

Key Points
-
•
Median OS in MPN-AP/BP is 0.86 years in a modern cohort without significant difference based on frontline treatment choice.
-
•
Median OS in those that underwent allo-HSCT is 2.3 years from time of allo-HSCT; response before allo-HSCT did not affect survival.
Visual Abstract
Abstract
Progression of myeloproliferative neoplasms (MPNs) to accelerated or blast phase is associated with poor survival outcomes. Since 2017 there have been several therapies approved for use in acute myeloid leukemia (AML); these therapies have been incorporated into the management of accelerated/blast-phase MPNs (MPN-AP/BP). We performed a multicenter analysis to investigate outcomes of patients diagnosed with MPN-AP/BP in 2017 or later. In total, 202 patients were identified; median overall survival (OS) was 0.86 years. We also analyzed patients based on first-line treatment; the 3 most common approaches were intensive chemotherapy (n = 65), DNA methyltransferase inhibitor (DNMTi)-based regimens (n = 65), and DNMTi + venetoclax–based regimens (n = 54). Median OS was not significantly different by treatment type. In addition, we evaluated response by 2017 European LeukemiaNet AML criteria and 2012 MPN-BP criteria in an effort to understand the association of response with survival outcomes. We also analyzed outcomes in 65 patients that received allogeneic hematopoietic stem cell transplant (allo-HSCT); median OS was 2.30 years from time of allo-HSCT. Our study demonstrates that survival among patients with MPN-AP/BP is limited in the absence of allo-HSCT even in the current era of therapeutics and underscores the urgent need for new agents and approaches.
Introduction
Philadelphia-chromosome–negative myeloproliferative neoplasms (MPNs) are a heterogeneous group of hematopoietic stem cell disorders characterized by proliferation of myeloid cells, activation of the JAK/STAT pathway, and a variable risk of progression to accelerated phase (AP) or blast phase (BP) that is influenced by disease phenotype and clinical, cytogenetic, and molecular features.1, 2, 3 AP is defined as 10% to 19% blasts in the peripheral blood or bone marrow whereas BP requires ≥20% blasts.4 Although the median overall survival (OS) of patients with chronic-phase MPN is several years, development of an AP/BP MPN (MPN-AP/BP) is associated with limited OS, particularly in the absence of allogeneic hematopoietic stem cell transplant (allo-HSCT). The median OS for patients with MPN-AP ranges from 12 to 18 months5, 6, 7 whereas the median OS of those with MPN-BP is 3 to 5 months.8, 9, 10, 11
Since 2017, there have been several novel therapies approved for use in acute myeloid leukemia (AML) that have led to significant evolution in the treatment approach in this disease.12,13 These treatment approaches are often applied to patients with MPN-AP/BP despite being molecularly and morphologically distinct from de novo AML.14, 15, 16, 17, 18 There are limited prospective data for the use of these therapies in MPN-AP/BP, and primarily real-world data have been analyzed to characterize their efficacy. There has been a particular focus on the outcomes of patients treated with venetoclax (VEN)-based therapies and isocitrate dehydrogenase (IDH) inhibitors. Median OS with VEN-based regimens ranges from 4 to 8 months19, 20, 21, 22 while utilization of IDH inhibition has demonstrated median OS ranging from 10 to 15 months.23, 24, 25 In addition, prospective efforts have investigated the use of DNA methyltransferase inhibitors (DNMTi’s) in combination with the JAK inhibitor ruxolitinib with reported median OS ranging from 7 to 9.5 months.26,27
Given the limited prospective data for therapies in MPN-AP/BP, there is heterogeneity and lack of consensus regarding treatment approach in the current era of myeloid therapies. Therefore, we aimed to analyze outcomes in adult patients with MPN-AP/BP diagnosed in 2017 or later using a large multicenter retrospective cohort to better understand the impact of treatment approach. We also investigated assessment of response by both AML-specific and MPN-BP specific criteria in relation to survival outcomes. In addition, we analyzed outcomes in the patients that underwent allo-HSCT for MPN-AP/BP.
Methods
All adult patients with MPN-AP/BP diagnosed in 2017 or later were identified at 9 participating academic centers. MPN-AP/BP was defined as the development of ≥10% blasts in the peripheral blood or bone marrow in patients with an underlying MPN. All participating centers obtained approval from their institutional review board. Patient demographics were collected including age at diagnosis, sex and self-reported race/ethnicity; 1 participating center was unable to contribute race/ethnicity data. Disease characteristics and treatment approaches for both chronic-phase MPN and MPN-AP/BP were collected. For patients with cytogenetic/molecular data available at the time of MPN-AP/BP diagnosis, a prognostic risk score was assigned using 2017 European LeukemiaNet (ELN) AML criteria.28 Response to therapies administered for treatment of MPN-AP/BP were assessed using both 2017 ELN AML response criteria and 2012 MPN-BP response criteria.28,29 Of note, 1 participating center was only able to characterize response using 2017 ELN criteria. All analyses were performed using SAS software version 9.4 (Cary, NC).
Each institution received approval from the institutional review board to conduct this retrospective project.
Results
Patient demographics and molecular information
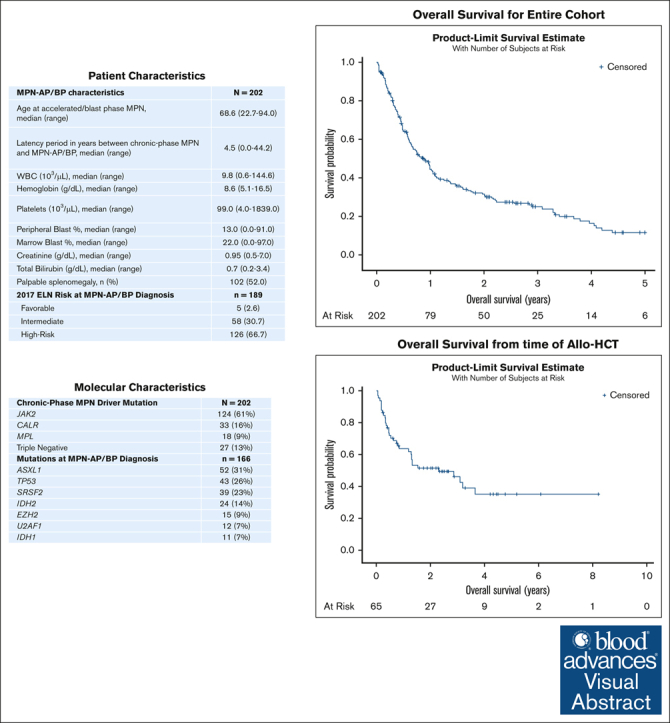
In total, 202 patients with MPN-AP/BP diagnosed during the specified period were identified; 140 patients had MPN-BP at time of progression whereas 62 had MPN-AP. The median age at time of MPN-AP/BP diagnosis was 68.6 years (range, 21-91) and 39.6% were women. The most common chronic-phase MPN in patients was primary myelofibrosis, which was noted in 33% of patients. When looking at underlying driver mutations at time of chronic-phase MPN, 61% of patients had JAK2-mutated disease, 16% had CALR-mutated disease, 9% had MPL-mutated disease, and 13% of patients had “triple-negative” disease. The most common therapies directed at chronic-phase MPN were hydroxyurea in 61% of patients and JAK inhibitor therapy in 36% of patients; 6 patients had previously undergone allo-HSCT for their chronic-phase MPN. Additional characteristics are summarized in Table 1.
Table 1.
Patient demographics and disease characteristics during chronic-phase MPN and at diagnosis of MPN-AP/BP
| Demographics | N = 202 |
|---|---|
| Age (y) at chronic-phase MPN diagnosis, median (range) | 61.9 (21.9-91.0) |
| Female, n (%) | 80 (39.6) |
| Race/ethnicity, n (%) | n = 158 |
| White | 142 (89.9) |
| Black | 9 (5.7) |
| Asian | 3 (1.9) |
| Other | 4 (2.5) |
| Hispanic ethnicity | 11 (7.0) |
| Chronic-phase MPN | N = 202 |
| Polycythemia vera | 40 (20%) |
| Essential thrombocythemia | 57 (28%) |
| Primary myelofibrosis | 67 (33%) |
| MPN-not otherwise specified/other | 38 (19%) |
| Driver mutation | N = 202 |
| JAK2 | 124 (61%) |
| CALR | 33 (16%) |
| MPL | 18 (9%) |
| Triple-negative | 27 (13%) |
| Therapies received for chronic-phase MPN | N = 202 |
| Hydroxyurea | 124 (61%) |
| JAK inhibitor | 72 (36%) |
| Interferon | 7 (4%) |
| DNMTi | 11 (5%) |
| Other | 39 (19%) |
| Allo-HSCT | 6 (3%) |
| MPN-AP/BP characteristics | N = 202 |
| Age at accelerated/blast phase MPN, median (range) | 68.6 (22.7-94.0) |
| Latency period in years between chronic-phase MPN and MPN-AP/BP, median (range) | 4.5 (0.0-44.2) |
| WBC (103/μL), median (range) | 9.8 (0.6-144.6) |
| Hemoglobin (g/dL), median (range) | 8.6 (5.1-16.5) |
| Platelets (103/μL), median (range) | 99.0 (4.0-1839.0) |
| Peripheral blast %, median (range) | 13.0 (0.0-91.0) |
| Marrow blast %, median (range) | 22.0 (0.0-97.0) |
| Creatinine (g/dL), median (range) | 0.95 (0.5-7.0) |
| Total bilirubin (g/dL), median (range) | 0.7 (0.2-3.4) |
| Palpable splenomegaly, n (%) | 102 (52.0) |
| 2017 ELN risk at MPN-AP/BP diagnosis | n = 189 |
| Favorable | 5 (2.6) |
| Intermediate | 58 (30.7) |
| High risk | 126 (66.7) |
| Mutations at MPN-AP/BP diagnosis | n = 166 |
| ASXL1 | 52 (31%) |
| TP53 | 43 (26%) |
| SRSF2 | 39 (23%) |
| IDH2 | 24 (14%) |
| EZH2 | 15 (9%) |
| U2AF1 | 12 7%) |
| IDH1 | 11 (7%) |
Median laboratory values at time of MPN-AP/BP diagnosis are characterized in Table 1. Of 189 patients with available data needed for 2017 ELN AML risk stratification, 67% had high-risk disease. In addition, 166 patients had next-generation sequencing performed at time of MPN-AP/BP diagnosis. Mutations with an incidence of ≥10% included ASXL (31%), TP53 (26%), SRSF2 (23%), TET2 (20%), RUNX1 (17%), IDH2 (14%), DNMT3A (11%), and CBL (10%). In addition, 7% and 4% of patients had disease with an IDH1 mutation and a FLT3 mutation, respectively.
Survival outcomes and response to therapy
The median OS for the entire 202 patient cohort was 0.86 years (Figure 1A); median OS for patients diagnosed with MPN-AP was 1.09 years whereas it was 0.67 years for those diagnosed with MPN-BP. Median follow-up time was 0.75 years; 51 patients were alive at time of data analysis. We also analyzed outcomes and responses based on the treatment approach for MPN-AP/BP. Of note, 1 patient proceeded directly to allo-HSCT and 7 patients received supportive care alone. Sixty-five patients (32%) received intensive chemotherapy (IC) as their initial therapy, 65 patients (32%) received DNMTi-based therapy, 54 patients (27%) received DNMTi + VEN–based therapy, 4 patients received a targeted inhibitor as monotherapy, and 6 patients received other therapies. The specific therapies are summarized in supplemental Table 1. Twenty-seven patients were treated in the context of a clinical trial. We analyzed differences in patient and disease characteristics among those treated with IC, DNMTi-based therapy, and DNMTi + VEN–based therapy (Table 2); patients treated with IC were significantly younger (P < .0001) whereas patients treated with DNMTi-based therapy had significantly lower marrow and peripheral blood blasts (P = .009 and P = .0006, respectively). Using Kaplan-Meier analysis, estimated OS based on treatment approach was also analyzed. Median OS for the IC group was 0.68 years, for the DNMTi + VEN–based group was 0.71 years, and 1.25 years for the DNMTi-based group (P = .47; Figure 1B). Of note, the median OS of the DNMTi-based group was not significantly different when compared with those treated with IC or DNMTI + VEN–based approaches (n = 119; median OS, 0.72 years; P = .80).
Figure 1.

Overall survival of patients diagnosed with accelerated/blast-phase myeloproliferative neoplasms from 2017 onward. (A) OS of patients diagnosed with MPN-AP/BP from 2017 onward. (B) OS of patients with MPN-AP/BP by frontline treatment approach.
Table 2.
Characteristics of patients with MPN-AP/BP treated with intensive chemotherapy, DNMTi-based therapy, and DNMTi + VEN–based therapy
| Variable, mean (SD) |
IC (n = 65) | DNMTi + VEN–based (n = 54) | DNMTi-based (n = 65) | P value |
|---|---|---|---|---|
| Age (y) at diagnosis of MPN-AP/BP | 62.9 (8.6) | 71.2 (7.4) | 69.7 (10.5) | <.0001∗ |
| WBC (103/μL) | 17.6 (21.5) | 23.5 (26.4) | 21.8 (33.1) | .48∗ |
| Platelet (103/μL) | 188.7 (287.4) | 184.4 (203.8) | 235.5 (330.1) | .54∗ |
| Hemoglobin (g/dL) | 9.3 (2.2) | 8.9 (1.9) | 8.8 (1.6) | .33∗ |
| Peripheral blasts (%) | 24.5 (22.9) | 21.0 (22.9) | 13.5 (14.4) | .009∗ |
| Marrow blasts (%) | 36.8 (22.1) | 34.6 (23.9) | 21.9 (19.0) | .0006∗ |
| Total bilirubin (g/dL) | 0.8 (0.6) | 0.8 (0.4) | 0.9 (0.5) | .70∗ |
| Creatinine (g/dL) | 1.0 (0.3) | 1.2 (0.5) | 1.0 (0.9) | .15∗ |
| Splenomegaly | 30 (48.4) | 21 (39.6) | 39 (61.9) | .052 |
| Chronic-phase MPN | ||||
| Polycythemia vera | 14 (21.5) | 9 (16.7) | 14 (21.5) | .22 |
| Essential thrombocythemia | 22 (33.9) | 15 (27.8) | 17 (26.2) | |
| Primary myelofibrosis | 13 (20.0) | 18 (33.3) | 26 (40.0) | |
| Other | 16 (24.6) | 12 (22.2) | 8 (12.3) | |
| Driver mutation | ||||
| JAK2 | 37 (56.9) | 38 (70.4) | 39 (60.0) | .30 |
| CALR | 9 (13.9) | 7 (13.0) | 13 (20.0) | .50 |
| MPL | 8 (12.3) | 6 (11.1) | 5 (7.7) | .67 |
| Triple-negative | 13 (20.0) | 4 (7.4) | 10 (15.4) | .15 |
| Mutations | ||||
| ASXL1 | 13 (20.0) | 16 (29.6) | 16 (24.6) | .48 |
| EZH2 | 4 (6.2) | 3 (5.6) | 7 (10.8) | .61† |
| SRSF2 | 7 (10.8) | 19 (35.2) | 8 (12.3) | .0008 |
| IDH1 | 3 (4.6) | 3 (5.6) | 4 (6.2) | 1.00† |
| IDH2 | 7 (10.8) | 6 (11.1) | 6 (9.2) | .94 |
| U2AF1 | 4 (6.2) | 5 (9.3) | 3 (4.6) | .66† |
| RUNX1 | 7 (10.8) | 11 (20.4) | 8 (12.3) | .28 |
| TP53 | 11 (15.9) | 13 (24.1) | 14 (21.5) | .62 |
Analysis of variance F-test.
Fisher exact test.
We also analyzed response to first-line treatment approach by both 2017 ELN and 2012 MPN-BP criteria, which is summarized in Table 3. The complete remission/complete remission with incomplete count recovery (CR/CRi) rate by 2017 ELN was 42% for the IC group, 41% for the DNMTi + VEN–based group, and 20% for the DNMTi-based group; when analyzing by 2012 MPN-BP response criteria the rate of acute leukemia response–complete (ALR-C) or better was 37% for the IC group, 39% for the DNMTi + VEN–based group, and 22% for the DNMTi-based group. Kaplan-Meier analysis of survival by 2017 ELN criteria demonstrated a median OS of 1.37 years for those that achieved a CR/CRi compared with 1.05 years for those that achieved partial remission (PR) or morphologic leukemia-free state (MLFS) and 0.59 years for those that achieved stable disease (SD) or treatment failure (TF; P = .0002; Figure 2A). Analyzing OS by 2012 MPN-BP response demonstrated a median OS of 1.75 years for those that achieved ALR-C or better compared with 0.74 years for those that achieved acute leukemia response–partial and 0.61 years for those that achieved SD or progressive disease (P = .006; Figure 2B).
Table 3.
Response to first-line therapies for MPN-AP/BP
| Therapy | 2017 ELN response | 2012 MPN-BP response |
|---|---|---|
| IC (n = 65) | CR: 28% CRi: 14% MLFS: 2% PR: 11% TF: 45% |
CCR: 8% ALR-C: 29% ALR-P: 20% SD: 10% PD: 17% |
| (n = 65 evaluable for ELN) | ||
| (n = 51 evaluable for MPN-BP) | ||
| DNMTi + VEN based (n = 54) | CR: 22% CRi: 19% MLFS: 6% PR: 4% SD: 17% TF: 32% |
CCR: 2% ALR-C: 37% ALR-P: 20% SD: 15% PD: 27% |
| (n = 54 evaluable for ELN) | ||
| (n = 42 evaluable for MPN-BP) | ||
| DNMTi based (n = 65) | CR: 9% CRi: 11% MLFS: 2% PR: 9% SD: 32% TF: 37% |
CMR: 6% CCR: 2% ALR-C: 14% ALR-P: 12% SD: 37% PD: 33% |
| (n = 65 evaluable for ELN) | ||
| (n = 49 evaluable for MPN-BP) | ||
| Targeted monotherapy (n = 4) | CRi: 25% SD: 50% TF: 25% |
ALR-P: 50% SD: 50% |
| (n = 4 evaluable for ELN) | ||
| (n = 4 evaluable for MPN-BP) | ||
| Other therapies (n = 6) | CRi: 17% PR: 17% SD: 17% TF: 30% |
ALR-C: 17% ALR-P: 17% SD: 33% PD: 33% |
| (n = 6 evaluable for ELN) | ||
| (n = 6 evaluable for MPN-BP) |
ALR-P, acute leukemia response–partial; CCR, complete cytogenetic response; CMR, complete molecular response; PD, progressive disease; PR, partial remission.
Figure 2.

Overall survival of patients with MPN-AP/BP stratifed by response. (A) OS of patients with MPN-AP/BP by 2017 ELN response criteria. (B) OS of patients with MPN-AP/BP by 2012 MPN-BP criteria. ALR-P, acute leukemia response–partial; CCR, complete cytogenetic response; CMR, complete molecular response; PD, progressive disease; PR, partial remission.
A total of 115 therapies were used in the second-line and beyond (2L+) setting for MPN-AP/BP with only 18 administered in the context of a clinical trial. The specific therapies are summarized in supplemental Table 2 whereas the response to therapy is in Table 4.
Table 4.
Response to second-line and beyond therapies for MPN-AP/BP
| Therapy | 2017 ELN response | 2012 MPN-BP response |
|---|---|---|
| DNMTi + VEN based (n = 38) | CR: 5% CRi: 5% MLFS: 16% PR: 5% SD: 13% TF: 55% |
CCR: 3% ALR-C: 22% ALR-P: 14% SD: 22% PD: 39% |
| (n = 38 evaluable for ELN) | ||
| (n = 36 evaluable for MPN-BP) | ||
| IC (n = 37) | CR: 16% CRi: 3% MLFS: 14% PR: 11% SD: 19% TF: 38% |
CCR: 6% ALR-C: 15% ALR-P: 29% SD: 21% PD: 29% |
| (n = 37 evaluable for ELN) | ||
| (n = 34 evaluable for MPN-BP) | ||
| DNMTi based (n = 16) | CR: 6% MLFS: 6% PR: 6% SD: 25% TF: 56% |
ALR-C: 14% ALR-P: 21% SD: 29% PD: 36% |
| (n = 16 evaluable for ELN) | ||
| (n = 14 evaluable for MPN-BP) | ||
| Targeted monotherapy (n = 11) | CR: 9% CRi: 18% MLFS: 18% SD: 18% TF: 36% |
CCR: 10% ALR-C: 30% ALR-P: 20% SD: 20% PD: 20% |
| (n = 11 evaluable for ELN) | ||
| (n = 10 evaluable for MPN-BP) | ||
| Other therapies (n = 13) | CR: 8% CRi: 8% PR: 23% SD: 38% TF: 23% |
ALR-C: 15% ALR-P: 31% SD: 31% PD: 23% |
| (n = 13 evaluable for ELN) | ||
| (n = 13 evaluable for MPN-BP) |
Abbreviations are explained in Table 3.
In an effort to better understand factors affecting OS beyond treatment choice, we performed a univariate analysis of several clinical and molecular/cytogenetic factors at time of MPN-AP/BP diagnosis, summarized in Table 5. Stratification of age, hemoglobin, white blood count (WBC), and platelet count was derived from the Mutation-enhanced International Prognostic Score System–Plus, version 2.0, whereas stratification of blast percentage was derived from definitions of chronic-phase, accelerated-phase, and blast-phase MPN.4,30 Stepwise Cox regression was performed to determine which factors from Table 5 were significant in a multivariate model (Table 6), and included age of >65 years (hazard ratio [HR], 1.87; 95% confidence interval [CI], 1.27-2.77), WBC of >25 × 103/μL (HR, 2.35; 95% CI, 1.59-3.49), hemoglobin of >10 g/dL vs <8 g/dL (HR, 0.44; 95% CI, 0.27-0.72), hemoglobin of 8 to 10 g/dL vs < 8 g/dL (HR, 0.63; 95% CI, 0.43-0.93), and TP53 mutation (HR, 2.15; 95% CI, 1.46-3.15). The stepwise Cox model included a P value for entry of 0.50 and a P value to remain of 0.15 for the potential factors.
Table 5.
Univariate analysis of factors affecting OS in MPN-AP/BP
| Variable | HR (95% CI) | P value |
|---|---|---|
| Age ≥ 65 y | 1.840 (1.271, 2.667) | .001 |
| WBC > 25 × 103/μL | 1.812 (1.267, 2.590) | .001 |
| Platelet > 100 × 103/μL | 0.687 (0.497, 0.947) | .022 |
| Hemoglobin > 10 g/dL vs < 8 g/dL | 0.380 (0.240, 0.603) | < .0001 |
| Hemoglobin 8-10 g/dL vs < 8g/dL | 0.626 (0.436, 0.899) | .011 |
| Peripheral blasts ≥ 20% vs < 10% | 1.414 (0.956, 2.094) | .08 |
| Peripheral blasts 10%-19% vs < 10% | 1.242 (0.826, 1.867) | .30 |
| Marrow blasts ≥ 20% vs <10% | 0.882 (0.456, 1.703) | .71 |
| Marrow blasts 10%-19% vs <10% | 0.805 (0.399, 1.626) | .55 |
| Total bilirubin | 0.896 (0.647, 1.242) | .51 |
| Creatinine | 1.164 (0.903, 1.502) | .24 |
| Splenomegaly | 1.259 (0.908, 1.746) | .17 |
| Driver mutation status | ||
| JAK2 | 0.970 (0.697, 1.351) | .86 |
| CALR | 1.056 (0.690, 1.617) | .80 |
| MPL | 1.848 (1.078, 3.166) | .03 |
| Triple negative | 0.722 (0.436, 1.197) | .21 |
| 2017 ELN risk | ||
| High risk vs intermediate/favorable | 1.859 (1.282, 2.696) | .001 |
| Mutation status | ||
| TP53 | 2.085 (1.456, 2.996) | < .0001 |
| IDH1 | 0.885 (0.433, 1.808) | .74 |
| IDH2 | 0.664 (0.389, 1.134) | .13 |
| ASXL1 | 0.897 (0.616, 1.307) | .57 |
| EZH2 | 1.076 (0.609, 1.901) | .80 |
| SRSF2 | 1.113 (0.742, 1.671) | .60 |
| RUNX1 | 0.774 (0.483, 1.241) | .29 |
| Chronic-phase MPN type | ||
| ET vs other | 1.134 (0.694, 1.853) | .62 |
| PMF vs other | 1.194 (0.737, 1.935) | .47 |
| PV vs other | 1.352 (0.803, 2.278) | .26 |
ET, essential thrombocythemia; PMF, primary myelofibrosis; PV, polycythemia vera.
Table 6.
Multivariate analysis of factors affecting OS in MPN-AP/BP
| Variable | HR (95% CI) | P value |
|---|---|---|
| Age ≥ 65 y | 1.870 (1.265, 2.765) | .002 |
| WBC > 25 × 109/L | 2.354 (1.589, 3.488) | < .0001 |
| Platelet > 100 × 109/L | 0.443 (0.272, 0.723) | .001 |
| Hemoglobin > 10 g/dL vs < 8 g/dL | 0.629 (0.425, 0.929) | .02 |
| TP53 mutation status | 2.146 (1.460, 3.153) | .0001 |
| Splenomegaly | 1.305 (0.925, 1.842) | .13 |
Outcomes in patients that received allo-HSCT
Overall, 65 patients from our cohort went onto receive allo-HSCT for MPN-AP/BP. Characteristics of patients that underwent allo-HSCT are summarized in supplemental Table 3. One patient proceeded directly to allo-HSCT after MPN-AP/BP diagnosis, 44 patients proceeded after first-line therapy, and 20 patients proceeded to allo-HSCT after 2L+ therapies. Fifty-one patients had next-generation sequencing performed at time of MPN-AP/BP diagnosis; nondriver mutations with an incidence of ≥10% were ASXL1 (29%), RUNX1 (24%), TP53 (20%), SRSF2 (20%), TET2 (18%), IDH1 (10%), and IDH2 (10%). To better understand how disease characteristics and treatment choices may have affected receipt of allo-HSCT, we performed univariate analysis of the variables summarized in supplemental Table 4A at time of MPN-AP/BP diagnosis. Stepwise logistic regression using the predictors shown in supplemental Table 4A resulted in the following statistically significant factors for receipt of allo-HSCT: age of ≥65 years, WBC of >25 × 103/μL, and hemoglobin of >10 g/dL vs < 8 g/dL (supplemental Table 4B). The stepwise logistic model included a P value for entry of 0.50 and a P value to remain of 0.15 for the potential factors.
Among 65 patients who received allo-HSCT, donor source was the following: 36 matched unrelated donors, 11 matched related donors, 11 haplo-identical donors, 6 mismatched unrelated donors, and 1 cord blood. Sixty patients received a reduced-intensity regimen whereas 15 patients received a myeloablative regimen. Twenty-five patients developed acute graft versus host disease whereas 16 developed chronic graft versus host disease. We analyzed survival outcomes using the Kaplan-Meier method. Median OS from the time of MPN-AP/BP diagnosis was 3.1 years and was 2.30 years from the time of allo-HSCT (Figure 3A-B). Thirty patients were alive at time of the data lock and among them 6 had relapsed MPN-AP/BP after allo-HSCT. Among 35 patients who died after allo-HSCT, the cause of death was noted as transplant related in 16, and relapse of MPN-AP/BP in 15 patients; 4 patients died of causes not related to allo-HSCT or MPN-AP/BP. The 2-year rate of relapse after allo-HSCT was 23% and 2-year nonrelapse-related mortality rate after allo-HSCT was 25%.
Figure 3.

Overall survival of patients with MPN-AP/BP that underwent allogeneic hematopoietic stem cell transplantation. (A) OS of patients with MPN-AP/BP that underwent allo-HSCT from time of diagnosis. (B) OS from time of allo-HSCT in patients with MPN-AP/BP. (C) OS from time of allo-HSCT in patients with MPN-AP/BP stratified by 2017 ELN response before allo-HSCT. PD, progressive disease.
We also analyzed several factors to try and identify what may correlate with improved OS in patients after allo-HSCT. We performed Kaplan-Meier analysis of OS from time of allo-HSCT based on 2017 ELN disease response going into allo-HSCT; median OS was 2.85 years in patients with CR/CRi, 2.30 years for those with PR or MLFS, and 0.74 years for those with SD or TF (P = .4994; Figure 3C). The median OS from time of allo-HSCT was significantly longer for those that achieved CR/CRi/PR/MLFS than for those that achieved SD/TF as best response before allo-HSCT (1.37 years vs 0.59 years, P < .0001). We also performed univariate analysis of the variables at time of MPN-AP/BP diagnosis, summarized in supplemental Table 5A, to identify potential factors associated with OS from time of allo-HSCT. Multivariate analysis identified age of ≥65 years, marrow blasts of 10% to 19% vs <10%, serum creatinine (g/dL), and TP53 mutation status as being significant factors (supplemental Table 5B). The stepwise logistic model included a P value for entry of 0.50 and a P value to remain of 0.15 for the potential factors.
Discussion
This series of 202 patients is, to our knowledge, the largest cohort of patients with MPN-AP/BP diagnosed and treated in the current era of myeloid therapies. Given the size of our cohort, heterogeneity in frontline treatment approaches, number of patients that proceeded to allo-HSCT, and response assessment using both AML and MPN-BP specific criteria, we were able to report on survival across a variety of dimensions. In addition, we were able to investigate the baseline factors that may influence frontline treatment choice and receipt of allo-HSCT.
The median OS in the entire cohort was 0.86 years, which is consistent with the survival reported in historical cohorts.8,9,31 There was no significant difference in OS based on front-line treatment approach. Patients that received IC were significantly younger whereas those that received DNMTi-based therapy had significantly lower marrow and peripheral blood blasts; these findings may help to elucidate some of the factors that underlie the selection of specific therapies. Interestingly, although CR rates were numerically higher with IC and DNMTi + VEN–based approaches, patients that received DNMTi-based approaches had numerically longer OS. Although comprehensive adverse event data were not collected, this may speak to the ability of patients to stay on DNMTi-based therapy with disease control even if a CR is not achieved.
Among the patients who received allo-HSCT in our cohort, the median OS was 2.3 years from time of allo-HSCT. The significant variables affecting receipt of allo-HSCT include age, WBC, hemoglobin, marrow blast percent, triple-negative disease status, and TP53 mutation status. When looking at disease control before allo-HSCT by 2017 ELN criteria, there was no significant association between response and OS after allo-HSCT. Of note, median OS was longer for patients that achieved CR/CRi/PR/MLFS before allo-HSCT than for those that had SD/TF. This suggests that reduction in blast burden may be of some benefit but the necessity of achieving CR/CRi is not clear. The allo-HSCT survival data reported in our cohort are consistent with previously reported data demonstrating that allo-HSCT seems to offer durable OS in patients with MPN-AP/BP32,33 but that the majority of patients do not receive allo-HSCT.9 Potential reasons for a minority of patients with MPN-AP/BP receiving allo-HSCT may be the relative lack of CRs achieved with current therapeutic options, age at diagnosis of disease, and defining pretransplant disease control using AML-specific criteria.
There is also significant discussion in the literature about the best way to gauge response in patients with MPN-AP/BP. When comparing outcomes of MPN-BP in remission at time of allo-HSCT compared with both de novo AML and AML arising from MDS, median OS is significantly shorter and the risk of relapse is significantly higher.34 This serves to highlight that typical AML response criteria may not appropriately capture the depth of response to therapy among patients with MPN-AP/BP. This observation may be because of the fact that even when therapies eradicate the AP/BP component of disease, features of the chronic phase MPN often persist, such as marrow fibrosis and circulating peripheral blasts. The 2012 MPN BP criteria29 were designed with these limitations in mind, but use of these criteria has not been widely adopted in studies focused on MPN-AP/BP. Our analysis of 2017 ELN response criteria demonstrated that achievement of CR/CRi was associated with longer OS when compared with other responses; similar findings were seen in patients that achieved an ALR-C or better by MPN-BP 2012 criteria. Median OS was 1.37 years in patients that achieved CR/CRi by 2017 ELN criteria whereas median OS was 1.75 years in those who achieved ALR-C or better by 2012 MPN-BP criteria. These data suggest that use of 2012 MPN-BP criteria when assessing therapeutic response may be as useful, and perhaps more relevant in this population, as the 2017 ELN AML-specific criteria. It remains to be determined how to best characterize disease control before allo-HSCT.
Lastly, our analysis sought to characterize the frequency of clinical trial enrollment in our cohort. Only 14% of patients in our cohort were treated in the context of a clinical trial for their frontline therapy; only 12% of 2L+ therapies were administered via clinical trial. Novel therapies are desperately needed for this patient group. An ongoing prospective trial investigating combined IDH2/JAK2 inhibition has noted promising preliminary results.35 In addition, preclinical work has identified other potential therapeutic targets including hypoxia-inducible factor and DUSP6; in addition, there may be a role for bromodomain and extra-terminal domain inhibition, lysine specific demethylase 1 inhibition, B-cell lymphoma-extra large, and cyclin-dependent kinase 9 inhibition in MPN-AP/BP as well.36, 37, 38, 39, 40, 41
Our study has limitations given its retrospective nature. Although this was a multicenter study, the centers included may not fully represent the breadth of patients with MPN-AP/BP. We were also unable to assess for both 2017 ELN response criteria and 2012 MPN-BP criteria in the entire cohort. In addition, there may be factors influencing treatment approach and receipt of allo-HSCT that were not adequately captured.
In summary, our study demonstrates that, even with new therapeutic approaches available for MPN-AP/BP, OS is quite limited. Only a minority of patients are treated in the context of a clinical trial, thus highlighting a significant need to not only develop novel and effective therapies but to also evaluate them in clinical trials inclusive of patients with MPN-AP/BP. Furthermore, prospective evaluation of both 2017 ELN and 2012 MPN-BP response criteria as well as other recently proposed MPN-AP/BP criteria is needed to ascertain the best method of response assessment.42
Conflict-of-interest disclosure: A.A.P. received honoraria from AbbVie and Bristol Myers Squibb, and research funding (institutional) from Pfizer and Kronos Bio. R.M.S. received honoraria from Bristol Myers Squibb, Kura Oncology, Gilead Sciences, Rigel, and Servier. E.C.C. received consulting fees from AbbVie and Rigel. S.G.I. received honoraria from Medical Logix (medical education) and reports an advisory board role with MorphoSys. R.K.R. received research funding from Incyte, Constellation, Zentalis, Stemline, and Ryyu, and received consulting fees from Celgene-Bristol Myers Squibb, Kartos, Zentalis, Karyopharm, Dainippon, GlaxoSmithKline-Sierra, Galecto, Pharmessentia, Incyte, CTI Biopharma, Servier, MorphoSys/Constellation, and Sumitumo. T.B. reports membership on advisory committee for Novartis, Geron Corporation, and Gilead, and speakers bureau for Novartis. Y.A. received research funding from Biomea, Curis, Biosight, and ALX Oncology Novartis, and honoraria from Servier, Pfizer, Bristol Myers Squibb, Kite, Astellas, and Rigel. J.S.G. received research funding from AbbVie, Genentech, New Wave, Pfizer, and Prelude, and served on steering committee/scientific advisory board for AbbVie, Bristol Myers Squibb, Genentech, and Servier. V.G. received research funding from AbbVie and Novartis; reports consulting fees from Novartis, Bristol Myers Squibb/Celgene, Keros, AbbVie, Constellation Biopharma, Pfizer, GlaxoSmithKline, and CTI Biopharma; reports honoraria from Novartis, Bristol Myers Squibb/Celgene, and AbbVie; and serves on the data safety monitoring or advisory board for Bristol Myers Squibb/Celgene, Roche, AbbVie, Pfizer, GlaxoSmithKline, and CTI Biopharma. K.M.P. reports consulting fees from Protagonist Therapeutics and AbbVie; received research funding from Protagonist Therapeutics, Merck, and AbbVie; and reports speakers bureau membership with Merck. O.O. reports consulting fees from AbbVie, Blueprint Medicines, Bristol Myers Squibb, CTI, Impact Biomedicines, Kymera, Novartis, SERVIER, Taiho Pharmaceutical, and Threadwell Therapeutics; and received research funding (to institution) from AbbVie, Agios, Aprea AB, Astex Pharmaceuticals, AstraZeneca, Bristol Myers Squibb, Celgene, CTI BioPharma Corp, Daiichi Sankyo, Incyte, Janssen Oncology, Kartos Therapeutics, Loxo, Novartis, NS Pharma, and OncoTherapy Science. The remaining authors declare no competing financial interests.
Acknowledgments
Authorship
Contribution: A.A.P. designed the study plan, performed data analysis, and wrote the manuscript; J.J.Y. offered input into the study plan, collected data, and reviewed/revised the manuscript; J.F.C. performed data analysis and reviewed/revised the manuscript; O.O. designed the study plan and reviewed/revised the manuscript; and all other authors collected data and reviewed/revised the manuscript.
Footnotes
Data are available on request from the corresponding author, Anand A. Patel (anand.patel@bsd.uchicago.edu).
The full-text version of this article contains a data supplement.
Supplementary Material
References
- 1.Patel AA, Odenike O. SOHO state of the art updates and next questions | accelerated phase of MPN: what it is and what to do about it. Clin Lymphoma Myeloma Leuk. 2023;23(5):303–309. doi: 10.1016/j.clml.2023.01.011. [DOI] [PubMed] [Google Scholar]
- 2.Dunbar AJ, Rampal RK, Levine R. Leukemia secondary to myeloproliferative neoplasms. Blood. 2020;136(1):61–70. doi: 10.1182/blood.2019000943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patel AA, Odenike O. Genomics of MPN progression. Hematology Am Soc Hematol Educ Program. 2020;2020(1):440–449. doi: 10.1182/hematology.2020000129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arber DA, Orazi A, Hasserjian RP, et al. International consensus classification of myeloid neoplasms and acute leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022;140(11):1200–1228. doi: 10.1182/blood.2022015850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shahin OA, Chifotides HT, Bose P, Masarova L, Verstovsek S. Accelerated phase of myeloproliferative neoplasms. Acta Haematol. 2021;144(5):484–499. doi: 10.1159/000512929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tam CS, Kantarjian H, Cortes J, et al. Dynamic model for predicting death within 12 months in patients with primary or post-polycythemia vera/essential thrombocythemia myelofibrosis. J Clin Oncol. 2009;27(33):5587–5593. doi: 10.1200/JCO.2009.22.8833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mudireddy M, Gangat N, Hanson CA, Ketterling RP, Pardanani A, Tefferi A. Validation of the WHO-defined 20% circulating blasts threshold for diagnosis of leukemic transformation in primary myelofibrosis. Blood Cancer J. 2018;8(6):57. doi: 10.1038/s41408-018-0095-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Odenike O. How I treat the blast phase of Philadelphia chromosome-negative myeloproliferative neoplasms. Blood. 2018;132(22):2339–2350. doi: 10.1182/blood-2018-03-785907. [DOI] [PubMed] [Google Scholar]
- 9.Tefferi A, Mudireddy M, Mannelli F, et al. Blast phase myeloproliferative neoplasm: Mayo-AGIMM study of 410 patients from two separate cohorts. Leukemia. 2018;32(5):1200–1210. doi: 10.1038/s41375-018-0019-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mesa RA, Li C-Y, Ketterling RP, Schroeder GS, Knudson RA, Tefferi A. Leukemic transformation in myelofibrosis with myeloid metaplasia: a single-institution experience with 91 cases. Blood. 2005;105(3):973–977. doi: 10.1182/blood-2004-07-2864. [DOI] [PubMed] [Google Scholar]
- 11.Kennedy JA, Atenafu EG, Messner HA, et al. Treatment outcomes following leukemic transformation in Philadelphia-negative myeloproliferative neoplasms. Blood. 2013;121(14):2725–2733. doi: 10.1182/blood-2012-10-464248. [DOI] [PubMed] [Google Scholar]
- 12.Roloff GW, Odenike O, Bajel A, Wei AH, Foley N, Uy GL. Contemporary approach to acute myeloid leukemia therapy in 2022. Am Soc Clin Oncol Educ Book. 2022;42(42):1–16. doi: 10.1200/EDBK_349605. [DOI] [PubMed] [Google Scholar]
- 13.Cooperrider JH, Shukla N, Nawas MT, Patel AA. The cup runneth over: treatment strategies for newly diagnosed acute myeloid leukemia. JCO Oncol Pract. 2023;19(2):74–85. doi: 10.1200/OP.22.00342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abdulkarim K, Girodon F, Johansson P, et al. AML transformation in 56 patients with Ph- MPD in two well defined populations. Eur J Haematol. 2009;82(2):106–111. doi: 10.1111/j.1600-0609.2008.01163.x. [DOI] [PubMed] [Google Scholar]
- 15.Rampal R, Ahn J, Abdel-Wahab O, et al. Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc Natl Acad Sci U S A. 2014;111(50):E5401–E5410. doi: 10.1073/pnas.1407792111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lasho TL, Mudireddy M, Finke CM, et al. Targeted next-generation sequencing in blast phase myeloproliferative neoplasms. Blood Adv. 2018;2(4):370–380. doi: 10.1182/bloodadvances.2018015875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Venton G, Courtier F, Charbonnier A, et al. Impact of gene mutations on treatment response and prognosis of acute myeloid leukemia secondary to myeloproliferative neoplasms. Am J Hematol. 2018;93(3):330–338. doi: 10.1002/ajh.24973. [DOI] [PubMed] [Google Scholar]
- 18.McNamara CJ, Panzarella T, Kennedy JA, et al. The mutational landscape of accelerated- and blast-phase myeloproliferative neoplasms impacts patient outcomes. Blood Adv. 2018;2(20):2658–2671. doi: 10.1182/bloodadvances.2018021469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tremblay D, Feld J, Dougherty M, et al. Venetoclax and hypomethylating agent combination therapy in acute myeloid leukemia secondary to a myeloproliferative neoplasm. Leuk Res. 2020;98 doi: 10.1016/j.leukres.2020.106456. [DOI] [PubMed] [Google Scholar]
- 20.King AC, Weis TM, Derkach A, et al. Multicenter evaluation of efficacy and toxicity of venetoclax-based combinations in patients with accelerated and blast phase myeloproliferative neoplasms. Am J Hematol. 2022;97(1):E7–E10. doi: 10.1002/ajh.26381. [DOI] [PubMed] [Google Scholar]
- 21.Gangat N, Ilyas R, Mc Cullough K, et al. Predictors of response to venetoclax plus hypomethylating agent therapy and survival in blast-phase myeloproliferative neoplasm. Haematologica. 2022;140(Suppl 1):9675–9676. doi: 10.3324/haematol.2022.282019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Masarova L, DiNardo CD, Bose P, et al. Single-center experience with venetoclax combinations in patients with newly diagnosed and relapsed AML evolving from MPNs. Blood Adv. 2021;5(8):2156–2164. doi: 10.1182/bloodadvances.2020003934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patel AA, Cahill K, Charnot-Katsikas A, et al. Clinical outcomes of IDH2-mutated advanced-phase Ph-negative myeloproliferative neoplasms treated with enasidenib. Br J Haematol. 2020;190(1):e48–e51. doi: 10.1111/bjh.16709. [DOI] [PubMed] [Google Scholar]
- 24.Chifotides HT, Masarova L, Alfayez M, et al. Outcome of patients with IDH1/2-mutated post-myeloproliferative neoplasm AML in the era of IDH inhibitors. Blood Adv. 2020;4(21):5336–5342. doi: 10.1182/bloodadvances.2020001528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gangat N, Ajufo H, Abdelmagid M, et al. IDH1/2 inhibitor monotherapy in blast-phase myeloproliferative neoplasms: a multicentre experience. Br J Haematol. 2023;203(3):e87–e92. doi: 10.1111/bjh.19027. [DOI] [PubMed] [Google Scholar]
- 26.Bose P, Verstovsek S, Cortes JE, et al. A phase 1/2 study of ruxolitinib and decitabine in patients with post-myeloproliferative neoplasm acute myeloid leukemia. Leukemia. 2020;34(9):2489–2492. doi: 10.1038/s41375-020-0778-0. [DOI] [PubMed] [Google Scholar]
- 27.Mascarenhas JO, Rampal RK, Kosiorek HE, et al. Phase 2 study of ruxolitinib and decitabine in patients with myeloproliferative neoplasm in accelerated and blast phase. Blood Adv. 2020;4(20):5246–5256. doi: 10.1182/bloodadvances.2020002119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–447. doi: 10.1182/blood-2016-08-733196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mascarenhas J, Heaney ML, Najfeld V, et al. Proposed criteria for response assessment in patients treated in clinical trials for myeloproliferative neoplasms in blast phase (MPN-BP): formal recommendations from the post-myeloproliferative neoplasm acute myeloid leukemia consortium. Leuk Res. 2012;36(12):1500–1504. doi: 10.1016/j.leukres.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tefferi A, Guglielmelli P, Lasho TL, et al. MIPSS70+ version 2.0: mutation and karyotype-enhanced international prognostic scoring system for primary myelofibrosis. J Clin Oncol. 2018;36(17):1769–1770. doi: 10.1200/JCO.2018.78.9867. [DOI] [PubMed] [Google Scholar]
- 31.Tefferi A, Guglielmelli P, Larson DR, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood. 2014;124(16):2507–2615. doi: 10.1182/blood-2014-05-579136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gagelmann N, Wolschke C, Salit RB, et al. Reduced intensity hematopoietic stem cell transplantation for accelerated-phase myelofibrosis. Blood Adv. 2022;6(4):1222–1231. doi: 10.1182/bloodadvances.2021006827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ortí G, Gras L, Zinger N, et al. Outcomes after allogeneic hematopoietic cell transplant in patients diagnosed with blast phase of myeloproliferative neoplasms: a retrospective study from the Chronic Malignancies Working Party of the European Society for Blood and Marrow Transplantation. Am J Hematol. 2023;98(4):628–638. doi: 10.1002/ajh.26833. [DOI] [PubMed] [Google Scholar]
- 34.Gupta V, Kim S, Hu Z-H, et al. Comparison of outcomes of HCT in blast phase of BCR-ABL1- MPN with de novo AML and with AML following MDS. Blood Adv. 2020;4(19):4748–4757. doi: 10.1182/bloodadvances.2020002621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bar-Natan M, Mascarenhas J, Gerds AT, et al. Molecularly targeted combination therapy for advanced phase myeloproliferative neoplasm: MPN-RC 119. Blood. 2022;140(Suppl 1):3988–3990. [Google Scholar]
- 36.Marinaccio C, Suraneni P, Celik H, et al. LKB1/STK11 is a tumor suppressor in the progression of myeloproliferative neoplasms. Cancer Discov. 2021;11(6):1398–1410. doi: 10.1158/2159-8290.CD-20-1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kong T, Laranjeira ABA, Yang K, et al. DUSP6 mediates resistance to JAK2 inhibition and drives leukemic progression. Nat Cancer. 2023;4(1):108–127. doi: 10.1038/s43018-022-00486-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saenz DT, Fiskus W, Manshouri T, et al. BET protein bromodomain inhibitor-based combinations are highly active against post-myeloproliferative neoplasm secondary AML cells. Leukemia. 2017;31(3):678–687. doi: 10.1038/leu.2016.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fiskus W, Mill CP, Nabet B, et al. Superior efficacy of co-targeting GFI1/KDM1A and BRD4 against AML and post-MPN secondary AML cells. Blood Cancer J. 2021;11(5):98. doi: 10.1038/s41408-021-00487-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fiskus W, Manshouri T, Birdwell C, et al. Efficacy of CDK9 inhibition in therapy of post-myeloproliferative neoplasm (MPN) secondary (s) AML cells. Blood Cancer J. 2022;12(1):23. doi: 10.1038/s41408-022-00618-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuusanmäki H, Dufva O, Vähä-Koskela M, et al. Erythroid/megakaryocytic differentiation confers BCL-XL dependency and venetoclax resistance in acute myeloid leukemia. Blood. 2023;141(13):1610–1625. doi: 10.1182/blood.2021011094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davidson MB, Kennedy JA, Capo-Chichi J-M, et al. Outcomes of intensive and nonintensive blast-reduction strategies in accelerated and blast-phase MPN. Blood Adv. 2024;8(5):1281–1294. doi: 10.1182/bloodadvances.2023011735. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.