acquired and inherited thrombophilia, trauma, etc) the underlying causes cannot be identified in 15% to 30% of cases.2, 3, 4,6
Several small series have suggested that CVST may represent the first clinical manifestation of Bcr/Abl-negative myeloproliferative neoplasms (MPNs), both in overt or “occult” MPNs (ie, without any biological/clinical signs of MPNs).7 The gain-of-function JAK2V617F mutation is the main molecular hallmark of MPNs, of which the prevalence is estimated to be between 2.6% and 3.9%.8,9 Hence, although JAK2V617F mutation is a leading cause of splanchnic veins thrombosis (25%-35%), another atypical location of venous thromboembolism, its relationship with CVST remains unclear and systematic screening in routine diagnostic workup is not recommended.5,9,10 Nevertheless, early diagnosis of MPNs during CVST could have critical therapeutic implications. In CVST, malignancies including MPNs are associated with an increased risk of thrombotic recurrence,11, 12, 13 ICH,14 and poor neurological outcome.2,4,15,16
Clinical studies have demonstrated that myeloid JAK2V617F expression is associated with increased risk for thrombotic complications and cardiovascular events.17 Accordingly, Jak2V617F mice have shown their propensity to thrombosis in previous venous thrombosis models such as FeCl3-injury to mesenteric veins,18 inferior vena cava ligation,19,20 or acute pulmonary embolism.21 JAK2V617F-driven pathophysiological mechanisms include abnormal cell interactions, changes in the inflammatory environment, and rheological properties that switch cells from a resting to a prothrombotic and proinflammatory phenotype.17 Numerous studies have provided evidence that the pathogenesis of MPN-related thrombosis involves platelet and neutrophil activation.22, 23, 24 Moreover, enhanced JAK2V617F neutrophil infiltration has been implicated in accelerated vascular remodeling.25, 26, 27
During CVST, although parenchymal lesions are critical for neurological outcome, predictive factors for their development, especially in MPNs, are still largely elusive.15,28 Using in vivo 2-photon imaging, Santisakultarm et al showed, in hematopoietic Jak2V617F transgenic mice, increased adhesion of blood cells in cerebral microcirculation, which mainly comprised platelet and neutrophil aggregates.29 These observations raise the possibility that hematopoietic expression of JAK2V617F enhance brain parenchymal injury, thus worsening CVST through an in vivo thromboinflammatory process. To date, human studies have mainly focused on JAK2V617F as a risk factor for CVST rather than on its pathophysiological-related process.
Using a mouse knockin (KI) model of MPNs with specific hematopoietic Jak2V617F expression, we investigated whether Jak2V617F expression contributes to CVST-related parenchymal injury. Specifically, we examined the role of thromboinflammation, focusing on neutrophils and platelets. We also investigated whether the presence of JAK2V617F mutation could affect clinical outcome and levels of circulating thromboinflammatory markers in a human cohort of CVST.
Methods
Animal experiments
We used a well characterized hematopoietic Jak2V617F KI model.18,30 Briefly, bone marrow (BM) cells (3 × 106 cells) from Jak2V617F KI/VavCre mice were engrafted into 6- to 8-week-old C57BL/6J mice that had undergone lethal total body irradiation thus resulting in polycythemia vera (PV)-like disease. Irradiated C57BL/6 mice transplanted with wild-type Jak2 (Jak2WT) BM cells (control littermate) were used as controls. Medullar reconstitution was allowed for 9 weeks before experiments were performed. This procedure ensures heterozygous expression of Jak2V617F in all hematopoietic cells and minimizes the risk of Jak2V617F expression in nonhematopoietic cells. Given that CVST is more likely to occur in women, we used female mice in all experiments. All animal procedures were carried out according guidelines formulated by the European Community for experimental animal use (L358-86/609EEC) under approval of the Committee on the Ethics of Animal Experiments (Paris Nord n°121, approval number n°14070). Animal experiments were performed using the animal facilities of the INSERM (APAFIS #2344-2016101015023392).
Mouse model for CVST
Superior sagittal sinus (SSS) thrombosis was performed using a thrombus-injected CVST model, as previously described.31 Briefly, standardized whole blood clots were formed 1 day before surgery using 1 UI/mL thrombin (Dade Behring Thrombin reagent, Siemens, Marburg, Germany) and 10 mM CaCl2. Mice were anesthetized using isoflurane gas in ambient air (4% for induction and 1.5% for maintenance). After craniotomy, the clot was injected into SSS after careful puncture. Clot injection was combined with bilateral external jugular vein ligation in the CVST group. The control (sham) group comprised age-matched mice with isolated craniotomy. All mice were subjected to a neurological sensory-motor evaluation 1 day before and the first day after surgery in a blinded fashion, as previously described.31 Neurological assessment and evolution at day 1 was expressed as a percentage of the basal neurological score.
Real-time intravital imaging of cortical circulation
Two hours after CVST, mice anesthetized using isoflurane gas (1.5% for maintenance) were injected with fluorescent tracers through the retro-orbital venous sinus, and placed under a fluorescence macroscope (MacroFluo; Leica Microsystems) equipped with a heating plate with a thermostat and connected to a scientific CMOS camera (ORCAFlash4.0; Hamamatsu Photonics, Hamamatsu City, Japan). All circulating leukocytes/platelets were stained with rhodamine 6G (3 mg/kg, Sigma, St Louis, MO). Specific neutrophils observation was performed using Alexa Fluor 647-conjugated rat anti-mouse Ly6G (0.1 mg/kg, clone 1A8, BD Pharmingen). The vascular network was visualized using 2000 kDa fluorescein isothiocyanate (FITC) dextran (10 mg/kg, Sigma). Images were acquired using MetaMorph software (Molecular Devices) and analyzed using ImageJ software (National Institutes of Health).
Magnetic resonance imaging (MRI)
On postoperative day 1, MRI was performed on a Pharmascan 7-Tesla small animal scanner equipped using a mouse brain volume coil (Bruker, Ettlingen, Germany). Ischemic and hemorrhagic volume were calculated using Horos software, as previously described (supplemental Material).31
Immunohistochemical analyses
On day 1 after surgery, mice were euthanized by transcardiac infusion of ice-cold phosphate-buffered saline (pH, 7.4) followed by zinc fixative. The brain was removed from the skull and divided into 4 segments. Macroscopic examination of the brain (adapted from Egashira et al32) was used to assign a grade from 0 to 2 depending on the severity of ICH, as follows: 0, no ICH; 1, localized/minimal ICH; and 2, diffuse/deep parenchymal ICH. Then, brains were preserved in zinc fixative for at least 24 hours before being processed and stained (supplemental Material).
Assessment of brain and plasma levels of thromboinflammatory markers
Plasma levels of soluble P-selectin, myeloperoxydase (MPO), and platelet factor 4 (PF4) were measured using enzyme-linked immunosorbent assay kits (R&D Systems, catalog no. MPS00, DY3667, and DY595, respectively). Adapted from Li et al,33 quantification of citrullinated histone H3–bound DNA (H3Cit-DNA) was performed using an anti-histone H3 (catalog no. 5103, Abcam) antibody followed by fluorescent Quant-iT Picogreen dsDNA assay (Invitrogen) to detect DNA. For measurements in brain homogenates, brains were recovered after intracardiac perfusion of ice-cold phosphate-buffered saline. MPO, PF4, immunoglobulin G (IgG), and hemoglobin were measured using different enzyme-linked immunosorbent assay kits (HycultBiotech, catalog no. HK210; R&D Systems, catalog no. DY595; Abcam, catalog no. ab151276 and catalog no. ab157715; respectively). A thromboinflammatory score, the systemic immune-inflammation index (SII index: [platelet, ×109/L × neutrophil, ×109/L]/[lymphocyte, ×109/L]) was calculated in mice from blood cell counts, as described previously.34
Assessment of platelet-neutrophil aggregates (PNA) and platelet activation by flow cytometry
PNA and platelet activation were measured in heparinized (15 UI/mL) unstimulated whole blood before, and 1 day after, CVST using a BD Accuri C6 Flow Cytometer (BD Biosciences, San Jose, CA). Combination of rat anti-mouse CD45-PerCP-Cy5.5 (30F11, Biolegend), Ly6G-AF555 (1A8, BD Biosciences), and CD11b-AF488 (M1 170, Biolegend) was used for labeling leukocytes and neutrophils. Rat anti-mouse CD42a-AF647 (GPIX; M051-0, Emfret) was used to label platelets. A minimum of 500 000 events were recorded. The percentage of CD45+/CD42a+ double-positive events representing platelet-leukocyte aggregates was recorded. Within platelet-leukocyte aggregates, PNAs were identified from Ly6G-positive events as the percentage of neutrophils.
Platelet activation was measured using surface exposure of P-selectin (rat anti-mouse CD62P-PE, RMP1, Biolegend). Activation of αIIbβ3 integrin (GPIIbIIIa) was detected using binding of FITC-labeled fibrinogen (30 μg/mL).
Human case-control study
On behalf of the prospective CVST French cohort study (FPCCVT) investigators,6 we retrospectively analyzed patients for whom peripheral blood DNA was available for JAK2V617F mutation screening. Genomic DNA was extracted using an automated standard procedure from whole blood collected by venipuncture on EDTA and stored at −80°C until analysis. JAK2V617F mutation was screened using high-resolution melting analysis on a LightCycler 480 instrument (Roche Diagnostics, Rotkreuz, Switzerland). Allelic burden was determined in JAK2V617F-positive cases using the Ipsogen JAK2 MutaQuant kit (Qiagen, Courtaboeuf, France) according to manufacturer’s instructions. JAK2V617F-positive MPNs were diagnosed according the 2022 World Health Organization diagnostic criteria.35
Demographic, clinical, imaging, and biological (including the SII index) data were compared between JAK2V617F-positive and -negative CVST at baseline and after 3 months of follow-up. In addition, plasma levels of MPO (no. DY3174, R&D Systems) were measured according to the manufacturer’s instruction.
Statistical analysis
Statistical analysis of differences between groups was performed using the appropriate test, as indicated. A P value <.05 was considered significant.
Results
Jak2V617F mutation exacerbates early vascular recruitment of neutrophils and platelets, 2 hours after CVST
We used a mouse model of MPN characterized by hematopoietic Jak2V617F expression resulting in a variant allele frequency (VAF) of 50% in all hematopoietic lineages, as previously described.30 Compared with Jak2WT, Jak2V617F mice displayed a PV-like phenotype, with higher hemoglobin level, neutrophil count (supplemental Table 2) and enlarged spleen (spleen weight, 296 ± 74 vs 66 ± 15 mg; P < .001).
Intravital microscopy was performed 2 hours after injection of standardized autologous thrombi into the SSS (Figure 1). Sham-operated mice (Jak2V617F and Jak2WT) exhibited sparse adherent leukocytes and platelets within the venous compartment (Figure 1B). After CVST, rolling and firm adhesion of leukocytes and platelets was observed along venous vessels (SSS, cortical veins, and venules). Adhesion of leukocytes and platelets appeared to be more pronounced in Jak2V617F mice compared with Jak2WT counterparts (Figure 1B-C; supplemental Video 1). Accordingly, quantification of intravascular rhodamine-associated fluorescence intensity was significantly higher in Jak2V617F than Jak2WT and sham-operated mice (Figure 1C). In Jak2V617F mice, Ly6G labeling indicated a greater level of neutrophil adhesion to the venous wall than Jak2WT mice (Figure 1D). At this stage, no extravasation of neutrophils or 2000 kDa FITC dextran reflecting blood-brain-barrier (BBB) leakage was observed into parenchyma.
Figure 1.

Jak2V617F mice showed an enhanced early endothelial adhesion of leukocyte and platelet in cerebral veins after CVST. (A) Intravital microscopy imaging of venous brain network from Jak2WT and Jak2V617F mice representing SSS and cortical venules (CtV) labeled with dextran 2 mKDa (green). Scale bar, 200 μm. (B) In sham-operated mice (left panel), sparse marginating leukocytes and platelets (rhodamine 6G, red) were found in Jak2V617F and Jak2WT mice. Two hours after CVST, adhesion and accumulation of platelets and leukocytes increased in both Jak2V617F and Jak2WT mice (right panel). Dextran imaging showed black segments within venules (white arrow), indicating a disruption in blood flow and formation of new thrombi. (C) Median of fluorescence intensity of rhodamine 6G–labeled platelets and leukocytes as a marker of thrombosis burden in CtV. Results are expressed as mean ± standard deviation (n = 5 per group). Data were analyzed using Kruskal-Wallis test; ∗P < .05; ∗∗P < .01. (D) Labeling of neutrophils (Ly6G, pink; lower panel) and vessels (dextran 2 mKDa, green; upper panel) showed that neutrophils were mainly involved in endothelial adhesion in Jak2V617F mice. Scale bars, 25 μm.
In all mice after CVST, dextran imaging showed black segments within venules, indicating a disruption in blood flow and formation of new thrombi at a distance from the original site (Figure 1B; supplemental Video 1). Venular occlusion appeared to be notably increased in Jak2V617F compared with in Jak2WT mice.
CVST specificities in Jak2V617F mice at day 1
The role of Jak2V617F mutation in CVST-related brain injury was assessed at day 1 after SSS thrombosis. MRI showed small brain ischemic infarcts, without any difference in volume between Jak2WT and Jak2V617F mice (Figure 2A-B). In contrast, Jak2V617F mice had larger MRI ICH volumes (P < .05) and higher brain hemoglobin levels (P < .05) than Jak2WT mice (Figure 2A-C). Accordingly, brain macroscopic examination showed that incidence of ICH was twofold higher in Jak2V617F mice (n = 13 of 17) than in Jak2WT mice (n = 5 of 12). Interestingly, Jak2V617F mice showed increased severity of ICH, compared with Jak2WT mice, ranging from localized (type 1) to deep parenchymal ICH (type 2; Figure 2D). Type 2 ICH, the most severe pattern, was observed in 41% of Jak2V617F mice (n = 7 of 17), and never in Jak2WT mice (Figure 2E). Among Jak2V617F mice, blood hemoglobin levels were not significantly different between mice with no, type 1, or type 2 ICH.
Figure 2.

Jak2V617F mice displayed more CVST–related hemorrhagic lesions at day 1. (A) Representative serial coronal T2-weighted images from Jak2WT and Jak2V617F mice, 1 day after CVST induction demonstrating localized ischemia (white asterisk) and/or ICH (black arrow) in cortical parenchyma. (B) Brain lesions volumes (ischemic and hemorrhagic) determined from coronal T2-weighted images from Jak2WT and Jak2V617F mice at day 1, n = 5 per group. (C) Quantification of hemoglobin brain content at day 1 after CVST; ∗P < .05 (t test), n = 6 per group. (D) Brain slices from Jak2V617F and Jak2WT mice showing different pattern of ICH severity after CVST (no ICH; type 1: localized ICH; type 2: diffuse ICH). (E) Incidence of severe ICH (i.e., type 2) was significantly increased in Jak2V617F mice compared with Jak2WT mice at day 1 after CVST (n = 17 and 12 per group, respectively). In Figure 2B,C, data were analyzed using Mann-Whitney U test. In Figure 2E, data were analyzed by χ2 test; ∗P < .05.
At day 1 after CVST, survival rate was significantly reduced in Jak2V617F mice (n = 13 of 17) compared with Jak2WT counterparts (n = 12 of 12; P = .044) although the neurological score did not differ between Jak2WT and Jak2V617F mice (−17.9% and −20.1%, compared from baseline, respectively). Sham-operated mice did not present any brain injury irrespective of Jak2 mutation status (data not shown).
Thrombosis expansion in Jak2V617F mice at day 1
Consistent with in vivo imaging, fluorescent staining of brain sections from Jak2WT and Jak2V617F mice confirmed the presence of fibrin deposits with a vascular pattern away from the SSS after CVST, suggesting new intravascular thrombi (supplemental Data; supplemental Figure 1). In addition, Jak2V617F mice showed more diffuse intravascular fibrin deposits compared with Jak2WT mice.
BBB disruption in Jak2V617F mice at day 1
Next, we assessed BBB integrity by measuring IgG in whole brain homogenates at day 1 (Figure 3). Although brain IgG levels in CVST were significantly increased in both types of mice compared with the sham-operated group, significantly higher IgG levels were observed in Jak2V617F than in Jak2WT mice (P < .05; Figure 3B). In brain sections, IgG extravasation was observed both in hemorrhagic and nonhemorrhagic areas around vessels but to a greater extent in Jak2V617F mice than in Jak2WT mice (Figure 3A).
Figure 3.

BBB leakage is exacerbated in Jak2V617F mice after CVST. (A) After CVST, staining for IgG (pink); blood vessel (GLUT-1, green); red blood cells (RBCs, white), and cell nuclei (4′,6-diamidino-2-phenylindole [DAPI], cyan) in brain sections from Jak2WT and Jak2V617F mice showed IgG extravasation in hemorrhagic and nonhemorrhagic areas. Scale bars, 25 μm. (B) Bar graph indicated quantification of IgG content (mean ± SD) in whole brain from Jak2WT and Jak2V617F mice in sham and CVST conditions at day 1, n = 5 to 6 per group. Data were analyzed using Kruskal-Wallis test followed by post hoc Dunn test; ∗P < .05.
At day 1 after CVST, Jak2V617F mutation is associated with brain neutrophil infiltration
We further investigated whether the Jak2V617F mutation affects neutrophil and platelet parenchymal infiltration at day 1 after CVST (Figure 4). No platelet accumulation was observed in either type of mice (data not shown). In both types of mice, CVST induced a parenchymal neutrophil infiltration, in nonhemorrhagic and hemorrhagic cortical areas, compared with in sham-operated mice, which was significantly increased in Jak2V617F mice compared with in Jak2WT mice (Figure 4A-C). We did not observe neutrophils stained positive for H3Cit (data not shown). PF4 and MPO in brain homogenates were measured as indicators of platelet and neutrophil infiltration, respectively (Figure 4D-E). At day 1 after CVST, only Jak2V617F mice had significantly elevated MPO levels (Figure 4E).
Figure 4.

Brain neutrophil infiltration is increased in Jak2V617F mice after CVST. (A) Representative images of coronal brain sections taken through the area of damage from Jak2WT and Jak2V617F mice after CVST at day 1. Hematoxylin and eosin (H&E) staining showed the presence of hemorrhagic (H) lesions and nonhemorrhagic cortical parenchyma (no H) in dotted rectangles. Scale bar, 500 μm. (B) Corresponding brain images of staining for MPO (red); cell nuclei (DAPI, cyan) and red blood cells (RBCs, pink) in “no H” (upper panel) and “H” area (lower panel) from Jak2WT and Jak2V617F mice after CVST at day 11. Scale bars, 50 μm. (C) Bar graph indicated number of neutrophils within “no H” cortical parenchyma in sham and CVST mice (Jak2WT and Jak2V617F), n = 10 per group. Bar graphs indicated quantification of PF4 (D) and MPO (E) contents in whole brain from Jak2WT and Jak2V617F mice at day 1 in sham-operated and CVST mice, n = 5 to 6 per group. All results are expressed as mean ± SD. Data were analyzed by Kruskal-Wallis test followed by post hoc Dunn test; ∗P < .05 and ∗∗∗P < .01.
In Jak2V617F mice, CVST was associated with higher circulating PNA levels and neutrophils activation
Because of the exacerbated neutrophils infiltration in brain parenchyma from Jak2V617F mice after CVST, we investigated neutrophil activation in whole blood at day 1 (Figure 5). In line with the MPN phenotype, circulating neutrophil count was increased in Jak2V617F mice compared with Jak2WT, irrespective of the experimental conditions (supplemental Table 2). Circulating PNA levels and neutrophil CD11b expression were significantly increased at baseline and day 1 after CVST in Jak2V617F mice compared with Jak2WT mice (Figure 5A-B). Next, we compared plasma MPO levels as a biomarker of neutrophil activation and degranulation (Figure 5C). Jak2V617F mice with CVST had significantly higher MPO levels than at baseline conditions but not compared with sham-operated Jak2V617F mice. After correction for neutrophil count, no significant difference in MPO levels was observed between Jak2WT and Jak2V617F mice with CVST or sham-operated mice (data not shown). Plasma levels of H3Cit-DNA, used as a biomarker for the release of neutrophil extracellular traps (NETs), tended to be higher in Jak2V617F mice than in Jak2WT mice after CVST (P = .06; Figure 5D).
Figure 5.

Increased formation of PNAs in Jak2V617F mice. (A) Circulating PNAs and (B) neutrophil CD11b expression are measured in Jak2WT and Jak2V617F mice at baseline, and at day 1 in sham-operated and CVST mice. (C) MPO and (D) H3Cit-DNA plasma levels are measured in the same conditions. PNAs were calculated from Ly6G-positive events as the percentage of neutrophils. Results are expressed as mean ± SD, n = 5 to 6 per group. Data were analyzed by Kruskal-Wallis test followed by post hoc Dunn test. ∗P < .05; ∗∗P < .01. D1, day 1.
In order to assess platelet activation, we measured surface P-selectin and activated αIIbβ3 integrin on circulating platelets (Figure 6). Although Jak2WT and Jak2V617F mice expressed similar level of activated αIIbβ3 integrin, P-selectin expression was significantly decreased in Jak2V617F mice compared with Jak2WT mice, with no difference between sham-operated and CVST mice (Figure 6B). Concurrently, soluble P-selectin levels were significantly higher in Jak2V617F mice than in their Jak2WT counterparts whereas platelet counts were similar (Figure 6C; supplemental Data; supplemental Table 2). The same pattern was observed using another biomarker of platelet α-granule, and soluble PF4, (supplemental Data; supplemental Figure 2). The thromboinflammatory marker, SII, was significantly increased from baseline to day 1 after CVST, regardless of Jak2 mutation status (supplemental Data; supplemental Table 2). However, after CVST, SII was significantly higher in Jak2V617F mice than in their Jak2WT counterparts (P < .01).
Figure 6.

Platelet activation in Jak2WT and Jak2V617F mice. Activation of αIIbβ3 integrin using binding of (A) labeled fibrinogen and (B) platelet P-selectin expression are measured in platelets using flow cytometry in both Jak2WT and Jak2V617F mice at baseline and at day 1 after sham-surgery or CVST. (C) Bar graph indicating plasma levels of soluble P-selectin (sP-Sel). Results are expressed as mean ± standard deviation, n = 7 to 8 per group. Data were analyzed using Kruskal-Wallis test followed by post hoc Dunn test. ∗P < .05; ∗∗P < .01.
JAK2V617F mutation specificities in a human cohort
To assess the clinical relevance of JAK2V617F mutation in CVST, we analyzed its prevalence in 216 patients with CVST from the prospective CVST French cohort study (FPCCVT).6 JAK2V617F mutation was detected in 4.2% of patients with CVST (ie, JAK2V617F-positive CVST; n = 9, all female) with a VAF ranging from 4% to 81% (supplemental Table 3). Clinical, biological, and radiological characteristics of patients with JAK2V617F-positive and -negative CVST are depicted in Table 1.
Table 1.
Characteristics of patients with CVST according to Jak2V617F mutation status
| JAK2V617F-negative CVST (n = 207) | JAK2V617F-positive CVST (n = 9) | P value | |
|---|---|---|---|
| Age, y | 39 [28-51] | 42 [24-75] | .45 |
| Male/female | 70/137 | 0/9 | .033 |
| BMI (kg/m2) | 25.6 [22.5-29] | 23.6 [21.5 27.2] | .27 |
| Mode of onset | |||
| Acute | 73/207 (35%) | 3/9 (33%) | .90 |
| Subacute | 118/207 (57%) | 6/9 (67%) | .56 |
| Chronic | 16/207 (8%) | 0/9 (0%) | 1 |
| Clinical findings | |||
| Isolated intracranial hypertension | 105/207 (50.7%) | 6/9 (66.6%) | .5 |
| Focal deficits | 87/207 (42%) | 2/9 (22.2%) | .31 |
| Encephalopathy | 13/207 (6.2%) | 1/9 (11.1%) | .46 |
| Acquired thrombophilia | |||
| Antiphospholipid syndrome | 1/81 (12%) | 0/9 (0%) | 1 |
| Hormonal (OC and/or pregnancy∗) | 101/207 (48.7%) | 4/9 (44.4%) | 1 |
| Systemic disorders† | 21/207 (10.1%) | 0/9 (0%) | .60 |
| Extracerebral neoplasia | 6/207 (2.8%) | 0/9 (0%) | 1 |
| Inherited thrombophilia | |||
| Protein C deficiency | 3/192 (1.6%) | 0/9 (0%) | 1 |
| Protein S deficiency | 8/191 (4.2%) | 0/9 (0%) | 1 |
| Antithrombin deficiency | 3/187 (1.6%) | 0/9 (0%) | 1 |
| Factor V Leiden | 15/142 (11.2%) | 0/6 (0%) | .6 |
| Factor II G20210A | 20/167 (11.9%) | 1/9 (11.1%) | .93 |
| Local risk factors‡ | 30/207 (14.4%) | 1/9 (11.1%) | 1 |
| Thrombosed sinuses/veins | |||
| SSS | 96/207 (46.3%) | 6/9 (66.6%) | .31 |
| Straight sinus | 26/207 (12.5%) | 3/9 (33.3%) | .10 |
| Lateral sinus | 119/207 (57.4%) | 6/9 (66.6%) | .73 |
| Deep venous system | 13/207 (6.2%) | 1/9 (11.1%) | .45 |
| Cortical veins | 86/207 (41.5%) | 5/9 (55.5%) | .49 |
| Number of sinuses involved | 2 [1-3] | 3 [1.5-4] | .23 |
| Combined sinus/vein | 74/207 (35.7%) | 6/9 (66.6%) | .079 |
| Parenchymal lesions | 78/207 (37.6%) | 4/9 (44.4%) | .73 |
| Ischemic | 26/78 (33.3%) | 2/4 (50%) | .60 |
| Hemorrhagic | 27/78 (34.6%) | 1/4 (25%) | 1 |
| Both | 25/78 (32%) | 1/4 (25%) | 1 |
| Biological findings | |||
| Hemoglobin, g/dL | 13.7 [12.7-15] | 14.1 [13.3-15.5] | .45 |
| Hematocrit, % | 41 [38.5-44] | 41.7 [40-53] | .23 |
| WBC count, ×109/L | 9 [7-11.3] | 13.3 [8.4-19.4] | .062 |
| Neutrophil count, ×109/L | 6.4 [4.5-8.7] | 8 [5.3-11.1] | .055 |
| Platelet count, ×109/L | 260 [208-317] | 319 [222-443] | .11 |
| D-dimers, ng/mL | 968 [553-1545] | 1205 [493-2043] | .91 |
| CRP | 12 [6-28] | 16 [7.5-35.5] | .45 |
| Fibrinogen, g/L | 4 [3.4-4.7] | 4.1 [3.5-4.9] | .74 |
| MPO (ng/mL) | 14.7 [9-22.4] | 19.7 [15-26] | .094 |
| SII index | 923 [531-1692] | 1664 [1066-1989] | .019 |
| Outcome (mRS, 3 mo follow-up) | |||
| <2 | 163/190 (85.7%) | 7/9 (77.7%) | .62 |
| ≥2 | 21/190 (11%) | 1/9 (11.1%) | 1 |
| Death | 6/190 (3.1%) | 1/9 (11.1%) | .28 |
Results are expressed as median [IQR, 25-75] or n (percentage,%).
BMI, body mass index; CRP, C-reactive protein; mRS, modified Rankin Scale; OC, estrogen contraceptive; WBC, white blood cell.
3 cases of pregnancy-related CVST.
Systemic disorders include Behçet disease, rheumatologic or connective tissue disease, vasculitis, and sarcoidosis.
Local risk factors include trauma, arteriovenous malformation, paracranial infection. Quantitative data were compared using Mann-Whitney t test. Qualitative data were analyzed by χ2 test.
Among patients with JAK2V617F-negative CVST, 24% had inherited thrombophilia, which did not significantly alter the thrombotic burden and outcome of CVST compared with in those without thrombophilia. Among patients with JAK2V617F-positive CVST, 5 had a “non-overt” MPN with normal blood counts, whereas the other 4 patients had abnormal blood cell counts (supplemental Table 3). None of these patients received antithrombotic or cytoreductive therapy at the time of enrollment. Compared with patients with JAK2V617F-negative CVST, those harboring JAK2V617F mutation tended to present a higher neutrophil count (P = .055). Apart from the use of estrogen contraceptives in 4 of 9 patients with JAK2V617F mutation, no other risk factor was observed in these patients. CVST thrombus load (ie, combined sinus and vein thrombosis) tended to increase in patients with JAK2V617F-positive CVST compared with in those with JAK2V617F-negative CVST (66.6% vs 35.7%; P = .079). In patients with JAK2V617F-positive CVST, the SII was significantly higher than in patients with JAK2V617F-negative CVST (P = .019). Because experimental data from Jak2V617F mice indicated enhanced neutrophil activation after CVST, we also measured plasma MPO levels in patients with CVST. A trend toward higher MPO levels was observed in JAK2V617F-positive CVST compared with JAK2V617F-negative CVST (Table 1).
Discussion
Using an original CVST mouse model and a human cohort of patients with CVST, our study provides new data pointing to a role for thromboinflammation in the pathophysiology of JAK2V617F-positive CVST. In Jak2V617F mice, CVST was characterized by widespread thrombosis, more severe ICH with higher mortality, increased BBB permeability, and higher circulating PNA aggregates. Interestingly, patients with JAK2V617F-positive CVST and Jak2V617F mice presented significantly higher levels of the SII thromboinflammatory biomarker than their counterparts.
From a mechanistic point of view, we observed that Jak2V617F mice had (1) increased neutrophil and platelet adhesion to the venous wall, (2) blood flow disturbance within venules, and (3) increased microvascular fibrin deposits remotely from the initial CVST site compared with Jak2WT mice. In line with previous venous thrombosis models, we show that Jak2V617F promotes propagation and extension of thrombosis.18, 19, 20, 21 In our model, although ischemic lesions were similar between mice, ICH was more frequent and severe in Jak2V617F mice than in Jak2WT mice. As reported in human CVST, ICH was likely responsible for the higher mortality observed in Jak2V617F mice.14,16,36 Together, these observations support the pathophysiological link between thrombosis and ICH in CVST.37,38 Indeed, as the thrombus expands in the venous circulation and its collaterals, intravenous pressure rises, potentially leading to BBB disruption, edema, and subsequent ICH.
Our study was based on a Jak2V617F KI mouse model in which the previously reported bleeding phenotype18 may have contributed to worsening ICH. Particularly, these mice presented a hemostatic defect characterized by (1) a mild collagen glycoprotein VI (GPVI) receptor deficiency, (2) low platelet activation response to GPVI, CLEC-2, and thrombin receptor agonists, (3) decreased content in large von Willebrand factor multimers responsible for acquired von Willebrand syndrome.18 In our study, no additional bleeding was observed in sham-operated Jak2V617F mice compared with Jak2WT mice, suggesting that hemostatic abnormalities alone do not explain the bleeding phenotype. These data support a scenario in which Jak2V617F promotes ICH through increased thrombotic burden of CVST rather than isolated hemostatic defect.
We hypothesize that dysregulated and excessive interplay between platelets and neutrophils may contribute to promote thrombus expansion that predispose Jak2V617F mice to develop vascular permeability and ICH. In line with this hypothesis, Jak2V617F mice had higher circulating PNA levels and upregulated membrane expression of neutrophil CD11b, than Jak2WT mice, after CVST. CD11b, a marker of neutrophil activation, is a member of β2-integrin (associated with CD18), which is known to interact with the platelet GPIb receptor. Our data strengthen previous findings, using in vivo mouse models, that showed abnormal activation of the β1/β2 integrin signaling pathway in Jak2V617F neutrophils, promoting their interaction with endothelium and platelets, as well as venous thrombosis.20,27 Moreover, we observed an increase in PF4 and soluble P-selectin levels, independent of CVST and platelet count, that likely indicates platelet activation in Jak2V617F mice as reported in patients with JAK2V617F.39 Together, these data suggest that increased interaction of neutrophils with platelets in Jak2V617F mice promote CVST expansion.24,39,40
Enhanced platelet-neutrophil interaction is also known to favor activation of the coagulation cascade through neutrophil activation, release of proteases, and NETs.41,42 In hematopoietic MPN models, it was previously reported that activated Jak2V617F neutrophils are more prone to form NETs, particularly after interaction with Jak2V617F platelets.19,21,43 In our study, plasma levels of H3Cit-DNA, a marker of NETs, tended to be higher in Jak2V617F mice than in Jak2WT mice only after CVST. No significant difference was observed in brain parenchyma. In line with upregulated CD11b expression, these results suggest that CVST induced a higher systemic neutrophil activation in Jak2V617F mice that could promote NETs formation. In a VavCre/Jak2V617F MPN model, higher levels of NETs and thrombosis rates were observed after partial ligation of inferior vena cava.19 Outside the context of MPN, prothrombotic effect of NETs has also been described in patients with CVST compared with controls via fibrinolytic resistance and procoagulant changes in the endothelium.44 Thus, the specificity of NETs and their role in the initiation and progression of CVST associated with Jak2V617F need to be addressed in further studies.
In Jak2V617F mice, PNA could also drive neutrophil migration across the endothelium, in which it can cause vascular injury through the release of reactive oxygen species, proteolytic enzymes, and chemokines.45, 46, 47, 48 Consistent with this important role of neutrophils, Nagai et al demonstrated that both CD18 immunoneutralization and neutropenia rescue BBB permeability in a FeCl3-injury CVST model.49 In our study, we observed the presence of extravasated IgG and neutrophils in nonhemorrhagic areas in parenchyma of Jak2V617F mice, suggesting that a compromised BBB is a putative precursor to ICH. Taken together, these data support a scenario in which enhanced neutrophil–platelet interaction, as part of the thromboinflammatory response, may predispose Jak2V617F mice to develop ICH by increasing the propensity for thrombus expansion and BBB permeability.
In the FPCCVT human cohort, we confirm that JAK2V617F mutation is associated to CVST in ∼4% of cases8,9 with a female preponderance and a heterogeneous biological profile ranging from overt to occult MPNs.8,50, 51, 52 As a result of this heterogeneity, the median blood count parameters were not significantly elevated in patients with JAK2V617F-positive CVST compared with patients with JAK2V617F-negative CVST. Conflicting data exist regarding the specificity of JAK2V617F-positive CVST, either indicating an increased thrombotic burden, risk of ICH and mortality,14,16,36,53 or a good prognosis.50,51 Consistent with our experimental data, patients with JAK2V617F-positive CVST tended to have a higher thrombosis load and had an increased SII compared with patients with JAK2V617F-negative CVST. In contrast to Li et al, we did not observe an association between higher SII and worse prognosis of CVST in these patients.34 Nevertheless, our findings suggest that an exacerbated thromboinflammatory response may predispose patients with JAK2V617F to extensive CVST with a higher risk of ICH. Thus, early curative anticoagulation at the onset of diagnosis may be critical in these patients to stop the thrombosis process and reduce the risk of ICH, as recommended.5,10
Nevertheless, some limitations need to be underlined. In the FPCCVT cohort, the rate of severe clinical presentation (ICH and coma) was lower than in previous studies, so it is possible that this latter cohort does not cover the full clinical spectrum of CVST.4,6 In addition, the small sample size of patients with JAK2V617F-positive CVST likely limits the power of statistical analysis. For experimental study, we used lethally irradiated recipient mice engrafted with BM from Jak2V617F KI/VavCre to restrict expression to hematopoietic cells but not endothelial cells (ECs). Thus, Jak2V617F mutation was not expressed in endothelial cells, which have been previously shown to be prothrombotic.54,55 In this model, the possibility that red blood cells could contribute to CVST expansion and ICH should be considered through increased (1) adhesion to endothelial laminin, (2) upregulation of pleckstrin 2, and (3) secretion of erythrocyte-derived microvesicules contributing to oxidative stress.56, 57, 58 Nevertheless, in Jak2V617F mice, we did not observe a relationship between hemoglobin levels and the incidence or severity of ICH. This suggests that increased viscosity and rheological changes may probably not play a major role in the development of ICH in our model.
Although the phenotype is also PV-like, this model results in a VAF of 50% in all hematopoietic lineages rather than a few hematopoietic stem cells as in the PF4iCre; Jak2V617F/WT model.59 To go further, competitive transplants of normal and Jak2V617F BM cells at different ratios could be performed to study the impact of increasing VAF in CVST, as observed in patients (4%-81%). Neutrophil- and platelet-specific MPN models without polycythemia (such as MRP8-iCre; Jak2V617F and PF4-Cre/JAK2V617F transgenic mice, respectively)43,60 or blockade strategy would also be helpful to confirm the mechanistic link between thromboinflammation and ICH during Jak2V617F-positive CVST.
Thromboinflammation processes represent a new biological target for innovative therapeutic strategies to reduce disability and mortality occurring during the course of JAK2V617F-related MPN diseases. Further studies are warranted to specifically address the respective roles of neutrophils and platelets in JAK2V617F-related CVST brain damage.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
A complete list of the members of FPCCVT study group investigators appears in “Appendix.”
Acknowledgments
The authors are grateful to the French Intergroup Myeloproliferative. The authors thank Odile Vandapel, Céline André, and Muriel Quillard-Muraine, members of INSERM CIC-CRB U1404, CHU de Rouen, for their help in receiving, controlling, preparing, preserving, and making available the human biological resources studied in this work, and the Direction de la Recherche Clinique et de l’Innovation of Rouen University Hospital for the clinical database of the FPCCVT. The authors also thank all investigators of the FPCCVT study.
This study was funded through a clinical research program (no. 2010/087/HP) of the French Ministry of Health. M-C.B. is the recipient of a grant poste d’accueil INSERM. This study was supported by INSERM.
Authorship
Contribution: M.-C.B., N.A., and M.M. conceptualized the study; M.-C.B. and N.A. conducted formal analysis; M.-C.B., M.S., C.J., C.F., and S.L.-C. performed investigation; I.P., J.-L.V., V.E., B.C., and E.V. provided resources; M.-C.B., N.A., and M.M. wrote the original draft of the manuscript; N.A., M.M., D.F., M.J.-P., and B.H.-T.-N. critically reviewed the manuscript; V.L.C.D. and A.T.B. were the main investigators of the FPCCVT study; and all authors have read and agreed to the published version of the manuscript.
Footnotes
N.A. and M.M. contributed equally to this work.
Data are available on request from the corresponding author, Marie-Charlotte Bourrienne (marie-charlotte.bourrienne@inserm.fr).
The full-text version of this article contains a data supplement.
Contributor Information
Marie-Charlotte Bourrienne, Email: marie-charlotte.bourrienne@inserm.fr.
FPCCVT Study Group:
Aude Triquenot Bagan, Véronique Le Cam Duchez, Isabelle Crassard, Ludovic Drouet, Marianne Barbieux-Guillot, Raphaël Marlu, Emmanuelle Robinet-Borgomino, Pierre-Emmanuel Morange, Valérie Wolff, Lelia Grunebaum, Frédéric Klapczynski, Elisabeth André-Kerneis, Fernando Pico, Brigitte Martin-Bastenaire, Emmanuel Ellie, Fanny Menard, François Rouanet, Geneviève Freyburger, Gaëlle Godenèche, Hong-An Allano, Thierry Moulin, Guillaume Mourey, Laurent Derex, Micheline Berruyer, Gwénaëlle Runavot, Catherine Trichet, Fausto Viader, Agnès Le Querrec, Thomas Tarek Husein, Sophie Cluet-Dennetiere, Francisco Macian-Montoro, Magali Donnard, Benoît Guillon, Catherine Ternisien, Mathieu Zuber, Sophie Laplanche, Philippe Tassan, Jean-Yves Peltier, Sandrine Canaple, Bertrand Roussel, Nicolas Gaillard, and Emilie Scavazza
Appendix
The members of the FPCCVT Study Group are: Aude Triquenot Bagan and Véronique Le Cam Duchez, CHU Rouen, Rouen France; Isabelle Crassard and Ludovic Drouet, Lariboisiere University Hospital, Paris, France; Marianne Barbieux-Guillot and Raphaël Marlu, Grenoble University Hospital, Grenoble, France; Emmanuelle Robinet-Borgomino and Pierre-Emmanuel Morange; Marseille University Hospital, Marseille, France; Valérie Wolff and Lelia Grunebaum, Strasbourg University Hospital, Strasbourg, France; Frédéric Klapczynski and Elisabeth André-Kerneis, Meaux Hospital, Meaux, France; Fernando Pico and Brigitte Martin-Bastenaire, Versailles Hospital, Versailles, France; Emmanuel Ellie and Fanny Menard, Bayonne Hospital, Bayonne, France; François Rouanet and Geneviève Freyburger, Bordeaux University Hospital, Bordeaux, France; Gaëlle Godenèche and Hong-An Allano, La Rochelle Hospital, La Rochelle, France; Thierry Moulin and Guillaume Mourey, Besançon University Hospital, Besançon, France; Laurent Derex and Micheline Berruyer, Lyon University Hospital, Lyon, France; Gwénaëlle Runavot and Catherine Trichet, Argenteuil Hospital, Argenteuil, France; Fausto Viader and Agnès Le Querrec, Caen University Hospital, Caen, France; Thomas Tarek Husein and Sophie Cluet-Dennetiere, Compiègne Hospital, Compiègne, France; Francisco Macian-Montoro and Magali Donnard, Limoges University Hospital, Limoges, France; Benoît Guillon and Catherine Ternisien, Nantes University Hospital, Nantes, France; Mathieu Zuber and Sophie Laplanche, Saint Joseph Hospital, Paris, France; Philippe Tassan and Jean-Yves Peltier, Poissy-Saint-Germain Hospital, Poissy, France; Sandrine Canaple and Bertrand Roussel, Amiens university Hospital, Amiens, France; Nicolas Gaillard and Emilie Scavazza, Perpignan Hospital, Perpignan, France.
Supplementary Material
References
- 1.Bousser M-G, Ferro JM. Cerebral venous thrombosis: an update. Lancet Neurol. 2007;6(2):162–170. doi: 10.1016/S1474-4422(07)70029-7. [DOI] [PubMed] [Google Scholar]
- 2.Duman T, Uluduz D, Midi I, et al. A multicenter study of 1144 patients with cerebral venous thrombosis: the VENOST study. J Stroke Cerebrovasc Dis. 2017;26(8):1848–1857. doi: 10.1016/j.jstrokecerebrovasdis.2017.04.020. [DOI] [PubMed] [Google Scholar]
- 3.Ferro JM, Lopes MG, Rosas MJ, Ferro MA, Fontes J, Cerebral Venous Thrombosis Portugese Collaborative Study Group Long-term prognosis of cerebral vein and dural sinus thrombosis. Cerebrovasc Dis. 2002;13(4):272–278. doi: 10.1159/000057855. [DOI] [PubMed] [Google Scholar]
- 4.Ferro JM, Canhão P, Stam J, Bousser M-G, Barinagarrementeria F, ISCVT Investigators Prognosis of cerebral vein and dural sinus thrombosis: results of the international study on cerebral vein and dural sinus thrombosis (ISCVT) Stroke. 2004;35(3):664–670. doi: 10.1161/01.STR.0000117571.76197.26. [DOI] [PubMed] [Google Scholar]
- 5.Saposnik G, Barinagarrementeria F, Brown RD, Jr., et al. Diagnosis and management of cerebral venous thrombosis. Stroke. 2011;42(4):1158–1192. doi: 10.1161/STR.0b013e31820a8364. [DOI] [PubMed] [Google Scholar]
- 6.Triquenot Bagan A, Crassard I, Drouet L, et al. Cerebral venous thrombosis: clinical, radiological, biological, and etiological characteristics of a French Prospective Cohort (FPCCVT)—comparison with ISCVT Cohort. Front Neurol. 2021;12 doi: 10.3389/fneur.2021.753110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Passamonti SM, Biguzzi E, Cazzola M, et al. The JAK2 V617F mutation in patients with cerebral venous thrombosis. J Thromb Haemost. 2012;10(6):998–1003. doi: 10.1111/j.1538-7836.2012.04719.x. [DOI] [PubMed] [Google Scholar]
- 8.Lamy M, Palazzo P, Agius P, et al. Should we screen for Janus kinase 2 V617F mutation in cerebral venous thrombosis? Cerebrovasc Dis. 2017;44(3-4):97–104. doi: 10.1159/000471891. [DOI] [PubMed] [Google Scholar]
- 9.Dentali F, Squizzato A, Brivio L, et al. JAK2V617F mutation for the early diagnosis of Ph− myeloproliferative neoplasms in patients with venous thromboembolism: a meta-analysis. Blood. 2009;113(22):5617–5623. doi: 10.1182/blood-2008-12-196014. [DOI] [PubMed] [Google Scholar]
- 10.Ferro JM, Bousser M-G, Canhão P, et al. European stroke organization guideline for the diagnosis and treatment of cerebral venous thrombosis - endorsed by the European academy of neurology. Eur Stroke J. 2017;2(3):195–221. doi: 10.1177/2396987317719364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miranda B, Ferro JM, Canhão P, et al. Venous thromboembolic events after cerebral vein thrombosis. Stroke. 2010;41(9):1901–1906. doi: 10.1161/STROKEAHA.110.581223. [DOI] [PubMed] [Google Scholar]
- 12.Lim HY, Ng C, Donnan G, Nandurkar H, Ho P. Ten years of cerebral venous thrombosis: male gender and myeloproliferative neoplasm is associated with thrombotic recurrence in unprovoked events. J Thromb Thrombolysis. 2016;42(3):423–431. doi: 10.1007/s11239-016-1362-3. [DOI] [PubMed] [Google Scholar]
- 13.Palazzo P, Agius P, Ingrand P, et al. Venous thrombotic recurrence after cerebral venous thrombosis: a long-term follow-up study. Stroke. 2017;48(2):321–326. doi: 10.1161/STROKEAHA.116.015294. [DOI] [PubMed] [Google Scholar]
- 14.Afifi K, Bellanger G, Buyck P-J, et al. Features of intracranial hemorrhage in cerebral venous thrombosis. J Neurol. 2020;267(11):3292–3298. doi: 10.1007/s00415-020-10008-0. [DOI] [PubMed] [Google Scholar]
- 15.Ferro JM, Bacelar-Nicolau H, Rodrigues T, et al. Risk score to predict the outcome of patients with cerebral vein and dural sinus thrombosis. Cerebrovasc Dis. 2009;28(1):39–44. doi: 10.1159/000215942. [DOI] [PubMed] [Google Scholar]
- 16.Porceddu E, Rezoagli E, Poli D, et al. Sex-related characteristics of cerebral vein thrombosis: a secondary analysis of a multicenter international cohort study. Thromb Res. 2020;196:371–374. doi: 10.1016/j.thromres.2020.09.023. [DOI] [PubMed] [Google Scholar]
- 17.Wolach O, Shacham Abulafia A. Can novel insights into the pathogenesis of myeloproliferative neoplasm-related thrombosis inform novel treatment approaches? Hemato. 2021;2(2):305–328. [Google Scholar]
- 18.Lamrani L, Lacout C, Ollivier V, et al. Hemostatic disorders in a JAK2V617F-driven mouse model of myeloproliferative neoplasm. Blood. 2014;124(7):1136–1145. doi: 10.1182/blood-2013-10-530832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolach O, Sellar RS, Martinod K, et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci Transl Med. 2018;10(436) doi: 10.1126/scitranslmed.aan8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edelmann B, Gupta N, Schnoeder TM, et al. JAK2-V617F promotes venous thrombosis through β1/β2 integrin activation. J Clin Invest. 2018;128(10):4359–4371. doi: 10.1172/JCI90312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Craver BM, Ramanathan G, Hoang S, et al. N-acetylcysteine inhibits thrombosis in a murine model of myeloproliferative neoplasm. Blood Adv. 2020;4(2):312–321. doi: 10.1182/bloodadvances.2019000967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barbui T, Finazzi G, Falanga A. Myeloproliferative neoplasms and thrombosis. Blood. 2013;122(13):2176–2184. doi: 10.1182/blood-2013-03-460154. [DOI] [PubMed] [Google Scholar]
- 23.Arellano-Rodrigo E, Alvarez-Larrán A, Reverter JC, Villamor N, Colomer D, Cervantes F. Increased platelet and leukocyte activation as contributing mechanisms for thrombosis in essential thrombocythemia and correlation with the JAK2 mutational status. Haematologica. 2006;91(2):169–175. [PubMed] [Google Scholar]
- 24.Falanga A, Marchetti M, Evangelista V, et al. Polymorphonuclear leukocyte activation and hemostasis in patients with essential thrombocythemia and polycythemia vera. Blood. 2000;96(13):4261–4266. [PubMed] [Google Scholar]
- 25.Sano S, Wang Y, Yura Y, et al. JAK2V617F-mediated clonal hematopoiesis accelerates pathological remodeling in murine heart failure. JACC Basic Transl Sci. 2019;4(6):684–697. doi: 10.1016/j.jacbts.2019.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kimishima Y, Misaka T, Yokokawa T, et al. Clonal hematopoiesis with JAK2V617F promotes pulmonary hypertension with ALK1 upregulation in lung neutrophils. Nat Commun. 2021;12(1) doi: 10.1038/s41467-021-26435-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang W, Liu W, Fidler T, et al. Macrophage inflammation, erythrophagocytosis and accelerated atherosclerosis in Jak2V617F mice. Circ Res. 2018;123(11):e35–e47. doi: 10.1161/CIRCRESAHA.118.313283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barboza MA, Chiquete E, Arauz A, et al. A practical score for prediction of outcome after cerebral venous thrombosis. Front Neurol. 2018;9:882. doi: 10.3389/fneur.2018.00882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Santisakultarm TP, Paduano CQ, Stokol T, et al. Stalled cerebral capillary blood flow in mouse models of essential thrombocythemia and polycythemia vera revealed by in vivo two-photon imaging. J Thromb Haemost. 2014;12(12):2120–2130. doi: 10.1111/jth.12738. [DOI] [PubMed] [Google Scholar]
- 30.Hasan S, Lacout C, Marty C, et al. JAK2V617F expression in mice amplifies early hematopoietic cells and gives them a competitive advantage that is hampered by IFNα. Blood. 2013;122(8):1464–1477. doi: 10.1182/blood-2013-04-498956. [DOI] [PubMed] [Google Scholar]
- 31.Bourrienne M-C, Loyau S, Benichi S, et al. A novel mouse model for cerebral venous sinus thrombosis. Transl Stroke Res. 2021;12(6):1055–1066. doi: 10.1007/s12975-021-00898-1. [DOI] [PubMed] [Google Scholar]
- 32.Egashira Y, Shishido H, Hua Y, Keep RF, Xi G. A new grading system based on magnetic resonance imaging in a mouse model of subarachnoid hemorrhage. Stroke. 2015;46(2):582–584. doi: 10.1161/STROKEAHA.114.007834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li M, Lin C, Leso A, Nefedova Y. Quantification of citrullinated histone H3 bound DNA for detection of neutrophil extracellular traps. Cancers. 2020;12(11):3424. doi: 10.3390/cancers12113424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li S, Liu K, Gao Y, et al. Prognostic value of systemic immune–inflammation index in acute/subacute patients with cerebral venous sinus thrombosis. Stroke Vasc Neurol. 2020;5(4):368–373. doi: 10.1136/svn-2020-000362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khoury JD, Solary E, Abla O, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia. 2022;36(7):1703–1719. doi: 10.1038/s41375-022-01613-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sales C, Wijeratne T, Lucero A. Is JAK2-mutation associated with extensive clot burden in cerebral venous thrombosis. Ann Clin Case Rep. 2021;6:1911. [Google Scholar]
- 37.Schaller B, Graf R. Cerebral venous infarction: the pathophysiological concept. Cerebrovasc Dis. 2004;18(3):179–188. doi: 10.1159/000079939. [DOI] [PubMed] [Google Scholar]
- 38.Capecchi M, Abbattista M, Martinelli I. Cerebral venous sinus thrombosis. J Thromb Haemost. 2018;16(10):1918–1931. doi: 10.1111/jth.14210. [DOI] [PubMed] [Google Scholar]
- 39.Guy A, Helzy K, Mansier O, et al. Platelet function studies in myeloproliferative neoplasms patients with Calreticulin or JAK2V617F mutation. Res Pract Thromb Haemost. 2023;7(2) doi: 10.1016/j.rpth.2023.100060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Falanga A, Marchetti M, Vignoli A, Balducci D, Barbui T. Leukocyte-platelet interaction in patients with essential thrombocythemia and polycythemia vera. Exp Hematol. 2005;33(5):523–530. doi: 10.1016/j.exphem.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 41.Swystun LL, Liaw PC. The role of leukocytes in thrombosis. Blood. 2016;128(6):753–762. doi: 10.1182/blood-2016-05-718114. [DOI] [PubMed] [Google Scholar]
- 42.Lisman T. Platelet–neutrophil interactions as drivers of inflammatory and thrombotic disease. Cell Tissue Res. 2018;371(3):567–576. doi: 10.1007/s00441-017-2727-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guy A, Garcia G, Gourdou-Latyszenok V, et al. Platelets and neutrophils cooperate to induce increased neutrophil extracellular trap formation in JAK2V617F myeloproliferative neoplasms. J Thromb Haemost. 2024;22(1):172–187. doi: 10.1016/j.jtha.2023.08.028. [DOI] [PubMed] [Google Scholar]
- 44.Jin J, Qiao S, Liu J, et al. Neutrophil extracellular traps promote thrombogenicity in cerebral venous sinus thrombosis. Cell Biosci. 2022;12(1) doi: 10.1186/s13578-022-00845-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ed Rainger G, Chimen M, Harrison MJ, et al. The role of platelets in the recruitment of leukocytes during vascular disease. Platelets. 2015;26(6):507–520. doi: 10.3109/09537104.2015.1064881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Meyer SF, Denorme F, Langhauser F, Geuss E, Fluri F, Kleinschnitz C. Thromboinflammation in stroke brain damage. Stroke. 2016;47(4):1165–1172. doi: 10.1161/STROKEAHA.115.011238. [DOI] [PubMed] [Google Scholar]
- 47.Desilles J-P, Syvannarath V, Ollivier V, et al. Exacerbation of thromboinflammation by hyperglycemia precipitates cerebral infarct growth and hemorrhagic transformation. Stroke. 2017;48(7):1932–1940. doi: 10.1161/STROKEAHA.117.017080. [DOI] [PubMed] [Google Scholar]
- 48.Jickling GC, Liu D, Ander BP, Stamova B, Zhan X, Sharp FR. Targeting neutrophils in ischemic stroke: translational insights from experimental studies. J Cerebr Blood Flow Metab. 2015;35(6):888–901. doi: 10.1038/jcbfm.2015.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ding J, Song B, Xie X, et al. Inflammation in cerebral venous thrombosis. Front Immunol. 2022;13:833490. doi: 10.3389/fimmu.2022.833490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dentali F, Ageno W, Rumi E, et al. Cerebral venous thrombosis and myeloproliferative neoplasms: results from two large databases. Thromb Res. 2014;134(1):41–43. doi: 10.1016/j.thromres.2014.03.040. [DOI] [PubMed] [Google Scholar]
- 51.Gangat N, Guglielmelli P, Betti S, et al. Cerebral venous thrombosis and myeloproliferative neoplasms: a three-center study of 74 consecutive cases. Am J Hematol. 2021;96(12):1580–1586. doi: 10.1002/ajh.26336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martinelli I, De Stefano V, Carobbio A, et al. Cerebral vein thrombosis in patients with Philadelphia-negative myeloproliferative neoplasms an European Leukemia Net study. Am J Hematol. 2014;89(11):E200–E205. doi: 10.1002/ajh.23809. [DOI] [PubMed] [Google Scholar]
- 53.Simaan N, Molad J, Honig A, et al. Characteristics of patients with cerebral sinus venous thrombosis and JAK2 V617F mutation. Acta Neurol Belg. 2023;123(5):1855–1859. doi: 10.1007/s13760-022-02077-x. [DOI] [PubMed] [Google Scholar]
- 54.Guadall A, Lesteven E, Letort G, et al. Endothelial cells harbouring the JAK2V617F mutation display pro-adherent and pro-thrombotic features. Thromb Haemost. 2018;118(9):1586–1599. doi: 10.1055/s-0038-1667015. [DOI] [PubMed] [Google Scholar]
- 55.Guy A, Gourdou-Latyszenok V, Le Lay N, et al. Vascular endothelial cell expression of JAK2V617F is sufficient to promote a pro-thrombotic state due to increased P-selectin expression. Haematologica. 2019;104(1):70–81. doi: 10.3324/haematol.2018.195321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.De Grandis M, Cambot M, Wautier M-P, et al. JAK2V617F activates Lu/BCAM-mediated red cell adhesion in polycythemia vera through an EpoR-independent Rap1/Akt pathway. Blood. 2013;121(4):658–665. doi: 10.1182/blood-2012-07-440487. [DOI] [PubMed] [Google Scholar]
- 57.Poisson J, Tanguy M, Davy H, et al. Erythrocyte-derived microvesicles induce arterial spasms in JAK2V617F myeloproliferative neoplasm. J Clin Invest. 2020;130(5):2630–2643. doi: 10.1172/JCI124566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao B, Mei Y, Cao L, et al. Loss of pleckstrin-2 reverts lethality and vascular occlusions in JAK2V617F-positive myeloproliferative neoplasms. J Clin Invest. 2018;128(1):125–140. doi: 10.1172/JCI94518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mansier O, Kilani B, Guitart AV, et al. Description of a knock-in mouse model of JAK2V617F MPN emerging from a minority of mutated hematopoietic stem cells. Blood. 2019;134(26):2383–2387. doi: 10.1182/blood.2019001163. [DOI] [PubMed] [Google Scholar]
- 60.Etheridge SL, Roh ME, Cosgrove ME, et al. JAK2V617F-positive endothelial cells contribute to clotting abnormalities in myeloproliferative neoplasms. Proc Natl Acad Sci U S A. 2014;111(6):2295–2300. doi: 10.1073/pnas.1312148111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.