

Summary
Prior research has suggested that GATA6+ pericardial macrophages may traffic to the myocardium to prevent interstitial fibrosis after myocardial infarction (MI), while subsequent literature claims that they do not. We demonstrate that GATA6+ pericardial macrophages are critical for preventing IL-33 induced pericarditis and attenuate trafficking of inflammatory monocytes and granulocytes to the pericardial cavity after MI. However, absence of GATA6+ macrophages did not affect myocardial inflammation due to MI or coxsackievirus-B3 induced myocarditis, or late-stage cardiac fibrosis and cardiac function post MI. GATA6+ macrophages are significantly less transcriptionally active following stimulation in vitro compared to bone marrow-derived macrophages and do not induce upregulation of inflammatory markers in fibroblasts. This suggests that GATA6+ pericardial macrophages attenuate inflammation through their interactions with surrounding cells. We therefore conclude that GATA6+ pericardial macrophages are critical in modulating pericardial inflammation, but do not play a significant role in controlling myocardial inflammation or fibrosis.
Subject areas: Natural sciences, Biological sciences, Biochemistry, Physiology, cell biology
Graphical abstract
Highlights
-
•
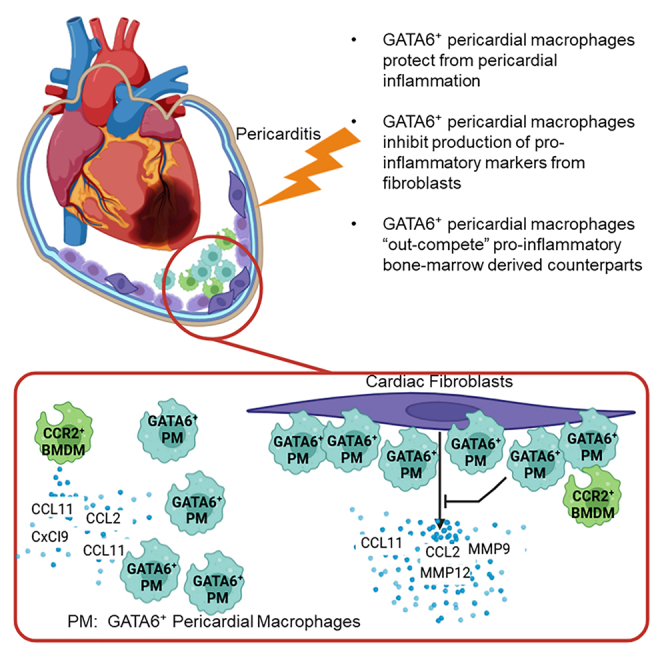
GATA6+ pericardial macrophages attenuate pericardial inflammation
-
•
GATA6+ pericardial macrophages minimally influence myocardial inflammation
-
•
GATA6+ macrophages inhibit fibroblasts from upregulating pro-inflammatory markers
Natural sciences; Biological sciences; Biochemistry; Physiology; Cell biology
Introduction
Serous cavities such as the cranial, pleural, pericardial, and peritoneal cavities serve critical functions by providing physical protection and lubrication to the tissues within the cavity and by hosting resident immune cells capable of responding to injury or infection.1,2,3,4 The pericardial cavity, the serous cavity that surrounds the heart, has long been studied for its role in maintaining cardiac health.5,6,7,8 The pericardium, which contains the pericardial cavity, is composed of three distinct layers: the parietal, visceral, and serous pericardium.7,8 The parietal pericardium, the outermost layer, consists primarily of a network of collagen fibers with smaller elastin fibers interspersed throughout to provide elasticity.7,9 The next innermost layer, the visceral pericardium, is a monolayer of mesothelial cells entrenched in a collagenous backing.1 The visceral pericardium forms an envelope comprising the innermost layer of the parietal pericardium and the outermost layer of the epicardium. This envelope contains the final layer of the pericardium, the serous pericardium or pericardial cavity. The serous pericardium contains the pericardial fluid. Most of the research has focused on the physical effects of either the fibrous parietal pericardium, or changes in the molecular contents of the pericardial fluid.1,2,10 This fluid is a plasma ultrafiltrate derived from the epicardial capillaries and interstitial fluid that fills the pericardial cavity and contains hundreds of molecules and cells with the potential for influencing disease progression.3,5,11,12 Much of the research on the pericardium during disease has focused on the non-cellular components such as adrenomedulin, IGF-1, MMP-2, asymmetric dmethylarginine, and FGF-25,6,11,12,13,14 and demonstrated that the molecular composition of the pericardium is significantly altered during disease. For example, pericardial fluid from ischemic heart disease patients is reported to induce apoptosis in cultured cardiomyocytes through the activation of the P38 MAPK pathway.15 Additionally, anterior myocardial infarction (MI) leads to a significant increase of adrenomedullin, IL-6 and IL-1β.16 MI also elevates pericardial fluid concentrations of MMP-2, MMP-9, IL-8, acidic fibroblast growth factor (FGF), and basic FGF.17,18,19
Despite these robust studies on the molecular composition of pericardial fluid and its relationship with cardiac disease, there is relatively less known with regards to how the pericardial cells themselves are able to influence disease progression. Pericardial interstitial cells, the dominant cell subtype in the healthy pericardium, include the pericardial fibroblasts and the pericardial mesothelial cells that make up the lining of the pericardial cavity.20,21,22,23,24 While these cells have been described in the healthy pericardium, little is known about their role during cardiac diseases. Recently, GATA6+ pericardial macrophages have emerged as a cell of interest during the development of MI.3,25 GATA6+ macrophages are a unique resident macrophage population primarily described by the transcription factor GATA6 and have been described in both the pleural and peritoneal cavities where they are thought to play important reparative roles during injury and disease progression.3,4,26
Serous cavities such as the peritoneum and pericardium contain two main macrophage phenotypes. There is a bone marrow-derived, small macrophage population that are referred to as small peritoneal macrophages (SPMs) in the peritoneum and a GATA6+ resident large macrophage population denoted as large peritoneal macrophages (LPMs) in the peritoneum.27 GATA6+ resident pericardial macrophages may be distinguished from other macrophages phenotypes within the cavity based on their expression of the transcription factor GATA6, the phosphatidylserine receptor TIM-4 and the marker CD73.28,29 GATA6+ resident serous cavity macrophages may also be distinguished from non-GATA6+ bone marrow-derived macrophages based changes in F4/80 and MHCII expression. GATA6+ resident macrophages express elevated F4/80 and lack MHCII, while bone marrow-derived non-resident serous cavity macrophages have a lower of F4/80 expression and elevated MHCII.27 The GATA6+ pericardial macrophages may also be distinguished from their myocardial counterparts based on their expression of GATA6.3 GATA6 is critical in these macrophages for metabolic regulation and survival. The myocardial tissue contains both resident and monocyte-derived macrophages.30,31 Both cardiac macrophages and monocytes may be CD64+F4/80+; however, monocytes may be distinguished from macrophages based on the expression of Ly6C on the monocytes.32 Macrophages may also be separated into resident and monocyte-derived through the expression of CCR2, where cardiac resident macrophages are CCR2− and monocyte-derived macrophages are CCR2+.33 The GATA6+ macrophages in other cavities, such as the pleural and peritoneal, are phenotypically similar to GATA6+ pericardial macrophages, leading to the assertion that they may play similar roles in the heart as peritoneal and pleural macrophages play in the peritoneum and lungs, respectively.3,34
Controversy has emerged over the role of GATA6+ pericardial macrophages during MI. A 2019 study found that GATA6+ pericardial macrophages migrate to the myocardium post-MI and prevent interstitial fibrosis. However, a separate 2022 publication discovered that these macrophages are incapable of such migration and therefore do not influence fibrosis post-MI.3,25 There is also no current literature on the role of GATA6+ pericardial macrophages in other cardiovascular diseases such as myocarditis or pericarditis. The goal of the research presented here is to fill in gaps in our understanding of how pericardial macrophages influence cardiac disease. We aim to understand how GATA6+ pericardial macrophages influence a variety of etiologies of cardiac inflammatory diseases to more comprehensively understand how these cells influence cardiac inflammation affecting the pericardium and myocardium. To this aim, we analyzed the role of GATA6+ macrophages in pericarditis, MI, and myocarditis. This allowed us to provide a comprehensive view of how GATA6+ macrophages behave during inflammation of both the pericardium and myocardium and their influence on cardiac health. This has also allowed us to determine not only how GATA6+ pericardial macrophages influence cardiac disease accompanied by inflammation, but also where they are most important for modulating inflammation around the heart: in the pericardium itself rather than the myocardial tissue. Through this we hope to illuminate important mechanisms by which GATA6+ pericardial macrophages influence MI, myocarditis, and pericarditis.
We have ordered these results such that we first examined inflammation during three major etiologies of cardiovascular disease (pericarditis, myocardial infarction, and myocarditis). Pericarditis was chosen first, as it is the home cavity of these cells. Myocardial infarction and myocarditis were both chosen as models of investigating how the GATA6+ pericardial macrophages would respond to injury in the myocardium. As the model of myocardial infarction involves direct injury to the pericardium, myocarditis was chosen as a secondary model of myocardial injury with no pericardial damage required. As the following results found that GATA6+ pericardial macrophages did not influence CVB3-induced myocarditis, we chose not to investigate the myocarditis disease model further. Once the influence of GATA6+ pericardial macrophages on the early inflammatory phase of myocardial infarction had been established, we chose to examine how pericardial macrophages influence specifically pericardial inflammation during disease. As the main outcome of myocardial infarction is late-stage remodeling leading to heart failure, we subsequently examined the influence of GATA6+ pericardial macrophages over this late-stage remodeling phase of myocardial infarction. Lastly, we utilized in vitro studies to explore the mechanism by which GATA6+ pericardial macrophages may be influencing these illnesses.
Results
GATA6+ pericardial macrophages protect against eosinophilic pericarditis
We first sought to examine how GATA6+ pericardial macrophages influence pericardial inflammation to better understand how these cells affect disease within the serous cavity. To this end we induced eosinophilic pericarditis in LysCre or LysCreGATA6fl/fl mice lacking pericardial macrophages using a previously established model to examine how GATA6+ pericardial macrophages influence pericardial inflammation35,36 (Figure 1A). We have previously demonstrated that this model of pericarditis induces clinically significant pericarditis in mice by IL-33 to innate lymphoid cells and eosinophils pathway.35 Briefly, IL-33 was injected intra-peritoneally at day 0, and every other subsequent day until mice were sacrificed at day 10 and hearts were examined by flow cytometry and histology. For histology images, IL-5 was injected concurrently with IL-33 in order to increase the number of eosinophils and severity of the pericarditis. We found that LysCre mice developed very mild pericardial inflammation while LysCreGATA6fl/fl mice developed significant pericarditis as evident by immune infiltration in the pericardium by histology (Figure 1B). To appropriately quantify the pericardial inflammation, the perimyocardial region most affected by pericardial inflammation was isolated from each sample (Figure 1C). These images were then fed into the MATLAB code where the nuclei were optically isolated and counted. An example output can be seen in Figure S2A. This analysis revealed that LysCreGATA6fl/fl perimyocardial regions contained significantly more nuclei, indicative of increased inflammation (Figure 1D). We also observed a trending increase in overall area of the selected region in LysCreGATA6fl/fl mice, corresponding to a significantly increased average pericardial thickness in these mice (Figures 1E and 1F). The average pericardial thickness was calculated by taking 12 representative measurements evenly distributed across the pericardium as shown in Figure S2B. There was no difference in the maximum thickness of the pericardium in either group, demonstrating that both groups did at least develop very mild disease, although disease did not affect as much of the pericardium in the LysCre controls based on the average thickness (Figure 1G).
Figure 1.

GATA6+ pericardial macrophages protect against eosinophilic pericarditis
(A) Schematic of experimental design.
(B) Representative histology of LysCre and LysCreGATA6fl/fl mice. Scale bar: 100 μm. Zoom: 10×.
(C) Histology showing isolated pericardium. Scale bar: 50 μm. Zoom: 20×.
(D) Nuclei present in each image of the pericardium (LysCren = 5, LysCreGATA6fl/fln = 4).
(E) Area of each pericardial histology section.
(F) Average thickness of the pericardium along the region of interest.
(G) Maximum thickness of the pericardium in the region of interest.
(H) Pie charts comparing major leukocyte compartments between LysCre and LysCreGATA6fl/fl mice (LysCren = 10, LysCreGATA6fl/fln = 10). Proportions are a percentage of CD45+ cells of whole heart with pericardium.
(I) Pie charts comparing Ly6C and CCR2 expression between LysCre and LysCreGATA6fl/fl mice. Proportions are a percentage of CD64+F4/80+ cells. ∗ indicates populations that are significantly different (p < 0.05).
(J) Ly6C+CCR2- cells as a proportion of CD64+F4/80+ cells.
(K) Ly6C−CCR2+ cells as a proportion of CD64+F4/80+ cells.
(L) Schematic of isolation of the pericardial tissue.
(M) Pie charts comparing major leukocyte compartments between LysCre and LysCreGATA6fl/fl mice in the pericardial tissue alone. Proportions are a percentage of CD45+ cells. Statistics: Student’s t test with Welch’s correction. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Next, to investigate which cells contributed to more severe pericarditis in Lys2CreGATA6fl/fl mice, we profiled the immune cells in the heart including both pericardium and myocardium using flow cytometry. We initially chose to examine the whole heart with the pericardial and myocardial tissues taken together. An example gating strategy may be found in Figure S1. We observed no significant difference in the total proportion of CD45+ leukocytes (Figure S2C); however, further analysis revealed that there were many significant differences in the composition of infiltrating immune cells in the hearts of LysCreGATA6fl/fl and LysCre mice as a percentage of total CD45+ leukocytes (Figure 1H). Namely, LysCreGATA6fl/fl mice had a lower proportion of CD19+ B cells, Ly6G+ neutrophils, and CD64+F4/80+ macrophages and monocytes. These mice also had a higher proportion of Ly6G+SiglecF+ granulocytes and SiglecF+ eosinophils. LysCreGATA6fl/fl mice also had a higher proportion of Ly6G+SiglecF+ granulocytes out of CD11b+ myeloid cells (Figure S2D). When we examined macrophages and monocytes as a proportion of their parent gate, the CD64+F4/80+ compartment specifically, LysCreGATA6fl/fl also had proportionally fewer Ly6C+CCR2− monocytes and more Ly6C−CCR2+ macrophages (Figures 1I–1K). Following this, we sought to determine whether these changes were specific to the pericardium or if there were any noticeable changes within the myocardium as well. We repeated the induction of pericarditis; however, we surgically separated pericardial and myocardial tissue during the sacrifice for separate flow cytometry analyses (Figures 1L and S2E). We found that there was no difference in the proportion of CD45+ cells between LysCreGATA6fj/fl mice and controls in the pericardial tissues (Figure S2F). We did, however, observe a significant increase in the proportion of Ly6G+ neutrophils, Ly6G+SiglecF+ granulocytes, and SiglecF+ eosinophils in the pericardium of LysCreGATA6fl/fl mice along with a decrease in the proportion of CD19+ B cells (Figure 1M). We observed no significant differences when examining the myocardium alone (Figures S2G and S2H). Taken together, these data indicate that GATA6+ pericardial macrophages prevent the recruitment of granulocytes specifically to the pericardial cavity and peri-myocardial area but not to the myocardium during IL-33 induced pericarditis. These results indicate that GATA6+ pericardial macrophages play an important role in preventing the accumulation of eosinophils within the pericardial cavity during IL-33 and IL-5 induced pericarditis mouse model.
GATA6+ pericardial macrophages do not attenuate myocardial inflammation in myocardial infarction or coxsackievirus-B3 myocarditis
Following the observation that GATA6+ pericardial macrophages appear to be protective against IL-33 induced pericarditis, we sought to examine whether GATA6+ pericardial macrophages had a role in protection from myocardial inflammation using two different models of myocardial inflammation, MI and in CVB3-induced myocarditis. We first examined myocardial inflammation during early-stage MI. The MI mouse model is characterized by three distinct phases: an early inflammatory phase peaking at day 3, a proliferative phase lasting from day 4 to day 10, and a final cardiac remodeling phase from day 11 onwards where the majority of ventricular remodeling occurs leading to heart failure.31,37 We induced infarction by surgically ligating the left anterior descending (LAD) artery (Figure 2A). At day 3 post-MI whole hearts, the pericardium and myocardium together, were collected for flow cytometry analysis. There was no difference in the proportion of total CD45+ cells between groups (Figure S3A). We found that the absence of GATA6+ pericardial macrophages lead to an increase in the proportion of Ly6C−CCR2+ macrophages and a decrease in the proportion of Ly6C+CCR2− monocytes out of total CD45+ cells (Figures 2B–2D). When we examined the macrophage and monocyte compartment exclusively, we found increases in the proportions of Ly6C−CCR2+ and Ly6C−CCR2- macrophages, while there was a decrease in the proportion of Ly6C+CCR2− monocytes in the LysCreGATA6fl/fl mice (Figure 2E). Overall, immune cell frequencies in the heart were minimally affected by the absence of GATA6+ pericardial macrophages during acute stage of MI.
Figure 2.

GATA6+ pericardial macrophages do not attenuate myocardial inflammation in myocardial infarction or coxsackievirus-B3 myocarditis
(A) Schematic of experimental design for MI.
(B) Pie charts comparing major leukocyte compartments between LysCre and LysCreGATA6fl/fl mice (LysCren = 8, LysCreGATA6fl/fln = 12) in the whole heart with pericardium and myocardium. Proportions are a percentage of CD45+ cells. ∗ indicates populations that are significantly different (p < 0.05).
(C) Ly6C+CCR2− cells as a proportion of CD45+ cells.
(D) Ly6C−CCR2+ cells as a proportion of CD45+ cells.
(E) Pie charts comparing Ly6C and CCR2 expression between LysCre and LysCreGATA6fl/fl mice. Proportions are a percentage of CD64+F4/80+ cells. ∗ indicates populations that are significantly different (p < 0.05).
(F) Schematic of experimental design for CVB3 myocarditis.
(G) Myocarditis score of mice exposed to CVB3 (LysCren = 9, LysCreGATA6fl/fln = 10). Score 1: <10% inflammation, Score 2: 10–20% inflammation, Score 3: 30–50% inflammation, Score 4: 50–90% inflammation.
(H) Representative histology slides of LysCre and LysCreGATA6fl/fl mice. Scale bar: 200 μm. Zoom: 1.25×.
(I) Pie charts comparing major leukocyte compartments between LysCre and LysCreGATA6fl/fl mice. Proportions are a percentage of CD45+ cells. Statistics: Student’s t test with Welch’s correction. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Since the MI model involves a surgical procedure that damages both the myocardium and pericardium, we chose to examine the role of GATA6+ pericardial macrophages in CVB3-induced myocarditis as well. We induced disease by injecting CVB3-virus intra-peritoneally and harvesting hearts at day 8 post-infection for histology and flow cytometry (Figure 2F). We found no differences in inflammation by myocarditis score as determined by histology (Figures 2G and 2H). Additionally, we observed no significant differences in the immune profile of hearts after infection by flow cytometry (Figures 2I and S3B). Additionally, we observed no indications of pericarditis in either group based on the histology results present in Figures 2G and 2H. Thus, the presence or absence of GATA6+ pericardial macrophages did not significantly alter the severity or type of immune cells infiltrating the heart during acute stage of CVB3-induced myocarditis. In summary, the absence of GATA6+ pericardial macrophages during acute MI and CVB3-induced myocarditis only led to minor changes in frequencies of immune cells infiltrating the myocardium, without affecting the severity of inflammation.
GATA6+ pericardial macrophages influence pericardial inflammation in response to myocardial tissue damage
While the absence of GATA6+ pericardial macrophages only induced minor changes to early inflammation within the myocardial and pericardial tissue, we next sought to analyze whether the GATA6+ pericardial macrophages significantly influenced inflammation within the pericardium specifically during myocardial infarction. We induced MI in LysCre or LysCreGATA6fl/fl mice and sacrificed the mice at day 3 post-MI. Controls received sham surgeries. At day 3 post-MI we isolated the pericardial tissue in the same manner as described previously (Figure 3A). There was no significant difference between LysCre or LysCreGATA6fl/fl mice in the changes in the percentage of CD45+ cells out of total live cells within the pericardial tissue of the test and control animals (Figure 3B). However, we found significant shifts in immune cell composition based on both injury (sham vs. MI) and genotype (the presence or absence of GATA6+ pericardial macrophages) (Figure 3C). We determined that LysCreGATA6fl/fl mice who received sham surgery had an elevated proportion of CD3+ T cells and SiglecF+ eosinophils, while exhibiting a significant decrease in the proportion of F4/80+ macrophages (Figures 3D–3F). Interestingly, there was a significant decrease in F4/80+ macrophages in LysCre mice who received infarctions, similar to the already described macrophage disappearance reaction in myocardium after MI.38 LysCreGATA6fl/fl who received infarctions only showed a trending decrease in F4/80+ macrophages compared to their LysCre counterparts, due to the reduction of macrophages in the LysCre mice compared to sham controls. LysCreGATA6fl/fl mice receiving infarction also showed a significant reduction in the proportion of CD19+ B cells and a significant increase in TCRγδ+ T cells and Ly6G+SiglecF+ granulocytes, similar to our findings in the pericarditis model (Figures 3G–3I). Thus, the absence of GATA6+ macrophages led to significant changes in immune cell composition in sham and MI mice, although it did not alter the severity of inflammation after MI.
Figure 3.

GATA6+ pericardial macrophages influence pericardial inflammation in response to myocardial tissue damage
(A) Schematic of experimental design.
(B) CD45+ leukocytes as a percentage of live cells (LysCre sham n = 4, LysCre infarct n = 9, LysCreGATA6fl/fl sham n = 4, LysCreGATA6fl/fln = 8) within the pericardium alone.
(C) Pie charts comparing major leukocyte compartments between LysCre and LysCreGATA6fl/fl mice based on surgical status of sham or infarction. Proportions are a percentage of CD45+ cells.
(D) CD3+ cells as a proportion of CD45+ cells.
(E) SiglecF+ cells as a proportion of CD45+ cells.
(F) F4/80+ cells as a percentage of CD45+ cells.
(G) CD19+ cells as a percentage of CD45+ cells.
(H) TCRγδ+ cells as a percentage of CD45+ cells.
(I) Ly6G+SiglecF+ cells as a percentage of CD45+ cells. Statistics: two-way ANOVA with Tukey’s multiple comparisons test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
GATA6+ pericardial macrophages minimally influence cardiac remodeling in myocardial infarction
While we observed only minor differences in acute inflammation between LysCre and LysCreGATA6fl/fl mice during MI, we examined whether the absence of GATA6+ pericardial macrophages would influence late-stage fibrosis and cardiac remodeling. Currently, there are contradictory findings on the role of GATA6+ pericardial macrophages in controlling post-infarction fibrosis. A 2019 publication found that GATA6+ pericardial macrophages migrated to the site of injury and were critical for preventing interstitial fibrosis, while a separate 2022 publication found that these macrophages did not migrate to the myocardium and do not influence fibrosis.3,25 We therefore sought to provide additional findings to help resolve this issue through an unbiased assessment of histology using MATLAB code coupled with echocardiography to examine cardiac function in addition to our findings in inflammation during pericarditis, myocarditis and the early inflammatory phase of MI. At day 21 post-infarction we found that overall fibrosis was significantly higher in LysCreGATA6fl/fl mice based on overall fibrotic area (Figures 4A and 4B). In order to more accurately assess fibrosis and remove researcher bias, images were run through a MATLAB code to isolate fibrotic and healthy tissue regions and determine the percent fibrotic area based on pixel counts. Example plots showing the separation of whole histology slices and smaller, zoomed in regions are displayed in Figures S4A and S4B. We also broke down the analysis into specific regions, similar to Deniset et al.3 The infarct zone was selected as the region in the middle of the left ventricle. We found no differences in fibrotic area in the infarct zone, both genotypes displayed significant fibrosis and interstitial fibrosis (Figures 4C and 4D). The border zone was chosen as the region where fibrosis from the infarct zone begins to enter healthy tissue. Again, we observed no significant differences in fibrotic area within this region (Figures 4E and 4F). The remote zone was chosen as a random tissue region outside the infarct zone. In contrast to previous reports, both LysCre and LysCreGATA6fl/fl mice exhibited very limited fibrosis in the remote zone, with the exception of one mouse in each group (Figures 4G and 4H). We also noted no significant change in the average or minimum left ventricular thickness at this late time point (Figures 4I and 4J). Additionally, we assessed cardiac function by echocardiography and found no difference in ejection fraction or left ventricular internal diameter end diastole (LVIDD) (Figures 4K and 4L). Similar results were obtained for fractional shortening, intraventricular septal thickness end systole (IVSS), intraventricular septal thickness end diastole (IVSD), left ventricular internal diameter end systole (LVIDS), left ventricular posterior wall thickness end systole (LVPWS), and left ventricular posterior wall end diastole (LVPWD) (Figures S5A–S5D). We also performed electrocardiography (EKG) on these mice and found no discernable differences in the DI waveforms based on the presence or absence of GATA6+ pericardial macrophages (Figures S5E and S5F). Overall, the absence of GATA6+ pericardial macrophages led to minor changes in overall fibrosis; however, this did not correspond to any meaningful changes in cardiac function or ventricular wall thickness.
Figure 4.

GATA6+ pericardial macrophages do not protect from cardiac remodeling in myocardial infarction
(A) Representative histology showing whole cross section between LysCre and LysCreGATA6fl/fl mice. Zoom: 1.25×, scale bar: 200 μm.
(B) Percent fibrotic area calculated via MATLAB (LysCren = 8, LysCreGATA6fl/fln = 10).
(C) Representative histology of the infarct zone.
(D) Percent fibrotic area of the infarct zone.
(E) Representative histology of the border zone.
(F) Percent fibrotic area of the border zone.
(G) Representative histology of the remote zone.
(H) Percent fibrotic area of the remote zone.
(I) Ejection fraction (EF).
(J) Intraventricular septal end diastole (IVSD).
(K) Left ventricular internal diameter end diastole (LVIDD).
(L) Left ventricular posterior wall end diastole (LVPWD). Zoomed in sections are 20× zoom, scale bar: 50 μm. Scale bars for zoomed in sections of (E) are applicable to zoomed in sections of (F) and (G). Statistics: Student’s t test with Welch’s correction. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
GATA6+ serous cavity macrophages uniquely respond to inflammatory stimuli in vitro
Our next step was to understand both how GATA6+ pericardial macrophages influence pericardial inflammation and why they do not appear to influence myocardial inflammation. Prior literature suggests that GATA6+ pericardial macrophages do not traffic to the myocardium post-infarction.25 Using imaging flow cytometry, we demonstrated that pericardial macrophages express a separate integrin dimer to myocardial macrophages (Figure 5A). GATA6+ pericardial macrophages primarily expressed the arginine-glycine-aspartic acid (RGD) receptor-binding integrin αVβ3, while myocardial macrophages expressed the laminin integrin receptor α6β1.39 We also noted differences in integrin expression by flow cytometry (Figure 5B). GATA6+ pericardial macrophages expressed the integrins β2, β3, and β5, while myocardial macrophages did not. GATA6+ pericardial macrophages also displayed greater heterogeneity, frequently showing a bimodal distribution, while myocardial macrophages typically displayed a monomodal distribution. These different integrins suggested that GATA6+ pericardial macrophages may have limited influence over the myocardial cavity due to a limited ability to migrate to and survive within the myocardial tissue.
Figure 5.

GATA6+ serous cavity macrophages uniquely respond to inflammatory stimuli in vitro
(A) Co-localization of major integrin dimers of pericardial macrophages (top) and myocardial macrophages (bottom).
(B) Histograms showing expression of integrin monomers on pericardial and myocardial macrophages.
(C) Schematic of in-vitro experiments.
(D) CCL2 mRNA fold change in macrophages (n = 3 for all groups).
(E) CCL11 mRNA fold change in macrophages.
(F) CxCL9 mRNA fold change in macrophages.
(G) IL-1β mRNA fold change in macrophages.
(H) CCL2 mRNA fold change in fibroblasts.
(I) CCL11 mRNA fold change in fibroblasts.
(J) GM-CSF mRNA fold change in fibroblasts.
(K) α-SMA mRNA fold change in fibroblasts. Statistics: for macrophages two-way ANOVA with Tukey’s multiple comparisons test was used. For fibroblasts one-way ANOVA with Tukey’s post-test was used. Lines between groups represent the result of Tukey’s post-test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Next, we examined a mechanism of how GATA6+ pericardial macrophages might influence pericardial inflammation. We performed in vitro experiments to determine how GATA6+ macrophages respond to inflammatory stimuli compared to their bone marrow-derived macrophage (BMDM) counterparts. As the number of GATA6+ pericardial macrophages needed for this experiment would have required a prohibitive number of mice, GATA6+ peritoneal macrophages were chosen as an analogue to GATA6+ pericardial macrophages since they are phenotypically similar.3,4,34 BMDMs were chosen as analogous to the small GATA6−, bone marrow-derived macrophages that are present within the serous cavity spaces.27 Peritoneal fibroblasts were also used as a feeder layer, as GATA6+ macrophages did not survive culture on their own (data not shown). The experimental workflow can be found in Figure 5C. In brief, peritoneal fibroblasts were obtained from wild-type (WT) B6 mice and co-cultured with either GATA6+ macrophages or BMDMs and stimulated with either 100 ng/mL lipopolysaccharide (LPS) or medium alone. Macrophages and fibroblasts were then separated via magnetic activated cell sorting (MACS) sorting using CD45 magnetic microbeads and processed for RT-PCR-based analysis of the expression levels of select genes.
BMDMs, but not GATA6+ macrophages, significantly upregulated CCL2, CCL11, CxCL9, and IL-1β in response to LPS stimulation (Figures 5D–5G). CCL2 is an important monocyte chemoattractant, while CCL11 and CxCl9 are important for attracting eosinophils. IL-1β is an important cytokine for multiple etiologies of inflammation and also plays a critical role in the recruitment of neutrophils. Similar results were observed for IL-6, IL-10, and MMP12 (Figures S6A–S6C). TIMP2 meanwhile, was significantly decreased in GATA6+ macrophages regardless of stimulation (Figure S5D). We also analyzed the fibroblasts that were subject to LPS stimulation and found that BMDMs but not GATA6+ macrophages stimulated fibroblasts to upregulate CCL2 and CCL11 (Figures 5H and 5I). GATA6+ macrophages did, however, cause a trending but not significant upregulation of granulocyte-macrophage colony stimulating factor (GM-CSF) and significant downregulation of α-SMA (Figures 5J and 5K). Additionally, we observed BMDMs lead to significant upregulation of MMP9 under LPS stimulation, while the expression of MMP9 induced by GATA6+ macrophages remained constant regardless of stimulation (Figure S6E). We also observed that BMDMs but not GATA6+ macrophages induced upregulation of MMP12 in fibroblasts (Figure S6F). Additionally, data are not shown for CCL24, MHCII, and IL-7 due to a lack of significant results. This demonstrates that GATA6+ macrophages respond uniquely to inflammatory stimuli. BMDMs stimulated with LPS led to upregulation of granulocyte chemoattractants while GATA6+ macrophages did not strongly respond to stimulation. Additionally, BMDMS but not GATA6+ macrophages induced upregulation of eosinophil and monocyte chemoattractants in fibroblasts. These effects may explain the previously described increase in granulocytes and CCR2+ macrophages during pericardial inflammation in mice lacking GATA6+ macrophages, as in the absence of GATA6+ macrophages, only non-GATA6+ macrophages will be present to interact with the surrounding stroma which may allow for accumulation of these pro-inflammatory markers. These results suggest that GATA6+ large pericardial macrophages may function primarily by out-competing or crowding out their non-GATA6+ small bone marrow-derived counterparts during homeostasis and disease.
Discussion
Myeloid cells have been shown to play important roles in many cardiovascular diseases.32,40,41,42 However, much of this work has focused on monocyte-derived macrophages and not the various subsets of resident macrophages that are associated with the heart. CD64+F4/80+ cardiac resident macrophages have emerged as a subset of cells of interest in cardiac inflammation.30,32 In addition to myocardial resident macrophages, a second population of resident macrophages within the pericardial cavity has been found that is phenotypically different from myocardial resident macrophages.3,34 Only recently have pericardial macrophages become a major topic of interest, where much of the research has been on the effects of pericardial macrophages on post-infarction cardiac remodeling or in clinical case studies.3,25,43,44 We therefore chose to examine the role of GATA6+ pericardial macrophages in multiple cardiovascular diseases pathologies in mice models to explore their contributions toward modulating inflammation in and around the heart.
We demonstrate here that the absence of GATA6+ pericardial macrophages is associated with more severe disease in a murine model of IL-33-induced pericarditis. This model has been previously associated with cardiac dysfunction via reduced ejection fraction, although this was not investigated in this study.35 The absence of GATA6+ cells is associated with increased trafficking of eosinophils and bone marrow-derived monocytes to the pericardium. The protective effects of GATA6+ pericardial macrophages appeared to be specific to the pericardium, as there was no effect on inflammation in the myocardial tissue during this model of pericarditis. These protective effects are reminiscent of a previously described effect where CCR2− cardiac macrophages inhibit the migration of monocytes to the site of injury.33 Cardiac macrophages have been assigned wide variety of roles during disease such as influencing the migration of cells to the site of injury, efferocytosis, phagocytic clearance of debris, or by promoting the production of anti-inflammatory factors.30,33,40,45,46 Prior research has also observed that macrophages prevent the accumulation of T cells within the tumor microenvironment based on experiments where macrophage depletion resulted in an increase in T cells within the tumor and an increase in Cxcl9 and CxCl10 production.47,48 However, while macrophages have been shown to interact with eosinophils and neutrophils, macrophages have not previously been shown to inhibit their trafficking.49,50,51 Our findings suggest that GATA6+ pericardial macrophages serve a similar function in the pericardium as CCR2− macrophages in the myocardium, with the GATA6+ macrophages primarily inhibiting granulocyte trafficking. By inhibiting the migration of granulocytes to the pericardium, the GATA6+ pericardial macrophages prevent IL-33 induced pericarditis in mice. We also observed a reduction in the proportion of Ly6C+CCR2− monocytes migrating to the myocardium in LysCreGATA6fl/fl mice, further demonstrating the similarity between GATA6+ pericardial macrophages and CCR2− myocardial resident macrophages.
There is currently significant disagreement on the role of GATA6+ pericardial macrophages during MI. One group has stated the GATA6+ pericardial macrophages traffic to the myocardium to prevent interstitial fibrosis and another group has showed these macrophages do not traffic to the myocardium and therefore should not affect interstitial fibrosis.3,25 We found that GATA6+ pericardial macrophages had minor but significant effects during the early inflammatory phase leading to a reduction in the proportion of Ly6C+CCR2− monocytes and an increase in the proportion of Ly6C−CCR2+ macrophages, similar to what was observed during IL-33 pericarditis. These CCR2+ macrophages are known to be pathogenic during cardiac inflammation and contribute to heart failure, fibrosis, and cardiac remodeling.52,53 We found only minor changes in cardiac remodeling with no significant alteration of cardiac function, despite the increase in CCR2+ macrophages. We also found a significant increase in fibrosis across the whole heart during MI. Deniset et al. found that pericardial macrophages prevented interstitial fibrosis in the remote region, the healthy myocardial tissue away from the site of injury.3 However, we were unable to replicate these results and found GATA6+ pericardial macrophages did not protect against fibrosis in any individual region of the myocardium, only across the whole cross-section of the heart. Despite the significant increase in fibrosis, we observed no significant change in left ventricle thickness or cardiac function based on echocardiography or EKG. This suggests that while GATA6+ pericardial macrophages minorly influence early inflammation and prevent more severe fibrosis post-MI, they are not able to influence overall ventricular structure, interstitial fibrosis or cardiac function. These results are reminiscent of recent controversy in the peritoneum. Prior research has claimed that peritoneal macrophages migrate to the liver during a liver injury model and prevent fibrosis in the liver; however, more recent literature has demonstrated that peritoneal macrophages do not deeply penetrate tissue and do not contribute to tissue repair or fibrosis in multiple models of liver inflammation.4,54 Similar results were found in the lungs, where pleural macrophages were unable to deeply penetrate the tissue and minimally contributed to tissue repair.54 This suggests our results fall into a larger trend wherein serous cavity macrophages as a whole do not appear to migrate into tissue in response to inflammatory stimulation and do not significantly contribute to tissue repair. However, these macrophages are critical in controlling inflammation within their respective cavities.55,56,57,58
To confirm the veracity of our claim that GATA6+ pericardial macrophages do not influence myocardial inflammation we chose to examine coxsackievirus-B3 induced myocarditis as a model of cardiac injury that did not require any surgical interaction with the pericardium or myocardium. Macrophages and monocytes have also been shown to play critical roles during the development of CVB3-induced myocarditis.59,60,61 However, we found GATA6+ macrophages did not protect the mice from cardiac inflammation, nor did they influence any cell phenotype infiltrating the myocardium in this model. In MI there is an inherent involvement of the pericardium while in CVB3-induced myocarditis, there is not.62 This may explain why we saw minor changes in inflammation during MI but no changes whatsoever in the myocarditis model.
While GATA6+ pericardial macrophages did not have a significant effect on myocardial inflammation, we found that the GATA6+ pericardial macrophages still affected the pericardial cavity during myocardial inflammation. Despite being characterized primarily by myocardial inflammation and remodeling, MI still directly involves the pericardium.62 In IL-33 pericarditis, the presence of GATA6+ pericardial macrophages appeared to prevent the accumulation of eosinophils within the pericardial cavity, while in MI the presence of GATA6+ pericardial macrophages correlated with a reduced influx of TCRγδ+ T cells and Ly6G+SiglecF+ granulocytes. It is possible these Ly6G+SiglecF+ granulocytes are either SiglecF+ neutrophils or Ly6G+ eosinophils, as both of these cell phenotypes have been previously described and SiglecF+ neutrophils specifically in MI.63,64,65,66 Additionally, both neutrophils and eosinophils play critical roles during MI.67,68 Based on previous reports we hypothesize that these cells are neutrophils; however, further phenotyping would be needed to confirm this.69 While prior literature found pericardial adipose tissue regulated granulopoiesis and fibrosis during MI we find that the GATA6+ pericardial macrophages also play a role in regulating granulopoiesis.70 However, these changes within the pericardial cavity did not affect changes within the myocardium, further supporting the hypothesis that GATA6+ pericardial macrophages only influence inflammation within the pericardial cavity.
We also noted that there was a macrophage disappearance reaction in the LysCre mice leading to an equal proportion of macrophages in the pericardium between the two genotypes. This is very similar to the macrophage disappearance reaction known to occur in the myocardium where cardiac resident macrophages are rapidly lost, while Ly6Chi monocytes traffic into the myocardium and differentiate into monocyte-derived macrophages.71,72,73 While Deniset et al. found that pericardial macrophages traffic into the myocardium, which would explain this disappearance, more recent research suggests that pericardial macrophages only accumulate at the mesothelial boundary between the epicardium and pericardium.3,25 This would mean the pericardial macrophages became embedded in the epicardium-facing section of the pericardium and were not collected with the remaining pericardial tissue.
We also found that differences in integrin structure may also help explain why pericardial macrophages do not appear to traffic into the myocardium. Integrins are dimerized receptors that play major roles in cell-cell adhesions and leukocyte trafficking.39 We found that myocardial macrophages primarily expressed the integrin dimer α6β1, a main binding receptor for laminin-111.74 Laminin is a major component of the cardiac basement membrane and is known to be elevated during cardiac fibrosis.75 In contrast, pericardial macrophages expressed the dimer αVβ3, which is the vitronectin receptor and binds to the RGD sequence (arg-gly-asp) common across many extracellular matrix proteins including fibronectin, vitronectin and fibrinogen.76,77 These differing extracellular matrix (ECM) binding affinities suggest that pericardial macrophages might not be attuned to the myocardial environment, lacking the primary integrins expressed by their myocardial counterpart therefore making them poorly suited to migrate to the myocardium.
To examine a potential mechanism by which GATA6+ pericardial macrophages affect pericardial inflammation we performed an in vitro experiment and found that BMDMs, but not GATA6+ macrophages, upregulated multiple inflammatory markers in response to LPS stimulation including CCL2, CCL11, and IL-1β. CCL2 is a potent chemoattractant required for the recruitment of monocytes and macrophages, CCL11 is responsible for the recruitment of eosinophils and IL-1β is a general inflammatory cytokine and has been tied to the recruitment of neutrophils.78,79,80,81,82 The dichotomy between GATA6+ macrophages and BMDMs is similar to a previously described difference between CCR2+ and CCR2− macrophages in a heart transplant model, where CCR2− macrophages inhibited monocyte trafficking and CCR2+ macrophages promoted it.33 Additionally, tumor associated macrophages were shown to inhibit CD8+ T cell migration into tumors in part by preventing the production of CxCL9.47,83 We observed an increase in CCR2+ macrophages, similar to those described in the aforementioned heart transplant model, in both pericarditis and MI. Based on our in vitro and in vivo findings we propose that GATA6+ pericardial macrophages prevent CCR2+ macrophages from accumulating in the pericardial cavity and releasing pro-inflammatory cytokines. When examining the fibroblasts that served as a feeder layer to the macrophages, BMDMs also led to the upregulation of inflammatory markers in fibroblasts while GATA6+ macrophages did not. Interestingly, the GATA6+ macrophages led to a reduction in the expression of α-SMA within fibroblasts. The expression of α-SMA in fibroblasts has previously been associated with the transition to myofibroblasts and increase production of ECM components leading to fibrosis.84,85,86 This may explain the changes in fibrosis we observed during MI, as an absence of GATA6+ macrophages lead to the surrounding stromal cells to acquiring a more pro-fibrotic phenotype. Fibroblast and macrophage interacting to promote inflammatory cytokines is a well-documented phenomenon where fibroblasts are able to contribute to heart failure.87 Fibroblasts are also a well-known component of the pericardial tissue and interact closely with GATA6+ pericardial macrophages.21,34 We therefore also hypothesize that GATA6+ pericardial macrophages, by preventing the influx of CCR2+ macrophages, may prevent CCR2+ macrophages from interacting with pericardial fibroblasts and thereby prevent fibroblasts from upregulating inflammatory markers. We therefore propose that GATA6+ large peritoneal macrophage-like pericardial macrophages function in part by outcompeting and outnumbering their bone marrow-derived, SPM-like counterparts thereby preventing the accumulation of pro-inflammatory factors within the cavity. GATA6+ pericardial macrophages may also prevent the interactions of SPM-like GATA6− pericardial macrophages with the surrounding stromal cells, preventing these stromal cells from acquiring a more pro-fibrotic or pro-remodeling phenotype.
In conclusion, we demonstrate that pericardial macrophages play a critical role in mitigating pericardial inflammation, but not myocardial inflammation. We show that GATA6+ pericardial macrophages minimize the infiltration of eosinophils during pericarditis and directly or indirectly attenuate pericardial inflammation during MI. We also demonstrate, however, that a lack of pericardial macrophages does not appear to alter myocardial inflammation during myocarditis and only minimally alters inflammation during MI. Additionally, minor changes in inflammation during MI corresponded only to minor changes in overall fibrosis with no change in ventricular thickness or cardiac function, indicating that these changes were not important for disease progression. We also propose that one of the potential mechanisms for peritoneal macrophages to attenuate pericardial inflammation is by remaining unresponsive to inflammatory stimulus and preventing the interaction of other macrophages with the pericardial stroma (Figure 6).
Figure 6.

GATA6+ pericardial macrophages prevent the accumulation of pro-inflammatory and pro-remodeling chemokines in response to pericardial injury
(A) When GATA6+ pericardial macrophages (represented as PM) are present in the pericardial cavity, they prevent the bone marrow-derived macrophages (BMDMs) from interacting with fibroblasts (shown in purple), thereby preventing the accumulation of pro-inflammatory and pro-remodeling markers. Additionally, the GATA6+ pericardial macrophages outnumber the BMDMs and remain less transcriptionally active in response to stimulation, thereby preventing the accumulation of pro-inflammatory markers.
(B) In the absence of GATA6+ pericardial macrophages, BMDMs are capable of interacting with fibroblasts within the pericardial cavity, leading to the upregulation of pro-inflammatory and pro-remodeling markers. BMDMs also represent a larger proportion of macrophages within the pericardial cavity, where they are free to produce pro-inflammatory markers.
Limitations of the study
Despite the advances in understanding how GATA6+ pericardial macrophages present in this work, there are some limitations to the study. In regards to the integrins study, it is unclear if the GATA6+ pericardial macrophages are capable of upregulating different integrins during inflammation which may allow them to migrate into the myocardium during inflammation. Additionally, there are still portions of the mechanism by which GATA6+ pericardial macrophages attenuate pericardial inflammation that are unclear. While our in vitro studies found that GATA6+ macrophages are unresponsive to inflammatory stimuli, additional tools such as multiplexing or single cell RNA-sequencing may be able to find upregulated genes that we did not examine during this study that may illuminate the behavior of GATA6+ pericardial macrophages more clearly. Moreover, we utilized GATA6+ peritoneal macrophages as an analogue to GATA6+ pericardial macrophages; however, there may be as yet unknown differences between these two populations which may influence their function. Lastly, the influence of GATA6+ pericardial macrophage over the recruitment and differentiation of CCR2+ monocytes and macrophages is not completely clear at this moment. These factors all require further investigation in the future.
STAR★Methods
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Daniela Cihakova (cihakova@jhmi.edu).
Materials availability
This study did not generate any new unique reagents.
Data and code availability
For inquiries on unprocessed data or code please e-mail the lead contact, Dr. Daniela Cihakova (cihakova@jhmi.edu)
Experimental model and study participant details
Mice
8–12-week-old male C57BL/6J wild-type (WT) (Jackson Laboratory, 000664) C57BL/6J LysCre (Jackson Laboratory, 004781) and C57BL/6J GATA6fl/fl developed by Sodhi et al. and gifted by Dr. Gwendalyn Randolph of Washington University were used.28,88,89 LysCre and GATA6fl/fl mice were crossed to develop LysCreGATA6fl/fl mice. LysCre mice were used as experimental controls. All animal protocols and methods were approved by the Animal Care and Use Committee of Johns Hopkins University. Mice were kept in the pathogen free facility at Johns Hopkins University. All procedures conform to NIH guidelines for the care and use of laboratory animals.
Induction of IL-33 induced pericarditis
IL-33 induced pericarditis was induced by injecting mice i.p . with 1 μg of IL-33 (BioLegend) at days 0, 2, 4, 6 and 8. Mice were sacrificed on day 10 and hearts were harvested for histology or flow cytometry. Due to difficulty in visualizing pericarditis in B6 mice, additional experiments were performed where 100 pmol/kg IL-5 (peprotec) was injected alongside IL-33.
Induction of myocardial infarction
Myocardial infarction was induced by ligating the left anterior descending (LAD) artery. Briefly, mice were anaesthetized with 4% isoflurane (Baxter), endotracheally intubated and mechanically ventilated with 1.5% isoflurane (Baxter) throughout the procedure by a model 845 MiniVent small animal ventilator (Harvard Apparatus). Pre-operational analgesics (1 mg/kg Buprenorphine, slow-release formulation, ZooPharm) and paralytics (1 mg/kg Succinylcholine, Henry Schein) were administered prior to the procedure via subcutaneous and I.P. injection respectively. Mice were subjected to left thoracotomy at either the third or fourth intercostal space. A 9-0 suture was advanced sub-epicardially, perpendicular to the LAD artery. Permanent occlusion was achieved by tightening the suture around the artery. Infarction was confirmed by myocardial bleaching and loss of motion below the ligation. The rib cage was closed with a 6-0 nylon suture and the skin was closed with a 5-0 silk suture. Sham-surgery animals received the same procedure without the ligation of the LAD artery. Mice were sacrificed at day 3 or 21 to assess inflammation and cardiac remodeling.
Induction of Coxsackievirus-B3 induced myocarditis
8–12-week-old mice were injected with 100 μL of 103 PFU of heart-passaged coxsackievirus-B3 (CVB3) in sterile PBS on day 0. Hearts were collected at day 8 post-infection. At sacrifice hearts were resected and cut longitudinally. One half was fixed in SafeFix II for histology. H&E histology slides were prepared by Histoserv, Inc. (German town, MD). The second half of the heart was processed for flow cytometry as described below. Histology was scored based on the proportion of inflammatory infiltrate that could be observed in the tissue based on H&E histology. Scores were assigned as: 0 – no inflammation present, 1 – < 10% inflammation, 2 – 10-30% inflammation, 3 – 30-50% inflammation, 4 – 50-90% inflammation, 5 - >90% inflammation.
Method details
Electrocardiography
Electrocardiography (EKG) was performed by anaesthetizing the mouse with 600uL of avertin injected I.P. The mouse was then placed in a supine position with paws outstretched such that the chest was tight. Electrodes (Intco Medical) were placed over the paws of the mouse with a small quantity of saline to provide electrical conductions. Leads were then attached to the electrodes via alligator clips and connected to a DigiMed Sinus Rhythm Analyzer. EKGs were then measured using DMSI-400. A representative EKG waveform section was then selected and printed to a PDF for analysis.
Processing of murine cardiac cells
Cells from murine myocardium were isolated by perfusing the heart with 1X PBS for 3 minutes via aortic perfusion. Hearts were then resected, minced and placed in GentleMACS C-tubes (Miltenyi Biotec) with 5mL of digestion buffer consisting of 5mL HBSS (Corning) with 5,000 U/mL Collagenase II and 500 U/mL DNase I (Worthington Biochemical). Samples were incubated for 37°C for 30 minutes with gentle agitation. Tissue was mechanically dissociated using GentleMACS system before and after the incubation. Cells were then filtered through a 40μm filter, washed with 1X PBS. Samples were then incubated in ACK Lysing Buffer (Quality Biologic) for 2 minutes and washed again with 1X PBs. Cells were then washed and resuspended in 1X PBS. For experiments where pericardial and myocardial tissue needed to be separated, mice were anaesthetized similar to the myocardial infarction surgery. A left thoracotomy was then performed, and the pericardium was carefully dissected. The mouse was then cervically dislocated, and the heart was resected. The myocardial tissue was then processed as described above. The pericardial tissue was then placed into a gentleMACS C-tube containing 5mL of HBSS with 1000U of collagenase II and 100U of DNase I. Samples were incubated for 37°C for 25 minutes with gentle agitation. Tissue was mechanically dissociated using gentleMACS system before and after the incubation. Cells were then filtered through a 70μm filter, washed with 1X PBS.
Flow cytometry
Flow cytometry was performed using a Cytek Aurora spectral flow cytometer or BD Fortessa. Imaging flow cytometry was performed using an Amnix Imagestream X MK II. LIVE/DEAD fixable aqua was used to determine cell viability (ThermoFisher). Prior to surface staining, cells were blocked by incubating with a 1:50 dilution of Anti-Mouse CD16/CD32 (Invitrogen). Antibodies were diluted in fluorescence activated cell sorting (FACS) buffer (2.5% BSA, 2mM EDTA in 1X PBS). For intranuclear staining, cells were fixed and permeabilized using FoxP3/Transcription Factor Staining Buffer Set (eBioscience). Antibodies and their concentrations can be found in Table S1. Compensation was performed using UltraComp eBeads Compensation Beads (Invitrogen). An example gating strategy is located in Figure S1.
Harvest of peritoneal macrophages
Peritoneal macrophages were harvested via peritoneal lavage. Mice were first anaesthetized with 600uL of Avertin injected I.P.. 5mL 1X phosphate buffered saline (PBS) containing 200U/mL collagenase II (Worthington) and 400U/mL hyaluronidase (Miltenyi) was injected into the peritoneal cavity of these mice. After 20 minutes mice were euthanized via cervical dislocation, the peritoneum was opened, and fluid was aspirated using a transfer pipette. F4/80+ cells were isolated using F4/80 positive selection via magnetic activated cell sorting (MACS) sorting with Miltenyi F4/80 mouse microbeads according to the manufacturers protocol. Cells were then transferred into complete Dulbecco’s modified eagle medium (DMEM) containing 1ng/mL M-CSF.
Isolation of bone marrow derived macrophages
Bone marrow derived macrophages were harvested from WT C57/B6 mice by first euthanizing the animal via I.P. injection of 600uL of Avertin followed by cervical dislocation. The femur was then removed and perfused with 1mL of sterile 1X PBS. Ammonium-Chloride-Potassium (ACK) lysing buffer was administered to remove erythrocytes and cells were once again washed with sterile 1X PBS. Cells were then resuspended in complete DMEM containing 1ng/mL M-CSF and supplemented with antibiotic/antimycotic solution and L-glutamine (Corning) in a T-25 plate for differentiation into macrophages.
Isolation of peritoneal fibroblasts
Murine peritoneal fibroblasts were obtained by first euthanizing the animal via I.P. injection of 600uL of Avertin and cervical dislocation. The skin was then removed from the peritoneum and a 1mm2 section of peritoneal tissue was excised. The peritoneal tissue was then minced and digested in gentleMACS C-tubes using Hank’s buffered salt solution (HBSS) containing 500U/mL deoxyribonuclease (DNase) I and 5000u/mL collagenase II. Cells were then transferred to a T-25 plate containing completed Roswell Park Memorial Institute (RPMI) media with 20% FBS and supplemented with antibiotic/antimycotic solution and L-glutamine (Corning). No additional selection was required to isolate fibroblasts, as the fibroblasts rapidly outgrew all other cell types by passage 2.
Co-culture of murine peritoneal macrophages and fibroblasts
Peritoneal Fibroblasts were transferred to 24-well plates at 30,000 cells/well in 1mL of complete RPMI with 20% FBS and supplemented with antibiotic/antimycotic solution and L-glutamine (Corning). After 24 hours media was aspirated and either peritoneal or bone marrow derived macrophages were added at 30,000 cells/well in 1mL of complete DMEM containing 1ng/mL of M-CSF. At 24 hours all media was aspirated and replaced with 1mL of completed DMEM containing 1ng/mL M-CSF and appropriate wells were given 100ng/mL of LPS. 16-hours post stimulation supernatant was removed and stored at -80°C for ELISA. Macrophages and fibroblasts were separated by MACS sorting using Miltenyi mouse CD45 microbeads. All cells were then placed in TRIzol for RNA extraction.
Quantitative PCR
RNA from cardiac tissue, MACS isolated cells or cultured cells was extracted in TRIzol and quantitated via a Spectramax Plus 384 Absorbance Plate Reader. Single-strand cDNA was synthesized using a Bio-Rad iScript cDNA synthesis kit. Expression levels of genes was detected using qPCR with iQ SYBR Green Master Mix and acquired on the Bio-rad CFX Opus 96 Real-Time PCR System. Gene expression was analyzed by the 2-ΔΔCt- method, normalizing threshold cycles to GAPDH expression. Primers are listed in Table S2.
Isolation of pericardial cells
Left thoracotomy was performed on mice as described above. The pericardium was carefully dissected and placed into a gentleMACS C-tube containing 5mL of HBSS with 1000U of collagenase II and 100U of DNase I. Samples were incubated for 37°C for 25 minutes with gentle agitation. Tissue was mechanically dissociated using gentleMACS system before and after the incubation. Cells were then filtered through a 70μm filter, washed with 1X PBS.
Imaging of histology slides
All histology slides were imaged using an Olympus BX43 microscope attached to an Olympus TL4 light source. All images were taken with a Nikon DS-Fi3 microscope camera with a 0.45 aperture. Whole heart cross-sections were obtained with a 1.25x objective with a 0.04 numerical aperture. Images of cardiac regions were obtained on a 10x or 20x objective with 0.30 and 0.50 numerical apertures respectively. Images were all saved as .tiff files. When required, the background of images was removed using PowerPoints built in background removal features. Images were annotated and measured using Fiji (ImageJ) and pericardial sections were isolated by cropping in Fiji. Histology images were analyzed by MATLAB processing. Images were first taken and appropriate Hue, Saturation and Value measurements were determined from MATLAB’s internal color thresholder application to separate either fibrosis from Mason’s Trichrome staining or nuclei from H&E staining. Values of 0.5 – 0.75 for hue, 0.2 – 1 for saturation and 0 – 1 for value were used to isolate fibrotic tissue while values of 0.75 – 1 for hue, 0.2 – 1 for saturation and 0 – 1 for value isolated healthy tissue from trichrome staining. For analyzing H&E staining values of 0.585 – 0.759 for hue, 0.096 – 1 for saturation and 0 – 1 for value were used to isolate nuclei while hue of 0.759 – 1, saturation of 0.096 – 1 and value of 0 – 1 isolated cardiac tissue. For fibrosis analysis, the percent fibrotic tissue was determined by counting the number of pixels of isolated fibrotic tissue and the number of pixels of isolated healthy tissue. For H&E analysis, the number of nuclei was counted by first converting the isolated nuclei image into a binary black and white image and running MATLAB’s bwlabel function on the resulting image.
Quantification and statistical analysis
All statistics were performed using GraphPad Prism 7. For comparisons between two groups a Student’s T-test with Welch’s Correction was performed when analyzing flow cytometry data, a Mann-Whitney U-test was performed for assessing myocarditis severity by histology. For comparisons between three groups two-way ANOVA with Tukey’s post-test for multiple comparisons was performed. A P-value of <0.05 was considered statistically significant. Statistical details for each experiment may be found in the figure legends.
Acknowledgments
This work was supported by the American Heart Association (AHA) 19TPA34910007 and 20TPA35490421, the National Institutes of Health (NIH)/National Heart, Lung, and Blood Institute (NHLBI) R01HL118183 and R01HL136586. T.W. was supported by the 2018 Rhett Lundy Memorial Research Fellowship from the Myocarditis Foundation. D.M.H. was funded by the NIH/NHLBI F31HL149328. V.M. is supported by AZV grants NU22-02-00161 and NU21-02-00402, and by the National Institute for Research of Metabolic and Cardiovascular Diseases (Program EXCELES, ID project no. LX22NPO5104)—funded by the European Union – Next Generation EU. We would like to thank Dr. Gwendalyn Randolph of Washington University School of Medicine in St. Louis for providing GATA6fl/fl mice for these experiments. We would also like to acknowledge Xiaoling Zhang and the Johns Hopkins University Ross Flow Cytometry Core for use of their instruments for our flow cytometry experiments. The Aurora at the Ross Flow Cytometry Core is funded by NIH S10OD026859. Figures were constructed with Biorender.
Author contributions
Conceptualization, D.M.H. and D.C.; methodology, D.M.H. and D.C.; investigation, D.M.H., T.W., M.V.T., H.M.K., W.B.-B., and D.C.; formal analysis, D.M.H.; resources, I.J., O.S., I.S., L.C., and V.M.; writing – original draft, D.M.H. and D.C.; writing – review & editing, D.M.H., D.C., T.W., M.V.T., H.M.K., I.J., O.S., I.S., L.C., V.M., W.B.-B., and D.C.; supervision, D.C.
Declaration of interests
The authors declare no competing interests.
Published: June 13, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.110244.
Supplemental information
References
- 1.Hoit B.D. Anatomy and Physiology of the Pericardium. Cardiol. Clin. 2017;35:481–490. doi: 10.1016/j.ccl.2017.07.002. [DOI] [PubMed] [Google Scholar]
- 2.Vogiatzidis K., Zarogiannis S.G., Aidonidis I., Solenov E.I., Molyvdas P.A., Gourgoulianis K.I., Hatzoglou C. Physiology of pericardial fluid production and drainage. Front. Physiol. 2015;6:62. doi: 10.3389/fphys.2015.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deniset J.F., Belke D., Lee W.Y., Jorch S.K., Deppermann C., Hassanabad A.F., Turnbull J.D., Teng G., Rozich I., Hudspeth K., et al. Gata6(+) Pericardial Cavity Macrophages Relocate to the Injured Heart and Prevent Cardiac Fibrosis. Immunity. 2019;51:131–140.e5. doi: 10.1016/j.immuni.2019.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang J., Kubes P. A Reservoir of Mature Cavity Macrophages that Can Rapidly Invade Visceral Organs to Affect Tissue Repair. Cell. 2016;165:668–678. doi: 10.1016/j.cell.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 5.Trindade F., Vitorino R., Leite-Moreira A., Falcão-Pires I. Pericardial fluid: an underrated molecular library of heart conditions and a potential vehicle for cardiac therapy. Basic Res. Cardiol. 2019;114:10. doi: 10.1007/s00395-019-0716-3. [DOI] [PubMed] [Google Scholar]
- 6.Corda S., Mebazaa A., Gandolfini M.P., Fitting C., Marotte F., Peynet J., Charlemagne D., Cavaillon J.M., Payen D., Rappaport L., Samuel J.L. Trophic effect of human pericardial fluid on adult cardiac myocytes. Differential role of fibroblast growth factor-2 and factors related to ventricular hypertrophy. Circ. Res. 1997;81:679–687. doi: 10.1161/01.res.81.5.679. [DOI] [PubMed] [Google Scholar]
- 7.Allen D.J., DiDio L.J., Zacharias A., Fentie I., McGrath A.J., Puig L.B., Pomerantzeff P.N., Zerbini E.J. Microscopic study of normal parietal pericardium and unimplanted Puig-Zerbini pericardial valvular heterografts. J. Thorac. Cardiovasc. Surg. 1984;87:845–855. [PubMed] [Google Scholar]
- 8.Talreja D.R., Edwards W.D., Danielson G.K., Schaff H.V., Tajik A.J., Tazelaar H.D., Breen J.F., Oh J.K. Constrictive pericarditis in 26 patients with histologically normal pericardial thickness. Circulation. 2003;108:1852–1857. doi: 10.1161/01.CIR.0000087606.18453.FD. [DOI] [PubMed] [Google Scholar]
- 9.Ishihara T., Ferrans V.J., Jones M., Boyce S.W., Roberts W.C. Structure of bovine parietal pericardium and of unimplanted Ionescu-Shiley pericardial valvular bioprostheses. J. Thorac. Cardiovasc. Surg. 1981;81:747–757. [PubMed] [Google Scholar]
- 10.Mebazaa A., Wetzel R.C., Dodd-o J.M., Redmond E.M., Shah A.M., Maeda K., Maistre G., Lakatta E.G., Robotham J.L. Potential paracrine role of the pericardium in the regulation of cardiac function. Cardiovasc. Res. 1998;40:332–342. doi: 10.1016/s0008-6363(98)00134-5. [DOI] [PubMed] [Google Scholar]
- 11.DeCoux A., Lindsey M.L., Villarreal F., Garcia R.A., Schulz R. Myocardial matrix metalloproteinase-2: inside out and upside down. J. Mol. Cell. Cardiol. 2014;77:64–72. doi: 10.1016/j.yjmcc.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ren J., Samson W.K., Sowers J.R. Insulin-like growth factor I as a cardiac hormone: physiological and pathophysiological implications in heart disease. J. Mol. Cell. Cardiol. 1999;31:2049–2061. doi: 10.1006/jmcc.1999.1036. [DOI] [PubMed] [Google Scholar]
- 13.Sibal L., Agarwal S.C., Home P.D., Boger R.H. The Role of Asymmetric Dimethylarginine (ADMA) in Endothelial Dysfunction and Cardiovascular Disease. Curr. Cardiol. Rev. 2010;6:82–90. doi: 10.2174/157340310791162659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang Z.S., Srisakuldee W., Soulet F., Bouche G., Kardami E. Non-angiogenic FGF-2 protects the ischemic heart from injury, in the presence or absence of reperfusion. Cardiovasc. Res. 2004;62:154–166. doi: 10.1016/j.cardiores.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 15.Iwakura A., Fujita M., Hasegawa K., Sawamura T., Nohara R., Sasayama S., Komeda M. Pericardial fluid from patients with unstable angina induces vascular endothelial cell apoptosis. J. Am. Coll. Cardiol. 2000;35:1785–1790. doi: 10.1016/s0735-1097(00)00651-3. [DOI] [PubMed] [Google Scholar]
- 16.Kiris I., Kapan S., Narin C., Ozaydın M., Cure M.C., Sutcu R., Okutan H. Relationship between site of myocardial infarction, left ventricular function and cytokine levels in patients undergoing coronary artery surgery. Cardiovasc. J. Afr. 2016;27:299–306. doi: 10.5830/CVJA-2016-027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kameda K., Matsunaga T., Abe N., Fujiwara T., Hanada H., Fukui K., Fukuda I., Osanai T., Okumura K. Increased pericardial fluid level of matrix metalloproteinase-9 activity in patients with acute myocardial infarction: possible role in the development of cardiac rupture. Circ. J. 2006;70:673–678. doi: 10.1253/circj.70.673. [DOI] [PubMed] [Google Scholar]
- 18.Yoneda T., Fujita M., Kihara Y., Hasegawa K., Sawamura T., Tanaka T., Inanami M., Nohara R., Sasayama S. Pericardial fluid from patients with ischemic heart disease accelerates the growth of human vascular smooth muscle cells. Jpn. Circ. J. 2000;64:495–498. doi: 10.1253/jcj.64.495. [DOI] [PubMed] [Google Scholar]
- 19.Fujita M., Komeda M., Hasegawa K., Kihara Y., Nohara R., Sasayama S. Pericardial fluid as a new material for clinical heart research. Int. J. Cardiol. 2001;77:113–118. doi: 10.1016/s0167-5273(00)00462-9. [DOI] [PubMed] [Google Scholar]
- 20.Liu X., Bai C., Gong D., Yuan Y., Han L., Lu F., Han Q., Tang H., Huang S., Xu Z. Pleiotropic effects of transforming growth factor-β1 on pericardial interstitial cells. Implications for fibrosis and calcification in idiopathic constrictive pericarditis. J. Am. Coll. Cardiol. 2011;57:1634–1635. doi: 10.1016/j.jacc.2010.10.054. [DOI] [PubMed] [Google Scholar]
- 21.Simionescu D.T., Kefalides N.A. The biosynthesis of proteoglycans and interstitial collagens by bovine pericardial fibroblasts. Exp. Cell Res. 1991;195:171–176. doi: 10.1016/0014-4827(91)90513-t. [DOI] [PubMed] [Google Scholar]
- 22.Horkay F., Szokodi I., Merkely B., Solti F., Gellér L., Kiss P., Selmeci L., Horváth I., Kékesi V., Juhász-Nagy A., Tóth M. Potential pathophysiologic role of endothelin-1 in canine pericardial fluid. J. Cardiovasc. Pharmacol. 1998;31:S401–S402. doi: 10.1097/00005344-199800001-00115. [DOI] [PubMed] [Google Scholar]
- 23.Szokodi I., Horkay F., Merkely B., Solti F., Gellér L., Kiss P., Selmeci L., Kékesi V., Vuolteenaho O., Ruskoaho H., et al. Intrapericardial infusion of endothelin-1 induces ventricular arrhythmias in dogs. Cardiovasc. Res. 1998;38:356–364. doi: 10.1016/s0008-6363(98)00018-2. [DOI] [PubMed] [Google Scholar]
- 24.Kuwahara M., Kuwahara M. Pericardial mesothelial cells produce endothelin-1 and possess functional endothelin ETB receptors. Eur. J. Pharmacol. 1998;347:329–335. doi: 10.1016/s0014-2999(98)00110-1. [DOI] [PubMed] [Google Scholar]
- 25.Jin H., Liu K., Huang X., Huo H., Mou J., Qiao Z., He B., Zhou B. Genetic Lineage Tracing of Pericardial Cavity Macrophages in the Injured Heart. Circ. Res. 2022;130:1682–1697. doi: 10.1161/CIRCRESAHA.122.320567. [DOI] [PubMed] [Google Scholar]
- 26.Cailhier J.F., Sawatzky D.A., Kipari T., Houlberg K., Walbaum D., Watson S., Lang R.A., Clay S., Kluth D., Savill J., Hughes J. Resident pleural macrophages are key orchestrators of neutrophil recruitment in pleural inflammation. Am. J. Respir. Crit. Care Med. 2006;173:540–547. doi: 10.1164/rccm.200504-538OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghosn E.E.B., Cassado A.A., Govoni G.R., Fukuhara T., Yang Y., Monack D.M., Bortoluci K.R., Almeida S.R., Herzenberg L.A., Herzenberg L.A. Two physically, functionally, and developmentally distinct peritoneal macrophage subsets. Proc. Natl. Acad. Sci. USA. 2010;107:2568–2573. doi: 10.1073/pnas.0915000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gautier E.L., Ivanov S., Williams J.W., Huang S.C.C., Marcelin G., Fairfax K., Wang P.L., Francis J.S., Leone P., Wilson D.B., et al. Gata6 regulates aspartoacylase expression in resident peritoneal macrophages and controls their survival. J. Exp. Med. 2014;211:1525–1531. doi: 10.1084/jem.20140570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosas M., Davies L.C., Giles P.J., Liao C.T., Kharfan B., Stone T.C., O'Donnell V.B., Fraser D.J., Jones S.A., Taylor P.R. The transcription factor Gata6 links tissue macrophage phenotype and proliferative renewal. Science. 2014;344:645–648. doi: 10.1126/science.1251414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Epelman S., Lavine K.J., Beaudin A.E., Sojka D.K., Carrero J.A., Calderon B., Brija T., Gautier E.L., Ivanov S., Satpathy A.T., et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity. 2014;40:91–104. doi: 10.1016/j.immuni.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peet C., Ivetic A., Bromage D.I., Shah A.M. Cardiac monocytes and macrophages after myocardial infarction. Cardiovasc. Res. 2020;116:1101–1112. doi: 10.1093/cvr/cvz336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hou X., Chen G., Bracamonte-Baran W., Choi H.S., Diny N.L., Sung J., Hughes D., Won T., Wood M.K., Talor M.V., et al. The Cardiac Microenvironment Instructs Divergent Monocyte Fates and Functions in Myocarditis. Cell Rep. 2019;28:172–189.e7. doi: 10.1016/j.celrep.2019.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bajpai G., Bredemeyer A., Li W., Zaitsev K., Koenig A.L., Lokshina I., Mohan J., Ivey B., Hsiao H.M., Weinheimer C., et al. Tissue Resident CCR2- and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circ. Res. 2019;124:263–278. doi: 10.1161/CIRCRESAHA.118.314028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buechler M.B., Kim K.W., Onufer E.J., Williams J.W., Little C.C., Dominguez C.X., Li Q., Sandoval W., Cooper J.E., Harris C.A., et al. A Stromal Niche Defined by Expression of the Transcription Factor WT1 Mediates Programming and Homeostasis of Cavity-Resident Macrophages. Immunity. 2019;51:119–130.e5. doi: 10.1016/j.immuni.2019.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi H.S., Won T., Hou X., Chen G., Bracamonte-Baran W., Talor M.V., Jurčová I., Szárszoi O., Čurnova L., Stříž I., et al. Innate Lymphoid Cells Play a Pathogenic Role in Pericarditis. Cell Rep. 2020;30:2989–3003.e6. doi: 10.1016/j.celrep.2020.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abston E.D., Barin J.G., Cihakova D., Bucek A., Coronado M.J., Brandt J.E., Bedja D., Kim J.B., Georgakopoulos D., Gabrielson K.L., et al. IL-33 independently induces eosinophilic pericarditis and cardiac dilation: ST2 improves cardiac function. Circ. Heart Fail. 2012;5:366–375. doi: 10.1161/CIRCHEARTFAILURE.111.963769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sutton M.G., Sharpe N. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation. 2000;101:2981–2988. doi: 10.1161/01.cir.101.25.2981. [DOI] [PubMed] [Google Scholar]
- 38.Gautier E.L., Ivanov S., Lesnik P., Randolph G.J. Local apoptosis mediates clearance of macrophages from resolving inflammation in mice. Blood. 2013;122:2714–2722. doi: 10.1182/blood-2013-01-478206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hynes R.O. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 40.Kubota A., Frangogiannis N.G. Macrophages in myocardial infarction. Am. J. Physiol. Cell Physiol. 2022;323:C1304–C1324. doi: 10.1152/ajpcell.00230.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bracamonte-Baran W., Čiháková D. Cardiac Autoimmunity: Myocarditis. Adv. Exp. Med. Biol. 2017;1003:187–221. doi: 10.1007/978-3-319-57613-8_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu B., Harb S.C., Cremer P.C. New Insights into Pericarditis: Mechanisms of Injury and Therapeutic Targets. Curr. Cardiol. Rep. 2017;19:60. doi: 10.1007/s11886-017-0866-6. [DOI] [PubMed] [Google Scholar]
- 43.Ooi A., Douds A.C., Kumar E.B., Nashef S.A.M. Boxer's pericardium. Eur. J. Cardio. Thorac. Surg. 2003;24:1043–1045. doi: 10.1016/s1010-7940(03)00579-7. [DOI] [PubMed] [Google Scholar]
- 44.Takahashi M., Kondo T., Yamasaki G., Sugimoto M., Asano M., Ueno Y., Nagasaki Y. An autopsy case report of aortic dissection complicated with histiolymphocytic pericarditis and aortic inflammation after mRNA COVID-19 vaccination. Leg. Med. 2022;59 doi: 10.1016/j.legalmed.2022.102154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luo P., Qiu B. The role of immune cells in pulmonary hypertension: Focusing on macrophages. Hum. Immunol. 2022;83:153–163. doi: 10.1016/j.humimm.2021.11.006. [DOI] [PubMed] [Google Scholar]
- 46.Jimenez J., Lavine K.J. The Dynamic Role of Cardiac Macrophages in Aging and Disease. Curr. Cardiol. Rep. 2022;24:925–933. doi: 10.1007/s11886-022-01714-4. [DOI] [PubMed] [Google Scholar]
- 47.Zhu Y., Knolhoff B.L., Meyer M.A., Nywening T.M., West B.L., Luo J., Wang-Gillam A., Goedegebuure S.P., Linehan D.C., DeNardo D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74:5057–5069. doi: 10.1158/0008-5472.CAN-13-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peranzoni E., Lemoine J., Vimeux L., Feuillet V., Barrin S., Kantari-Mimoun C., Bercovici N., Guérin M., Biton J., Ouakrim H., et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc. Natl. Acad. Sci. USA. 2018;115:E4041–E4050. doi: 10.1073/pnas.1720948115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu L., Yang Y., Wen Y., Jeong J.M., Emontzpohl C., Atkins C.L., Sun Z., Poulsen K.L., Hall D.R., Steve Bynon J., et al. Hepatic recruitment of eosinophils and their protective function during acute liver injury. J. Hepatol. 2022;77:344–352. doi: 10.1016/j.jhep.2022.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.da Silva Marques P., da Fonseca-Martins A.M., Carneiro M.P.D., Amorim N.R.T., de Pão C.R.R., Canetti C., Diaz B.L., de Matos Guedes H.L., Bandeira-Melo C. Eosinophils increase macrophage ability to control intracellular Leishmania amazonensis infection via PGD(2) paracrine activity in vitro. Cell. Immunol. 2021;363 doi: 10.1016/j.cellimm.2021.104316. [DOI] [PubMed] [Google Scholar]
- 51.Kim J., Bae J.S. Tumor-Associated Macrophages and Neutrophils in Tumor Microenvironment. Mediat. Inflamm. 2016;2016 doi: 10.1155/2016/6058147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frangogiannis N.G., Dewald O., Xia Y., Ren G., Haudek S., Leucker T., Kraemer D., Taffet G., Rollins B.J., Entman M.L. Critical role of monocyte chemoattractant protein-1/CC chemokine ligand 2 in the pathogenesis of ischemic cardiomyopathy. Circulation. 2007;115:584–592. doi: 10.1161/CIRCULATIONAHA.106.646091. [DOI] [PubMed] [Google Scholar]
- 53.Wong N.R., Mohan J., Kopecky B.J., Guo S., Du L., Leid J., Feng G., Lokshina I., Dmytrenko O., Luehmann H., et al. Resident cardiac macrophages mediate adaptive myocardial remodeling. Immunity. 2021;54:2072–2088.e7. doi: 10.1016/j.immuni.2021.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jin H., Liu K., Tang J., Huang X., Wang H., Zhang Q., Zhu H., Li Y., Pu W., Zhao H., et al. Genetic fate-mapping reveals surface accumulation but not deep organ invasion of pleural and peritoneal cavity macrophages following injury. Nat. Commun. 2021;12:2863. doi: 10.1038/s41467-021-23197-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Benard A., Podolska M.J., Czubayko F., Kutschick I., Klosch B., Jacobsen A., Naschberger E., Brunner M., Krautz C., Trufa D.I., et al. Pleural Resident Macrophages and Pleural IRA B Cells Promote Efficient Immunity Against Pneumonia by Inducing Early Pleural Space Inflammation. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.821480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leendertse M., Willems R.J.L., Giebelen I.A.J., Roelofs J.J.T.H., van Rooijen N., Bonten M.J.M., van der Poll T. Peritoneal macrophages are important for the early containment of Enterococcus faecium peritonitis in mice. Innate Immun. 2009;15:3–12. doi: 10.1177/1753425908100238. [DOI] [PubMed] [Google Scholar]
- 57.Zindel J., Peiseler M., Hossain M., Deppermann C., Lee W.Y., Haenni B., Zuber B., Deniset J.F., Surewaard B.G.J., Candinas D., Kubes P. Primordial GATA6 macrophages function as extravascular platelets in sterile injury. Science. 2021;371 doi: 10.1126/science.abe0595. [DOI] [PubMed] [Google Scholar]
- 58.Capobianco A., Cottone L., Monno A., Manfredi A.A., Rovere-Querini P. The peritoneum: healing, immunity, and diseases. J. Pathol. 2017;243:137–147. doi: 10.1002/path.4942. [DOI] [PubMed] [Google Scholar]
- 59.Fairweather D., Frisancho-Kiss S., Njoku D.B., Nyland J.F., Kaya Z., Yusung S.A., Davis S.E., Frisancho J.A., Barrett M.A., Rose N.R. Complement receptor 1 and 2 deficiency increases coxsackievirus B3-induced myocarditis, dilated cardiomyopathy, and heart failure by increasing macrophages, IL-1beta, and immune complex deposition in the heart. J. Immunol. 2006;176:3516–3524. doi: 10.4049/jimmunol.176.6.3516. [DOI] [PubMed] [Google Scholar]
- 60.Bao J., Sun T., Yue Y., Xiong S. Macrophage NLRP3 inflammasome activated by CVB3 capsid proteins contributes to the development of viral myocarditis. Mol. Immunol. 2019;114:41–48. doi: 10.1016/j.molimm.2019.07.012. [DOI] [PubMed] [Google Scholar]
- 61.Miteva K., Pappritz K., El-Shafeey M., Dong F., Ringe J., Tschöpe C., Van Linthout S. Mesenchymal Stromal Cells Modulate Monocytes Trafficking in Coxsackievirus B3-Induced Myocarditis. Stem Cells Transl. Med. 2017;6:1249–1261. doi: 10.1002/sctm.16-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Agrawal A., Zabad M.N., Dayanand S., Lygouris G., Witzke C. Pericardium: The Forgotten Space During Acute Myocardial Infarction. J. Emerg. Med. 2018;55:e85–e91. doi: 10.1016/j.jemermed.2018.07.013. [DOI] [PubMed] [Google Scholar]
- 63.Limkar A.R., Mai E., Sek A.C., Percopo C.M., Rosenberg H.F. Frontline Science: Cytokine-mediated developmental phenotype of mouse eosinophils: IL-5-associated expression of the Ly6G/Gr1 surface Ag. J. Leukoc. Biol. 2020;107:367–377. doi: 10.1002/JLB.1HI1019-116RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Percopo C.M., Brenner T.A., Ma M., Kraemer L.S., Hakeem R.M.A., Lee J.J., Rosenberg H.F. SiglecF+Gr1hi eosinophils are a distinct subpopulation within the lungs of allergen-challenged mice. J. Leukoc. Biol. 2017;101:321–328. doi: 10.1189/jlb.3A0416-166R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ryu S., Shin J.W., Kwon S., Lee J., Kim Y.C., Bae Y.S., Bae Y.S., Kim D.K., Kim Y.S., Yang S.H., Kim H.Y. Siglec-F-expressing neutrophils are essential for creating a profibrotic microenvironment in renal fibrosis. J. Clin. Invest. 2022;132 doi: 10.1172/JCI156876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vafadarnejad E., Rizzo G., Krampert L., Arampatzi P., Arias-Loza A.P., Nazzal Y., Rizakou A., Knochenhauer T., Bandi S.R., Nugroho V.A., et al. Dynamics of Cardiac Neutrophil Diversity in Murine Myocardial Infarction. Circ. Res. 2020;127:e232–e249. doi: 10.1161/CIRCRESAHA.120.317200. [DOI] [PubMed] [Google Scholar]
- 67.Horckmans M., Ring L., Duchene J., Santovito D., Schloss M.J., Drechsler M., Weber C., Soehnlein O., Steffens S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 2017;38:187–197. doi: 10.1093/eurheartj/ehw002. [DOI] [PubMed] [Google Scholar]
- 68.Liu J., Yang C., Liu T., Deng Z., Fang W., Zhang X., Li J., Huang Q., Liu C., Wang Y., et al. Eosinophils improve cardiac function after myocardial infarction. Nat. Commun. 2020;11:6396. doi: 10.1038/s41467-020-19297-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Calcagno D.M., Zhang C., Toomu A., Huang K., Ninh V.K., Miyamoto S., Aguirre A.D., Fu Z., Heller Brown J., King K.R. SiglecF(HI) Marks Late-Stage Neutrophils of the Infarcted Heart: A Single-Cell Transcriptomic Analysis of Neutrophil Diversification. J. Am. Heart Assoc. 2021;10 doi: 10.1161/JAHA.120.019019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Horckmans M., Bianchini M., Santovito D., Megens R.T.A., Springael J.Y., Negri I., Vacca M., Di Eusanio M., Moschetta A., Weber C., et al. Pericardial Adipose Tissue Regulates Granulopoiesis, Fibrosis, and Cardiac Function After Myocardial Infarction. Circulation. 2018;137:948–960. doi: 10.1161/CIRCULATIONAHA.117.028833. [DOI] [PubMed] [Google Scholar]
- 71.Nahrendorf M., Swirski F.K., Aikawa E., Stangenberg L., Wurdinger T., Figueiredo J.L., Libby P., Weissleder R., Pittet M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007;204:3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rizzo G., Gropper J., Piollet M., Vafadarnejad E., Rizakou A., Bandi S.R., Arampatzi P., Krammer T., DiFabion N., Dietrich O., et al. Dynamics of monocyte-derived macrophage diversity in experimental myocardial infarction. Cardiovasc. Res. 2023;119:772–785. doi: 10.1093/cvr/cvac113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Heidt T., Courties G., Dutta P., Sager H.B., Sebas M., Iwamoto Y., Sun Y., Da Silva N., Panizzi P., van der Laan A.M., et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 2014;115:284–295. doi: 10.1161/CIRCRESAHA.115.303567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dao L., Blaue C., Franz C.M. Integrin α2β1 as a negative regulator of the laminin receptors α6β1 and α6β4. Micron. 2021;148 doi: 10.1016/j.micron.2021.103106. [DOI] [PubMed] [Google Scholar]
- 75.Wang J., Xie L., Chen X., Lyu P., Zhang Q. Changes in Laminin in Acute Heart Failure. Int. Heart J. 2022;63:454–458. doi: 10.1536/ihj.21-769. [DOI] [PubMed] [Google Scholar]
- 76.Horton M.A. The alpha v beta 3 integrin "vitronectin receptor". Int. J. Biochem. Cell Biol. 1997;29:721–725. doi: 10.1016/s1357-2725(96)00155-0. [DOI] [PubMed] [Google Scholar]
- 77.Bellis S.L. Advantages of RGD peptides for directing cell association with biomaterials. Biomaterials. 2011;32:4205–4210. doi: 10.1016/j.biomaterials.2011.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Siebert H., Sachse A., Kuziel W.A., Maeda N., Brück W. The chemokine receptor CCR2 is involved in macrophage recruitment to the injured peripheral nervous system. J. Neuroimmunol. 2000;110:177–185. doi: 10.1016/s0165-5728(00)00343-x. [DOI] [PubMed] [Google Scholar]
- 79.Dewald O., Zymek P., Winkelmann K., Koerting A., Ren G., Abou-Khamis T., Michael L.H., Rollins B.J., Entman M.L., Frangogiannis N.G. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ. Res. 2005;96:881–889. doi: 10.1161/01.RES.0000163017.13772.3a. [DOI] [PubMed] [Google Scholar]
- 80.Kalayci M., Gul E. Eotaxin-1 Levels in Patients with Myocardial Infarction. Clin. Lab. 2022;68 doi: 10.7754/Clin.Lab.2021.210806. [DOI] [PubMed] [Google Scholar]
- 81.Coffelt S.B., Kersten K., Doornebal C.W., Weiden J., Vrijland K., Hau C.S., Verstegen N.J.M., Ciampricotti M., Hawinkels L.J.A.C., Jonkers J., de Visser K.E. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522:345–348. doi: 10.1038/nature14282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Weber A., Wasiliew P., Kracht M. Interleukin-1beta (IL-1beta) processing pathway. Sci. Signal. 2010;3 doi: 10.1126/scisignal.3105cm2. [DOI] [PubMed] [Google Scholar]
- 83.Gyori D., Lim E.L., Grant F.M., Spensberger D., Roychoudhuri R., Shuttleworth S.J., Okkenhaug K., Stephens L.R., Hawkins P.T. Compensation between CSF1R+ macrophages and Foxp3+ Treg cells drives resistance to tumor immunotherapy. JCI Insight. 2018;3 doi: 10.1172/jci.insight.120631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shinde A.V., Humeres C., Frangogiannis N.G. The role of alpha-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. Biochim. Biophys. Acta, Mol. Basis Dis. 2017;1863:298–309. doi: 10.1016/j.bbadis.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hinz B. Myofibroblasts. Exp. Eye Res. 2016;142:56–70. doi: 10.1016/j.exer.2015.07.009. [DOI] [PubMed] [Google Scholar]
- 86.Gibb A.A., Lazaropoulos M.P., Elrod J.W. Myofibroblasts and Fibrosis: Mitochondrial and Metabolic Control of Cellular Differentiation. Circ. Res. 2020;127:427–447. doi: 10.1161/CIRCRESAHA.120.316958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Buechler M.B., Fu W., Turley S.J. Fibroblast-macrophage reciprocal interactions in health, fibrosis, and cancer. Immunity. 2021;54:903–915. doi: 10.1016/j.immuni.2021.04.021. [DOI] [PubMed] [Google Scholar]
- 88.Clausen B.E., Burkhardt C., Reith W., Renkawitz R., Förster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 89.Sodhi C.P., Li J., Duncan S.A. Generation of mice harbouring a conditional loss-of-function allele of Gata6. BMC Dev. Biol. 2006;6:19. doi: 10.1186/1471-213X-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
For inquiries on unprocessed data or code please e-mail the lead contact, Dr. Daniela Cihakova (cihakova@jhmi.edu)