

Abstract
Current strategies to understand the molecular basis of Marek’s disease virus (MDV) virulence primarily consist of cataloging divergent nucleotides between strains with different phenotypes. However, most comparative genomic studies of MDV rely on previously published consensus genomes despite the confirmed existence of MDV strains as mixed viral populations. To assess the reliability of interstrain genomic comparisons relying on published consensus genomes of MDV, we obtained two additional consensus genomes of vaccine strain CVI988 (Rispens) and two additional consensus genomes of the very virulent strain Md5 by sequencing viral stocks and cultured field isolates. In conjunction with the published genomes of CVI988 and Md5, this allowed us to perform three-way comparisons between multiple consensus genomes of the same strain. We found that consensus genomes of CVI988 can vary in as many as 236 positions involving 13 open reading frames (ORFs). By contrast, we found that Md5 genomes varied only in 11 positions involving a single ORF. Notably, we were able to identify 3 single-nucleotide polymorphisms (SNPs) in the unique long region and 16 SNPs in the unique short (US) region of CVI988GenBank.BAC that were not present in either CVI988Pirbright.lab or CVI988USDA.PA.field. Recombination analyses of field strains previously described as natural recombinants of CVI988 yielded no evidence of crossover events in the US region when either CVI988Pirbright.lab or CVI988USDA.PA.field were used to represent CVI988 instead of CVI988GenBank.BAC. We were also able to confirm that both CVI988 and Md5 populations were mixed, exhibiting a total of 29 and 27 high-confidence minor variant positions, respectively. However, we did not find any evidence of minor variants in the positions corresponding to the 19 SNPs in the unique regions of CVI988GenBank.BAC. Taken together, our findings suggest that continued reliance on the same published consensus genome of CVI988 may have led to an overestimation of genomic divergence between CVI988 and virulent strains and that multiple consensus genomes per strain may be necessary to ensure the accuracy of interstrain genomic comparisons.
Keywords: intrastrain, diversity, population, recombination, MDV, CVI988, Md5, variation
Introduction
Marek’s disease virus (MDV) is an oncogenic, lymphoproliferative, and neuropathic alphaherpesvirus affecting chickens (Mardivirus gallidalpha 2, genus Mardivirus; family Herpesviridae). It was first reported in 1907 by Hungarian veterinarian József Marek (Marek 1907). Successful isolation of MDV in culture was only realized six decades later, closely followed by the development of the first vaccine, HPRS-16/Att (Churchill and Biggs 1967, Witter et al. 1968, Churchill et al. 1969). HPRS-16/Att was soon replaced by a better replicating, equally protective vaccine derived from the strain FC126 of herpesvirus of turkeys (HVT) (Witter et al. 1970). The HVT vaccine became the first widely used commercial vaccine for Marek’s disease (MD) (Okazaki et al. 1970). HVT was successful in reducing MD incidence in commercial flocks until the late 1970s when reports of strains with increased virulence that were able to bypass vaccine-induced immunity started to emerge across the USA (Witter et al. 1980, Schat et al. 1982). A bivalent vaccine consisting of HVT with MDV serotype 2 strain SB-1 was introduced in response. However, a similar shift in virulence occurred in just over a decade (Witter 1982, 1997). Outside the USA, a vaccine known as CVI988 had been in use since the mid-1970s (Rispens et al. 1972). Developed in the Netherlands by a group of veterinary scientists under Dr Bart Rispens, CVI988 offered the best protection against emerging variants, and it quickly became the “gold standard” vaccine around the world (Schat 2016). CVI988 is colloquially known today as the “Rispens” vaccine for MDV. Although CVI988 has mostly kept MD losses at bay for the past 30 years, there are now reports of strains potentially able to bypass CVI988-conferred immunity, and the most recent estimate places MDV-associated losses in the poultry industry at around US $1–2 billion per annum (Schumacher et al. 2002, Davison and Venugopal 2004, Zhang et al. 2015, Sun et al. 2017).
Comparative genomics has been a driving force in efforts to understand the molecular basis of MDV-induced virulence ever since the first viral genomes were sequenced over two decades ago (Tulman et al. 2000, Lee et al. 2000a). MDV has a DNA genome with large unique regions—named for their length as unique long (UL) and unique short (US)—that are flanked by large structural repeats. The current approach to MDV comparative genomics has focused on identifying nucleotide positions that differ between published consensus genomes of MDV strains (Spatz et al. 2007a, Hildebrandt et al. 2014, Trimpert et al. 2017, Bell et al. 2019, Dunn et al. 2019, Zhang et al. 2022). These MDV strains are categorized on the basis of their ability to break through vaccine protection, or their “pathotype” (Dunn et al. 2019). There are four officially recognized pathotypes of MDV: mild (m), virulent (v), very virulent (vv), and very virulent plus (vv+) (Dunn et al. 2019). The main goal of conducting interstrain comparisons in this way is to identify the genes and polymorphisms associated with virulence. However, the degree to which published consensus genomes of MDV represent the average for each strain has never been assessed, despite the fact that herpesviruses are now known to exist as mixed populations. Several reports suggest that herpesviruses such as HSV, VZV, and HCMV possess extensive genetic variability (Renzette et al. 2014, Depledge et al. 2018, Renner et al. 2018, Cudini et al. 2019). In the specific case of MDV, population diversity has been shown to positively correlate with virulence (Trimpert et al. 2019). In addition, all available consensus genomes for MDV are low-coverage genomes that only account for the major variants (≥50%) present in the original viral population from which they were sequenced. The lack of information on minor variants (≤50%) can be problematic, due to previous reports suggesting that the diversity within a given viral population contributes to the overall pathotype of a strain (Gimeno et al. 1999, Spatz 2010, Spatz et al. 2012, Hildebrandt et al. 2014). Although the extent of this contribution is still unknown, this nonetheless suggests that failing to account for minor variants can negatively impact our ability to correlate sequencing data to pathotypes (Dunn et al. 2019). Finally, many of the available reference genomes underwent cloning steps prior to sequencing. This includes the only available CVI988 consensus genome, which was sequenced from a bacterial artificial chromosome (BAC) copy of the viral genome, followed by plasmid-based cloning thereof (Spatz et al. 2007a). The Md5 consensus genome was obtained from plasmid-based cloning of viral DNA amplified in cell culture (Tulman et al. 2000). The use of BAC and plasmid cloning approaches generates an artificially homogenous population of viral genomes, further moving away from the real-world biology of herpesvirus strains. Moreover, by randomly selecting one viral haplotype from a population, these methods inherently reduce the chances of obtaining a representative consensus genome.
To address these issues, it is necessary to test the reliability of published MDV consensus genomes and assess the genomic heterogeneity of MDV populations using next-generation sequencing (NGS) approaches. Whole-genome resequencing is a simple yet powerful approach that consists of comparing newly sequenced individuals against a reference (Stratton 2008). Here, we have adapted this strategy for viruses by sequencing multiple populations of the same strain and determining whether these yield a consistent consensus genome under the assumption that large shifts in genomic variant frequencies will manifest as consensus-level differences. We independently sequenced two samples of the attenuated vaccine strain CVI988 and two samples of the very virulent prototype strain Md5 from a combination of viral stocks and cultured field isolates. The newly sequenced genomes were compared with previously published consensus genomes using whole-genome pairwise alignments. Using this approach, we were able to detect differences between consensus genomes of each strain. Notably, we found 19 single-nucleotide polymorphisms (SNPs) in the unique regions of CVI988GenBank.BAC that were not present in CVI988Pirbright.lab or CVI988USDA.PA.field. These included 16 SNPs in the unique short region impacting posited recombination breakpoints in MDV strains previously described as natural vaccine recombinants (He et al. 2018, 2020, Zhang et al. 2022). We then assessed the genomic heterogeneity of CVI988 and Md5 by analyzing ultra-deep Illumina sequencing data (>1000× depth) and identified positions where at least 2% of the reads supported an alternative allele (Lauring 2020). This method enabled detection of minor allele frequency variants and allowed us to confirm that the ultra-deep sequenced samples of CVI988 and Md5 were mixed viral populations. These findings highlight the need to sequence multiple consensus genomes of each MDV strain in order to ensure the accuracy of interstrain genomic comparisons and enhance our ability to link viral genotypes to virulence phenotypes.
Materials and methods
Viral culture and DNA isolation of CVI988USDA.PA.field and Md5USDA.lab
The virulence phenotypes and targeted (amplicon-based) sequencing of USDA CVI988USDA.PA.field and Md5USDA.lab have been previously described by Dunn et al. (2019). The sample we refer to as “CVI988USDA.PA.field” was collected as a field sample in 2010 in Pennsylvania (MDV 709B_v_2010_PA), and “Md5USDA.lab” was collected in Maryland in 1977 (MDV Md5_vv_1977_MD) (Dunn et al. 2019). Duck embryo fibroblasts (DEFs) were used to culture these virus stocks. Viral DNA was isolated from a 5-day DEF infection initiated using CVI988USDA.PA.field at Passage 6 (titer 5.8 × 105 PFU/ml) or a 6-day DEF infection initiated using Md5USDA.lab at Passage 7 (titer 1.2 × 106 PFU/ml). DEF cultures were maintained in a 1:1 mixture of Leibovitz’s L-15 and McCoy’s 5A (LM) media supplemented with fetal bovine serum (FBS), 200 U/ml penicillin, 20 μg/ml streptomycin, and 2 μg/ml amphotericin B in a 37°C, 5% CO2 incubator. Cells were plated with 4% FBS LM media and maintained in 1% FBS LM media. For storage as viral stocks, infected cells were suspended in freezing media composed of 10% DMSO, 45% FBS, and 45% LM media and kept in liquid nitrogen. Viral DNA was isolated using the Gentra Puregene DNA isolation kit (Qiagen).
DNA library preparation and sequencing of CVI988USDA.PA.field and Md5USDA.lab
DNA libraries for CVI988USDA.PA.field and Md5USDA.lab were prepared for next-generation sequencing using an Illumina Miseq platform as previously described (Bowen et al. 2016, Pandey et al. 2016, Bell et al. 2019). Briefly, genetic material, including viral nucleocapsid DNA, was quantified by Qubit (Invitrogen, Carlsbad, CA, USA) and by a virus-specific qPCR. Total DNA was then acoustically sheared using a Covaris M220, with settings at 60-s duration, peak power 50, 10% duty cycle, at 4°C. DNA was then processed through the Illumina TruSeq Nano DNA prep kit, using the manufacturer recommendations, and further quantified by Qubit (Invitrogen, Carlsbad, CA, USA), Bioanalyzer (Agilent), and library-specific qPCR (KAPA Biosystems). Libraries were then diluted to 17 pM per the manufacturer’s recommendation and sequenced using version 3 paired-end (2 × 300 bp length) chemistry.
Viral culture and DNA isolation of CVI988Pirbright.lab and Md5Pirbright.lab
Primary chick embryo fibroblasts (CEFs) were prepared from 10-day old embryos and maintained in M199 medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 5% FBS (Sigma-Aldrich, Darmstadt, Germany), 100 units/ml of penicillin and streptomycin (Thermo Fisher Scientific), and 10% tryptose phosphate broth (Sigma).
The CVI988 stock we refer to as “CVI988Pirbright.lab” was obtained from the Pirbright Institute and prepared from commercial CVI988 vaccine, sourced in the UK (Poulvac Marek CVI vaccine; Zoetis), following two passages in CEF at a multiplicity of infection of 0.01. The infected CEFs were harvested 3 days later. DNA was prepared from ∼5 × 106 cells using the DNeasy-96 kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions, and eluted in DNase-free water.
The Md5 stock we refer to as “Md5Pirbright.lab” was also obtained from the Pirbright Institute and derived from a seventh DEF passage stock kindly provided by Dr A. M. Fadly [Avian Disease and Oncology Laboratory (ADOL), USA]. To amplify this stock, 5-day-old Rhode Island Red chickens were inoculated intra-abdominally with 1000 plaque forming units of virus. Lymphocytes isolated from spleens harvested at 14 days post-infection were cultured with CEF, the cell-associated virus was harvested at 7 days when cytopathic effect was clearly visible, and then further passed in CEF every 5–6 days to produce virus stocks. DNA was prepared from approximately 5 × 106 cells of the ninth passage CEF stock using the DNeasy-96 kit and eluted in DNase-free water.
Oligo-bait-based enrichment, DNA library preparation, and sequencing of CVI988Pirbright.lab and Md5Pirbright.lab
DNA isolated from each MDV culture was quantified for total DNA by Qubit (Invitrogen), and the amount of either Md5 DNA or CVI988 DNA was confirmed by a previously described qPCR assay (Kennedy et al. 2017). Samples were then acoustically sheared using Covaris and high-throughput, deep sequencing DNA library preparations were produced using the KAPA HyperPrep Library Kit (Roche) as per the manufacturer’s protocol, with 14 PCR cycles. Then, samples were processed through a previously described oligo-enrichment procedure, where MDV-specific probes (Trimpert et al. 2017) (myBaits, Arbor Biosciences) were used with the myBaits Target Capture Kit (Arbor Biosciences) to enrich for MDV DNA (Shipley et al. 2020). An additional PCR amplification (14 cycles) yielded libraries that were assessed for quality by Qubit, as well as a KAPA-specific qPCR, which allowed for accurate dilutions of libraries to 4 pM before sequencing. Finally, libraries were sequenced using v3 chemistry 300 bp × 300 bp paired-end sequencing on an Illumina MiSeq, as per the manufacturer’s instruction.
Processing of sequencing reads and genome assembly
Viral reads were identified by aligning raw reads in FASTQ files against a local database of 97 MDV genomes using BLASTN with an E-value cutoff of 1 × 10−2 (see Supplementary Table S1 for strain names and accession numbers). The selected reads were then subjected to the quality control and preprocessing step (Step 1) of our published viral genome assembly (VirGA) workflow (Parsons et al. 2015). These properly paired reads were then used for de novo assembly using metaSPAdes v3.14.0 (Nurk et al. 2017). The resulting file containing the metaSPAdes scaffolds in FASTA format served as input for the remaining steps of VirGA (Steps 3 and 4), which include genome linearization, annotation, and post-assembly quality assessments. For the reference-guided contig-ordering step, we used a trimmed version (TRL and TRS regions removed) of the published consensus genome of each strain (Table 1). New consensus genomes were also trimmed for downstream analyses. Tandem repeats were identified in trimmed consensus genomes using Tandem Repeats Finder (TRF) v4.09 (Benson 1999). Manual verification and masking of tandem repeats were conducted using Geneious Prime v2023.0.4. To resolve meq at the consensus level, reads were mapped to modified reference genomes possessing different isoforms of meq. Local sequencing coverages were then used to determine the consensus-level isoform.
Table 1.
Sequencing statistics for new and previously published CVI988 and Md5 consensus genomes.
| MDV strain | Number of MDV-specific reads | Number of reads used for assembly | Coverage | GenBank accession | Genome length (bp) | Prior citation |
|---|---|---|---|---|---|---|
| CVI988GenBank.BAC | N/A | N/A | 6× | DQ530348 | 178 311 | Spatz et al. (2007a) |
| CVI988USDA.PA.field | 227 487 | 138 122 | 149× | PP032833 | 176 946 | – |
| CVI988Pirbright.lab | 11 997 583a | 9 730 216 | 10 055× | PP032835 | 176 532 | – |
| Md5Genbank | N/A | N/A | 6× | NC_002229 | 177 874 | Tulman et al. (2000) |
| Md5USDA.lab | 1 030 131 | 727 124 | 793× | PP032832 | 176 724 | – |
| Md5Pirbright.lab | 10 138 498a | 8 296 132 | 8530× | PP032834 | 178 276 | – |
The UK viral stock populations were subjected to oligo-bait-based enrichment to increase the yield of viral DNA (Trimpert et al. 2017, Shipley et al. 2020).
Pairwise identity and genetic distance comparisons
Trimmed consensus genomes with masked tandem repeats were aligned using MAFFT v7.505 with default settings (Katoh et al. 2002). The resulting alignment file in clustal format was imported into Geneious to visualize variable positions (SNPs and Indels) between the genomes. Published MDV consensus genomes sequenced from field samples or cultured stocks with PubMed Identifiers (n = 33) and all consensus genomes used for comparative analyses in the present study (n = 6) were aligned using MAFFT (see Supplementary Table S1 for details). A neighbor-joining (NJ) tree was constructed from the multi-genome alignment file in clustal format using the Geneious Tree Builder tool with a Tamura-Nei genetic distance model and no outgroup.
Recombination analyses
GenBank nucleotide sequences (see Supplementary Table S1 for accession numbers) were trimmed using Geneious and aligned using MAFFT. Detection of potential recombination breakpoints via bootscanning was performed using SimPlot v3.5.1 (Lole et al. 1999). Informative sites were identified using the FindSites option provided by SimPlot.
Identification of minor variant positions
A custom script was used to identify positions with at least 2% read support for an alternative allele—i.e. a minor variant—in genomes with >1000× coverage. Positions in tandem repeats and homopolymers were excluded. All remaining minor variant positions were verified through manual inspection of binary alignment map (BAM) files using the Integrative Genomics Viewer v2.12.3 (Robinson et al. 2011).
Nucleotide sequence accession numbers
GenBank accession numbers for the four new and two prior viral genomes are listed in Table 1. The data underlying this article are available in GenBank and and can be accessed with the accession numbers in Table 1.
Results
Resequencing viral stocks and field isolates of CVI988 and Md5 using Illumina whole-genome sequencing
To enable intrastrain whole-genome comparisons, we first obtained two additional consensus genomes of both vaccine strain CVI988 (mild pathotype) and prototype very virulent strain Md5 (vv pathotype) (Table 1). Md5USDA.lab and Md5Pirbright.lab were sequenced from standard lab stocks (i.e. a virus population initiated from a prior viral stock and expanded in host cells). CVI988Pirbright.lab was likewise derived from lab stock. By contrast, CVI988USDA.PA.field was isolated from a sample collected at a Pennsylvania farm, before isolation and expansion in host cells in the laboratory. Targeted, amplicon-based sequencing of portions of the Md5USDA.lab (Md5_vv_1977_MD) and CVI988USDA.PA.field (709B_v_2010_PA) genomes has been previously reported (Dunn et al. 2019). All viral stocks were propagated similarly for viral DNA isolation and sequencing library preparation (see the “Materials and methods” section for details). CVI988Pirbright.lab and Md5Pirbright.lab were additionally enriched using published methods for oligonucleotide-bait-based enrichment in solution (Trimpert et al. 2017), which can enable a greater sequencing depth by enriching viral sequences from the overall milieu of DNA. Viral consensus genomes for each stock were determined using a previously published combination of de novo and reference-guided assembly methods (Table 1; see the “Materials and methods” section for details).
The MDV genome has a double-stranded DNA structure, with a full length of approximately 175–180 kb. The genomic layout is split into two large unique regions known as UL and US, each flanked by inverted repeat sequences [internal repeat long (IRL) and internal repeat short (IRS)] and terminal repeat sequences [terminal repeat long (TRL) and terminal repeat short (TRS)] (Fig. 1a, center). The α-type packaging sequences are located at the genomic termini and at the IRL/IRS junction. None of the new genomes contained any truncated or missing proteins. The average G + C content of all genomes was 44.0%. The average sequencing coverage of Illumina sequencing data exceeded 100× for all viral genomes, and it exceeded 1000× for the two genomes subjected to oligonucleotide-bait-based enrichment (Table 1).
Figure 1.
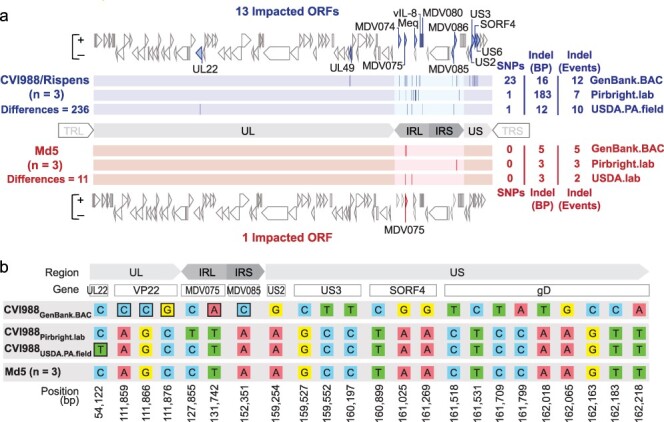
Comparative analysis of CVI988 and Md5 consensus genomes. (a) A total of three trimmed consensus genomes (TRL and TRS removed) were obtained for each strain (CVI988 = Blue, Md5 = Red) and aligned using MAFFT. Consensus-level differences including SNPs and Indels are highlighted (dark lines) based on their relative locations in each respective genome. The total number of differences between genomes of the same strain is indicated on the left, with a breakdown for each individual genome indicated on the right; these loci are all listed in detail in Supplementary Tables S2 and S3. For each strain, ORFs with consensus-level differences in at least one of the three genomes belonging to that strain are highlighted and labeled (with alternative names when possible). The total number of ORFs with consensus-level differences is 13 for CVI988 and 1 for Md5. (b) ORF-associated SNPs were found between consensus genomes of CVI988. Genomic positions are indicated relative to CVI988GenBank.BAC, which accounted for 21 out of 23 ORF-associated SNPs. Completely unique SNPs that were not found in any of the other 38 MDV genomes included in the study are highlighted (black borders).
Pairwise alignment of same-strain consensus genomes reveals greater intrastrain variation in CVI988 than Md5
For each strain, trimmed consensus genomes with masked tandem repeats were aligned and visualized. Consensus-level differences corresponding to SNPs, insertions, and deletions (Indels) were identified and verified through manual inspection of sequence-read alignment files (i.e. BAM files). A total of 236 differences were detected in the three consensus genomes of vaccine strain CVI988, impacting a total of 13 open reading frames (ORFs). These include the genes UL22/gH (MDV034), UL49/VP22 (MDV062), MDV074, MDV075, Meq/RLORF7 (MDV076/MDV005), vIL-8/RLORF2 (MDV078/MDV003), MDV080, MDV085, MDV086, US2 (MDV091), US3 (MDV092), SORF4/S4 (MDV093), and US6/gD (MDV094) (Fig. 1a, Supplementary Table S2). Outside of regions associated with Indels and repetitive elements, CVI988Pirbright.lab and CVI988USDA.PA.field were found to be nearly identical (>99.99% identity), being distinguished only by a single synonymous SNP in UL22. By contrast, CVI988GenBank.BAC possessed 23 unique SNPs and was the only CVI988 genome to exhibit variation in the US subgenomic region. On the other hand, the three consensus genomes of the very virulent strain Md5 showed a total of 11 differences, with only a single gene, MDV075, being impacted (Supplementary Table S3).
The meq oncogene contributes significantly to overall intrastrain variation in consensus genomes of CVI988
Out of the 236 differences between CVI988 consensus genomes (Supplementary Table S2), 214 were found in internal repeat long (n = 209 bp) and internal repeat short (n = 5 bp). A large portion of these differences (n = 177 bp) corresponded to a 59 amino acid (AA) insertion in the meq oncogene, which was found in the CVI988GenBank.BAC and CVI988USDA.PA.field consensus genomes but absent in the CVI988Pirbright.lab consensus genome (Fig. 2). For meq, previous studies have suggested the existence of up to three isoforms: Meq, long Meq (L-Meq), and short Meq (S-Meq) (Lee et al. 2000b, Renz et al. 2012, Murata et al. 2021, Sato et al. 2022). Meq is the standard isoform and is 339 AA in length. L-Meq is a longer version of Meq that is characterized by a 59-AA insertion in the transactivation domain (TAD), which is often accompanied by a 3-nucleotide CCA deletion immediately upstream from the insertion site. S-meq is a shorter version of Meq that exhibits a 41-AA deletion in the TAD. The CCA deletion that is typically associated with the presence of the L-meq isoform was confirmed to exist as a subpopulation of considerable size (∼40%) in the UK CVI988 sample, but it did not appear to be linked to the presence of the 177-nucleotide (59-AA) insertion (Bell et al. 2019). All three Md5 genomes exhibited the standard isoform of meq (Fig. 2), again indicating less diversity when compared to CVI988.
Figure 2.
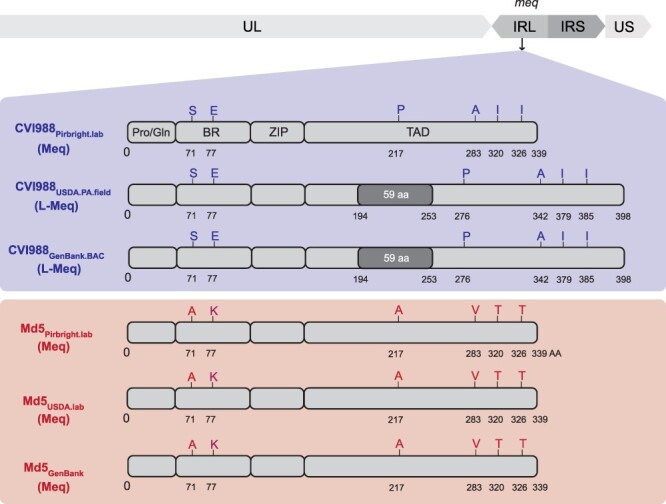
Comparison of the meq oncogene in Md5 and CVI988 consensus genomes. Schematic representation of the meq isoforms found in the repeat long regions (IRL shown here) in CVI988 (blue/top panel) and Md5 (red/bottom panel) consensus genomes. The meq oncogene is comprised of the proline/glutamine (Pro/Gln)-rich domain, the basic region (BR), the leucine zipper (ZIP), and the transactivation domain (TAD) at the C-terminal region. Differences in the AA sequence of the two strains are highlighted and labeled (A = Alanine, E = Glutamic acid, I = Isoleucine, K = Lysine, P = Proline, S = Serine, T = Threonine, V = Valine). The Meq protein is 339 AA in length. The L-Meq isoform contains a 59-AA insertion in the TAD, resulting in a total length of 398 AA.
Increased intrastrain variation in structural repeat regions is associated with greater homopolymer instability
Outside of differences associated with meq isoforms, 28 of the 37 remaining differences found in the internal repeat regions of CVI988 consensus genomes were associated with variation in homopolymer lengths (Supplementary Table S2). By contrast, out of the 22 differences found across the unique regions of CVI988 consensus genomes, 20 were ORF-associated SNPs, with an even distribution between synonymous (n = 10) and non-synonymous (n = 10). In Md5 consensus genomes, all 11 differences were found in either internal repeat long (n = 6) or internal repeat short (n = 5) and were all exclusively associated with variation in homopolymer lengths (Supplementary Table S3).
Intrastrain variation in unique regions is associated with 19 SNPs in the published CVI988 consensus genome
Out of the 20 ORF-associated SNPs found in unique regions of CVI988 consensus genomes, 19 belonged to CVI988GenBank.BAC, including 3 SNPs in UL and 16 SNPs in US (Fig. 1b). Phylogenomic analyses confirmed divergence of CVI988GenBank.BAC from CVI988Pirbright.lab and CVI988USDA.PA.field (Fig. 3). Additionally, none of the three SNPs found in UL of CVI988GenBank.BAC were present in any of the other 38 MDV genomes included in this study. By contrast, all 16 SNPs in US of CVI988GenBank.BAC were found in subsets across six different strains (EU-1, J-1, 814, GX0101, ATE2539, and Kgs-c1). However, no other strain apart from CVI988GenBank.BAC individually possessed all 16 SNPs in the US region. The nucleotides found across all 19 positions in CVI988Pirbright.lab and CVI988USDA.PA.field were also present in the three Md5 genomes (Fig. 1b) and in 27 of the 33 additional MDV genomes used for phylogenomic analyses.
Figure 3.
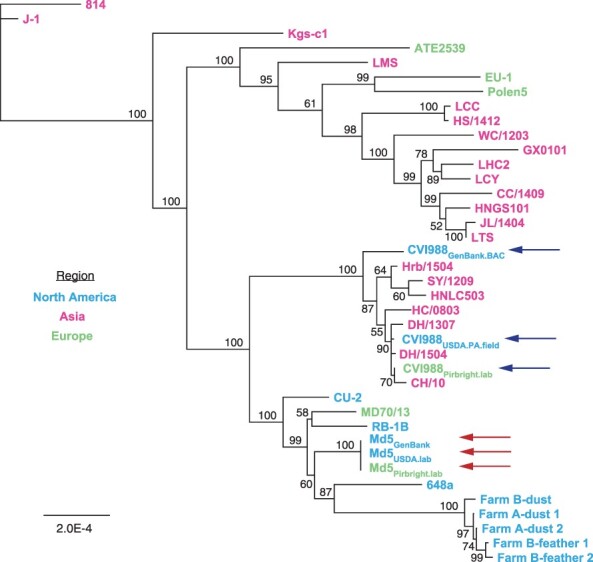
Neighbor-joining tree of previously published MDV strains and the new consensus genomes of CVI988 and Md5. Consensus genomes of CVI988, Md5, and 33 additional MDV strains from North America, Asia, and Europe were aligned using MAFFT (Supplementary Table S1 for full list of comparison strains and accession IDs). The resulting MSA was used to infer a neighbor-joining tree using Geneious Prime v2023.0.4. CVI988 (blue/upper set of arrows) and Md5 (red/lower set of arrows) genomes used for comparative analyses are highlighted. “CVI988-like” strains such as HC/0803 that cluster near the CVI988 strain genomes are further illustrated in Fig. 4.
Differences between CVI988 consensus genomes coincide with previously proposed recombination breakpoints in “CVI988-like” variants
A total of seven isolates from China have been described as natural recombinants of CVI988, or “CVI988-like” variants, including CH/10, DH/1307, DH/1504, HC/0803, HNLC503, Hrb/1504, and SY/1209 (He et al. 2018, 2020, Zhang et al. 2022). Bootscan analyses of the US region of HC/0803 revealed that recombination events were only detected when the CVI988GenBank.BAC consensus genome was used to represent CVI988 (Fig. 4a). No recombination events were detected when using either CVI988Pirbright.lab or CVI988USDA.PA.field to represent CVI988. This same pattern was observed for CH/10, DH/1307, DH/1504, HNLC503, Hrb/1504, and SY/1209 (Supplementary Fig. S1). Inspection of the 30 informative sites associated with these recombination events revealed that CVI988Pirbright.lab and CVI988USDA.PA.field shared 100% sequence identity with all seven “CVI988-like” variants (Fig. 4b). These 30 informative sites also included 15 out of the 16 SNPs found exclusively in US of CVI988GenBank.BAC.
Figure 4.
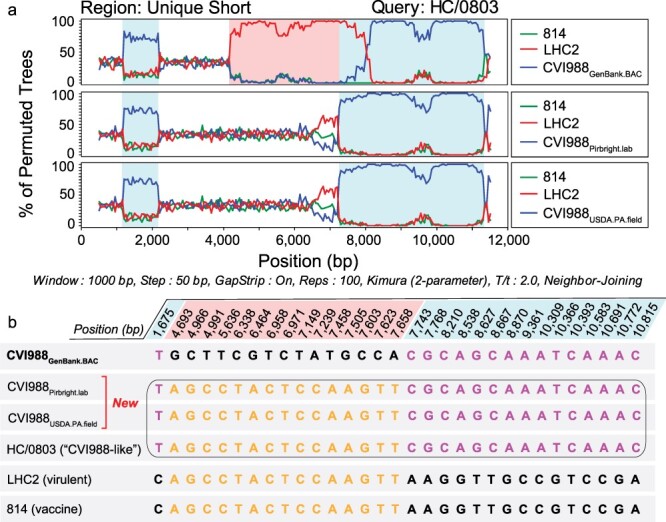
Recombination analyses of MDV strains previously described as natural recombinants of CVI988. (a) Bootscan analyses of the unique short region of “CVI988-like” MDV strain HC/0803 using SimPlot. Additional strains include 814 (vaccine strain), LHC2 (virulent strain; both from China), CVI988GenBank.BAC (top), CVI988Pirbright.lab (middle), and CVI988USDA.PA.field (bottom). Nucleotide positions are indicated relative to the US region of CVI988GenBank.BAC. (b) The FindSites tool in SimPlot was used to identify permuted trees and informative sites associated with recombination events in the US region of HC/0803. When CVI988GenBank.BAC was used for the analysis, two permuted trees were detected. Each permuted tree was associated with 15 informative sites (shaded light blue or light red, depending on their respective permuted tree). By contrast, both CVI988Pirbright.lab and CVI988USDA.PA.field shared 100% identity with “CVI988-like” variants across all 30 positions (black border). This resulted in only a single permuted tree and no recombination events being detected. See Supplementary Fig. S1 for related analyses of additional strains.
Ultra-deep sequencing reveals minor variants of CVI988 and Md5
In addition to consensus-level comparisons, we performed minor variant analysis on genomes with a sequencing depth exceeding 1000× (Fig. 5, see the “Materials and methods” section for details). This included CVI988Pirbright.lab (>10 000×) and Md5Pirbright.lab (>8500×; Table 1). A total of 29 minor variant positions were detected in the original CVI988Pirbright.lab sample across a combination of coding and regulatory regions. These minor variants impacted a total of nine ORFs: MDV011, UL13 (MDV025), UL26 (MDV038), MDV069, MDV072, meq (MDV076), MDV077, ICP4 (MDV084), and US3 (MDV092) (Supplementary Table S4). Similarly, a total of 27 minor variants were detected in the UK Md5 sample. These impacted a total of 10 ORFs: UL19 (MDV031), UL41/VHS (MDV054), UL44/gC (MDV057), UL48/VP16 (MDV061), UL52 (MDV066), MDV069, MDV072, MDV078, MDV079, and MDV084 (Supplementary Table S5). Twelve of the 14 minor variant positions found across MDV069 and MDV072 were common to both strains. No minor variants associated with the US region were identified in the Md5Pirbright.lab sample. Additionally, we verified that no minor variant positions coincided with the 19 unique SNPs in CVI988GenBank.BAC (Fig. 1b).
Figure 5.
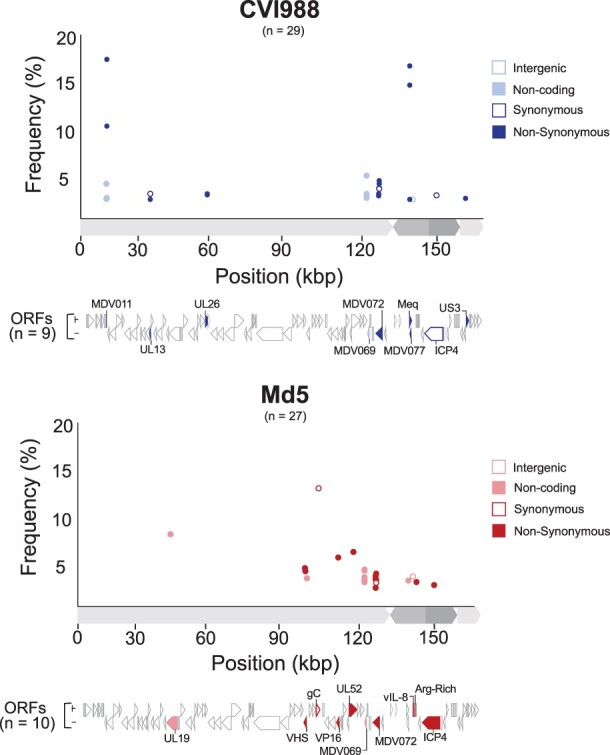
Minor variant distribution in Pirbright CVI988 and Md5 populations. For each strain (CVI988 = blue/top panel, Md5 = red/bottom panel), a single viral population was ultra-deep sequenced (>1000×) to enable identification of minor variants (Table 1). Positions outside of tandem repeats and homopolymers where the minor allele was present in ≥2% of the reads were identified as minor variants. Each minor variant was classified depending on its relative location to ORFs and its impact on the resulting amino-acid sequence (when applicable). ORFs associated with minor variants are labeled and highlighted beneath the graph using the same criteria.
Discussion
The goal of comparative genomics is to provide a link between genotype and phenotype. The accuracy with which these two elements are represented is therefore directly proportional to our ability to make biologically relevant conclusions through the use of comparative genomic methods. DNA viruses have long been considered to be inherently stable, especially when compared to RNA viruses, which are now often described as mutant swarms, or “quasispecies” (Domingo and Perales 2019, Singh et al. 2023). This has led to the assumption that viral consensus genomes, which represent the majority nucleotide at any given position, can accurately represent the genotype of MDV strains such as vaccine strain CVI988 and the prototype very virulent strain Md5. As demonstrated here and in prior studies, these and other herpesvirus populations can harbor minor variants both in culture and in vivo (Loncoman et al. 2017, Renner et al. 2018, Shipley et al. 2018, Trimpert et al. 2019). Here, by sequencing additional viral stocks and field isolates of these strains, we have sought to evaluate the impact of conducting interstrain comparisons without accounting for the intrinsic genomic heterogeneity of MDV strain populations.
CVI988 is an attenuated vaccine strain that has long been known to exist as a mixed population due to undergoing multiple passages in vitro, in different laboratories, and in multiple vaccine manufacturing companies. There have also been reports suggesting that the effectiveness of CVI988 vaccines can vary depending on the manufacturer and that even different batches from the same manufacturer can vary in effectiveness (Dambrine et al. 2016, López de Juan Abad et al. 2019). By contrast, Md5 is a virulent field strain of MDV that is currently used as the prototype very virulent (vv) strain in the USDA-ARS ADOL pathotype classification system (Witter et al. 2005). As such, the fact that we found 235 differences between consensus genomes of CVI988 while only finding 11 differences between consensus genomes of Md5 is consistent with their history and real-world biology. Whether the larger number of variant loci in CVI988 reflects this strain’s attenuation through serial passage, or if it is a feature of this strain’s genetic background, remains to be determined. In addition, the fact that we were able to detect 29 minor variant positions in CVI988Pirbright.lab and 27 minor variant positions in Md5Pirbright.lab confirms that both strains exist as mixed viral populations. Over time, minor variant differences may expand during passage(s) in culture or during in vivo infections, leading to consensus-level differences (Trimpert et al. 2019, Kuny et al. 2020). Minor variants may thus provide an explanation for the differences observed between consensus genomes of each strain.
Having access to the minor variant profiles of CVI988 and Md5 has the added advantage of providing insight into the potential effects of intrastrain variation in loci of high interest, such as pp38 (MDV073). Currently, available qPCR assays to distinguish between CVI988 and virulent MDV strains are based on an SNP located at position 320 of the pp38 gene (Baigent et al. 2016). Our results show pp38 to be a highly conserved gene between different populations of CVI988 and Md5, with no minor variant positions coinciding with this gene. This not only confirms the robustness of qPCR assays based on the SNP #320 of pp38 but also establishes pp38 as a locus with high levels of within-strain conservation. Overall, our study provides a solid starting point for assessing intrastrain diversity in MDV strains. However, access to additional consensus genomes of CVI988 and Md5 will still be required in order to obtain the complete picture of MDV intrastrain diversity, as well as resequencing other virulent and attenuated MDV strains.
Among our most significant findings was the discovery of 19 SNPs present in the unique regions of CVI988GenBank.BAC but absent in the two new CVI988 consensus genomes sequenced as part of this study. These 19 SNPs were also not present as minor variants in the CVI988Pirbright.lab viral population, where these positions were sequenced at an average depth of >10 000×. CVI988GenBank.BAC was sequenced by Spatz et al. (2007a) from a BAC clone of CVI988 at 6× coverage (Table 1). This BAC clone was derived by Petherbridge et al. (2003) from a commercial vaccine supplied by Fort Dodge Animal Health. As part of the cloning process, a BAC vector was inserted into US2, and the missing region was later reconstructed using PCR and wild-type CVI988 DNA. While explaining the origin of these 19 SNPs in CVI988GenBank.BAC is beyond the scope of this study, our results suggest that consensus genomes of CVI988 obtained from cultured stocks need not differ from virulent strains such as Md5, 648a, or RB-1B in any of these positions.
The implications of these findings are multiple. First, these 19 SNPs have been reported as differences between CVI988 and virulent strains like Md5, RB-1B, GA, and Md11 by many prior studies, starting with the original publication by Spatz and Silva (2007), Spatz et al. (2007b), and Spatz and Rue (2008). Second, the 16 SNPs in US of CVI988GenBank.BAC impacting US2, US3, SORF4, and US6/gD have been previously proposed as evidence of naturally occurring recombination between CVI988 and virulent strains (He et al. 2018). To date, a total of seven isolates from China have been described as natural recombinants of CVI988, or “CVI988-like” variants, largely on the basis of these 16 SNPs (He et al. 2018, 2020, Zhang et al. 2022). The two new consensus genomes of CVI988 generated as part of this study share the same nucleotides in all 16 positions with these isolates, calling into question whether they are in fact natural recombinants of CVI988. An alternative explanation could be that these isolates are examples of circulating CVI988 that have regained some level of virulence (Zhang et al. 2022). In such a scenario, the remaining nucleotide differences between these isolates and our two new CVI988 consensus genomes could yield valuable information regarding loci associated with virulence. On the other hand, these isolates could also correspond to vaccine CVI988 reisolated from vaccinated birds. Either way, confirming the status of these seven isolates will require additional consensus genomes of CVI988, ideally from vaccine samples currently being distributed in China. Overall, these findings suggest that a heavy reliance on the published CVI988 consensus genome may have led to a historical overestimation of genomic divergence between CVI988 and virulent strains.
Finally, it is worth mentioning that current Illumina-based approaches do not provide insight into certain regions in MDV genomes where additional intrastrain variation is likely to be found. Specifically, tandem repeats have been suggested to be an important source of diversity in herpesviruses, yet they remain mostly inaccessible to NGS strategies at both the consensus and sub-consensus levels (van Belkum et al. 1998, Renner et al. 2018). The use of long-read technologies (e.g. Oxford Nanopore and Pacific Biosciences) could potentially help to overcome these limitations and make it possible to reliably assess intrastrain variation at tandem repeats and homopolymers (Rhoads and Au 2015, Wang et al. 2021). In addition, the use of long reads could enhance our ability to link minor variant positions into proper haplotypes and help us to better understand the real-world biology of MDV strains.
In conclusion, by sequencing multiple viral stocks and field isolates of the vaccine strain CVI988 and the very virulent strain Md5, we have shown that consensus genomes of these strains can differ in multiple positions across all genomic regions. We have also established that these differences are most likely made possible by fluctuations in a mixed population of viral genomes, the existence of which we have demonstrated using ultra-deep Illumina sequencing in combination with oligo-bait-based enrichment of MDV. In addition, we have shown that not accounting for the intrinsic genomic heterogeneity of MDV strains may have led to a historical overestimation of divergence between CVI988 and virulent strains, providing an alternative explanation for sequencing data that were previously used to suggest naturally occurring recombination between the two. In doing so, we have demonstrated the value of working with multiple consensus genomes per strain and challenged prevailing notions regarding the genomic stability of DNA viruses.
Supplementary Material
Acknowledgements
The authors thank members of the Szpara, Kennedy, and Nair labs for helpful feedback and discussion. We thank Matthew J. Jones and Andrew S. Bell for their contributions to the early preparation of two samples for this study.
Contributor Information
Alejandro Ortigas-Vasquez, Department of Biology, Center for Infectious Disease Dynamics, Huck Institutes of the Life Sciences, Pennsylvania State University, University Park, PA 16802, USA.
Utsav Pandey, Department of Biochemistry and Molecular Biology, Center for Infectious Disease Dynamics, Huck Institutes of the Life Sciences, Pennsylvania State University, University Park, PA 16802, USA.
Daniel W Renner, Department of Biology, Center for Infectious Disease Dynamics, Huck Institutes of the Life Sciences, Pennsylvania State University, University Park, PA 16802, USA.
Chris D Bowen, Department of Biology, Center for Infectious Disease Dynamics, Huck Institutes of the Life Sciences, Pennsylvania State University, University Park, PA 16802, USA.
Susan J Baigent, Viral Oncogenesis Group, The Pirbright Institute, Woking GU24 0NF, UK.
John Dunn, United States Department of Agriculture, Agricultural Research Service, US National Poultry Research Center, Southeast Poultry Research Laboratory, Athens, GA 30605, USA.
Hans Cheng, United States Department of Agriculture, Agricultural Research Service, US National Poultry Research Center, Avian Disease and Oncology Laboratory, East Lansing, MI 48823, USA.
Yongxiu Yao, Viral Oncogenesis Group, The Pirbright Institute, Woking GU24 0NF, UK.
Andrew F Read, Department of Biology, Center for Infectious Disease Dynamics, Huck Institutes of the Life Sciences, Pennsylvania State University, University Park, PA 16802, USA.
Venugopal Nair, Viral Oncogenesis Group, The Pirbright Institute, Woking GU24 0NF, UK.
Dave A Kennedy, Department of Biology, Center for Infectious Disease Dynamics, Huck Institutes of the Life Sciences, Pennsylvania State University, University Park, PA 16802, USA.
Moriah L Szpara, Department of Biology, Center for Infectious Disease Dynamics, Huck Institutes of the Life Sciences, Pennsylvania State University, University Park, PA 16802, USA; Department of Biochemistry and Molecular Biology, Center for Infectious Disease Dynamics, Huck Institutes of the Life Sciences, Pennsylvania State University, University Park, PA 16802, USA.
Author contributions
A.O.-V., M.L.S., and D.A.K. (Conceptualization); U.P., D.W.R., and A.O.-V. (Data curation); A.O.-V. (Formal analysis); D.A.K., V.N., M.L.S., and A.F.R. (Funding acquisition); U.P., C.D.B., S.J.B., Y.Y., J.D., and H.C. (Investigation); U.P. and C.D. (Methodology); M.L.S. and D.A.K. (Project administration); J.D., H.C., S.J.B., Y.Y., V.N., D.W.R., and A.F.R. (Resources); U.P., D.W.R., and A.O.-V. (Software); M.L.S. and D.A.K. (Supervision); C.D.B., D.W.R., and A.O.-V. (Validation); A.O.-V., M.L.S., and D.A.K. (Visualization); A.O.-V. (Writing); and A.O.-V., M.L.S., and D.A.K. (Writing—review & editing).
Supplementary data
Supplementary data is available at VEVOLU Journal online.
Conflict of interest:
None declared.
Funding
This work was supported by a United States National Science Foundation (NSF) and National Institutes of Health (NIH) joint Ecology and Evolution of Infectious Diseases (EEID) award, 1 R01 GM140459 (DK, MS, VN), and the following United Kingdom Biotechnology and Biological Sciences Research Council (BBSRC) grants: BBS/E/I/00007039, BBS/E/PI/23NB0003, BB/CCG2250/1 and BB/V017748/1.
References
- Baigent SJ, Nair VK, Le Galludec H. Real-time PCR for differential quantification of CVI988 vaccine virus and virulent strains of Marek’s disease virus. J Virol Methods 2016;233:23–36.doi: 10.1016/j.jviromet.2016.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell AS, Kennedy DA, Jones MJ. et al. Molecular epidemiology of Marek’s disease virus in central Pennsylvania, USA. Virus Evol 2019;5:vey042.doi: 10.1093/ve/vey042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 1999;27:573–80.doi: 10.1093/nar/27.2.573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen CD, Renner DW, Shreve JT. et al. Viral forensic genomics reveals the relatedness of classic herpes simplex virus strains KOS, KOS63, and KOS79. Virology 2016;492:179–86.doi: 10.1016/j.virol.2016.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill AE, Biggs PM. Agent of Marek’s disease in tissue culture. Nature 1967;215:528–30.doi: 10.1038/215528a0 [DOI] [PubMed] [Google Scholar]
- Churchill AE, Payne LN, Chubb RC. Immunization against Marek’s disease using a live attenuated virus. Nature 1969;221:744–47.doi: 10.1038/221744a0 [DOI] [PubMed] [Google Scholar]
- Cudini J, Roy S, Houldcroft CJ. et al. Human cytomegalovirus haplotype reconstruction reveals high diversity due to superinfection and evidence of within-host recombination. Proc Natl Acad Sci 2019;116:5693–98.doi: 10.1073/pnas.1818130116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dambrine G, Labaille J, Boissel É. et al. Marek’s disease herpesvirus: a model of vaccine-dependent adaptation. Virol Montrouge Fr 2016;20:273–86. [DOI] [PubMed] [Google Scholar]
- Davison F, Venugopal N. Marek’s Disease: An Evolving Problem, 1st edn. Amsterdam; Boston: Elsevier, 2004. [Google Scholar]
- Depledge DP, Cudini J, Kundu S. et al. High viral diversity and mixed infections in cerebral spinal fluid from cases of varicella zoster virus encephalitis. J Infect Dis 2018;218:1592–601.doi: 10.1093/infdis/jiy358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo E, Perales C. Viral quasispecies. PLoS Genet 2019;15:e1008271.doi: 10.1371/journal.pgen.1008271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn JR, Black Pyrkosz A, Steep A. et al. Identification of Marek’s disease virus genes associated with virulence of US strains. J Gen Virol 2019;100:1132–39.doi: 10.1099/jgv.0.001288 [DOI] [PubMed] [Google Scholar]
- Gimeno IM, Witter RL, Reed WM. Four distinct neurologic syndromes in Marek’s disease: effect of viral strain and pathotype. Avian Dis 1999;43:721–37.doi: 10.2307/1592741 [DOI] [PubMed] [Google Scholar]
- He L, Li J, Peng P. et al. Genomic analysis of a Chinese MDV strain derived from vaccine strain CVI988 through recombination. Infect Genet Evol 2020;78:104045. [DOI] [PubMed] [Google Scholar]
- He L, Li J, Zhang Y. et al. Phylogenetic and molecular epidemiological studies reveal evidence of recombination among Marek’s disease viruses. Virology 2018;516:202–09.doi: 10.1016/j.virol.2018.01.019 [DOI] [PubMed] [Google Scholar]
- Hildebrandt E, Dunn JR, Perumbakkam S. et al. Characterizing the molecular basis of attenuation of Marek’s disease virus via in vitro serial passage identifies de novo mutations in the helicase-primase subunit gene UL5 and other candidates associated with reduced virulence. J Virol 2014;88:6232–42.doi: 10.1128/JVI.03869-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K. et al. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 2002;30:3059–66.doi: 10.1093/nar/gkf436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy DA, Cairns C, Jones MJ. et al. Industry-wide surveillance of Marek’s disease virus on commercial poultry farms. Avian Dis 2017;61:153–64.doi: 10.1637/11525-110216-Reg.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuny CV, Bowen CD, Renner DW. et al. In vitro evolution of herpes simplex virus 1 (HSV-1) reveals selection for syncytia and other minor variants in cell culture. Virus Evol 2020;6:veaa013.doi: 10.1093/ve/veaa013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauring AS. Within-host viral diversity: a window into viral evolution. Annu Rev Virol 2020;7:63–81.doi: 10.1146/annurev-virology-010320-061642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee LF, Wu P, Sui D. et al. The complete unique long sequence and the overall genomic organization of the GA strain of Marek’s disease virus. Proc Natl Acad Sci U S A 2000a;97:6091–96.doi: 10.1073/pnas.97.11.6091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SI, Takagi M, Ohashi K. et al. Difference in the meq gene between oncogenic and attenuated strains of Marek’s disease virus serotype 1. J Vet Med Sci 2000b;62:287–92.doi: 10.1292/jvms.62.287 [DOI] [PubMed] [Google Scholar]
- Lole KS, Bollinger RC, Paranjape RS. et al. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol 1999;73:152–60.doi: 10.1128/JVI.73.1.152-160.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loncoman CA, Vaz PK, Coppo MJ. et al. Natural recombination in alphaherpesviruses: insights into viral evolution through full genome sequencing and sequence analysis. Infect Genet Evol 2017;49:174–85. [DOI] [PubMed] [Google Scholar]
- López de Juan Abad BA, Cortes AL, Correa M. et al. Evaluation of factors that influence dose variability of Marek’s disease vaccines. Avian Dis 2019;63:591–98.doi: 10.1637/aviandiseases-D-19-00097 [DOI] [PubMed] [Google Scholar]
- Marek J. Multiple Nervenentzündung (Polyneuritis) bei Hühnern. Dtsch Tierärztl Wochenschr 1907;15:417–21. [Google Scholar]
- Murata S, Yamamoto E, Sakashita N. et al. Research note: characterization of S-Meq containing the deletion in Meq protein’s transactivation domain in a Marek’s disease virus strain in Japan. Poult Sci 2021;100:101461.doi: 10.1016/j.psj.2021.101461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk S, Meleshko D, Korobeynikov A. et al. metaSPAdes: a new versatile metagenomic assembler. Genome Res 2017;27:824–34.doi: 10.1101/gr.213959.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki W, Purchase HG, Burmester BR. Protection against Marek’s disease by vaccination with a herpesvirus of turkeys. Avian Dis 1970;14:413–29.doi: 10.2307/1588488 [DOI] [PubMed] [Google Scholar]
- Pandey U, Bell AS, Renner DW. et al. DNA from dust: comparative genomics of large DNA viruses in field surveillance samples. mSphere 2016;1:e00132-16.doi: 10.1128/mSphere.00132-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons LR, Tafuri YR, Shreve JT. et al. Rapid genome assembly and comparison decode intrastrain variation in human alphaherpesviruses. mBio 2015;6:e02213-14.doi: 10.1128/mBio.02213-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petherbridge L, Howes K, Baigent SJ. et al. Replication-competent bacterial artificial chromosomes of Marek’s disease virus: novel tools for generation of molecularly defined herpesvirus vaccines. J Virol 2003;77:8712–18.doi: 10.1128/JVI.77.16.8712-8718.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner DW, Szpara ML, Glaunsinger BA. The impacts of genome-wide analyses on our understanding of human herpesvirus diversity and evolution. J Virol 2018;92:e00908-17.doi: 10.1128/JVI.00908-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renz KG, Cooke J, Clarke N. et al. Pathotyping of Australian isolates of Marek’s disease virus and association of pathogenicity with meq gene polymorphism. Avian Pathol 2012;41:161–76.doi: 10.1080/03079457.2012.656077 [DOI] [PubMed] [Google Scholar]
- Renzette N, Gibson L, Jensen JD. et al. Human cytomegalovirus intrahost evolution—a new avenue for understanding and controlling herpesvirus infections. Curr Opin Virol 2014;8:109–15.doi: 10.1016/j.coviro.2014.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoads A, Au KF. PacBio sequencing and its applications. Genomics Proteomics Bioinf 2015;13:278–89.doi: 10.1016/j.gpb.2015.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rispens BH, van Vloten H, Mastenbroek N. et al. Control of Marek’s disease in the Netherlands. I. Isolation of an avirulent Marek’s disease virus (strain CVI 988) and its use in laboratory vaccination trials. Avian Dis 1972;16:108–25.doi: 10.2307/1588905 [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W. et al. Integrative genomics viewer. Nat Biotechnol 2011;29:24–26.doi: 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato J, Murata S, Yang Z. et al. Effect of insertion and deletion in the Meq protein encoded by highly oncogenic Marek’s disease virus on transactivation activity and virulence. Viruses 2022;14:382.doi: 10.3390/v14020382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schat KA. History of the first-generation Marek’s disease vaccines: the science and little-known facts. Avian Dis 2016;60:715–24.doi: 10.1637/11429-050216-Hist [DOI] [PubMed] [Google Scholar]
- Schat KA, Calnek BW, Fabricant J. Characterisation of two highly oncogenic strains of Marek’s disease virus. Avian Pathol J WVPA 1982;11:593–605.doi: 10.1080/03079458208436134 [DOI] [PubMed] [Google Scholar]
- Schumacher D, Tischer BK, Teifke J-P. et al. Generation of a permanent cell line that supports efficient growth of Marek’s disease virus (MDV) by constitutive expression of MDV glycoprotein E. J Gen Virol 2002;83:1987–92.doi: 10.1099/0022-1317-83-8-1987 [DOI] [PubMed] [Google Scholar]
- Shipley MM, Rathbun MM, Szpara ML. Oligonucleotide enrichment of HSV-1 genomic DNA from clinical specimens for use in high-throughput sequencing. Methods Mol Biol Clifton NJ 2020;2060:199–217. [DOI] [PubMed] [Google Scholar]
- Shipley MM, Renner DW, Ott M. et al. Genome-wide surveillance of genital herpes simplex virus type 1 from multiple anatomic sites over time. J Infect Dis 2018;218:595–605.doi: 10.1093/infdis/jiy216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K, Mehta D, Dumka S. et al. Quasispecies nature of RNA viruses: lessons from the past. Vaccines 2023;11:308.doi: 10.3390/vaccines11020308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spatz SJ. Accumulation of attenuating mutations in varying proportions within a high passage very virulent plus strain of Gallid herpesvirus type 2. Virus Res 2010;149:135–42.doi: 10.1016/j.virusres.2010.01.007 [DOI] [PubMed] [Google Scholar]
- Spatz SJ, Petherbridge L, Zhao Y. et al. Comparative full-length sequence analysis of oncogenic and vaccine (Rispens) strains of Marek’s disease virus. J Gen Virol 2007a;88:1080–96.doi: 10.1099/vir.0.82600-0 [DOI] [PubMed] [Google Scholar]
- Spatz SJ, Rue CA. Sequence determination of a mildly virulent strain (CU-2) of Gallid herpesvirus type 2 using 454 pyrosequencing. Virus Genes 2008;36:479–89.doi: 10.1007/s11262-008-0213-5 [DOI] [PubMed] [Google Scholar]
- Spatz SJ, Silva RF. Sequence determination of variable regions within the genomes of gallid herpesvirus-2 pathotypes. Arch Virol 2007;152:1665–78.doi: 10.1007/s00705-007-0992-3 [DOI] [PubMed] [Google Scholar]
- Spatz SJ, Volkening JD, Gimeno IM. et al. Dynamic equilibrium of Marek’s disease genomes during in vitro serial passage. Virus Genes 2012;45:526–36.doi: 10.1007/s11262-012-0792-z [DOI] [PubMed] [Google Scholar]
- Spatz SJ, Zhao Y, Petherbridge L. et al. Comparative sequence analysis of a highly oncogenic but horizontal spread-defective clone of Marek’s disease virus. Virus Genes 2007b;35:753–66.doi: 10.1007/s11262-007-0157-1 [DOI] [PubMed] [Google Scholar]
- Stratton M. Genome resequencing and genetic variation. Nat Biotechnol 2008;26:65–66.doi: 10.1038/nbt0108-65 [DOI] [PubMed] [Google Scholar]
- Sun G-R, Zhang Y-P, H-c L. et al. A Chinese variant Marek’s disease virus strain with divergence between virulence and vaccine resistance. Viruses 2017;9:E71.doi: 10.3390/v9040071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimpert J, Groenke N, Jenckel M. et al. A phylogenomic analysis of Marek’s disease virus reveals independent paths to virulence in Eurasia and North America. Evol Appl 2017;10:1091–101.doi: 10.1111/eva.12515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimpert J, Groenke N, Kunec D. et al. A proofreading-impaired herpesvirus generates populations with quasispecies-like structure. Nat Microbiol 2019;4:2175–83.doi: 10.1038/s41564-019-0547-x [DOI] [PubMed] [Google Scholar]
- Tulman ER, Afonso CL, Lu Z. et al. The genome of a very virulent Marek’s disease virus. J Virol 2000;74:7980–88.doi: 10.1128/JVI.74.17.7980-7988.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Belkum A, Scherer S, van Alphen L. et al. Short-sequence DNA repeats in prokaryotic genomes. Microbiol Mol Biol Rev MMBR 1998;62:275–93.doi: 10.1128/MMBR.62.2.275-293.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhao Y, Bollas A. et al. Nanopore sequencing technology, bioinformatics and applications. Nat Biotechnol 2021;39:1348–65.doi: 10.1038/s41587-021-01108-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witter RL. Protection by attenuated and polyvalent vaccines against highly virulent strains of Marek’s disease virus. Avian Pathol J WVPA 1982;11:49–62.doi: 10.1080/03079458208436081 [DOI] [PubMed] [Google Scholar]
- Witter RL. Increased virulence of Marek’s disease virus field isolates. Avian Dis 1997;41:149–63.doi: 10.2307/1592455 [DOI] [PubMed] [Google Scholar]
- Witter RL, Burgoyne GH, Solomon JJ. Preliminary studies on cell cultures infected with Marek’s disease agent. Avian Dis 1968;12:169–85.doi: 10.2307/1588098 [DOI] [PubMed] [Google Scholar]
- Witter RL, Calnek BW, Buscaglia C. et al. Classification of Marek’s disease viruses according to pathotype: philosophy and methodology. Avian Pathol J WVPA 2005;34:75–90.doi: 10.1080/03079450500059255 [DOI] [PubMed] [Google Scholar]
- Witter RL, Nazerian K, Purchase HG. et al. Isolation from turkeys of a cell-associated herpesvirus antigenically related to Marek’s disease virus. Am J Vet Res 1970;31:525–38. [PubMed] [Google Scholar]
- Witter RL, Sharma JM, Fadly AM. Pathogenicity of variant marek’s disease virus isolants in vaccinated and unvaccinated chickens. Avian Dis 1980;24:210–32.doi: 10.2307/1589781 [DOI] [Google Scholar]
- Zhang Y, Lan X, Wang Y. et al. Emerging natural recombinant Marek’s disease virus between vaccine and virulence strains and their pathogenicity. Transbound Emerg Dis 2022;69:e1702–09.doi: 10.1111/tbed.14506 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Li Z, Bao K. et al. Pathogenic characteristics of Marek’s disease virus field strains prevalent in China and the effectiveness of existing vaccines against them. Vet Microbiol 2015;177:62–68.doi: 10.1016/j.vetmic.2014.12.020 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.