

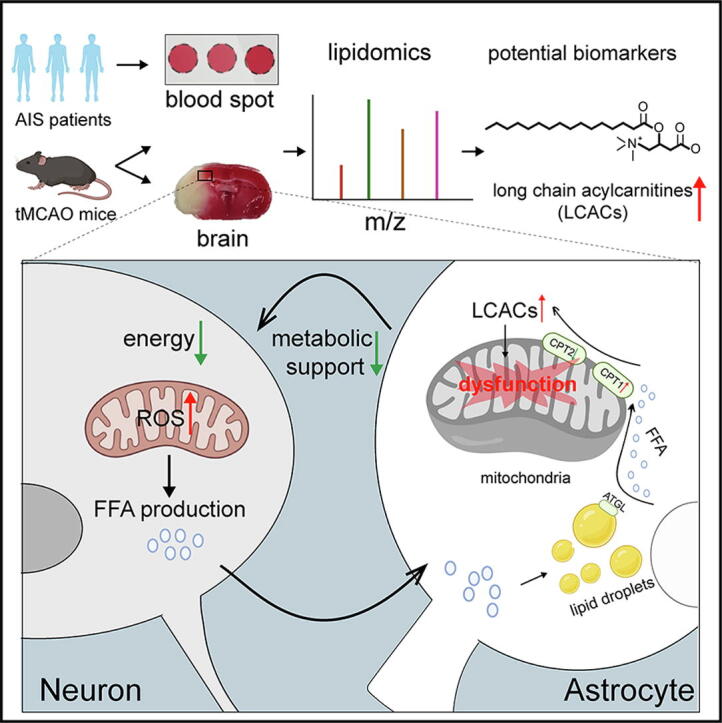
Graphical abstract
Keywords: Long-chain acylcarnitine, Ischemic stroke, Biomarker, Astrocytic mitochondrial, Lipid droplet
Highlights
-
•
LCACs are novel promising diagnostic and prognostic biomarkers for AIS.
-
•
Increased LCACs amplified while decreased LCACs ameliorated stroke injury.
-
•
Astrocytes liberate FFAs from lipid droplets to mitochondria to form LCACs.
-
•
LCACs induce astrocytic mitochondrial damage and thereby aggravated neuronal damage.
Abstract
Introduction
Lipid metabolism dysfunction is widely involved in the pathological process of acute ischemic stroke (AIS). The coordination of lipid metabolism between neurons and astrocytes is of great significance. However, the full scope of lipid dynamic changes and the function of key lipids during AIS remain unknown. Hence, identifying lipid alterations and characterizing their key roles in AIS is of great importance.
Methods
Untargeted and targeted lipidomic analyses were applied to profile lipid changes in the ischemic penumbra and peripheral blood of transient middle cerebral artery occlusion (tMCAO) mice as well as the peripheral blood of AIS patients. Infarct volume and neurological deficits were assessed after tMCAO. The cell viability and dendritic complexity of primary neurons were evaluated by CCK8 assay and Sholl analysis. Seahorse, MitoTracker Green, tetramethyl rhodamine methyl ester (TMRM), 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) and MitoSOX were used as markers of mitochondrial health. Fluorescent and isotopic free fatty acid (FFA) pulse-chase assays were used to track FFA flux in astrocytes.
Results
Long-chain acylcarnitines (LCACs) were the lipids with the most dramatic changes in the ischemic penumbra and peripheral blood of tMCAO mice. LCACs were significantly elevated on admission in AIS patients and associated with poor outcomes in AIS patients. Increasing LCACs through a bolus administration of palmitoylcarnitine amplified stroke injury, while decreasing LCACs by overexpressing carnitine palmitoyltransferase 2 (CPT2) ameliorated stroke injury. Palmitoylcarnitine aggravated astrocytic mitochondrial damage after OGD/R, while CPT2 overexpression in astrocytes ameliorated cocultured neuron viability. Further study revealed that astrocytes stimulated by OGD/R liberated FFAs from lipid droplets into mitochondria to form LCACs, resulting in mitochondrial damage and lowered astrocytic metabolic support and thereby aggravated neuronal damage.
Conclusion
LCACs could accumulate and damage neurons by inducing astrocytic mitochondrial dysfunction in AIS. LCACs play a crucial role in the pathology of AIS and are novel promising diagnostic and prognostic biomarkers for AIS.
Introduction
Acute ischemic stroke (AIS) is one of the leading causes of disability and death worldwide [1]. Intravenous thrombolytic therapy within 4.5 h and endovascular thrombectomy in patients with salvageable brain tissue within 24 h serve as the only two approved treatments for AIS [2]. However, only a small proportion of patients can benefit from these therapies owing to the narrow time window and their side effects [3], [4]. An understanding of the distinct mechanism underlying AIS will pave the way for therapeutic interventions albeit with little success. Thus, there is an urgent need to investigate additional mechanisms involved in AIS. Among them is lipid metabolism disorder, a well-known risk factor for stroke [5], [6]. Lipids and lipid intermediates, constituting 50 % of the brain dry weight, exert significant roles in maintaining the normal structure and function of the brain. Alterations in phosphatidylcholine acyl remodeling in neurons, the sphingolipid pathway in microglia and free fatty acid (FFA)-lipid droplet coupling between neurons and astrocytes are implicated in the pathogenesis of ischemic stroke [7], [8], [9], [10]. Although large-scale profiling of lipid species is available due to advancements in mass spectrometry (MS) technology, the full scope of lipid changes in the ischemic brain remains unknown. Hence, identifying novel lipid biomarkers and characterizing their role in AIS is of great importance.
Perturbations in energy metabolism pathways may cause the accumulation of long-chain acylcarnitines (AcCas) (LCACs, >12 carbon atoms), intermediates of fatty acid oxidation (FAO) and transporters of long-chain fatty acids through the mitochondrial membranes. LCACs are commonly used markers of incomplete FAO to diagnose inherited fatty acid oxidation disorders (FAODs) and type 2 diabetes mellitus [11], [12]. LCACs are also elevated in the peripheral blood of ischemic stroke patients and in the brain tissues of mouse models of traumatic brain injury [13], [14], indicating that dysregulated FAO metabolism is involved in acute brain injury. However, little is known about the role of LCACs in brain energy metabolism.
Although the major energy substrate in the brain is glucose or its derivative, lactate, astrocytic FAO contributes approximately 20 % of the brain’s energy [15]. Intracellular long-chain fatty acids (LCFAs, >12 carbon atoms) are esterified to carnitine by carnitine palmitoyltransferase 1 (CPT1) to form LCACs that are able to be transported into the mitochondria. LCACs in the mitochondrial matrix are metabolized to LCFA-coenzyme A by CPT2 and then enter the FAO pathway [11]. The stimulation of astrocytic FAO has been proven to be protective against AIS injury [16]. Instead, the overload of LCACs causes oxidative stress and toxicity in glioblastoma and cardiac mitochondria [17], [18] and participates in the neurotoxicity induced by reactive astrocytes [19]. However, the dynamic changes in LCACs and their regulation and roles in stroke are largely unknown.
In this study, we first performed untargeted lipidomic profiling in the ischemic penumbra of tMCAO-induced stroke model mice, and the most prominent changes in LCACs were further validated using targeted lipidomic analysis in the ischemic penumbra as well as the peripheral blood of tMCAO mice. Additionally, we recruited AIS patients to investigate the potential of LCACs as novel diagnostic and prognostic biomarkers. Then, we investigated the impacts of LCACs on stroke in vitro and in vivo. Furthermore, we revealed that the neuronal toxicity of LCACs was associated with astrocytic mitochondrial dysfunction. Therefore, we hypothesized that LCACs not only serve as a byproduct of FAO but also play an important role in ischemic stroke pathogenesis and progression.
Methods
Ethics statement
All of the animal experimental procedures were approved by the Institutional Animal Ethics Committee of Nanjing Medical University (approval number: IACUC-1912026). We followed the ARRIVE (Animal Research: Reporting In Vivo Experiments) ethical guidelines. Written informed consent was obtained from participants or their legal representatives for the use of patients’ blood before inclusion in the study, and the protocol was approved by the Ethical Committee of the First Affiliated Hospital of Nanjing Medical University, Nanjing, China (approval ID: 2021-SR-187).
tMCAO
C57BL/6J male mice between 6 and 8 weeks of age (20–25 g) at the beginning of experiments were used. The tMCAO model was established as previously described [20]. (Supplemental Methods).
Intravenous administration of AcCa(C16)
Palmitoylcarnitine (AcCa(C16), 100 μM, Sigma, P1645) was injected via the tail vein at the reperfusion of tMCAO mice [21]. The ischemic penumbra was isolated 1 day after injection, and LCACs were quantified by LC-MS/MS.
Neurological deficits and behavioral tests
The modified neurological severity score (mNSS) test, corner test, and latency to move test were performed as previously described [22], [23], [24]. (Supplemental Methods).
2,3,5-Triphenyltetrazolium chloride (TTC) staining and infarct volume analysis
Triphenyltetrazolium chloride (T8857-25 g, Sigma, America) was used to measure the infarct volume 1 and 3 days after tMCAO as previously described [25] (Supplemental Methods).
Nissl staining and atrophy volume analysis
Using a Leica CT1000S vibratome and coronal slices that were 40 μm thick, the paraffinized brains were sectioned and then Nissl stained in accordance with the manufacturer's instructions (KGMP0185, KeyGEN BioTECH, Nanjing). The contralateral area less the ipsilateral area was used to calculate the atrophy volume.
Virus infection
The adenovirus vector CMV-MCS-3FLAG-SV40-EGFP (3 × 1010 PFU/mL, GeneChem, Shanghai, China) was generated and administered. The adenovirus encompassing the Mus musculus carnitine palmitoyltransferase 2 (CPT2) gene was named “Ad-CPT2”, and GFP-expressing adenovirus (Ad-GFP) was used as a negative control. Each aliquot of the adenovirus (10–20 μl) was stored at -80 °C until use. Three days before tMCAO, Ad-CPT2 or Ad-GFP microinjection was conducted as previously described [26]. (Supplemental Methods).
Lipidomic analysis
Details of the untargeted and targeted lipidomic analyses of brain tissues and dried blood spot (DBS) samples are provided in the Supplemental Methods.
Subjects and sample collection
AIS patients and healthy controls were recruited from the First Affiliated Hospital of Nanjing Medical University, Nanjing, China. This study was approved by the ethics committees at the First Affiliated Hospital of Nanjing Medical University, Nanjing, China (approval ID: 2021-SR-187). The following were the inclusion criteria used in this study: 1) middle cerebral artery M1/M2 segment occlusions and 2) age > 30 years. Patients who had brain hemorrhage detected on noncontrast computed tomography, active malignancy, hematological disease, or inflammatory or infectious disorders were excluded from the study. The severity of stroke was assessed using the National Institutes of Health Stroke Scale (NIHSS) at the time of admission, and the functional outcome was assessed using the modified Rankin scale (mRS) 3 months later. Irreversibly infarcted tissues (infarct volume) were defined as the region with relative cerebral blood flow (CBF) < 30 % contralateral normal brain tissues. Hypoperfusion volume was defined as tissue with a Tmax value > 6 s. Automatic volumes of ischemic core and hypoperfusion were generated by using these thresholds. The mismatch volume (penumbra volume) was calculated as hypoperfusion volume minus volume of ischemic core [27], [28].
DBS samples were collected from healthy controls (HC, n = 33) and patients who did not receive drug therapy within 24 h after stroke onset (n = 41). To construct the diagnostic model, AIS patients with diabetes mellitus (DM) (n = 8) were excluded. The DBS samples of patients subjected to mechanical thrombectomy were collected before surgery and at 24 h after mechanical thrombectomy (n=18). Whole blood was dropped on DBS paper and stored at 4 °C in the dark for subsequent analysis after the blood spot was allowed to dry naturally. All subjects provided informed consent for sample collection.
Fuzzy C-Means clustering
AcCa species from 5 stages during tMCAO (1 h before tMCAO and 6 h, 1 d, 3 d and 7 d after tMCAO) were grouped into different clusters according to their expression profiles by the Mfuzz package in R [29].
Immunostaining
Procedures for the immunostaining and lipid droplet staining of brain sections and cells were conducted as previously described [25], [30]. (Supplemental Methods).
Transmission electron microscopy (TEM)
TEM was performed as previously described [31]. Briefly, tissue blocks about 1 mm3 in size were excised from the ischemic penumbra of tMCAO mice. The blocks were fixed in 2.5 % glutaraldehyde at 4 °C for 48 h and then 1 % osmium tetroxide. Following dehydration, the sections were cut by an ultrathin slicer and stained with uranyl acetate and lead citrate. The sctions were observed with a FEI Tecnai Spirit Biotwin electron microscope.
Triglyceride extraction and quantification
TGs were extracted from the ipsilateral hemisphere and corresponding areas of the sham group and then quantified using a TG Colorimetric Assay Kit (Cayman Chemicals, 10010303) according to the manufacturer's instructions.
CPT1 and CPT2 enzyme activity assay
Mitochondria were isolated from the ipsilateral hemisphere of tMCAO mice and the corresponding area of sham mice and then CPT1 and CPT2 enzyme activity were measured using quantitative assay kits based on enzymatic colorimetric method according to the manufacturer’s instructions, respectively (QIYI biological technology (Shanghai, China), QYS-239013 for CPT1 enzyme activity assay, GMS50118.2.3v.A for CPT2 enzyme activity assay) [32], [33]. Through the action of CPT1 and CPT2, acyl-CoA is produced and sulfhydrylcoenzyme A (CoA-SH) is released. After reacting with Ellman reagent (5,5′-Dithiobis (2-nitrobenzoic acid), DTNB], the 5-thio-2-nitrobenzoic acid (TNB) is produced and measured at 412-nm wavelength using a microplate reader (BioTek Cytation 5, USA). We also determined mitochondria protein concentrations using BCA protein assay method (KGP903, KeyGEN BioTECH, China) on the same sample. CPT1 activity was expressed as nM TNB/min/mg mitochondria protein and CPT2 activity was expressed as μM CoA /min/mg mitochondria.
Cell culture and reagents
Primary neuron, astrocyte and microglia cultures were performed as previously described, and these cells were isolated from 1‐ to 2‐day‐old postnatal C57BL/6J mice [20]. (Supplemental Methods).
Western blotting
Western blotting was performed as previously described and detail procedures are provided in the Supplemental Methods [34].
Oxygen–glucose deprivation and reoxygenation (OGD/R)
The OGD/R protocol was performed as described previously [35], [36]. (Supplemental Methods).
Fluorescent fatty acid pulse-chase assay
Primary cultured astrocytes were incubated in complete media containing 1 mM BODIPY 558/568 C12 (Red-C12, Life Technologies, D3835) for 16 h. Astrocytes were exposed to OGD for 6 h. After that, astrocytes were chased in normal medium with or without AGTLi for 24 h. Mitochondria were labeled with 100 nM MitoTracker Green FM (Life Technologies, M7514) for 30 min, and LDs were labeled with BODIPY 493/503 (Life Technologies, D3922) for 15 min prior to imaging to determine Red C12 localization. Coloc 2 Plugin and Fiji were used to calculate Pearson's correlation coefficient.
Isotopic fatty acid pulse-chase assay and lipidomic analysis of cells
Lipidomic experiments were performed as described previously [37].
Cell health assay
Protocols for the cell viability assay; the Seahorse assay; the measurement of intracellular and mitochondrial reactive oxygen species (ROS) levels, mitochondrial membrane potential (MMP), and mitochondrial mass; and mitochondrial morphology analysis are provided in the supplemental methods.
Statistics
Software R3.6.0 and GraphPad Prism 8.1.1 were used to conduct statistical analysis. Data are presented as the mean ± standard deviation (SD). Before statistical analysis, the Shapiro-Wilk test or Kolmogorov-Smirnov test was used to check the data distribution. Differences between two groups were analyzed using Student’s t test or a Mann-Whitney test for nonnormally distributed data. The Wilcoxon matched-pairs signed rank test or the t test, depending on the situation, were used for pairwise comparisons between two groups. Comparisons between multiple groups were analyzed using one/two-way ANOVA and a post hoc test (Tukey’s or Dunnett’s post hoc test) [38]. In detail, Tukey’s multiple-comparison test was used to analyze differences between the means of two or more independent groups and Dunnet’s multiple-comparison test was used to compare distinct groups against control group. Specifically, AcCa data obtained from DBS tests of tMCAO mice at multiple repeated time points were analyzed by repeated-measures ANOVA followed by Dunnett’s test for parametric analysis or Friedman’s test followed by Dunnett’s test for nonparametric analysis. Behavioral data collected at multiple repeated time points were analyzed using two-way repeated-measures ANOVA, followed by Tukey’s post hoc tests. The least absolute shrinkage and selection operator (LASSO) regression algorithm followed by assessment in a multivariate stepwise regression model was applied to select the significant diagnostic and prognostic features using 10-fold cross-validation. Areas under the receiver-operating characteristic (ROC) curve (AUC) for selected features were used to assess the classification performance of the model, and statistical significance between different models was calculated by the DeLong test. The statistical analyses of different experiments are indicated in each figure legend. P < 0.05 was considered significantly different.
Results
Lipid profiles of the ischemic penumbra and lipidomic analyses of significantly changed lipid species
Through untargeted lipidomic analysis, we provided comprehensive identification and quantification of lipid compositions in the ischemic penumbra (d1-ip, d2-ip, d3-ip) and the corresponding contralateral area (d1-con, d2-con, d3-con) at 1, 2, and 3 days after tMCAO followed by bioinformatic analysis to identify potential lipid biomarkers of AIS (Fig. 1A). First, the stability and reproducibility of liquid chromatography MS (LC–MS) for large-scale sample analyses were observed in total ion chromatograms (Supplementary Fig. 1Aa) and the principal component analysis (PCA) plot (Supplementary Fig. 1Ab). The response strength and retention time of each chromatographic peak overlapped, indicating that the experiment was reproducible. Then, the peaks extracted from all the experimental samples and all seven quality control (QC) samples were further analyzed by PCA after Pareto scaling. As shown in Supplementary Fig. 1Ab, QC samples were clustered together and located in the middle of each group, verifying the reproducibility of the results of this project. Second, a total of 1011 lipid species were identified through the integration of positive and negative electrospray ionization tandem MS. Lipid species were grouped into 30 lipid classes and 6 lipid categories according to their chemical structures (Supplementary Fig. 1B). Each lipid class contained a different number of lipid species. For example, triglycerides (TGs) consisted of 49 lipid species (Supplementary Fig. 1C). Finally, the unsupervised PCA method was performed to display the trends of separation between distinct groups. The presence of one outlier (d1-con-4) was highlighted, which was removed as well as its corresponding ipsilateral sample (d1-ip-4) (Supplementary Fig. 1D). The score plots illustrated good classification of the ischemic penumbra and its corresponding contralateral area over 3 days (Supplementary Fig. 1E, F).
Fig. 1.

OPLS-DA and WGCNA identified 16 significantly changed lipid species in the ischemic penumbra. (A) Diagram showing the experimental design. (B) The plot depicting separation of the d1-con (d2-con, d3-con) and d1-ip (d2-ip, d3-ip) utilizing the OPLS-DA model. (C) Venn diagram of significantly changed lipid species between ischemic penumbra and corresponding contralateral area among 3 days. Lipid species with VIP > 1.0 in the OPLS-DA model, fold change (FC) > 1.5 and statistical significance (p < 0.05, t-test) between ischemic penumbra and corresponding contralateral area are considered as significantly regulated lipid species. (D) The soft-thresholding powers selection of WGCNA analysis. The left panel shows the scale-free fit index (y-axis) as a function of soft-thresholding power (x-axis). The red line represents the y-axis value and here the adjacency matrix is defined using soft-thresholds with beta = 7. The right panel shows the mean connectivity (y-axis) as a function of soft-thresholding powers (x-axis). (E) Clustering dendrograms of lipid species, with dissimilarity based on topological overlap, together with assigned module colors. (F) The module-group relationship heatmap. Each column corresponds to a group. Each row corresponds to a module, labeled with a color as in Fig. 1E. The first line in every square is the corresponding correlation coefficient and the second line is the p-value. The left side of heatmap indicates the module name and the number of lipid species is indicated. The right side of heatmap indicates the colors of correlation (blue represents negatively correlated and red represents positively correlated). Modules with significantly changed lipid species are indicated with red underlines. (G) Venn diagram displaying the overlapping lipid species in WGCNA modules (magenta, greenyellow and pink modules, Fig. 1F) and OPLS-DA models (Fig. 1C). (H) Heatmap showing the abundance of 16 overlapping lipid species in ischemic penumbra and corresponding contralateral area among 3 days. (I) ROC curve for individual lipid specie in (Fig. 1H) to separate ischemic penumbra from corresponding contralateral area. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Next, orthogonal partial least squares discriminant analysis (OPLS-DA) was performed to minimize the possible contribution of intergroup variability and to further improve the separation between the two groups. The score plots from OPLS-DA models for Day 1, Day 2, and Day 3 showed a clear differentiation between the ischemic penumbra and the corresponding contralateral area (Fig. 1B). The model parameters for the explained variation, R2, and the predictive capability, Q2, were significantly high (R2, Q2 > 0.5) over 3 days, indicating excellent models (Supplementary Fig. 2A). Important lipid species driving separation between the groups were identified using high variable importance in projection (VIP) scores (which estimate the relative importance of a variable in the OPLS-DA model). Given that a major aim of this study was to find a series of stable biomarkers in the early stage of ischemic stroke, we next defined potential lipid biomarkers as significantly regulated lipid species with a VIP score > 1, p < 0.05, and fold change > 1.5 over 3 days, and 47 lipid species were found (Fig. 1C and visualized in a heatmap(Supplementary Fig. S2B), a large number of which were phosphatidylethanolamine (PE, 17/47), acylcarnitine (AcCa, 7/47), ceramides (Cers, 6/47), TG (4/47), and sphingomyelin (SM, 4/47).
Weighted gene coexpression network analysis (WGCNA) is a systematic biological method for describing the patterns of metabolite association among different samples [39], [40]. It can be used to identify highly synergistic metabolite sets, candidate biomarkers, and metabolic targets according to the connectivity of the metabolite sets. The relative abundance of 1011 identified lipid species in six groups (d1-con/d1-ip, d2-con/d2-ip, d3-con/d3-ip) was used to develop coexpression modules with the WGCNA package. We used the filter (cut height = 50, red line) to remove outlier samples in the sampleTree, and the d1-con-7, d1-ip-1, d2-ip-8, and d3-ip-4 samples were excluded after removing outliers in the sample based on the lipidomic data (Supplementary Fig. S2C). When the power value was equal to 7, the scale R2 reached 0.85 (Fig. 1D). Therefore, we defined the adjacency matrix using soft thresholding with beta = 7 to construct and identify distinct coexpression lipid modules. The identified coexpression modules were distributed in different colors (Fig. 1E). Notably, 3 of 12 coexpression modules were composed of lipid species that were significantly upregulated in at least one ischemic penumbra group (Fig. 1F). The pink module consisted of 39 lipid species specific to the ischemic penumbra over 3 days. The greenyellow module, with 5 lipid species, was highly and positively associated with d2-ip and d3-ip, and the magenta module (26 lipid species) was highly and positively associated with d3-ip.
To further investigate the significance of the lipid species, we utilized both a priori (OPLS-DA method) and agnostic approaches (WGCNA method). We then defined potential lipid biomarkers as lipid species that were identified in both WGCNA modules (magenta, greenyellow, and pink modules, Fig. 1F) and OPLS-DA models (Fig. 1C), and we found 16 overlapping lipid species (Fig. 1G). Validation using unsupervised clustering revealed perfect classification for the ischemic penumbra and corresponding contralateral area (Fig. 1H). Additionally, receiver operating characteristic (ROC) analysis indicated that the 16 potential lipid biomarkers had high area under the curve (AUC) values (Fig. 1I).
AcCas markedly accumulate in the ischemic penumbra and peripheral blood of tMCAO mice
LCACs, which accounted for 5 lipid species among the 16 potential lipid biomarkers, are helpful for the early differential diagnosis of AIS from patients with vertigo [13]. In our initial analysis, only relative quantification of LCACs was performed; thus, we performed ultrahigh-performance liquid chromatography–tandem MS (UHPLC-MS/MS) analysis. Forty-two AcCas, including short chain AcCas (SCACs) (C2–C5), medium chain AcCas (MCACs) (C6–C12), and LCACs (>C12), were successfully identified and quantified from the brain samples. First, PCA showed clear differentiation between the ischemic penumbra (d1-ip, d2-ip, and d3-ip) and the corresponding contralateral area (d1-con, d2-con, d3-con) (Fig. 2A). The sum of AcCas, SCACs, MCACs, and LCACs was elevated in the ischemic penumbra, with the greatest increase in the d3-ip group (Fig. 2B, C). As shown in Fig. 2D, the marked elevation in the LCAC pattern was dominated by AcCa(C16), AcCa(C16:1), AcCa(C18), and AcCa(C18:1).
Fig. 2.

Acylcarnitine species accumulate markedly in the ischemic penumbra and peripheral blood in tMCAO mice. (A) PCA plot depicting separation of the ischemic penumbra (d1-ip, d2-ip, d3-ip) and corresponding contralateral area (d1-con, d2-con, d3-con) utilizing the first two components of the PCA model. (B) Concentration of total AcCa, SCAC, MCAC and LCAC in the ischemic penumbra and corresponding contralateral area among 3 days as measured by UHPLC-MS/MS. Data are presented as median ± max/min (N = 5 each group). Significant tMCAO effects (two-way ANOVA): &&&p < 0.001. Significant time effects (two-way ANOVA): #p < 0.05, ##p < 0.01, ###p < 0.001. Significant tMCAO-time interaction effects: $p < 0.05, $$p < 0.01, $$$p < 0.001. Significant tMCAO effects within each day (unpaired, two-tailed t test): *p < 0.05, **p < 0.01, ***p < 0.001. (C) Heatmap displaying the abundance of the 42 AcCa species in ischemic penumbra and corresponding contralateral area among 3 days. N = 5 each group. (D) Significantly accumulated AcCa species in the ischemic penumbra compared with corresponding contralateral area (p < 0.05 and FC > 1.5 within each day). Size of the circles indicates fold changes and colour of the circles indicates subgroups of AcCa. (E) PCA plot depicting separation of time-course lipidomic data from DBS samples of tMCAO mice 1 h before tMCAO as well as 6 h, 1 d, 3 d and 7 d after tMCAO. (F-G) Fuzzy c-means clustering identified 6 distinct temporal patterns of AcCa expression and AcCa species in each cluster were displayed. (H) Concentration of AcCa(C16), AcCa(C16:1), AcCa(C18), and AcCa(C18:1). Data are presented as mean ± SD (N = 8 each group). *p < 0.05, **p < 0.01, repeated measures one-way ANOVA followed by Dunnett's multiple comparisons test for AcCa(C16) and AcCa(18:1) and Friedman’s test for AcCa(C16:1) and AcCa(C18). (I) Relative level of AcCa(C8/C10) and AcCa(C3/C16). ***p < 0.01, repeated measures one-way ANOVA followed by Dunnett's multiple comparisons test.
Next, we examined the level of AcCas in the peripheral blood of tMCAO mice (before and after surgery). We performed direct infusion MS, a clinically well-established and fast method that can be performed within 1–2 min of DBS sampling instead of the complicated and time-consuming procedures of UHPLC-MS/MS [13]. We applied the fuzzy c-means algorithm [41] to cluster AcCa (including 16 ratios among the quantified AcCa species) expression profiles at different stages during tMCAO. In total, we observed six distinct clusters of temporal patterns (Fig. 2E, F). AcCa species in Clusters 1, 2, 5, and 6 increased after tMCAO and subsequently declined thereafter (Fig. 2G). Consistent with our previous results, all the LCACs fell into these clusters, indicating incomplete FAO after stroke. As shown in Fig. 2H, AcCa(C16) and AcCa(C18) significantly increased on day 3 after tMCAO and declined to similar level with pre-tMCAO on day 7 after tMCAO. AcCa(C18:1) increased significantly on 6 h after tMCAO while no significant change was observed in AcCa(C16:1) after tMCAO. In addition, variables in Cluster 3 and 4 mainly consist of ratios of AcCa species. Variables in cluster 3 decreased on day 1 after tMCAO and subsequently increase thereafter while variables in cluster 4 subsequently increase after tMCAO and persist until day 7 after tMCAO (Fig. 2I).
LCACs are elevated in the peripheral blood of AIS patients
We then performed direct infusion MS to quantify AcCa levels in AIS patients (AIS) and healthy controls (HC). The sociodemographic and clinical characteristics of the subjects are listed in Supplementary Table 1. The score plots from OPLS-DA models showed clear differentiation between AIS patients and healthy controls (Fig. 3A, Supplementary Fig. 3A). Twenty-four significantly changed AcCas were found (Fig. 3B). Potential lipid biomarkers were defined as AcCas with a VIP > 1, p < 0.05, and FC > 1.2 or FC < 0.833, and 14 AcCas were found (Supplementary Fig. 3B). However, it is rather complicated for clinical use with 14 AcCas and we constructed a multivariate regression model based on 14 potential biomarkers and LASSO logistic regression classifiers based on all AcCas to find the simplest combination of AcCas with satisfactory diagnostic performance. Of 14 potential biomarkers assessed in the multivariate regression model, only 4 AcCas including AcCa(C2), AcCa(C12:1), AcCa(C14), and AcCa(C18:1-OH) were retained and the AUC of the 4 combined AcCas was 0.916 (Supplementary Fig. 3C). The results of LASSO logistic regression classifier are shown in Supplementary Fig. 3D and a panel of only 3 AcCas including AcCa(C4DC + C5-OH), AcCa(C12:1), and AcCa(C14-OH) achieved better AUC values (0.943) and may serve as a promising biomarker for AIS diagnosis (Fig. 3C). To explore the dynamic change in AcCa levels during ischemic stroke, eighteen AIS patients receiving mechanical thrombectomy were enrolled, and DBS samples were collected before and 1 day after mechanical thrombectomy. As shown in Fig. 3D and Fig. 3E, the concentrations of MCACs and LCACs decreased significantly after mechanical thrombectomy. We further investigated whether the baseline levels of AcCas were correlated with stroke outcome assessed by 3-month mRS. The sociodemographic and clinical characteristics of the subjects are listed in Supplementary Table 2. We applied a LASSO regression algorithm based on profiling AcCa levels from DBS samples (Fig. 3F), and 3 variables (AcCa(C3DC) + AcCa(C4-OH), AcCa(C3)/AcCa(C16), AcCa(C8)/AcCa(C10)) with nonzero coefficients were retained. The three retained variables were used for multivariate stepwise regression analysis. Among these, AcCa(C3)/AcCa(C16) and AcCa(C8)/AcCa(C10) were independent predictors for 3-month mRS of AIS patients. A significant difference in the baseline AcCa(C3)/AcCa(C16) and AcCa(C8)/AcCa(C10) levels between AIS patients with good outcomes and those with poor outcomes is shown in Fig. 3G. In addition, by adding the two variables into a simple clinical model (including age, sex and NIHSS score at admission), the AUC value was significantly increased from 0.781 to 0.883 (Fig. 3H, Supplementary Table 3). Furthermore, Spearman correlation analysis was used to investigate whether AcCa profiles were correlated with clinical phenotypes of AIS patients including demographic characteristics (age, sex), comorbidities (DM, high blood pressure (HBP), smoke, hyperlipidemia (HL)), stroke severity (infarct volume, penumbra volume and NIHSS score at admission), etiology of AIS and hemorrhagic transformation(Supplementary Fig. 4). Several SCACs including AcCa(C4DC-OH), AcCa(C5) and AcCa(C5DC-OH) were associated with DM, HBP and HL. There was a significant positive correlation between AcCa(C5:1) and infarct volume. Positive correlation between AcCa(C6DC) and penumbra volume was also observed. For LCACs, we found that AcCa(C14:2) and AcCa(C14-OH) negatively correlated with smoke history. AcCa(C16) was positively correlated with HL. Interestingly, we found AcCa(C18-OH) negatively correlated with penumbra volume. In addition, no significant correlations between LCACs and age, sex, DM, HBP, infarct volume, NIHSS score at admission, stroke etiology and HT were found in our cohort.
Fig. 3.

LCAC is upregulated in the peripheral blood ofAISpatients. (A) The plot depicting separation of HC group and AIS group utilizing the OPLS-DA model of the AcCa profiling. (B) Heatmap displaying the abundance of significantly regulated AcCa species in HC and AIS patients. (N = 33 in HC group and N = 33 in AIS group, all p < 0.05, unpaired, two-tailed t-test for AcCa(C2), AcCa(C3),AcCa(C16), AcCa(18), AcCa(C18:1) and unpaired, Mann Whitney test for other AcCas. (C) ROC curve for individual AcCa(C4DC + C5-OH), AcCa(C12:1), AcCa(C14-OH) and the combination of the three AcCas to separate AIS group from HC group. (D) The concentration of SCAC, MCAC, and LCAC of AIS patients before mechanical thrombectomy versus the day 1 after surgery. (two-tailed Student’s t-test for pairwise comparisons) (E) The concentration of significantly changed MCAC and LCAC of AIS patients before mechanical thrombectomy versus the day 1 after surgery. (N = 18 each group, all p < 0.05, paired, two-tailed t-test for AcCa(C16), AcCa(C18), AcCa(C18:1), AcCa(C18:2) or Wilcoxon matched-pairs signed rank test for pairwise comparisons for other AcCas) (F) LASSO logistic regression for AcCa features selection and signature construction. On the basis of one standard error of the minimum criteria for the least cross-validation binominal deviance, a tuning parameter (λ) was selected via 10-fold cross-validation. The vertical line indicates the optimal λ value (λ = 0.108, log (λ) = − 2.230) resulting in three features (AcCa(C3DC) + AcCa(C4-OH), AcCa(C3)/AcCa(C16), AcCa(C8)/AcCa(C10)) with nonzero coefficients (LASSO coefficient profiles) according to 10-fold cross-validation. (G) The ratio of AcCa(C3)/AcCa(C16) and AcCa(C8)/AcCa(C10) on the 1st day in AIS patients with good (mRS 0 to 2) outcomes versus poor (mRS 3 to 6) outcomes 3 months later. (H) ROC curve for Model 1 (include age, sex and NIHSS) and Model 2 (include Model 1 and the ratio of AcCa(C3)/AcCa(C16) and AcCa(C8)/AcCa(C10)) to separate AIS patients with good outcomes from poor outcomes 3 months later.
CPT2 overexpression decreases LCAC levels in vitro and in vivo
An accumulation of LCACs is also a hallmark of FAODs, especially CPT2 deficiency [42]. Therefore, we determined whether CPTs, including CPT1, the rate-limiting enzyme for mitochondrial long-chain fatty acid oxidation, and CPT2, the only enzyme responsible for LCAC metabolization, contribute to the accumulation of LCACs during stroke. As shown in Fig. 4A, B, CPT1 activity was increased on Days 1 and 3 after tMCAO; meanwhile, CPT2 activity was significantly decreased on Days 1 and 3 after tMCAO, suggesting that CPT dysregulation participated in LCAC accumulation in stroke. Thus, we surmised that CPT2 overexpression might have therapeutic potential for reducing LCAC accumulation.
Fig. 4.

CPT2 overexpression decreases LCAC levels in vitro and in vivo. (A-B) CPT1 and CPT2 activity were determined in isolated mitochondria extracted from ischemic penumbra in tMCAO mice and corresponding area in sham mice. Data are presented as mean ± SD (N = 7 each group). *p < 0.05, ***p < 0.001, one-way ANOVA followed by Dunnett's multiple comparisons test. (C) Relative abundance of the AcCa(C16), AcCa(C18), AcCa(C18:1) and LCAC (the sum of AcCa(C16), AcCa(C18), and AcCa(C18:1)) in ischemic penumbra after 3 days in tMCAO mice or corresponding area in sham mice. Data are presented as mean ± SD (N = 5–6 each group). *p < 0.05, **p < 0.01, one-way ANOVA followed by Tukey's multiple comparisons test. (D, G, J) Microglia (D), neuron (G), astrocyte (J) cultures were immunostained for Iba-1, MAP2, GFAP to label microglia, neuron, astrocyte, respectively. scale bar = 50 μm. (E, H, K) PCA plot depicting separation of the control and OGD/R microglia (E), neuron (H) and astrocytes (K) utilizing the first two components of the PCA model. (F, I, L) Heatmap of detected AcCa in microglia (F), neuron (I) and astrocytes (L). (M) Relative abundance of the AcCa(C16), AcCa(C18), AcCa(C18:1) and LCAC (the sum of AcCa(C16), AcCa(C18), and AcCa(C18:1)) in the control and OGD/R astrocytes. Data are presented as mean ± SD (N = 5 each group). *p < 0.05, **p < 0.01, ***p < 0.001, unpaired, two-tailed t test. (N) Relative abundance of the AcCa(C16), AcCa(C18), AcCa(C18:1) and LCAC (the sum of AcCa(C16), AcCa(C18), and AcCa(C18:1)) in the OGD/R astrocytes with Ad-GFP or Ad-CPT2 infection. Data are presented as mean ± SD (N = 5–6 each group).
We injected adult mice with adenovirus containing green fluorescent protein (Ad-GFP)-CPT2 or adenovirus expressing GFP alone (Ad-GFP) 3 days before tMCAO. First, the efficacy of Ad-CPT2 in the cortex was verified by immunofluorescence staining and Western blotting (Supplementary Fig. 5A, B). We found that GFP colocalized with the astrocyte marker GFP rather than the neuronal marker MAP2 and microglia marker TMEM119 (Supplementary Fig. 5C, D), which supports that fatty acid oxidation in the brain occurs exclusively in astrocytes [43], [44]. As expected, tMCAO mice with Ad-GFP microinjection exhibited significantly increased LCACs compared with sham mice, which was partly reversed by Ad-CPT2 microinjection (Fig. 4C).
Then, we determined which cell type was responsible for the increased LCAC level detected in the ischemic penumbra. Different types of primary cultured cells from the brain including astrocytes, microglia, and neurons, were isolated and exposed to OGD/R, the in vitro model of stroke, and then their AcCa profiles were identified (Fig. 4D-L). The PCA plot revealed a clear separation of LCAC levels between normoxic control astrocytes and OGD/R astrocytes (Fig. 4J-L), and the concentration of LCACs was significantly increased during OGD/R (Fig. 4L, M). However, there was no significant difference in LCAC levels in neurons or microglia (Fig. 4F, I), suggesting that astrocytes were responsible for the accumulation of LCACs in the ischemic penumbra. Next, we determined whether overexpressed CPT2 was able to lower LCAC levels during OGD/R in astrocytes. First, efficient CPT2 overexpression in astrocytes was validated by Western blotting and immunostaining staining (Supplementary Fig. 5E, F). As shown in Fig. 4N, Ad-CPT2 caused a trend of decreased LCAC levels in OGD/R astrocytes. Therefore, overexpressed CPT2 was capable of decreasing LCAC levels in vitro and in vivo.
Decreasing LCACs by overexpressing CPT2 is neuroprotective against stroke in vitro and in vivo
Next, we examined the effect of LCACs on stroke injury by intravenously injecting AcCa(C16), the potential predictor for outcome of AIS patients and mostly changed LCAC in the ischemic penumbra of tMCAO mice and peripheral blood of tMCAO mice as well as AIS patients, into tMCAO mice at reperfusion to increase LCAC levels and by microinjecting adult mice with Ad-CPT2 3 days before tMCAO to decrease LCAC levels (Fig. 5A). As shown in Fig. 5B, intravenous administration of AcCa(C16) successfully led to an increase in LCAC levels in the ischemic penumbra at Day 1 after tMCAO, indicating the penetration of AcCa(C16) into the brain. Moreover, AcCa(C16)-treated mice displayed increased infarct volume (Fig. 5C) and worsened neurological deficits (Fig. 5D).
Fig. 5.

Decreased LCAC by overexpressing CPT2 is neuroprotective after stroke in vivo. (A) Diagram showing the experimental design. (B) Relative abundance of the AcCa(C16), AcCa(C18), AcCa(C18:1) and LCAC (the sum of AcCa(C16), AcCa(C18), and AcCa(C18:1)) in ischemic penumbra in tMCAO mice or corresponding area in sham mice. Data are presented as mean ± SD (N = 5 each group). *p < 0.05, **p < 0.01, one-way ANOVA followed by Dunnett's multiple comparisons test. (C) Infarct volume was quantified in TTC-stained brain sections at 24 h after tMCAO. Data are presented as mean ± SD (N = 8 each group). **p < 0.01, unpaired, two-tailed t test. Scale bar = 1 cm. (D) Neurological deficits were measured at 24 h after tMCAO by mNSS. Data are presented as mean ± SD (N = 8 in each group). *p < 0.05, unpaired, two-tailed t test. (E) Infarct volume was quantified in TTC-stained brain sections at 3 d after tMCAO. Data are presented as mean ± SD (N = 8–9 each group). *p < 0.05, unpaired, two-tailed t test. Scale bar = 1 cm. (F) Neurological deficits were measured at 1–3 d after tMCAO by mNSS. Data are presented as mean ± SD (N = 12 in each group). *p < 0.05, **p < 0.01, two-way repeated-measures ANOVA followed by Tukey's multiple comparisons test. (G) Atrophy volume was quantified in Nissl-stained brain sections at 28 d after tMCAO. Data are presented as mean ± SD (N = 7–8 in each group). *p < 0.05, unpaired, two-tailed t test. (H) Neurological deficits were measured at 7 d, 14 d, 21 d, and 28 d after tMCAO by mNSS. Data are presented as mean ± SD (N = 7–8 in each group). *p < 0.05, **p < 0.01, two-way repeated-measures ANOVA followed by Tukey's multiple comparisons test. (I) Percentage of right turn of 10 times tests were analyzed at 7 d, 14 d, 21 d, and 28 d after tMCAO. Data are presented as mean ± SD (N = 7–8 in each group). *p < 0.05, **p < 0.01, two-way repeated-measures ANOVA followed by Tukey's multiple comparisons test. (J) Latency to move a body length were measured at 1w, 2w, 3w, 4w after tMCAO. Data are presented as mean ± SD (N = 7–8 in each group). *p < 0.05, **p < 0.01, **p < 0.01, two-way repeated-measures ANOVA followed by Tukey's multiple comparisons test.
Then, we injected adult mice with Ad-GFP-CPT2 or Ad-GFP 3 days before tMCAO to decrease the LCAC levels. TTC staining and mNSS tests at 1–3 days after tMCAO were used to evaluate the infarct volume and neurological deficits. Compared to tMCAO mice with Ad-GFP pretreatment, tMCAO mice with Ad-CPT2 pretreatment exhibited significantly decreased infarct volume at 3 days after tMCAO (Fig. 5E) and improved neurological function at 1–3 days after tMCAO (Fig. 5F). Furthermore, we investigated whether Ad-CPT2 exerts long-term protection in tMCAO mice. Our results showed that tMCAO mice pretreated with Ad-CPT2 exhibited attenuated brain atrophy at 28 days after tMCAO, improved mNSS test scores, and enhanced motor recovery, including turner test score values and latency (Fig. 5G-J).
Furthermore, we investigated the neuroprotective mechanism of the effects of Ad-CPT2 in vitro. Similar to the in vivo findings, CPT2 overexpression exerted neuroprotective effects in vitro. The results of the CCK8 assay showed concentration-dependent effects of Ad-CPT2 on astrocytes at different MOIs, and an MOI of 12.5 showed the largest increase in Ad-CPT2 astrocytes compared with Ad-GFP astrocytes under OGD/R conditions, which was used for subsequent in vitro studies (Fig. 6A). Moreover, Ad-CPT2 reversed the decrease in cell viability caused by AcCa(C16) (Fig. 6B). We determined the potential of Ad-CPT2 astrocytes to protect neuronal survival using the neuron-astrocyte coculture system (Fig. 6C). OGD/R astrocytes pretreated with Ad-GFP inhibited the protective effects of astrocytes under normoxic condition on neuronal viability, whereas OGD/R astrocytes pretreated with Ad-CPT2 partly reversed the neuroprotective effects (Fig. 6D). The neuroprotective effect of Ad-CPT2 was also confirmed by MAP2 immunostaining. A significant decrease in total dendritic length and reduced morphological complexity were observed in OGD/R neurons and OGD/R neurons cocultured with OGD/R astrocytes pretreated with Ad-GFP, and these effects were ameliorated in OGD/R neurons cocultured with OGD/R astrocytes pretreated with Ad-GFP (Fig. 6E–G).
Fig. 6.

Decreased LCAC by overexpressing CPT2is neuroprotective after stroke in vitro. (A) Astrocytes were subject to OGD/R after 2 days infection with Ad-GFP or Ad-CPT2 and then cell viability was measured using the CCK8 assay. Data are presented as mean ± SD (N = 5 each group). *p < 0.05, **p < 0.01, one-way ANOVA followed by Dunnett's multiple comparisons test. (B) Astrocytes were subject to OGD for 6 h and incubated with vehicle or AcCa(C16) for 24 h at the initiation of reoxygenation after 2 days infection with Ad-GFP or Ad-CPT2 and then cell viability was measured using the CCK8 assay. Data are presented as mean ± SD (N = 4 each group). **p < 0.01, one-way ANOVA followed by Dunnett's multiple comparisons test. (C) Schematic diagram of neuron-astrocyte co-culture system to determine the effect of astrocytes with Ad-CPT2 overexpression on neuron viability and morphology. (D) Cell viability of neurons in co-cultured system exposed to OGD/R. (E-G) Representative images of primary neurons (E), sholl analysis (F), and total dendritic length (G) from neurons in co-cultured system. Data are presented as mean ± SD (N = 35–50 cells from 5 coverslips per group.). *p < 0.05, ***p < 0.001, one-way ANOVA followed by Tukey's multiple comparisons test (F); ***p < 0.001: significant differences were observed at 20 –240 pixels from the soma, ##p < 0.01: significant differences were observed at 40 –110 pixels from the soma, two-way ANOVA followed by Tukey’s post hoc multiple-comparison tests (G). Scale bar = 20 μm.
Excess AcCa(C16:0) aggravates astrocytic mitochondrial damage in vitro
We determined whether the overloading of AcCa(C16) in astrocytes alone was sufficient to induce mitochondrial toxicity and cell death during stroke. Supplementation with AcCa(C16) led to dose-dependent cell death in primary astrocytes (Fig. 7A). LCAC-induced cell toxicity recapitulated mitochondrial damage, including increased total ROS levels (>25 μM) and total mitochondrial ROS levels and decreased MMP (>50 μM) (Fig. 7B–D). Astrocyte metabolic health was also examined by the Seahorse XFe96 analyzer. Real-time measurement of the mitochondrial oxygen consumption rate (OCR) showed no significant difference in basal mitochondrial respiration, ATP production, or maximal mitochondrial respiration between AcCa(C16)- and vehicle-treated normoxic astrocytes (Fig. 7E). However, there was a significant decrease in basal mitochondrial respiration, ATP production, and maximal mitochondrial respiration in 100 μM AcCa(C16)-treated OGD/R astrocytes (Fig. 7F). Moreover, 25 μM AcCa(C16) tended to disrupt mitochondrial structural integrity with increased fragmented mitochondria in OGD/R astrocytes (Fig. 7G, H). The ultrastructure of astrocytic mitochondria in the ischemic penumbra was further visualized using transmission electron microscopy, which showed a typical morphology with a dense cytoplasmic matrix and an augmented number of glycogen, and lipofuscin granules [45]. We observed that the astrocytic mitochondria in the tMCAO + saline group presented with swelling and crest fracture, which was more severe in the tMCAO + AcCa(C16) group (Fig. 7I). Moreover, the downregulation of LCACs by Ad-CPT2 administration reversed the decreased MMP caused by OGD/R and AcCa(C16) (Fig. 7J, K). Altogether, these results indicate that excess AcCa(C16) aggravates stroke injury and mitochondrial damage.
Fig. 7.

Excess AcCa(C16:0) amplifiesastrocytic mitochondrial damage from OGD/R. (A) Cell viability astrocytes exposed to AcCa(C16) under control or OGD/R conditions. Data are presented as mean ± SD (N = 5 each group). Significant OGD/R treatment (two-way ANOVA): ###p < 0.001. Significant LCAC treatment in OGD/R group (two-way ANOVA followed by Dunnett's multiple comparisons test, *p < 0.05, **p < 0.01, ***p < 0.001) (B-D) Total ROS using DCFH-DA (B) (N = 5 each group), mitochondrial ROS levels using MitoSOX (C) (N = 5 each group), mitochondrial membrane potential using TMRM (D) (N = 5 each group) were determined 24 h after initiation of reoxygenation with vehicle or AcCa(C16) after OGD 6 h. Data are presented as mean ± SD. Significant OGD/R treatment (two-way ANOVA): ###p < 0.001. Significant LCAC treatment in OGD/R group (two-way ANOVA followed by Dunnett's multiple comparisons test, *p < 0.05, **p < 0.01, ***p < 0.001). (E-F) Oxygen consumption rate (OCR) in control (E) or OGD/R astrocytes (F) treated with vehicle or AcCa(C16). Oligo, oligomycin; FCCP, carbonyl cyanide 4-trifluoromethoxy-phenylhydrazone; Rot, rotenone. Data are presented as mean ± SD (N = 10 wells from 3 independent experiments). *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA followed by Tukey's multiple comparisons test. (G-H) Representative photomicrographs of astrocytes (G) and mitochondrial mean length of rods/branches (H) and at 24 h after initiation of reoxygenation with vehicle or AcCa(C16) after OGD 6 h. Data are presented as mean ± SD (N = 22–28 mitochondria from 3 independent experiments, each point indicates a single mitochondrion). **p < 0.01, unpaired, two-tailed t test. Significant OGD/R treatment (two-way ANOVA): ###p < 0.001. Scale bar = 10 μm. (I) Representative images of astrocytic mitochondria in the ischemic penumbra of tMCAO mice. (J) TMRM of control astrocytes and OGD/R astrocytes infected with Ad-GFP or Ad-CPT2 were determined 24 h after initiation of reoxygenation. Data are presented as mean ± SD (N = 3–4). *p < 0.05, ***p < 0.001, one-way ANOVA followed by Tukey's multiple comparisons test. (K) TMRM of control astrocytes and OGD/R astrocytes infected with Ad-GFP or Ad-CPT2 were determined 24 h after initiation of reoxygenation with AcCa(C16) after OGD 6 h. Data are presented as mean ± SD (N = 3 each group). *p < 0.05, ***p < 0.001, one-way ANOVA followed by Tukey's multiple comparisons test.
Astrocytes liberate FFAs from LDs into mitochondria to form LCACs during OGD/R
Elevated LCACs, coupled with mitochondrial dysfunction and oxidative stress, are reportedly caused by LD disturbance [17], [37]. In our initial untargeted lipidomic analysis, not only LCAC species but also several TG species were identified as potential biomarkers (Fig. 1H, I). Next, we explored how LD metabolism is correlated with increased LCACs in astrocytes during OGD/R.
We first quantified TG levels in the ischemic penumbra during tMCAO/R, which were significantly increased on Days 3 and 7 after tMCAO (Fig. 8A). BD493 and oil red O staining also identified numerous lipid droplets in the peri-infarct area in tMCAO mice (Fig. 8B). The LD membrane protein TIP47 or BD493 colocalized with the astrocyte marker GFAP (Fig. 8C) [17], indicating LD formation in astrocytes following tMCAO. Consistently, astrocytes induced numerous LDs during OGD/R (Fig. 8D). Using the neuron-astrocyte coculture system, which enables Red C12, a fluorescent FFA exchange but not physical contact, we found that OGD induced neurons to transfer FFAs to astrocytes, which then formed into LDs in astrocytes (Supplementary Fig. 6). Next, we determined whether astrocytic LDs are mobilized into FFAs through lipolysis or lipophagy during OGD/R. ATGL, the rate-limiting cytoplasmic lipase, rather than LAMP1 or LC3, the main lipophagic machinery, localized to LDs in OGD/R astrocytes (Supplementary Fig. 7A). Additionally, LDs were significantly increased with the ATGL inhibitor (ATGLi) atglistatin (Supplementary Fig. 7B); however, LD number was not changed with the lysosome inhibitor chloroquine (Supplementary Fig. 7C), indicating that astrocytes utilize lipolysis rather than lipophagy to break down LDs during OGD/R. Furthermore, we used a Red C12 pulse-chase labeling assay to track FFAs in relation to LDs and mitochondria in astrocytes during OGD/R and the effect of inhibiting lipase activity with ATGLi treatment [46] (Fig. 8E). OGD/R astrocytes showed a significant increase in colocalization between the Red C12 signal and mitochondria compared with normoxic control astrocytes; however, the Red C12 signal remained in the LDs instead of mitochondria with ATGLi treatment of OGD/R astrocytes (Fig. 8F, G). To more specifically track FFA flux, we employed isotopic palmitate tracing [37]. Isotopically labeled palmitate (13C4-FFA) was added 24 h before OGD/R, and 13C4-FFA-incorporated LCACs were analyzed by LC-MS (Fig. 8H). As shown in Fig. 8I, ATGLi treatment significantly reduced 13C4-FFA incorporation into AcCa(C18:1) and LCACs (the sum of detected d4-incorporated AcCa(C14), AcCa(C16), AcCa(C18), and AcCa(C18:1)). Together, these results indicate that lipolysis is responsible for FFA delivery into the mitochondria to form LCACs during OGD/R in astrocytes, thus causing astrocytic mitochondrial damage to impair the energy supply for neurons.
Fig. 8.

Astrocytes liberateFFAs from LDs into mitochondria to form LCAC during OGD/R. (A) Total TG levels in ischemic penumbra and corresponding areas in sham or tMCAO mice (1 d, 3 d and 7 d after reperfusion). **p < 0.01, one-way ANOVA followed by Dunnett's multiple comparisons test. (B) Representative imaging of LDs stained with BD493 or oil red in the ischemic penumbra or and corresponding areas from sham or tMCAO mice (1 d, 3 d and 7 d after reperfusion). Scale bar = 500 μm. (C) Ipsilateral hemisphere from tMCAO/R 7 d mice was immunostained with astrocytes and TIP47 or BD493. Scale bar = 100 μm. (D) Astrocytes were exposed to OGD/R for the indicated times and BD493 was used to visualize LDs, and LD number was determined. Data are presented as mean ± SD (N = 6 each group). *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA followed by Tukey's multiple comparisons test. scale bar = 10 μm. (E) Schematic representation of the Red C12 pulse-chase assay: astrocytes were pulsed with Red C12 for 16 h, washed, and exposed to OGD 6 h/R 24 h with or without ATGLi at reoxygenation. Astrocytes were then imaged with BD493 or Mitotracker to determine the subcellular localization of the Red C12. (F-G) Astrocytes were assayed as described in Fig. 3E (F) and quantification of the correlation between Red C12 signal and mitochondria (upper panel) or LDs (lower panel). Data are presented as mean ± SD (N = 3 each group). *p < 0.05, **p < 0.01, one-way ANOVA followed by Tukey's multiple comparisons test. scale bar = 10 μm. (H-I) Schematic representation of the 13C4-FFA FFA pulse-chase assay: Astrocytes were first incubated with 13C4-FFA complexed with 0.5 % BSA for 24 h and exposed to OGD 6 h/R 24 h with or without ATGLi at reoxygenation (H) and relative abundance of d4-incooperated AcCa(C14), AcCa(C16), AcCa(C18), AcCa(C18:1) and LCAC (the sum of AcCa(C14), AcCa(C16), AcCa(C18), AcCa(C18:1)) was detected (I). Data are presented as mean ± SD (N = 5 each group). *p < 0.05, unpaired, two-tailed t test. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Discussion
The major findings in this study were as follows. 1) Increased LCACs were found in the ischemic penumbra of tMCAO mice and peripheral blood of both tMCAO mice and AIS patients. LCACs have the potential to serve as new diagnostic and prognostic biomarkers in AIS. 2) LCACs act as an important pathogenic driver rather than byproducts in FAO to induce astrocytic mitochondrial dysfunction to amplify stroke injury. Lowering LCACs or stimulating astrocytic FAO is capable of ameliorating stroke injury. 3) OGD/R stimulated neurons to transfer FFAs into astrocytes to form LDs, which could be liberated through lipolysis into mitochondria to form LCACs, thus causing mitochondrial dysfunction.
Lipidomics, the profiling of lipid species on a broad scale, has been used in a variety of metabolic stress conditions, including cancer, diabetes mellitus, and cardiovascular diseases; however, lipidomic profiles have been far less studied in neurological diseases, especially stroke [12], [47], [48]. In this study, we used untargeted lipidomic analysis to identify 1011 lipid species belonging to 30 lipid classes and identified 47 differentially expressed lipid species by OPLS-DA and WGCNA methods between the ischemic penumbra and contralateral hemisphere from Days 1 to 3 after tMCAO. Our data confirmed the presence of previously reported lipids after stroke, such as TGs, PEs, and Cers [9], [49], [50]. Among the 16 potential lipid biomarkers identified by untargeted lipidomic analysis of the ischemic penumbra of tMCAO mice, four are TGs. Yang et al. and Zheng et al. also reported that TGs were increased in the brain of tMCAO mice as well as peripheral blood of AIS patients [9], [50]. However, it remains uncertain whether higher or lower TGs are associated with severer stroke [51], [52]. One possible explanation for these controversial findings is that these investigations focused on total TG levels rather than individual TG species. Investigating the roles of TG species in AIS could bring new insights. Three PE species were identified in our untargeted lipidomic analysis. Increased PEs were also found in the ischemic brain of tMCAO mice and peripheral blood in AIS patients. However, the role of PE in general or specific PE species in stroke remains largely unknown. Cers are associated with neuroinflammation and mitochondrial dysfunction in the brain [53], [54]. Consistent with previous studies that Cers are increased in AIS patients and positively associated with stroke severity [55], the current study also found significantly increased Cers in the ischemic penumbra in tMCAO mice. More importantly, we showed for the first time that LCACs were sharply increased in the ischemic penumbra after stroke, which was then verified using targeted lipidomics. In vitro lipidomics further showed that astrocytes were responsible for the increased LCACs in the brain. Apart from FAODs, increased LCACs were also found in ischemic hearts and traumatic brains, indicating the universal role of LCACs in ischemic and mechanical injury-exposed tissues [14], [18].
Until now, several studies have applied metabolomic or lipidomic method for diagnosis of AIS [50], [56], [57], [58]. Compared with them, mostly of which depend on the typical LC-MS/MS-based metabolomic/lipidomic platform, the direct-infusion MS platform is able to measure 30 AcCas within several minutes and make it a promising tool for fast AIS diagnosis [59]. In this study, we collected DBS samples from clinical cohorts of AIS patients and found significantly increased AcCa levels, especially LCACs. The levels of AcCa(C16), AcCa(C18), AcCa(C18-OH) and AcCa(C18:1-OH) were also significantly increased in AIS patients compared with patients with vertigo in a previous metabolomics study [13]. Moreover, AcCa(C18:1) was also reported to be decreased to normal levels at 8 days after the occurrence of AIS. Interestingly, we also found that the levels of multiple AcCas, especially LCACs such as AcCa(C16), AcCa(C18), and AcCa(C18:1), were restored to normal levels after mechanical thrombectomy, suggesting partial recovery from AIS injury. Furthermore, our study revealed that decreased AcCa(C8)/AcCa(C10) and AcCa(C3)/AcCa(C16) were correlated with poor outcomes in AIS patients. Consistently, several clinical studies conducted in large numbers of subjects revealed that both MCACs and LCACs were associated with cardiovascular events independent of known risk factors of cardiovascular diseases [60], [61], [62]. To the best of our knowledge, this is the first study to highlight the predictive significance of LCACs for AIS patients. Our findings provided preliminary support for LCACs to develop into a novel diagnostic and prognostic biomarker that warrants further study.
It is also interesting to compare the findings of lipidomics from ischemic penumbra of tMCAO mice and peripheral blood of tMCAO mice as well as AIS patients. In the ischemic penumbra of tMCAO mice, the marked elevation of LCAC pattern was dominated by AcCa(C16), AcCa(C16:1), AcCa(C18), and AcCa(C18:1). These LCACs were all significantly increased in the peripheral blood of AIS patients while only AcCa(C16), AcCa(C18) and AcCa(C18:1) showed significantly increase in the peripheral blood of tMCAO mice. However, these LCACs did not retain in the final diagnosis panel constructed by lasso regression for AIS, which may be related to the high correlations between AcCa(C12:1) and AcCa(C16), AcCa(C16:1), AcCa(C18), and AcCa(C18:1). Thus, it is not surprising that LCACs could not completely overlap between different samples due to species difference, complexity of clinical trials, insufficient clinical sample size and statistical methods we used. Interestingly, we found that the prognostic variables identified in clinical patients (AcCa(C3/C16) and AcCa(C8/C10)) was in cluster 4 and AcCa(C3/C16) significantly increased on day 7 after tMCAO (Fig. 2). It is believed that functional rehabilitation process has occurred on day 7 after tMCAO in mice and increased AcCa(C3/C16) suggest a recovery of brain injury after stroke, which is consistent with our findings in AIS patients. Actually, SCACs with odd numbers of carbons like AcCa(C3) are branched-chain amino acids derived and associated with gut microbiota homeostasis, improved exercise capacity and oxygen consumption in chronic heart failure patients [63]. Thus, combination of AcCa(C3) and AcCa(C16) would provide more information about the prediction of outcome of AIS patients. No research has been reported the association of AcCa(C3) and AIS yet and further researches about the role of AcCa(C3) in AIS would be of interest. In summary, representative LCACs such as AcCa(C16) and AcCa(C18) were significantly elevated in the ischemic penumbra and peripheral blood of tMCAO mice and peripheral blood of AIS patients, and AcCa(C16) may predict the poor prognosis of AIS patients.
LCAC accumulation is associated with cancers and ischemic diseases [17], [64], [65]. Here, for the first time, we determined the role of LCACs in stroke injury by decreasing LCACs by overexpressing CPT2 before tMCAO and increasing LCACs via intravenous administration of AcCa(C16) during reperfusion in tMCAO. An infarct volume and neurological deficits study showed that increased LCACs amplified stroke injury, while decreased LCACs ameliorated stroke injury in the acute phase. Consistently, decreasing LCACs by overexpressing CPT2 was capable of attenuating brain atrophy, thus improving neurological deficits in the chronic phase of stroke. Excessive LCACs led to mitochondrial dysfunction and the induction of cell death in astrocytes. Severer astrocytic mitochondrial injury was also found after AcCa(C16) administration to tMCAO mice. Accordingly, decreasing LCACs by overexpressing CPT2 in astrocytes protected mitochondrial function, thus maintaining the viability of the cocultured neurons. These findings broaden a new vision for the role of LCACs in stroke.
Recently, it was discovered that FFA metabolism during neuron stimulation was coupled between neurons and astrocytes, suggesting that neurons had a self-protective mechanism while under stress [8], [66]. After delivery to astrocytes, FFAs are formed into LDs to avoid FFA overload and toxicity [67]. Using fluorescent and isotopic tracing of FFA flux during OGD/R in astrocytes, we showed that FFAs could be transported from neurons to astrocytes after OGD and revealed that astrocytic LDs liberated FFAs into LDs by lipolysis to form LCACs, which was ATGL-dependent. It is interesting to note that another study also supported the idea that, following activation, astrocytes prefer lipolysis over lipophagy for the purpose of LD degradation [8]. One reason may be related to the membrane contact of LDs with mitochondria via lipolysis, thus avoiding FFA overload in the cytoplasm via lipophagy [37], [46]. However, insufficient FAO during stroke led to the accumulation of other harmful lipid species, including LCACs, and the subsequent dysfunction of the mitochondria. These data suggest an interrelationship between stroke and LCACs: incomplete FAO due to mitochondrial dysfunction after ischemic stroke rapidly triggers LCAC accumulation, which in turn exacerbates mitochondrial dysfunction.
Notably, we found that numerous LDs were found in astrocytes in vitro and in vivo during stroke. Nevertheless, the functions of LDs in stroke are unclear. LDs may play protective roles, such as reducing toxicity caused by lipids and oxidative stress [8], [68]. The detrimental roles of LDs in glia, such as inducing oxidative stress, were also reported in neurogenerative diseases and the aging brain [69], [70]. In this study, astrocytic LDs liberated FFAs into mitochondria to form LCACs during OGD/R, supporting that LDs are antioxidative in stroke. Similarly, the inhibition of LD biogenesis by targeting DGAT1 during starvation resulted in an increase in LCACs and subsequent mitochondrial dysfunction [17], [37]. In addition to astrocytes, other cells also accumulate LDs, which play divergent roles after stroke. For example, LD-accumulating microglia exhibit decreased phagocytic ability and proinflammatory status in the aging brain [70]. A reshaping of cholesterol content in LDs was also found in endothelial cells, while its role in the neurovascular unit remains unknown [71], [72]. Thus, further studies are required to determine lipid alterations in other cell types and their distinct roles in stroke.
The current study has some limitations. First, only male mice were used in this study. Both male and female mice should be included in future preclinical studies. Second, long-term AcCa profiles of ischemic penumbra of tMCAO mice are required as predictors for the outcome of AIS and comparison with the findings in AIS patients. Third, the lipidomic analyses of AIS patients are tempered by small samples and prospective cohort studies with larger sample sizes are warranted to clarify the diagnostic and prognostic value of LCACs for AIS.
Taken together, the results of this study demonstrated that astrocytes liberated FFAs from LDs into mitochondria to form LCACs, which then damaged astrocytic mitochondria to amplify stroke injury. These findings suggest that LCACs have the potential to be develop into new diagnostic or prognostic biomarkers as well as novel therapeutic targets for AIS.
All of the animal experimental procedures were approved by The Institutional Animal Ethics Committee of Nanjing Medical University (approval number: IACUC- 1912026). We followed the ARRIVE (Animal Research: Reporting in Vivo Experiments) ethical guidelines. Written informed consent was obtained from participants or legal representatives for the use of patients’ blood before inclusion in the study, and the protocol was approved by the Ethical Committee of The First Affiliated Hospital of Nanjing Medical University, Nanjing, China (approval ID: 2021-SR-187).
Ethics
All of the animal experimental procedures were approved by The Institutional Animal Ethics Committee of Nanjing Medical University (approval number: IACUC- 1912026). We followed the ARRIVE (Animal Research: Reporting in Vivo Experiments) ethical guidelines. Written informed consent was obtained from participants or legal representatives for the use of patients’ blood before inclusion in the study, and the protocol was approved by the Ethical Committee of The First Affiliated Hospital of Nanjing Medical University, Nanjing, China (approval ID: 2021-SR-187).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This study was supported by the National Key R&D Program of China (No.2021ZD0202900), the National Natural Science Foundation of China (Nos. 81973301, 81971613 and 82003732), Medical Research Project of Jiangsu Commission of Health (No. ZDA2020006), Priority Academic Program Development of Jiangsu Higher Education Institutions, and the Postgraduate Research and Practice Innovation Program of Jiangsu Province (No.KYCX21_1585).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jare.2023.08.007.
Contributor Information
Sheng Liu, Email: liusheng@njmu.edu.cn.
Xiu-Lan Sun, Email: xiulans@njmu.edu.cn.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- 1.Feigin V.L., et al. Global, regional, and national burden of stroke and its risk factors, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 2021;20:795–820. doi: 10.1016/S1474-4422(21)00252-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ma H., et al. Thrombolysis Guided by Perfusion Imaging up to 9 Hours after Onset of Stroke. N Engl J Med. 2019;380:1795–1803. doi: 10.1056/NEJMoa1813046. [DOI] [PubMed] [Google Scholar]
- 3.Powers WJ et al., 2018 Guidelines for the Early Management of Patients With Acute Ischemic Stroke: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. (2018).
- 4.M. D, et al., Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation133 (2016). [DOI] [PubMed]
- 5.Jové M., et al. Metabolomics predicts stroke recurrence after transient ischemic attack. Neurology. 2015;84:36–45. doi: 10.1212/WNL.0000000000001093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi J.-Y., et al. High free fatty acid level is associated with recurrent stroke in cardioembolic stroke patients. Neurology. 2014;82:1142–1148. doi: 10.1212/WNL.0000000000000264. [DOI] [PubMed] [Google Scholar]
- 7.Xue T., et al. Sphingosine-1-phosphate, a novel TREM2 ligand, promotes microglial phagocytosis to protect against ischemic brain injury. Acta Pharm Sin B. 2022;12:1885–1898. doi: 10.1016/j.apsb.2021.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ioannou M.S., et al. Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell. 2019;177:1522–1535.e14. doi: 10.1016/j.cell.2019.04.001. [DOI] [PubMed] [Google Scholar]
- 9.Zheng L., et al. An imbalanced ratio between PC(16:0/16:0) and LPC(16:0) revealed by lipidomics supports the role of the Lands cycle in ischemic brain injury. J Biol Chem. 2021;296 doi: 10.1074/jbc.RA120.016565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramos-Lopes J., et al. Phospholipase A2 and Ischemic Stroke Etiology. Neurologist. 2021;26:32–35. doi: 10.1097/NRL.0000000000000329. [DOI] [PubMed] [Google Scholar]
- 11.McCoin C.S., Knotts T.A., Adams S.H. Acylcarnitines-old actors auditioning for new roles in metabolic physiology. Nat Rev Endocrinol. 2015;11:617–625. doi: 10.1038/nrendo.2015.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun L., et al. Early prediction of developing type 2 diabetes by plasma acylcarnitines: A population-based study. Diabetes Care. 2016;39:1563–1570. doi: 10.2337/dc16-0232. [DOI] [PubMed] [Google Scholar]
- 13.Sun R., Li Y., Cai M., Cao Y., Piao X. Discovery of a New Biomarker Pattern for Differential Diagnosis of Acute Ischemic Stroke Using Targeted Metabolomics. Front Neurol. 2019;10:1–7. doi: 10.3389/fneur.2019.01011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mallah K., et al. Matrix-Assisted Laser Desorption/Ionization-Mass Spectrometry Imaging of Lipids in Experimental Model of Traumatic Brain Injury Detecting Acylcarnitines as Injury Related Markers. Anal Chem. 2019;91:11879–11887. doi: 10.1021/acs.analchem.9b02633. [DOI] [PubMed] [Google Scholar]
- 15.Ebert D., Haller R.G., Walton M.E. Energy contribution of octanoate to intact rat brain metabolism measured by 13C nuclear magnetic resonance spectroscopy. J Neurosci. 2003;23:5928–5935. doi: 10.1523/JNEUROSCI.23-13-05928.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sayre N.L., et al. Stimulation of astrocyte fatty acid oxidation by thyroid hormone is protective against ischemic stroke-induced damage. J Cereb Blood Flow Metab. 2017;37:514–527. doi: 10.1177/0271678X16629153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng X., et al. Targeting DGAT1 Ameliorates Glioblastoma by Increasing Fat Catabolism and Oxidative Stress. Cell Metab. 2020;32:229–242.e8. doi: 10.1016/j.cmet.2020.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liepinsh E., et al. Long-chain acylcarnitines determine ischaemia/reperfusion-induced damage in heart mitochondria. Biochem J. 2016;473:1191–1202. doi: 10.1042/BCJ20160164. [DOI] [PubMed] [Google Scholar]
- 19.Guttenplan K.A., et al. Neurotoxic reactive astrocytes induce cell death via saturated lipids. Nature. 2021;599:102–107. doi: 10.1038/s41586-021-03960-y. [DOI] [PubMed] [Google Scholar]
- 20.Li L., et al. Oridonin prevents oxidative stress-induced endothelial injury via promoting Nrf-2 pathway in ischaemic stroke. J Cell Mol Med. 2021;25:9753–9766. doi: 10.1111/jcmm.16923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simcox J., et al. Global Analysis of Plasma Lipids Identifies Liver-Derived Acylcarnitines as a Fuel Source for Brown Fat Thermogenesis. Cell Metab. 2017;26:509–522.e6. doi: 10.1016/j.cmet.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y., et al. Intrastriatal transplantation of bone marrow nonhematopoietic cells improves functional recovery after stroke in adult mice. J Cereb Blood Flow Metab. 2000;20:1311–1319. doi: 10.1097/00004647-200009000-00006. [DOI] [PubMed] [Google Scholar]
- 23.Lubjuhn J., et al. Functional testing in a mouse stroke model induced by occlusion of the distal middle cerebral artery. J Neurosci Methods. 2009;184:95–103. doi: 10.1016/j.jneumeth.2009.07.029. [DOI] [PubMed] [Google Scholar]
- 24.Yang L., et al. Extracellular Vesicle-Mediated Delivery of Circular RNA SCMH1 Promotes Functional Recovery in Rodent and Nonhuman Primate Ischemic Stroke Models. Circulation. 2020;142:556–574. doi: 10.1161/CIRCULATIONAHA.120.045765. [DOI] [PubMed] [Google Scholar]
- 25.Wu F., et al. Circular RNA TLK1 Aggravates Neuronal Injury and Neurological Deficits after Ischemic Stroke via miR-335-3p/TIPARP. J Neurosci. 2019;39:7369–7393. doi: 10.1523/JNEUROSCI.0299-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang P., Qin Y., Zhang W., Bian Z., Wang R. Sensorimotor Cortex Injection of Adeno-Associated Viral Vector Mediates Knockout of PTEN in Neurons of the Brain and Spinal Cord of Mice. J Mol Neurosci. 2015;57:470–476. doi: 10.1007/s12031-015-0610-x. [DOI] [PubMed] [Google Scholar]
- 27.Campbell B.C.V., et al. Cerebral blood flow is the optimal CT perfusion parameter for assessing infarct core. Stroke. 2011;42:3435–3440. doi: 10.1161/STROKEAHA.111.618355. [DOI] [PubMed] [Google Scholar]
- 28.S. shan Lu, et al., Comparison of CT angiography collaterals for predicting target perfusion profile and clinical outcome in patients with acute ischemic stroke. Eur. Radiol.29, 4922–4929 (2019). [DOI] [PubMed]
- 29.Kumar L., Futschik M.E. Mfuzz: A software package for soft clustering of microarray data. Bioinformation. 2007;2:5–7. doi: 10.6026/97320630002005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schott M.B., et al. Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes. J Cell Biol. 2019;218:3320–3335. doi: 10.1083/jcb.201803153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo W., et al. Iptakalim alleviates synaptic damages via targeting mitochondrial ATP-sensitive potassium channel in depression. FASEB J. 2021;35:1–18. doi: 10.1096/fj.202100124RR. [DOI] [PubMed] [Google Scholar]
- 32.Li X., et al. SREBP-1c overexpression induces triglycerides accumulation through increasing lipid synthesis and decreasing lipid oxidation and VLDL assembly in bovine hepatocytes. J Steroid Biochem Mol Biol. 2014;143:174–182. doi: 10.1016/j.jsbmb.2014.02.009. [DOI] [PubMed] [Google Scholar]
- 33.Peffer P.L., et al. Ontogeny of carnitine palmitoyltransferase I activity, carnitine-K m, and mRNA abundance in pigs throughout growth and development. J Nutr. 2007;137:898–903. doi: 10.1093/jn/137.4.898. [DOI] [PubMed] [Google Scholar]
- 34.Ji J., et al. Antagonizing peroxisome proliferator-activated receptor γ facilitates M1-to-M2 shift of microglia by enhancing autophagy via the LKB1–AMPK signaling pathway. Aging Cell. 2018;17:1–16. doi: 10.1111/acel.12774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song X., et al. Baicalin combats glutamate excitotoxicity via protecting glutamine synthetase from ROS-induced 20S proteasomal degradation. Redox Biol. 2020;34 doi: 10.1016/j.redox.2020.101559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang J., et al. Neuronal extracellular vesicle derived miR-98 prevents salvageable neurons from microglial phagocytosis in acute ischemic stroke. Cell Death Dis. 2021;12 doi: 10.1038/s41419-020-03310-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nguyen T.B., et al. DGAT1-Dependent Lipid Droplet Biogenesis Protects Mitochondrial Function during Starvation-Induced Autophagy. Dev Cell. 2017;42:9–21.e5. doi: 10.1016/j.devcel.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kornberg M.D., et al. Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science (80-) 2018;360:449–453. doi: 10.1126/science.aan4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Su Y., et al. Metabolomic and network analysis of astaxanthin-producing Haematococcus pluvialis under various stress conditions. Bioresour Technol. 2014;170:522–529. doi: 10.1016/j.biortech.2014.08.018. [DOI] [PubMed] [Google Scholar]
- 40.Bhargava P., Fitzgerald K.C., Calabresi P.A., Mowry E.M. Metabolic alterations in multiple sclerosis and the impact of vitamin D supplementation. JCI Insight. 2017;2:1–13. doi: 10.1172/jci.insight.95302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Futschik M.E., Carlisle B. Noise-robust soft clustering of gene expression time-course data. J Bioinform Comput Biol. 2005;3:965–988. doi: 10.1142/s0219720005001375. [DOI] [PubMed] [Google Scholar]
- 42.Boufroura F.Z., et al. A new AMPK activator, GSK773, corrects fatty acid oxidation and differentiation defect in CPT2-deficient myotubes. Hum Mol Genet. 2018;27:3417–3433. doi: 10.1093/hmg/ddy254. [DOI] [PubMed] [Google Scholar]
- 43.Bernier L.-P., et al. Microglial metabolic flexibility supports immune surveillance of the brain parenchyma. Nat Commun. 2020;11:1559. doi: 10.1038/s41467-020-15267-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jernberg J.N., Bowman C.E., Wolfgang M.J., Scafidi S. Developmental regulation and localization of carnitine palmitoyltransferases (CPTs) in rat brain. J Neurochem. 2017;142:407–419. doi: 10.1111/jnc.14072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Castejón O.J. Electron microscopy of astrocyte changes and subtypes in traumatic human edematous cerebral cortex: A review. Ultrastruct Pathol. 2013;37:417–424. doi: 10.3109/01913123.2013.831157. [DOI] [PubMed] [Google Scholar]
- 46.Rambold A.S., Cohen S., Lippincott-Schwartz J. Fatty Acid Trafficking in Starved Cells: Regulation by Lipid Droplet Lipolysis, Autophagy, and Mitochondrial Fusion Dynamics. Dev Cell. 2015;32:678–692. doi: 10.1016/j.devcel.2015.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tham Y.K., et al. Lipidomic Profiles of the Heart and Circulation in Response to Exercise versus Cardiac Pathology: A Resource of Potential Biomarkers and Drug Targets. Cell Rep. 2018;24:2757–2772. doi: 10.1016/j.celrep.2018.08.017. [DOI] [PubMed] [Google Scholar]
- 48.Lin L., et al. Functional lipidomics: Palmitic acid impairs hepatocellular carcinoma development by modulating membrane fluidity and glucose metabolism. Hepatology. 2017;66:432–448. doi: 10.1002/hep.29033. [DOI] [PubMed] [Google Scholar]
- 49.Chao H.-C., et al. Sphingolipidomics Investigation of the Temporal Dynamics after Ischemic Brain Injury. J Proteome Res. 2019 doi: 10.1021/acs.jproteome.9b00370. [DOI] [PubMed] [Google Scholar]
- 50.Yang L., et al. Lipidomic analysis of plasma in patients with lacunar infarction using normal-phase/reversed-phase two-dimensional liquid chromatography–quadrupole time-of-flight mass spectrometry. Anal Bioanal Chem. 2017;409:3211–3222. doi: 10.1007/s00216-017-0261-6. [DOI] [PubMed] [Google Scholar]
- 51.Kloska A., Malinowska M., Gabig-Cimińska M., Jakóbkiewicz-Banecka J. Lipids and lipid mediators associated with the risk and pathology of ischemic stroke. Int J Mol Sci. 2020;21:1–26. doi: 10.3390/ijms21103618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li Z., Zhang J., Luo Y. Impact of triglyceride playing on stroke severity correlated to bilirubin. Medicine (Baltimore) 2020;99:e21792. doi: 10.1097/MD.0000000000021792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mohamud Yusuf A., Hagemann N., Hermann D.M. The Acid Sphingomyelinase/ Ceramide System as Target for Ischemic Stroke Therapies. Neurosignals. 2019;27:32–43. doi: 10.33594/000000184. [DOI] [PubMed] [Google Scholar]
- 54.Novgorodov S.A., et al. SIRT3 deacetylates ceramide synthases: Implications for mitochondrial dysfunction and brain injury. J Biol Chem. 2016;291:1957–1973. doi: 10.1074/jbc.M115.668228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.kun Gui Y, et al., Plasma levels of ceramides relate to ischemic stroke risk and clinical severity. Brain Res. Bull.158, 122–127 (2020). [DOI] [PubMed]
- 56.Au A. 1st Ed. (Elsevier Inc.; 2018. Metabolomics and Lipidomics of Ischemic Stroke. [DOI] [PubMed] [Google Scholar]
- 57.Liu P., et al. Discovery of Metabolite Biomarkers for Acute Ischemic Stroke Progression. J Proteome Res. 2017;16:773–779. doi: 10.1021/acs.jproteome.6b00779. [DOI] [PubMed] [Google Scholar]
- 58.Shin T.H., et al. Metabolome Changes in Cerebral Ischemia. Cells. 2020;9:1630. doi: 10.3390/cells9071630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gucciardi A., Pirillo P., Di Gangi I.M., Naturale M., Giordano G. A rapid UPLC-MS/MS method for simultaneous separation of 48 acylcarnitines in dried blood spots and plasma useful as a second-tier test for expanded newborn screening. Anal Bioanal Chem. 2012;404:741–751. doi: 10.1007/s00216-012-6194-1. [DOI] [PubMed] [Google Scholar]
- 60.Rizza S., et al. Metabolomics signature improves the prediction of cardiovascular events in elderly subjects. Atherosclerosis. 2014;232:260–264. doi: 10.1016/j.atherosclerosis.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 61.Guasch-Ferré M., et al. Plasma acylcarnitines and risk of cardiovascular disease: Effect of Mediterranean diet interventions. Am J Clin Nutr. 2016;103:1408–1416. doi: 10.3945/ajcn.116.130492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kalim S., et al. A plasma long-chain acylcarnitine predicts cardiovascular mortality in incident dialysis patients. J Am Heart Assoc. 2013;2:1–11. doi: 10.1161/JAHA.113.000542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ferrari R., et al. Therapeutic effects of L-carnitine and propionyl-L-carnitine on cardiovascular diseases: A review. Ann N Y Acad Sci. 2004;1033:79–91. doi: 10.1196/annals.1320.007. [DOI] [PubMed] [Google Scholar]
- 64.Tang M., et al. CPT1A-mediated fatty acid oxidation promotes cell proliferation via nucleoside metabolism in nasopharyngeal carcinoma. Cell Death Dis. 2022;13:331. doi: 10.1038/s41419-022-04730-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McCoin C.S., Knotts T.A., Adams S.H. Acylcarnitines–old actors auditioning for new roles in metabolic physiology. Nat Rev Endocrinol. 2015;11:617–625. doi: 10.1038/nrendo.2015.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Qi G., et al. ApoE4 Impairs Neuron-Astrocyte Coupling of Fatty Acid Metabolism. Cell Rep. 2021;34:1–39. doi: 10.1016/j.celrep.2020.108572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rambold A.S., et al. Fatty Acid Trafficking in Starved Cells : Regulation by Lipid Droplet Lipolysis, Autophagy, and Article Fatty Acid Trafficking in Starved Cells : Regulation by Lipid Droplet Lipolysis, Autophagy, and Mitochondrial Fusion Dynamics. Dev Cell. 2015;678–692 doi: 10.1016/j.devcel.2015.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bailey A.P., et al. Antioxidant Role for Lipid Droplets in a Stem Cell Niche of Drosophila. Cell. 2015;163:340–353. doi: 10.1016/j.cell.2015.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu L., et al. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell. 2015;160:177–190. doi: 10.1016/j.cell.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Marschallinger J., et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat Neurosci. 2020;23:194–208. doi: 10.1038/s41593-019-0566-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lonati E., et al. Lipid reshaping and lipophagy are induced in a modeled ischemia-reperfusion injury of blood brain barrier. Int J Mol Sci. 2019;20 doi: 10.3390/ijms20153752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kuo A., Lee M.Y., Sessa W.C. Lipid Droplet Biogenesis and Function in the Endothelium. Circ Res. 2017;120:1289–1297. doi: 10.1161/CIRCRESAHA.116.310498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.