

Abstract
Objectives
The GOSR2 gene is a Golgi vesicle transport gene that encodes for the Golgi SNAP receptor complex member 2 protein. This protein mediates transport between the medial and trans-Golgi compartments. The homozygous missense variant in the GOSR2 gene, c.430G>T, has been associated with progressive myoclonus epilepsy (PME). There have been reports suggesting that compound heterozygous GOSR2 variants are associated with the congenital muscular dystrophy (CMD) phenotype.
Methods
In this article, we report a pediatric case with congenital hypotonia, motor delay, elevated creatine kinase, and abnormal muscle biopsy consistent with CMD who subsequently developed PME. Whole-exome sequencing identified pathogenic compound heterozygous variants in the GOSR2 gene, one of which was the previously described PME-related c.430G>T(p.Gly144Trp), and a novel variant, c.22dup(p.Thr8fs).
Result
To our knowledge, this is a novel case of compound heterozygous variants in GOSR2 associated with both CMD and PME phenotypes.
Discussion
This case adds to the expanding clinical phenotype of GOSR2-related neurologic diseases.
Introduction
The GOSR2 gene encodes for the Golgi SNAP receptor complex member 2 protein (GOSR/Membrin) that functions in intracellular trafficking of COPII vesicles from the endoplasmic reticulum to cis-Golgi.1 An increasing number of pathogenic variants in the GOSR2 gene have been associated with a spectrum of neurologic conditions.
Homozygosity for c.430G>T(p.Gly144Trp) in GOSR2 is associated with a rare type of progressive myoclonus epilepsy (PME), also referred to as “North Sea PME” (NS-PME) because of the ancestry of first patients described. NS-PME presents with ataxia around the age of 2 years followed by myoclonus and seizures, and despite extensive motor involvement, cognition remains relatively preserved. On examination, patients have areflexia and mildly elevated creatine kinase (CK), without apparent signs of myopathy.2,3 In animal models, knockdown of Membrin (the GOSR2 ortholog) in glial cells leads to a heat-sensitive epilepsy phenotype like what is observed in patients with NS-PME.4 Neurophysiology studies have shown anterior horn cell changes and sensory neuronopathy, suggesting peripheral nervous system involvement.5,6 Biallelic variants of GOSR2 were also reported in patients with developmental delay, seizures, and atypical slow progression of movement disorder with tremor starting in adolescence, moving on to myoclonus, ataxia, and dystonia.7
Compound heterozygous variants in GOSR2 are associated with congenital muscular dystrophy (CMD).8-10 Larson et al. reported 2 sisters with infantile onset hypotonia, elevated CK, muscle weakness, and absence of seizures in early childhood.9 Muscle biopsy showed dystrophic changes and hypoglycosylation of alpha-dystroglycan. Both had the C.430 G>T(p.Gly144Trp) variant and a novel variant c.2T>G(p.Met1Arg).9 Henige et al. reported a patient with congenital hypotonia and elevated CK who had the c.430 G>T(p.Gly144Trp) variant and a novel c.82C>T(p.Gln28Ter) variant.10 Tsai et al. described a patient with the c.430G>T variant and a novel c.336+1G>A variant who presented with CMD and seizures at age 28 months.11
In this report, we present a patient with compound heterozygous variants in GOSR2 presenting with both CMD and PME phenotypes.
Methods
Written parental consent for publication was obtained. Parents requested the patient's face to be shown.
Results
Clinical Presentation
A female patient was born by vaginal delivery at term with a weight of 3 kg to a 26-year-old G1P1 mother and a 39-year-old father. Prenatal ultrasounds noted mild oligohydramnios at 36 weeks. Fetal movement was normal. Both parents were of Caucasian European descent, without consanguinity and with no family history of neurologic diseases.
At birth, she had bilateral club feet and feeding difficulties. She passed hearing screen at birth and at age 3 months. She developed plagiocephaly and torticollis at age 4 months. Neurologic examination at 6 months of age showed generalized weakness, adducted thumbs, and areflexia.
CK was elevated at 3,428 unit/L (ref: 50–270 unit/L). Serum amino acids, lactate, pyruvate, acylcarnitine, and urine organic acids were normal. Brain MRI showed mild enlargement of the lateral ventricles with mildly delayed myelination (Figure 1A). Echocardiograms and electrocardiograms were normal.
Figure 1. Brain MRI and EEG.

(A) MRI T1-weighted gradient echo pulse sequence axial image showing mildly enlarged lateral ventricles and delayed myelination for age. (B) Near-continuous high-amplitude 2–3-Hz occipital intermittent rhythmic activity with occasional intermixed spikes in the right occipital region.
Muscle biopsy of the quadriceps showed moderate variation in fiber size with scattered atrophic fibers, along with degenerating and regenerating fibers and fibrofatty tissue replacement. Trichrome stain showed multiple fibers with magenta-colored granular subsarcolemmal and sarcoplasmic inclusions. The granular inclusions stained positive with mitochondrial-specific DPNH, SDH, and COX stains. Electron microscopy revealed subsarcolemmal, perinuclear, and occasional sarcoplasmic accumulations of mitochondria with abnormal shapes and occasional enlarged size, which correlated with the granular inclusions seen under a light microscope. Findings seem consistent with CMD with secondary mitochondrial abnormal proliferation (Figure 2).
Figure 2. Muscle Biopsy Findings.
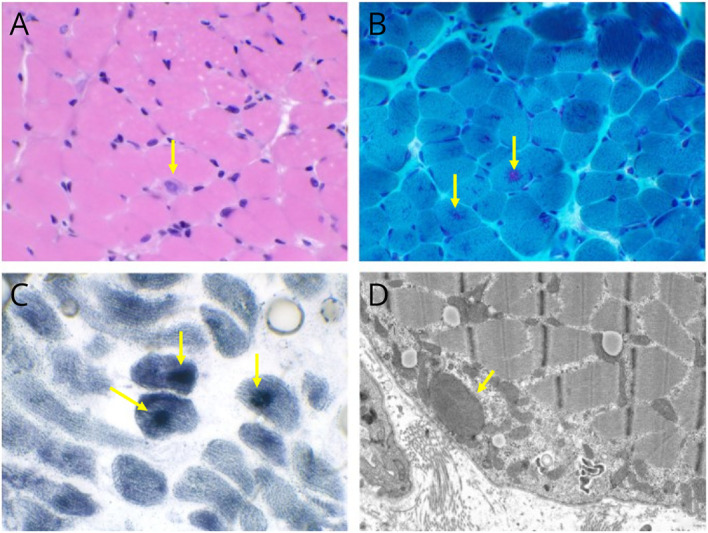
(A) Moderate fiber size variation and a regenerating fiber are noted on hematoxylin and eosin–stained sections (H&E, ×400). (B) Magenta granular subsarcolemmal and sarcoplasmic inclusions (Gomori trichrome, ×400). (C) Inclusions are densely stained for succinate dehydrogenase (SDH, ×400). (D) Ultrastructural analysis revealed abnormal-shaped and enlarged mitochondria correlated with the granular inclusions seen under a light microscope.
At 20 months of age, she had a seizure with behavioral arrest and generalized jerking. EEG showed nearly continuous occipital intermittent rhythmic delta activity with occasional intermixed epileptiform discharges with right-sided predominance (Figure 1B). Levetiracetam was started with good initial seizure control. At age 22 months, she was rolling over, grabbing objects, and using single words. She had left apex curvature of the thoracolumbar spine and x-ray confirmed scoliosis (Figure 3).
Figure 3. Photographs of Physical Examination Findings and Spine X-Ray.

(A) Poor head control needed extra assistance while sitting in the tripod position. (B) Notable truncal hypotonia and scoliosis. (C) Marked hypertonia and generalized weakness with ability to flex elbows, flex and extend knees against gravity with more weakness distally, holding ankles plantar flexed and inverted. (D) X-ray showing scoliosis.
Myoclonic jerks developed around age 2 years, which started around the mouth, then hands, and legs, evolving to multifocal myoclonus confirmed by EEG. Seizure recurred at age 27 months with an estimated 1 to 2 unprovoked seizures per month. Parents reported cognitive decline. There was no detectable ataxia or changes in fine motor control on examination at 27 months, although assessments were limited because of limited baseline motor function. Because of significant global delays, neuropsychological evaluation was unable to be performed. Topiramate was added but seizures continued, and a ketogenic diet was started at age 3 with improvement of seizure control.
Genetic Analysis
High-resolution chromosome analysis (karyotype) was normal. TK2 testing (Prevention Genetics, Marshfield, WI) and mitochondrial whole-genome analysis including next-generation sequencing and deletion/duplication testing (CCHMC Molecular Genetics Laboratory) were negative. Clinical whole-exome sequencing identified a maternally inherited novel variant in the GOSR2 gene (NM_001321133.1), c.22dup(Thr8fs), in addition to a paternally inherited PME-related variant, c.430G>T(p.Gly144Trp) (CCHMC Molecular Genetics Laboratory).
The c.22dup variant comprises a single base-pair duplication that is predicted to introduce a frame shift starting at codon 8, substituting the threonine at this position with an asparagine and resulting in a premature STOP codon 54 codons downstream. This variant has not been reported in the Exome Sequencing Project, Human Gene Mutation database, or medical literature. It was detected in the heterozygous state in 6 individuals in the Genome Aggregation Database (gnomAD) and listed with a minor allele frequency of 0.008% in the non-Finnish European population (rs746855352). It has been reported in ClinVar as a variant of uncertain clinical significance (RCV000468851.1). Functional studies for this variant are not available; however, it meets the criteria for pathogenic classification based on guidelines published by the American College of Medical Genetics and Genomics: a predicted null variant where loss-of-function is a known disease mechanism for GOSR2; occurrence in trans with a pathogenic variant; and presence in the setting of a highly specific clinical phenotype.12
Discussion
This case report describes a combined PME-CMD phenotype associated with GOSR2 variants. Our case expands the genotype-phenotype spectrum of GOSR2-related disorders.
The c.430G>T(p.Gly144Trp) variant has been described in association with progressive myoclonus ataxia with the epilepsy phenotype.2,3 Compared with the previous reported cases, our patient also developed myoclonus and seizures earlier in life. Her motor function was severely affected in that she never achieved independent sitting. She did not have apparent ataxia, although her complex motor phenotype might make detection of ataxia challenging.
The previously described patients with NS-PME had mildly elevated CK that was not correlating with frequency of myoclonic jerks, suggesting that it could represent a subclinical myopathy.3 Muscle biopsies on most reported patients with NS-PME have been normal. However, there were findings of subsarcolemmal collections of mitochondria on electron microscopy in some cases. One patient had vacuolated mitochondria and reduced complex III levels while another patient had subsarcolemmal collections of mitochondria without paracrystalline inclusion.4 These findings suggest that the GOSR2 gene may be involved in mitochondrial structure formation and proliferation. The abnormal mitochondria accumulation and large-sized mitochondria seen in muscle biopsy of our case were most consistent with findings secondary to GOSR2-related CMD.
Our patient developed epilepsy at age 20 months, followed by worsening of seizures and development of myoclonic jerks, clinically consistent with PME. We speculate that the novel c.22dup(Thr8fs) allele being a truncating variant, in conjunction with the c.430G>T(p.Gly144Trp) allele, led to disruption of the genetic function responsible for pathophysiology of both PME and CMD. Further molecular studies would help shed light on the underlying mechanism of this novel variant.
Acknowledgment
The authors thank the patient and family for permission to share this case report; parent consent was obtained for publication. Parents specifically requested patient's face to be shown in the figure containing patient's photograph.
Appendix. Authors
| Name | Location | Contribution |
| Monica S. Arroyo, MD | Division of Neurology, Joe DiMaggio Children's Hospital; Division of Neurology, Cincinnati Children's Hospital Medical Center | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data |
| Christine Fuller, MD | Division of Pathology, Upstate Medical University; Division of Pathology, Cincinnati Children's Medical Center; | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data |
| Elizabeth K. Schorry, MD, BS | Division of Human Genetics, Cincinnati Children's Hospital Medical Center; Department of Pediatrics, University of Cincinnati College of Medicine | Drafting/revision of the manuscript for content, including medical writing for content; analysis or interpretation of data |
| Elizabeth Ulm, LGC | Division of Human Genetics, Cincinnati Children's Hospital Medical Center | Drafting/revision of the manuscript for content, including medical writing for content; analysis or interpretation of data |
| Cuixia Tian, MD | Division of Neurology, Cincinnati Children's Hospital Medical Center; Department of Pediatrics, University of Cincinnati College of Medicine | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data |
Study Funding
The authors report no targeted funding.
Disclosure
The authors report no relevant disclosures. Go to Neurology.org/NG for full disclosures.
References
- 1.Lowe SL, Peter F, Subramaniam VN, Wong SH, Hong W. A SNARE involved in protein transport through the Golgi apparatus. Nature. 1997;389(6653):881-884. doi: 10.1038/39923 [DOI] [PubMed] [Google Scholar]
- 2.Corbett MA, Schwake M, Bahlo M, et al. A mutation in the Golgi Qb-SNARE gene GOSR2 causes progressive myoclonus epilepsy with early ataxia. Am J Hum Genet. 2011;88(5):657-663. doi: 10.1016/j.ajhg.2011.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boissé Lomax L, Bayly MA, Hjalgrim H, et al. North Sea' progressive myoclonus epilepsy: phenotype of subjects with GOSR2 mutation. Brain. 2013;136(Pt 4):1146-1154. doi: 10.1093/brain/awt021 [DOI] [PubMed] [Google Scholar]
- 4.Lambrechts RA, Polet SS, Hernandez-Pichardo A, et al. North Sea progressive myoclonus epilepsy is exacerbated by heat, a phenotype primarily associated with affected Glia. Neuroscience. 2019;423:1-11. doi: 10.1016/j.neuroscience.2019.10.035 [DOI] [PubMed] [Google Scholar]
- 5.van Egmond ME, Weijenberg A, van Rijn ME, et al. The efficacy of the modified Atkins diet in North Sea Progressive Myoclonus Epilepsy: an observational prospective open-label study. Orphanet J Rare Dis. 2017;12(1):45. doi: 10.1186/s13023-017-0595-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Polet SS, Anderson DG, Koens LH, et al. A detailed description of the phenotypic spectrum of North Sea Progressive Myoclonus Epilepsy in a large cohort of seventeen patients. Parkinsonism Relat Disord. 2020;72:44-48. doi: 10.1016/j.parkreldis.2020.02.005 [DOI] [PubMed] [Google Scholar]
- 7.Hentrich L, Parnes M, Lotze TE, et al. Novel genetic and phenotypic expansion in GOSR2-related progressive myoclonus epilepsy. Genes (Basel). 2023;14(10):1860. doi: 10.3390/genes14101860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Praschberger R, Balint B, Mencacci NE, et al. Expanding the phenotype and genetic defects associated with the GOSR2 gene. Movement Disord Clin Pract 2015;2(3):271-273. doi: 10.1002/mdc3.12190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Larson AA, Baker PR, Milev MP, et al. TRAPPC11 and GOSR2 mutations associate with hypoglycosylation of α-dystroglycan and muscular dystrophy. Skeletal Muscle. 2018;8(1):17. doi: 10.1186/s13395-018-0163-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henige H, Kaur S, Pappas K. Compound heterozygous variants in GOSR2 associated with congenital muscular dystrophy: a case report. Eur J Med Genet. 2021;64(4):104184. doi: 10.1016/j.ejmg.2021.104184 [DOI] [PubMed] [Google Scholar]
- 11.Tsai L, Schwake M, Corbett MA, Gecz J, Berkovic S, Shieh PB. GOSR2: a novel form of congenital muscular dystrophy. Neuromuscul Disord. 2013;23(9-10):748. doi: 10.1016/j.nmd.2013.06.404 [DOI] [Google Scholar]
- 12.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]