

Abstract
In the past few years, chimeric antigen receptor (CAR) T-cell therapy has emerged as a promising treatment for cancers that failed standard treatments. Such therapies have already been approved in several blood cancers, such as B-cell leukemia and lymphoma. Despite this progress, a significant proportion of patients experience primary or secondary resistance to CAR T-cell therapy. Here, we review the mechanisms by which CAR T cells eliminate their target and how cancer cells may be insensitive to such killing (here referred to as intrinsic resistance). Recent studies suggest that the activation of apoptosis through death receptor signaling is responsible for a major part of CAR T-cell cytotoxicity in vivo. Indeed, cancer cells harboring aberrant apoptotic machinery may be insensitive to CAR T-cell killing. This intrinsic resistance of cancer cells to CAR T-cell killing could be responsible for a significant portion of treatment failure. Finally, we discuss strategies that may be envisioned to overcome such resistance to enhance CAR T-cell efficacy.
Introduction
In the past several years, adoptive immunotherapies have gained unprecedented interest in oncology with the development of chimeric antigen receptors (CAR) T cells (1, 2). CAR T cells are T cells that are genetically engineered to express a CAR that includes a single-chain variable fragment (scFv) recognition domain specific for a tumor antigen linked to the signaling machinery of the T-cell receptor (TCR; CD3zeta) and a costimulatory domain (3). Unlike natural T cells, which recognize their target through major histocompatibility complex (MHC) presentation, CAR T cells can kill tumor cells in an MHC-independent manner (4). This approach demonstrated remarkable efficacy in relapsed or refractory (R/R) CD19 B-cell malignancies, leading to the FDA approval of four CD19-directed CAR T cells, in pediatric B-cell acute lymphoblastic leukemia (B-ALL; ref. 5), diffuse large B-cell lymphoma (DLBCL; refs. 6, 7), mantle cell lymphoma (8), and more recently follicular lymphoma (9). Despite these remarkable results, a significant number of patients treated with CAR T cells may experience primary (no response) or secondary (initial response followed by relapse/escape) resistance. Efforts are being made to understand the underlying mechanisms of resistance to CAR T-cell therapy to develop alternative strategies that would overcome these limitations. Three main mechanisms of resistance have been described thus far: (i) antigen escape; (ii) defective CAR T-cell function; and (iii) immunosuppressive microenvironment (10). However, recent data suggest that cancer cells may also survive CAR T-cell therapy due to intrinsic resistance mechanisms that render tumor cells insensitive to CAR T-cell killing (Fig. 1; ref. 11). To understand this novel mechanism of resistance, it is important to understand how CAR T cells kill their target. The apoptotic pathways are highly complex and involve a cascade of molecular events (12). These interactions are even more complex due to the tumor heterogeneity. Like natural cytotoxic T lymphocytes (CTL), CAR T cells can engage two distinct pathways to mediate tumor cell death: (i) the perforin/granzyme pathway and (ii) the death receptor pathway. These signaling cascades have in common the same terminal effector pathway, converging to the cleavage of caspase-3, finally triggering cell apoptosis through DNA damage. Moreover, similar to CTLs, CAR T cells are able to secrete cytokines that can act as effectors for target cell killing. The perforin/granzyme axis has been considered as the most important pathway for CD8+ and CD4+ CTLs cytotoxic functions and for CAR T cells by extension (13). However, recent data suggest that the death receptor axis and cytokine-mediated killing should not be overlooked when deciphering CAR T-cell–mediated killing (Figs. 2 and 3). Here, we review the data that support the critical role of the intrinsic mechanisms of resistance and discuss the therapeutic implications that could be employed to overcome this resistance. CAR T-cell resistance is a complex and dynamic process. Intrinsic resistance as described in this review is one of the many resistance mechanisms potentially involved in CAR T-cell failure.
Figure 1.
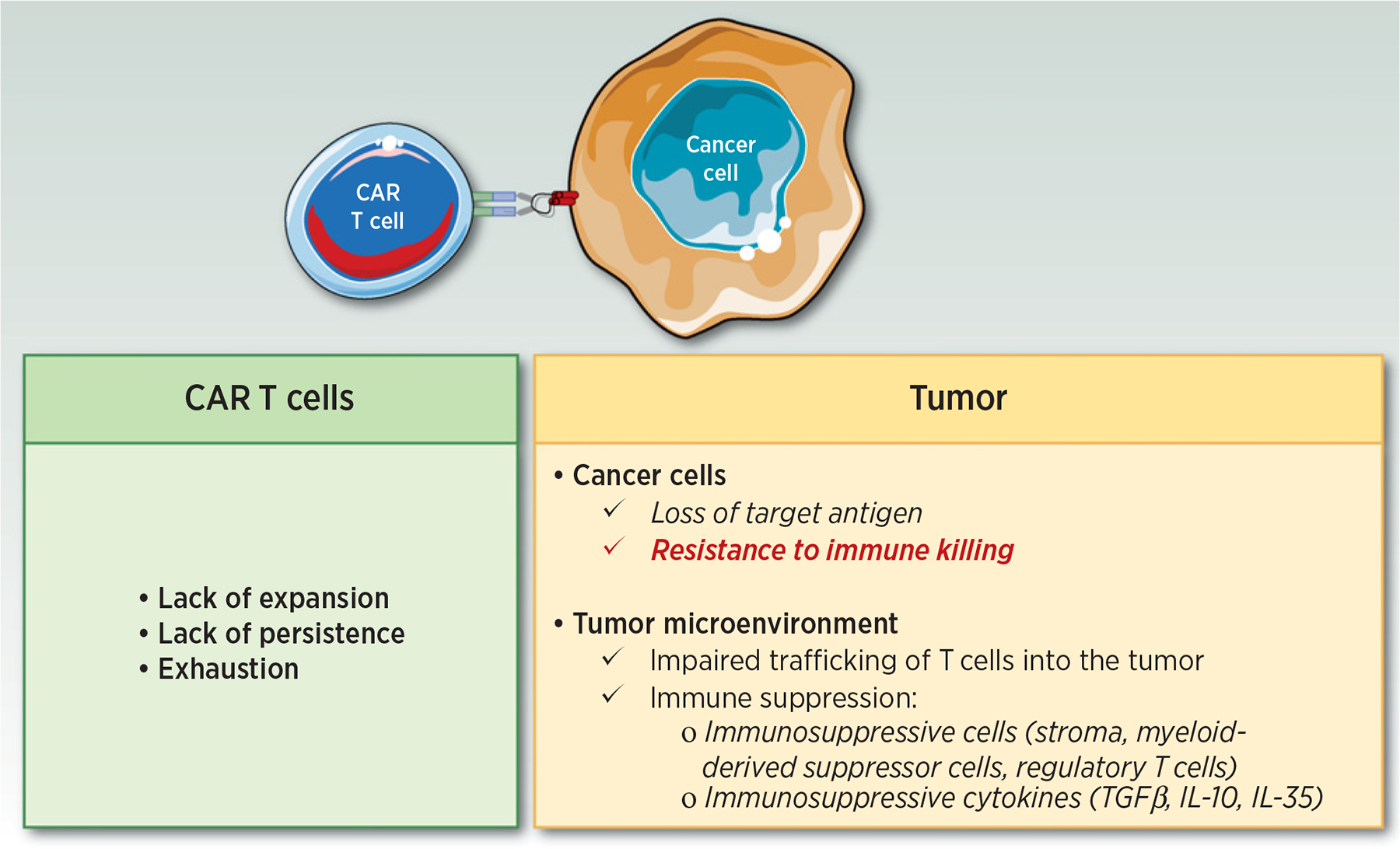
Mechanisms of resistance to CAR T-cell therapy.
Figure 2.
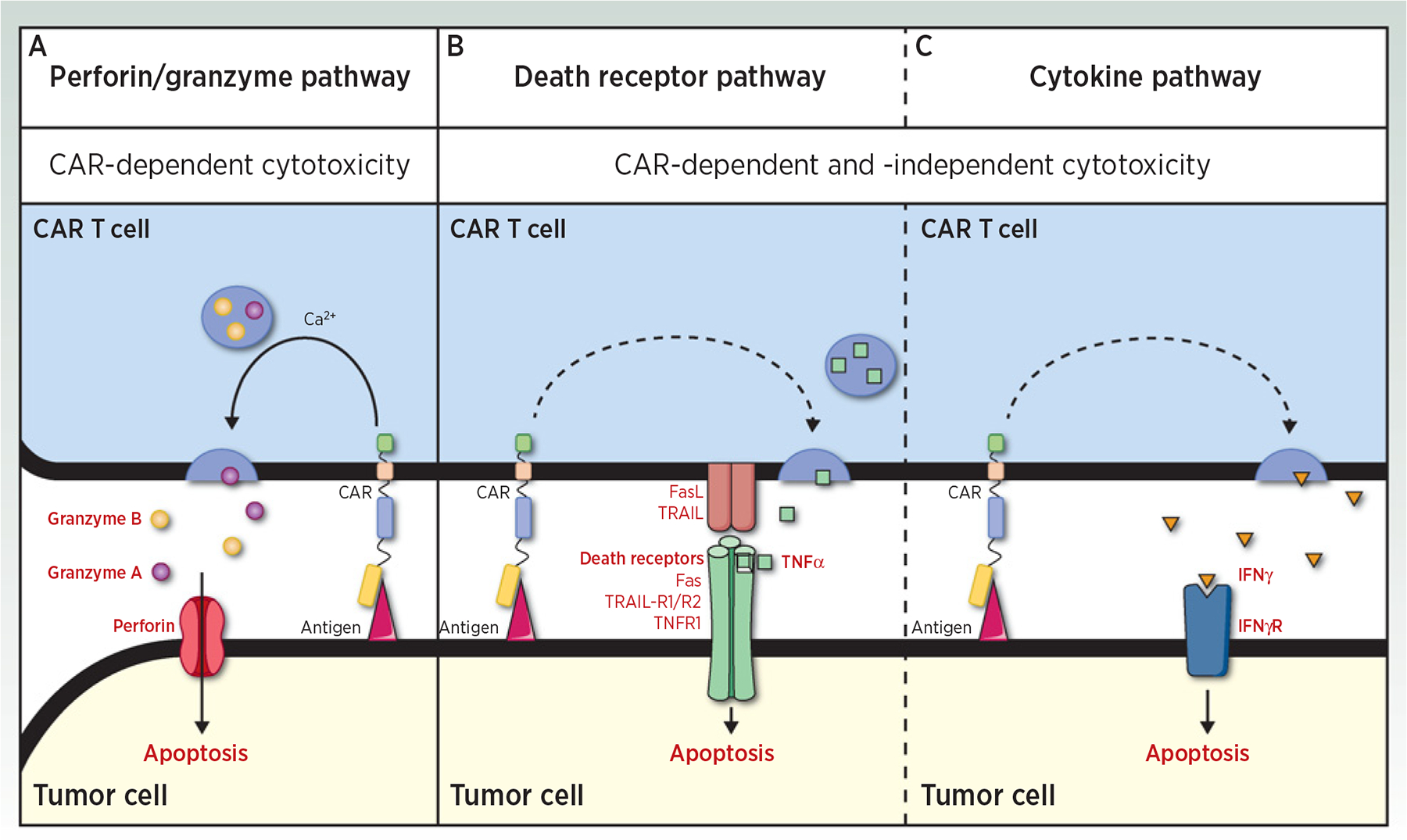
CAR-dependent and CAR-independent killing mechanisms of CAR T cells. Three pathways may be used by CAR T cells to kill their target. A–C, The perforin/granzyme pathway (granzyme/perforin; A), the death receptor pathway (FasL/Fas, TRAIL/TRAIL-R1/2, and TNFα/TNFR1; B), and the cytokine pathway (IFNγ; C). The perforin/granzyme pathway is dependent on target antigen recognition by CAR, whereas the death receptor and the cytokine pathways could mediate cytotoxicity independently of this specific recognition (dashed arrow). Indeed, CAR T cells may be activated by antigen-positive tumor cells and then induce bystander killing of antigen-negative tumor cells through the death receptor and the cytokine pathways.
Figure 3.
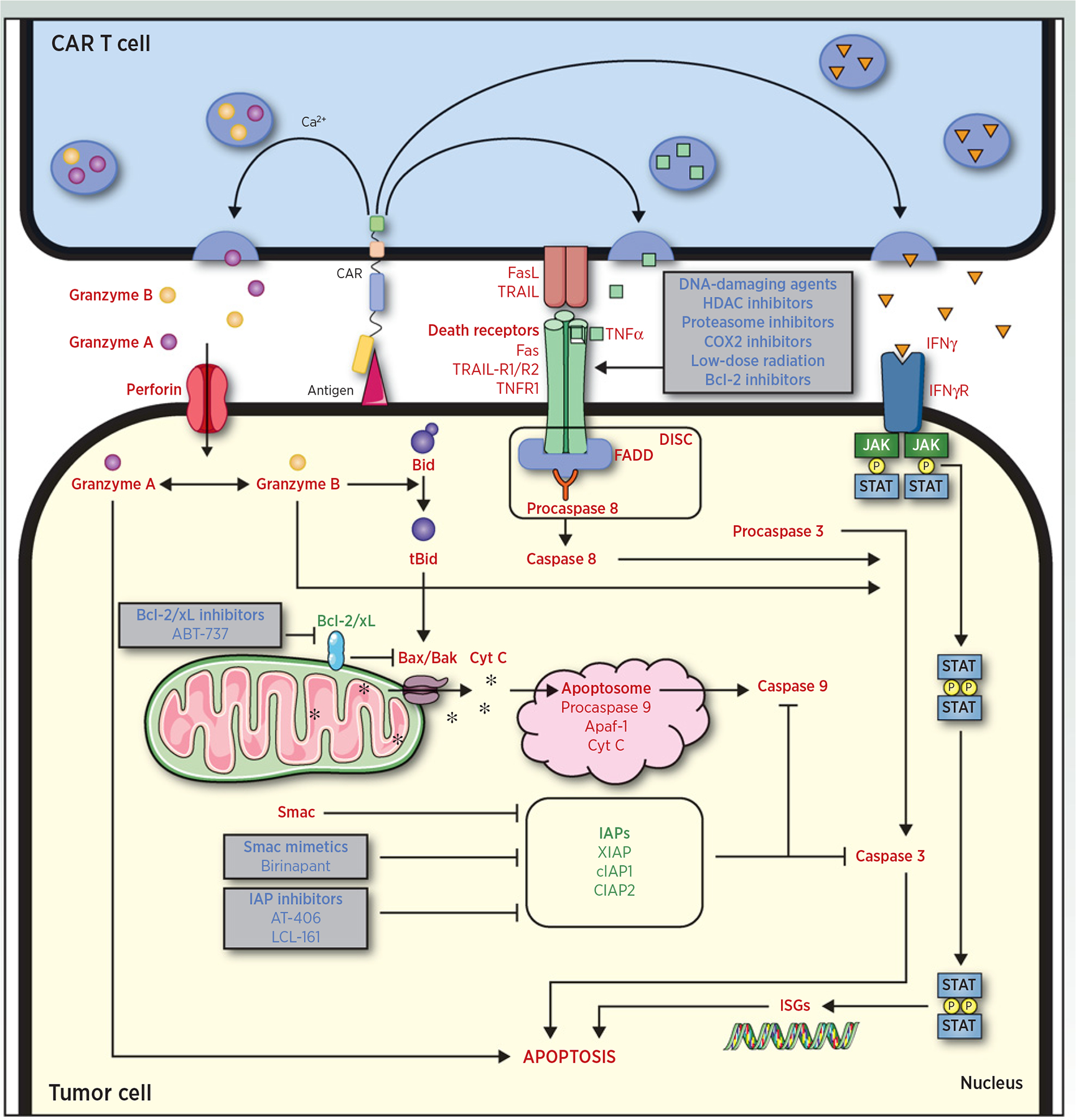
Killing mechanisms of CAR T cells and therapeutic strategies to overcome intrinsic resistance and/or enhance sensitivity to CAR T-cell killing. Three pathways may be used by CAR T cells to kill their target: (i) the perforin/granzyme pathway (granzyme/perforin); (ii) the death receptor pathway (FasL/Fas, TRAIL/TRAIL-R1/2, and TNFα/TNFR1); and (iii) the cytokine pathway (IFNγ). The perforin/granzyme pathway target cytochrome C releases from the mitochondrial intermembrane space through activation of Bax and Bak by tBid. Once released, cytochrome C organizes the apoptosome platform and targets the terminal effector pathway of apoptosis. The death receptor and cytokine pathways target tumor cell apoptosis independently from the mitochondria. Using therapeutic agents affecting different steps of the apoptosis cascade, tumor cell sensitivity to apoptosis can be enhanced, leading to CAR T-cell–mediated killing improvement. Proapoptotic molecules (red), antiapoptotic molecules (green), therapeutic agents to modulate apoptosis sensitivity (blue).
Perforin and Granzymes
Cytotoxic T cells
CTLs can exert their cytotoxic function on tumor cells by secreting preformed granules containing perforin, a pore-forming protein, and granzymes, which are serine proteases (14). Upon T-cell receptor engagement, CTLs link to their target cells, establishing the immune synapse (IS), a highly organized structure that acts as a signaling platform for bidirectional interactions (15). After TCR-mediated target-cell recognition, cytotoxic granule contents are released in a calcium-dependent manner into the IS. When released, perforin induces channel formation on the target cell facilitating proapoptotic granzymes entry. Thus far, granzyme B has received the most attention because it activates the caspase-dependent pathway of apoptosis through (i) caspase-10 activation; (ii) facilitation of cytochrome C release from the mitochondrial intermembrane space; and (iii) direct caspase-3 targeting (16). Moreover, granzyme B–induced cell death is not abolished by complete caspase inhibition, unraveling the ability of granzyme B to induce cell death in a caspase-independent manner (17). Indeed, one important function of granzyme B is to cleave Bid (BH3-interacting domain death agonist), a member of the proapoptotic Bcl-2 (B-cell lymphoma 2) family, generating truncated Bid (tBid). tBid translocates to the mitochondria and activates Bax (Bcl-2 homologous antagonist killer) and Bak (Bcl-2 associated X protein), two proapoptotic members of the Bcl-2 family, which results in the release of cytochrome C. Once released in the cytosol, cytochrome C binds to and activates apoptotic protease activating factor 1 (Apaf-1) and procaspase-9. This molecular platform called “apoptosome” enables the activation of functional caspase 9 (18). Granzyme B–mediated release of cytochrome C is inhibited by Bcl-2 and Bcl-xL, two antiapoptotic mitochondria localized molecules. Granzyme B is also able to directly disrupt the mitochondrial transmembrane potential in a caspase- and Bid-independent manner (19). In parallel, the release of second mitochondrial-derived activator of caspases (Smac) promotes apoptosis by inhibiting the activity of the inhibitor of apoptosis proteins (IAP; refs. 20, 21). Indeed, IAP represent a family of caspase inhibitors that bind to and inhibit caspases 3 and 9, thereby preventing apoptosis (22). Among IAP family members, XIAP (X-linked IAP), cIAP1, and cIAP2 are the most studied. Finally, the granzyme B signaling cascade converges toward the execution pathway through the activation of the effector caspase-3 and leads to cell death. Finally, another granzyme molecule, granzyme A, that is contained in cytotoxic granules induces cell death in a caspase-independent manner (23). This pathway is still not completely understood, as downstream caspase substrates are not involved, and cytochrome C is not released.
CAR T cells
Unlike CTLs, CAR T cells can recognize antigens without the need for MHC presentation. Although CAR T cells utilize, at least in part, the conventional TCR signaling machinery, substantial differences in the IS structure have been observed (24). This nonclassical CAR T-cell target cell IS is capable of accelerating the delivery of lytic granules resulting in faster killing of the target cells (25). In vitro coculture settings of CAR T cells with targeted antigen-expressing cells demonstrated upregulation of CD107a, a degranulation reporter, and secretion of perforin and granzyme B, suggesting the involvement of the exocytosis pathway (25–27). Moreover, blocking perforin release with egtazic acid (EGTA), a calcium ion chelator, or using perforin-deficient CAR T cells was shown to significantly decrease CAR T-cell mediated killing in vitro (28, 29). In vivo experiments reported an involvement of the exocytosis pathway for CAR T cells mediated killing and tumor eradication that was only decreased and not abolished using perforin-deficient CAR T cells (30–32). Lack of perforin negatively affected both CD8+ and CD4+ CD19-directed CAR T-cell cytotoxicity and resulted in incomplete B-cell aplasia (31). These observations suggest that CAR T-cell killing could be mediated by other signaling pathways, including the death receptor pathway (Figs. 2 and 3; Table 1).
Table 1.
Preclinical and clinical studies addressing the mechanisms CAR T-cell killing.
| Reference | Experiments | Results | |
|---|---|---|---|
| Perforin/granzymes | Davenport et al., 2015 (25) | Imaging killing assay coculturing HER2 CAR T with HER2-expressing tumor cells | Confocal microscopy showing PI blush after CAR T-targeted cell contact, suggesting the formation of perforin pores |
| Koehler et al., 2007 (26) | Cytotoxic assay coculture CEA CAR T with CEA-expressing colon carcinoma cell lines | Granzyme B secretion by CAR T cells in the presence of targeted antigen | |
| Kumaresan et al., 2014 (27) | Dectin-1 CAR cocultured with Aspergillus hyphae | Threefold upregulation of CD107a and secretion of perforin/granzyme B | |
| Darcy et al., 2000 (28) | Chrome release assay coculturing CEA CAR T with CEA-expressing colon carcinoma cell lines | Chrome-release abolished when using perforin-deficient CAR T cells | |
| Mamonkin et al., 2015 (29) | Cytotoxic assay coculture CD5 CAR T with Jurkat cells Perforin/granzyme blocking by EGTA |
Decreased cytotoxicity upon perforin/granzyme blockade | |
| Haynes et al., 2002 (30) | In vivo CEA-expressing colon carcinomas growth monitoring after CEA CAR adoptive transfer | Antitumoral efficacy decreased with perforin-deficient CAR T cells | |
| Ishii et al., 2020 (31) | Treatment with CD19 CAR T cells of leukemia-bearing mice | Antitumoral efficacy decreased with perforin-deficient CAR T cells | |
| Li et al., 2020 (32) | Glypican 3 CAR T treatment for orthotopic hepatocellular carcinoma in mice | Perforin and granzyme B-mediated apoptosis and tumor eradication | |
| Death receptors | Mamonkin et al., 2015 (29) | Cytotoxic assay coculture CD5 CAR T with Jurkat cells Anti-FasL blocking antibodies |
Decreased cytotoxicity upon FasL blockade |
| Upadhyay et al., 2020 (11) | Cytotoxic assay coculture CD19 CAR T cells with CD19+ or CD19− lymphoma cells Adoptive transfer of CD19 CAR T cells in CD19+ or CD19+/CD19− lymphomas bearing mice |
Fas deficiency moderately decreased CD19 CAR T-cell cytotoxicity against CD19+ lymphoma cells and nearly abolished CD19 CAR T-cell bystander cytotoxicity against CD19− lymphoma cells CD19 CAR T prolonged survival of both CD19+ and CD19+/CD19− lymphoma-bearing mice FasL blockade impaired on-target and bystander CD19 CAR T-cell efficacy in vivo |
|
| Darcy et al., 1998 (43) | Cytotoxic assay coculture CEA CAR T with CEA-expressing colon carcinoma cell lines | Decreased cytotoxicity with Fas-deficient CEA-expressing target cells | |
| DeSelm et al., 2018 (44) | Sialyl Lewis-A (sLeA) CAR adoptive transfer on sLeA+ and sLeA− heterogeneous tumor-bearing mice | Low-dose radiation sensitizes both on-target and bystander tumor cell killing by CAR T-cell via the TRAIL axis | |
| Hong et al., 2018 (45) | Cytotoxic assay coculture CD30 CAR T with CD30+ and CD30− tumor cells | Fas/FasL-dependent killing of CD30− tumor cells if CAR T cells were preactivated with CD30+ tumor cells | |
| Singh et al., 2020 (47) | Unbiased genome-wide loss-of-function screen in B-ALL cell lines under CD19 CAR T-cell pressure | Disruption of genes associated with proapoptotic death receptor signaling pathway (FADD, BID, CASP8, and TNFRSF10B) confers resistance to CAR-T cell killing Knockout of antiapoptotic genes (CFLAR, TRAF2, and BIRC2) led to an increased susceptibility to CAR-T killing |
|
| Dufva et al., 2020 (48) | Unbiased genome-wide loss-of-function screen in B-ALL and DLBCL cells under CD19 CAR T cells pressure associated with high-throughput drug screening | Death receptor signaling (FADD, TNFRSF10B) is a key mediator of CAR T-cell cytotoxicity Increased CAR T-cell cytotoxicity in presence of SMAC mimetics and IAP antagonists |
|
| Cytokines | Kumaresan et al., 2014 (27) | Dectin-1 CAR cocultured with Aspergillus hyphae | IFNγ production when exposed to their target antigen |
| Haynes et al., 2002 (30) | In vivo CEA-expressing colon carcinomas growth monitoring after CEA CAR adoptive transfer | Antitumoral efficacy decreased with IFNγ deficient CAR T | |
| Ishii et al., 2020 (31) | Treatment with CD19 CAR T cells of leukemia-bearing mice | Antileukemia activity despite the absence of perforin Compensatory overexpression of IFNγ IFNγ blockade decreased antileukemia activity |
|
| Arenas et al., 2021 (71) | HER2-driven cell lines and patient-derived xenografts treated with HER2 CAR T cells | Antitumoral efficacy decreased with IFNγ deficient CAR T, notably through the downregulation of JAK2 |
Death Receptor Pathway
Cytotoxic T cells
Together with calcium-dependent granule exocytosis, calcium-independent death receptor-mediated killing is a major axis by which target cells are lysed by CTLs. The death receptor signaling pathway of apoptosis involves interactions between death receptors expressed on the target cells and their ligands on effector cells (33). These death receptors are upregulated upon T-cell activation caused by target cell recognition. They are members of the TNF receptor superfamily and are characterized by their intracytoplasmic death domains (34). These domains are responsible for the death signaling transmission from the cell surface to the intracellular signaling pathways upon receptor trimerization by its ligands. Multiple ligand/death receptor couples have been described. The best-characterized are FasL/Fas, TRAIL (TNF-related apoptosis including ligand)/TRAIL-R1/2, and TNα/TNFR1 (35–37). Interaction between death receptors and their ligands results in the activation of caspase-8, mediated by the adapter protein FADD (Fas-associated protein) and pro–caspase-8, which form the death-inducing signaling complex (DISC; refs. 38–40). Ultimately, caspase-8 activation leads to the execution phase of apoptosis through caspase-3 activation.
Recently, Upadhyay and colleagues established an in vitro model in which GFP-specific CTLs were cocultured with both GFP-expressing (GFP+) and GFP-nonexpressing (GFP–) lymphoma cells (11). They showed that GFP+ cells eradication by GFP-specific CTLs was abrogated under Fas knockout in target cells despite CTL proliferation, granzyme B production, and degranulation. Moreover, CTLs exerted bystander killing of GFP– lymphoma cells in a Fas-dependent manner. Consistent with these results, they observed earlier recurrence of Fas–/– GFP+ tumors compared with the WT counterparts and blunted response to the therapy for tumors with Fas–/– GFP– bystander cells upon GFP-specific CTLs adoptive transfer in vivo. They demonstrated that bystander cytotoxicity was geographically restricted and dependent on colocalized GFP+ and GFP– tumor cells. Fas-mediated on-target and bystander apoptosis was increased after treatment with inhibitors of proteins with known modulatory activity within the Fas pathway, the Smac mimetic birinapant and the Bcl-2/xL inhibitor ABT-737. In line with these results, death receptor–mediated apoptosis was shown to be crucial for tumor cell sensitivity to T-cell killing in patients with cancer treated with immune checkpoint blockers (41, 42).
CAR T cells
In vitro cytotoxic assays with death receptor pathway disruption using anti-FasL antibodies or Fas-deficient target cells resulted in reduced CAR T-cell antigen-dependent triggered apoptosis (11, 29, 43). Thus, this apoptosis pathway is required for full CAR T-cell effector functions. Interestingly, it has been shown that CAR T cells are also able to mediate tumor cell bystander killing in an antigen-independent manner via a contact-dependent Fas/FasL interaction or via the TRAIL axis (11, 44, 45). Preactivation of CAR T cells upon antigen exposure was necessary to observe such alternative killing mechanism. Indeed, CAR engagement induces upregulation of FasL and TRAIL expression enabling the CAR T cells to kill tumor cells using the death receptor pathway in both an antigen-dependent and independent manner. Antigen-independent killing offers a path that could be exploited to circumvent relapses due to antigen loss or when degranulation is poor or hampered (46). In vivo, CD19 CAR T-cell efficacy was similar against homogeneous CD19+ tumors and heterogeneous tumors composed with both CD19+ and CD19– cells (11). In these murine models, FasL blockade severely impaired CAR T-cell functions reaffirming death receptor pathway dependence of on-target CAR T-cell cytotoxicity. Moreover, upon FasL blockade, mice bearing heterogeneous tumors had a significantly worse survival than their counterparts bearing homogeneous tumors, suggesting that the Fas–FasL axis was critical for CAR T-cell bystander cytotoxicity against CD19-loss variants.
Recently, Singh and colleagues addressed the question of tumor-intrinsic resistance to CAR T cells by performing an unbiased genome-wide loss-of-function screen in B-ALL cell lines under CD19 CAR T-cell pressure (47). Using CRISPR–Cas9 technology, they found that disruption of genes associated with proapoptotic death receptor signaling pathway such as FADD, BID, CASP8, and TNFRSF10B conferred resistance to CAR T-cell killing. Conversely, knock out of antiapoptotic genes such as CFLAR, TRAF2, and BIRC2 (encoding for cIAP1) led to an increased susceptibility to CAR T-cell killing. Moreover, CRISPR targeting of the genes encoding for FasL or TRAIL in CAR T cells resulted in a decreased killing, confirming that the death receptor pathway is crucial for CAR T-cell cytotoxicity. In their model, impairing apoptosis by targeting death receptors or their downstream signaling partners led to CAR T-cell dysfunction. In fact, loss of death receptor signaling is responsible for decreased tumor killing and persistent antigen exposure, which in turn causes CAR T-cell exhaustion through a global reprogramming of CAR T cells at the transcriptomic and epigenetic levels. Using samples from two multi-center clinical trials for relapsed pediatric B-ALL, they found a significantly higher death receptor signaling signature in samples from patients who had achieved complete remissions (CR) compared with patients who did not respond although they retained CD19 expression (NR). No difference was seen between CRs and NRs in the intrinsic apoptosis signature. This study unravels a novel mechanism of resistance to CAR T cells that is antigen-independent and relies on death receptor pathway disruption in tumor cells.
Dufva and colleagues validated death receptor genes FADD and TNFRSF10B as major contributors of tumor-intrinsic resistance to CAR T-cell cytotoxicity on leukemia and lymphoma cell lines (48). In agreement with Singh and colleagues, disruption of these genes resulted in decreased tumor sensitivity to CAR T-cell cytotoxicity. To further identify crucial pathways for CAR T-cell function and candidate molecules to modulate cytotoxicity, they carried out a high-throughput drug screen using a coculture assay with CD19-directed CAR T cells CD19 NALM6 B-ALL. Strikingly, the three drugs that most significantly enhanced CAR T-cell cytotoxicity all belonged to the same class, namely Smac mimetics or IAPi (birinapant, AT-406, and LCL-161), consistent with results obtained with CTLs (11).
These studies unravel a critical role for the death receptor pathway of apoptosis and highlight the link between a death receptor signaling signature and sensitivity to CAR T-cell therapy. These observations suggest the potential benefit of strategies aimed to sensitize tumor cells to this type of apoptosis. Lowering the threshold necessary to induce death receptor-mediated apoptosis may indeed overcome intrinsic tumor cell resistance to CAR T-cell therapy. It is well known that cancer therapeutic agents can modulate death receptor expression by tumor cells in p53-dependent and p53-independent manners (49). Multiple cancer therapeutic families can increase Fas, TRAIL-R1/2, or both on tumor cells, including DNA-damaging agents (50), histone deacetylase (HDAC) inhibitors (51), proteasome inhibitors (52), and cyclooxygenase-2 (COX2) inhibitors (53). HDAC inhibitors (SAHA and LBH589) and COX2 inhibitors (celexoxib) were able to partially reverse CD19 CAR T-cell resistance of non-Hodgkin lymphoma cell lines due to altered death receptor pathway machinery (54). In these cell lines, pretreatment with HDAC inhibitors or COX2 inhibitors enhanced tumor cell sensitivity to TRAIL-mediated apoptosis to overcome acquired intrinsic resistance despite CD19 expression. Similarly, low-dose radiation could sensitize tumor cells to CAR T-cell killing by targeting gene sets associated with sensitivity to TRAIL-mediated death, without increasing target antigen expression (44). This strategy increased both on-target and bystander CAR T-cell cytotoxicity, enabling tumoral control even in the case of target antigen loss variants. In vivo, the combination of IAP inhibition by birinapant with CAR T-cell therapy increased tumor cell death in a perforin-independent and TNF-dependent manner (55). In a human-HER2 transgenic mouse model, this combination was required to achieve antitumor activity, whereas each treatment alone failed to control tumor growth, demonstrating that the addition of birinapant enhanced CAR T-cell antitumor efficacy. Under these conditions, CAR T cells expanded >100-fold upon Smac mimetic exposure in vitro, showing lack of overt toxicity of IAP inhibition on CAR T cells.
Bcl-2 is an inhibitor of the intrinsic apoptosis pathway by blocking Bax and Bak and thus cytochrome C release, potentially causing treatment resistance. Thus, the combination of Bcl-2 inhibitors with CAR T cells appears to be an attractive strategy. In a B-ALL and CD19 CAR T-cell coculture model, it has been shown that CAR T-cell mediated caspase-3 targeting in tumor cells could be improved through Bcl-2 inhibition. Bcl-2 inhibition was responsible for a change of tumor cell phenotype with increased levels of death receptors. Moreover, Bcl-2 inhibitors are responsible for the upregulation of TRAIL-R2 by tumor cells and render them more sensitive to death receptor–mediated apoptosis (56). Thus, Bcl-2 inhibition decreases the apoptosis threshold and enhances CAR T-cell cytotoxic functions. However, one may keep in mind that, physiologically, Bcl-2 protects T cells from activation-induced cell death (AICD) by inhibiting death receptor-mediated apoptosis (57). This raises the concern Bcl-2 inhibitors may negatively impact CAR T-cell survival. First, Mamonkin and colleagues showed high expression levels of Bcl-2 in CAR T cells, which seemed sufficient to protect them from apoptosis by Bcl-2 inhibitors. Second, Bcl-2 inhibitors were able to presensitize tumor cells to CAR T-cell therapy, enabling their use only before the adoptive transfer.
Cytokine-Mediated Killing
Cytotoxic T cells
CTLs also produce several cytokines, such as TNα and IFNs that have cytotoxic function when secreted in the vicinity of target cells. TNFα is the soluble ligand of the death receptor TNFR1 and is implicated in the death receptor pathway of apoptosis as described above. IFNs are naturally secreted glycoproteins produced by almost every cell type as a mechanism of defense, possibly in response to tumor cells (58). IFNs are classified into three subfamilies. Type I IFNs composed of multiple members, including IFNα, type II IFNs contain IFNγ as the only member, and type III IFNs (59, 60). Functionally, when IFNs bind to the extracellular domain of their receptor, the intracellular portion triggers activation of the Janus kinase (JAK) through its phosphorylation. In turn, activated JAK phosphorylates signal transducer and activator of transcription proteins (STAT). Activated STATs dimers localize from the cytosol to the nucleus and initiate transcription of IFN-stimulated genes (ISG; refs. 61, 62). As IFNs are able to regulate the expression of approximately 2,000 ISGs, their biological effects are pleiotropic and depend on the IFN transcriptional signature expressed by the target cell in response to IFNs exposure, which can be modulated by other signaling pathways. Thus, IFNs are able to sensitize some cells to apoptosis by lowering the threshold of the intrinsic death pathway (63). However, not all tumor cells are susceptible to the apoptotic effects of IFNs (64). Conversely, some ISGs have prosurvival functions. For example, unphosphorylated STAT1 (U-STAT1) can drive the expression of ISGs with survival functions and be responsible for cancer resistance to cancer therapies (65, 66). Studies in tumor-bearing mice indicate that the IFNγ signaling plays an essential role in antitumor immune response mediated by immune checkpoint blockers (67, 68). Consistent with this observation, melanoma patients who are refractory to immune checkpoint blockers harbor a much higher rate of genomic defects in the IFN-γ signaling pathway as compared with responders (68, 69). Interestingly, the cytokines TNα and IFNγ produced by CTL increase the sensitivity of tumor cells to CTL killing by both granule- and FasL-mediated death pathways, unravelling synergistic effect of cytotoxic mechanisms (70).
CAR T cells
CAR T cells are known for their ability to secrete a high amount of IFNγ when exposed to target cells in vitro (27). In vivo, tumor control after adoptive transfer in colon carcinoma–bearing mice was less effective with IFNγ-deficient CAR T cells compared with wild-type (WT) CAR T cells (30). Recently, Ishii and colleagues established a syngeneic murine model to compare perforin-deficient and WT CD19 CAR T-cell therapy against B-ALL (31). Perforin-deficient CAR T cells demonstrated inferior in vitro cytotoxicity with slower kinetics of leukemia cell killing compared with WT CAR T cells. In parallel, perforin-deficient CAR T cells produced significantly higher amounts of IFNγ and TNFα. Consistent with these in vitro observations, perforin-deficient CAR T cells were less efficient at eradicating leukemia in vivo, although this difference could be overcome with a higher cell dose. Moreover, they showed that IFNγ neutralization negatively impacted the ability of perforin-deficient CAR T cells to eradicate leukemia in vitro and in vivo, suggesting that the IFNγ pathway may take over the exocytosis pathway in case of dysfunction. Finally, a recent study by Arenas and colleagues demonstrated that the disruption of IFNγ signaling, notably through the downmodulation of JAK2, conferred resistance to killing of cancer cells by CAR T cells directed against HER2 in preclinical models (HER2-driven cell lines and patient-derived xenografts; ref. 71). Given the potential contribution of IFNγ in CAR T-cell killing, strategies that aim to increase IFNγ secretion by CAR T cells may enhance their efficacy. This may be achieved by using 4th generation CAR T cells called TRUCKs for “T cells redirected for universal cytokine-mediated killing”, which can secrete cytokines and increase IFNγ production directly or indirectly (72). Avanzi and colleagues designed armored, IL18-secreting CAR T cells, which increased IFNγ secretion and tunned the tumor microenvironment toward an IFNγ signature (73). These IL18-secreting CAR T cells were able to modulate the tumor microenvironment and enhance the endogenous antitumor immune response. IL12 is another proinflammatory cytokine, which has been shown to increase the antitumor efficacy of CAR T cells (74). In a syngeneic tumor model, IL12 promoted IFNγ secretion by CAR T cells in an autocrine fashion. This secretion rendered CAR T cells resistant to the immunosuppression induced by regulatory T cells and myeloid-derived suppressor cells, and thus improved CAR T-cell cytotoxicity. These armored CAR T cells, by shaping the cytokine landscape within the tumor, represent a promising strategy to improve the efficacy of adoptive T-cell therapy. However, these approaches may be limited by the toxicity of high concentrations of IFNγ.
Conclusion
Learning how CAR T cells mediate tumor cell killing is a crucial step toward a better understanding of the mechanisms of primary and acquired resistance. Such understanding offers the opportunity to envision strategies to overcome intrinsic resistance of tumor cells to CAR T-cell killing. For a long time, it was believed that CAR T cells exerted their cytotoxic function mostly through degranulation involving the perforin/granzyme axis. However, recent studies suggest that the death receptor pathway of apoptosis, which is triggered by death receptors and their ligands, is crucial for CAR T-cell functions in vivo. These studies demonstrated that tumor cells harboring aberrant apoptotic machinery that renders them insensitive to the death receptor signaling are intrinsically resistant to CAR T-cell killing. From a clinical point of view, patients experiencing durable clinical responses upon CAR T-cell treatment have a significantly higher sensitivity to the death receptor pathway of apoptosis (6, 11). Thus, lowering the threshold necessary for CAR T cells to trigger tumor cell killing through the death receptor pathway may enhance tumor eradication. Among candidate molecules, IAP inhibitors, Smac mimetics, and Bcl-2 inhibitors appear promising. Furthermore, these approaches may enable the eradication of antigen-defective subsets of tumor cells by CAR T cells through an antigen-independent death receptor–dependent bystander killing. Although intrinsic resistance of cancer cells to immune killing may be important, other mechanisms may also contribute to CAR T-cell resistance, including loss of target antigen and immune suppression of CAR T cells by the tumor microenvironment. These alternative mechanisms may limit the clinical impact of strategies meant to increase or restore the sensitivity of tumor cells to apoptosis.
Authors’ Disclosures
M. Ruella reports grants and other support from Novartis and Tmunity; grants and personal fees from Abclon; personal fees from GSK, BMS, Bayer, and NanoString; and other support from viTToria Biotherapeutics during the conduct of the study, as well as grants and other support outside the submitted work and a patent issued, licensed, and with royalties paid. No disclosures were reported by the other authors.
References
- 1.Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res 2006;12:6106–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lamers CHJ, Willemsen R, van Elzakker P, van Steenbergen-Langeveld S, Broertjes M, Oosterwijk-Wakka J, et al. Immune responses to transgene and retroviral vector in patients treated with ex vivo–engineered T cells. Blood 2011; 117:72–82. [DOI] [PubMed] [Google Scholar]
- 3.Ruella M, Kalos M. Adoptive immunotherapy for cancer. Immunol Rev 2014; 257:14–38. [DOI] [PubMed] [Google Scholar]
- 4.Harris DT, Kranz DM. Adoptive T cell therapies: a comparison of T cell receptors and chimeric antigen receptors. Trends Pharmacol Sci 2016;37:220–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. N Engl J Med 2018;378:439–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-Cell therapy in refractory large B-Cell lymphoma. N Engl J Med 2017;377:2531–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chong EA, Ruella M, Schuster SJ, Lymphoma program investigators at the university of pennsylvania. five-year outcomes for refractory B-Cell lymphomas with CAR T-Cell therapy. N Engl J Med 2021;384:673–4. [DOI] [PubMed] [Google Scholar]
- 8.Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-Cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2020;382:1331–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacobson C, Chavez JC, Sehgal AR, William BM, Munoz J, Salles G, et al. Primary analysis of zuma-5: a phase 2 study of axicabtagene ciloleucel (Axi-Cel) in patients with relapsed/refractory (R/R) indolent non-hodgkin lymphoma (iNHL). Blood 2020;136:40–1. [Google Scholar]
- 10.Brown CE, Mackall CL. CAR T cell therapy: inroads to response and resistance. Nat Rev Immunol 2019;19:73–4. [DOI] [PubMed] [Google Scholar]
- 11.Upadhyay R, Boiarsky JA, Pantsulaia G, Svensson-Arvelund J, Lin MJ, Wroblewska A, et al. A critical role for fas-mediated off-target tumor killing in T cell immunotherapy. Cancer Discov 2021;11:599–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol 2007;35: 495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yasukawa M, Ohminami H, Arai J, Kasahara Y, Ishida Y, Fujita S. Granule exocytosis, and not the Fas/Fas ligand system, is the main pathway of cytotoxicity mediated by alloantigen-specific CD4+ as well as CD8+ cytotoxic T lymphocytes in humans. Blood 2000;95:2352–5. [PubMed] [Google Scholar]
- 14.Lieberman J. The ABCs of granule-mediated cytotoxicity: new weapons in the arsenal. Nat Rev Immunol 2003;3:361–70. [DOI] [PubMed] [Google Scholar]
- 15.Monks CR, Freiberg BA, Kupfer H, Sciaky N, Kupfer A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature 1998;395: 82–6. [DOI] [PubMed] [Google Scholar]
- 16.Darmon AJ, Nicholson DW, Bleackley RC. Activation of the apoptotic protease CPP32 by cytotoxic T-cell-derived granzyme B. Nature 1995;377:446–8. [DOI] [PubMed] [Google Scholar]
- 17.Thomas DA, Du C, Xu M, Wang X, Ley TJ. DFF45/ICAD can be directly processed by granzyme B during the induction of apoptosis. Immunity 2000;12: 621–32. [DOI] [PubMed] [Google Scholar]
- 18.Chinnaiyan AM. The apoptosome: heart and soul of the cell death machine. Neoplasia 1999;1:5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.MacDonald G, Shi L, Vande Velde C, Lieberman J, Greenberg AH. Mitochondria-dependent and -independent regulation of granzyme B-induced apoptosis. J Exp Med 1999;189:131–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salvesen GS, Duckett CS. IAP proteins: blocking the road to death’s door. Nat Rev Mol Cell Biol 2002;3:401–10. [DOI] [PubMed] [Google Scholar]
- 21.Zamzami N, Kroemer G. The mitochondrion in apoptosis: how Pandora’s box opens. Nat Rev Mol Cell Biol 2001;2:67–71. [DOI] [PubMed] [Google Scholar]
- 22.Schimmer AD. Inhibitor of apoptosis proteins: translating basic knowledge into clinical practice. Cancer Res 2004;64:7183–90. [DOI] [PubMed] [Google Scholar]
- 23.Beresford PJ, Xia Z, Greenberg AH, Lieberman J., Granzyme A loading induces rapid cytolysis and a novel form of DNA damage independently of caspase activation. Immunity 1999;10:585–94. [DOI] [PubMed] [Google Scholar]
- 24.Roda-Navarro P, Álvarez-Vallina L. Understanding the spatial topology of artificial immunological synapses assembled in T Cell-redirecting strategies: a major issue in cancer immunotherapy. Front Cell Dev Biol 2019;7:370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davenport AJ, Jenkins MR, Cross RS, Yong CS, Prince HM, Ritchie DS, et al. CAR-T cells inflict sequential killing of multiple tumor target cells. Cancer Immunol Res 2015;3:483–94. [DOI] [PubMed] [Google Scholar]
- 26.Koehler H, Kofler D, Hombach A, Abken H. CD28 Costimulation overcomes transforming growth factor-b–mediated repression of proliferation of redirected human CD4þ and CD8þ T Cells in an antitumor cell attack. Cancer Res 2007;67: 2265–73. [DOI] [PubMed] [Google Scholar]
- 27.Kumaresan PR, Manuri PR, Albert ND, Maiti S, Singh H, Mi T, et al. Bioengineering T cells to target carbohydrate to treat opportunistic fungal infection. Proc Natl Acad Sci U S A 2014;111:10660–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Darcy PK, Haynes NM, Snook MB, Trapani JA, Cerruti L, Jane SM, et al. Redirected perforin-dependent lysis of colon carcinoma by ex vivo genetically engineered CTL. J Immunol 2000;164:3705–12. [DOI] [PubMed] [Google Scholar]
- 29.Mamonkin M, Rouce RH, Tashiro H, Brenner MK. A T-cell–directed chimeric antigen receptor for the selective treatment of T-cell malignancies. Blood 2015; 126:983–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haynes NM, Trapani JA, Teng MWL, Jackson JT, Cerruti L, Jane SM, et al. Rejection of syngeneic colon carcinoma by CTLs expressing single-chain antibody receptors codelivering CD28 costimulation. J Immunol 2002;169: 5780–6. [DOI] [PubMed] [Google Scholar]
- 31.Ishii K, Pouzolles M, Chien CD, Erwin-Cohen RA, Kohler ME, Qin H, et al. Perforin-deficient CAR T cells recapitulate late-onset inflammatory toxicities observed in patients. J Clin Invest 2020;130:5425–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li D, Li N, Zhang Y-F, Fu H, Feng M, Schneider D, et al. Persistent polyfunctional chimeric antigen receptor T Cells that target glypican 3 eliminate orthotopic hepatocellular carcinomas in mice. Gastroenterology 2020;158:2250–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fu Q, Fu T-M, Cruz AC, Sengupta P, Thomas SK, Wang S, et al. Structural basis and functional role of intramembrane trimerization of the Fas/CD95 death receptor. Mol Cell 2016;61:602–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell 2001;104:487–501. [DOI] [PubMed] [Google Scholar]
- 35.Ashkenazi A. Death receptors: signaling and modulation. Science 1998;281: 1305–8. [DOI] [PubMed] [Google Scholar]
- 36.Peter ME, Krammer PH. Mechanisms of CD95 (APO-1/Fas)-mediated apoptosis. Curr Opin Immunol 1998;10:545–51. [DOI] [PubMed] [Google Scholar]
- 37.Suliman A, Lam A, Datta R, Srivastava RK. Intracellular mechanisms of TRAIL: apoptosis through mitochondrial-dependent and -independent pathways. Oncogene 2001;20:2122–33. [DOI] [PubMed] [Google Scholar]
- 38.Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell 1995;81:495–504. [DOI] [PubMed] [Google Scholar]
- 39.Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, et al. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J 1995;14:5579–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wajant H. The fas signaling pathway: more than a paradigm. Science 2002;296: 1635–6. [DOI] [PubMed] [Google Scholar]
- 41.Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity 2018;48:434–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vredevoogd DW, Kuilman T, Ligtenberg MA, Boshuizen J, Stecker KE, de Bruijn B, et al. Augmenting immunotherapy impact by lowering tumor TNF cytotoxicity threshold. Cell 2019;178:585–99. [DOI] [PubMed] [Google Scholar]
- 43.Darcy PK, Kershaw MH, Trapani JA, Smyth MJ. Expression in cytotoxic T lymphocytes of a single-chain anti-carcinoembryonic antigen antibody. Redirected Fas ligand-mediated lysis of colon carcinoma. Eur J Immunol 1998;28:1663–72. [DOI] [PubMed] [Google Scholar]
- 44.DeSelm C, Palomba ML, Yahalom J, Hamieh M, Eyquem J, Rajasekhar VK, et al. Low-dose radiation conditioning enables CAR T Cells to mitigate antigen escape. Mol Ther J Am Soc Gene Ther 2018;26:2542–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hong LK, Chen Y, Smith CC, Montgomery SA, Vincent BG, Dotti G, et al. CD30-Redirected chimeric antigen receptor T Cells target CD30+ and CD30− embryonal carcinoma via antigen-dependent and Fas/FasL interactions. Cancer Immunol Res 2018;6:1274–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hassin D, Garber OG, Meiraz A, Schiffenbauer YS, Berke G. Cytotoxic T lymphocyte perforin and Fas ligand working in concert even when Fas ligand lytic action is still not detectable: CTL perforin and FasL complementation. Immunology 2011;133:190–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Singh N, Lee YG, Shestova O, Ravikumar P, Hayer KE, Hong SJ, et al. Impaired death receptor signaling in leukemia causes antigen-independent resistance by inducing CAR T-cell dysfunction. Cancer Discov 2020;10:552–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dufva O, Koski J, Maliniemi P, Ianevski A, Klievink J, Leitner J, et al. Integrated drug profiling and CRISPR screening identify essential pathways for CAR T-cell cytotoxicity. Blood 2020;135:597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Elrod HA, Sun S-Y. Modulation of death receptors by cancer therapeutic agents. Cancer Biol Ther 2008;7:163–73. [DOI] [PubMed] [Google Scholar]
- 50.Müller M, Wilder S, Bannasch D, Israeli D, Lehlbach K, Li-Weber M, et al. p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J Exp Med 1998;188:2033–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fandy TE, Shankar S, Ross DD, Sausville E, Srivastava RK. Interactive effects of HDAC inhibitors and TRAIL on apoptosis are associated with changes in mitochondrial functions and expressions of cell cycle regulatory genes in multiple myeloma. Neoplasia N Y N 2005;7:646–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.He Q, Huang Y, Sheikh MS. Proteasome inhibitor MG132 upregulates death receptor 5 and cooperates with Apo2L/TRAIL to induce apoptosis in Bax-proficient and -deficient cells. Oncogene 2004;23:2554–8. [DOI] [PubMed] [Google Scholar]
- 53.Huang Y, He Q, Hillman MJ, Rong R, Sheikh MS. Sulindac sulfide-induced apoptosis involves death receptor 5 and the caspase 8-dependent pathway in human colon and prostate cancer cells. Cancer Res 2001;61:6918–24. [PubMed] [Google Scholar]
- 54.Torres-Collado AX, Jazirehi AR. Overcoming resistance of human non-hodgkin’s lymphoma to CD19-CAR CTL therapy by celecoxib and histone deacetylase inhibitors. Cancers 2018;10:200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Michie J, Beavis PA, Freeman AJ, Vervoort SJ, Ramsbottom KM, Narasimhan V, et al. Antagonism of IAPs enhances CAR T-cell efficacy. Cancer Immunol Res 2019;7:183–92. [DOI] [PubMed] [Google Scholar]
- 56.Song JH, Kandasamy K, Kraft AS. ABT-737 induces expression of the death receptor 5 and sensitizes human cancer cells to TRAIL-induced apoptosis. J Biol Chem 2008;283:25003–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krammer PH. CD95’s deadly mission in the immune system. Nature 2000;407: 789–95. [DOI] [PubMed] [Google Scholar]
- 58.Young CW. Interferon induction in cancer: with some observations on the clinical effects of poly 1:C. Med Clin North Am 1971;55:721–8. [DOI] [PubMed] [Google Scholar]
- 59.Kontsek P, Karayianni-Vasconcelos G, Kontseková E. The human interferon system: characterization and classification after discovery of novel members. Acta Virol 2003;47:201–15. [PubMed] [Google Scholar]
- 60.Vilcek J. Novel interferons. Nat Immunol 2003;4:8–9. [DOI] [PubMed] [Google Scholar]
- 61.Kotredes KP, Gamero AM. Interferons as inducers of apoptosis in malignant cells. J Interferon Cytokine Res 2013;33:162–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stark GR, Darnell JE. The JAK-STAT pathway at twenty. Immunity 2012;36:503–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rosebeck S, Leaman DW. Mitochondrial localization and pro-apoptotic effects of the interferon-inducible protein ISG12a. Apoptosis Int J Program Cell Death 2008;13:562–72. [DOI] [PubMed] [Google Scholar]
- 64.Trubiani O, Bosco D, Di Primio R. Interferon-gamma (IFN-gamma) induces programmed cell death in differentiated human leukemic B cell lines. Exp Cell Res 1994;215:23–7. [DOI] [PubMed] [Google Scholar]
- 65.Khodarev NN, Beckett M, Labay E, Darga T, Roizman B, Weichselbaum RR. STAT1 is overexpressed in tumors selected for radioresistance and confers protection from radiation in transduced sensitive cells. Proc Natl Acad Sci U S A 2004;101:1714–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Luszczek W, Cheriyath V, Mekhail TM, Borden EC.Combinations of DNA methyltransferase and histone deacetylase inhibitors induce DNA damage in small cell lung cancer cells: correlation of resistance with IFN-stimulated gene expression. Mol Cancer Ther 2010;9:2309–21. [DOI] [PubMed] [Google Scholar]
- 67.Fu T, He Q, Sharma P. The ICOS/ICOSL pathway is required for optimal antitumor responses mediated by anti-CTLA-4 therapy. Cancer Res 2011;71: 5445–54. [DOI] [PubMed] [Google Scholar]
- 68.Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 2016; 167:397–404.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 2017;127:2930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Seki N, Brooks AD, Carter CRD, Back TC, Parsoneault EM, Smyth MJ, et al. Tumor-specific CTL kill murine renal cancer cells using both perforin and fas ligand-mediated lysis in vitro, but cause tumor regression in vivo in the absence of perforin. J Immunol 2002;168:3484–92. [DOI] [PubMed] [Google Scholar]
- 71.Arenas EJ, Martínez-Sabadell A, Rius Ruiz I, Román Alonso M, Escorihuela M, Luque A, et al. Acquired cancer cell resistance to T cell bispecific antibodies and CAR T targeting HER2 through JAK2 down-modulation. Nat Commun 2021;12:1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology 2015;4:e994446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Avanzi MP, Yeku O, Li X, Wijewarnasuriya DP, van Leeuwen DG, Cheung K, et al. Engineered tumor-targeted T cells mediate enhanced anti-tumor efficacy both directly and through activation of the endogenous immune system. Cell Rep 2018;23:2130–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood 2012;119: 4133–41. [DOI] [PMC free article] [PubMed] [Google Scholar]