

Executive summary
Despite substantial progress in reducing the global impact of many non-communicable diseases, including heart disease and cancer, morbidity and mortality due to chronic respiratory disease continues to increase. This increase is driven primarily by the growing burden of chronic obstructive pulmonary disease (COPD), and has occurred despite the identification of cigarette smoking as the major risk factor for the disease more than 50 years ago. Many factors have contributed to what must now be considered a public health emergency: failure to limit the sale and consumption of tobacco products, unchecked exposure to environmental pollutants across the life course, and the ageing of the global population (partly as a result of improved outcomes for other conditions). Additionally, despite the heterogeneity of COPD, diagnostic approaches have not changed in decades and rely almost exclusively on post-bronchodilator spirometry, which is insensitive for early pathological changes, underused, often misinterpreted, and not predictive of symptoms. Furthermore, guidelines recommend only simplistic disease classification strategies, resulting in the same therapeutic approach for patients with widely differing conditions that are almost certainly driven by variable pathophysiological mechanisms. And, compared with other diseases with similar or less morbidity and mortality, the investment of financial and intellectual resources from both the public and private sector to advance understanding of COPD, reduce exposure to known risks, and develop new therapeutics has been woefully inadequate.
In this Lancet Commission on COPD, our objective is to set the course to eliminate the disease by challenging accepted dogma and generating debate. We recognise that many of our recommendations could not be used as the foundation for evidenced-based guidelines. However, that is not our goal. We believe that a wholesale rethinking of COPD is needed. In general, we believe that the traditional incremental approach to advancing understanding of disease and developing new methods for diagnosis and treatment has failed. In particular, we advocate for: broader understanding of risk factors (including the devastating effects of global poverty) and the preventive measures necessary to avoid future cases of COPD, disruptive approaches to diagnosis that are not solely based on spirometric airflow limitation but also involve identification of early pathological changes that are more amenable to reversal, classification of the disease into types that share pathophysiological similarities and could lead to novel preventive and therapeutic approaches, and a new approach to the diagnosis and assessment of exacerbations of COPD that focuses on disease mechanisms. We also advocate for a coordinated plan to combat the disease through increased financial investment, broad public policy initiatives, regulatory reform, and the alignment of health-care systems, which will enable a path towards prevention and cure rather than crisis management.
The most efficient way to reduce the burden of COPD is to ban cigarette smoking in all its forms. We strongly advocate for this ban, and support the associated financial, technological, and retraining investments that would be necessary to prevent economic disaster among people dependent on the tobacco industry for their livelihood. However, risk factors unrelated to tobacco are increasingly responsible for the burden of COPD, and are likely to surpass the risk attributable to smoking within the next two decades. These risks include many underappreciated factors that span the life course, from in-utero exposures to maternal factors leading to preterm birth or low birthweight, early-life infections, and indoor and outdoor pollution. Poverty increases the prevalence of these risk factors but decreases societal efforts to control them. Messaging targeted at medical professionals, government officials, private corporations, and the general public should emphasise these risk factors and drive preventive strategies. Otherwise the tobacco-related COPD crisis will be replaced by a COPD crisis driven by one or more of the other factors.
The definition of COPD requires the presence of spirometric airflow limitation, which all but eliminates the possibility that the disease could be cured or eliminated globally because the pathological changes required for airflow limitation are almost certainly permanent. We advocate for a broader definition of COPD that includes people with airflow limitation detected by more sensitive pulmonary function tests or pathological changes detected by imaging techniques. This broader definition will enable detection of patients with earlier pathological changes, which would enhance the possibility of understanding the mechanistic pathways driving disease inception and could thus lead to the development of effective treatments to interrupt and reverse the course of COPD. In low-income settings, we also advocate for the use of risks and symptoms to identify a population with probable COPD, in whom preventive measures and low risk non-pharmacological and pharmacological treatments could be beneficial. Throughout the Commission, we have provided similar options for alternative approaches when technology, the health-care system, or the personnel available limit the implementation of our recommendations.
Closely tied to the inadequacy of current diagnostic criteria is the failure to classify COPD in a way that could help to identify new approaches to prevention and treatment. We recommend that COPD be classified into types based on five main risk factors: genetics, early-life events, pulmonary infections, tobacco smoke exposure, and pollution. This approach mirrors that developed for pulmonary hypertension in the 1970s, which has revolutionised understanding of the disease and led to numerous novel therapeutics targeting individual classes of the disorder. We recognise that the risks factors leading to airflow limitation are numerous, and that many patients are affected by more than one. However, this is also true for patients with pulmonary hypertension, who can have more than one mechanism driving their disease (eg, heart failure and coexistent lung disease). Our proposal is not perfect and, like the WHO classification of pulmonary hypertension, will require iterative refinement, but it is far more likely to yield new therapeutics than a system that relies solely on the presence of spirometric airflow limitation and a patient’s level of dyspnoea and exacerbation frequency (the extent of current attempts to classify COPD).
An acute worsening of COPD is termed an exacerbation, and such episodes account for a substantial proportion of the attributable cost of the disease and are associated with accelerated lung function loss, prolonged impairments in quality of life, and similar prognosis to many stage III or IV solid organ malignancies. Yet exacerbations tend to be imprecisely defined, and their severity is judged according to the site of treatment rather than the extent of underlying physiological derangement. Few exacerbations are thoroughly investigated to establish the underlying trigger (despite the availability of many diagnostic tools). Reflexive treatment with corticosteroids or antibiotics, or both, is prescribed in almost all cases—a therapeutic approach that has not changed in more than 30 years and in some cases is almost certainly harmful. We call for an objective definition of COPD exacerbations, a standardised assessment, and a precision approach to treatment (such a treatment approach will take time, because the development of new therapeutics will be contingent on the implementation of the new definition).
In the almost 4 years since this Commission was formed, the devastating COVID-19 pandemic has led to unprecedented efforts to align health authorities, regulatory agencies, private corporations, and the public in the fight against the disease. Although many parts of the world do not yet have access to the vaccines and treatments that were rapidly developed to combat COVID-19, the availability of these therapeutics in many high-income countries has shown that barriers preventing a coordinated response to a global crisis can be overcome and that a rapid response is possible. We argue that such a response is needed for the many chronic diseases that cause far greater yearly morbidity and mortality than COVID-19, including COPD. This response will require substantially increased investment in public health policies to prevent exposure to risk factors, implementation of our proposals to capture COPD earlier and enable phenotyping of patients, and research and development of precision therapeutics.
The ultimate objective of this Lancet Commission is to set the course to eliminate COPD, which will require novel and more effective approaches to preventing new cases and reversing the disease in people who are currently affected. We propose six core strategies to achieve this ambitious goal: (1) a broadened understanding of the multiple and interacting risk factors for COPD; (2) classification of the disease into types based on underlying causative mechanisms, including genetics, early-life events, respiratory infections, and tobacco and other environmental exposures; (3) implementation of a more inclusive diagnosis of COPD, allowing for the detection of mild disease before irreversible pathological changes have occurred; (4) development of personalised prevention and treatment strategies for both stable disease and exacerbations that are informed by a holistic assessment of COPD pathophysiology, symptoms, and patients’ needs, capabilities, and preferences; (5) investment in the development of curative and regenerative therapies that go beyond the largely symptomatic treatment options available; and (6) deployment of public health preventive strategies for banning smoking and maintaining clean air. We are fully cognisant that the scientific evidence to support some of our proposals is lacking. However, it is the intention of the Commission to generate discourse and debate, catalyse momentum, and provide a much-needed new vision to set the course towards the elimination of COPD.
Introduction
The lungs are constantly exposed to environmental factors throughout life and are vulnerable to a range of insults including tobacco smoke, infections, and pollutants.1 Even in utero, environmental exposures and maternal factors can increase the fetus’s risk for respiratory disease later in life.2 Tremendous progress has been made towards understanding the pathogenesis of chronic obstructive pulmonary disease (COPD) and risk factors for developing the disease (including non-tobacco-related factors). However, the prevalence of chronic respiratory disease—of which COPD is the most prevalent type—increased by almost 40% between 1990 and 2017, and by 2017 chronic respiratory disease had become the third leading cause of death globally.3
COPD accounted for about 55% of all chronic respiratory diseases in both men and women in 2017, a relative increase of 5·9% since 1990.3 Globally, COPD affects more than 300 million people and, in 2019, it resulted in 3·3 million deaths, ranking as the eighth leading cause of years of life lost. COPD is associated with an increase in disability-adjusted life years and years of life lost across the life course (figure 1). This substantial and increasing global burden makes COPD a public health problem that requires urgent attention.
Figure 1: DALYs (A) and years of life lost (B) to COPD.
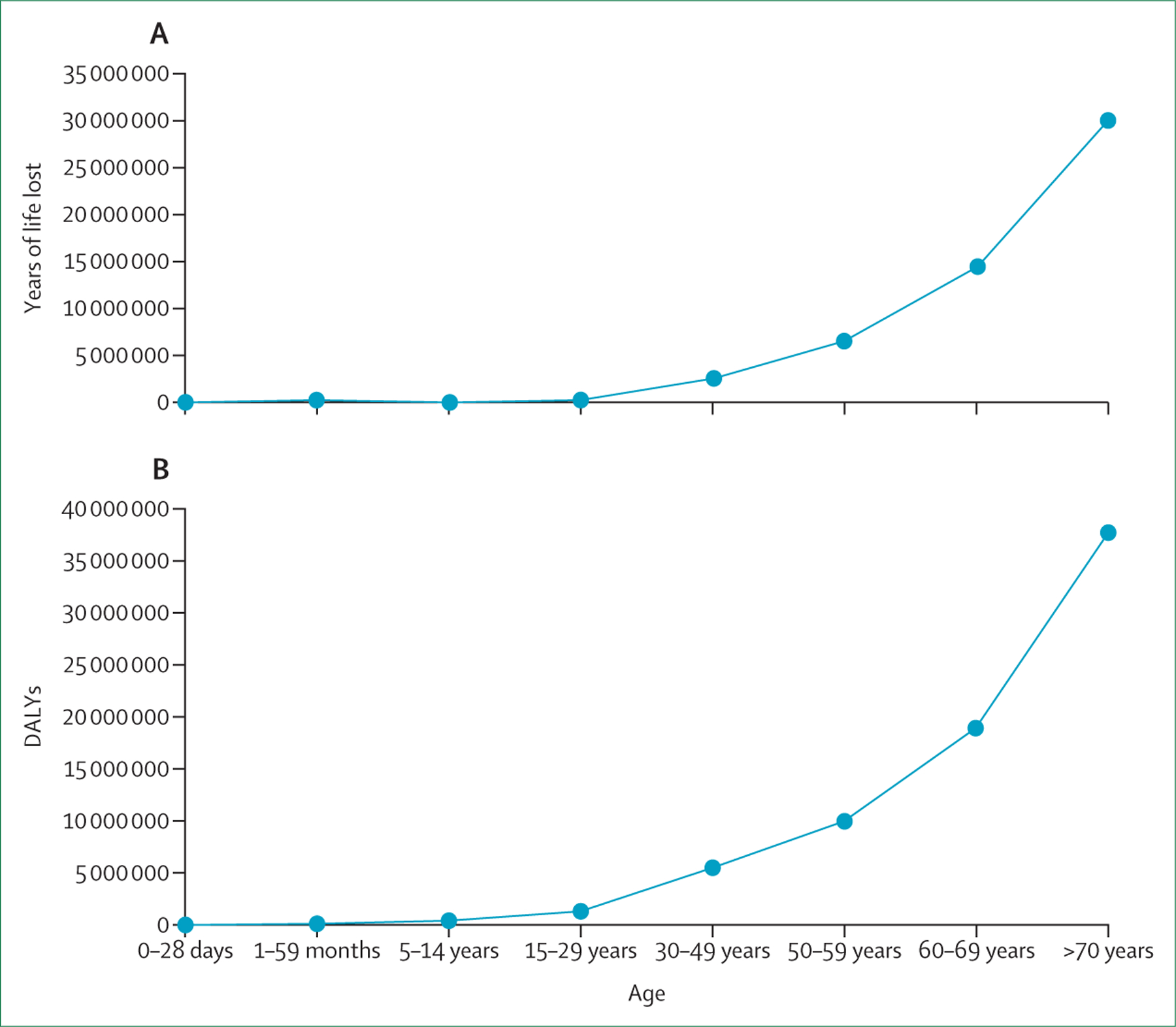
In 2019, COPD was the seventh leading cause of DALYs globally and the eighth leading cause of years of life lost.4 DALYs=disability-adjusted life-years. COPD=chronic obstructive pulmonary disease.
COPD is a heterogeneous respiratory condition characterised by a combination of injury to, and remodelling of, the airways, lung parenchyma, and lung vasculature. These lung insults result in progressive airflow limitation, leading to increased dyspnoea, disability, and premature death.5 Common symptoms of COPD include dyspnoea, chronic cough (often associated with phlegm), exercise intolerance, and episodic flare-ups of respiratory symptoms, which have been termed exacerbations.6 The disease also causes substantial social and economic consequences for both individual patients and health systems. Limitations in normal daily activities due to COPD-related breathlessness—and the subsequent psychological implications for social life, family life, and overall quality of life, leading to fear, anxiety, and social isolation—are often underappreciated. COPD affects people from all countries, socioeconomic classes, and age groups, but the magnitude of its effects varies: elderly people and people in low-income and middle-income countries (LMICs), where 80% of COPD deaths occur, are disproportionately affected.7
As the prevalence of COPD has steadily increased, understanding of the disease has also advanced. Tobacco products are responsible for most of the disease burden worldwide, and their continued production and distribution is unconscionable. However, COPD can no longer be thought of solely as a self-inflicted disease caused by smoking. Rather, it is a complex condition caused by multiple risk factors that can interact and coexist throughout life—as evidenced by the fact that not all smokers develop COPD and at least 20–30% of people with COPD have never smoked.8 Indeed, it is very likely that a substantial proportion of COPD is preventable, and there is an opportunity to greatly reduce the burden of the disease through prompt, coordinated, and pragmatic efforts from the medical community, governmental bodies, and society as a whole.
COPD: a global health crisis
COPD has become a global health crisis partly as a result of the failure of governmental, medical, and scientific agencies, as well as private enterprises, to eliminate active cigarette smoking and to recognise and eliminate other environmental exposures. This Commission9 aims to change the course of this global emergency by demanding urgent action targeting the factors most responsible for the uncontrolled burden of COPD. We will focus on the greatest difficulties to overcome, which lie in these key areas: (1) the effects of multiple overlapping risk factors, (2) scarce and insensitive diagnostic tools, (3) complex, unpredictable, and inadequate treatments, (4) disproportionate risk and financial burden in lower income populations, (5) misalignment and poor coordination of health-care systems, and (6) inadequate research funding (panel 1).
Multiple, overlapping, and interacting risk factors
Smoking is the main and most characterised cause of COPD, but other important and overlooked risk factors include genetics, early-life events (including those occurring in utero), infections, and detrimental environmental exposures.8,10 Premature births are associated with the development of COPD11 and are increasing as a result of inadequate maternal and prenatal health care in low-income settings, advances in neonatal care (leading to improving survivorship among extremely preterm infants), changes in maternal age, and an increase in multiple births due to assisted reproductive technologies. In many parts of the world, prevalence of tuberculosis and HIV remain very high, and both are associated with airflow limitation. In addition, pneumonia accounts for more than 2 million deaths in children annually; survivors of pneumonic infections often do not attain maximum lung function as young adults, and thus are predisposed to COPD.12 The increasing burden of COPD is also closely tied to environmental exposures in both industrial and non-industrial settings. Unfortunately, legislation passed to limit environmental exposures, as well as efforts to ban or restrict the use of cigarettes and other inhaled substances, have not been sufficient or adequately enforced. Despite the establishment in 1983 of the Energy Sector Management Assistance Program, a global knowledge and technical assistance programme that aimed to mitigate household cooking exposures and improve clean energy access, more than 40% of the world’s population is still exposed to high levels of indoor air pollution.13 This pollution results from the use of biomass fuels in inefficient and poorly ventilated stoves for cooking and heating in low-income countries, and from the use of gas for cooking and heating without extractor fans in middle-income and high-income countries. Occupational exposures to dusts, gases, and fumes are also a major risk factor for COPD and are common in many industries, including manufacturing, mining, steelworks, farming, and automotive repair.14 In some occupations, respiratory protection standards are inadequate and poorly enforced and there is often a dangerous lag between the identification of harmful exposures and implementation of protective measures. Absurdly, some legislation allows employers to assess the risks, set the standards for safe levels of airborne pollutants, and enact protective measures without any external regulatory oversight.15
Insensitive diagnostic tools, delayed recognition, and late presentation
COPD is currently defined as incompletely reversible limitation of expiratory airflow (as detected by spirometry) due to a combination of small airway remodelling and emphysematous destruction of the lung parenchyma.5,16 It is underdiagnosed and typically diagnosed late, partly because it results from accumulated damage to the respiratory system over the life course. Spirometry forms the foundation of diagnosis of COPD. The presence of spirometric expiratory airflow limitation as defined by a ratio of the forced expiratory volume in 1 s (FEV1) to the forced vital capacity (FVC) of less than 0·7 (or less than the adjusted lower limit of normal) is the gold standard. This approach to disease detection has facilitated communication in the medical community and empowered epidemiological investigation and development of symptomatic treatments, but it provides an overly simplified view of COPD. Spirometry is a poor predictor of symptoms, exercise capacity, and overall quality of life and does not capture the heterogeneity of the disease.17 Moreover, the emphasis on spirometric obstruction precludes detection of very early disease, which might be more amenable to interception—or even reversal and cure—and has led to therapies that primarily target moderate-to-severe disease. It is misleading to equate the absence of spirometric expiratory airflow limitation to the absence of COPD, because airway and parenchymal changes are clearly present in many individuals with normal spirometry results.18 In addition, even people with compromised and worsening lung health but surplus functional reserve might have unremarkable spirometric results compared with the population-based norm, with thresholds for disease crossed only after irreversible pathological changes have occurred in the lung.19 COPD can also sometimes arise early in life due to the failure of normal lung development (usually, lung function peaks around age 25 years in men and age 18–20 years in women).20 These patients are rarely if ever assessed for respiratory disease and seldom undergo spirometry, and better biomarkers are needed to identify them.
Definition of COPD based on spirometric criteria alone is flawed and dangerous because early airway changes and emphysematous destruction of the lung parenchyma do not reliably translate to airflow limitation as measured by spirometry. This erroneous notion increasingly hinders progress and is partly responsible for the lack of biomarker-based assessments to signal early compromise in lung health.19 Additionally, there is often a delay in diagnosis of COPD until the lung damage is irreversible because of the under-recognition of respiratory symptoms and the fact that prolonged periods of disease activity and worsening lung health can be associated with no or only minimal symptoms (figure 2).
Figure 2: The importance of early diagnosis to eliminate COPD.
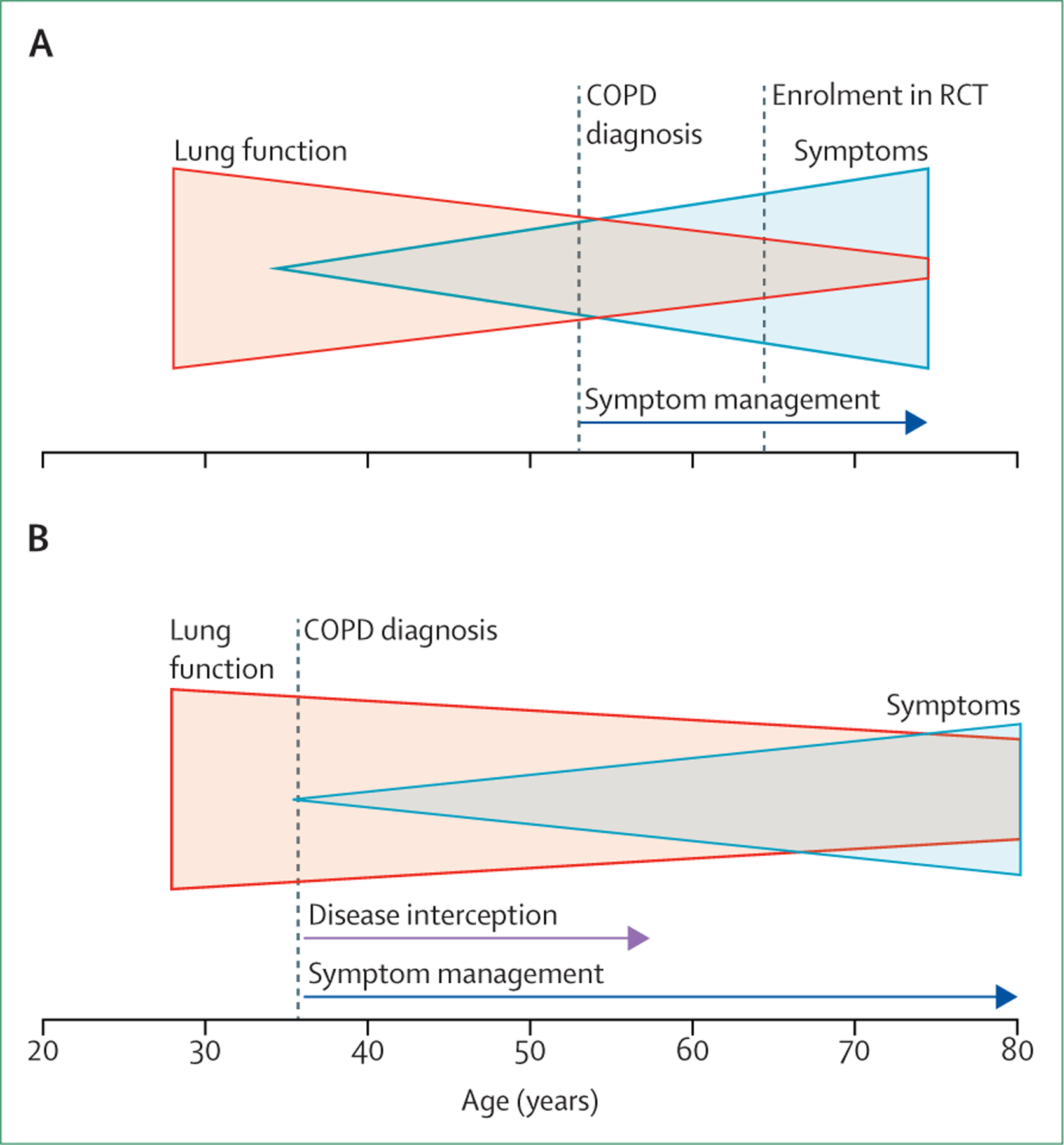
(A) Currently, COPD is diagnosed at a stage when pathological changes are irreversible. This late diagnosis is due to a combination of factors, including the lack of predictive biomarkers, under-recognised clinical symptoms, a long period of disease activity associated with no or minimal symptoms, and reliance on spirometry, an insensitive diagnostic tool. (B) Implementation of a more inclusive diagnosis of COPD allows for the detection of early disease before irreversible pathological changes have occurred and could lead to disease interception. COPD=chronic obstructive pulmonary disease. RCT=randomised controlled trial.
Furthermore, despite the years-long emphasis on the crucial role of spirometry in the diagnosis of COPD, this simple technology is still not universally available, much less routinely used worldwide (figure 3, table 1). Global attempts to define COPD preceding the development of spirometric airflow limitation, including the concept of pre-COPD,21 have raised awareness of the complexity of the problem but have not yet translated to improved diagnosis or outcomes. A new approach to diagnosis will require both consideration of the predictive utility of respiratory symptoms alone22 and expansion of diagnostic criteria based on non-spirometric lung function assessments and imaging. In a survey that we did through the European Respiratory Society, we found that chest CT was as available as or more available than spirometry in many countries (figure 3, table 1).
Figure 3: Proportion of global population with access to spirometry (A), chest CT (B), and chest radiography (C) for diagnosis of chronic obstructive pulmonary disease.
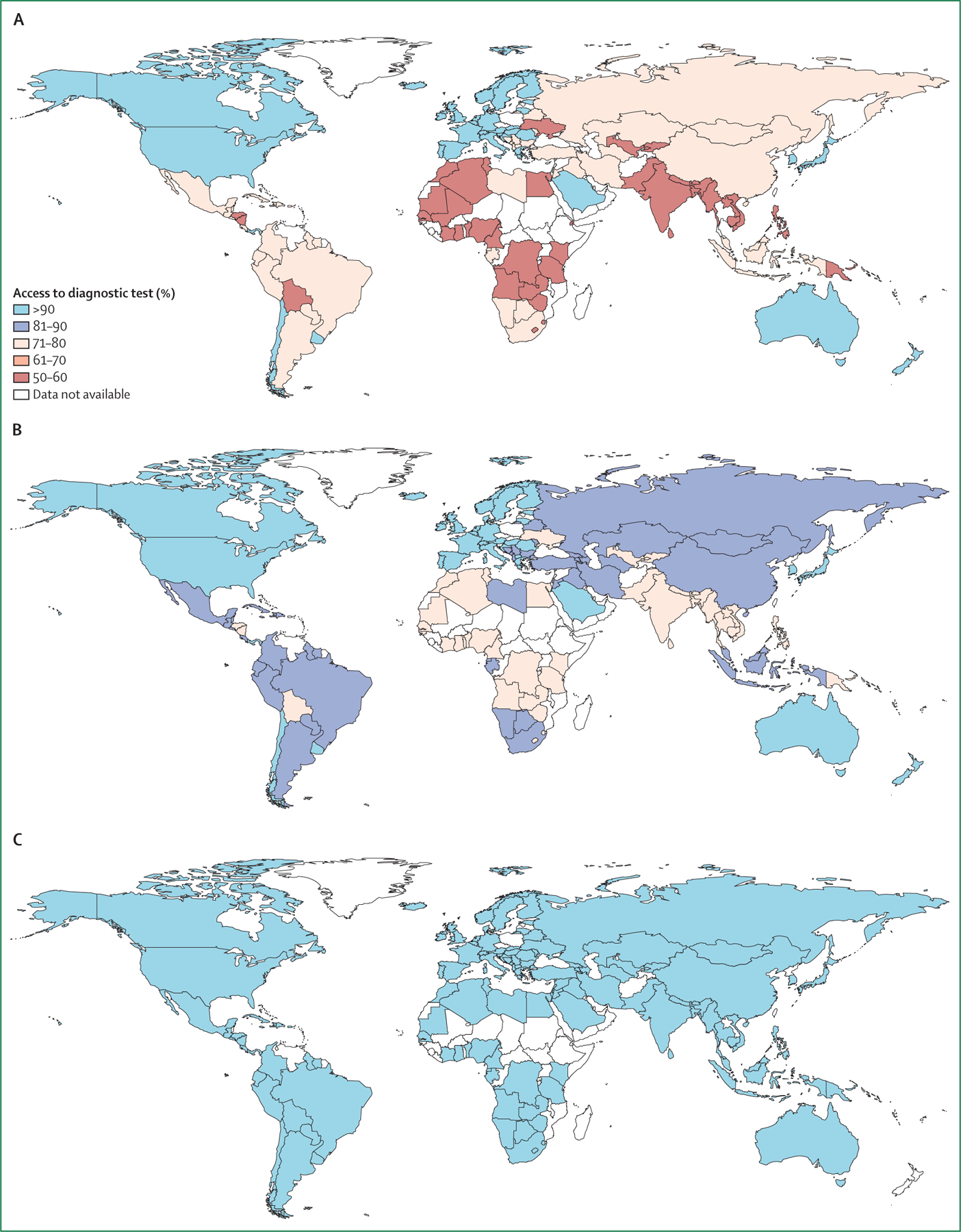
Data were obtained from the survey done by the Commission (appendix pp 1–5).
Table 1:
Availability of tests considered relevant in the diagnosis of chronic obstructive pulmonary disease, by country income group
| Primary and specialist care | Primary care only | Specialist care only | Limited* availability in primary or specialist care, or both | Not available | |
|---|---|---|---|---|---|
| Lower-middle-income countries (n=11) | |||||
| Spirometry | 2 (18%) | 0 | 4 (36%) | 5 (45%) | 0 |
| Reversibility testing | 1 (9%) | 0 | 6 (55%) | 4 (36%) | 0 |
| Whole body plethysmography | 0 | 0 | 3 (27%) | 5 (45%) | 3 (27%) |
| Diffusion capacity measurement | 0 | 0 | 3 (27%) | 5 (45%) | 3 (27%) |
| Arterial blood gas analysis | 0 | 0 | 6 (55%) | 5 (45%) | 0 |
| Chest radiography | 6 (55%) | 3 (27%) | 0 | 2 (18%) | 0 |
| Chest CT | 1 (9%) | 0 | 7 (64%) | 3 (27%) | 0 |
| Upper-middle-income countries (n=15) | |||||
| Spirometry | 3 (20%) | 2 (13%) | 6 (40%) | 4 (27%) | 0 |
| Reversibility testing | 2 (13%) | 1 (7%) | 8 (53%) | 4 (27%) | 0 |
| Whole body plethysmography | 0 | 0 | 5 (33%) | 9 (60%) | 1 (7%) |
| Diffusion capacity measurement | 0 | 0 | 6 (40%) | 8 (53%) | 1 (7%) |
| Arterial blood gas analysis | 1 (7%) | 2 (13%) | 8 (53%) | 4 (27%) | 0 |
| Chest radiography | 8 (53%) | 4 (27%) | 3 (20%) | 0 | 0 |
| Chest CT | 3 (20%) | 0 | 10 (67%) | 2 (13%) | 0 |
| High-income countries (n=17) | |||||
| Spirometry | 12 (71%) | 1 (6%) | 3 (18%) | 1 (6%) | 0 |
| Reversibility testing | 8 (47%) | 0 | 9 (53%) | 0 | 0 |
| Whole body plethysmography | 0 | 0 | 16 (94%) | 1 (6%) | 0 |
| Diffusion capacity measurement | 1 (6%) | 0 | 14 (82%) | 2 (12%) | 0 |
| Arterial blood gas analysis | 1 (6%) | 0 | 15 (88%) | 1 (6%) | 0 |
| Chest radiography | 12 (71%) | 0 | 4 (24%) | 1 (6%) | 0 |
| Chest CT | 4 (24%) | 0 | 12 (71%) | 1 (6%) | 0 |
Data are n (%). We sent an online survey to more than 100 national and regional respiratory societies worldwide, including the American Thoracic Society, European Respiratory Society, the Pan African Thoracic Society, and the Asociación Latinoamericana de Tórax. We received responses from societies representing 43 of the 120 countries approached. Countries were categorised into the four World Bank income groups based on the 2019 gross national income per person. The lower-middle-income countries that responded were Algeria, India, Kenya, Kyrgyzstan, Moldova, Nepal, Pakistan, Philippines, Sri Lanka, Tunisia, and Viet Nam. The upper-middle-income countries were Albania, Brazil, Bulgaria, Costa Rica, Iran, Jordan, Kazakhstan, Lebanon, Malaysia, Mexico, North Macedonia, Serbia, South Africa, Thailand, and Türkiye. The high-income countries were Australia, Austria, Belgium, Canada, Finland, France, Germany, Lithuania, Norway, Poland, Portugal, Slovenia, Spain, Switzerland, Taiwan, Uruguay, and the USA. No surveys were returned from physicians in low-income countries (see appendix for additional methods).
The availability of a diagnostic test was defined as “limited” if it was available to a maximum of 15% of patients.
These challenges highlight the need to seek alternative methods with increased sensitivity that can detect the pathological processes associated with COPD at an early stage. Diagnostic methods that show lung destruction or airway remodelling either directly (eg, CT) or indirectly (eg, by showing reductions in gas exchange [diffusion capacity] or impaired respiratory mechanics [ forced oscillation]) should be considered as complementary to spirometry.
Insufficient treatment options
In addition to inadequate diagnostics, treatment of COPD is suboptimal for several reasons. The most prominent therapeutic shortcoming is the absence of curative therapies. Furthermore, available pharmacological treatments largely target symptoms (and often have only limited effects, including on cough and sputum production), and none reverse the underlying airway remodelling, emphysema, and vascular abnormalities characteristic of the disease. We should not settle for this state of affairs, and rather should strive for treatments that halt, reverse, and cure the disease. However, such therapies will require dedicated and substantial research investment commensurate with the scope of the problem. The type of approach necessary is exemplified by the development of CFTR modulators that successfully target the underlying molecular defect driving cystic fibrosis and have revolutionised treatment of that disease (although we argue that the price of CFTR modulators should be lowered to increase net health benefits to society). Although the genetics of COPD are far more complicated than those of cystic fibrosis, which means that ground-breaking treatments will almost certainly be more difficult to identify, the goal should still be to reverse pathology and normalise physiology.
Few markers reliably predict responsiveness to available COPD treatments. Circulating blood biomarkers such as C-reactive protein and procalcitonin are associated with response to antibiotic therapy at the time of exacerbations.23 Blood eosinophil counts seem related to response to inhaled corticosteroids in stable disease and could predict response to systemic corticosteroids during exacerbations.24,25 However, although other blood-based and radiological biomarkers have been suggested, few have proven clinically useful and there is an urgent need for novel, robust predictors of therapeutic response and adverse effects. In other chronic and progressive diseases, the identification of meaningful surrogate and intermediate endpoints—eg, glucose and glycated haemoglobin in diabetes, cholesterol in ischaemic heart disease—has been instrumental in controlling morbidity and mortality. Such biomarkers can be used to identify early disease and measure responsiveness to treatment when there is still time to delay, prevent, or reverse organ damage.
Furthermore, lung function impairment, respiratory symptoms, and exacerbations (and the associated morbidity, mortality and health-care costs) are only partly mitigated by available treatments. Typically, the degree of improvement in these outcomes in randomised trials is 15–25% irrespective of the therapy tested.26 The heterogeneity of COPD, combined with the fact that many trials enrol patients based only on the presence of spirometric obstruction, means that the potential benefits of treatments suitable for only a subgroup of patients can be overlooked. This situation could be avoided if greater phenotypic or endotypic characterisation were used to define more homogeneous populations whose disease was driven by the specific pathway targeted by the drug under investigation. The development of monoclonal antibodies aimed at specific underlying biological mechanisms has led to substantial reductions in symptoms and exacerbations (and associated hospital admissions) in some subsets of patients with severe refractory asthma.27 This precision approach has been widely discussed but infrequently tested in patients with COPD.
COPD is associated with numerous systemic manifestations and comorbidities, and thus symptoms can reflect not only the extent of lung function impairment but also the effects of deconditioning, obesity, anaemia, anxiety, depression, and congestive heart failure, among other disorders. As a consequence, the classification of patients with COPD solely according to respiratory symptoms could lead to overestimation of the severity of pulmonary disease—and thus of the potential benefit of bronchodilators and inhaled corticosteroids—and underestimation of the effect of managing related conditions via different treatment approaches. Because of shared risks factors, including ageing and tobacco smoking, and perhaps also shared pathobiology, the proportion of patients with COPD who also have multimorbidity is higher than that of patients with many other chronic diseases; thus a multi-dimensional and patient-centred approach to prevention, diagnosis, and treatment is required.28
Non-adherence or incorrect administration of inhaled medications is common in COPD because of the number and complexity of delivery devices, a problem that is not shared with most other chronic conditions.29 Devices include dry powder inhalers, pressurized metred-dose inhalers, and soft mist inhalers, each of which requires different inhalation techniques that are often not taught to patients (or are taught incorrectly). Determinants of poor inhaler technique include older age and the number of devices used, both of which become more problematic as the disease progresses.30
Monitoring of treatment responses is often challenging, particularly in patients who do not exhibit early symptomatic clinical responses. It takes time to establish whether exacerbation frequency improves with initial or modified treatment, and such improvements might not be perceived by patients or reliably detected by health-care providers. Increases in lung function are more easily identified, but are not always accompanied by clinically relevant improvements in patient-reported outcomes or reduced exacebation risk. Similar patterns are apparent for the effects of pulmonary rehabilitation in COPD: effects on exercise capacity and symptoms during daily activities differ both between and within patients over time. Treatment responses would be best monitored according to composite outcome measures, but these are complex, time consuming, and difficult to administer.
Disproportionate effects on marginalised populations
Almost all risk factors for COPD are associated with low socioeconomic status, which also predicts poor clinical outcomes. These disparities are most pronounced when LMICs are compared with high-income countries, but are also a major cause for concern within high-income countries, where poverty and neighbourhood-level socio-economic disadvantage are independent predictors of COPD and associated respiratory morbidity.31 Furthermore, individual-level and neighbourhood-level socio-economic factors partly explain racial health disparities in COPD,32 probably through clustering of risk factors such as tobacco smoking, poor indoor and outdoor air quality, substandard housing, and limited access to healthy food and health care.
As discussed previously, premature birth is a risk factor for COPD.33 Global data from 2017 showed that less than two-thirds of pregnant women received the recommended four or more antenatal visits and nearly 20% of women had no health insurance before pregnancy, both of which increase the risk for premature birth.34 Household air pollution is associated with an increased risk of COPD. More than 2·4 billion people, generally among the world’s poorest, are estimated to rely directly upon biomass fuel for their heating and cooking needs.35 Tobacco smoking is also increasing in LMICs (although it is declining in high-income countries), partly because there is little or no regulation of tobacco sales and partly because of the dependence of farmers on the tobacco industry for their livelihood.
In 2019, an estimated 1·1 billion people smoked worldwide.36 Although various legislative and social measures decreased the number of cigarette smokers globally by approximately 60 million between 2000 and 2018, population growth eliminated any overall benefit associated with this decline.36 Tobacco companies have also begun to produce alternatives to cigarettes, including electronic nicotine delivery systems, such as vaping devices and e-cigarettes. Aggressive and unregulated marketing of these devices is directed at young and vulnerable people and is highly successful, as evidenced by the substantially increased prevalence of smoking in children and the fact that the initial age of smoking onset is lower than ever before.37
The worldwide economic burden of COPD is high, not only because of the disease’s prevalence, chronic course, and high treatment costs, but also because of the high costs of informal caregivers, productivity loss due to premature withdrawal from the workforce, and increased absenteeism due to illness. For obvious reasons, poor, disadvantaged, and vulnerable people are particularly affected by these factors.
Limited access to therapeutics results in suboptimal care and is of particular concern in LMICs. An online survey done by the Commission through national respiratory societies in 43 countries showed that the availability of pharmacological and non-pharmacological interventions was highly variable (figure 4; appendix pp 1–5). Access to advanced treatments such as endobronchial valves, lung volume reduction surgery, and lung transplantation was restricted even in many high-income countries, and there was almost no access in LMICs. More concerning was the very low availability of standard inhaled treatments for COPD in LMICs. Our survey suggested that access to pharmacotherapy was also affected by out-of-pocket costs: 50% of patients in low-income and lower-middle-income countries, and 33% of those in upper-middle-income countries, were unable to afford COPD pharmacotherapies.
Figure 4: Availability of pharmacological therapies (A) and non-pharmacological therapies (B) for chronic obstructive pulmonary disease in lower-middle-income, upper-middle-income, and high-income countries.
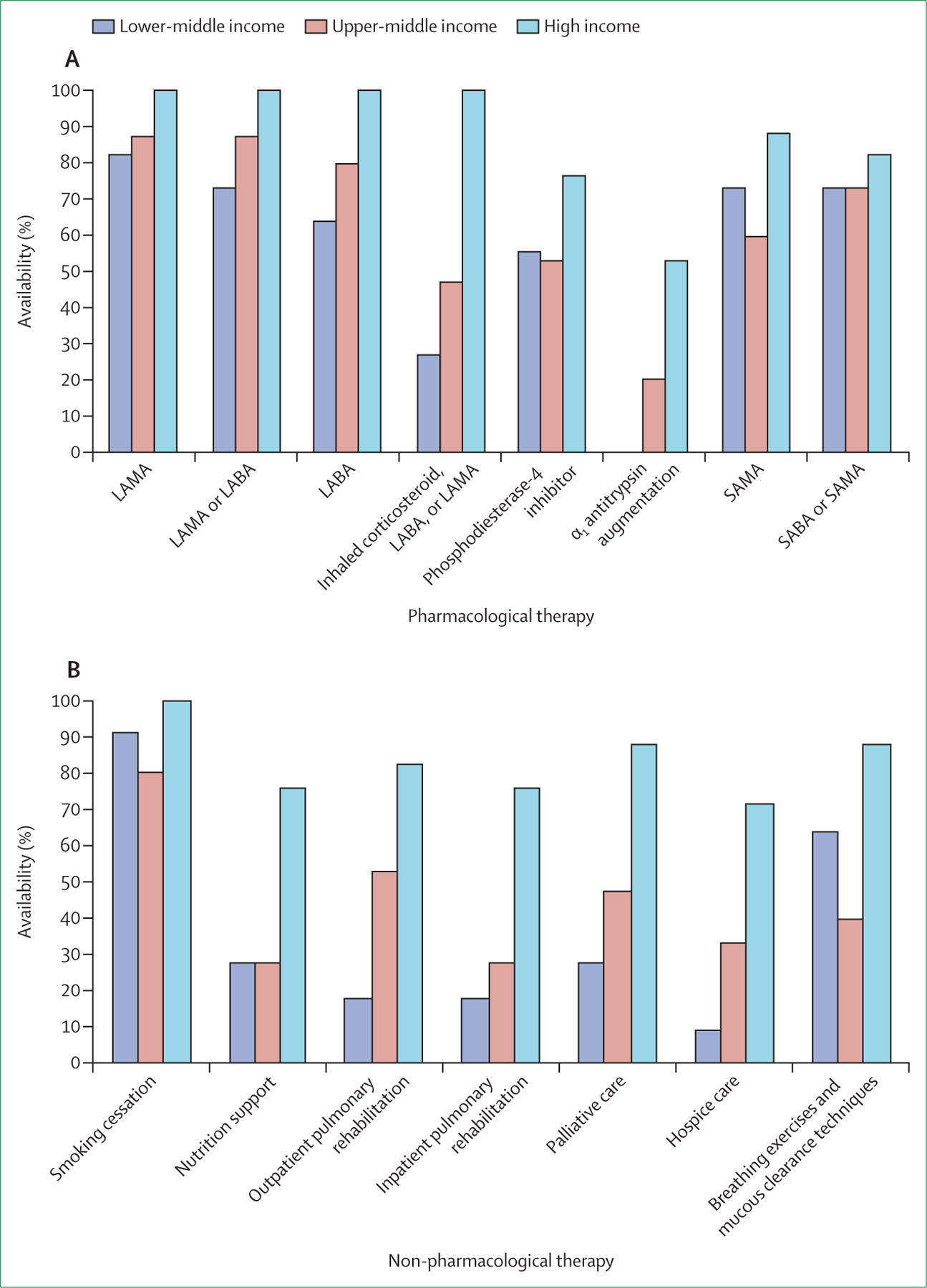
LAMA=long-acting muscarinic antagonist. LABA=long-acting β agonist. SAMA=short-acting muscarinic antagonist. SABA=short-acting β agonist.
Shortcomings of health-care systems
The financial challenges of COPD care are amplified by the complexity and poor coordination of many health-care systems and the misalignment between reimbursement for components of care and their relative efficacy. In many countries, most patients with COPD are managed in primary care because there are few incentives to seek specialist input and a shortage of pulmonologists. Care needs to be provided by clinicians who have sufficient training: pulmonologists or chest physicians or primary care practitioners who have interest, training, and expertise in COPD and updated knowledge of the treatments and procedures necessary for optimal care. In high-income countries, people with severe COPD commonly see specialists and the substantial proportion of patients with multi-morbidity tend to see many health-care providers.28,38 Unfortunately, coordination of care and communication between these providers is often poor and systems are prone to inefficiencies such as duplication of tests and services, late recognition of progression and acute events, and overtreatment and undertreatment; patients are also at risk of the potentially harmful effects of drug interactions due to polypharmacy.39 In addition, decisions about management are based on guidelines that are informed by clinical studies done in populations that do not represent the general COPD population. For many patients, even those in high-income countries with good overall access to health care, the system tends to be fragmented and difficult to navigate.
In most countries, health-care systems react to acute illnesses and are reimbursed well for those efforts, but little support is available for the prevention of chronic diseases. This is perhaps best illustrated by the failure to deliver low-cost, effective smoking cessation treatments or interventions to reduce residential biomass fuel exposure in low-income countries. The costs of care associated with smoking account for about 15% of the aggregate health-care expenditure in high-income countries and are estimated to be US$500 billion worldwide.40 In 2018, the US Medicaid programme covered the cost of smoking cessation treatments in only 15 states—and in only two states were all barriers to patient access removed, including requirements for co-payment, previous authorisation and counselling for the medication, limited treatment duration, and limited numbers of quit attempts per year and per lifetime.41 Most countries in Europe, the Western Pacific, and southeast Asia offer nicotine replacement therapy or some cessation services, or both, but the costs tend to be covered for only one type of therapy (and are sometimes only partly covered).42
Lack of innovation and inadequate research funding
Over the past 25 years, there have been few innovations in either pharmacological or non-pharmacological management of COPD, and the disease lags far beyond other conditions with high morbidity and mortality in terms of the development of precision medicine approaches. The pharmacological innovations that have reached the market are primarily long-acting, dual and triple combinations of well-established inhaled bronchodilators and corticosteroids. Furthermore, the use of digital information technologies to identify early COPD or COPD exacerbations, or to support remote monitoring and self-management, is not widespread. Multiple factors, including a lack of investment and resources in the studies necessary to generate the evidence for these approaches, structural barriers to embedding digital tools, and the older age and lower socioeconomic background of the COPD population, are implicated in the scarcity of these approaches.
Furthermore, there is substantial misalignment between the burden of COPD and the priority afforded to the disease by governmental and other funding agencies. Similar to the low resources dedicated to smoking cessation, the US National Institutes of Health’s funding for COPD-specific research is low compared with that for other chronic diseases, such as cardiovascular disease, cancer, and diabetes (figure 5).43,44 COPD research is also substantially underfunded compared with asthma: asthma received $338 million in funding from the National Institutes of Health in 2020 ($81,500 per US death) whereas COPD received $121 million (less than $800 per US death). The reasons for this discrepancy are complex and difficult to rationalise. Possible explanations include the substantial social stigma that inaccurately characterises COPD as a self-inflicted disease, which is less evident in cardiovascular disease and type 2 diabetes, even though lifestyle factors are important contributors to both illnesses. There has been substantial recent research investment in lung cancer, for which smoking is the most important risk factor, for the development of biologics and immunotherapies, which in many cases offer only an incremental survival advantage. This survival advantage is accepted by regulators as sufficient proof of benefit and thus incentivises investment from the pharmaceutical industry. For a chronic disease such as COPD, incremental or overall survival advantages are far more difficult to show because of the prolonged trajectory of the illness.
Figure 5: Research expenditure per death by the US National Institutes of Health, by disease, 2020.
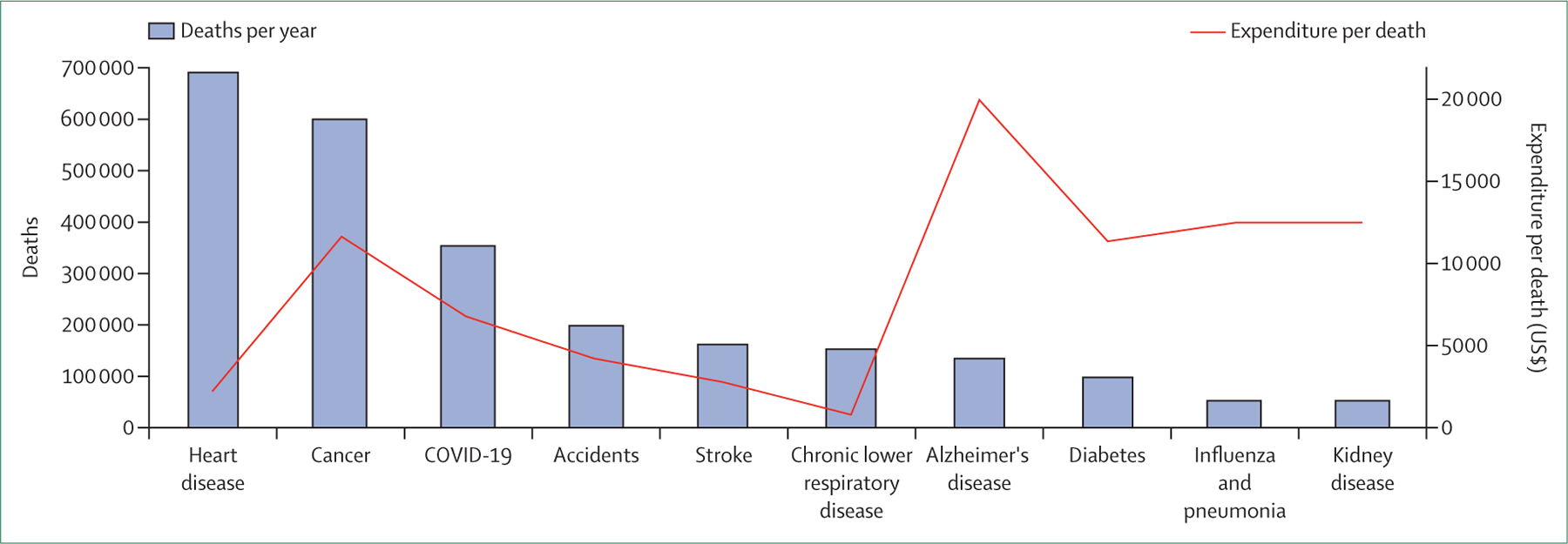
The bars depict the deaths per year per disease in the USA. In terms of US dollars committed per death from each disease, chronic obstructive pulmonary disease receives the lowest funding.43,44
In addition to hampering diagnosis and causing confusion about the goals of COPD treatment, over-reliance on spirometry has also complicated drug development. Despite knowledge that FEV1 correlates poorly with individual patient-centred outcomes such as dyspnoea, risk of exacerbation, and mortality, regulatory bodies continue to emphasise FEV1 as the primary outcome by which to assess novel treatments.45 FEV1 is also an inadequate surrogate marker for many key COPD features because it correlates only weakly (if at all) with morphological and imaging endpoints such as emphysema and small airways disease.46 Regulatory bodies have been slow to accept patient-reported outcomes as primary endpoints in registration trials in favour of spirometric measures. Neither exercise tolerance tests, which are approved in other respiratory disorders, nor emphysema assessment by CT, which is arguably more reproducible in clinical trial settings than spirometry, have been approved as endpoints for COPD. New therapies that reduce emphysema progression could plausibly have little effect on FEV1 but substantial value in altering the disease course. However, the regulatory environment renders the risk of testing these potential therapies prohibitive. Ultimately, what matters to patients are quality of life and survival; all other endpoints are surrogates. The US Food and Drug Administration articulates this concept by requiring endpoints in therapeutic trials to measure how a patient feels, functions, and survives. FEV1 does not qualify based on this criterion and is at best a weak and imperfect surrogate.
Under the current regulatory standards, studies large enough to show reductions in mortality are expensive, although some such studies have been done.47,48 However, these studies have enrolled heterogeneous patient populations solely according to spirometric criteria and perceived exacerbation risk and not on the basis of the underlying pathophysiology driving the disease. Therefore, it is not surprising that until 2018, mortality advantages for inhaled therapies were difficult to show.47,48 Furthermore, instruments that have been validated in COPD suggest that therapies such as bronchodilators and inhaled corticosteroids have only moderate effects on quality of life, despite the detectable effects of these treatments on FEV1 and exacerbations.47,48
As a multidisciplinary group of Commissioners, we have a unique opportunity to present ideas that can reframe the problems facing the field and challenge accepted dogma that could be hampering progress. We hope to foster discussion and debate, advocate for policy change, and influence the research agenda to steer the global COPD community and its stakeholders towards a path that leads to COPD elimination. The objective of the Lancet Commission on COPD is to identify the factors contributing to the unrelenting burden of the disease and to propose actionable recommendations to transform our approach to prevention, management, and research.
COPD: beyond tobacco smoking
Real-world observations call into question Fletcher and Peto’s traditional conceptual definition of COPD as a self-inflicted disease caused by tobacco smoking that is associated with accelerated loss of lung function and eventually results in persistent respiratory symptoms.49 This definition, which was formulated in 1976, has increasingly been undermined by evidence of risk factors for COPD other than smoking.8 These risk factors can have distinct pathophysiological mechanisms and clinical manifestations, which can in turn affect diagnosis and treatment. Not all smokers develop COPD and not all patients with COPD smoke or exhibit accelerated decline in lung function with age.50 In fact, about half of patients with COPD seem to have a normal rate of lung function decline, but never reached the expected healthy peak of lung function in early adulthood.20 There is mounting evidence that other factors, such as genetic predisposition, in-utero events and prematurity, early-life events, early or recurrent respiratory infections, and exposure to air pollution and biomass fuel smoke, account for a large proportion of COPD cases worldwide.51 Respiratory morbidity in early childhood can affect respiratory morbidity in late adulthood. Early-life events can alter the respiratory and immune system in ways that render the lungs more vulnerable, leading to greater susceptibility to adult risk factors—eg, maternal smoking accentuates the effects of later-in-life smoking and occupational exposures in the infant, heightening the risk of airflow limitation.52,53
Although the clinical consequences of disease caused by these other risk factors might be similar to that of tobacco smoke exposure, peculiarities in the pathophysiological mechanisms related to each of these exposures could plausibly translate into distinct diagnostic, prognostic, and therapeutic considerations. Non-smoking patients with COPD tend to be younger and have better lung function than patients who develop COPD secondary to smoking.54 More in-depth investigation into these COPD subtypes is essential to advance precision COPD management. With this in mind, we propose a novel COPD classification. This approach defines five types that capture the variation in the causes of COPD and in turn link to the underlying molecular endotypes driving pathophysiology (figure 6). Although we acknowledge that there are unmodifiable risk factors for COPD and potentially new risk factors that have yet to be identified, many COPD cases (and the associated disease burden) are preventable. To prevent new cases of COPD and ultimately eliminate the disease, it is necessary to heighten awareness of the non-tobacco risk factors while still acknowledging the terrible toll of tobacco smoking. We propose a model that challenges accepted dogma. We concede that some aspects of our proposal are speculative. However, disruptive efforts to reclassify diseases have been successful in the past. For example, the 1975 classification of pulmonary hypertension was initially controversial, but the resulting WHO system has since led to substantial progress in understanding of the disease.55 Pulmonary hypertension has not been eliminated, and the 1975 classification system has required substantial refinement over the years, but the model has still been instrumental in improving identification of people affected by the disease and in focusing research efforts that have yielded life-prolonging therapeutics. The optimal approach to diagnose and classify COPD is uncertain, but the status quo is not working. Given that most COPD is preventable, development of a system that prioritises risk factors will hopefully set a course towards elimination.
Figure 6: Proposed classification of COPD according to major risk factors.
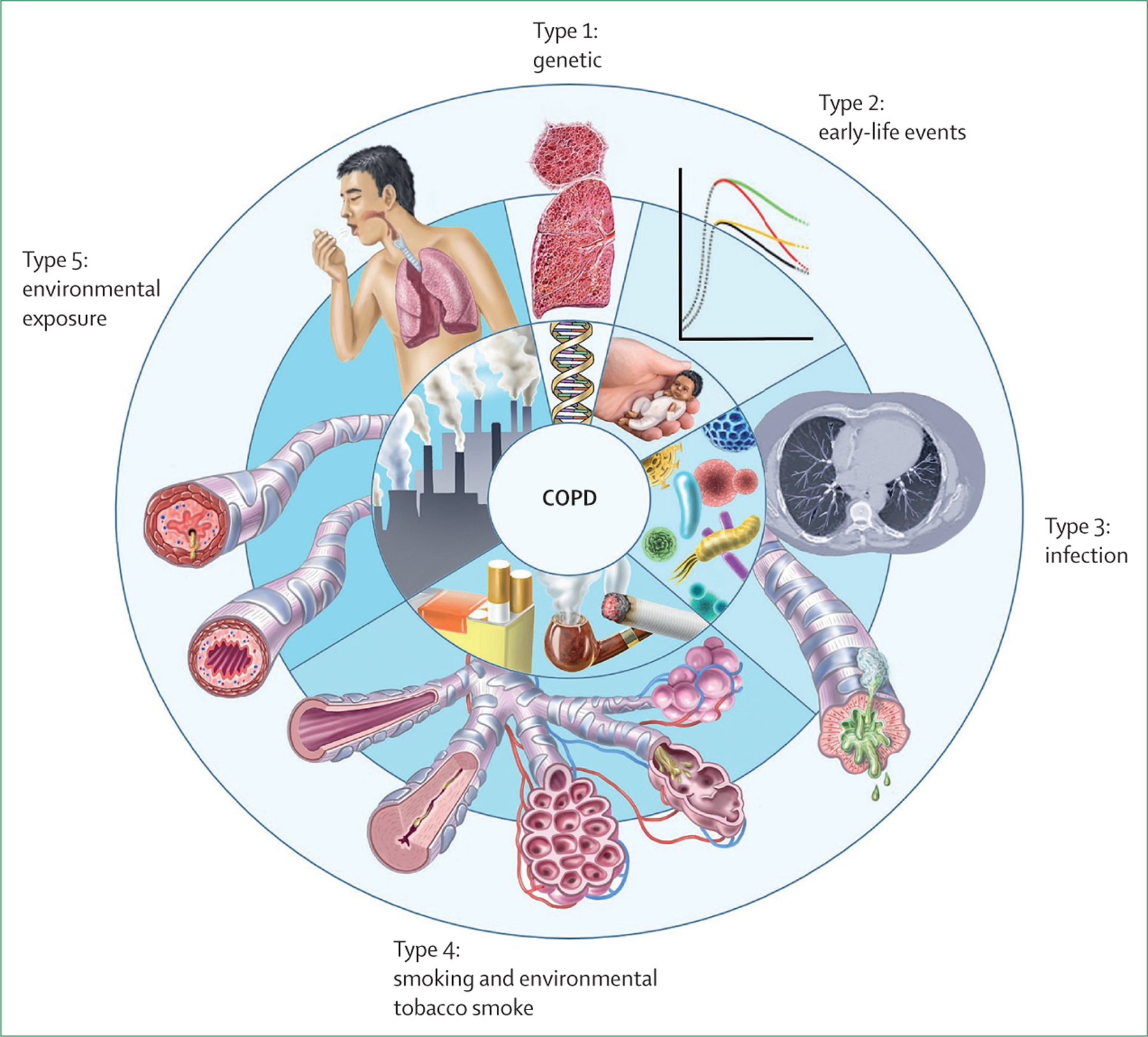
The five proposed types are related to genetics, early-life events, infections, exposure to tobacco smoke, and environmental exposures. We remain cognisant, however, that individuals are prone to multiple exposures throughout life, which could cause additive or interactive damage to lung health. COPD=chronic obstructive pulmonary disease.
Type 1: genetically determined COPD
Genome-wide association studies focusing on disease phenotypes defined by spirometry suggest that the genetic heritability of COPD is moderate.56 Although about 40% of the variability in airflow limitation and up to 60% of the risk for COPD related to smoking is attributable to genetics,57 α1 antitrypsin deficiency and telomerase reverse transcriptase mutation are the only two monogenetic variants that have been clearly shown to have a causative role in the disease. The role of epigenetic changes resulting from gene–environment interactions, such as DNA methylation and histone modifications, which could influence the development of COPD in adulthood, is incompletely explored.56,58 In addition, smoking behaviour could have genetic origins.59
α1 antitrypsin deficiency
α1 antitrypsin is a glycoprotein mainly produced in hepatocytes that inhibits neutrophil elastase and thus protects the lungs from the neutrophil-elastase-mediated development of emphysema. Deficiencies in α1 antitrypsin are noted in 1–2% of white patients with COPD.60 The most frequently observed abnormal genotype is PI*ZZ, which is also associated with more severe disease than other genotypes, including the PI*S allele. Homozygosity for PI*ZZ is associated with the development of COPD irrespective of smoking status, whereas heterozygosity is associated with a moderate deficiency of α1 antitrypsin that increases the risk of developing COPD in smokers (figure 7). Current guidelines recommend that all patients with COPD be tested for α1 antitrypsin deficiency.5 This guideline has become even more pertinent since the publication of data showing the cumulative effect of multiple α1 antitrypsin (SERPINA1) variants on lung function.61
Figure 7: Thoracic CT of a 72-year-old patient with α1 antitrypsin deficiency.
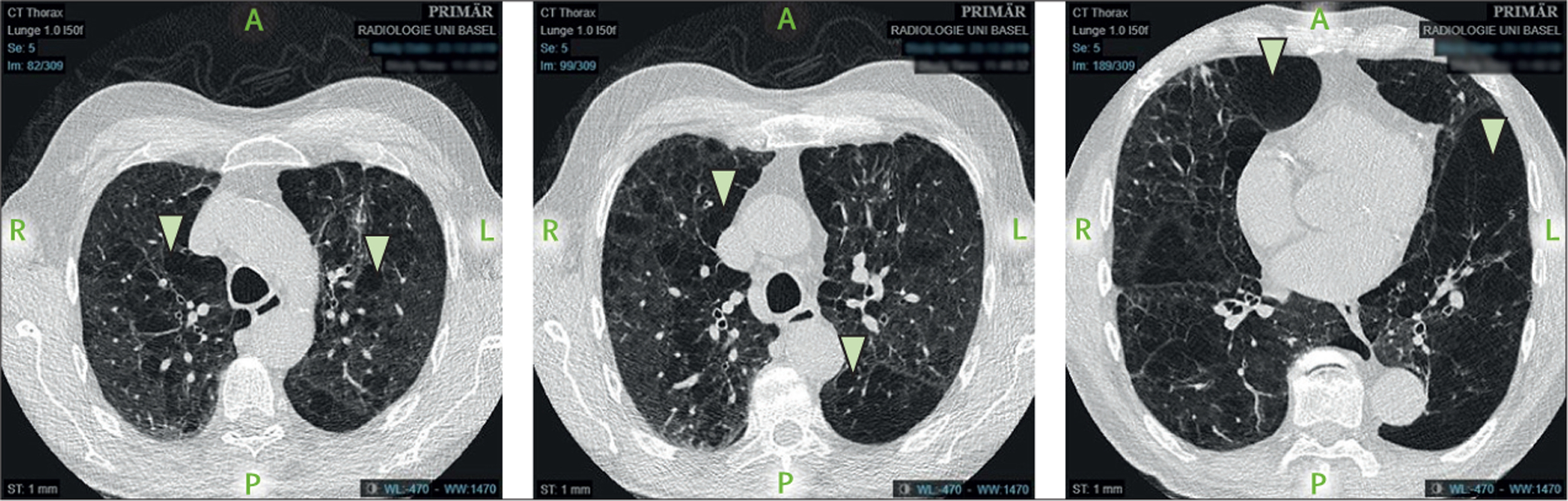
The arrows show emphysematous areas in the lung. The patient is an ex-smoker with a 40-pack-year history of tobacco use, a COPD assessment test score of 21 (range 0–40; higher scores suggest greater disease effects), and a modified medical research council dyspnoea score of 3 (0–4; higher scores suggest greater dyspnoea). COPD=chronic obstructive pulmonary disease.
Patients with α1 antitrypsin deficiency are similar to patients with COPD in that they are prone to dyspnoea, chronic and episodic bronchitis, and upper respiratory tract infections (URTIs), although they might present at a younger age than people with COPD that is unrelated to α1 antitrypsin deficiency.60 The rate of decline of FEV1 in α1 antitrypsin deficiency is associated with smoking and environmental exposures, male sex, age, FEV1 at diagnosis, the presence of chronic bronchitis, and bronchodilator responsiveness. Although the emphysema distribution patterns and airway changes are broadly similar between α1 antitrypsin deficiency-related COPD and COPD unrelated to α1 antitrypsin deficiency, patients with α1 antitrypsin deficiency tend to have greater basilar emphysema.62 The proportion of patients experiencing exacerbations of COPD is similar between α1 antitrypsin deficiency and non-α1 antitrypsin deficiency populations.
TERT mutations
Telomerase enzymes protect against telomere shortening during every cell division, but lose activity as people age.63 Mutations in telomerase genes result in increased telomere shortening, stem-cell exhaustion, premature ageing, cell senescence, and increased secretion of pro-inflammatory cytokines from the senescent cells.64 The resulting chronic inflammatory state leads to lung tissue destruction and remodelling. Although these pathophysiological changes become more common with age even without mutations in TERT (the gene that encodes for telomerase reverse transcriptase), mutations can exaggerate these processes and are predominantly found in women and in 1% of patients with severe early-onset COPD.65–67 The same TERT mutation can result in differing patterns of lung damage according to the environment, leading to fibrosis in non-smokers and emphysema in smokers.68
Type 2: COPD related to early-life events
Prematurity
Around 10–12% of all deliveries are premature births,69 resulting in approximately 15 million premature infants worldwide yearly, some born as early as 23 or 24 weeks’ gestation. Prematurity is associated with low birthweight, nutritional problems, susceptibility to respiratory infections, and poor lung function early in life. Together with insults such as exposure to tobacco smoke in the second half of pregnancy, prematurity can cause substantial impairment in alveolar, vascular, and airway development, leading to reduced lung maturation postnatally, particularly in infants born before 28 weeks’ gestation.70 These pathological changes increase susceptibility to COPD later in life—thus, prevention of prematurity is key to the elimination of COPD.
Respiratory failure is the most common morbidity associated with extremely preterm birth. As a result of advancements in neonatal and perinatal care, infant survival is higher than ever before. Chronic lung disease of infancy can occur together with prematurity and be worsened by other risk factors, such as neonatal infection, persistent ductus arteriosus, and fluid overload. The production of free radical species from supplemental oxygen, together with barotrauma and pressure from mechanical ventilation, induces additional lung injury and scarring. Chronic lung disease of infancy is characterised by lung fibrosis, emphysema due to poorly developed alveolar spaces, disturbed vascular growth with pulmonary hypertension and vascular hyperreactivity, airflow limitation, and bronchial hyper-responsiveness.
Severe lung injury associated with prematurity can also result in the clinically well recognised bronchopulmonary dysplasia.71 The term bronchopulmonary dysplasia is often used interchangeably with chronic lung disease of prematurity. The American Thoracic Society guidelines suggest that chronic lung disease of infancy is the final common pathway of a heterogeneous group of pulmonary disorders that start in the neonatal period and include both bronchopulmonary dysplasia and chronic lung disease of prematurity.72 We are not suggesting that bronchopulmonary dysplasia should be considered a subtype of COPD. Rather, we are noting that pulmonary insults during the prenatal and early-life periods lead to lower peak lung function in adulthood, which in turn contributes to the development of COPD in some individuals.2,73
Lung function anomalies due to prematurity persist through childhood73 and into adulthood, with clear associations between prematurity and airflow limitation in the early thirties and early fifties.74,75 Long-term CT studies also show that the lungs of people born prematurely who have chronic lung disease (particularly those born during the pre-surfactant period) have many pathological similarities to those of people with COPD.11,76
Although there are gradual improvements in indices used as surrogates for quantifying lung function, most individuals born prematurely with low birthweight or who are small for gestational age never attain age-appropriate levels of pulmonary function and are at increased risk of persistent airflow limitation and COPD later in life.74,77 Air pollution exposure during pregnancy can also affect postnatal lung function in preterm infants more severely than infants born at term.78
Childhood asthma
Asthma is the most frequently diagnosed chronic airway inflammatory disorder in childhood. Whether asthma is a single disease or encompasses several diseases as an umbrella term is arguable. Whether early-life asthma is a cause or consequence of poor lung development is debatable because a child with abnormal lung development due to any of the mechanisms discussed previously is likely to have symptoms that mimic those of asthma. Therefore, it is quite likely that such a child would end up being diagnosed with, and treated for, asthma. Conversely, longitudinal observations suggest that some early-life wheeze phenotypes slow the growth of adolescent lung function.79 Furthermore, early-life asthma adversely affects later lung function independent of early-life lung function.80
Together, these findings suggest that, at minimum, a diagnosis of early-life asthma has an adverse effect on lung development. Evidence that early-life asthma is associated with airway remodelling also supports this notion.81 Teasing out the relative contributions of early-life risk profiles from childhood lung function in the development of poor adult lung function and COPD is highly complex. However, Bui and colleagues80 reported that the effect of childhood asthma on development of COPD and risk of low lung function in middle age is largely transmitted through active adult asthma (and to a lesser degree through reduced childhood lung function), highlighting the importance of controlling asthma throughout life. Overall, the balance of evidence suggests that controlling early-life asthma could be crucial to the prevention of COPD, although definitive data for the beneficial effects of asthma control on long-term lung function, especially in relation to incidence of COPD, are not available.82
Type 3: infection-related COPD
Pulmonary and systemic infections are among the most important non-smoking related risk factors for chronic lung disease and COPD, especially in LMICs.4,7,8 An estimated 10–42% of people with early-life pneumonia, tuberculosis, or HIV develop COPD.83–85
Childhood infections
Multiple factors influence childhood respiratory health, but respiratory infections are of particular concern. Respiratory syncytial virus is one of the best-described pathogens causing pneumonia and other lower respiratory tract infections (LRTIs).86 Impaired lung function at birth, which is influenced by maternal nutrition,87 pre-pregnancy body-mass index (BMI),88 and maternal smoking, could predispose to LRTIs. Estimates suggest that childhood LRTIs account for 33% of COPD in non-smokers.54 Frequent LRTIs in the first seven years of life are associated with pulmonary vasculature abnormalities, airway remodelling, parenchymal destruction, and impairment in mucociliary clearance and local immune defences.89 Impaired airway host defences create an environment that is susceptible to both viral and bacterial infection and chronic colonisation of the lower respiratory tract.90 Chronic colonisation can induce a chronic inflammatory response, with bacterial antigens causing hypersensitivity that enhances airway hyperreactivity and lung damage.
Tuberculosis-associated COPD
Tuberculosis remains a major health problem in many countries. There is a strong association between a history of tuberculosis and the development of COPD in non-smoking patients.91 Despite successful treatment of tuberculosis, impaired lung function often persists.92 The risk of airflow limitation in patients with a history of tuberculosis is more than twice that in patients with no history of tuberculosis. The chronic inflammatory response and long-term tuberculosis-associated structural alterations, such as airway remodelling with scar formation, stenosis, and bronchiectasis, are the pathological basis for the development of COPD. Patients with tuberculosis-related COPD have higher air trapping, more fibrosis and emphysema, and an altered immunological and inflammatory profile (fewer neutrophils) compared with patients with COPD with no history of tuberculosis.93 Lung parenchymal inflammation in tuberculosis is mediated heavily by matrix metalloproteinases, which have a substantial role in the accelerated destruction of the pulmonary extracellular matrix noted in COPD.93
HIV-associated COPD
During the first decade of the AIDS epidemic, opportunistic infections were the most common reason for pulmonary impairment in patients with HIV. However, despite limitations in their methods, cross-sectional studies report a prevalence of emphysema as high as 65% in people with HIV, which is in turn associated with early mortality.94 Other features, including airflow limitation, diffusion impairment, and pulmonary vascular abnormalities, are also present to varying degrees. The advent of highly active antiretroviral therapy resulted in an improvement in HIV outcomes, but increased respiratory symptoms and decreased lung function have been noted in people with HIV and are independent of other risk factors such as smoking and intravenous drug use.95 In people with HIV, the prevalence of COPD can be as high as 23%, and people with HIV and COPD experience more rapid declines in lung function than HIV-negative people with COPD.96 Age, BMI, and CD4 cell count are independently associated with HIV-related COPD97 and impaired diffusing capacity of the lungs for carbon monoxide (DLCO) is also highly prevalent in patients with HIV (figure 8). COPD, like other chronic illnesses, is underdiagnosed in patients with HIV and the implications of this underdiagnosis are incompletely recognised and understood.94
Figure 8: Thoracic CT of a patient aged 56 years diagnosed with HIV in 1995.
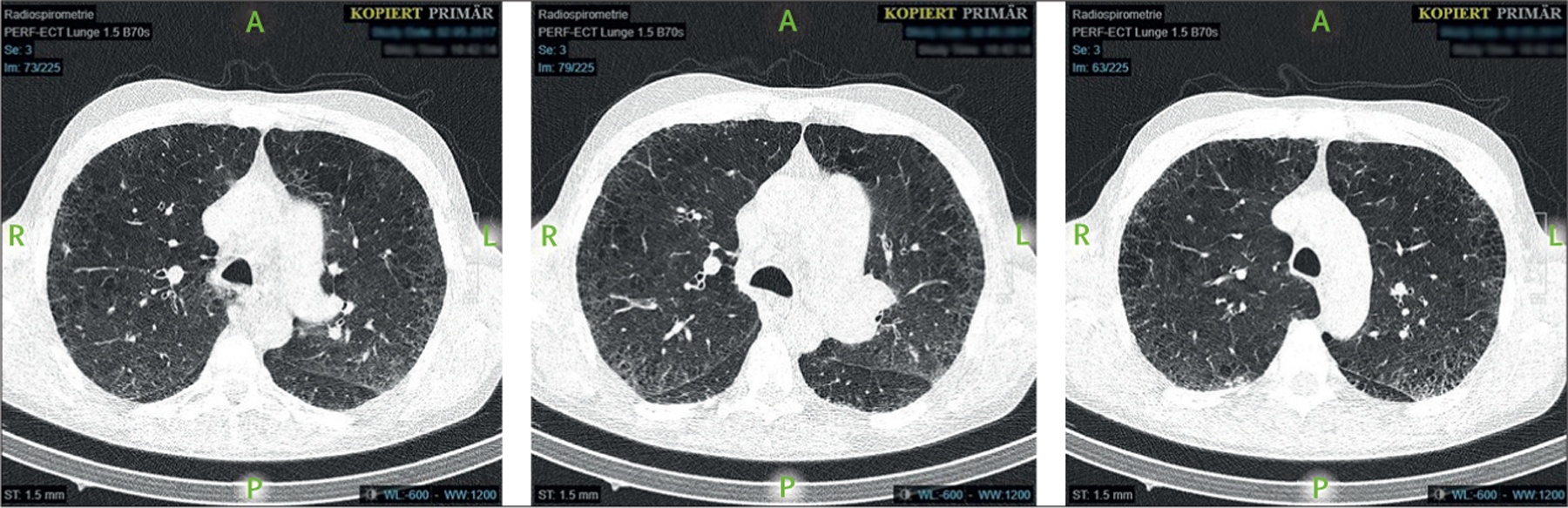
At the time of the CT, the patient was an active smoker and had a diffusing capacity of the lungs for carbon monoxide of 32·2% of predicted, severe pulmonary hypertension, and cachexia (body-mass index 18·9 kg/m2).
Emerging data suggest that the pathological changes present in infection-related causes of COPD are highly variable (table 2). This variability suggests the possibility that the underlying molecular pathways driving COPD could also be different, which in turn has implications for treatment and cure.
Table 2:
Importance of pathophysiological features associated with infection-related causes of chronic obstructive pulmonary disease
| Childhood infections | Tuberculosis | HIV | |
|---|---|---|---|
| Hypertrophy of airway smooth muscle | Sometimes occurs normally, but can be mildly important | Moderately important | Mildly important |
| Thickening of basement membrane | Sometimes occurs normally, but can be mildly or moderately important | Very important | Mildly important |
| Inflammation* | Mildly to moderately important | Moderately important | Mildly important |
| Change in turnover of extracellular matrix | Moderately important | Very important | Moderately important |
| Damage to mucociliary clearance or mucus hypersecretion | Moderately important | Moderately important | Moderately important |
Neutrophilic, eosinophilic, or paucigranulocytic inflammation.
Type 4: COPD related to smoking or vaping
The clinical and radiographic characteristics associated with tobacco smoking are well known and account for most knowledge about COPD. Maternal smoking, second-hand and third-hand smoking (ie, exposure to pollutants that settle on surfaces when tobacco is smoked indoors), and vaping or e-cigarette smoking also have substantial implications for lifelong lung health.
In-utero and early-life exposure to tobacco smoke
Smoking during pregnancy is associated with serious adverse outcomes, including preterm birth, low birthweight, birth defects, and infant death.98 In 2016, roughly 7·2% of mothers in the US smoked during pregnancy; the greatest prevalence of smoking was in mothers aged 20–24 years (10·7%).98 Long-lasting respiratory impairment is evident in children of mothers who smoked during pregnancy, and these effects can be amplified in offspring who also smoke as adults: reduced lung function at birth limits the ability to reach healthy peak lung function, and tobacco smoke then triggers early and accelerated lung function decline.2 Maternal smoking during pregnancy is also associated with increased respiratory symptoms, including wheezing and chronic bronchitis.99 Notably, a study100 has suggested that exposure to smoking among parents even before conception—especially paternal exposure to smoke around puberty—can affect lung function in offspring.
Infants whose mothers smoke during the prenatal and postnatal periods have thicker large-airway walls than infants born to non-smokers, and this increased wall thickness has been associated with sudden infant death syndrome.101 Among children in grades 4 (average age 10 years), 7 (average age 13 years), and 10 (average age 16 years), a history of in-utero exposure to tobacco smoke was associated with reduced forced expiratory flow and peak expiratory flow rate.102 Airflow limitation in offspring exposed to maternal smoke could be due to reduced alveolarisation and augmented lung damage as a result of increased oxidative stress from nicotine exposure.103 Glutathione S-transferase genes have a crucial role in managing oxidative stress in the lungs. A study104 of US children aged 6–10 years showed an association between maternal smoking and reduced lung function, which was more common in children whose mothers were carriers of a GSTM1 deletion (and thus had no functional enzyme). An Australian study105 also showed that carriers of null mutations in glutathione S-transferase genes and GSTP1 genes with Ile/Ile alleles have an increased likelihood of developing lung function deficits by early-to-late adolescence when exposed to tobacco smoke in early life.
Animal models have also shed light on the adverse respiratory effects of prenatal and early-life exposure to smoke. Neonatal mice exposed to cigarette smoke over 4 days had increased neutrophils and macrophages in bronchial fluid and displayed increased mucus production, goblet cell hyperplasia, and evidence of epithelial and vascular remodelling.106 Offspring of rhesus monkeys exposed to nicotine during their pregnancy had increased airway resistance, increased collagen deposition, decreased lung weight, and fixed lung volumes compared with offspring whose mothers were not exposed to nicotine.107
Passive smoking
The EU-funded TackSHS project concluded that the respiratory burden attributable to second-hand smoke exposure is still substantial and mainly due to exposure in the home.108 Around 1·5% of deaths in children aged 0–14 years and 0·6% of deaths in non-smoking adults are attributable to exposure to second-hand smoke at home.107 Exposure to second-hand smoke as an adult in the workplace is also associated with increased respiratory symptoms, including cough, phlegm, and frequent colds.109
Similar to the effects of active smoking, passive smoking can contribute to increased airway inflammation and mucus secretion.110 Independent of their current smoking status, adults whose mothers smoked during their childhood had lower FEV1 and higher risk of post-bronchodilator airflow limitation than those who were not exposed to smoke by their mothers during childhood.53 Maternal smoking during early childhood could also have an additive effect on the negative consequences of the individual smoking habits of offspring, particularly airflow limitation.111 The radiographic changes noted in COPD are also evident in adults with and without COPD who have a history of exposure to environmental tobacco smoke in childhood.112
Vaping or e-cigarette use
Use of e-cigarettes impairs the body’s ability to fight infections by reducing the antimicrobial activity of alveolar macrophages, neutrophils, and epithelial cells and by reducing the expression of immune-related genes.113 Use of e-cigarettes alters more than 200 proteins in airway epithelial cells; the effects on 113 of these proteins are unique to e-cigarette use.114
E-cigarette use can promote lung inflammation and is associated with respiratory symptoms such as cough, sputum production, and wheezing.115 In an analysis116 in which combustible tobacco use was controlled for, e-cigarette use was independently associated with chronic respiratory diseases, including asthma and COPD. The risk of developing COPD from combustible cigarettes is doubled when combined with e-cigarette use, and use of e-cigarettes in never-tobacco-smokers doubles or triples the risk of self-reported COPD, emphysema, or chronic bronchitis.117 Additional studies are needed to confirm the direct effect of e-cigarettes on the development of COPD, but the available data are compelling and concerning.
Cannabis
The prevalence of cannabis smoking is rapidly increasing. In Canada, for instance, where cannabis use was legalised in 2018, 27% of the population surveyed reported having used cannabis in 2020, an increase of 25% from 2019.118 Only 17% of cannabis smokers also regularly consume tobacco cigarettes, and the remainder never or almost never smoked both tobacco and cannabis.118 Although the respiratory health effects of cannabis smoking are incompletely understood, emerging evidence suggests probable harm. A 2018 systematic review119 showed that cannabis use was associated with increased risk for cough (risk ratio [RR] 2·0), sputum production (3·8), wheezing (2·8), and dyspnoea (1·6) compared with not using cannabis or tobacco. The effect of cannabis smoking on lung function and particularly on lung function decline over time is controversial. In the largest study120 of its kind, the Canadian Cohort of Obstructive Lung Disease followed up 1285 participants aged 65 years for an average of 6 years. The findings suggested that heavy cannabis use (defined as more than 20 joint-years [the number of joints per day, multiplied by years]) was associated with a steeper decline in FEV1 compared with non-smokers (–40·20 mL per year vs –10·75 mL per year; p=0·0007). The effects of cannabis on a molecular and cellular level are poorly understood. Endobronchial biopsies have shown that cannabis smokers have a greater degree of squamous metaplasia, goblet cell hyperplasia, and cellular disorganisation than non-smokers, at rates similar to those among tobacco smokers; the alveolar macrophages of cannabis smokers also show diminished phagocytic and antimicrobial activity.121,122
Type 5: environmental exposure-related COPD
Environmental exposure encompasses exposure to particles and gases from indoor fuel use, wildfire smoke, air pollution or smog, and occupational exposure. Emerging data suggest that different environmental exposures can lead to differing pathological processes leading to COPD (table 3).
Table 3:
Importance of pathophysiological features associated with environmental exposure-related causes of chronic obstructive pulmonary disease
| Indoor pollutant exposure | Ambient air pollution or smog | Wildfire smoke | Occupational exposure | |
|---|---|---|---|---|
| Clinical manifestations | ||||
| Chronic cough | Moderately important | Moderately important | Moderately important | Very important |
| Sputum production | Moderately important | Moderately important | Mildly important | Moderately important |
| Dyspnoea | Moderately important | Mildly important | ·· | Moderately important |
| Wheezing | Mildly important | Mildly important | ·· | Moderately important |
| Exacerbations | Moderately important | Moderately important | Moderately important | Moderately important |
| Systemic manifestations | Moderately important | Mildly important | ·· | Mildly important |
| Airway remodelling and inflammation | ||||
| Hypertrophy of airway smooth hypertrophy | Very important | ·· | ·· | Very important |
| Thickening of basement membrane | Moderately important | ·· | ·· | Moderately important |
| Eosinophilic inflammation | Moderately important | ·· | ·· | Moderately important |
| Neutrophilic inflammation | Mildly important | Mildly important | Mildly important | ·· |
| Pauci immune inflammation | Mildly important | ·· | ·· | ·· |
| Change in turnover of extracellular matrix | Moderately important | ·· | ·· | ·· |
| Persistent airflow limitation | ||||
| FEV1/FVC <0·70 | Very important | Moderately important | ·· | Very important |
| Ratio of total to effective resistance | Very important | ·· | ·· | Very important |
| Difference in oscillation resistance at 5 Hz and 19 Hz | Very important | Moderately important | ·· | Very important |
| Reactance area | Very important | Moderately important | ·· | Very important |
| Parenchymal destruction | ||||
| CT emphysema | Mildly important | Mildly important | ·· | Moderately important |
| Decreased DLCO | Mildly important | ·· | ·· | Moderately important |
FEV1=forced expiratory volume in 1 s. FVC=forced vital capacity. DLCO=diffusing capacity of the lungs for carbon monoxide.
Exposure to indoor pollutants
Roughly 3 billion people, mainly in LMICs, use biomass fuel for cooking and heating.123 Particulate matter generated by inefficient and poorly ventilated stoves burning wood, crop waste, dung, or coal is a major source of indoor pollution. The World Bank estimated that the COVID-19 pandemic would push between 88 and 115 million people into extreme poverty, which could mean increased use of biomass fuels (as a result of being unable to afford more expensive alternatives).124 Exposure to smoke from biomass fuels and other indoor air pollutants results in altered lung function, increased respiratory infections, and pronounced airway inflammation, all of which increase the risk of COPD.125,126 COPD caused by biomass fuel smoke could have distinct features and demographic characteristics compared with that associated with tobacco smoking. COPD induced by exposure to biomass fuel pollution is more likely to affect older women born in rural areas and is associated with a higher prevalence of chronic bronchitis and bronchial hyperreactivity and milder airflow limitation and slower declines in lung function than cigarette smoke-induced COPD.125,126 Exposure to biomass smoke leads to an airway-predominant COPD phenotype with increased anthracosis, airway thickening, air trapping, small airway fibrosis, and pulmonary arteriole intimal thickening and less emphysema and goblet cell hyperplasia compared with COPD associated with tobacco smoking.127 Evidence also suggests that exposures to indoor pollutants—even to concentrations lower than the WHO recommended limits—can lead to reduced lung function, particularly when the exposure occurs during early life.128
Ambient air pollution and smog
Globally, the population exposed to air pollution levels above the WHO Air Quality Guideline safe limit declined modestly between 2010 and 2016; however, this decline was driven by improvements in North America and Europe, whereas levels in Asia remain extremely high.129 New WHO global air quality guidelines provide clear evidence of the damage that air pollution inflicts on human health at even lower concentrations than previously thought.130 Exposure to low-to-moderate environmental air pollution during pregnancy and in the first two years of life is associated with decreased lung function after birth131 and up to the sixth year of life.132 Importantly, exposure to ambient pollution up to the sixth year of life was associated with reduced lung function not only in cross-sectional studies but also in longitudinal studies, in which children exposed to air pollution had a lower rate of lung growth than those not exposed to pollution.133 Yet in the USA, roughly one in every 11 community schools lies within 150 m of a highway, truck route, or other road with substantial traffic.134 Air pollution also has adverse effects in adults: the SAPALDIA study showed that living within 20 m of a main street increased the risk of sputum production and wheezing in never smokers.135 Exposure to ambient air pollutants, particularly ozone and particulate matter, decreases FVC and FEV1, increases respiratory symptoms, and increases the risk of emphysema in adults, irrespective of smoking status.135
Wildfire smoke
As a result of the hotter and drier conditions resulting from climate change, the number of wildfires has decreased but the intensity of the fires and the acreage destroyed have increased.136 Smoke from wildfires is formed from a mixture of carbon monoxide, benzene, aldehydes, and polycyclic aromatic hydrocarbons.137 A study138 in the USA between 2004 and 2009 showed that racial and ethnic minorities older than 65 years and those living in poverty were particularly susceptible to developing lung disease from the air particulates in wildfire smoke. Increased respiratory symptoms and physician visits have also been reported in children exposed to the southern California wildfires during 2003.139 Human airway epithelial cells exposed to wildfire smoke extract in vitro showed greater inhibition of autophagy and increased barrier dysfunction compared with cells exposed to cigarette smoke.140
Occupational exposures
Studies have suggested that occupational exposures to vapours, gases, dusts, or fumes are associated with the development of COPD3 and emphysema and airway disease as measured by quantitative CT.141 Such exposure occurs in a broad range of occupations, including—but not limited to—production plant workers, woodworkers, hairdressers, food service workers, and construction workers. A meta-analysis142 showed that people occupationally exposed to airborne pollutants have a 22% increased risk of COPD and that 15% of all COPD cases in high-income countries are attributed to occupational exposures. Dust exposure from other occupations including cement, primary coffee processing, street sweeping, and quarry mines can also lead to decreased lung function and increased respiratory symptoms, especially cough and sputum production.
Why revisit the diagnostic criteria for COPD?
It is well accepted that COPD is a complex and heterogeneous disease: complex in that the disease has several distinct components and is driven by both genetic and environmental factors that dynamically interact over time, heterogeneous in that all components are not present in all individuals at any given timepoint and can vary in severity over time.143 Diagnostic criteria for COPD capture none of this complexity nor the variation in underlying pathophysiology. Rather, they rely exclusively on the presence of spirometric airflow limitation in at-risk patients, as assessed by the post-bronchodilator ratio of FEV1 to FVC. Unfortunately, spirometric airflow limitation is detectable only at later pathophysiological stages after the expression of genetic effects, early-life events, infections, and exposure to tobacco smoke or environmental factors have led to major damage to the lung. Thus the spirometric definition of COPD misses early pathological changes, which means that COPD is almost always diagnosed in patients with largely irreversible lung damage. Expansion of diagnostic criteria to encompass COPD heterogeneity will increase sensitivity for early disease and could lead to the discovery of new preventive measures and therapeutic approaches.
Varied pulmonary and extra-pulmonary COPD manifestations
The heterogeneity of COPD is shown by the substantial variation in respiratory symptoms and morbidity and the range of systemic consequences of the disease (and its common comorbid conditions). Dyspnoea upon modest exertion and chronic cough have been extensively explored in smokers with spirometrically defined COPD because they greatly affect patients and are typically the reason for seeking medical attention (although they are only weakly related to measures of lung function).144 Challenges arise when clinicians see patients with no respiratory symptoms but with airflow limitation, or vice versa. Although the importance of respiratory symptoms is generally acknowledged, which symptoms to consider or how severe symptoms should be before they are considered pathological remains unclear.
Acute respiratory exacerbations are common in patients with COPD but also occur in individuals with spirometrically normal lung function.18 Characterised by increased cough, sputum production, and breathlessness, exacerbations are an important cause of accelerated decline in spirometric lung function, absence from work, hospital admissions, and mortality.5 As we will discuss later in this Commission, the definition of an exacerbation is imprecise, which makes clustering of patients based on the predilection to exacerbations or the frequency of exacerbations challenging. Yet guidelines5 use the term “frequent exacerbator” to define a group of COPD patients who experience at least two exacerbations per year and to define treatment for these patients. These patients have a worse prognosis than patients with fewer exacerbations, but the phenotype is not stable. Reducing the frequency of exacerbations is paramount, but permanent classification of patients according to an outcome of illness is counterproductive, because treatment could improve or prevent the outcome and thus change the patient’s phenotype. As Lopez-Campos and colleagues have stated, “a clinical phenotype should be a natural manifestation of the disease and the phenotypic feature should be stable over time”.145
The presence and severity of pulmonary hypertension in patients with COPD are also highly variable. In a study146 in which CT and clinical features were used to identify airway remodelling and emphysema associated with COPD-related pulmonary hypertension, pulmonary hypertension was present in 34–57% of patients. No differences in age, dyspnoea, and or degree of emphysema or spirometric lung function measurements were noted between COPD patients with and without pulmonary hypertension. Patients with COPD and moderate pulmonary hypertension, however, had a shorter 6-min walk distance and lower oxygen partial pressure than those without pulmonary hypertension. Significant differences in airway remodelling and pulmonary artery diameter were also noted between patients with COPD with and without pulmonary hypertension, as measured by CT.
Cardiovascular, musculoskeletal, and mental disorders and diabetes are the most prevalent comorbidities among patients with COPD. The associations between these comorbidities or systemic manifestations and COPD can be classified into three clusters with different clinical, functional, and prognostic consequences. First, some conditions share risk factors and pathological pathways with COPD, including ageing, senescence, and exposures. This link is best exemplified by the fact that smoking is the primary risk factor for both lung cancer and COPD. Second, some chronic comorbidities aggravate COPD, causing further deterioration or intensification of symptoms—eg, depression or cardiovascular disease can lead to reduced physical activity, weight-gain, and decreased lung function. Finally, some acute illnesses that require treatment can cause adverse effects on respiratory symptoms or pulmonary function.
Cardiovascular effects associated with COPD are not limited to the pulmonary vasculature. Patients with COPD have more cardiovascular disease than current and former smokers without COPD.147,148 This increased burden of disease could be because of shared pathophysiology, such as increased oxidative stress, systemic inflammation, hypoxia, and physical inactivity.148 Cardiovascular disease worsens the prognosis of COPD because it is implicated in the pathogenesis of exacerbations, and vice versa.149 Indeed, most patients with mild-to-moderate COPD die of cardiovascular disease, not of respiratory failure.150 It would be sensible to explore the extent to which the treatment of comorbidities affects COPD control and clinical outcomes, but such studies have not yet been done.
In addition to vascular effects, COPD is associated with other debilitating systemic manifestations such as cachexia and muscle dysfunction (eg, muscle weakness, sarcopenia). Cachexia is defined by the Cachexia Consensus Working Group as a complex metabolic syndrome that is associated with underlying illness and characterised by muscle loss.151 It is diagnosed on the basis of weight loss of at least 5% in 12 months or less in the presence of an underlying disease, plus three of decreased muscle strength, fatigue, anorexia, low fat-free mass index, and abnormal biochemistry (increased C-reactive protein or interleukin 6, anaemia, or low serum albumin). Because of the challenges in obtaining this detailed information, the pragmatic diagnosis of cachexia in patients with COPD has been simplified to unintentional weight loss of greater than 5% in 6 months and a fat-free mass index less than 17 kg/m2 in men and less than 15 kg/m2 in women.152 The prevalence of cachexia in patients with COPD attending outpatient clinics ranges between 4·6% and 10·4%, depending on the definition used; cachexia is associated with decreased FEV1 and exercise capacity, more exacerbations per year, and increased mortality, irrespective of the definition used.152
Depression and anxiety in COPD are under-recognised and undertreated. Many patients experience anxiety or depression, and a bidirectional relationship between COPD and mental disorders is probably operational: individuals with mental disorders are more likely to smoke, which increases the risk of developing COPD, and the social isolation and disability associated with COPD can contribute to new-onset depressive symptoms. Additionally, mental comorbidities influence self-management strategies. Depression might be more strongly associated with patient-reported outcomes in COPD than FEV1.153 Although depression in patients with COPD has been associated with low treatment compliance, poor quality of life, extended hospital stays, increased hospital admissions, and increased mortality, it is often untreated or incorrectly treated.154
Type 2 diabetes is a strong predictor of mortality in COPD and affects 16% of patients.155 Several features associated with COPD have been identified as risk factors for type 2 diabetes, including physical inactivity and bodyweight. Use of inhaled corticosteroids in COPD increases the risk of new-onset type 2 diabetes and worsens glycaemic control.156 Furthermore, low-grade systemic inflammation in COPD has been associated with altered glucose metabolism and the development of insulin resistance.157
Like COPD, lung cancer is primarily caused by smoking tobacco. In 2020, more than 2·2 million people were diagnosed with lung cancer.158 COPD increases the risk of developing lung cancer, even in never-smokers.159 The landmark National Lung Screening Trial160 found that low-dose CT screening reduced mortality from lung cancer by 20%. In 2013, the US Preventive Services Task Force recommended yearly screening for current smokers and for people aged 50–80 years who had quit in the past 15 years and had at least a 30-pack-year smoking history. However, uptake of lung cancer screening in high-risk individuals in the USA (including those with COPD) remains low, particularly among poor and uninsured people.161 Adoption of lung cancer screening practices is also low in LMICs because of lack of infrastructure, costs, social disparities, and cultural barriers.162 Screening in COPD is complicated by the fact that although the risk of lung cancer increases as FEV1 declines, the potential for curative treatment is reduced. In addition, patients with COPD are at increased risk of complications related to invasive diagnostic procedures.163 However, an analysis164 of the National Lung Screening Trial suggested a mortality advantage for those with mild to moderate airflow limitation, but not those with severe or very severe disease.
Varied lung function trajectories
There are various lung function trajectories throughout life, and more than one of these trajectories could lead to COPD. Recognition of these trajectories and the fact that not all patients develop COPD on the basis of accelerated loss of lung function could help to identify new preventive and therapeutic approaches. These variable lung function trajectories are due to the differential effects of multiple individual and environmental factors that influence lung function growth, peak lung function attained, duration of the plateau phase, and the subsequent rate of lung function decline.2 Experimental identification depends on the frequency of lung function measurements, total duration of follow-up, and the number of participants in the study. To date, epidemiological evidence suggests that most lung function changes over the life course can be grouped into six major trajectories, resulting from lifelong, dynamic, or cumulative gene–environment interactions: persistently high lung function, average lung function, below average but stable lung function, persistently low lung function, below average lung function in early life followed by accelerated decline, and low lung function in early life followed by accelerated growth and average decline (figure 9).165 Importantly, some children who initially follow the low lung function trajectory can regain normal lung function.165 Understanding the mechanisms underlying this trajectory is essential to the discovery of therapeutic interventions that can potentially reverse or cure the disease.
Figure 9:
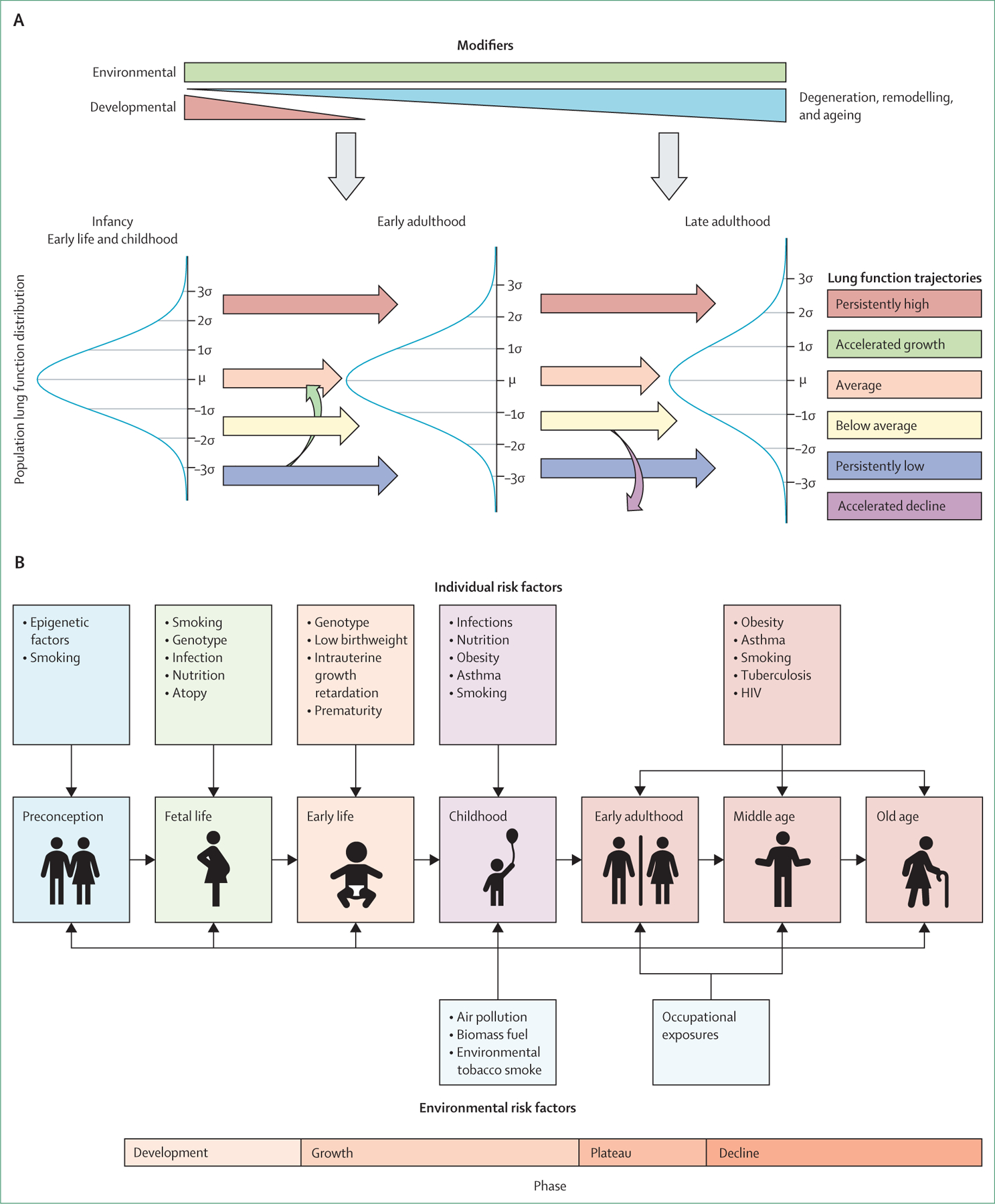
Lung function trajectories across the lifecourse (A) and environmental and individual risk factors that affect lung function trajectories and lung health from before conception to old age (B)
The long-term presence of low lung function before a clinical diagnosis of COPD (which generally does not occur until patients are in their 50s or 60s) presents an opportunity to identify high-risk individuals at a time (eg, infancy, adolescence, early adulthood) when interventions could reduce risk of developing the disease. Early detection of people at risk and targeted prevention strategies are important to slow and perhaps arrest the development of COPD. Importantly, patients who develop COPD subsequent to different lung function trajectories could have different underlying pathological mechanisms, which could in turn lead to different clinical presentations, prognosis, and treatment needs. A study20 showed that a similar proportion of people with normal peak lung function followed by an accelerated decline in FEV1 developed COPD to those with low peak lung function and a normal decline in FEV1. It seems highly unlikely that the pathological processes underlying these two trajectories are the same. However, a disadvantage of using lung function trajectories to define heterogeneity in COPD is that this approach is completely dependent on spirometry, which is rarely done in young people. Identification of novel biomarkers that distinguish the various lung function trajectories in the early decades of life is thus essential.
Patients have been characterised with early COPD on the basis of their age and a post-bronchodilator ratio of FEV1 to FVC of less than 0·70.21 This diagnostic approach does not consider early lung changes or account for symptomatic patients with normal spirometry. Some evidence suggests the potential importance of preserved ratio impaired spirometry, which is defined as reduced FEV1 with a preserved FEV1 to FVC ratio.166 Use of the term pre-COPD has also been suggested.21 Progress based on these classifications will be hindered by the tremendous heterogeneity of the patients included, partly because those with differing risk factors and underlying pathological processes are grouped together. Our suggestion for classification based on five COPD types, which admittedly can overlap, and new diagnostic criteria for these types, acknowledges the concept of pre-COPD and preserved ratio impaired spirometry. However, we specifically aim to reduce heterogeneity by grouping patients according to the cause of their COPD and possibly similar pathophysiology.
Varied histopathological changes
The heterogeneity of COPD pathology is evident in the wide variation in the extent of airway remodelling, including smooth muscle alteration and fibrosis, airspace enlargement as noted in emphysema, inflammation, and vascular changes across patients grouped on the basis of the presence of airflow limitation (figure 10). Many of these pathophysiological changes are not included in current diagnostic algorithms.
Figure 10: Comparison of physiology between healthy lungs and lungs affected by COPD.
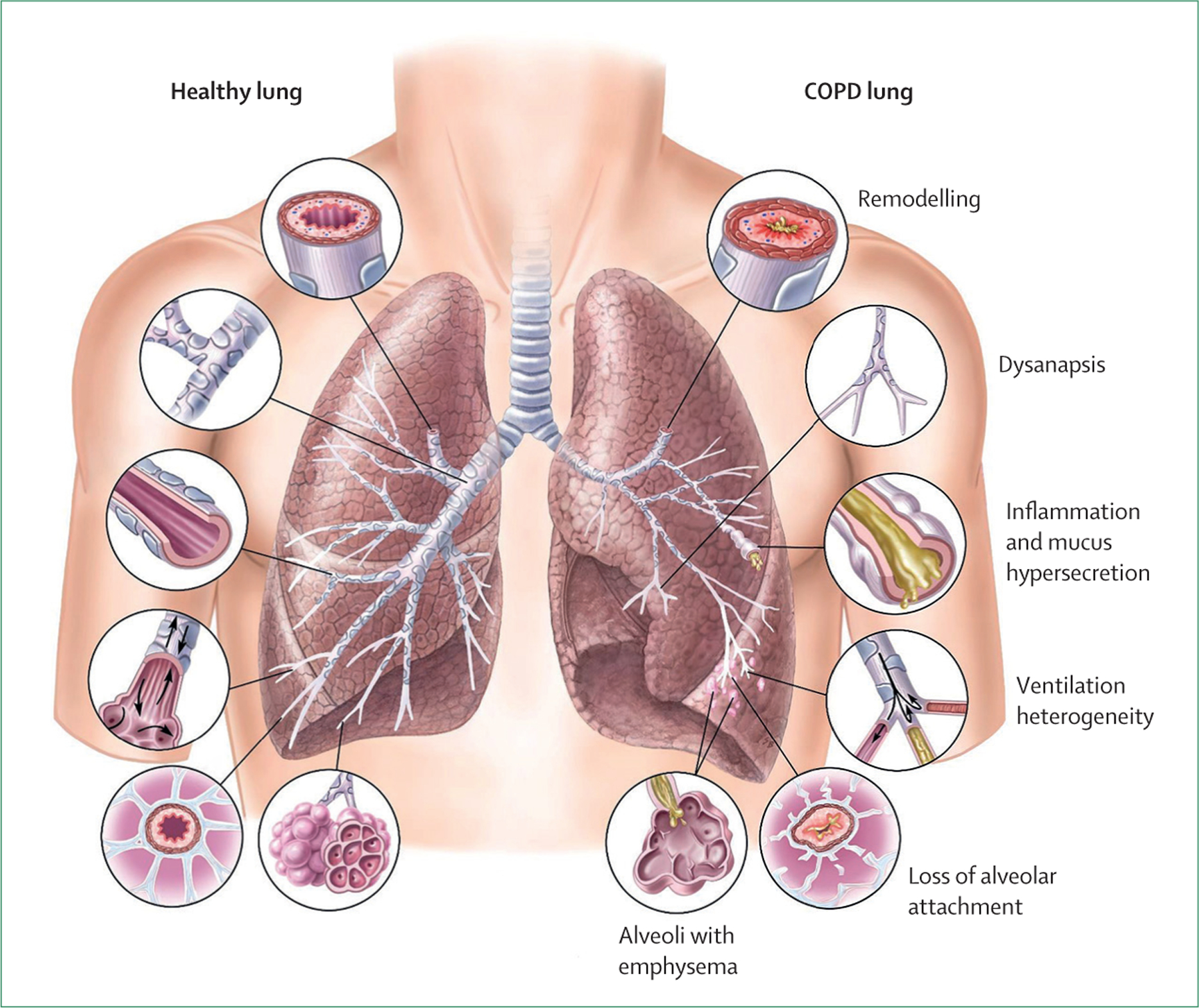
COPD=chronic obstructive pulmonary disease.
Airway remodelling is characterised by differences that include the fractional area of elastic fibres, as well as variations in the extent of epithelial changes, such as goblet-cell metaplasia and squamous metaplasia.167 Moreover, airway smooth muscle content is increased in COPD, albeit to a lesser extent than in asthma, but is highly variable between patients. Emphysema is another hallmark of COPD that is driven by numerous complex pathological changes, including impaired tissue repair and regeneration, but its severity varies substantially between patients. In addition, inflammatory cells lead to apoptosis and degradation of the extracellular matrix due to the release of proteinases, oxidants, and cytotoxic products but the intensity of these effects also varies. Collagen degradation is upregulated, particularly during exacerbations, and the non-uniformity of this degradation within the lung affects disease presentation.168,169
Although an abnormal inflammatory response is central to COPD, the types of inflammation are heterogeneous: patients can have neutrophil-dominant, eosinophildominant, or pauci-inflammatory phenotypes based on sputum analyses. Treatment guidelines5 suggest that a subgroup of patients with high circulating eosinophil counts respond better to inhaled corticosteroids, although there are challenges in the identification of a threshold, because a large proportion of patients with COPD have varying blood eosinophil counts.170 A study171 combining data from 11 clinical trials found that blood eosinophils are not a reliable biomarker for predicting future COPD exacerbations.
Although expanded knowledge of the importance of eosinophils could prove useful, most exacerbations are mediated by neutrophilic inflammation. There is increased presence of neutrophils in the airways of patients with COPD, and in biopsies taken from both the airway wall and parenchyma.172 The degree of neutrophilic inflammation correlates with important clinical outcomes such as the rate of lung function decline and exacerbation events.173
The microbiome is important for lung health and perturbations in the normal microbiome, even in early life, can predispose individuals to COPD.174 The microbiome is shaped shortly after birth and is affected by important early-life events such as antibiotic exposure and respiratory infections. Resident microbiota can also affect the risk of subsequent childhood respiratory infections, particularly viral infections, and childhood respiratory infections can in turn affect the resident microbiota. Thus, the microbiome is associated with many of the key factors influencing the development of maximal lung function in early adulthood and could potentially modulate the immune response upon exposure to cigarette smoke or environmental pollution, thereby contributing to the development of COPD. In stable COPD, patients with neutrophil-dominant inflammation mainly had a balanced bacterial microbiome, but some subgroups’ microbiomes were dominated by either Haemophilus, Moraxella, or Streptococcus species.175 Loss of microbial diversity, as measured by the richness and balance of microbial communities, is noted in patients with COPD with lower FEV1, more frequent exacerbations, and increased mortality compared with healthy controls or smokers without COPD.176 In patients whose inflammatory profiles are not neutrophil dominant, bacterial microbiomes are balanced during stable COPD.177 Fungal sequences have also been detected in sputum samples from patients with COPD, and increased fungal diversity and load is associated with frequent exacerbations.178 Treatment of COPD with inhaled corticosteroids affects the lung microbiome and the expression of epithelial genes involved in tight junction promotion, interleukin 17, and tumour necrosis factor inflammatory pathways.179
One suggested approach to simplify and manage the heterogeneity of COPD is to remove all diagnostic labels such as COPD, asthma, and bronchitis, and to focus instead on individual therapeutic goals and clinical, physiological, or biological characteristics that are quantifiable with biomarkers (commonly referred to as treatable traits).180 This approach has proven useful in some settings, but its full validation and implementation are dependent on further evidence.
A proposal for diagnosis of COPD
As discussed, diagnostic guidelines do not account for the heterogeneity of the pathophysiological processes underlying COPD, which are in turn driven by the genetic determinants and cumulative lifelong environmental exposures leading to the disease. The elimination of COPD will require both prevention of new cases and cure for those with existing disease. Thus, a novel approach to diagnosis that moves beyond spirometric criteria alone (which identifies disease only after irreversible airway damage has already occurred) is required. Current diagnostic approaches substantially underestimate the burden and morbidity of COPD and potentially prevent the development of curative treatments. Although spirometry has a long, proven history for many purposes, including epidemiological surveillance and drug development, there is clear evidence of pronounced lung disease in those without spirometric airflow limitation that might be more amenable to treatment, including reversal of disease and cure.
Lowe and colleagues181 used four criteria to categorise patients as having possible, probable, or definite COPD. This approach increased the COPD population by 36%.181 The four criteria used were exposure (only to tobacco smoking), CT (≥5% emphysematous involvement, average wall thickness for a hypothetical airway with a 10 mm lumen perimeter ≥2·5 mm, or 15% air trapping), symptoms (modified medical research council scale score ≥2 or chronic bronchitis defined as chronic cough and phlegm), and spirometry (FEV1 <80% of predicted or a FEV1 to FVC ratio <0·70). Lowe and colleagues’ diagnostic criteria are an advance on standard guidelines, but several aspects can be further improved. First, COPD is a lifelong disease, and it is crucial to account for early life and other events that impair the ability to attain maximal lung function, such as prematurity, predisposing genetics, recurrent childhood respiratory illnesses, and other infections such as HIV. Second, additional emphasis on exacerbations or acute worsening of respiratory symptoms is essential. In the group of patients with exposure only (ie, the reference group), 1% of the participants had severe exacerbations, but no data were published for mild or moderate exacerbations, which should also be considered symptoms of COPD. Therefore, the reference group already includes participants who belong in a different group (ie, the exposure plus symptoms group—possible COPD). Third, persistent airflow limitation was assessed using spirometry only, and we believe that a broader definition of airflow limitation is required.
On the basis of our current understanding of the heterogeneity of COPD, a range of exposures, symptoms, and physiological, radiographic, and pathological abnormalities could be considered in a new diagnostic approach (panel 2), although not all of the supporting testing would be required in all patients. We propose a model based on this broader range of factors that is useful and feasible in primary-care settings (including low-income settings), and also applicable in tertiary and quaternary care centres where advanced testing is available (figure 11). We advocate that the initial emphasis in the assessment process should be on identifying individuals with symptoms of cough, dyspnoea, and recurrent events concerning for exacerbations. We argue that a diagnosis of COPD should be considered even in individuals with minimal symptoms, who can still have clinically significant pathological changes. We recommend a personalised assessment strategy based on a thorough history and physical examination to identify specific high-risk features. The proposed diagnostic algorithm also suggests a category of probable COPD based solely on the presence of substantial respiratory symptoms, although this diagnosis could be complemented with inexpensive pulmonary testing, including peak flow measurements.
Figure 11: Proposed diagnostic algorithm for COPD.
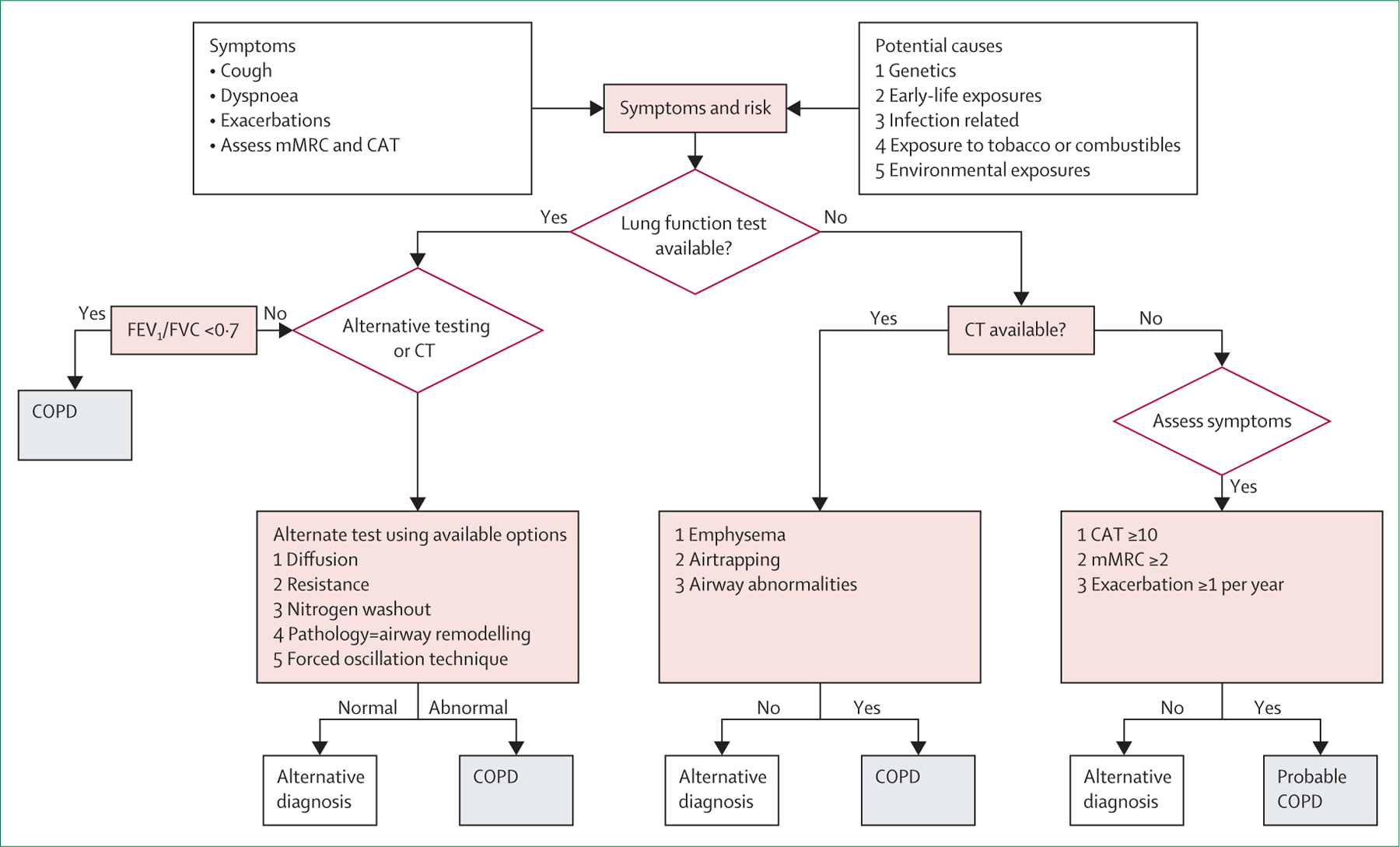
COPD=chronic obstructive pulmonary disease. mMRC=modified medical research council scale. CAT=COPD assessment test. FEV1=forced expiratory volume in 1 s. FVC=forced vital capacity.
Although the proposed algorithm cannot be fully supported by available data, we believe that it will improve recognition of early disease, increase awareness of risk factors, and foster research around each of the COPD types. As previously discussed, these types refer to the main causative factors responsible for COPD and might be linked to specific, targetable pathophysiological processes. The emphasis on disease types allows for more targeted screening of high-risk individuals and increases recognition of underappreciated risks. The type-based approach and the diagnostic testing criteria will need to be applied sensibly and will require clinical judgment. For instance, the level of exposure to air pollution that would put someone at risk for type 5 COPD is debatable, because there is no definitive safe threshold. If data were available to estimate the risk of disease against a reference standard, the development of a weighted model could refine risk stratification, but such sophisticated models are yet to be developed. However, we still believe that clinicians can use the proposed model to approach potential disease.
Utility of symptoms and types in diagnosis
We believe that identification of symptoms is essential for prompting assessment and as a diagnostic tool even in settings where lung function testing or imaging are unavailable. Our model focuses on the identification of individuals with cough, dyspnoea, and a history of exacerbation events. After recognition of symptoms, we advocate for assessment of symptom burden potentially related to COPD using the modified medical research council scale or the COPD assessment test. The Commission places a high value on the recognition of symptoms, which are prognostically important and could themselves define disease. We examined the usefulness of symptoms alone, and in combination with spirometry and CT data, to predict exacerbation risk in 10 000 patients enrolled in the COPDGene study (appendix p 6). We found that models including only demographics and symptoms did well in predicting exacerbation events and that the addition of spirometric or CT data only moderately improved the model (figure 12).
Figure 12: Receiver operating characteristic curves (A) and calibration curves (B) for prediction of exacerbations of chronic obstructive pulmonary disease.
Analyses that include lung function use pre-bronchodilator measurements. Higher AUC indicates better discrimination, whereas lower ICI indicates better calibration. For more information, see the appendix (p 6). AUC=area under the receiver operating curve. ICI=integrated calibration index.
These models were designed to assess exacerbations and not as a diagnostic tool, but this information highlights the importance of assessing symptoms along with the appropriate exposures in identifying clinically significant COPD. Although we do not advocate that objective physiological or imaging examinations be routinely omitted, our data suggest that in low-income settings, the combination of demographics, smoking history, and respiratory symptoms can be used to prognosticate and potentially select patients for treatment. Inexpensive pulmonary testing, including peak flow measurements, could also be included in a diagnostic algorithm for low-income areas.
As discussed previously, there are crucial causative factors other than exposure to tobacco smoke that are highly predictive of COPD risk. The establishment of COPD types promotes increased recognition of these factors by researchers and clinicians. It is also increasingly evident that these types might be driven by different pathophysiological processes, or by varying severity of shared processes, and that each can result in clinical COPD (figure 13). Although more research is needed in this area, we believe that this framework will support the accelerated development of therapies that target the underlying pathophysiology central to specific types of COPD.
Figure 13: Association between proposed COPD types, endotypes, and phenotypes.
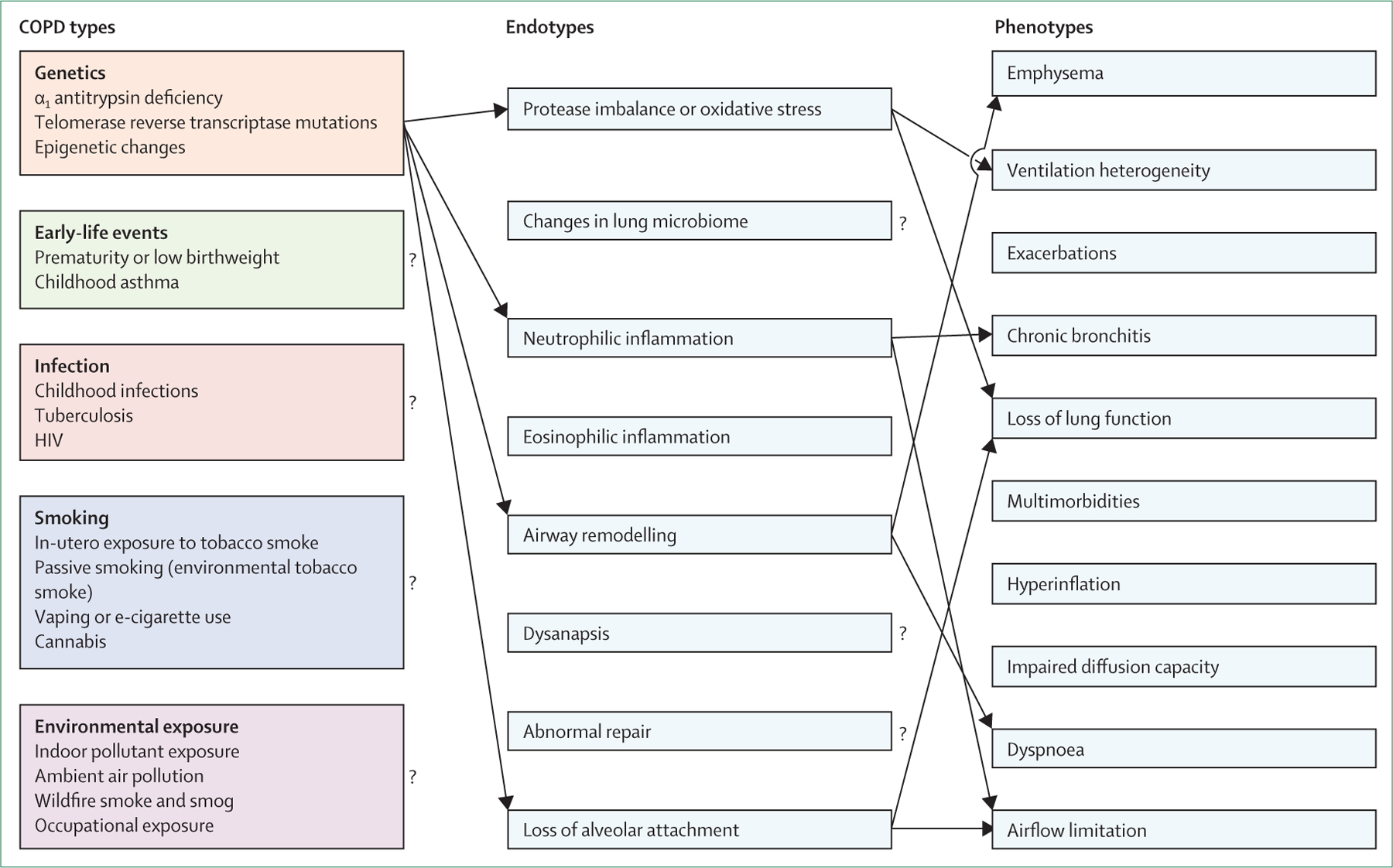
The proposed COPD types probably result from varying molecular mechanisms (endotypes) that in turn result in the observable phenotypes of the disease.
COPD=chronic obstructive pulmonary disease
Diagnostics in symptomatic people with COPD types
Extensive efforts have been made to drive the uptake of spirometry in diagnosis of COPD. Use of spirometry is common practice, and guidelines recommend spirometric measurements of lung function before and after administration of a short-acting bronchodilator to diagnose COPD (based on the post-bronchodilator measurement). However, use of post-bronchodilator testing to establish the presence of airflow limitation adds little predictive utility beyond the information gained from pre-bronchodilator testing, and indeed, no other chronic condition requires a similar diagnostic approach.182 Bronchodilator response was historically thought to help to differentiate asthma from COPD; however, it is well established that not all people with asthma have fully reversible expiratory airflow limitation and that many patients with COPD have a noteworthy bronchodilator response. Therefore, bronchodilator response is neither sensitive nor specific enough to differentiate asthma from COPD, does not stratify COPD severity, and has debatable predictive value for clinical outcomes.183 The Commission’s stance is not that spirometry is good or bad, but rather that spirometry is too insensitive and that new methods (or combinations of methods) are required to adequately detect limitation of expiratory airflow and pathological correlates and to diagnose COPD (figure 11).
Alternative lung function testing
Substantial pathological changes have already occurred before limitation of expiratory airflow can be detected by spirometry. Decrements in lung function have clear prognostic value through developmental stages of life and early adulthood, even in the absence of spirometric evidence of airflow limitation. Current diagnostic algorithms fail the population of patients with COPD who do not have detectable airflow limitation. Research in observational cohorts2,51 has showed that low peak lung function or rapid subsequent decline in lung function, or both, is associated with increased risk of future lung disease. Low peak lung function in youth also predicts future extra-pulmonary conditions, such as heart disease, cachexia, muscle loss, depression, and renal disease.184,185 Although the reasons for these relationships are not fully understood, their existence strongly supports the widespread implementation of routine lung function testing in young adults. Up to now, lung function has been based mainly on spirometric measurements. Some lung function tests that show promise as potential tools for identifying individuals with COPD include DLCO, resistance testing, nitrogen washout, and forced oscillation techniques (FOT). Unfortunately, none of these testing modalities have been studied as extensively as spirometry. As such, the ideal cutoffs for distinguishing normal from abnormal lung function has yet to be established for these tests.
Specific effective airway resistance (sReff) reflects the patency of airways. In a study with nearly 1000 participants,186 sReff differentiated asthma from COPD. In another study (unpublished), sReff distinguished current smokers or ex-smokers from never smokers without spirometric airflow limitation (ie, a normal FEV1 to FVC ratio): even though the current or former smokers did not have spirometrically defined obstruction, they still had a higher sReff than non-smokers (figure 14; appendix pp 6, 7). As expected, patients with spirometrically defined airflow limitation have a higher sReff than those without limitation. The increase in sReff evident in current or former smokers with a preserved FEV1 to FVC ratio could be used to identify the presence of COPD at an earlier stage than spirometry.
Figure 14: Specific effective airway resistance in patients with COPD with and without airflow limitation (A); and in current and former smokers vs never smokers without airflow limitation (B) and with airflow limitation (C).
Specific effective airway resistance is pathological if it is ≥1·2 kPa per s, which is further classified as mild (1·2–2·0 kPa per s), moderate (2·1–4·0 kPa per s), or severe (>4·0 kPa per s). It is higher in current or former smokers than in non-smokers, irrespective of the presence or absence of spirometrically determined airflow limitation.
COPD=chronic obstructive pulmonary disease.
The difference in oscillation resistance at 5 Hz and 19 Hz (R5–19) and area of reactance are the main outcomes measured with FOT, but use of a range of frequencies could improve the sensitivity of the technique.187,188 FOT is not complicated and can be used in children and elderly people. Children with respiratory symptoms have higher oscillation resistance at 5 Hz and R5–19 than children with no respiratory symptoms, oscillation resistance is increased in children exposed to maternal smoking, and R5 may be more sensitive than the FEV1 to FVC ratio for the detection in children of lung damage caused by air pollution.189 A European Respiratory Society technical standards document188 outlines new reference equations to establish pathology. The lung clearance index, which is measured via a multiple-breath nitrogen washout, detects ventilation inhomogeneity in central and peripheral airways. The convection-dependent conductive ventilation heterogeneity index represents ventilation inhomogeneity in pre-acinar airways, and the diffusion-convection-dependent acinar ventilation heterogeneity index represents ventilation inhomogeneity close to the entrance of acinar airways.190 Either calculated Z scores or the absolute values can be compared between patients. The slope of phase 3 of a single-breath nitrogen washout test shows the ventilation inhomogeneity of peripheral airways. In addition to associations with lung function parameters such as percentage predicted FEV1, the ratio of residual volume to total lung capacity, and DLCO, this value was also associated with dyspnoea, exercise-induced desaturation, and exercise capacity.191,192
Another diagnostic tool that merits further consideration is pathological analysis of airway remodelling. Airway remodelling, including hypertrophy of airway smooth muscle cells, thickening of the basement membrane, increased airway inflammation, and pathological changes in extracellular matrix turnover, can lead to persistent airflow limitation.167 In patients with COPD, airway smooth muscle mass increases in the small airways, and squamous epithelial metaplasia and airway wall fibrosis are present.167,193 Basement membrane thickening was thought to be a feature only of asthma but also occurs in 40% of patients with COPD.193 Airway smooth muscle cells contribute to airway remodelling by producing inflammatory cytokines, proteases, and growth factors. An increase in inflammatory cells such as neutrophils and T lymphocytes results in increased secretion of metalloproteinases, elastases, hyaluronidases, and chondritinases, which degrade the extracellular matrix of the lung.194 Elastin is the main target, but collagen degradation is also upregulated and serum fragments of these core components of the extracellular matrix are associated with COPD disease severity, time to exacerbation, and overall prognosis.168,194 COPD is also characterised by neutrophilic inflammation, but a subset of patients have eosinophilic inflammation.195 Patients with COPD who have low blood eosinophil counts also have altered adaptive immunity, as shown by changes in concentrations of IgA, IgG, and IgM in bronchoalveolar lavage samples.196
As with inflammatory bowel diseases, in which endoscopies with biopsies are required for disease diagnosis and assessment of progression, a bronchoscopy, including endobronchial biopsies, should be considered a complementary approach to the diagnosis and characterisation of COPD.197 Induced or spontaneous sputum can also be used for classification of the inflammatory profile of COPD. Eosinophilic inflammation is defined as eosinophils accounting for more than 2·5% of total cells in sputum. Neutrophilic inflammation is defined as neutrophils making up more than 61% of total sputum cells, whereas pauci granulocytic inflammation is defined as eosinophils making up no more than 2·5%, and neutrophils no more than 61%, of the total cells in the sputum sample. A mixed inflammatory profile is defined as eosinophils accounting for more than 2·5%, and neutrophils for more than 61%, of total sputum cells. However, it should be noted that inflammatory patterns can vary over time in COPD.
This expanded set of potential diagnostic tools could help to identify early COPD before spirometric airflow obstruction is detected. This list is not exhaustive, but includes methods for which some supporting evidence has been published. As uptake of our type-based classification of COPD increases, new research will refine the appropriate tools and cutoffs to increase diagnostic precision.
An expanded role for CT
In many settings, CT is routinely available and accessible, whereas spirometric testing can be challenging to obtain, is often interpreted incorrectly, and is insensitive for early disease (even when done correctly). An estimated 85 million thoracic CT scans are done annually in the USA alone, for reasons ranging from routine clinical care to lung cancer screening in high-risk populations.198 Yet these images are not routinely used to assess the presence of chronic lung disease. The addition of routine radiologist or computer-assisted readings of these images could substantially increase detection of COPD, including early abnormalities, and should be immediately implemented. We also advocate that CT-detected emphysema, air trapping, and airway remodelling should be considered diagnostic of COPD, even in the absence of confirmatory spirometry. In places where spirometry is challenging to obtain but CT is available, we similarly advocate that thoracic CT be used to diagnose COPD.
Advancements in CT have led to the development of other novel metrics, including the assessment of dysanapsis and parametric response mapping, which both inform understanding of COPD.199,200 Dysanapsis refers to the mismatch between airway tree calibre and lung growth, which was previously believed to be a physiological aberration without clinical implications. However, dysanapsis is now known to be associated with relevant outcomes and spirometrically defined COPD in older adults (mean age 63–69 years).199 Parametric response mapping is a CT biomarker that compares functional small airway disease and emphysema with healthy parenchyma, thus helping to identify terminal bronchiole pathology. These measures, along with others in development, could prove useful in defining disease mechanisms in patients with different types of COPD, including in those with very early disease, and could ultimately lead to novel preventive and therapeutic strategies. We should embrace rather than resist this potential.
In summary, we propose a model for diagnosing COPD that captures the heterogeneous clinical and pathophysiological characteristics of the disease, reflects the importance of a range of risk factors, and expands diagnostic criteria beyond spirometry.
A crucial moment in COPD: revisiting exacerbations
Exacerbations of COPD are a major cause of morbidity, and account for more than 1 million hospitalisations annually in Europe.201 Reducing and ultimately eliminating exacerbations is thus a crucial step on the pathway to elimination of COPD. Substantial progress in understanding of exacerbations has been made in the past few decades, but therapeutic approaches have not advanced in tandem. We aim to offer a new way forward to catalyse progress.
What is in a name?
In a European study,202 411 (65%) of 633 patients never or rarely used the term “exacerbation” when speaking with a nurse or physician, and physicians were unaware of its morbid implications. This problem is related to the term itself, the lack of patient and physician education about how to recognise exacerbations (and their importance), and difficulties in defining what constitutes an event. Patients with COPD and health-care providers often ignore or minimise increases in respiratory symptoms, which leads to underinvestigation of potential exacerbations and ultimately to under-reporting, underdiagnosis, and undertreatment.203 A survey supported by the European Lung Foundation showed that 404 (64%) of 633 patients with COPD think that “crisis” is an appropriate description of how they feel during an exacerbation.202 In the Netherlands, COPD exacerbations have been renamed COPD lung attacks; the term was introduced in a national action plan that also included an integrated care plan to reduce hospitalisations due to lung attacks and provided guidelines for diagnosis and treatment.204
What is in a definition?
Although many patients recognise exacerbations when they happen, an operational definition that can be used in both clinical care and research has proved challenging. An exacerbation of COPD is defined as “a sustained worsening of the patient’s condition, from the stable state and beyond normal day-to-day variations that is acute in onset and may warrant additional treatment in a patient with underlying COPD”.5 Unfortunately, this definition captures none of the underlying processes driving the event, does not include subtyping or the use of biomarkers to categorise the event, and can lead to misdiagnosis and misinterpretation, hindering advancement in the field. Novel approaches have been proposed6 but none have been widely adopted. By contrast, the definition of an acute myocardial infarction has undergone four revisions during the past decade and includes both objective clinical and biochemical criteria and five defined subtypes.205 The definition of an exacerbation of COPD includes five terms that are at best ambiguous and at worst misleading (table 4). Furthermore, event-based and symptom-based definitions of exacerbations are not closely associated. The Anthonisen classification system has been used as an alternative definition for COPD exacerbations, but these criteria were created to capture symptoms of an acute exacerbation of chronic bronchitis that would respond to antibiotics as part of a trial design.206
Table 4:
Ambiguous terms used to define exacerbations of chronic obstructive pulmonary disease
| Ambiguity | |
|---|---|
| Sustained | What duration of symptoms defines “sustained”? |
| Worsening | Which components of disease are getting worse? |
| Beyond | Occurring after how much time? |
| Acute | How quickly does a symptom have to emerge to be judged “acute”? |
| Might warrant | Does it require or not require treatment? |
We propose that exacerbations be defined as an augmentation of the pathophysiological process underlying COPD in individuals with increased respiratory symptoms after exclusion of alternative diagnoses. The uncertainties in identifying and characterising an exacerbation are not only patient-specific but also can be affected by cultural and regulatory differences across countries and health systems, which can have implications for both practice and clinical trials. In the IMPACT study207 (figure 15), for example, rates of exacerbation and pneumonia varied widely according to local definitions, standards of management and reimbursement, and national health-care organisations. In addition to the limitations of the definition of exacerbation in general, characterisation of exacerbation severity is based on the pharmacotherapy used to treat the event and the treatment setting. This approach is problematic because it is dependent on decision making by the provider and the health-care setting instead of the underlying disease process. It is therefore essential that the definition and severity assessments of exacerbations are rewritten to improve understanding of their epidemiology and to personalise management.
Figure 15: Maps of the incidence of moderate or severe exacerbations (A) and pneumonia (B) in patients with chronic obstructive pulmonary disease.
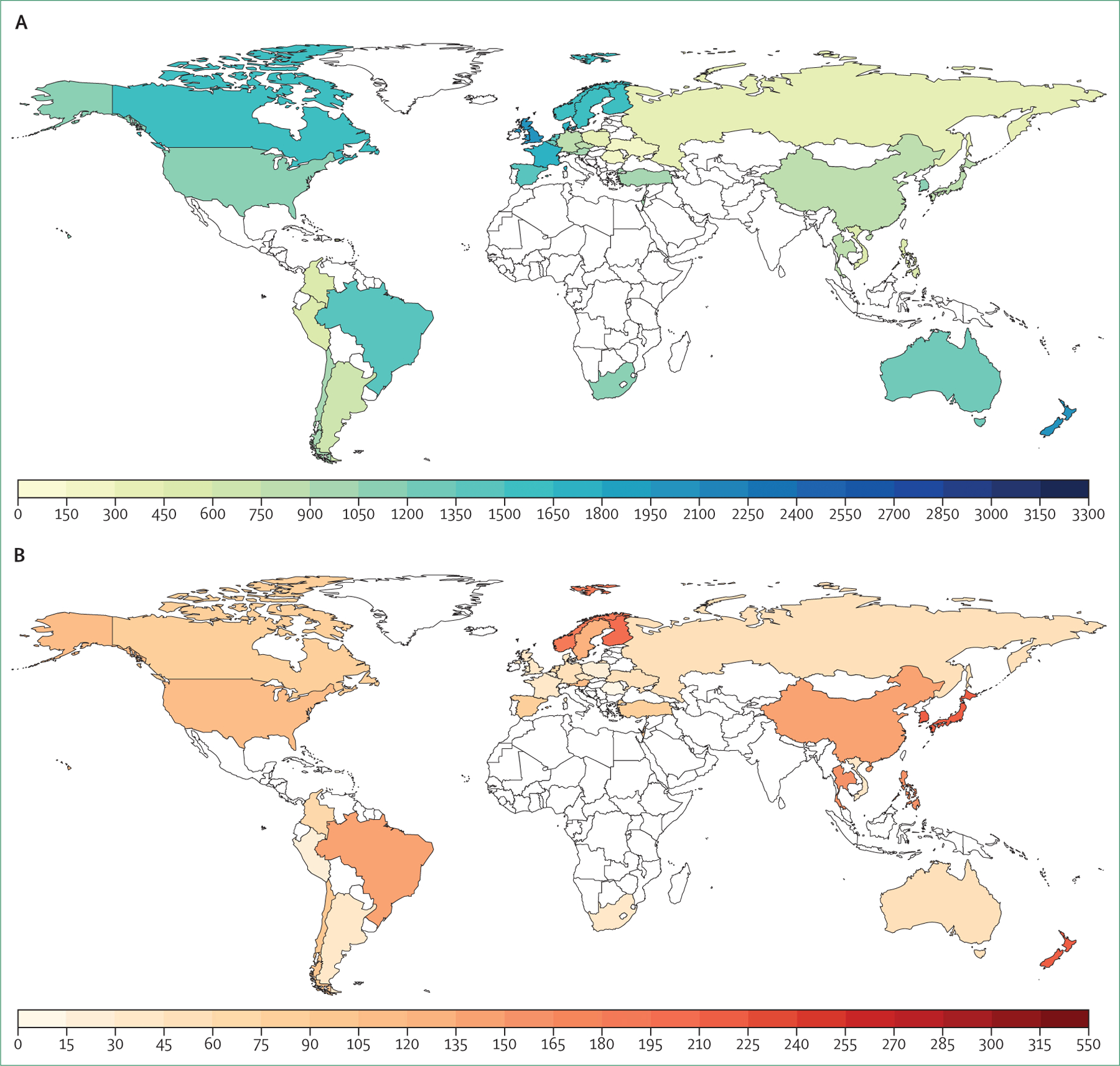
Data sourced from the IMPACT study.207 This figure is redrawn with permission from GlaxoSmithKline.
Why revisit diagnostic criteria for exacerbations?
Classically, a COPD exacerbation is a diagnosis achieved by excluding other possible causes for increased respiratory symptoms (eg, pulmonary embolism, heart failure, pneumothorax). However, there are no clear standardised recommendations for how to exclude these conditions in guidelines, and misdiagnosis of an exacerbation at the time of increased respiratory symptoms can lead to ineffective treatment. For example, the estimated prevalence of pulmonary embolism in patients with COPD who report worsening symptoms of dyspnoea is as high as 20%.208 In addition, patients with COPD can experience late detection of a cardiac event, even though 25% present with increased troponin concentrations indicative of myocardial injury.209 Failure to recognise and treat pulmonary embolism and myocardial infarction in patients with COPD has deleterious consequences. Additionally, some URTIs are associated with decreased quality of life, spirometric impairment, and increased scores on the COPD assessment test and the modified medical research council scale,210 and yet are not classified as exacerbations in the current guidelines.
Comprehensive study of patients during an exacerbation can be difficult. Many mild events go unreported. Most moderate events are treated at home and thus blood, sputum, or other biological specimens are not obtained. Some patients with exacerbations can also be very ill, which can limit collection of detailed respiratory, physiological, biological, or radiological data. The resulting inability to appropriately classify the event hinders identification of potential triggers and the development of novel treatment targets.
The frequency of severe exacerbations (ie, requiring hospital admission) has decreased by roughly 50% since the beginning of the COVID-19 pandemic.211 This reduction is probably attributable to social isolation and distancing, increased hand hygiene, and particularly the use of facemasks, which decisively decrease viral transmission and suggest that the main underlying cause of exacerbations is directly or indirectly virus-related. Assessment of the continued use of these measures in high-risk patients with COPD will be important after the resolution of the pandemic. For many years, exacerbations have been thought to be preceded by a URTI. A 2019 study212 showed that URTIs led to exacerbation in 16% of cases, and for about a third of these events the same virus was detected at both the onset of the URTI and during the exacerbation. However, establishing whether a microbe is the causative agent for an exacerbation and the importance of targeting that microbe as part of treatment is complicated. The tools used for microbial detection are limited by slow turnaround times (3 days for sputum culture and 48 h for qualitative viral detection), which reduces their usefulness. Rapid PCR analysis is much faster and has consistently increased detection of microbes compared with standard culture techniques, but not all detected pathogens are clinically relevant—many patients with COPD have chronic infections with associated inflammation.213 There is therefore no practical way to establish if the detected microbe is driving changes in airway resistance or hyperinflation or if treatment targeting that microbe will improve physiology. Furthermore, no antiviral medications have been shown to be effective in the treatment of exacerbations.
The degrees to which inflammation (pulmonary and systemic), airway resistance, changes in airway calibre, or neural respiratory drive contribute to an exacerbation are not fully elucidated. Exacerbations are clustered according to the underlying predominant inflammatory profile.214 These exacerbation clusters are difficult to distinguish clinically but have been defined on the basis of unsupervised statistical analysis of airway mediators and include a pro-inflammatory endotype (bacteria-predominant), a T-helper-2 (Th2) endotype (eosinophil-predominant), a T-helper-1 (Th1) endotype (virus-predominant), and a low-inflammatory profile (pauci inflammatory). Identification of these endotypes of exacerbation is possible based on sputum markers (interleukin 1β) for pro-inflammatory endotypes and blood markers for Th2 and Th1 endotypes (percentage eosinophil count and CXCL10, respectively). The lack of rapid sputum and blood assays limits progress in profiling exacerbations on the basis of these biomarkers. However, the finding that some of these endotypes (eg, pro-inflammatory bacteria-associated and Th2-eosinophilic-associated exacerbations) are predictable from a stable state provides insight into underlying mechanisms and potential treatment planning.25,215
Identification of a single biomarker to inform treatment of an exacerbation of COPD has proven very difficult (although the fact that C-reactive protein and procalcitonin concentrations remain low in exacerbations that do not require antibiotic therapy has facilitated antibiotic stewardship).23 It is increasingly likely that a panel of biomarkers aimed at detecting both inflammation and bacterial or viral infections will prove useful for the identification of the underlying exacerbation cause.
The systemic ramifications of exacerbations are poorly characterised. An increase in lung inflammation during an exacerbation could have important spillover effects on systemic inflammation. Surges in systemic inflammation could in turn have important negative consequences for the heart, peripheral vasculature, and gastrointestinal, renal, musculoskeletal, and bone marrow systems. Additionally, the effects of exacerbation treatments on non-pulmonary organ function are poorly characterised. Understanding the ramifications of the systemic consequences of these acute events could expedite the development of pulmonary and non-pulmonary therapeutic targets.
New diagnostic criteria for COPD exacerbations
Exacerbations should be framed as an increase in respiratory symptoms due to an augmentation of the pathophysiological process underlying COPD in the absence of an alternative diagnosis. To operationalise this, we suggest that an exacerbation should be defined as an increase in cough, dyspnoea, or sputum production and at least one of an increase in airflow limitation or ventilation heterogeneity, an increase in airway or systemic inflammation, or evidence of bacterial or viral infection, in the absence of evidence of acute cardiac ischaemia, congestive heart failure, or pulmonary embolism.
Bacterial exacerbations requiring antibiotic treatment occur across a continuum that includes infection limited to the airways and varying degrees of parenchymal invasion that often meet diagnostic criteria for pneumonia. In fact, pneumonic exacerbations occur in 20–36% of patients with COPD.216 However, not all such episodes of parenchymal invasion are detectable by conventional chest radiography and chest CT might be required. Use of CT to detect parenchymal infection in patients with exacerbations should be considered to improve diagnostic precision and potentially guide alternative treatments, although we acknowledge that CT is unavailable in some health-care systems (and that costs can limit use even if it is available).
Clinicians responsible for management of exacerbations need to specify a temporal and causal relationship. A systems biology approach, which captures the effects on the lungs and other organs as well as the potential pathogens or other triggers implicated, should be advocated. All clinicians managing patients with COPD should establish the inflammatory profile of exacerbations and at a minimum relate this profile to eosinophilic or neutrophilic inflammatory subtypes and confirm whether it is bacterial dependent or independent.
To define and endotype a COPD exacerbation and delineate the underlying cause, a standardised investigation panel is required. We have proposed one such approach (table 5), for which we strongly support prospective testing and iterative improvement. Patient follow-up after an exacerbation should include reassessment of the clinical, physiological, and biological features of the event and should occur within a reasonable timeframe. We acknowledge that resources vary across countries and that the proposed panel will not be feasible in all health-care systems.
Table 5:
Standard investigations when patients with chronic obstructive pulmonary disease seek medical attention for suspected disease exacerbation
| Essential | Dependent on context or presentation | |
|---|---|---|
| Inflammation and infection | ||
| Full blood count | X | |
| C-reactive protein or procalcitonin | X | |
| Airway microscopy and culture | X | |
| Airway cytology (eosinophils) | X | |
| Airway molecular testing for pathogens | X | |
| Fractional exhaled nitric oxide | X | |
| Hypoxia, hypercapnia, and metabolic status | ||
| pH of venous blood gas | X | |
| Arterial oxygen saturation | X | |
| General physiology | ||
| Breathing rate | X | |
| Electrocardiography | X | |
| Lung function | X | |
| Imaging | ||
| Chest radiography, ultrasonography, or CT | X | |
| Systemic measurements | ||
| D-dimer | X | |
| Renal function | X | |
| Troponin | X | |
| B-type natriuretic peptide | X | |
Assessment of exacerbation severity
The severity of an exacerbation should not be judged in terms of the type of treatment or the setting where treatment is provided, but rather on the basis of an objective categorisation of the degree of clinical, biological, and physiological deterioration. The Commission proposes to eliminate the definitions for mild or moderate exacerbations and to instead diagnose only severe exacerbations or non-severe exacerbations. Panel 3 details our proposal for the diagnostic and severity criteria for a severe exacerbation.
Telemedical management of deteriorations
During the COVID-19 pandemic, many clinics used telemedicine services to care for patients with COPD to comply with social distancing recommendations and prevent spread of infection to a high-risk population. Telemedicine allows medical personnel to reach patients with poor access to care, particularly in underserved populations, and it has the potential to reduce health-care costs. When patients experience an increase in respiratory symptoms indicative of an exacerbation, they frequently contact health-care practitioners by telephone. Evidence suggests that telemedicine-based symptom reporting promotes early detection of exacerbations,217 yet there is no clear guidance on the management of patients who report exacerbation symptoms by phone. Although most telemedicine research focuses on high-income countries, telemedicine could have a greater effect in LMICs, where resources including health-care facilities and health-care providers can be scarce. Telemedicine has been used in LMICs to treat hypertension and diabetes, and to diagnose post-operative infections.218–20 Soon after the national COVID-19 lockdown in India, use of telemedicine rapidly escalated: the government-provided telemedicine platform logged 3 million consults in 7 months.221 For patients with COPD who live in LMICs, telemedicine services could aid in the management of stable disease and the diagnosis and treatment of exacerbations. The mandatory diagnostic tools needed to assess an exacerbation include an assessment of vitals (respiratory rate and oxygen saturation), chest imaging, and blood tests (full blood count and measurement of C-reactive protein, procalcitonin, and D-dimer concentrations; table 5). To eliminate health disparities and move towards equitable access to care globally, efforts should be made to make these diagnostic tools readily available universally. This access could be accomplished through point-of-care testing or mobile diagnostic units that have basic imaging and laboratory capabilities. We suggest a COPD management algorithm, which incorporates the use of telemedicine to assess patients with suspected acute exacerbations of COPD (figure 16). Although the algorithm does include in-person appointments, if there are barriers to in-person care or times when outpatient visits are limited (such as in the COVID-19 pandemic), video virtual visits can replace in-person visits. Many examination findings that would prompt an emergency department visit can be assessed by visual inspection over camera.
Figure 16: Flowchart to assess the status of patients with chronic obstructive pulmonary disease with new or worsening respiratory symptoms who contact medical personnel by telephone.
*Concerning symptoms. †Alarming symptoms. ‡Mandatory diagnostics include respiratory rate, oxygen saturation, chest image, full blood count, and measurement of C-reactive protein and D-dimer.
Recurrent exacerbations and disease control
As with many chronic diseases, COPD can be classified as controlled or uncontrolled. Management of most chronic diseases is based on monitoring of intermediate outcomes—eg, blood pressure and hypercholesterolaemia in heart disease, glycaemia (as indicated by glycated haemoglobin A1C) in diabetes. Identification of the equivalent meaningful intermediate endpoints in COPD is crucial because so far the disease has been classified only as stable or exacerbated. A patient with controlled COPD experiences mild or no symptoms, no effects on quality of life or exercise capacity, no exacerbations, and no decreased life expectancy. The term uncontrolled COPD refers to progressive or prolonged respiratory symptoms with or without recurrent exacerbations that are associated with reduced quality of life and decreased life expectancy. The patient group now referred to as frequent exacerbators constitute part of the uncontrolled COPD population, but so do patients with persistent symptoms and impaired health status despite maintenance treatment. Patients with uncontrolled COPD should have their therapeutic regimens intensified. Additional pharmacological and non-pharmacological treatments should be considered, and there should be less acceptance of ongoing symptoms.
In patients with chronic airway disease (including asthma and COPD), day-to-day lung function can vary.222 In the BIOAIER cohort,222 patients were stratified into four clusters on the basis of FEV1 fluctuation over 1 year. Fluctuation clusters were partly independent of clinical diagnosis; however, COPD was mainly associated with the most severe cluster (ie, the most severe fluctuation phenotype). Strong differences in mechanical impairment of the lung (eg, airway obstruction, reversibility, diffusion capacity) independent of type 2 inflammation and substantial differences in exacerbation frequency were noted among the four clusters.222 Consistent with these findings, a study223 in patients with severe asthma indicated that airway remodelling as detected by CT was associated with acceleration in age-related lung function decline and with exacerbation frequency. Similar findings are likely to be found for COPD. A lung mechanical model was proposed to explain how alterations in lung mechanics and remodelling can lead to increased exacerbation frequency independent of the underlying inflammatory process.224 The model predicts that altered structure–function interaction could contribute to a self-perpetuating persistent exacerbation pattern, which almost certainly requires novel therapeutic strategies.
Difficulties in treating COPD exacerbations
Randomised clinical trials of treatments for COPD exacerbations have largely focused on pharmacotherapy with antibiotics, bronchodilators, and corticosteroids, oxygen and non-invasive ventilation to tackle respiratory compromise, and pulmonary rehabilitation to tackle both the pulmonary and the systemic consequences.5 Use of bronchodilators, systemic corticosteroids, and antibiotics as the gold standard for treatment of exacerbations is largely independent of the patient’s presentation and the pathobiology driving the event. As a result, there are no specific disease-modifying therapies targeting exacerbations or the contributing pathways, including hypoxic tissue injury, apoptosis, and necrosis. This absence contrasts with the innovative treatments available for acute myocardial or neurological infarctions, which target tissue hypoxia, prevent cell death, and foster cell regeneration.225,226
Disappointingly for patients and health-care practitioners, there are no dedicated efforts by relevant stakeholders to change the acute treatment of an exacerbation. Furthermore, despite poor treatment responses in many patients25 and the substantial burden of harm,227 guidelines continue to advise the use of systemic corticosteroids for most exacerbations, an approach that was first assessed more than 40 years ago.228 Moreover, national and international guidelines and Cochrane systematic meta-analyses229 have stated that no further evidence is needed to establish the role of corticosteroids in exacerbations. Thus, trials of novel treatments have been complicated, because they have to include corticosteroids as standard of care in the comparator group, the efficacy of which is questionable in many cases. The use of self-management strategies, including so-called rescue packs of corticosteroids and antibiotics, is widespread and is associated with overtreatment and no clear evidence of benefit.230 Furthermore, most COPD therapies developed by the pharmaceutical industry and marketed for reducing exacerbations are based on results of short-term clinical trials of inhaled medications directed only at the pulmonary component of exacerbations with narrow patient populations that are enriched to show treatment efficacy but not treatment effectiveness. These respiratory-centric treatments have limited effects on the multisystem pathobiological manifestations of COPD exacerbations.
Outcomes after exacerbations
The inescapable truth is that outcomes after COPD exacerbations that lead to hospitalisation have remained poor worldwide. An index event could be associated with inpatient mortality of roughly 10%, and all-cause mortality of up to 50% 2 years later.5,231 Many factors contribute to these poor outcomes, including inadequate definition and classification of exacerbation severity, poor understanding of the underlying biological processes (augmented by a lack of standardisation in capturing the pulmonary and systemic manifestations), and the scarceness of effective treatments. Outcomes after COPD exacerbations are reported in terms of readmissions, mortality, and treatment failure, which can include readmission and mortality within 30 days of the primary event. These outcomes do not attempt to differentiate physiological, biochemical, or structural changes and instead reflect poor responses to the limited treatments tried at the time of an exacerbation.
The detrimental effect of exacerbations on disease progression and quality of life are known and the reduction and eventual elimination of exacerbations should thus be prioritised. Urgent action is needed to establish a standard assessment at the time of an exacerbation, explore and implement endotype-specific treatments, and monitor physiological and patient-centred outcomes after exacerbations.
How can COPD therapy be more effective?
Currently, COPD care starts after the presence of persistent airflow limitation is spirometrically confirmed in a person with dyspnoea, exercise limitation, cough, or sputum production. Available pharmacological therapies focus on symptom relief, improvement of lung function, and reduction of the risk and frequency of exacerbations. Some data suggest that pharmacological treatments are associated with improvements in survival, but only in subsets of patients with severe disease—and even then some scepticism surrounds the results.47,48 COPD drug development has largely focused on two main therapeutic classes (figure 17): bronchodilators (short-acting, long-acting, and ultra-long-acting, given either alone or in-combination) and inhaled corticosteroids. These treatments are given to different patient groups according to the severity of airflow limitation and previous exacerbation history. Even though COPD is the third leading cause of death worldwide, there are currently only 780 ongoing therapeutic clinical trials related to the disease, compared with more than 41 000 trials of cancer treatments. This lack of research partly explains the fact that only one new drug class has been approved for COPD in the past three decades.232
Figure 17: Timeline of medications entering the market for COPD vs cardiovascular disease.
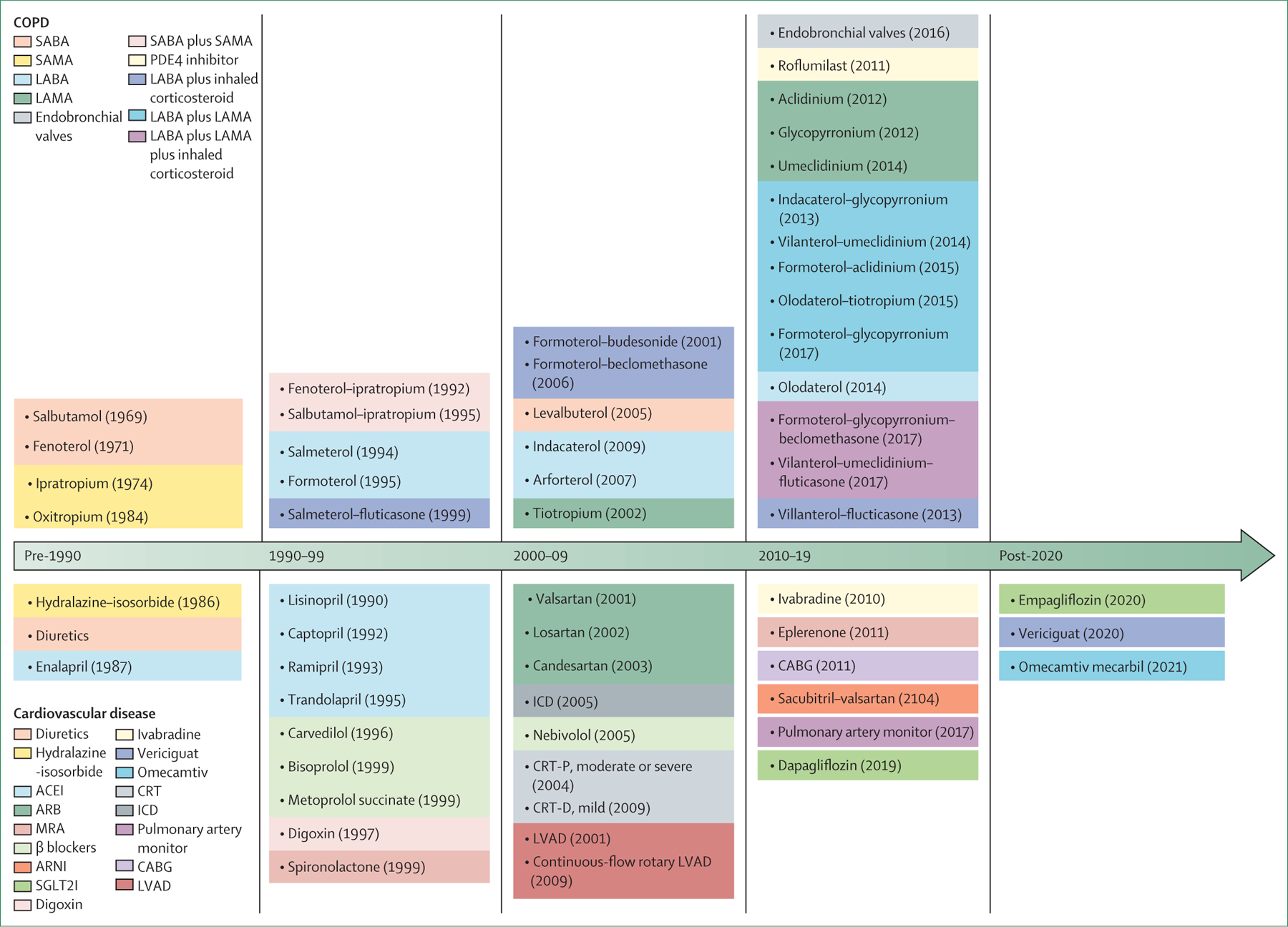
COPD=chronic obstructive pulmonary disease. SABA=short-acting β agonist. SAMA=short-acting muscarinic antagonist. LABA=long-acting β agonist. LAMA=long-acting muscarinic antagonist. PDE4=phosphodiesterase-4. ACEI=angiotensin-converting enzyme inhibitor. ARB=angiotensin receptor blocker. MRA=mineralcorticoid receptor antagonists. ARNI=angiotensin receptor–neprilysin inhibitor. SGLT2I=sodium-glucose linked transporter 2 inhibitor. CRT=cardiac resynchronisation therapy. ICD=implantable cardioverter defibrillator. CABG=coronary artery bypass graft. LVAD=left-ventricular assisted device. CRT-P=cardiac resynchronisation therapy pacemaker. CRT-D=cardiac resynchronisation therapy defibrillator..
A global analysis233 in 2019 also showed that only 31% of molecular targeted therapies under investigation in COPD were classified as first in class, compared with an average of more than 40% across all other indications. The scarcity of new drugs is due to not only an underpopulated pipeline, but also consistent failure to translate promising experimental and early clinical data into positive phase 2 and 3 trials.234,235 A possible explanation for this failure is the unrealistic expectation that one blockbuster drug will lead to lung function improvement or to a reduction in exacerbations in all patients defined primarily by the presence of airflow limitation on spirometry. It is important to remember that COPD is heterogeneous, with many endotypes and phenotypes reflecting diverse pathophysiological mechanisms, and that outcomes other than airflow limitation and exacerbation are needed, especially in early disease.
The reluctance of drug manufacturers to invest in new drugs that target a restricted patient population is perhaps understandable in view of the cost of phenotyping, the complexity of study design given the lack of surrogate markers, and the myopic view of regulatory agencies about endpoints suitable to support drug approvals. This situation must change. The outcomes that define treatment success have a major influence on which drugs will ultimately enter clinical practice. Research into COPD treatments has been dominated by a focus on lung function (especially FEV1) to define the disease, indicate severity and prognosis, and assess the efficacy of treatments. As a result, in most studies done in the past 20 years FEV1 is either the primary endpoint or a key secondary endpoint. In view of this FEV1-centric approach to therapeutic development, it is not surprising that nearly all drugs licensed by regulatory authorities in the USA or EU are bronchodilators. Now is the time to embrace innovative approaches, including imaging endpoints and digital technologies such as monitors that assess physical activity, cough, dyspnoea, and other symptoms. Use of such technologies would allow daily and remote monitoring of COPD to establish how patients feel, function, and survive and thus to establish the effectiveness of a therapy in real-world settings. The five COPD types we proposed could plausibly serve as a further phenotyping tool for grouping patients with similar pathophysiology who are thus likely to respond to similar therapeutic options. Only relevant clinical trials in these COPD types could prove or refute this hypothesis, although similar approaches have proved useful in other conditions, including pulmonary hypertension, heart failure, diabetes, and cancer. In lung cancer, for example, patients are grouped according to the pathophysiology of their disease—ie, non-small cell lung cancer versus small cell lung cancer (and also based on targetable mutations)—and treatment is adapted accordingly. For example, patients with type 3 COPD (ie, infection-related COPD) might have overwhelmingly neutrophilic airway inflammation and therefore benefit more from an anti-neutrophilic medication, whereas those with type 4 COPD (associated with environmental exposure) might have more eosinophilic airway inflammation and would therefore not benefit from such treatment.
A curative approach that reverses organ damage would ultimately be needed to eliminate COPD, but before such treatment is available, patients should at the very least be able to achieve disease control in the short-term. Available therapies have enabled this goal to be partly achieved in some patients. However, pharmacological and non-pharmacological therapies have differing efficacy and many limitations.
Available COPD treatments
The most used therapeutics for COPD include anticholinergics, β2 agonists, inhaled corticosteroids, macrolides, mucolytics, and phospdiesterase-4 inhibitors, all of which have differing mechanisms of action (table 6). Non-pharmacological treatments of COPD complement pharmacotherapy to bring about improvements in symptoms and quality of life, prevent disease progression, and improve survival.5 They include behavioural changes, such as smoking cessation and self-management, pulmonary rehabilitation, comprehensive, integrated care by a multidisciplinary team, surgical intervention, medical devices such as endobronchial valves, home oxygen therapy, non-invasive ventilation, and palliative care (table 7). Unfortunately, many pharmacological and non-pharmacological therapies are not available in LMICs. If they were deployed in LMICs, there could be major benefits at the patient and public health levels.
Table 6:
Established pharmacological treatments for chronic obstructive pulmonary disease
| Mechanism of action | |
|---|---|
| Anticholinergics | Block the muscarinic acetylcholine receptor to reduce bronchoconstriction |
| β2 agonists | Stimulate β2 receptors, thereby increasing cAMP and relaxing bronchial smooth muscle |
| Inhaled corticosteroids | Inhibit the recruitment and survival of inflammatory cells in airways |
| Macrolides | Inhibit bacterial RNA-dependent protein synthesis |
| Mucolytics | Reduce viscosity of mucous secretions; potential anti-inflammatory effects |
| Phosphodiesterase-4 inhibitors | Inhibit phosphodiesterase-4, thereby reducing breakdown of cAMP, which leads to bronchodilation and decreased inflammation |
cAMP=cyclic adenosine monophosphate.
Table 7:
Established non-pharmacological treatments for COPD
| Comments | |
|---|---|
| Smoking cessation | Combined counselling and pharmacotherapy are required to optimise quitting success; pharmacotherapies have differing mechanisms of action |
| Pulmonary rehabilitation | Improves respiratory and other muscle strength, cardiovascular function, mental health, self-efficacy, and adherence to improvements in physical activity and overall quality of life |
| Endobronchial valves | One-way valves placed in a target lobe that cause lung deflation, improving mechanics of breathing and expiratory airflow |
| Lung volume reduction surgery | Resection of emphysematous lung reduces overall lung and thoracic volume, improving mechanics of breathing and expiratory airflow |
| Non-invasive ventilation | Reduces the work of breathing, allowing a larger tidal volume for a given respiratory effort, which improves alveolar ventilation |
| Oxygen supplementation | Ameliorates tissue hypoxia by increasing blood oxygen saturation, thereby improving exercise tolerance |
| Targeted lung denervation | Selective denervation of cholinergic nerves surrounding the main bronchi by targeted administration of radiofrequency waves via bronchoscopy resulting in bronchodilation and reduced mucus secretion |
| Lung transplantation | <5000 procedures annually worldwide; new transplantation guidelines do not favour patients with COPD |
COPD=chronic obstructive pulmonary disease.
What is required to achieve COPD control?
For maximum effect and to ultimately eliminate the disease, new COPD therapies must be precise and target the specific molecular pathways or endotypes responsible for disease expression. These therapies might be more effective versions of the inhaled or orally administered small molecules that are already available, but could also include stem-cell or other regenerative approaches, nanotechnology delivery systems, or gene therapy. The prospects for the development of these treatments (and the associated timeline) are uncertain, although all are under active investigation. Unfortunately, although available treatments improve some important disease manifestations, several needs remain unmet, including the elimination of respiratory symptoms and tissue inflammation, reversal of airway remodelling, lung regeneration, and control of secondary systemic pathology.
Elimination of respiratory symptoms
Cough and sputum production
Mucociliary dysfunction is an important part of the pathogenesis of COPD and dysregulation of airway mucins is a key pathological process.236 Mucociliary dysfunction also results in chronic bronchitis, one of the historical defining features of COPD. Targeting mucociliary clearance has proved challenging, despite evidence that sputum production is linked to both poor quality of life and survival in COPD.236 The only mucoactive drugs in use are cysteine derivatives (eg, carbocisteine, erdosteine) and roflumilast, although the benefits of these drugs are debated and none are available globally. Novel treatments under investigation include bronchoscopic delivery of liquid nitrogen to destroy and denude the epithelial layer (including hyperplastic goblet cells).237 The epithelium regenerates rapidly after the procedure and matures into a more normal epithelium within 3 or 4 weeks. An alternative approach is rheoplasty, in which non-thermal pulsed electrical fields are used to ablate the epithelium, which again regenerates as normal epithelium. CFTR modulators have revolutionised care and outcomes in patients with cystic fibrosis, and studies238–42 suggest that some patients with COPD might also have acquired CFTR dysfunction, which could be amenable to similar treatments. However, it is unclear whether these drugs will progress to phase 3 trials, because their effect on lung function is modest and thus the risks of investing in further development are substantial in the current regulatory environment.242
Dyspnoea
The level of dyspnoea experienced by patients with COPD is a better indicator of health-related quality of life than is FEV1 and does not necessarily correlate to any objective measure of lung function, hypoxaemia, or exercise capacity. Morphine is used as a palliative treatment for chronic dyspnoea in COPD but social stigma and concerns about real and perceived side-effects, including respiratory depression and dependence, limit access to it in clinical practice.243 Novel and safer approaches are needed. Some evidence suggests that acupuncture point stimulation could improve exertional dyspnoea in COPD, perhaps through the release of endogenous opiates.244
Exacerbations
Viral and bacterial infections are common in individuals admitted to hospital with COPD exacerbations. Some studies212,245 have suggested worse outcomes in exacerbations triggered by viruses than in those triggered by bacteria. Rhinoviruses, influenza viruses, and respiratory syncytial viruses are the most common viruses detected, but few antiviral agents are available.246 Some evidence suggests that targeting intercellular adhesion molecule-1 (which facilitates rhinoviral infection of the respiratory epithelium), the immunoproteasome (which governs viral related inflammation), or the interleukin 33–interleukin 13 axis (which drives many viral infections) could be beneficial, but more work is needed.247–49 During most exacerbations that occur after viral URTIs, the original virus that caused the URTI can no longer be detected.212 It is tempting to hypothesise that the virus catalyses inflammatory processes associated with the exacerbation and thus that anti-inflammatory treatments should be explored in addition to antivirals. One strategy is to counteract changes in the extracellular matrix by giving glycosaminoglycans such as hyaluronic acid or heparan sulfate. A placebo-controlled randomised trial250 showed that high-molecular-weight hyaluronan ameliorated respiratory failure in patients with severe COPD exacerbations. Several antivirals are also in the pipeline, including a dry-powder formulation of ribavirin.251 Modest evidence suggests that influenza vaccination could reduce COPD exacerbations.252 COVID-19 vaccines are likely to reduce infections in patients with COPD, but no vaccines are available for rhinovirus or respiratory syncytial virus, which would probably have more substantial benefits.
Stratification of treatment based on Th2 biology and the degree of eosinophilic inflammation at the onset of an acute exacerbation was first investigated in a single-centre placebo-controlled pilot study published in 2012.25 In one group, systemic treatment with prednisolone was given at the onset of an exacerbation (standard treatment) whereas in the other only patients in whom more than 2% of peripheral blood cells were eosinophils were given prednisolone (the other patients in the group were given placebo). The primary outcome of the study, non-inferiority, was met, which suggests that prednisolone prescriptions could be safely reduced by 50%.25 Furthermore, in secondary analysis, patients who received prednisolone without the biological endotype (low blood eosinophils) experienced worsened symptoms and treatment was more likely to be unsuccessful compared with patients with the same endotype who were given placebo.25 The results of a multicentre trial253 of eosinophil-directed systemic corticosteroid treatment in patients hospitalised with severe COPD exacerbations also showed that stratification of treatment based on blood eosinophils reduced corticosteroid exposures without increasing treatment failures. Studies of the use of anti-eosinophil monoclonal antibodies at the time of exacerbation are underway.
Elimination of inflammation
Neutrophilic inflammation
Neutrophilic inflammation is a dominant endotype in COPD and both animal and human data suggest that an imbalance between neutrophil-derived proteases and anti-proteases is central to disease pathogenesis.254 Nonetheless, targeting neutrophilic inflammation therapeutically has been challenging not only in COPD but also in other conditions driven even more clearly by neutrophils, including cystic fibrosis and non-cystic fibrosis bronchiectasis, and no drug has yet been licensed. Drugs targeting neutrophil function—including CXCR2 receptor antagonists, inhaled PI3k delta inhibitors, and oral neutrophil elastase inhibitors—have all been tested in COPD and in general results have been disappointing, partly because of their minimal effects on lung function and an increased risk of infection.255 Encouraging results have been reported in a phase 2 study256 of a novel dipeptidyl peptidase-1 inhibitor, which inhibits neutrophil serine protease activity including neutrophil elastase. It prolonged time to first exacerbation in patients with bronchiectasis but has not yet been tested for COPD.
Eosinophilic inflammation
Eosinophilic lung inflammation in patients with COPD is heterogeneous. Eosinophils can be detected in biopsy specimens, bronchoalveolar lavage samples, and sputum, and often comprise more than 3% of total cells, the cutoff that is widely used to define eosinophilic inflammation.257 Blood eosinophil counts correlate moderately with sputum eosinophil counts,258 which has led to extensive investigation of eosinophilic inflammation as a prognostic biomarker and therapeutic target. Response to inhaled corticosteroids can be predicted on the basis of blood eosinophil counts: greater reductions in exacerbations are noted in patients with higher eosinophil counts.259 The licensing of anti-interleukin-5, anti-interleukin-5-receptor, and interleukin-4–interleukin-13-targeting monoclonal antibodies for asthma has led to interest in the potential effectiveness of these treatments in the eosinophilic endotype of COPD.260,261 Trials262 done to date have not shown consistent reductions in exacerbations, although post-hoc analyses suggest that a subgroup defined by more frequent previous exacerbation and higher eosinophil counts might be more likely to benefit.
Thymic stromal lymphopoietin (TSLP) and interleukin 33 are among a family of signalling molecules sometimes referred to as alarmins, which are released by epithelial cells after inflammatory stimulation. Release of TSLP is stimulated by a broad range of triggers, including allergens, bacteria, and cigarette smoke, and data for severe asthma suggest that tezepelumab, a monoclonal antibody that targets TSLP, improves lung function and symptoms and reduces exacerbations compared with placebo.263 Interleukin 33 is an upstream signalling molecule that activates both innate and adaptive immune cells, and a genome-wide association study264 showed that it is associated with risk of asthma. A trial265 in patients with asthma showed a clear benefit with the anti-interleukin 33 monoclonal antibody itepekimab compared with placebo, and there could be subpopulations of patients with COPD, particularly former smokers, who might also benefit. In addition to potential reductions in the frequency of exacerbations, anti-interleukin-22 monoclonal antibodies could have disease-modifying effects leading to changes in the histopathological features of Th2-high inflammation, such as hypertrophy of the airway smooth muscle and thickening of the basal membrane. However, these studies264,265 also show the challenges in translating the beneficial effects of highly specific therapies that are beneficial in asthma to the more complex and heterogeneous inflammatory milieu that is characteristic of COPD.
Lung microbiome
Modulation of the microbiome is also a potential novel therapeutic approach in COPD that could reduce chronic inflammation. Modulation could perhaps be achieved through direct approaches similar to fecal transplantation to restore the microbiota in the gut (which is used as a treatment for Clostridium difficile infection), but more intriguing is the potential to indirectly modulate the lung microbiome by the avoidance of unnecessary antibiotics early in life, which could reduce the selective pressure for pathogenic organisms.266 It is also possible that the benefits of azithromycin in patients with COPD could be due to the drug’s effects on the microbiome in addition to increases in the concentrations of several bacterial metabolites with anti-inflammatory effects.267
Reversal of airway remodelling
In addition to biologic treatments targeting Th2-associated inflammation, other agents could have the potential to improve airway remodelling, including G protein-coupled receptor modulators, mitogen-activated protein kinase inhibitors, receptor tyrosine kinase inhibitors, non-receptor tyrosine kinase inhibitors, other kinase inhibitors, and phosphodiesterase inhibitors.268 The reduction of hyaluronic acid in the lung caused by tobacco smoke results in increased elastolysis, which can also lead to remodelling of the airway and parenchyma.269 Hyaluronic acid aerosol has shown promising results in mice and in early clinical trials in humans.250
Lung regeneration
The tissue destruction in COPD cannot be cured or reversed at present, and thus there is a large unmet medical need for novel treatment options. Emerging pulmonary regenerative medicine efforts focus on several ways to achieve lung regeneration, including activation of the endogenous repair capacity of the lungs and the development of exogenous regeneration through tissue engineering, bioartificial scaffolds, or the application of healthy progenitor or stem cells to the lungs.270–72
Endogenous lung tissue regeneration
Maintenance and repair upon injury of the highly complex lung structure relies on progenitor cell populations in the lung epithelium, which are tightly spatiotemporally regulated by signalling pathways.272 Basal cells, club cells, and alveolar type II cells repopulate distal airways and alveolar epithelium after severe injury in several mouse models.272–75 The endogenous regenerative mechanisms of the lung are severely compromised in COPD, and failure to activate core developmental pathways after injury causally contributes to the disease.276 Therefore, activation of these pathways, which are essential for lung development and generation, could boost suboptimal repair processes, alveolarisation, and lung function.
Although many of these studies were done in animal models, the Wnt–β catenin signalling pathway initiated lung epithelial cell progenitor growth in human lung organoids and activation of alveolar repair mechanisms in lung tissue cultures derived from patients with COPD.273 The potential to use pharmacological compounds to foster proper functioning of healthy cells and activate endogenous repair also provides an opportunity to treat COPD at much earlier stages and to potentially avoid long-term treatment that could be harmful. Early intervention could prevent structural changes and enhance the possibility of achieving lung regeneration, thus halting or even reversing the disease.
Stem-cell therapy
Mesenchymal stem cells are immunomodulatory and therefore could affect several aspects of COPD, including airway remodelling and tissue regeneration. Clinical trials277,278 have shown that mesenchymal stem cells originating from bone marrow, umbilical cords, or adipose tissue are safe but no trials have shown efficacy in COPD. This absence of efficacy could be due to effects of the COPD microenvironment on the stem cells and further highlights that additional aspects of the pathophysiological changes in COPD need to be targeted simultaneously.279
Three-dimensional printing
Breakthroughs in bioengineering and three-dimensional organ bioprinting open novel avenues for potential COPD therapies. In a proof-of-concept study,280 extracellular-matrix-reinforced biolinks were used to print human airways that had primary human epithelial progenitor and smooth muscle cells capable of differentiating into mature cells. The airways remained stable and the cells were viable for 1 month in vitro. Use of biolink-containing extracellular matrices derived from decellularised tissue is a promising new approach for generating functional human tissue and can potentially be combined with other regenerative therapy approaches to target different COPD phenotypes.
Control of secondary systemic pathology
Exercise
When properly prescribed in terms of frequency, intensity, type, and time, exercise training through pulmonary rehabilitation is a powerful physiological stimulus to restore many of the non-respiratory consequences of COPD, including deconditioning, skeletal muscle dysfunction, cardiovascular risk, and mental problems.281 These benefits are more pronounced when exercise is combined with long-acting bronchodilators, but rates of referral to, and uptake of, pulmonary rehabilitation are low.282 Alternative scalable methods to deliver rehabilitation in non-traditional settings, including at home via remote technologies, could substantially improve access even in rural or underserved areas.283
Androgenic anabolic steroids and testosterone
Hypogonadism is prevalent in 22–69% of patients with COPD and could be the result of ageing, smoking, obesity, systemic inflammation, hypoxaemia, hypercapnia, or glucocorticoid use.284 Additionally, a link has been noted between low serum testosterone concentrations and both reduced lung function and accelerated lung function decline in both men and women.285 Experimental evidence of the relationship between sex steroid receptors and signalling in the airways and immune system suggests that a link between sex steroid hormones and lung health is biologically plausible. In a rat model of COPD, testosterone replacement therapy attenuated pulmonary epithelial inflammation.286 Various clinical trials of androgenic anabolic steroids in patients with COPD showed improvements in fat-free mass (ie, muscle mass), lean body mass, arm muscle circumference, weight, walking test results, and sexual quality of life compared with placebo, but none showed improvements in lung function as assessed by spirometry.287 As is the case for other therapeutic approaches that do not improve lung function, enthusiasm for further investigation of modification of sex hormones or other endocrine pathways has been limited.
Nutrition supplementation
Poor nutritional status has been associated with reduced lung growth and accelerated lung function decline and is a clear risk factor for the development of COPD.288–91 Poor nutritional status could be a particularly important contributor to COPD burden in LMICs, where nutritional deficiencies are more common, and there is evidence that correction of nutrient deficiencies could improve respiratory outcomes. For example, vitamin A is important in the regulation of early lung development and alveolar formation, and in a Nepali population with chronic vitamin A deficiency, the offspring of mothers who received supplementation with vitamin A before, during, and after pregnancy had improved lung function compared with the offspring of mothers who received placebo.288 Other data have shown associations between intake of vitamin C289 and vitamin E290 and improved lung function. In a systematic review and meta-analysis,292 vitamin D supplementation reduced exacerbation frequency in patients with a baseline serum calcifediol concentration of less than 10 ng/mL, and the GOLD guidelines5 recommend that patients hospitalised for COPD exacerbations should be assessed for vitamin D deficiency. Other data also suggest that dietary modifications might reduce COPD morbidity. Increased intake of omega-3 fatty acids, which promote resolution of inflammation, are associated with fewer exacerbations and better quality of life than increased intake of omega-6 fatty acids, which are pro-inflammatory.293 Increased intake of the antioxidant coenzyme Q and conjugated linoleic acid both improved exercise capacity compared with placebo in patients with COPD.294,295
BMI is perhaps the best studied marker of the nutritional status of an individual. An estimated 25–40% of patients with COPD are underweight and 35% have severely low muscle mass.296 Increased protein intake reduces loss of muscle mass but data for the effects of oral nutrition supplementation are inconclusive, because studies have usually combined supplementation with pulmonary rehabilitation and the amount of supplementation prescribed varied greatly. In hospitalised, malnourished patients with COPD, treatment with nutritional supplementation decreased mortality risk and increased bodyweight compared with placebo within 90 days of hospital discharge.297 Further investigation into the effectiveness of dietary modification in reducing COPD risk and improving outcomes is clearly indicated, particularly given the low cost, safety, and broad applicability of these interventions.
At a minimum, COPD therapy should lead to disease control and the available therapies have enabled some progress towards that goal. However, many patients receiving standard-of-care treatment continue to experience symptoms, exacerbations, lung function loss, and early mortality. Novel agents will be required to achieve disease control for all patients. Ideally, treatments would intercept COPD at an early stage and allow reversal of the accumulated lung damage leading to cure. These new therapeutics will not be useful in all patients with COPD as defined by the presence of spirometric airflow limitation, and their development will rely on testing in COPD subpopulations. We believe that clustering of patients by COPD type could promote the discovery of these more potent treatments of the future.
Towards the elimination of COPD
Advocacy efforts in COPD have largely focused on raising public awareness of the disease. Such efforts include World COPD Day, which has occurred since 2002 and is typically the third Thursday in November. World COPD Day has been highly successful in mobilising COPD organisations and helping patients to find their voice. However, to prevent and eliminate COPD, fundamental changes in approaches to advocacy are required.
The Commission’s proposal to classify COPD on the basis of the primary risk factor provides an opportunity for the disease to be recognised broadly—not only as a disease of smokers. People with COPD have long lived with the stigma of having a self-inflicted disease due to smoking. However, if the varied underlying causes of COPD can be better communicated, understood, and recognised, then compassion, support, and research funding could be increased.
All stakeholders in the public health domain need to recognise that the potential effects of primary prevention are immense and incomparable to any treatment that could be offered to patients with established COPD. Therefore, true elimination of COPD relies predominantly on the immediate and complete elimination of risk factors for lung disease, particularly exposure to anything other than clean air (including early-life exposures affecting lung growth and remodelling). Importantly, elimination of risk factors will not only prevent COPD; it will also help to maintain respiratory health throughout life and to promote healthy ageing. To achieve this ambitious goal, strict international and national legislation is required to eliminate exposure to irritants and toxins (eg, cigarette smoking, vaping, cannabis, air pollution, occupational exposures) throughout the life span (and prenatally), to promote improvement in maternal and perinatal care, and to minimise severe childhood respiratory infections. Furthermore, efforts to arrest disease progression and improve the lives of individuals with established disease should focus on refinement of diagnostic tools and early disease identification, increases in the efficacy and effectiveness of treatments, and equality in access to therapy. A boost in investment in research and innovation from the pharmaceutical and non-pharmaceutical sectors will be necessary to expedite progress. Finally, prevention and care of COPD requires streamlined health systems and urgent public policy actions at a global level. Here we summarise the actions needed to progress towards the elimination of COPD.
Appreciation of overlapping and interacting risk factors
A major barrier to the advancement of COPD care is the underappreciation of important risk factors beyond smoking. Targeted efforts to reduce these risk factors and to increase knowledge and understanding of the effects of these exposures on lung growth, disease initiation, and lung function decline will pave the way to innovative treatment strategies and perhaps the possibility to eliminate COPD. Many of these risk factors for COPD are closely linked to poverty, and although beyond the remit of the Commission, elimination of global poverty would clearly contribute to the elimination of COPD.
Early-life events
To safeguard the respiratory health of future generations, policies need to be implemented to tackle early-life risk factors and improve maternal, fetal, and childhood respiratory health. Action plans should be adopted, such as those put forward by WHO to reduce preterm birth.298 These actions could include provision of family planning services and improvements to antenatal care, patient and provider education, and the prevention and management of preterm labour. The importance of controlling childhood asthma needs to be well communicated to parents and health-care providers.
Respiratory Infections
Key actions to reduce the effects of childhood respiratory infections globally include vaccinations, adequate nutrition, and breastfeeding. Vaccination of the general population, and particularly of vulnerable groups, is extremely important and requires the involvement of all stakeholders. Major side-effects of vaccinations are very rare, and this message should be effectively communicated to the respiratory patient community and the public at large. Initiatives to encourage health-care professionals to vaccinate should be supported by professional medical organisations.
Inhalation of tobacco, drugs, and other combustible substances
The effects of exposure to tobacco smoke are clear, and targeted efforts to address environmental tobacco smoke have the potential to bring about considerable change. Exposure to environmental tobacco smoke is pronounced in the home and in private vehicles, but varies according to race and socioeconomic status, and urban versus rural residence.299 Legislation aimed at smoke-free policies for private transportation have been implemented, but smoke-free home policies that reduce exposure to environmental tobacco smoke are not legislation-driven but dependent on the individual smoker.300 There are also vast differences in smoke-free legislations between and within countries, even though such legislation is beneficial to child health.301 Understanding of exposure to e-cigarette smoke is growing, and it is now clear that vaping is harmful. Due to a lack of legislation and conflicting approaches to e-cigarette use in the medical field,302 e-cigarettes and vapes can often be used in smoke-free areas and public confusion persists regarding their harmful effects.
Combatting tobacco use, especially among young people, will require a multipronged approach that focuses on advocacy and public education, more stringent anti-tobacco legislation, and improved enforcement of legislation. If all countries adopt smoking bans, health warnings, and advertising bans at the strictest level, and raise cigarette prices, there could be 142 218 fewer deaths due to COPD, 725 578 fewer years of life lived with disability, and 2 419 035 life-years gained worldwide yearly (figure 18).303 In addition to adopting new and more rigorous legislation, the medical community should be proactive in informing smokers about the benefits of smoke-free homes and smoke-free transportation. A study304 in the Netherlands showed that only 27 (11%) of 245 physicians consistently addressed passive smoking in children and only 61 (25%) had received training on the deleterious effects of passive smoking. Patient assessments should concentrate not only on cigarette smoking but should also relate to all types of smoking (e-cigarettes, cannabis, and water-pipe smoking, and environmental exposure to tobacco smoke).
Figure 18: Estimation of the number of deaths avoided (A), years lived with disability avoided (B), and years of life gained (C) after the implementation of more stringent tobacco control policies.
Such policies include increased cigarette prices, protections from environmental tobacco smoke, health warnings, and bans on tobacco advertising, promotion, and sponsorship. Data are broken down by WHO region. The appendix (pp 7–8) contains full details on the methods.
Indoor pollutants
The health effects of indoor air pollution related to indoor energy sources are substantial, and improved management of this lifelong risk factor will help to eliminate COPD. Wood-burning stoves reduce heating costs and are easily accessible, but wood smoke adversely affects respiratory health and disproportionately affects women and children in low-income settings. Global initiatives are needed to reduce the effects of this exposure. Increased public awareness will be an important factor for driving change.
Given that LMICs account for more than 80% of deaths and illness episodes related to COPD,7 any mission to eliminate COPD globally should include strategies targeted towards these settings. Installation of kitchen chimneys and improved biomass stoves reduce the effects of indoor air pollution.305 Improved stove and ventilation system designs also attenuate lung function decline and reduce the incidence of COPD.306 However, several previous studies of interventions to replace biomass stoves with clean fuel (biogas or liquefied petroleum gas) stoves were unsuccessful in the long-term because of financial and infrastructure problems, low awareness, and sociocultural barriers.307 Biomass is the most affordable fuel for domestic energy production due to its availability, and interventions to move towards cleaner fuels are not always feasible in rural communities or for those with low socioeconomic status. The FRESH AIR project has helped to identify factors that are essential to the successful implementation of health interventions in LMICs.308 One of the key learnings from the project was that strategies need to be tailored to the local context (culturally and with awareness of regional health and political infrastructure). When fuel sources cannot be changed, improving ventilation and reducing the proximity of children to smoke within the home are key factors that can be improved via public awareness.
A study309 showed that use of air cleaners with high-efficiency particulate air and carbon filters in the homes of former smokers with COPD was associated with reductions in indoor particulate matter concentrations in a US setting. Although the study did not reach statistical significance for the primary outcome (6-month change in scores on the St George’s Respiratory Questionnaire) in the intention-to-treat analysis, portable high-efficiency particulate air cleaner use significantly improved several respiratory outcomes. Environmental interventions that improve air quality represent a potentially novel approach to reducing respiratory morbidity in patients with COPD, although larger studies are needed to confirm health benefits.
Ambient air pollution
Many organisations promote the concept that access to clean air is a right and not a privilege. In 2018, the first WHO conference on air quality and health emphasised the need to include health arguments in advocacy efforts to improve air quality globally. In 2021, WHO revised its air quality standards and declared that there are no thresholds for health effects: anything but clean air is harmful.130 A commitment by stakeholders to effect change agreed upon by the conference has led to the development of a commitment paper in Europe signed by more than 50 medical societies and patient organisations representing different diseases.310 The organisations committed to include air quality in their educational programmes, to ensure that clinical guidelines include specific consideration of the effects of air quality, to provide information for health-care professionals to help advise patients, to invest in research on the link between air quality and health, and to raise awareness to make reduction of air pollution a priority for all.
WHO’s BreatheLife campaign encourages cities to improve air quality through multiple measures. Healthy Air is an American Lung Association (ALA) initiative that aims to improve the quality of both indoor and outdoor air by reducing vehicle idling near schools and optimising city planning of new schools, among other initiatives. The US Environmental Protection Agency (EPA), in partnership with the ALA, has designed an Indoor Air Quality Tools for Schools programme that, if widely adopted, has the potential to improve air quality for children at school.311 As part of the Healthy Air initiative, the ALA also analyses data from official air-quality monitors and releases an annual State of the Air report that allows citizens to look up air quality in their community.
In 1970, the USA adopted the Clean Air Act, which gave the EPA the power to take action to fight environmental pollution. EPA officials then established the National Ambient Air Quality Standards (NAAQS): all states, cities, and towns in the USA have to maintain levels of pollutants beneath the thresholds set by the NAAQS or face substantial fines and penalties. From 1980 to 2015, total emissions of the major air pollutants regulated by the NAAQS dropped by 63%, despite concomitant increases in the population.312 Unfortunately, pollutant levels remain above NAAQS standards in many parts of the USA, and organisations such as the ALA continue to push the EPA to enforce stronger pollution limits and Congress to protect the Clean Air Act from those seeking to weaken clean-air protections. In many countries, including China and India, there are no governmental efforts to reduce the exponential growth in ambient air pollution and the full respiratory effects of this pollution have not yet manifested.
Particular attention should be paid to the effects of air pollution on infants and children, because air pollution impairs lung function and growth. Although the individual effect size of air pollution on lung function and growth might be small, the number of attributable cases of COPD in whom lung function is below normal within the population is very large.
The long-term effects of global warming on respiratory health are not certain. However, emerging evidence suggests that global warming could have major implications for the control of the burden of COPD, because ambient heat is closely linked to the risk of hospitalisation in patients with COPD.313
Occupational exposure
In the EU, exposure to chemicals in the workplace is regulated by occupational health and safety legislation and EU chemicals legislation. Employers are required to identify all chemical hazards in the workplace, to do exposure and risk assessments, and to act on the findings of these assessments. In the USA, the Occupational Safety and Health Administration within the US Department of Labor oversees respiratory health measures in the workplace. The revised Respiratory Protection Standard (1988) outlines respiratory protection standards for general, maritime, and construction industries. A revision of the Respiratory Protection Standards is due because the workforce is ageing and data for long-term acceptable levels of exposure are needed. In the absence of agreed standards, the precautionary principle to keep levels as low as reasonably achievable should be implemented.
Compounding the situation, many respiratory health-care professionals have little knowledge of, or training in, the recognition of occupational exposures or the relationship between these exposures and lung disease. Clearly, more research and education of both the public and respiratory health professionals is needed to reduce and prevent chronic lung diseases related to occupational exposures. To this end, the European Lung Foundation has developed an evidence-based digital occupational tool available in Dutch, English, French, Portuguese, and German that targets at-risk populations. The tool aims to help people to understand whether a lung problem is related to their workplace and to provide protection advice. Social media has had a major role in targeting the at-risk audience. For example, via Facebook advertising individuals tag their friends and colleagues who they believe could also be at risk. If issues arise, the person using the tool is encouraged to visit a health-care professional and talk about their work environment. To date, more than 22 000 individuals have used the tool. Around 80% of the people who used the tool came via Facebook, and roughly 50% were recommended to consult a health-care professional on the basis of their answers. More than 70% of respondents report having received no training in the workplace on protecting their lung health, and more than 90% report that they had to leave a previous job because of respiratory symptoms.
Improving predictive and diagnostic tools for early disease identification
People with COPD most commonly present with advanced disease. Yet in most people COPD has a prolonged asymptomatic phase during which preventative strategies are likely to be more effective. Therefore, screening and case-finding for COPD is a promising way forward. However, the available tools provide only a list of risk indicators—they do not quantify the risk. Furthermore, many health agencies recommend against screening. This approach is in stark contrast to that in heart disease, for which there are risk calculators based on algorithms derived by prediction models to educate the public about their risks and flag the requirement for confirmatory tests. There is a pressing need for targeted and innovative approaches to improve the prediction of COPD.
It is time to move beyond a diagnostic algorithm for COPD based solely on spirometric lung function abnormalities and to adopt a more comprehensive approach. Emphasis should be placed on early diagnosis and type-specific classification based on algorithms that capture early symptoms and biomarkers of risk, complemented by a range of pulmonary function testing and imaging (particularly CT).
In low-income settings, early diagnosis of COPD is challenging due to limited access to health care, including spirometry. Hence, interest is increasing in establishing new case-finding instruments for COPD that can be implemented in low-income settings (including use of symptoms and peak flow meters).314 An example of a successful case-finding method implemented by WHO’s acute lower respiratory infection programme was the use of respiratory rate and chest in-drawing for early diagnosis of pneumonia in children by primary health-care workers.315 The simplification and broad deployment of this approach to early diagnosis and treatment of pneumonia have enabled substantial reductions in mortality in LMICs, where access to paediatricians is limited. Such innovative diagnostic approaches are crucial to handling the high COPD burden in low-income settings.
Increased treatment efficacy and effectiveness and cure
In a survey316 of 1102 US patients with COPD, 82% were satisfied with their treatment plan, but only 12% stated that their disease was completely controlled. Further surveys by the COPD Foundation have suggested that most patients desire treatments that have a greater effect on their symptoms or that halt disease progression.317 The drug pipeline needs to be more robust, and there needs to be a shift of focus from blockbuster drugs aimed at most or all patients with moderate to severe disease to drugs targeting specific molecular pathways in subgroups, particularly patients with early disease. Increased public and private financial investment into the basic research necessary to identify targetable pathways among these subgroups is urgently needed, as is faster translation of these findings to the clinic. Faster translation will require testing of novel therapeutics in complex tissue models, including cell-based organoids that mimic the pathophysiology of specific COPD types, and a new approach to clinical trials whereby more homogeneous populations defined according to disease mechanism and not spirometry are enrolled.
Low availability of, inadequate health insurance coverage for, poor referral rate to, and low adherence to pulmonary rehabilitation preclude substantial effects at the population level.318 Alternative points of access, improved training of professionals, and consensus between health-care professionals and payers on the definition of pulmonary rehabilitation could improve the situation. An American Thoracic Society taskforce has defined the essential components of a rehabilitation process, which in turn facilitates the development of novel approaches such as tele-rehabilitation.281 Although the effects of exercise training last longer than those of respiratory pharmacotherapy, the benefits of exercise progressively diminish over time and thus maintenance programmes are essential to conserve and extend the benefits.281
Individual, personalised care of patients with COPD is important. It is imperative that patients are approached holistically, with a focus on individual needs, capabilities, preferences, and resources. Patients’ broader living environments should also be covered in assessments. Thereafter, an individualised interdisciplinary care plan should be designed that specifically targets the priorities jointly set by the patient and their formal and informal care providers. Besides pharmacological treatment, inhalaion instruction, exacerbation management, and physical exercise, such a care programme might also include healthy lifestyle, psychological, self-management, and social care support, which are commonly not disease specific.
Eliminating disparities
A growing body of evidence shows race and gender disparities in COPD outcomes, but social and socioeconomic factors almost certainly drive most of these inequities.31,32 WHO estimates that at least 250 million children younger than five years are in danger of poor development in LMICs due to poverty.34 In parts of India and Africa, growth stunting affects one in three children,319 and it will be many years before the full effects on lung health and the prevalence of COPD are understood. Although agencies such as UNICEF, WHO, the World Food Programme, and the International Fund for Agricultural Development are helping to drive global efforts to end malnutrition, the burden of the problem is massive and will require intervention at the country and regional level. Furthermore, given the racial, regional, and socioeconomic disparities in COPD outcomes,31,32,320 individual countries will ultimately need to develop local initiatives tailored to specific regions to address differences in food access, maternal care, preventive health measures, access to primary and specialty care, and availability of pharmacological and non-pharmacological treatments.
Advancing research and development
Funding for COPD research has been disproportionately low relative to the impact of the disease for many decades, and has declined even further since the inception of this Commission. For no other chronic disease is there such a mismatch between funding and public health burden. However, efforts are underway to address this situation.
In 2017, the US National Institutes of Health launched a COPD national action plan that provided 203 grant awards by 2019. One of the grants was for the first ever lung health cohort, which will examine factors influencing lung health over the life course.321 A cohort of patients with early COPD is also being developed in Europe and the USA. The Lung Foundation in the Netherlands has launched the Accelerate project to focus on innovative solutions for lung health, including lung regeneration. By 2023, Accelerate aims to have funded internal collaborations to fully understand lung regeneration, to have an organoid that mimics the biology of the lung, and to have begun the first clinical studies of lung stem cells that repair damaged lung tissue.
We also advocate for better funding and support for non-pharmacological interventions in COPD. Of 1685 individuals living with chronic lung disease, 1549 (92%) thought that pulmonary rehabilitation should be part of the health-care services available to all patients.322 However, 1015 (60%) reported challenges in accessing pulmonary rehabilitation.322 As part of the Rehabilitation 2030 initiative, WHO is building evidence-based packages for rehabilitation that should foster global expansion of access for all patients with COPD.323
When calling for the development of innovative treatment options, we acknowledge that the health system is given finite resources. Money can be spent only once, and spending it on a new treatment could displace treatment (and thus benefits) elsewhere in the system. Therefore, payers require evidence of cost-effectiveness before taking the decision to reimburse a new treatment. Development of personalised and precision medicine in COPD means that new treatments will probably be suitable only in target subgroups of patients, which will cause treatment costs to go up. The cost-effectiveness of these new treatments might therefore be difficult to prove.
The cost-effectiveness of interventions in COPD is generally estimated with health economic models. In the past, these models were mainly Markov models in which COPD severity was classified on the basis of airflow limitation only, and COPD exacerbation frequencies, costs, and utilities were then assigned to these severity states. These models generate only a few outcome measures, including lung function decline, exacerbation frequency, quality-adjusted life-years, and costs. Pharmaceutical companies, academics, and consultants have each constructed their own models, which makes comparison of results challenging. As treatments are increasingly personalised, comprehensive patient-level simulation models that include many patient and disease characteristics, comorbidities, and a greater diversity of outcomes are needed. In one patient-level simulation model of this type, the cost-effectiveness of interventions that reduced lung function decline, increased time to a COPD exacerbation, improved physical activity, and reduced the probability of having symptoms was compared.324 Although the model is based on data from more than 35 000 people with COPD, all the data come from one pharmaceutical company. What is really needed to inform decision makers is an open-access core model of COPD that allows estimation of the cost-effectiveness of many different interventions with the same model across datasets.
People-centred integrated care models, especially for vulnerable people with multimorbidities, aim to improve outcomes beyond health, including wellbeing (eg, social relationships and participation, resilience, enjoyment of life, autonomy) and experience with care (eg, continuity of care, person-centredness, feeling safe). Such outcomes are not captured by quality-adjusted life-years, so there is a need to broaden the assessment framework and explore the potential of multi-criteria decision analyses,325 which combine measurement of a broader set of benefits with the weighting of these benefits to generate a single overall score that allows promotion of an economy of wellbeing.
Conclusions and recommendations
It is quite possible that the elimination of COPD is not achievable, much as it might be impossible to eliminate other highly complex diseases with high morbidity and mortality, such as cancer, heart disease, and diabetes. However, our approach to date has lacked clarity, focus, and urgency and the global burden of COPD continues to increase. Although the decline in tobacco smoking in high-income countries is welcome, failure to control other risk factors jeopardises the gains that might ultimately be realised. In this Commission, we have attempted to reset the conversation about COPD to emphasise the importance of non-tobacco risk factors and primary prevention and the need to promote lung health across the life course beginning at conception, to improve disease classification and expand tools for diagnosis, and to develop better treatments aimed primarily at reversal and cure. Table 8 details our urgent recommendations to help drive transformational change and to assess progress along this new path towards the elimination of COPD. We are committed to tracking this progress, and ask the respiratory community to join us.
Table 8:
Recommendations for the elimination of COPD
| Practical goals | |
|---|---|
| Prohibit all kinds of smoking including—but not restricted to—cigarette smoking, water-pipe smoking, e-cigarette smoking (vaping), cannabis smoking, and smoking of other combustible substances | 50% of countries to ban smoking by 2035 |
| Eliminate environmental exposures to anything but clean air, including indoor and outdoor pollution, wildfire smoke, and occupational exposures to toxic fumes and gasses; regulatory authorities should strengthen legislation governing acceptable levels of exposure to inhalable particulate matter and ozone | 50% of countries to introduce annual limits (lower than those recommended by WHO) for exposure to inhalable particulate matter <2·5 μm in diameter, particulate matter <10 μm in diameter, and ozone |
| Support measures associated with improved and sustained general health, including reductions in global poverty and improvements in nutrition, vaccination, prenatal care, physical activity, and mental health | At least a 50% reduction in people living below the poverty-line by 2035; all countries should provide free vaccinations and mobilise educational campaigns to inform at-risk individuals; free or low-cost health care for all |
| Diagnose COPD based on expanded criteria, including the presence of respiratory symptoms, personal history of risk factors, and persistent airflow limitation or ventilatory heterogeneity (as assessed by spirometry, other pulmonary function testing, or CT) | By 2035, the proportion of patients diagnosed with mild spirometric airflow obstruction should increase to at least 50% of the total |
| Research and development should focus on treatment of early disease | By 2030, 75% of published clinical trials should be focused on patients with early or mild disease |
| COPD should be classified into one of five types on the basis of the predominant risk factor present to increase awareness of risk factors, improve detection of people with non-smoking-related COPD and those with early disease, and foster research into therapies targeting specific disease mechanisms | COPD diagnosis by type should be included in the International Classification of Diseases coding system By 2035, at least one specific pharmacological or non-pharmacological therapy should be approved for each type of COPD |
| Diagnosis of exacerbations should be based on a standard assessment confirmed by evidence of worsening airflow limitation or ventilatory heterogeneity, airways or systemic inflammation, or lung infection in a patient with increased respiratory symptoms (after exclusion of other disorders that mimic this presentation) | Exacerbation frequency should be similar worldwide by 2035 as a result of the establishment of a standard definition and assessment |
| Effective pharmacological and non-pharmacological therapies should be made available worldwide; development of new therapies should focus on underlying pathophysiology and take into account disease heterogeneity (including COPD type) | By 2035, at least 80% of patients with COPD should have disease control, as evidenced by the absence of respiratory symptoms and exacerbations and normal or near-normal quality of life, exercise capacity, and life expectancy |
| Definitions of treatment effectiveness should take patient-reported outcomes into account | By 2030, 75% of studies should include a patient-centred outcome as a primary outcome |
| Regulatory agencies should regularly revisit and update endpoints for clinical trials of treatments for different COPD types | By 2035, 75% of new therapies should be approved on the basis of non-spirometric criteria |
| Funding agencies should increase financial investments to adapt to the worldwide burden of COPD | By 2030, the total public and private global research and development expenditures for COPD should increase by 50% |
COPD=chronic obstructive pulmonary disease.
Supplementary Material
Key messages.
Chronic obstructive pulmonary disease (COPD) is a global health emergency that affects people from all countries, socioeconomic classes, and age groups, although it disproportionately affects poor and disadvantaged people.
A substantial proportion of the COPD burden is preventable. Prohibiting all kinds of smoking and eliminating exposure to any form of air pollution would greatly reduce the burden of the disease.
COPD is a complex and heterogeneous disease, and its pathophysiology implicates varying degrees of airway remodelling, inflammation, and tissue destruction. This heterogeneity manifests in the wide variations in respiratory symptoms, systemic consequences, and comorbid conditions among patients.
Throughout the life course, there are multiple lung function trajectories, several of which can lead to COPD. Recognition of these varied trajectories, and the fact that not all patients develop COPD as a result of accelerated loss of lung function, could lead to new preventive and therapeutic approaches.
COPD should be classified into five types on the basis of the predominant risk factor driving the disease: genetics, early-life events, respiratory infections, tobacco exposure, or other environmental exposures. Pathophysiological mechanisms related to each of these exposures could translate into distinct diagnostic, prognostic, and therapeutic considerations.
Diagnosis should be based on expanded criteria including the presence of respiratory symptoms, personal history of risk factors, and persistent airflow limitation or ventilatory heterogeneity as assessed by spirometry, other pulmonary function testing, or CT. Airflow limitation based on spirometry alone is insufficient to diagnose COPD because spirometry does not reliably capture early airway changes or emphysematous destruction of the lung parenchyma, and probably only detects irreversible disease.
Reducing the frequency of COPD exacerbations is a crucial step on the path towards the elimination of COPD. Diagnosis of exacerbation should be based on standardised assessments and should be confirmed by evidence of worsening airflow limitation or ventilatory heterogeneity, airway or systemic inflammation, or lung infection in a patient with increased respiratory symptoms (after exclusion of other disorders with similar presentations).
The severity of a COPD exacerbations should be judged not in terms of the type of treatment or the setting in which care is provided, but rather based on objective categorisation of the degree of clinical, biological, and physiological deterioration. The Commission proposes the elimination of the definitions for mild or moderate exacerbations, such that exacerbations would be either severe or not severe.
Current pharmacological and non-pharmacological therapies improve respiratory symptoms and quality of life, but are not available to many people with COPD. There is a moral imperative to improve access to effective treatment. Additionally, the development of curative and regenerative therapies for COPD is more likely to succeed if focused on early disease and pathophysiological differences underlying the specific disease types.
Elimination of COPD requires consistent and coordinated action and substantially greater investment of financial and intellectual resources from all stakeholders, including medical professionals, governmental health and regulatory agencies, private industry, and the general public.
Panel 1: Challenges in the prevention and cure of chronic obstructive pulmonary disease.
- Multiple, overlapping, and interacting risk factors for disease
- Genetics
- Early-life events
- Respiratory infections
- Inhalation of tobacco, drugs, and other combustible substances
- Environmental exposures (eg, indoor pollutants, ambient air pollution, occupational exposures)
- Insensitive diagnostic tools, delayed recognition, and late presentation
- Dependence on spirometry, which is unsuitable for the detection of mild disease
- Poor availability of other diagnostic tools
- Insufficient or inefficient treatment options
- No curative treatments
- Inadequate disease control in most patients
- Unpredictable treatment responses and complex treatment delivery
- Disproportionate burden among poor and disadvantaged people
- Increased prevalence of risk factors for compromising lung health
- Unequal social and financial burdens
- Limited access to treatment in low-income and middle-income countries
- Misalignment of health-care system
- Scarcity of multidisciplinary and specialist care
- Mixed availability of, and support for, smoking cessation and pulmonary rehabilitation
Lack of innovation in management and inadequate research funding
Panel 2: Diagnostic considerations in chronic obstructive pulmonary disease.
- Chronic respiratory symptoms*
- Dyspnoea
- Cough
- Sputum production
Acute worsening of respiratory symptoms*
- History
- Premature birth
- Childhood respiratory infections (pneumonia, viral bronchiolitis, tuberculosis)
- Childhood respiratory illnesses (asthma, chronic lung disease of infancy)
- Exposure
- Tobacco smoking, environmental tobacco smoke, vaping
- Environmental exposure (indoor fuel fumes, ambient air pollution or smog, wildfire smoke, occupational exposure)
- Genetics
- α1 antitrypsin deficiency
- Telomerase polymorphism (TERT gene)
- Family history in non-smokers
- Airway remodelling*
- Airway smooth muscle cell hypertrophy
- Thickening of basement membrane
- Chronic airway inflammation
- Pathological turnover of the extracellular matrix
- Increased airway wall thickness on CT
- Persistent airflow limitation on lung function tests*
- Forced expiratory volume in 1 s/forced vital capacity
- Specific effective airway resistance
- Lung clearance index
- Reduced peak flow
- Nitrogen washout measures of heterogeneity
- Forced oscillometry (difference in oscillation resistance at 5 Hz and 19 Hz, reactance area)
- Parenchymal abnormality*
- ≥ 5% emphysematous involvement (defined as areas of lung density less than –950 HFU) on CT
- Destruction of terminal bronchioles visible on CT
- Decreased diffusion capacity
Panel 3: Classification criteria for severe exacerbations of chronic obstructive pulmonary disease.
Use of accessory respiratory muscles or paradoxical chest wall movements, or both
Clinically significant hypoxaemia and new or worsening hypercapnia or respiratory acidosis
Reduced alertness (eg, confusion, lethargy, coma)
Failure to respond to initial medical management
Right heart failure, cardiac ischaemia, haemodynamic instability, or clinically significant arrhythmia
The presence of any one of these criteria is sufficient to define an exacerbation as severe. A total severity score (range 1–5) can be calculated based on the total number of criteria that are met.
Acknowledgments
The Commission has been supported by the University Hospital Basel, Switzerland, the Swiss Respiratory Society, and the Lung Health Center, Heersink School of Medicine, University of Alabama at Birmingham. The European Respiratory Society provided logistic support for meetings during annual conferences. The US National Institutes of Health provided logistic support and space for a meeting of the Commission. We thank the European Lung Foundation for participation, and Sonja Burger for her help with illustrations.
Declaration of interests
DS reports a grant from the Swiss National Foundation (SNF 320030_189280), and unrestricted grants from Curetis, AstraZeneca, and Boston Scientifics (paid to their institution); honoraria for participation in data safety monitoring or advisory boards or talks for CSL Behring, Berlin-Chemie Menarini, Novartis, GlaxoSmithKline, AstraZeneca, Vifor, Merck, Sanofi, Merck Sharp & Dohme, Boehringer Ingelheim, and Chiesi; and is the current Global Initiative for Chronic Obstructive Lung Disease (GOLD) representative for Switzerland, the immediate past Education Council Chair of the European Respiratory Society, and President of the Education Committee of the Swiss Respiratory Society. SYA reports grants from the US National Institutes of Health (K08HL145118) and the Pulmonary Fibrosis Foundation (the I M Rosenzweig Junior Investigator Award), and is an owner of Quantitative Imaging Solutions. AA reports unrestricted research grants from GlaxoSmithKline and AstraZeneca; consulting fees from GlaxoSmithKline, AstraZeneca, Sanofi and Merck Sharp & Dohme; and payment for lectures and presentations from GlaxoSmithKline, AstraZeneca, Chiesi, and Menarini. MH reports personal fees from GlaxoSmithKline, AstraZeneca, Boehringer Ingelheim, Cipla, Chiesi, Novartis, Pulmonx, Teva, Verona, Merck, Mylan, Sanofi, DevPro, Aerogen, Polarian, Regeneron, United Therapeutics, UpToDate, Altesa Biopharma, Medscape, NACE, and Integrity; has received either in-kind research support or funds paid to their institution from the US National Institutes of Health, Novartis, Sunovion, Nuvaira, Sanofi, AstraZeneca, Boehringer Ingelheim, Gala Therapeutics, Biodesix, the COPD Foundation, and the American Lung Association; has participated in data safety monitoring boards for Novartis and Medtronic (funds paid to their institution); and has received stock options from Meissa Vaccines and Altesa Biopharma. TMP reports an early career development grant (K23HL153672) from the US National Heart, Lung, and Blood Institute. MRvM’s department received €2000 from the Clinic for Respiratory Medicine and Pulmonary Cell Research, University Hospital Basel (Basel, Switzerland) for calculating the smoking-attributable burden of COPD reported in the Commission. Her department also received an unrestricted grant of €198 000 from Boehringer Ingelheim to develop a health economic cost-effectiveness model of COPD. BS is supported by a National Institutes of Health grant (U01 HL-139466). JDC reports grants from or contracts with AstraZeneca, GlaxoSmithKline, Boehringer Ingelheim, Gilead Sciences, Grifols, Insmed, and Novartis, and consulting fees from AstraZeneca, GlaxoSmithKline, Boehringer Ingelheim, Janssen, Grifols, Zambon, Pfizer, Novartis, Chiesi, and Insmed. NMH reports grants from AstraZeneca, and payment or honoraria for presentations, speakers’ bureaus, or participation on advisory boards from AstraZeneca, Novartis, and BI-Lilley. MTD reports grants or contracts from the American Lung Association, the US Department of Defense, and the US National Institutes of Health, consulting fees from AstraZeneca, GlaxoSmithKline, Novartis, Pulmonx, and Teva, and support for attending meetings from Pulmonx. YS has received support from the Science and Technology Commission of Shanghai Municipility (200Z2261200). TW has served as an advisory board member or received honoraria for lectures from AstraZeneca, Berlin-Chemie, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, and Novartis, and has received research grants from the German Ministry for Research and Education, GlaxoSmithKline, and AstraZeneca. FMEF reports institutional study grants from AstraZeneca and personal fees for consultancy or presentations from AstraZeneca, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, Merck Sharp & Dohme, and Novartis. SCD holds investigator-initiated grants from AstraZeneca and GlaxoSmithKline. DDS reports honoraria for speaking engagements for AstraZeneca, Boehringer Ingelheim, and GlaxoSmithKline. MB reports grants or contracts (to their institution) from AstraZeneca and Roche and consulting fees (paid to their institution) from AstraZeneca, Sanofi, and Roche; honoraria for lectures, presentations, speakers’ bureaus, manuscript writing, or educational events from AstraZeneca, Sanofi, Chiesi, and GlaxoSmithKline; participation on an advisory board from AstraZeneca; and scientific advisor work for ProAxsis and Albushealth. NNH reports grants or contracts (to their institution) from the National Instiutes of Health, AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, and the COPD Foundation; and participation on data safety monitoring boards or advisory boards for AstraZeneca and GlaxoSmithKline. RK reports grants from the National Heart, Lung, and Blood Institute, the Respiratory Health Association, PneumRx, Spiration, and AstraZeneca, and personal fees from AstraZeneca, CVS Caremark, GlaxoSmithKline, CSA Medical, and Boehringer Ingelheim. GJC has received personal fees from Almirall, Amgen, AstraZeneca, Boehringer Ingelheim, Broncus Medical, Chiesi, CSA Medical, Eolo, Gala Therapeutics, GlaxoSmithKline, Helios Medical, Merck, Medtronic, Mereo BioPharma, NGM Biopharmaceuticals, Novartis, Nuvaira, Olympus, Philips, Pulmonx, Respironics, Respivant Sciences, the Implementation Group, Sanofi, Regeneron, Gilead, and Verona. GRW has been supported by the National Heart, Lung, and Blood Institute (grants R01 HL116473 and R01 HL122464). All other authors declare no competing interests.
Footnotes
Not explained by another condition.
Contributor Information
Daiana Stolz, Clinic of Respiratory Medicine and Pulmonary Cell Research, University Hospital Basel, Basel, Switzerland; Department of Clinical Research, University Hospital Basel, Basel, Switzerland; Clinic of Respiratory Medicine and Faculty of Medicine, University of Freiburg, Freiburg, Germany.
Takudzwa Mkorombindo, Lung Health Center, Division of Pulmonary, Allergy, and Critical Care Medicine, Department of Medicine, University of Alabama at Birmingham, Birmingham, AL, USA.
Desiree M Schumann, Clinic of Respiratory Medicine and Pulmonary Cell Research, University Hospital Basel, Basel, Switzerland.
Alvar Agusti, Respiratory Institute-Hospital Clinic, University of Barcelona IDIBAPS, CIBERES, Barcelona, Spain.
Samuel Y Ash, Division of Pulmonary and Critical Care Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA.
Mona Bafadhel, School of Immunology and Microbial Sciences, Faculty of Life Sciences and Medicine, King’s College London, London, UK; Department of Respiratory Medicine, Nuffield Department of Medicine, University of Oxford, Oxford, UK.
Chunxue Bai, Department of Pulmonary and Critical Care Medicine, Zhongshan Hospital, Fudan University, Shanghai, China.
James D Chalmers, Scottish Centre for Respiratory Research, University of Dundee, Dundee, UK.
Gerard J Criner, Department of Thoracic Medicine and Surgery, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, USA.
Shyamali C Dharmage, Centre for Epidemiology and Biostatistics, School of Population and Global health, University of Melbourne, Melbourne, VIC, Australia.
Frits M E Franssen, Department of Research and Education, CIRO, Horn, Netherlands; Department of Respiratory Medicine, Maastricht University Medical Centre, Maastricht, Netherlands.
Urs Frey, University Children’s Hospital Basel, Basel, Switzerland.
MeiLan Han, Department of Internal Medicine, University of Michigan, Ann Arbor, MI, USA.
Nadia N Hansel, Pulmonary and Critical Care Medicine, School of Medicine, Johns Hopkins University, Baltimore, MD, USA.
Nathaniel M Hawkins, Centre for Cardiovascular Innovation, University of British Columbia, Vancouver, BC, Canada.
Ravi Kalhan, Department of Preventive Medicine and Division of Pulmonary and Critical Care Medicine, Department of Medicine, Northwestern University Feinberg School of Medicine, Chicago, IL, USA.
Melanie Konigshoff, Division of Pulmonary, Allergy and Critical Care Medicine, School of Medicine, University of Pittsburgh, Pittsburgh, PA, USA.
Fanny W Ko, The Chinese University of Hong Kong, Hong Kong Special Administrative Region, China.
Trisha M Parekh, Lung Health Center, Division of Pulmonary, Allergy, and Critical Care Medicine, Department of Medicine, University of Alabama at Birmingham, Birmingham, AL, USA.
Pippa Powell, European Lung Foundation, Sheffield, UK.
Maureen Rutten-van Mölken, Erasmus School of Health Policy & Management and Institute for Medical Technology Assessment, Erasmus University Rotterdam, Rotterdam, Netherlands.
Jodie Simpson, Priority Research Centre for Healthy Lungs, Faculty of Health and Medicine, University of Newcastle, Newcastle, NSW, Australia.
Don D Sin, Centre for Heart Lung Innovation and Division of Respiratory Medicine, Department of Medicine, University of British Columbia, St Paul’s Hospital, Vancouver, BC, Canada.
Yuanlin Song, Department of Pulmonary and Critical Care Medicine, Zhongshan Hospital and National Clinical Research Center for Aging and Medicine, Huashan Hospital, Fudan University, Shanghai, China; Shanghai Respiratory Research Institute, Shanghai, China; Jinshan Hospital of Fudan University, Shanghai, China.
Bela Suki, Department of Biomedical Engineering, Boston University, Boston, MA, USA.
Thierry Troosters, Department of Rehabilitation Sciences, Research Group for Rehabilitation in Internal Disorders, KU Leuven, Leuven, Belgium.
George R Washko, Division of Pulmonary and Critical Care Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA.
Tobias Welte, Department of Respiratory Medicine, Hannover Medical School, Hannover, Germany; Biomedical Research in Endstage and Obstructive Lung Disease, German Center for Lung Research, Hannover, Germany.
Mark T Dransfield, Lung Health Center, Division of Pulmonary, Allergy, and Critical Care Medicine, Department of Medicine, University of Alabama at Birmingham, Birmingham, AL, USA; Birmingham VA Medical Center, Birmingham, AL, USA.
References
- 1.Polverino F, Celli B. The challenge of controlling the COPD epidemic: unmet needs. Am J Med 2018; 131: 1–6. [DOI] [PubMed] [Google Scholar]
- 2.Agustí A, Faner R. Lung function trajectories in health and disease. Lancet Respir Med 2019; 7: 358–64. [DOI] [PubMed] [Google Scholar]
- 3.Soriano JB, Kendrick PJ, Paulson KR, et al. Prevalence and attributable health burden of chronic respiratory diseases, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Respir Med 2020; 8: 585–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.WHO Department of Data and Analytics. Global health estimates 2019: disease burden by cause, age, sex, by country and by region, 2000–2019. Geneva: World Health Organization, 2020. [Google Scholar]
- 5.Singh D, Agusti A, Anzueto A, et al. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease: the GOLD science committee report 2019. Eur Respir J 2019; 53: 1900164. [DOI] [PubMed] [Google Scholar]
- 6.Celli BR, Fabbri LM, Aaron SD, et al. An updated definition and severity classification of chronic obstructive pulmonary disease exacerbations: the Rome proposal. Am J Respir Crit Care Med 2021; 204: 1251–58. [DOI] [PubMed] [Google Scholar]
- 7.Adeloye D, Song P, Zhu Y, et al. Global, regional, and national prevalence of, and risk factors for, chronic obstructive pulmonary disease (COPD) in 2019: a systematic review and modelling analysis. Lancet Respir Med 2022; 10: 447–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang IA, Jenkins CR, Salvi SS. Chronic obstructive pulmonary disease in never-smokers: risk factors, pathogenesis, and implications for prevention and treatment. Lancet Respir Med 2022; 10: 497–511. [DOI] [PubMed] [Google Scholar]
- 9.Dransfield M, Stolz D, Kleinert S, Lancet COPD Commissioners. Towards eradication of chronic obstructive pulmonary disease: a Lancet Commission. Lancet 2019; 393: 1786–88. [DOI] [PubMed] [Google Scholar]
- 10.Chang HY, Chang JH, Chi H, et al. Reduced lung function at preschool age in survivors of very low birth weight preterm infants. Front Pediatr 2020; 8: 577673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Urs R, Kotecha S, Hall GL, Simpson SJ. Persistent and progressive long-term lung disease in survivors of preterm birth. Paediatr Respir Rev 2018; 28: 87–94. [DOI] [PubMed] [Google Scholar]
- 12.Gray DM, Turkovic L, Willemse L, et al. Lung function in African infants in the Drakenstein Child Health Study. Impact of lower respiratory tract illness. Am J Respir Crit Care Med 2017; 195: 212–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balmes JR. Household air pollution from domestic combustion of solid fuels and health. J Allergy Clin Immunol 2019; 143: 1979–87. [DOI] [PubMed] [Google Scholar]
- 14.Silver SR, Alarcon WA, Li J. Incident chronic obstructive pulmonary disease associated with occupation, industry, and workplace exposures in the Health and Retirement Study. Am J Ind Med 2021; 4: 26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Matteis S, Heederik D, Burdorf A, et al. Current and new challenges in occupational lung diseases. Eur Respir Rev 2017; 26: 170080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McDonough JE, Yuan R, Suzuki M, et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med 2011; 365: 1567–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Agusti A, Calverley PM, Celli B, et al. Characterisation of COPD heterogeneity in the ECLIPSE cohort. Respir Res 2010; 11: 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woodruff PG, Barr RG, Bleecker E, et al. Clinical significance of symptoms in smokers with preserved pulmonary function. N Engl J Med 2016; 374: 1811–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reyfman PA, Washko GR, Dransfield MT, Spira A, Han MK, Kalhan R. Defining impaired respiratory health. A paradigm shift for pulmonary medicine. Am J Respir Crit Care Med 2018; 198: 440–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lange P, Celli B, Agustí A, et al. Lung-function trajectories leading to chronic obstructive pulmonary disease. N Engl J Med 2015; 373: 111–22. [DOI] [PubMed] [Google Scholar]
- 21.Han MK, Agusti A, Celli BR, et al. From GOLD 0 to pre-COPD. Am J Respir Crit Care Med 2021; 203: 414–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kalhan R, Dransfield MT, Colangelo LA, et al. Respiratory symptoms in young adults and future lung disease. The CARDIA lung study. Am J Respir Crit Care Med 2018; 197: 1616–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stolz D, Christ-Crain M, Bingisser R, et al. Antibiotic treatment of exacerbations of COPD: a randomized, controlled trial comparing procalcitonin-guidance with standard therapy. Chest 2007; 131: 9–19. [DOI] [PubMed] [Google Scholar]
- 24.Pascoe S, Barnes N, Brusselle G, et al. Blood eosinophils and treatment response with triple and dual combination therapy in chronic obstructive pulmonary disease: analysis of the IMPACT trial. Lancet Respir Med 2019; 7: 745–56. [DOI] [PubMed] [Google Scholar]
- 25.Bafadhel M, McKenna S, Terry S, et al. Blood eosinophils to direct corticosteroid treatment of exacerbations of chronic obstructive pulmonary disease: a randomized placebo-controlled trial. Am J Respir Crit Care Med 2012; 186: 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Criner GJ, Bourbeau J, Diekemper RL, et al. Prevention of acute exacerbations of COPD: American College of Chest Physicians and Canadian Thoracic Society Guideline. Chest 2015; 147: 894–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buhl R, Bel E, Bourdin A, et al. Effective management of severe asthma with biologic medications in adult patients: a literature review and international expert opinion. J Allergy Clin Immunol Pract 2022; 10: 422–32. [DOI] [PubMed] [Google Scholar]
- 28.Vanfleteren LE, Spruit MA, Franssen FM. Tailoring the approach to multimorbidity in adults with respiratory disease: the NICE guideline. Eur Respir J 2017; 49: 1601696. [DOI] [PubMed] [Google Scholar]
- 29.Blackstock FC, ZuWallack R, Nici L, Lareau SC. Why don’t our patients with chronic obstructive pulmonary disease listen to us? The enigma of nonadherence. Ann Am Thorac Soc 2016; 3: 317–23. [DOI] [PubMed] [Google Scholar]
- 30.Vanoverschelde A, van der Wel P, Putman B, Lahousse L. Determinants of poor inhaler technique and poor therapy adherence in obstructive lung diseases: a cross-sectional study in community pharmacies. BMJ Open Respir Res 2021; 8: e000823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raju S, Keet CA, Paulin LM, et al. Rural residence and poverty are independent risk factors for chronic obstructive pulmonary disease in the United States. Am J Respir Crit Care Med 2019; 199: 961–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ejike CO, Woo H, Galiatsatos P, et al. Contribution of individual and neighborhood factors to racial disparities in respiratory outcomes. Am J Respir Crit Care Med 2021; 203: 987–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bui DS, Perret JL, Walters EH, et al. Association between very to moderate preterm births, lung function deficits, and COPD at age 53 years: a prospective cohort study over six decades. Lancet Respir Med 2022; 10: 478–84. [DOI] [PubMed] [Google Scholar]
- 34.WHO. Global strategy for women’s, children’s and adolescents’ health (2016–2030) 2018 monitoring report: current status and strategic priorities. Geneva: World Health Organization, 2018. [Google Scholar]
- 35.Siddharthan T, Grigsby MR, Goodman D, et al. Association between household air pollution exposure and chronic obstructive pulmonary disease outcomes in 13 low- and middle-income country settings. Am J Respir Crit Care Med 2018; 197: 611–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reitsma MB, Kendrick PJ, Ababneh E. Spatial, temporal, and demographic patterns in prevalence of smoking tobacco use and attributable disease burden in 204 countries and territories, 1990–2019: a systematic analysis from the Global Burden of Disease Study 2019. Lancet 2021; 397: 2337–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evans-Polce R, Veliz P, Boyd CJ, McCabe VV, McCabe SE. Trends in E-cigarette, cigarette, cigar, and smokeless tobacco use among US adolescent cohorts, 2014–2018. Am J Public Health 2020; 110: 163–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leijten FRM, Hoedemakers M, Struckmann V, et al. Defining good health and care from the perspective of persons with multimorbidity: results from a qualitative study of focus groups in eight European countries. BMJ Open 2018; 8: e021072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Han MK, Martinez CH, Au DH, et al. Meeting the challenge of COPD care delivery in the USA: a multiprovider perspective. Lancet Respir Med 2016; 4: 473–526. [DOI] [PubMed] [Google Scholar]
- 40.Ekpu VU, Brown AK. The economic impact of smoking and of reducing smoking prevalence: review of evidence. Tob Use Insights 2015; 8: 1–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DiGiulio A, Jump Z, Babb S, et al. State Medicaid coverage for tobacco cessation treatments and barriers to accessing treatments—United States 2008–2018. MMWR Morb Mortal Wkly Rep 2020; 69: 155–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.WHO. WHO report on the global tobacco epidemic 2019: offer help to quit tobacco use. Geneva: World Health Organization, 2019. [Google Scholar]
- 43.US National Institutes of Health. Estimates of funding for various research, condition, and disease categories (RCDC) 2022. report.nih.gov/funding/categorical-spending#/ (accessed Aug 16, 2022).
- 44.US Centers for Disease Control and Prevention. Data brief 427. Mortality in the United States. 2021. https://www.cdc.gov/nchs/data/databriefs/db427-tables.pdf#4 (accessed Aug 16, 2022).
- 45.Donohue JF, Jones PW, Bartels C, et al. Correlations between FEV1 and patient-reported outcomes: a pooled analysis of 23 clinical trials in patients with chronic obstructive pulmonary disease.Pulm Pharmacol Ther 2018; 49: 11–19. [DOI] [PubMed] [Google Scholar]
- 46.Bodduluri S, Reinhardt JM, Hoffman EA, Newell JD Jr, Bhatt SP. Recent advances in computed tomography imaging in chronic obstructive pulmonary disease. Ann Am Thorac Soc 2018; 15: 281–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lipson DA, Barnhart F, Brealey N, et al. Once-daily single-inhaler triple versus dual therapy in patients with COPD. N Engl J Med 2018; 378: 1671–80. [DOI] [PubMed] [Google Scholar]
- 48.Rabe KF, Martinez FJ, Ferguson GT, et al. Triple inhaled therapy at two glucocorticoid doses in moderate-to-very-severe COPD.N Engl J Med 2020; 383: 35–48. [DOI] [PubMed] [Google Scholar]
- 49.Fletcher C, Peto R. The natural history of chronic airflow obstruction. BMJ 1977; 1: 1645–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Postma DS, Bush A, van den Berge M. Risk factors and early origins of chronic obstructive pulmonary disease. Lancet 2015; 385: 899–909. [DOI] [PubMed] [Google Scholar]
- 51.Agustí A, Noell G, Brugada J, Faner R. Lung function in early adulthood and health in later life: a transgenerational cohort analysis. Lancet Respir Med 2017; 5: 935–45. [DOI] [PubMed] [Google Scholar]
- 52.Magnus MC, Henderson J, Tilling K, Howe LD, Fraser A. Independent and combined associations of maternal and own smoking with adult lung function and COPD. Int J Epidemiol 2018; 47: 1855–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perret JL, Walters H, Johns D, et al. Mother’s smoking and complex lung function of offspring in middle age: a cohort study from childhood. Respirology 2016; 21: 911–19. [DOI] [PubMed] [Google Scholar]
- 54.Thomsen M, Nordestgaard BG, Vestbo J, Lange P. Characteristics and outcomes of chronic obstructive pulmonary disease in never smokers in Denmark: a prospective population study. Lancet Respir Med 2013; 1: 543–50. [DOI] [PubMed] [Google Scholar]
- 55.Hassoun PM. Pulmonary arterial hypertension. N Engl J Med 2021; 385: 2361–76. [DOI] [PubMed] [Google Scholar]
- 56.Cho MH, Hobbs BD, Silverman EK. Genetics of chronic obstructive pulmonary disease: understanding the pathobiology and heterogeneity of a complex disorder. Lancet Respir Med 2022; 10: 485–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou JJ, Cho MH, Castaldi PJ, Hersh CP, Silverman EK, Laird NM. Heritability of chronic obstructive pulmonary disease and related phenotypes in smokers. Am J Respir Crit Care Med 2013; 188: 941–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Agustí A, Melén E, DeMeo DL, Breyer-Kohansal R, Faner R. Pathogenesis of chronic obstructive pulmonary disease: understanding the contributions of gene–environment interactions across the lifespan. Lancet Respir Med 2022; 10: 512–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.El-Boraie A, Tyndale RF. The role of pharmacogenetics in smoking. Clin Pharmacol Ther 2021; 110: 599–606. [DOI] [PubMed] [Google Scholar]
- 60.Strange C Alpha-1 antitrypsin deficiency associated COPD. Clin Chest Med 2020; 41: 339–45. [DOI] [PubMed] [Google Scholar]
- 61.Ortega VE, Li X, O’Neal WK, et al. The effects of rare SERPINA1 variants on lung function and emphysema in SPIROMICS. Am J Respir Crit Care Med 2020; 201: 540–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Konietzke P, Jobst B, Wagner WL, et al. Similarities in the computed tomography appearance in α1-antitrypsin deficiency and smoking-related chronic obstructive pulmonary disease in a smoking collective. Respiration 2018; 96: 231–39. [DOI] [PubMed] [Google Scholar]
- 63.Hoffman TW, van Moorsel CHM, Borie R, Crestani B. Pulmonary phenotypes associated with genetic variation in telomere-related genes. Curr Opin Pulm Med 2018; 24: 269–80. [DOI] [PubMed] [Google Scholar]
- 64.Amsellem V, Gary-Bobo G, Marcos E, et al. Telomere dysfunction causes sustained inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2011; 184: 1358–66. [DOI] [PubMed] [Google Scholar]
- 65.Ding Y, Li Q, Wu C, et al. TERT gene polymorphisms are associated with chronic obstructive pulmonary disease risk in the Chinese Li population. Mol Genet Genomic Med 2019; 7: e773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kordinas V, Ioannidis A, Chatzipanagiotou S. The telomere/telomerase system in chronic inflammatory diseases. Cause or effect? Genes (Basel) 2016; 7: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stanley SE, Merck SJ, Armanios M. Telomerase and the genetics of emphysema susceptibility. implications for pathogenesis paradigms and patient care. Ann Am Thorac Soc 2016; 13 (suppl 5): S447–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stanley SE, Chen JJL, Podlevsky JD, et al. Telomerase mutations in smokers with severe emphysema. J Clin Invest 2015; 125: 563–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Blencowe H, Cousens S, Oestergaard MZ, et al. National, regional, and worldwide estimates of preterm birth rates in the year 2010 with time trends since 1990 for selected countries: a systematic analysis and implications. Lancet 2012; 379: 2162–72. [DOI] [PubMed] [Google Scholar]
- 70.Baraldi E, Filippone M. Chronic lung disease after premature birth. N Engl J Med 2007; 357: 1946–55. [DOI] [PubMed] [Google Scholar]
- 71.Jobe AH, Bancalari E. Bronchopulmonary dysplasia. Am J Respir Crit Care Med 2001; 163: 1723–29. [DOI] [PubMed] [Google Scholar]
- 72.Allen J, Zwerdling R, Ehrenkranz R, et al. Statement on the care of the child with chronic lung disease of infancy and childhood. Am J Respir Crit Care Med 2003; 168: 356–96. [DOI] [PubMed] [Google Scholar]
- 73.Allen JL. Airway function throughout the lifespan: pediatric origins of adult respiratory disease. Pediatr Investig 2019; 3: 236–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Simpson SJ, Turkovic L, Wilson AC, et al. Lung function trajectories throughout childhood in survivors of very preterm birth: a longitudinal cohort study. Lancet Child Adolesc Health 2018; 2: 350–59. [DOI] [PubMed] [Google Scholar]
- 75.Bui DS, Lodge CJ, Perret JL, et al. Trajectories of asthma and allergies from 7 years to 53 years and associations with lung function and extrapulmonary comorbidity profiles: a prospective cohort study. Lancet Respir Med 2021; 9: 387–96. [DOI] [PubMed] [Google Scholar]
- 76.Aukland SM, Halvorsen T, Fosse KR, Daltveit AK, Rosendahl K. High-resolution CT of the chest in children and young adults who were born prematurely: findings in a population-based study. AJR Am J Roentgenol 2006; 187: 1012–18. [DOI] [PubMed] [Google Scholar]
- 77.Filippone M, Bonetto G, Cherubin E, Carraro S, Baraldi E. Childhood course of lung function in survivors of bronchopulmonary dysplasia. JAMA 2009; 302: 1418–20. [DOI] [PubMed] [Google Scholar]
- 78.Decrue F, Gorlanova O, Salem Y, et al. Increased impact of air pollution on lung function in preterm versus term infants: the BILD study. Am J Respir Crit Care Med 2022; 205: 99–107. [DOI] [PubMed] [Google Scholar]
- 79.Lodge CJ, Lowe AJ, Allen KJ, et al. Childhood wheeze phenotypes show less than expected growth in FEV1 across adolescence. Am J Respir Crit Care Med 2014; 189: 1351–58. [DOI] [PubMed] [Google Scholar]
- 80.Bui DS, Walters HE, Burgess JA, et al. Childhood respiratory risk factor profiles and middle-age lung function: a prospective cohort study from the first to sixth decade. Ann Am Thorac Soc 2018; 15: 1057–66. [DOI] [PubMed] [Google Scholar]
- 81.Saglani S, Malmström K, Pelkonen AS, et al. Airway remodeling and inflammation in symptomatic infants with reversible airflow obstruction. Am J Respir Crit Care Med 2005; 171: 722–27. [DOI] [PubMed] [Google Scholar]
- 82.Bui DS, Burgess JA, Lowe AJ, et al. Childhood lung function predicts adult chronic obstructive pulmonary disease and asthma-chronic obstructive pulmonary disease overlap syndrome. Am J Respir Crit Care Med 2017; 196: 39–46. [DOI] [PubMed] [Google Scholar]
- 83.Perret JL, Lodge CJ, Lowe AJ, et al. Childhood pneumonia, pleurisy and lung function: a cohort study from the first to sixth decade of life. Thorax 2020; 75: 28–37. [DOI] [PubMed] [Google Scholar]
- 84.Menezes AM, Hallal PC, Perez-Padilla R, et al. Tuberculosis and airflow obstruction: evidence from the PLATINO study in Latin America. Eur Respir J 2007; 30: 1180–85. [DOI] [PubMed] [Google Scholar]
- 85.Gingo MR, Nouraie M, Kessinger CJ, et al. Decreased lung function and all-cause mortality in HIV-infected individuals. Ann Am Thorac Soc 2018; 15: 192–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sarna M, Lambert SB, Sloots TP, et al. Viruses causing lower respiratory symptoms in young children: findings from the ORChID birth cohort. Thorax 2018; 73: 969–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Heyob KM, Mieth S, Sugar SS, et al. Maternal high-fat diet alters lung development and function in the offspring. Am J Physiol Lung Cell Mol Physiol 2019; 317: L167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rajappan A, Pearce A, Inskip HM, et al. Maternal body mass index: relation with infant respiratory symptoms and infections. Pediatr Pulmonol 2017; 52: 1291–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Holloway RA, Donnelly LE. Immunopathogenesis of chronic obstructive pulmonary disease. Curr Opin Pulm Med 2013; 19: 95–102. [DOI] [PubMed] [Google Scholar]
- 90.Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med 2008; 359: 2355–65. [DOI] [PubMed] [Google Scholar]
- 91.Yakar HI, Gunen H, Pehlivan E, Aydogan S. The role of tuberculosis in COPD. Int J Chron Obstruct Pulmon Dis 2017; 12: 323–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Amaral AF, Coton S, Kato B, et al. Lung function defects in treated pulmonary tuberculosis patients. Eur Respir J 2016; 47: 352–53. [DOI] [PubMed] [Google Scholar]
- 93.Guiedem E, Pefura-Yone EW, Ikomey GM, et al. Cytokine profile in the sputum of subjects with post-tuberculosis airflow obstruction and in those with tobacco related chronic obstructive pulmonary disease. BMC Immunol 2020; 21: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Drummond MB, Kunisaki KM, Huang L. Obstructive lung diseases in HIV: a clinical review and identification of key future research needs. Semin Respir Crit Care Med 2016; 37: 277–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sampériz G, Guerrero D, López M, et al. Prevalence of and risk factors for pulmonary abnormalities in HIV-infected patients treated with antiretroviral therapy. HIV Med 2014; 15: 321–29. [DOI] [PubMed] [Google Scholar]
- 96.Bigna JJ, Kenne AM, Asangbeh SL, Sibetcheu AT. Prevalence of chronic obstructive pulmonary disease in the global population with HIV: a systematic review and meta-analysis. Lancet Glob Health 2018; 6: e193–202. [DOI] [PubMed] [Google Scholar]
- 97.Triplette M, Attia EF, Akgün KM, et al. A low peripheral blood CD4/CD8 ratio is associated with pulmonary emphysema in HIV. PLoS One 2017; 12: e0170857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Drake P, Driscoll AK, Matthews TJ. Cigarette smoking during pregnancy: United States, 2016. 2018. https://www.cdc.gov/nchs/data/databriefs/db305.pdf (accessed Jan 9, 2020). [PubMed]
- 99.Vardavas CI, Hohmann C, Patelarou E, et al. The independent role of prenatal and postnatal exposure to active and passive smoking on the development of early wheeze in children. Eur Respir J 2016; 48: 115–24. [DOI] [PubMed] [Google Scholar]
- 100.Accordini S, Calciano L, Johannessen A, et al. Prenatal and prepubertal exposures to tobacco smoke in men may cause lower lung function in future offspring: a three-generation study using a causal modelling approach. Eur Respir J 2021; 58: 2002791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Elliot J, Vullermin P, Robinson P. Maternal cigarette smoking is associated with increased inner airway wall thickness in children who die from sudden infant death syndrome. Am J Respir Crit Care Med 1998; 158: 802–06. [DOI] [PubMed] [Google Scholar]
- 102.Gilliland FD, Berhane K, McConnell R, et al. Maternal smoking during pregnancy, environmental tobacco smoke exposure and childhood lung function. Thorax 2000; 55: 271–76.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Orhon FS, Ulukol B, Kahya D, Cengiz B, Başkan S, Tezcan S. The influence of maternal smoking on maternal and newborn oxidant and antioxidant status. Eur J Pediatr 2009; 168: 975–81. [DOI] [PubMed] [Google Scholar]
- 104.Chen X, Abdulhamid I, Woodcroft K. Maternal smoking during pregnancy, polymorphic CYP1A1 and GSTM1, and lung-function measures in urban family children. Environ Res 2011; 111: 1215–21. [DOI] [PubMed] [Google Scholar]
- 105.Dai X, Dharmage SC, Bowatte G, et al. Interaction of glutathione S-transferase M1, T1, and P1 genes with early life tobacco smoke exposure on lung function in adolescents. Chest 2019; 155: 94–102. [DOI] [PubMed] [Google Scholar]
- 106.Jia J, Conlon TM, Ballester Lopez C, et al. Cigarette smoke causes acute airway disease and exacerbates chronic obstructive lung disease in neonatal mice. Am J Physiol Lung Cell Mol Physiol 2016; 311: L602–10. [DOI] [PubMed] [Google Scholar]
- 107.Sekhon HS, Keller JA, Benowitz NL, Spindel ER. Prenatal nicotine exposure alters pulmonary function in newborn rhesus monkeys. Am J Respir Crit Care Med 2001; 164: 989–94. [DOI] [PubMed] [Google Scholar]
- 108.Carreras G, Lachi A, Cortini B, et al. Burden of disease from exposure to secondhand smoke in children in Europe. Pediatr Res 2021; 90: 216–22. [DOI] [PubMed] [Google Scholar]
- 109.Ho SY, Lam TH, Chung SF, Lam TP. Cross-sectional and prospective associations between passive smoking and respiratory symptoms at the workplace. Ann Epidemiol 2007; 17: 126–31. [DOI] [PubMed] [Google Scholar]
- 110.Liu Y, Di YP. Effects of second hand smoke on airway secretion and mucociliary clearance. Front Physiol 2012; 3: 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Upton MN, Smith GD, McConnachie A, Hart CL, Watt GC. Maternal and personal cigarette smoking synergize to increase airflow limitation in adults. Am J Respir Crit Care Med 2004; 169: 479–87. [DOI] [PubMed] [Google Scholar]
- 112.Lovasi GS, Diez Roux AV, Hoffman EA, Kawut SM, Jacobs DR Jr, Barr RG. Association of environmental tobacco smoke exposure in childhood with early emphysema in adulthood among nonsmokers: the MESA-lung study. Am J Epidemiol 2010; 171: 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Martin EM, Clapp PW, Rebuli ME, et al. E-cigarette use results in suppression of immune and inflammatory-response genes in nasal epithelial cells similar to cigarette smoke. Am J Physiol Lung Cell Mol Physiol 2016; 311: L135–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ghosh A, Coakley RC, Mascenik T, et al. Chronic e-cigarette exposure alters the human bronchial epithelial proteome. Am J Respir Crit Care Med 2018; 198: 67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bhatta DN, Glantz SA. Association of e-cigarette use with respiratory disease among adults: a longitudinal analysis. Am J Prev Med 2020; 58: 182–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Osei AD, Mirbolouk M, Orimoloye OA, et al. Association between e-cigarette use and chronic obstructive pulmonary disease by smoking status: behavioral risk factor surveillance system 2016 and 2017. Am J Prev Med 2020; 58: 336–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Perez MF, Atuegwu NC, Mead EL, Oncken C, Mortensen EM. Adult e-cigarettes use associated with a self-reported diagnosis of COPD. Int J Environ Res Public Health 2019; 16: 3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Government of Canada. Canadian cannabis survey 2020: summary. 2021. https://www.canada.ca/en/health-canada/services/drugs-medication/cannabis/research-data/canadian-cannabis-survey-2020-summary.html (accessed Dec 10, 2021).
- 119.Ghasemiesfe M, Ravi D, Vali M, et al. Marijuana use, respiratory symptoms, and pulmonary function: a systematic review and meta-analysis. Ann Intern Med 2018; 169: 106–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tan WC, Bourbeau J, Aaron SD, et al. The effects of marijuana smoking on lung function in older people. Eur Respir J 2019; 54: 1900826. [DOI] [PubMed] [Google Scholar]
- 121.Fligiel SE, Roth MD, Kleerup EC, Barsky SH, Simmons MS, Tashkin DP. Tracheobronchial histopathology in habitual smokers of cocaine, marijuana, and/or tobacco. Chest 1997; 112: 319–26. [DOI] [PubMed] [Google Scholar]
- 122.Baldwin GC, Tashkin DP, Buckley DM, Park AN, Dubinett SM, Roth MD. Marijuana and cocaine impair alveolar macrophage function and cytokine production. Am J Respir Crit Care Med 1997; 156: 1606–13. [DOI] [PubMed] [Google Scholar]
- 123.WHO. Residential heating with wood and coal: health impacts and policy options in Europe and North America. Copenhagen: World Health Organization, 2015. [Google Scholar]
- 124.Lakner C, Yonzan N, Mahler DG, Castaneda Aguilar RA, Wu H, Fleury M. Updated estimates of the impact of COVID-19 on global poverty: the effect of new data. 2020. https://www.un.org/development/desa/dspd/wp-content/uploads/sites/22/2021/05/Mahler_Paper.pdf (accessed Aug 3, 2022).
- 125.Pérez-Padilla R, Ramirez-Venegas A, Sansores-Martinez R. Clinical characteristics of patients with biomass smoke-associated COPD and chronic bronchitis, 2004–2014. Chronic Obstr Pulm Dis 2014; 1: 23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Camp PG, Ramirez-Venegas A, Sansores RH, et al. COPD phenotypes in biomass smoke-versus tobacco smoke-exposed Mexican women. Eur Respir J 2014; 43: 725–34. [DOI] [PubMed] [Google Scholar]
- 127.Zhao D, Zhou Y, Jiang C, Zhao Z, He F, Ran P. Small airway disease: a different phenotype of early stage COPD associated with biomass smoke exposure. Respirology 2018; 23: 198–205. [DOI] [PubMed] [Google Scholar]
- 128.Hansel NN, McCormack MC, Belli AJ, et al. In-home air pollution is linked to respiratory morbidity in former smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2013; 187: 1085–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shaddick G, Thomas ML, Mudu P, Ruggeri G, Gumy S. Half the world’s population are exposed to increasing air pollution. NPJ Clim Atmos Sci 2020; 3: 23. [Google Scholar]
- 130.WHO. WHO global air quality guidelines: particulate matter (PM2·5 and PM10), ozone, nitrogen dioxide, sulfur dioxide and carbon monoxide. Geneva: World Health Organization, 2021. [PubMed] [Google Scholar]
- 131.Latzin P, Röösli M, Huss A, Kuehni CE, Frey U. Air pollution during pregnancy and lung function in newborns: a birth cohort study. Eur Respir J 2009; 33: 594–603. [DOI] [PubMed] [Google Scholar]
- 132.Usemann J, Decrue F, Korten I, et al. Exposure to moderate air pollution and associations with lung function at school-age: a birth cohort study. Environ Int 2019; 126: 682–89. [DOI] [PubMed] [Google Scholar]
- 133.Gauderman WJ, Avol E, Gilliland F, et al. The effect of air pollution on lung development from 10 to 18 years of age. N Engl J Med 2004; 351: 1057–67. [DOI] [PubMed] [Google Scholar]
- 134.Smith Hopkins J The invisible hazard afflicting thousands of schools. The Center for Public Integrity, 2017. publicintegrity.org/environment/the-invisible-hazard-afflicting-thousands-of-schools/ (accessed Aug 14, 2022).
- 135.Ackermann-Liebrich U, Leuenberger P, Schwartz J, et al. Lung function and long term exposure to air pollutants in Switzerland. Study on air pollution and lung diseases in adults (SAPALDIA) team. Am J Respir Crit Care Med 1997; 155: 122–29. [DOI] [PubMed] [Google Scholar]
- 136.National Centers for Environmental Information. June 2022 wildfires report. 2022. https://www.ncei.noaa.gov/access/monitoring/monthly-report/fire/202206 (accessed Aug 5, 2022).
- 137.Groot E, Caturay A, Khan Y, Copes R. A systematic review of the health impacts of occupational exposure to wildland fires. Int J Occup Med Environ Health 2019; 32: 121–40. [DOI] [PubMed] [Google Scholar]
- 138.Liu JC, Wilson A, Mickley LJ, et al. Who among the elderly is most vulnerable to exposure to and health risks of fine particulate matter from wildfire smoke? Am J Epidemiol 2017; 186: 730–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Künzli N, Avol E, Wu J, et al. Health effects of the 2003 Southern California wildfires on children. Am J Respir Crit Care Med 2006; 174: 1221–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Roscioli E, Hamon R, Lester SE, Jersmann HPA, Reynolds PN, Hodge S. Airway epithelial cells exposed to wildfire smoke extract exhibit dysregulated autophagy and barrier dysfunction consistent with COPD. Respir Res 2018; 19: 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Paulin LM, Smith BM, Koch A, et al. Occupational exposures and computed tomographic imaging characteristics in the SPIROMICS cohort. Ann Am Thorac Soc 2018; 15: 1411–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sadhra S, Kurmi OP, Sadhra SS, Lam KB, Ayres JG. Occupational COPD and job exposure matrices: a systematic review and meta-analysis. Int J Chron Obstruct Pulmon Dis 2017; 12: 725–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Agusti A The path to personalised medicine in COPD. Thorax 2014; 69: 857–64. [DOI] [PubMed] [Google Scholar]
- 144.Jones P, Miravitlles M, van der Molen T, Kulich K. Beyond FEV₁ in COPD: a review of patient-reported outcomes and their measurement. Int J Chron Obstruct Pulmon Dis 2012; 7: 697–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lopez-Campos JL, Bustamante V, Muñoz X, Barreiro E. Moving towards patient-centered medicine for COPD management: multidimensional approaches versus phenotype-based medicine—a critical view. COPD 2014; 11: 591–602. [DOI] [PubMed] [Google Scholar]
- 146.Dournes G, Laurent F, Coste F, et al. Computed tomographic measurement of airway remodeling and emphysema in advanced chronic obstructive pulmonary disease. Correlation with pulmonary hypertension. Am J Respir Crit Care Med 2015; 191: 63–70. [DOI] [PubMed] [Google Scholar]
- 147.Greulich T, Weist BJD, Koczulla AR, et al. Prevalence of comorbidities in COPD patients by disease severity in a German population. Respir Med 2017; 132: 132–38. [DOI] [PubMed] [Google Scholar]
- 148.Chen W, Thomas J, Sadatsafavi M, FitzGerald JM. Risk of cardiovascular comorbidity in patients with chronic obstructive pulmonary disease: a systematic review and meta-analysis. Lancet Respir Med 2015; 3: 631–39. [DOI] [PubMed] [Google Scholar]
- 149.MacDonald MI, Shafuddin E, King PT, Chang CL, Bardin PG, Hancox RJ. Cardiac dysfunction during exacerbations of chronic obstructive pulmonary disease. Lancet Respir Med 2016; 4: 138–48. [DOI] [PubMed] [Google Scholar]
- 150.Balbirsingh V, Mohammed AS, Turner AM, Newnham M. Cardiovascular disease in chronic obstructive pulmonary disease: a narrative review. Thorax 2022; published online June 30. DOI: 10.1136/thoraxjnl-2021-218333. [DOI] [PubMed] [Google Scholar]
- 151.Evans WJ, Morley JE, Argilés J, et al. Cachexia: a new definition. Clin Nutr 2008; 27: 793–99. [DOI] [PubMed] [Google Scholar]
- 152.McDonald MN, Wouters EFM, Rutten E, et al. It’s more than low BMI: prevalence of cachexia and associated mortality in COPD. Respir Res 2019; 20: 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.O’Toole J, Woo H, Putcha N, et al. Comparative impact of depressive symptoms and FEV1% on chronic obstructive pulmonary disease. Ann Am Thorac Soc 2022; 19: 171–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Iyer AS, Parekh TM, O’Toole J, et al. Clinically significant and comorbid anxiety and depression symptoms predict severe respiratory exacerbations in smokers: a post hoc analysis of the COPDGene and SPIROMICS cohorts. Ann Am Thorac Soc 2022; 19: 143–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Miller J, Edwards LD, Agustí A, et al. Comorbidity, systemic inflammation and outcomes in the ECLIPSE cohort. Respir Med 2013; 107: 1376–84. [DOI] [PubMed] [Google Scholar]
- 156.Ernst P, Saad N, Suissa S. Inhaled corticosteroids in COPD: the clinical evidence. Eur Respir J 2015; 45: 525–37. [DOI] [PubMed] [Google Scholar]
- 157.Bolton CE, Evans M, Ionescu AA, et al. Insulin resistance and inflammation—a further systemic complication of COPD. COPD 2007; 4: 121–26. [DOI] [PubMed] [Google Scholar]
- 158.Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2021; 71: 209–49. [DOI] [PubMed] [Google Scholar]
- 159.Park HY, Kang D, Shin SH, et al. Chronic obstructive pulmonary disease and lung cancer incidence in never smokers: a cohort study. Thorax 2020; 75: 506–09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Aberle DR, Adams AM, Berg CD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011; 365: 395–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yong PC, Sigel K, Rehmani S, Wisnivesky J, Kale MS. Lung cancer screening uptake in the United States. Chest 2020; 157: 236–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Edelman Saul E, Guerra RB, Edelman Saul M, et al. The challenges of implementing low-dose computed tomography for lung cancer screening in low- and middle-income countries. Nature Cancer 2020; 1: 1140–52. [DOI] [PubMed] [Google Scholar]
- 163.Criner GJ, Agusti A, Borghaei H, et al. Chronic obstructive pulmonary disease and lung cancer: a review for clinicians. Chronic Obstr Pulm Dis 2022; 9: 454–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Young RP, Hopkins RJ. Chronic obstructive pulmonary disease (COPD) and lung cancer screening. Transl Lung Cancer Res 2018; 7: 347–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Bui DS, Lodge CJ, Burgess JA, et al. Childhood predictors of lung function trajectories and future COPD risk: a prospective cohort study from the first to the sixth decade of life. Lancet Respir Med 2018; 6: 535–44. [DOI] [PubMed] [Google Scholar]
- 166.Wan ES, Castaldi PJ, Cho MH, et al. Epidemiology, genetics, and subtyping of preserved ratio impaired spirometry (PRISm) in COPDGene. Respir Res 2014; 15: 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Karakioulaki M, Papakonstantinou E, Stolz D. Extracellular matrix remodelling in COPD. Eur Respir Rev 2020; 29: 190124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Schumann DM, Leeming D, Papakonstantinou E, et al. Collagen degradation and formation are elevated in exacerbated COPD compared with stable disease. Chest 2018; 154: 798–807. [DOI] [PubMed] [Google Scholar]
- 169.Papakonstantinou E, Roth M, Klagas I, Karakiulakis G, Tamm M, Stolz D. COPD exacerbations are associated with proinflammatory degradation of hyaluronic acid. Chest 2015; 148: 1497–507. [DOI] [PubMed] [Google Scholar]
- 170.Schumann DM, Tamm M, Kostikas K, Stolz D. Stability of the blood eosinophilic phenotype in stable and exacerbated COPD. Chest 2019; 156: 456–65. [DOI] [PubMed] [Google Scholar]
- 171.Singh D, Wedzicha JA, Siddiqui S, et al. Blood eosinophils as a biomarker of future COPD exacerbation risk: pooled data from 11 clinical trials. Respir Res 2020; 21: 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Pesci A, Majori M, Cuomo A, et al. Neutrophils infiltrating bronchial epithelium in chronic obstructive pulmonary disease. Respir Med 1998; 92: 863–70. [DOI] [PubMed] [Google Scholar]
- 173.Stănescu D, Sanna A, Veriter C, et al. Airways obstruction, chronic expectoration, and rapid decline of FEV1 in smokers are associated with increased levels of sputum neutrophils. Thorax 1996; 51: 267–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Man WH, van Houten MA, Mérelle ME, et al. Bacterial and viral respiratory tract microbiota and host characteristics in children with lower respiratory tract infections: a matched case-control study. Lancet Respir Med 2019; 7: 417–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wang Z, Locantore N, Haldar K, et al. Inflammatory endotype associated airway microbiome in COPD clinical stability and exacerbations—a multi-cohort longitudinal analysis. Am J Respir Crit Care Med 2021; 203: 1488–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Dicker AJ, Huang JTJ, Lonergan M, et al. The sputum microbiome, airway inflammation, and mortality in chronic obstructive pulmonary disease. J Allergy Clin Immunol 2021; 147: 158–67. [DOI] [PubMed] [Google Scholar]
- 177.Mayhew D, Devos N, Lambert C, et al. Longitudinal profiling of the lung microbiome in the AERIS study demonstrates repeatability of bacterial and eosinophilic COPD exacerbations. Thorax 2018; 73: 422–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Tiew PY, Dicker AJ, Keir HR, et al. A high-risk airway mycobiome is associated with frequent exacerbation and mortality in COPD. Eur Respir J 2020; 57: 2002050. [DOI] [PubMed] [Google Scholar]
- 179.Ramsheh MY, Haldar K, Esteve-Codina A, et al. Lung microbiome composition and bronchial epithelial gene expression in patients with COPD versus healthy individuals: a bacterial 16S rRNA gene sequencing and host transcriptomic analysis. Lancet Microbe 2021; 2: e300–10. [DOI] [PubMed] [Google Scholar]
- 180.Agusti A, Bel E, Thomas M, et al. Treatable traits: toward precision medicine of chronic airway diseases. Eur Respir J 2016; 47: 410–19. [DOI] [PubMed] [Google Scholar]
- 181.Lowe KE, Regan EA, Anzueto A, et al. COPDGene 2019: redefining the diagnosis of chronic obstructive pulmonary disease. Chronic Obstr Pulm Dis 2019; 6: 384–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Bhatt SP, Balte PP, Schwartz JE, et al. Discriminative accuracy of FEV1:FVC thresholds for COPD-related hospitalization and mortality. JAMA 2019; 321: 2438–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Albert P, Agusti A, Edwards L, et al. Bronchodilator responsiveness as a phenotypic characteristic of established chronic obstructive pulmonary disease. Thorax 2012; 67: 701–08. [DOI] [PubMed] [Google Scholar]
- 184.Sumida K, Kwak L, Grams ME, et al. Lung function and incident kidney disease: the atherosclerosis risk in communities (ARIC) study. Am J Kidney Dis 2017; 70: 675–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Cuttica MJ, Colangelo LA, Dransfield MT, et al. Lung function in young adults and risk of cardiovascular events over 29 years: the CARDIA study. J Am Heart Assoc 2018; 7: e010672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Topalovic M, Derom E, Osadnik CR, Troosters T, Decramer M, Janssens W. Airways resistance and specific conductance for the diagnosis of obstructive airways diseases. Respir Res 2015; 16: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Bhattarai P, Myers S, Chia C, et al. Clinical application of forced oscillation technique (FOT) in early detection of airway changes in smokers. J Clin Med 2020; 9: 2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.King GG, Bates J, Berger KI, et al. Technical standards for respiratory oscillometry. Eur Respir J 2020; 55: 1900753. [DOI] [PubMed] [Google Scholar]
- 189.Bellisario V, Piccioni P, Bugiani M, et al. Tobacco smoke exposure, urban and environmental factors as respiratory disease predictors in Italian adolescents. Int J Environ Res Public Health 2019; 16: 4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Nyilas S, Singer F, Kumar N, et al. Physiological phenotyping of pediatric chronic obstructive airway diseases. J Appl Physiol 2016; 121: 324–32. [DOI] [PubMed] [Google Scholar]
- 191.Boeck L, Gensmer A, Nyilas S, et al. Single-breath washout tests to assess small airway disease in COPD. Chest 2016; 150: 1091–100. [DOI] [PubMed] [Google Scholar]
- 192.Olofson J, Bake B, Bergman B, Vanfleteren L, Svärdsudd K. Prediction of COPD by the single-breath nitrogen test and various respiratory symptoms. ERJ Open Res 2021; 7: 0383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Papakonstantinou E, Savic S, Siebeneichler A, et al. A pilot study to test the feasibility of histological characterisation of asthma–COPD overlap. Eur Respir J 2019; 53: 1801941. [DOI] [PubMed] [Google Scholar]
- 194.Stolz D, Leeming DJ, Kristensen JHE, et al. Systemic biomarkers of collagen and elastin turnover are associated with clinically relevant outcomes in COPD. Chest 2017; 151: 47–59. [DOI] [PubMed] [Google Scholar]
- 195.Wen Y, Reid DW, Zhang D, Ward C, Wood-Baker R, Walters EH. Assessment of airway inflammation using sputum, BAL, and endobronchial biopsies in current and ex-smokers with established COPD. Int J Chron Obstruct Pulmon Dis 2010; 5: 327–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Southworth T, Higham A, Kolsum U, et al. The relationship between airway immunoglobulin activity and eosinophils in COPD. J Cell Mol Med 2021; 25: 2203–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Wells JM, Arenberg DA, Barjaktarevic I, et al. Safety and tolerability of comprehensive research bronchoscopy in chronic obstructive pulmonary disease. Results from the SPIROMICS bronchoscopy substudy. Ann Am Thorac Soc 2019; 16: 439–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Slovis BH, Lowry T, Delman BN, et al. Patient crossover and potentially avoidable repeat computed tomography exams across a health information exchange. J Am Med Inform Assoc 2017; 24: 30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Smith BM, Kirby M, Hoffman EA, et al. Association of dysanapsis with chronic obstructive pulmonary disease among older adults. JAMA 2020; 323: 2268–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Bhatt SP, Soler X, Wang X, et al. Association between functional small airway disease and FEV1 decline in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2016; 194: 178–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Bafadhel M, Criner G, Dransfield MT, et al. Exacerbations of chronic obstructive pulmonary disease: time to rename. Lancet Respir Med 2020; 8: 133–35. [DOI] [PubMed] [Google Scholar]
- 202.Mwasuku C, Krassowska K, Coleman C, Powell P, Bafadhel M. Rethinking COPD exacerbations: a global patient perspective. Eur Respir J 2021; 58: PA1805. [Google Scholar]
- 203.Frei A, Siebeling L, Wolters C, et al. The inaccuracy of patient recall for COPD exacerbation rate estimation and its implications: results from central adjudication. Chest 2016; 150: 860–68. [DOI] [PubMed] [Google Scholar]
- 204.Holverda S, Rutgers MR, Kerstjens HAM. Time to rename COPD exacerbations: implementing the term lung attack. Lancet Respir Med 2020; 8: e25. [DOI] [PubMed] [Google Scholar]
- 205.Thygesen K, Alpert JS, Jaffe AS, et al. Fourth universal definition of myocardial infarction (2018). Circulation 2018; 138: e618–51. [DOI] [PubMed] [Google Scholar]
- 206.Anthonisen NR, Manfreda J, Warren CP, Hershfield ES, Harding GK, Nelson NA. Antibiotic therapy in exacerbations of chronic obstructive pulmonary disease. Ann Intern Med 1987; 106: 196–204. [DOI] [PubMed] [Google Scholar]
- 207.Dransfield MT, Crim C, Criner GJ, et al. Risk of exacerbation and pneumonia with single-inhaler triple versus dual therapy in IMPACT. Ann Am Thorac Soc 2021; 18: 788–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Rizkallah J, Man SFP, Sin DD. Prevalence of pulmonary embolism in acute exacerbations of COPD: a systematic review and metaanalysis. Chest 2009; 135: 786–93. [DOI] [PubMed] [Google Scholar]
- 209.Rothnie KJ, Smeeth L, Pearce N, et al. Predicting mortality after acute coronary syndromes in people with chronic obstructive pulmonary disease. Heart 2016; 102: 1442–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Stolz D, Hirsch HH, Schilter D, et al. Intensified therapy with inhaled corticosteroids and long-acting β(2)-agonists at the onset of upper respiratory tract infection to prevent chronic obstructive pulmonary disease exacerbations. A multicenter, randomized, double-blind, placebo-controlled trial. Am J Respir Crit Care Med 2018; 197: 1136–46. [DOI] [PubMed] [Google Scholar]
- 211.Tan JY, Conceicao EP, Wee LE, Sim XYJ, Venkatachalam I. COVID-19 public health measures: a reduction in hospital admissions for COPD exacerbations. Thorax 2021; 76: 512–13. [DOI] [PubMed] [Google Scholar]
- 212.Stolz D, Papakonstantinou E, Grize L, et al. Time-course of upper respiratory tract viral infection and COPD exacerbation. Eur Respir J 2019; 54: 1900407. [DOI] [PubMed] [Google Scholar]
- 213.Barker BL, Haldar K, Patel H, et al. Association between pathogens detected using quantitative polymerase chain reaction with airway inflammation in COPD at stable state and exacerbations. Chest 2015; 147: 46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Bafadhel M, McKenna S, Terry S, et al. Acute exacerbations of chronic obstructive pulmonary disease: identification of biologic clusters and their biomarkers. Am J Respir Crit Care Med 2011; 184: 662–71. [DOI] [PubMed] [Google Scholar]
- 215.Kim VL, Coombs NA, Staples KJ, et al. Impact and associations of eosinophilic inflammation in COPD: analysis of the AERIS cohort. Eur Respir J 2017; 50: 1700853. [DOI] [PubMed] [Google Scholar]
- 216.Crisafulli E, Manco A, Ferrer M, et al. Pneumonic versus nonpneumonic exacerbations of chronic obstructive pulmonary disease. Semin Respir Crit Care Med 2020; 41: 817–29. [DOI] [PubMed] [Google Scholar]
- 217.Cordova FC, Ciccolella D, Grabianowski C, et al. A telemedicine-based intervention reduces the frequency and severity of COPD exacerbation symptoms: a randomized, controlled trial. Telemed J E Health 2016; 22: 114–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Hoffer-Hawlik M, Moran A, Zerihun L, Usseglio J, Cohn J, Gupta R. Telemedicine interventions for hypertension management in low- and middle-income countries: a scoping review. PLoS One 2021; 16: e0254222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Correia JC, Meraj H, Teoh SH, et al. Telemedicine to deliver diabetes care in low- and middle-income countries: a systematic review and meta-analysis. Bull World Health Organ 2021; 99: 209–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Sandberg CEJ, Knight SR, Qureshi AU, Pathak S. Using telemedicine to diagnose surgical site infections in low- and middle-income countries: systematic review. JMIR Mhealth Uhealth 2019; 7: e13309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Dash S, Aarthy R, Mohan V. Telemedicine during COVID-19 in India—a new policy and its challenges. J Public Health Policy 2021; 42: 501–09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Delgado-Eckert E, James A, Meier-Girard D, et al. Lung function fluctuation patterns unveil asthma and COPD phenotypes unrelated to type 2 inflammation. J Allergy Clin Immunol 2021; 148: 407–19. [DOI] [PubMed] [Google Scholar]
- 223.Krings JG, Goss CW, Lew D, et al. Quantitative CT metrics are associated with longitudinal lung function decline and future asthma exacerbations: results from SARP-3. J Allergy Clin Immunol 2021; 148: 752–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Winkler T, Frey U. Airway remodeling: shifting the trigger point for exacerbations in asthma. J Allergy Clin Immunol 2021; 148: 710–12. [DOI] [PubMed] [Google Scholar]
- 225.Dvir T, Bauer M, Schroeder A, et al. Nanoparticles targeting the infarcted heart. Nano Lett 2011; 11: 4411–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Candelario-Jalil E Injury and repair mechanisms in ischemic stroke: considerations for the development of novel neurotherapeutics. Curr Opin Investig Drugs 2009; 10: 644–54. [PubMed] [Google Scholar]
- 227.Waljee AK, Rogers MA, Lin P, et al. Short term use of oral corticosteroids and related harms among adults in the United States: population based cohort study. BMJ 2017; 357: j1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Albert RK, Martin TR, Lewis SW. Controlled clinical trial of methylprednisolone in patients with chronic bronchitis and acute respiratory insufficiency. Ann Intern Med 1980; 92: 753–58. [DOI] [PubMed] [Google Scholar]
- 229.Walters JA, Tan DJ, White CJ, Gibson PG, Wood-Baker R, Walters EH. Systemic corticosteroids for acute exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2014; 9: CD001288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Lenferink A, van der Palen J, van der Valk P, et al. Exacerbation action plans for patients with COPD and comorbidities: a randomised controlled trial. Eur Respir J 2019; 54: 1802134. [DOI] [PubMed] [Google Scholar]
- 231.Hartl S, Lopez-Campos JL, Pozo-Rodriguez F, et al. Risk of death and readmission of hospital-admitted COPD exacerbations: European COPD audit. Eur Respir J 2016; 47: 113–21. [DOI] [PubMed] [Google Scholar]
- 232.Martinez FJ, Calverley PMA, Goehring U-M, Brose M, Fabbri LM, Rabe KF. Effect of roflumilast on exacerbations in patients with severe chronic obstructive pulmonary disease uncontrolled by combination therapy (REACT): a multicentre randomised controlled trial. Lancet 2015; 385: 857–66. [DOI] [PubMed] [Google Scholar]
- 233.van Haarst A, McGarvey L, Paglialunga S. Review of drug development guidance to treat chronic obstructive pulmonary disease: US and EU perspectives. Clin Pharmacol Ther 2019; 106: 1222–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Rennard SI, Dale DC, Donohue JF, et al. CXCR2 antagonist MK-7123. A phase 2 proof-of-concept trial for chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2015; 191: 1001–11. [DOI] [PubMed] [Google Scholar]
- 235.Rennard SI, Fogarty C, Kelsen S, et al. The safety and efficacy of infliximab in moderate to severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2007; 175: 926–34. [DOI] [PubMed] [Google Scholar]
- 236.Boucher RC. Muco-obstructive lung diseases. N Engl J Med 2019; 380: 1941–53. [DOI] [PubMed] [Google Scholar]
- 237.Hartman JE, Garner JL, Shah PL, Slebos DJ. New bronchoscopic treatment modalities for patients with chronic bronchitis. Eur Respir Rev 2021; 30: 200281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Raju SV, Tate JH, Peacock SK, et al. Impact of heterozygote CFTR mutations in COPD patients with chronic bronchitis. Respir Res 2014; 15: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Raju SV, Jackson PL, Courville CA, et al. Cigarette smoke induces systemic defects in cystic fibrosis transmembrane conductance regulator function. Am J Respir Crit Care Med 2013; 188: 1321–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Raju SV, Lin VY, Liu L, et al. The cystic fibrosis transmembrane conductance regulator potentiator ivacaftor augments mucociliary clearance abrogating cystic fibrosis transmembrane conductance regulator inhibition by cigarette smoke. Am J Respir Cell Mol Biol 2017; 56: 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Dransfield MT, Wilhelm AM, Flanagan B, et al. Acquired cystic fibrosis transmembrane conductance regulator dysfunction in the lower airways in COPD. Chest 2013; 144: 498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Dransfield M, Rowe S, Vogelmeier CF, et al. Cystic fibrosis transmembrane conductance regulator: roles in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2022; 205: 631–40. [DOI] [PubMed] [Google Scholar]
- 243.Verberkt CA, van den Beuken-van Everdingen MHJ, Schols J, Hameleers N, Wouters EFM, Janssen DJA. Effect of sustained-release morphine for refractory breathlessness in chronic obstructive pulmonary disease on health status: a randomized clinical trial. JAMA Intern Med 2020; 180: 1306–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Tsai CL, Lan CC, Wu CW, et al. Acupuncture point stimulation treatments combined with conventional treatment in chronic obstructive pulmonary disease: a systematic review and network meta-analysis. Front Med 2021; 8: 586900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Alotaibi NM, Chen V, Hollander Z, et al. Phenotyping and outcomes of hospitalized COPD patients using rapid molecular diagnostics on sputum samples. Int J Chron Obstruct Pulmon Dis 2019; 14: 311–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Mohan A, Chandra S, Agarwal D, et al. Prevalence of viral infection detected by PCR and RT-PCR in patients with acute exacerbation of COPD: a systematic review. Respirology 2010; 15: 536–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Shukla SD, Shastri MD, Vanka SK, et al. Targeting intercellular adhesion molecule-1 (ICAM-1) to reduce rhinovirus-induced acute exacerbations in chronic respiratory diseases. Inflammopharmacology 2022; 30: 725–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Donovan C, Bourke JE, Vlahos R. Targeting the IL-33/IL-13 axis for respiratory viral infections. Trends Pharmacol Sci 2016; 37: 252–61. [DOI] [PubMed] [Google Scholar]
- 249.Dimasuay KG, Sanchez A, Schaefer N, Polanco J, Ferrington DA, Chu HW. Immunoproteasomes as a novel antiviral mechanism in rhinovirus-infected airways. Clin Sci 2018; 132: 1711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Galdi F, Pedone C, McGee CA, et al. Inhaled high molecular weight hyaluronan ameliorates respiratory failure in acute COPD exacerbation: a pilot study. Respir Res 2021; 22: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Dumont EF, Oliver AJ, Ioannou C, et al. A novel inhaled dry-powder formulation of ribavirin allows for efficient lung delivery in healthy participants and those with chronic obstructive pulmonary disease in a phase 1 study. Antimicrob Agents Chemother 2020; 64: e02267–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Martinez-Baz I, Casado I, Navascues A, et al. Chronic obstructive pulmonary disease and influenza vaccination effect in preventing outpatient and inpatient influenza cases. Sci Rep 2022; 12: 4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Sivapalan P, Lapperre TS, Janner J, et al. Eosinophil-guided corticosteroid therapy in patients admitted to hospital with COPD exacerbation (CORTICO-COP): a multicentre, randomised, controlled, open-label, non-inferiority trial. Lancet Respir Med 2019; 7: 699–709. [DOI] [PubMed] [Google Scholar]
- 254.Voynow JA, Shinbashi M. Neutrophil elastase and chronic lung disease. Biomolecules 2021; 11: 1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Vogelmeier C, Aquino TO, O’Brien CD, Perrett J, Gunawardena KA. A randomised, placebo-controlled, dose-finding study of AZD9668, an oral inhibitor of neutrophil elastase, in patients with chronic obstructive pulmonary disease treated with tiotropium. COPD 2012; 9: 111–20. [DOI] [PubMed] [Google Scholar]
- 256.Chalmers JD, Haworth CS, Metersky ML, et al. Phase 2 trial of the DPP-1 inhibitor brensocatib in bronchiectasis. N Engl J Med 2020; 383: 2127–37. [DOI] [PubMed] [Google Scholar]
- 257.Agustí A, Bafadhel M, Beasley R, et al. Precision medicine in airway diseases: moving to clinical practice. Eur Respir J 2017; 50: 1701655. [DOI] [PubMed] [Google Scholar]
- 258.Pignatti P, Visca D, Cherubino F, et al. Do blood eosinophils strictly reflect airway inflammation in COPD? Comparison with asthmatic patients. Respir Res 2019; 20: 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Bafadhel M, Peterson S, De Blas MA, et al. Predictors of exacerbation risk and response to budesonide in patients with chronic obstructive pulmonary disease: a post-hoc analysis of three randomised trials. Lancet Respir Med 2018; 6: 117–26. [DOI] [PubMed] [Google Scholar]
- 260.Pavord ID, Chanez P, Criner GJ, et al. Mepolizumab for eosinophilic chronic obstructive pulmonary disease. N Engl J Med 2017; 377: 1613–29. [DOI] [PubMed] [Google Scholar]
- 261.Criner GJ, Celli BR, Brightling CE, et al. Benralizumab for the prevention of COPD exacerbations. N Engl J Med 2019; 381: 1023–34. [DOI] [PubMed] [Google Scholar]
- 262.Donovan T, Milan SJ, Wang R, Banchoff E, Bradley P, Crossingham I. Anti-IL-5 therapies for chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2020; 12: CD013432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Menzies-Gow A, Corren J, Bourdin A, et al. Tezepelumab in adults and adolescents with severe, uncontrolled asthma. N Engl J Med 2021; 384: 1800–09. [DOI] [PubMed] [Google Scholar]
- 264.Rabe KF, Celli BR, Wechsler ME, et al. Safety and efficacy of itepekimab in patients with moderate-to-severe COPD: a genetic association study and randomised, double-blind, phase 2a trial. Lancet Respir Med 2021; 9: 1288–98. [DOI] [PubMed] [Google Scholar]
- 265.Wechsler ME, Ruddy MK, Pavord ID, et al. Efficacy and safety of itepekimab in patients with moderate-to-severe asthma. N Engl J Med 2021; 385: 1656–68. [DOI] [PubMed] [Google Scholar]
- 266.Taylor SL, Simpson JL, Rogers GB. The influence of early-life microbial exposures on long-term respiratory health. Paediatr Respir Rev 2021; 40: 15–23. [DOI] [PubMed] [Google Scholar]
- 267.Segal LN, Clemente JC, Wu BG, et al. Randomised, double-blind, placebo-controlled trial with azithromycin selects for anti-inflammatory microbial metabolites in the emphysematous lung. Thorax 2017; 72: 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Brasier AR. Therapeutic targets for inflammation-mediated airway remodeling in chronic lung disease. Expert Rev Respir Med 2018; 12: 931–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Carro LM, Martínez-García MA. Use of hyaluronic acid (HA) in chronic airway diseases. Cells 2020; 9: 2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Rock J, Königshoff M. Endogenous lung regeneration: potential and limitations. Am J Respir Crit Care Med 2012; 186: 1213–19. [DOI] [PubMed] [Google Scholar]
- 271.Calle EA, Ghaedi M, Sundaram S, Sivarapatna A, Tseng MK, Niklason LE. Strategies for whole lung tissue engineering. IEEE Trans Biomed Eng 2014; 61: 1482–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Vaughan AE, Brumwell AN, Xi Y, et al. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature 2015; 517: 621–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Nabhan AN, Brownfield DG, Harbury PB, Krasnow MA, Desai TJ. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 2018; 359: 1118–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Hogan BL, Barkauskas CE, Chapman HA, et al. Repair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell 2014; 15: 123–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Zuo W, Zhang T, Wu DZ, et al. p63(+)Krt5(+) distal airway stem cells are essential for lung regeneration. Nature 2015; 517: 616–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Staudt MR, Buro-Auriemma LJ, Walters MS, et al. Airway basal stem/progenitor cells have diminished capacity to regenerate airway epithelium in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2014; 190: 955–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Weiss DJ, Casaburi R, Flannery R, LeRoux-Williams M, Tashkin DP. A placebo-controlled, randomized trial of mesenchymal stem cells in COPD. Chest 2013; 143: 1590–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Squassoni SD, Sekiya EJ, Fiss E, et al. Autologous infusion of bone marrow and mesenchymal stromal cells in patients with chronic obstructive pulmonary disease: phase I randomized clinical trial. Int J Chron Obstruct Pulmon Dis 2021; 16: 3561–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Elowsson Rendin L, Löfdahl A, Kadefors M, et al. Harnessing the ECM microenvironment to ameliorate mesenchymal stromal cell-based therapy in chronic lung diseases. Front Pharmacol 2021; 12: 645558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.De Santis MM, Alsafadi HN, Tas S, et al. Extracellular-matrix-reinforced bioinks for 3D bioprinting human tissue. Adv Mater 2021; 33: e2005476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Holland AE, Cox NS, Houchen-Wolloff L, et al. Defining modern pulmonary rehabilitation. An official American Thoracic Society workshop report. Ann Am Thorac Soc 2021; 18: e12–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Troosters T, Maltais F, Leidy N, et al. Effect of bronchodilation, exercise training, and behavior modification on symptoms and physical activity in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2018; 198: 1021–32. [DOI] [PubMed] [Google Scholar]
- 283.Bhatt SP, Baugh D, Hitchcock J, et al. Video telehealth pulmonary rehabilitation for chronic obstructive pulmonary disease is associated with clinical improvement similar to center-based pulmonary rehabilitation. Ann Am Thorac Soc 2022; 19: 331–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Balasubramanian V, Naing S. Hypogonadism in chronic obstructive pulmonary disease: incidence and effects. Curr Opin Pulm Med 2012; 18: 112–17. [DOI] [PubMed] [Google Scholar]
- 285.Mohan SS, Knuiman MW, Divitini ML, et al. Higher serum testosterone and dihydrotestosterone, but not oestradiol, are independently associated with favourable indices of lung function in community-dwelling men. Clin Endocrinol (Oxf) 2015; 83: 268–76. [DOI] [PubMed] [Google Scholar]
- 286.Wang X, Huang L, Jiang S, et al. Testosterone attenuates pulmonary epithelial inflammation in male rats of COPD model through preventing NRF1-derived NF-κB signaling. J Mol Cell Biol 2021; 13: 128–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Velema MS, Kwa BH, de Ronde W. Should androgenic anabolic steroids be considered in the treatment regime of selected chronic obstructive pulmonary disease patients? Curr Opin Pulm Med 2012; 18: 118–24. [DOI] [PubMed] [Google Scholar]
- 288.Checkley W, West KP, Jr., Wise RA, et al. Maternal vitamin A supplementation and lung function in offspring. N Engl J Med 2010; 362: 1784–94. [DOI] [PubMed] [Google Scholar]
- 289.McKeever TM, Scrivener S, Broadfield E, Jones Z, Britton J, Lewis SA. Prospective study of diet and decline in lung function in a general population. Am J Respir Crit Care Med 2002; 165: 1299–303. [DOI] [PubMed] [Google Scholar]
- 290.Hanson C, Lyden E, Furtado J, et al. Serum tocopherol levels and vitamin E intake are associated with lung function in the normative aging study. Clin Nutr 2016; 35: 169–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Kim T, Choi H, Kim J. Association between dietary nutrient intake and chronic obstructive pulmonary disease severity: a nationwide population-based representative sample. COPD 2020; 17: 49–58. [DOI] [PubMed] [Google Scholar]
- 292.Jolliffe DA, Greenberg L, Hooper RL, et al. Vitamin D to prevent exacerbations of COPD: systematic review and meta-analysis of individual participant data from randomised controlled trials. Thorax 2019; 74: 337–45. [DOI] [PubMed] [Google Scholar]
- 293.Lemoine C, Brigham E, Woo H, et al. Relationship between omega-3 and omega-6 fatty acid intake and chronic obstructive pulmonary disease morbidity. Ann Am Thorac Soc 2020; 17: 378–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Matin S, Nemati A, Ghobadi H, Alipanah-Moghadam R, Rezagholizadeh L. The effect of conjugated linoleic acid on oxidative stress and matrix metalloproteinases 2 and 9 in patients with COPD. Int J Chron Obstruct Pulmon Dis 2018; 13: 1449–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Gutierrez-Mariscal FM, Arenas-de Larriva AP, Limia-Perez L, Romero-Cabrera JL, Yubero-Serrano EM, López-Miranda J. Coenzyme Q(10) supplementation for the reduction of oxidative stress: clinical implications in the treatment of chronic diseases. Int J Mol Sci 2020; 21: 7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Keogh E, Mark Williams E. Managing malnutrition in COPD: a review. Respir Med 2021; 176: 106248. [DOI] [PubMed] [Google Scholar]
- 297.Deutz NE, Ziegler TR, Matheson EM, et al. Reduced mortality risk in malnourished hospitalized older adult patients with COPD treated with a specialized oral nutritional supplement: sub-group analysis of the NOURISH study. Clin Nutr 2021; 40: 1388–95. [DOI] [PubMed] [Google Scholar]
- 298.WHO. Preterm birth. 2018. https://www.who.int/news-room/fact-sheets/detail/preterm-birth#cms (accessed May 31, 2022).
- 299.Mantey DS, Omega-Njemnobi O, Barroso CS. Secondhand smoke exposure at home and/or in a vehicle: differences between urban and non-urban adolescents in the United States, from 2015 to 2018. Nicotine Tob Res 2021; 23: 1327–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Li D, Shi H, Xie Z, et al. Home smoking and vaping policies among US adults: results from the Population Assessment of Tobacco and Health (PATH) study, wave 3. Prev Med 2020; 139: 106215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Faber T, Kumar A, Mackenbach JP, et al. Effect of tobacco control policies on perinatal and child health: a systematic review and meta-analysis. Lancet Public Health 2017; 2: E420–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Tattan-Birch H, Hartmann-Boyce J, Kock L, et al. Heated tobacco products for smoking cessation and reducing smoking prevalence. Cochrane Database Syst Rev 2022; 1: CD013790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Flor LS, Reitsma MB, Gupta V, Ng M, Gakidou E. The effects of tobacco control policies on global smoking prevalence. Nat Med 2021; 27: 239–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Hutchinson SG, Kuijlaars JS, Mesters I, et al. Addressing passive smoking in children. PLoS One 2014; 9: e9322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Hartinger SM, Commodore AA, Hattendorf J, et al. Chimney stoves modestly improved indoor air quality measurements compared with traditional open fire stoves: results from a small-scale intervention study in rural Peru. Indoor Air 2013; 23: 342–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Zhou Y, Zou Y, Li X, et al. Lung function and incidence of chronic obstructive pulmonary disease after improved cooking fuels and kitchen ventilation: a 9-year prospective cohort study. PLoS Med 2014; 11: e1001621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Pope D, Bruce N, Dherani M, Jagoe K, Rehfuess E. Real-life effectiveness of ‘improved’ stoves and clean fuels in reducing PM(2.5) and CO: systematic review and meta-analysis. Environ Int 2017; 101: 7–18. [DOI] [PubMed] [Google Scholar]
- 308.van Gemert F, de Jong C, Kirenga B, et al. Effects and acceptability of implementing improved cookstoves and heaters to reduce household air pollution: a FRESH AIR study. NPJ Prim Care Respir Med 2019; 29: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Hansel NN, Putcha N, Woo H, et al. Randomized clinical trial of air cleaners to improve indoor air quality and COPD health: results of the CLEAN AIR study. Am J Respir Crit Care Med 2021; 205: 421–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Hoffmann B, Boogaard H, de Nazelle A, et al. WHO air quality guidelines 2021—aiming for healthier air for all: a joint statement by medical, public health, scientific societies and patient representative organisations. Int J Public Health 2021; 66: 1604465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.US Environmental Protection Agency. Indoor air quality tools for schools action kit. https://www.epa.gov/iaq-schools/indoor-air-quality-tools-schools-action-kit (accessed Aug 1, 2022).
- 312.US Environmental Protection Agency. National air quality: status and trends of key air pollutants. https://www.epa.gov/air-trends (accessed Aug 1, 2022).
- 313.Zhao Q, Li S, Coelho M, et al. Ambient heat and hospitalisation for COPD in Brazil: a nationwide case-crossover study. Thorax 2019; 74: 1031–36. [DOI] [PubMed] [Google Scholar]
- 314.Siddharthan T, Wosu AC, Pollard SL, et al. A Novel case-finding instrument for chronic obstructive pulmonary disease in low-and middle-income country settings. Int J Chron Obstruct Pulmon Dis 2020; 15: 2769–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.WHO. Acute respiratory infections. 1990. https://apps.who.int/iris/handle/10665/61939 (accessed Aug 1, 2022).
- 316.BusinessWire. COPD Foundation releases groundbreaking COPE survey results: low patient awareness about COPD exacerbations poses barrier to effective management. 2014. https://www.businesswire.com/news/home/20140617005140/en/COPD-Foundation-Releases-Groundbreaking-COPE-Survey-Results-Low-Patient-Awareness-About-COPD%C2%A0Exacerbations-Poses-Barrier-to-Effective-Management (accessed Aug 1, 2022).
- 317.Gruß I, McCreary GM, Ivlev I, et al. Developing a patient-driven chronic obstructive pulmonary disease (COPD) research agenda in the US. J Patient Rep Outcomes 2021; 5: 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Rochester CL, Vogiatzis I, Holland AE, et al. An official American Thoracic Society/European Respiratory Society policy statement: enhancing implementation, use, and delivery of pulmonary rehabilitation. Am J Respir Crit Care Med 2015; 192: 1373–86. [DOI] [PubMed] [Google Scholar]
- 319.UNICEF WHO, World Bank Group. Levels and trends in child malnutrition: key findings of the 2018 edition of the Joint Child Malnutrition Estimates. 2018. https://data.unicef.org/wp-content/uploads/2018/05/JME-2018-brochure-.pdf (accessed Aug 4, 2022).
- 320.Reyfman PA, Khuder B, Pierce JB, et al. Urban–rural mortality disparities from chronic lower respiratory diseases in the United States, 1999–2019. Am J Respir Crit Care Med 2021; 203: 1435–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Reyfman PA, Sugar E, Hazucha H, et al. Study protocol for a national cohort of adults focused on respiratory health: the American Lung Association Lung Health Cohort (ALA-LHC) Study. BMJ Open 2021; 11: e053342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Rochester CL, Vogiatzis I, Powell P, Masefield S, Spruit MA. Patients’ perspective on pulmonary rehabilitation: experiences of European and American individuals with chronic respiratory diseases. ERJ Open Res 2018; 4: 00085–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Negrini S, Arienti C, Patrini M, Kiekens C, Rauch A, Cieza A. Cochrane collaborates with the World Health Organization to establish a package of rehabilitation interventions based on the best available evidence. Eur J Phys Rehabil Med 2021; 57: 478–80. [DOI] [PubMed] [Google Scholar]
- 324.Hoogendoorn M, Corro Ramos I, Baldwin M, Gonzalez-Rojas Guix N, Rutten-van Mölken M. Broadening the perspective of cost-effectiveness modeling in chronic obstructive pulmonary disease: a new patient-level simulation model suitable to evaluate stratified medicine. Value Health 2019; 22: 313–21. [DOI] [PubMed] [Google Scholar]
- 325.Rutten-van Mölken M, Leijten F, Hoedemakers M, et al. Strengthening the evidence-base of integrated care for people with multi-morbidity in Europe using multi-criteria decision analysis (MCDA). BMC Health Serv Res 2018; 18: 576. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.