

Abstract
G protein-coupled receptors (GPCRs) are responsible for most cytoplasmic signaling in response to extracellular ligands with different efficacy profiles. Various spectroscopic techniques have identified that agonists exhibiting varying efficacies can selectively stabilize a specific conformation of the receptor. However, the structural basis for activation of the GPCR-G protein complex by ligands with different efficacies is incompletely understood. To better understand the structural basis underlying the mechanisms by which ligands with varying efficacies differentially regulate the conformations of receptors and G proteins, we determined the structures of β2AR-Gαs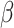 γ bound with partial agonist salbutamol or bound with full agonist isoprenaline using single-particle cryo-electron microscopy at resolutions of 3.26 Å and 3.80 Å, respectively. Structural comparisons between the β2AR-Gs-salbutamol and β2AR-Gs-isoprenaline complexes demonstrated that the decreased binding affinity and efficacy of salbutamol compared with those of isoprenaline might be attributed to weakened hydrogen bonding interactions, attenuated hydrophobic interactions in the orthosteric binding pocket and different conformational changes in the rotamer toggle switch in TM6. Moreover, the observed stronger interactions between the intracellular loop 2 or 3 (ICL2 or ICL3) of β2AR and Gαs with binding of salbutamol versus isoprenaline might decrease phosphorylation in the salbutamol-activated β2AR-Gs complex. From the observed structural differences between these complexes of β2AR, a mechanism of β2AR activation by partial and full agonists is proposed to provide structural insights into β2AR desensitization.
γ bound with partial agonist salbutamol or bound with full agonist isoprenaline using single-particle cryo-electron microscopy at resolutions of 3.26 Å and 3.80 Å, respectively. Structural comparisons between the β2AR-Gs-salbutamol and β2AR-Gs-isoprenaline complexes demonstrated that the decreased binding affinity and efficacy of salbutamol compared with those of isoprenaline might be attributed to weakened hydrogen bonding interactions, attenuated hydrophobic interactions in the orthosteric binding pocket and different conformational changes in the rotamer toggle switch in TM6. Moreover, the observed stronger interactions between the intracellular loop 2 or 3 (ICL2 or ICL3) of β2AR and Gαs with binding of salbutamol versus isoprenaline might decrease phosphorylation in the salbutamol-activated β2AR-Gs complex. From the observed structural differences between these complexes of β2AR, a mechanism of β2AR activation by partial and full agonists is proposed to provide structural insights into β2AR desensitization.
Keywords: cryo-EM structure, G protein-coupled receptor (GPCR), partial and full agonists, conformational change, desensitization
INTRODUCTION
G protein-coupled receptors (GPCRs) regulate a wide variety of physiological functions in response to extracellular stimuli. The varying efficacies of agonists binding to the receptor mediate distinct interaction networks in the orthosteric site, and preferentially stabilize different active conformational states of GPCRs [1–4]. The different conformations of receptors promote binding and activation of different downstream signaling effectors, such as G proteins and β-arrestins, leading to a wide range of intracellular signaling profiles, referred to as efficacy profiles [5–7]. Biophysical studies have indicated that ligands with different efficacy profiles stabilize distinct receptor conformations [8–10], but these conformations and the mechanism by which ligands induce them have not been fully understood.
Notably, GPCR conformational changes caused by binding of agonists with varying efficacies not only reflect the efficacy of the agonist but also induce GPCR desensitization [11], that is, decreased receptor responses to continuous agonist stimulation [12,13]. Numerous studies have shown that the process of GPCR desensitization involves multiple steps, including protein kinase A (PKA)-mediated receptor phosphorylation of intracellular loop 3 (ICL3), G protein receptor kinase (GRK)-mediated receptor phosphorylation in the intracellular loops and the C-terminal tail (C-tail), β-arrestin binding to the receptor, and receptor endocytosis or recycling [13–16]. Among these mechanisms, phosphorylation of ICL3 was observed to induce uncoupling of the receptor from the Gs complex [12], eventually leading to desensitization. Functional and biophysical studies demonstrated that partial agonist binding caused less GPCR desensitization than full agonist binding [14,17,18]. Notably, GPCR desensitization plays crucial roles in modulating receptor activation, which is also essential for analyzing the pharmacokinetics of drugs targeting GPCR. However, the structural basis of GPCR desensitization induced by partial or full agonists is still elusive and needs to be addressed.
The β2 adrenergic receptor (β2AR) is a prototypical family A GPCR. Salbutamol (albuterol) is a rapid-onset, short-acting, selective partial agonist of β2AR over β1AR, which is located in the heart; thus, its cardiac toxicity is minimized [19,20] (Fig. 1a). More interestingly, salbutamol is a functionally selective β2AR partial agonist that is biased toward Gs over arrestin [21], which may prevent arrestin-dependent proinflammatory effects. These pharmacological properties of salbutamol have contributed to its successful use in treating asthma and chronic obstructive pulmonary disease (COPD). Isoprenaline is a full agonist that has shown higher intrinsic efficacy and a stronger bias toward β-arrestin recruitment than salbutamol (Fig. 1a). Interestingly, continuous agonist stimulation induces GPCR desensitization [13]. A strong correlation was found between the coupling efficiencies of the agonists and their ability to induce desensitization; for example, compared with the full agonist isoprenaline, the partial agonist salbutamol caused greater reductions in the initial rates of phosphorylation and β-arrestin recruitment and significantly reduced desensitization [14,22]. In addition, recent studies have revealed that G protein and β-arrestin compete for overlapping binding sites in the GPCR transmembrane core [11]. To illustrate the structural foundation of β2AR activation and desensitization upon binding of partial or full agonists, we sought to determine the three-dimensional structures of the β2AR-Gαs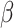 γ complex bound with a partial agonist salbutamol or a full agonist isoprenaline via single-particle cryo-EM.
γ complex bound with a partial agonist salbutamol or a full agonist isoprenaline via single-particle cryo-EM.
Figure 1.

Cryo-EM structure of salbutamol or isoprenaline-bound β2AR-Gs complex. (a) Chemical structures of salbutamol and isoprenaline. (b) Size exclusive chromatography and SDS-PAGE profile of the purified salbutamol-β2AR-Gs complex. (c) Cryo-EM density map and ribbon diagram representation of the cryo-EM structure of salbutamol (yellow), β2AR (blue), Gαs Ras-like (grey), Gβ (gold), Gγ (red), and Nb35 (cyan). (d) Cryo-EM density map and ribbon diagram representation of the cryo-EM structure of isoprenaline (cyan), β2AR (magenta), Gαs Ras-like (grey), Gβ (green), Gγ (orange), and Nb35 (blue).
RESULTS
Structures of the salbutamol- and isoprenaline-bound β2AR-Gs complexes
The cryo-EM structures of the partial agonist salbutamol- and the full agonist isoprenaline-bound β2AR-Gs complexes were determined at 3.26 Å and 3.80 Å resolution, respectively (Fig. 1 and Figs S1–S7). The partial agonist (salbutamol) or full agonist (isoprenaline) was clearly identified in the orthosteric binding site of β2AR. The global folds of salbutamol–bound β2AR-Gs and isoprenaline–bound β2AR-Gs were similar (Fig. 2a and b). Comparison of these structures with that of the inactive-state β2AR, which was bound with the antagonist carazolol, revealed outward movement of TM6, suggesting that both salbutamol- and isoprenaline-bound β2AR are in an active state (Fig. 4a) [23,24].
Figure 2.

Comparison of agonist-binding modes of partial agonists and full agonists. Side views of the orthosteric binding pockets in the salbutamol-bound (yellow) (a), isoprenaline-bound (purple) β2AR-Gs complex (b), salmeterol-bound (magenta) β2AR-Nb71 complex (c) and BI167107-bound (green) β2AR-Gs complex (d). Residues within 4 Å of all ligands are shown as sticks and the hydrogen bond interactions are represented by dotted lines. Red and blue sticks represent oxygen and nitrogen, respectively. The hydrophobic amino acid residues are shown in red.
Figure 4.

The notable conformational changes of the transmembrane helical bundle. (a) Comparison of the cytoplasmic view of transmembrane helical bundle conformation in the carazolol-bound β2AR (green), the salmeterol-bound β2AR-Nb71 (gray), the salbutamol (blue) and the isoprenaline- (magenta) bound β2AR-Gs complex. The red arrow shows the position of TM6 relative to the helical bundle (the Cα of Glu2686.30 as a reference). (b) Comparison of the side view of transmembrane domain conformation in the carazolol-bound β2AR (green), the salmeterol-bound β2AR-Nb71 (gray), the salbutamol (blue) and the isoprenaline- (magenta) bound β2AR-Gs complex. Enlarged view of the conserved core of the receptors (right), the rotamer toggle switch (b-i) and ionic lock (b-ii) are presented, respectively. (c and d) cAMP accumulation assay of F290A in the rotamer toggle switch in the salbutamol-bound and isoprenaline-bound β2AR.
Notable differences in interactions within the orthosteric binding site of partial agonists and full agonists
Different binding interfaces of the partial or full agonists in the orthosteric binding site of β2AR might indicate different activation mechanisms [25]. The β2AR-Gs complex structure (3SN6), activated by the ultrahigh affinity agonist BI167107, represents the fully active state of β2AR [23]. To better understand different agonist-induced conformational changes, we compared the structures of the ligand binding pockets of β2AR bound to the full agonists BI167107 or isoprenaline with those of β2AR bound to the partial agonists salbutamol or salmeterol. All four agonists bound in similar orthosteric sites. Moreover, all the head groups of the agonists can form hydrogen bonds with S2035.42 and S2075.46 [26], and all of the β-hydroxyl groups on the agonists form hydrogen bonds with D1133.32 (Fig. 2). Notably, three major differences were observed in the agonist binding pocket when the full agonists were bound compared to when the partial agonists were bound. First, structural differences in S2045.43 and N2936.55 were observed (Fig. 2). Specifically, N2936.55 forms a hydrogen bond with S2045.43 in isoprenaline-bound β2AR, and this hydrogen bond plays an important role in stabilizing ligand binding (Fig. 2b). However, this hydrogen bond interaction is not observed in the salbutamol-bound β2AR-Gs complex, apparently because of the rotameric conformational change in S2045.43 in salbutamol-bound β2AR (Fig. 2a). In addition, a hydrogen bond is observed between N2936.55 and the meta-hydroxyl of isoprenaline, but is not present between N2936.55 and salbutamol. The cAMP accumulation functional assay combined with alanine mutagenesis revealed that mutation of residues S2045.43 or S2936.55 substantially reduced isoprenaline potency and signaling but had little effect on salbutamol function (Fig. S8, Table S3). The observed result is consistent with previous functional studies of these residues, which verified that mutation of S2045.43 and N2936.55 induced decreases in Gs activation and β-arrestin recruitment [27]. These indicated that the attenuated hydrogen bond interactions in the salbutamol-bound β2AR structure might be responsible for the reduced affinity and desensitizing effect of the partial agonist salbutamol compared with the full agonist isoprenaline (Fig. 2a and b). Secondly, a significant difference between the partial and full agonists is that attenuated hydrophobic interactions are formed only between the aromatic ring of salbutamol/salmeterol and residues of β2AR through V1173.36 and F193ECL2 (Fig. 2a and c), while hydrophobic interactions are formed between isoprenaline/BI167107 and residues of β2AR through V1143.33 and V1173.36 in TM3, F193 in ECL2 and F2906.52 in TM6 (Fig. 2b and d). Therefore, the decreased interaction of salbutamol versus isoprenaline with residues in the binding pocket of β2AR might cause the weakened binding affinity and reduced activation, resulting from salbutamol binding to β2AR compared with isoprenaline binding to β2AR. The structural comparisons of the ligand binding pockets of β2AR for salbutamol and salmeterol demonstrated highly similar interactions, verifying our observed interactions between salbutamol and residues in the orthosteric binding site of β2AR (Fig. 2a and c and Fig. S9). Thirdly, K3057.32 in the salbutamol-bound β2AR forms a hydrogen bond with D192ECL2. However, K3057.32 in the BI167107-bound β2AR-Gs complex trades its salt bridge with D192ECL2 for an interaction with the backbone carbonyl of F193ECL2, stabilizing its movement toward Y3087.35 to form a lid over the orthosteric binding site (Fig. S10) [28]. The lid obstructs ligand association and dissociation (Fig. 2d). The distance between Y3087.35 and F193ECL2 in the salbutamol-bound β2AR-Gs complex is longer than that in the BI167107-bound state, further suggesting the low affinity and partial activation effect of salbutamol (Fig. S10). Although both isoprenaline and BI167107 are full agonists, BI167107 interacts more strongly than the isoprenaline with β2AR (Fig. 2b and d). In isoprenaline-bound β2AR, the side chain of K3057.32 moves but still interacts with F192ECL2, and it did not cause Y3087.35 to move toward the ligand (Fig. S10). The cAMP accumulation assay revealed that alanine substitution of residues K305 and F193 in the isoprenaline binding pocket decreased the potency of isoprenaline (Fig. S10b, Table S3), which confirmed that these residues played important roles in the isoprenaline-mediated cAMP signaling pathway.
Most strikingly, salbutamol exhibits a selectivity of approximately 20-fold for β2AR over β1AR [19]. We compared the structure of the salbutamol-bound β2AR-Gs complex with that of the salbutamol-bound β1AR-Nb80 complex (PDB : 6H7M) [29]. In the salbutamol-bound β1AR-Nb80 complex, W182ECL2 interacts with F201ECL2 and causes F201ECL2 to move away from F3257.35 (Fig. 3). However, in the salbutamol-bound β2AR-Gs complex, F193ECL2 is within the van der Waals distance of Y3087.35 on the opposite side of the entrance to the orthosteric binding pocket, which had a major effect on decreasing the rates of ligand association and dissociation. Moreover, an electrostatic interaction is formed between D192ECL2 and K3057.32 in β2AR, which was not observed in β1AR (Fig. 3a and b). Because of these interactions, dissociation of salbutamol from β2AR is more difficult.
Figure 3.
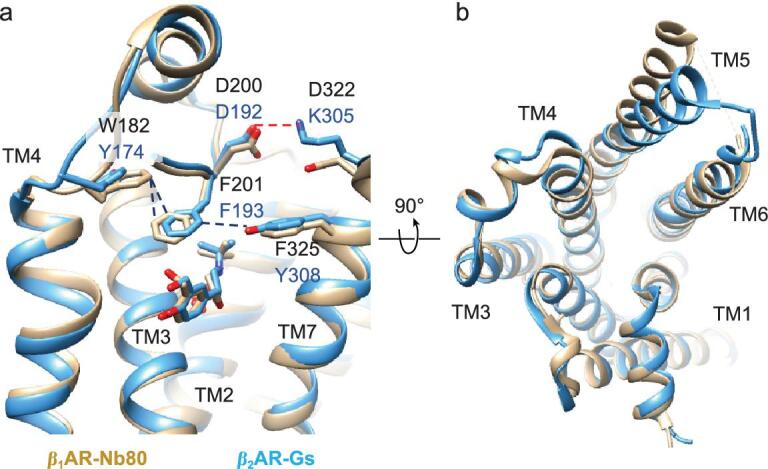
Comparison of the salbutamol-bound β1AR and β2AR. Side views (a) and intracellular views (b) of the salbutamol-bound β1AR-Nb80 (gold) and β2AR-Gs (blue) complex. Interactions between amino acids are indicated by dotted lines.
Conformational variations in the intracellular side of salbutamol- and isoprenaline-bound β2AR
The intracellular side of the receptor in the salbutamol- and isoprenaline-bound β2AR-Gs structures exhibit marked conformational differences relative to the structures of the inverse agonist carazolol-bound β2AR complex and the highly potent partial agonist salmeterol-bound β2AR complex. As shown in Fig. 4, relative to the inactive state carazolol-β2AR structure, the displacement of the cytoplasmic end of TM6 in the salbutamol-bound β2AR-Gs structure (14.1 Å) is larger than in the salmeterol-bound β2AR-Nb71 structure (8 Å), but slightly smaller than that in the isoprenaline-bound β2AR structure (14.6 Å) when measured at the Cα carbon of E2686.30 (Fig. 4a). Two molecular switches have been reported
to be associated with receptor activation and to be responsible for the movement of TM6. The first one is the rotamer toggle switch, referred to as the rotamer configurations of Cys2856.47, Trp2866.48 and Phe2906.52, which are coupled and modulate the bend angle of TM6 around the highly conserved proline kink at Pro2886.50, leading to movement of the cytoplasmic end of TM6 upon activation [30,31]. The other molecular switch that plays a decisive role in TM6 movement is the ionic lock, which is defined on the cytoplasmic end of the receptor [32]. In the inactive state, R1313.50 on TM3 forms a salt bridge with D1303.49 and E2686.30 on TM6, which is disrupted upon agonist binding, causing TM6 to be released and to move away from TM3 (Fig. 4b) [23]. The side chains of Phe2906.52 in the isoprenaline-β2AR structure undergo significant rotation relative to their positions in the carazolol-β2AR and salbutamol-β2AR structure to form the bending angle of Pro2886.50, and the side chains of R1313.50 and E2686.30 in isoprenaline-bound β2AR move up and disrupt the ionic lock (Fig. S11). Thus, both the rotamer toggle switch and the ionic lock switch exist in isoprenaline-β2AR, which is a hallmark of GPCR activation, leading to the largest movement of TM6 (14.6 Å) and contributing to its high efficacy. However, in salbutamol-bound β2AR, salbutamol does not trigger the rotamer toggle switch in TM6 but only disrupts the ionic lock between TM3 and TM6, thus leading to a smaller movement of TM6 (14.1 Å) and contributing to its lower efficacy. The local density maps of the rotamer toggle switch in the salbutamol or isoprenaline-bound β2AR-Gs complex are shown in Fig. S11 a and b. The cAMP accumulation assay revealed that mutation of the residue F290 to Ala reduced isoprenaline activated signaling but had little effect on salbutamol function (Fig. 4c and d, Table S3). In the salmeterol-bound β2AR-Nb71 structure (PDB : 6MXT), the rotamer toggle is not triggered, and the ionic lock still exists, resulting in the smallest movement of TM6 (8 Å), which indicates the importance of Gs protein binding for β2AR activation (Fig. 4a and b).
Intracellular loops mediate different G protein-activated conformations upon partial agonist and full agonist binding
Structural comparisons between the salbutamol-β2AR-Gs and isoprenaline-β2AR-Gs complexes also demonstrated that binding of partial or full agonists to β2AR led to different conformations in the intracellular region and different interaction interfaces with the Gαsβγ complex. Our cryo-EM density map allowed us to define the interaction between ICL3 and Gαs (Fig. 5). As shown in Fig. 5a, F240 in ICL3 in salbutamol-bound β2AR forms a hydrophobic interaction with L346 in the Gα-α4 helix. The side chain of R239 in ICL3 is 3.2 Å away from D343 in the α4 helix of the G protein and can form electrostatic interactions. In the structure of the isoprenaline-β2AR-Gs complex, D343 flips, which keeps D343 away from R239 of ICL3 (Fig. 5c). When the partial agonist salbutamol binds to β2AR, β2AR-ICL3 interacts more strongly with the G protein than when the full agonist isoprenaline binds. Moreover, β2AR-ICL2 becomes more tightly bound to the Gs protein. In addition to the hydrophobic pockets formed by F139ICL2 and F376 in α5 helix that have been observed in the isoprenaline-β2AR-Gs complex, residue Q35 in the αN helix of Gαs forms a polar interaction with S143ICL2 in the receptor in the salbutamol-β2AR-Gs complex. Residue R38 in αN helix also forms a polar interaction with the main chain carbonyl oxygen of Q142 in ICL2, which was not observed in the isoprenaline-β2AR-Gs complex (Fig. 5c and d and Fig. S12). These structural characteristics indicate that the interaction between the β2AR-Gs interface in the partial agonist salbutamol-bound β2AR-Gs complex is enhanced compared with that in the full agonist isoprenaline-bound β2AR-Gs complex. The stronger interaction between the receptor and G protein in the salbutamol-β2AR-Gs complex might make it difficult to expose the phosphorylation site in the loop, which probably contributes to the decreased desensitizing effect of salbutamol compared with isoprenaline.
Figure 5.
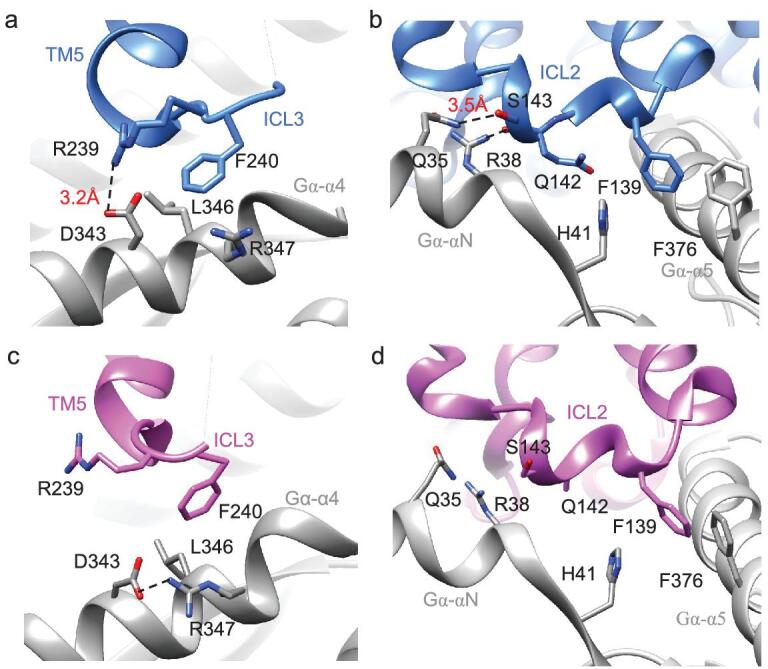
Structural comparison of intracellular loops conformation between salbutamol- and isoprenaline-bound β2AR-Gs. The interaction interface between ICL3 (a), ICL2 (b) of β2AR and Gs protein in the salbutamol-bound β2AR-Gs complex. The interaction interface between ICL3 (c), ICL2 (d) of β2AR and Gs protein in the isoprenaline-bound β2AR-Gs complex.
DISCUSSION
Functional and biophysical approaches, such as nuclear magnetic resonance spectroscopy and single-molecule fluorescence technology, have demonstrated that partial and full agonists induce distinct active conformations of GPCRs [10,33,34]. Our work provides the structural basis for the different conformational changes in the GPCR-G protein complex evoked by partial or full agonists. We propose two significant determinants that affect the difference in the agonist efficacy between the partial agonist salbutamol and the full agonist isoprenaline: the weakening of agonist interactions with the orthosteric binding site for salbutamol, and the less successful induction of conformational changes involving the rotamer toggle switch and the ionic lock switch on the intracellular side of salbutamol-bound β2AR. Although in the absence of G protein, full agonists and partial agonists will cause different degrees of conformational changes at the C-terminus of TM6 of the receptor. However, when the receptor binds to the G protein, it can make the TM6 of the receptor reach a fully activated state, no matter whether combined with full agonists or partial agonists.
GPCR desensitization was previously proposed through phosphorylation of ICL3 and the C-tail on the GPCR, uncoupling of G proteins, binding of β-arrestin to the receptor, and GPCR internalization or endocytosis [13–16] (Fig. 6). Accumulated previous studies have illustrated that ICL3 could play an important role in G protein coupling and receptor phosphorylation [13,35,36]. Therefore, we hypothesized a model for the mechanism by which partial agonists induced less desensitization based on the structures of the salbutamol- and isoprenaline-bound β2AR-Gs complexes. Specifically, notable differences, including attenuated hydrogen bonds and hydrophobic interactions, were observed in the ligand binding pockets following treatment with the partial agonist salbutamol compared to the full agonist isoprenaline. A recent study reported that residues in the allosteric ligand binding pocket regulate GPCR interactions with β-arrestin [37]. Herein, it is observed that β2AR-ICL3 could interact more tightly with the G protein during binding of the partial agonist salbutamol, than binding of the full agonist isoprenaline. Therefore, phosphorylation of ICL3 could be more difficult in the salbutamol-β2AR-Gs complex, which might contribute to the decreased desensitization, triggered by salbutamol binding versus isoprenaline binding.
Figure 6.
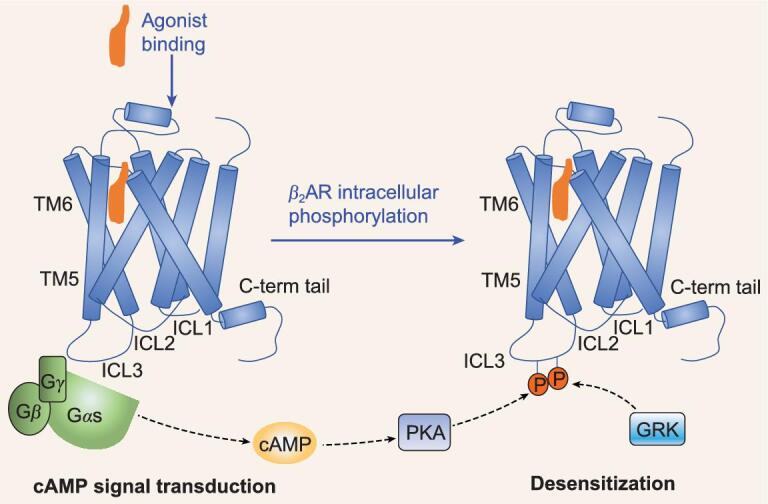
Diagrammatic model representation of receptor desensitization regulated by agonist.
A previous study indicated that agonists promote GPCR phosphorylation not only at ICL3 and the C-tail but also at the first and second intracellular loops [38]. In addition, the formoterol-bound β1AR-arrestin complex demonstrates that both the ICL1 and ICL2 loops in β1AR engage in the interaction with arrestin [39]. Relative to its position in the β1AR-arrestin/formoterol complex, the amino acid in ICL2 that is involved in the binding of both the G protein and arrestin is in a different conformational state. This observation further illustrates the role of ICL2 in receptor activation and desensitization (Fig. S13). However, as the C-tail of β2AR was truncated at residue K348 in this expression construct, we could not observe the configuration around consensus substrate sites phosphorylated by GRKs. Herein, combinations of the structural comparisons and function assays of β2AR-Gs complex bound with isoprenaline or salbutamol indicate that the increased interaction interface between the β2AR and Gs protein in the salbutamol-β2AR-Gs structure attenuates agonist-dependent receptor phosphorylation, which could lead to the reduced β2AR desensitizing effect of the partial agonist salbutamol. Therefore, structural and cAMP assays in this work suggest a framework for different extents of β2AR desensitization upon binding of partial or full agonist. Further structural and functional studies are required to elucidate detailed mechanisms of arrestin-mediated desensitization upon partial or full agonist binding.
CONCLUSION
We report the cryo-EM structures of the β2AR-Gs complex bound to the partial agonist salbutamol or the full agonist isoprenaline. Comparison of salbutamol-bound β2AR with isoprenaline-bound β2AR revealed notable differences in the ligand binding pockets. First, the interaction between S2045.43 and N2936.55 is eliminated and a hydrogen bond is formed between salbutamol and N2936.55 in the salbutamol-β2AR structure relative to the isoprenaline-β2AR structure. Second, hydrophobic interactions between the salbutamol aromatic ring and β2AR are attenuated compared with those between isoprenaline and β2AR. We speculate that these collective structural differences in ligand binding pockets might account for the decreased affinity of the partial agonist salbutamol compared with the full agonist isoprenaline. Moreover, unlike isoprenaline, salbutamol does not trigger the rotamer toggle switch in TM6 but only disrupts the ionic lock between TM3 and TM6, contributing to its lower efficacy. In addition, the stronger interactions between the β2AR-Gs protein binding interface in the partial agonist salbutamol-bound β2AR-Gs complex might decrease phosphorylation in the salbutamol-activated β2AR-Gs complex, contributing to weaker β-arrestin binding and lower desensitization. Thus, this work provides structural insights into the differences in GPCR activation between the partial agonist salbutamol and the full agonist isoprenaline and extends knowledge of agonist-induced desensitization, which is important for drug development and disease treatment.
METHODS
Expression and purification of human β2AR
The human β2AR truncated at the C-terminal from residue 348 was optimized as described previously [40,41]. The construct with FLAG tag at N-terminal and 10 × His at C-terminal was synthesized by GenScript and then cloned into pFastBac1 vector and was expressed in Spodoptera frugiperda Sf9 insect cells using the baculovirus method. The mutation E122W was introduced to improve thermostability of the receptor. The receptor was extracted from insect cell membranes with 1% n-dodecyl-β-D-maltopyranoside (DDM) and purified by TALON Metal Affinity Resin (Clontech). The eluted protein was concentrated and further purified by size-exclusion chromatography on a Superdex 200 10/300 GL column (GE Healthcare) (Supplementary data).
Expression, purification of Nb35, Gαs, Gβγ and Gs complex reconstitution
Nanobody35 (Nb35) [32] was cloned into pET22b vector. The human Gαs was cloned into pET28a vector. They were expressed in E. coli (BL21(Gold)). The bovine Gβ1-C68S and N-terminal 6 × His tagged Gγ2 were cloned into pFastBac-Dual vector and expressed in Sf9 insect cells (Supplementary data).
β2AR-Gs complex preparation
The β2AR and Gs complex proteins were mixed at a molar ratio 1 : 1.2. The mixed sample was incubated at room temperature for 1.5 h, then Apyrase was added. The mixture was incubated with 1% L-MNG to exchange the detergent, and Nb35 was added to further maintain the stability of the receptor-G protein complex. The protein complex was concentrated and further purified by size-exclusion chromatography on a Superdex 200 10/300 GL column (GE Healthcare).
Cryo-EM sample preparation and data collection
An aliquot of 2.5 μL of the sample (0.5 mg/mL) was applied to plasma-treated (H2/O2, 10 s) grids (Quantifoil R1.2/1.3300-mesh Au Holey Carbon). The grids were blotted for 6 s at 100% humidity and 4°C.
Cryo-EM images were recorded on a Gatan K2 Summit direct electron detector in an FEI Titan Krios electron microscope at 300 kV. Serial-EM was used for automated data collection [42]. Movies were collected at a nominal magnification of 29 000 × in counting mode, corresponding to a pixel size of 1.014 Å.
Image processing
For salbutamol-bound β2AR-Gs complex, a total of 7026 micrograph stacks were collected and subjected to motion correction using motioncor2 [43]. Contrast transfer function parameters were estimated with Gctf [44]. A 50 Å low-pass filtered 3D initial model de novo from the 2D average particles was generated using the stochastic gradient descent (SGD) algorithm in Relion-3.0 [45]. The 455 803 particles from the best-looking class were selected for 3D auto-refinement. By post-processing and particle polishing, the final resolution was improved to 3.26 Å. Map resolution was estimated with the gold-standard Fourier shell correction 0.143 criterion. Local resolution was estimated using Resmap [46].
For isoprenaline-bound β2AR-Gs complex, 702 049 particles from well-defined 2D averages were selected from 6217 micrographs. A selected subset of 231 827 particles was used to obtain the final map. The global resolution of this map was estimated to be 3.8 Å based on the gold-standard Fourier shell correlation (FSC).
Details on ‘Model building and refinement’ and ‘Functional analysis of cAMP assay’ are available in the Supplementary data.
Density maps and structure coordinates have been deposited in the Electron Microscopy Database and the Protein Data Bank with accession numbers 7DHI and 7DHR.
Supplementary Material
Acknowledgements
We are grateful to the Center of Cryo-Electron Microscopy, University of Science and Technology of China, and Center of Cryo-Electron Microscopy, Zhejiang University for collection of the cryo-EM data.
Contributor Information
Fan Yang, Hefei National Laboratory of Physical Sciences at Microscale and School of Life Sciences, University of Science and Technology of China, Hefei 230026, China.
Shenglong Ling, Hefei National Laboratory of Physical Sciences at Microscale and School of Life Sciences, University of Science and Technology of China, Hefei 230026, China.
Yingxin Zhou, Hefei National Laboratory of Physical Sciences at Microscale and School of Life Sciences, University of Science and Technology of China, Hefei 230026, China.
Yanan Zhang, Hefei National Laboratory of Physical Sciences at Microscale and School of Life Sciences, University of Science and Technology of China, Hefei 230026, China.
Pei Lv, Hefei National Laboratory of Physical Sciences at Microscale and School of Life Sciences, University of Science and Technology of China, Hefei 230026, China.
Sanling Liu, Hefei National Laboratory of Physical Sciences at Microscale and School of Life Sciences, University of Science and Technology of China, Hefei 230026, China.
Wei Fang, Hefei National Laboratory of Physical Sciences at Microscale and School of Life Sciences, University of Science and Technology of China, Hefei 230026, China.
Wenjing Sun, Hefei National Laboratory of Physical Sciences at Microscale and School of Life Sciences, University of Science and Technology of China, Hefei 230026, China.
Liaoyuan A Hu, Amgen Asia R&D Center, Amgen Research, Shanghai 201210, China.
Longhua Zhang, Hefei National Laboratory of Physical Sciences at Microscale and School of Life Sciences, University of Science and Technology of China, Hefei 230026, China.
Pan Shi, Hefei National Laboratory of Physical Sciences at Microscale and School of Life Sciences, University of Science and Technology of China, Hefei 230026, China.
Changlin Tian, Hefei National Laboratory of Physical Sciences at Microscale and School of Life Sciences, University of Science and Technology of China, Hefei 230026, China; High Magnetic Field Laboratory, Chinese Academy of Sciences, Hefei 230030, China.
FUNDING
This work was supported by the National Key Research and Development Project of China (2016YFA0400903 to C.L. and 2017YFA0505400 to P.S.), the National Natural Science Foundation of China (21825703 to C.L. and 31971152 to P.S.), and the Innovative Program of Development Foundation of Hefei Center for Physical Science and Technology (2018CXFX004). Dr Sanling Liu was a recipient of the Amgen Postdoc Fellowship (China) at the time the research was conducted.
AUTHOR CONTRIBUTIONS
S.-L.L. (Shenglong Ling) and Y.-N.Z. developed sample preparation protocols assisted by W.F. and Y.-X.Z.. F.Y. prepared cryo-EM grids, collected and processed cryo-EM data, solved the structures, built and refined models with S.-L.L.(Sanling Liu). F.Y., S.-L.L. (Shenglong Ling), P.S., P.L. and L.-Y.H. analyzed structures. Y.-X.Z. and S.W. performed functional assays. L.-H.Z., S.-L.L. (Shenglong Ling), P.S. and C.-L.T. supervised the project and co-wrote the manuscript.
Conflict of interest statement. None declared.
REFERENCES
- 1.Furness SG, Liang YL, Nowell CJet al. Ligand-dependent modulation of G protein conformation alters drug efficacy. Cell 2016; 167: 739–49. 10.1016/j.cell.2016.09.021 [DOI] [PubMed] [Google Scholar]
- 2.Ghanouni P, Gryczynski Z, Steenhuis JJet al. Functionally different agonists induce distinct conformations in the G protein coupling domain of the beta 2 adrenergic receptor. J Biol Chem 2001; 276: 24433–6. 10.1074/jbc.C100162200 [DOI] [PubMed] [Google Scholar]
- 3.Hilger D, Masureel M, Kobilka BK. Structure and dynamics of GPCR signaling complexes. Nat Struct Mol Biol 2018; 25: 4–12. 10.1038/s41594-017-0011-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thal DM, Glukhova A, Sexton PMet al. Structural insights into G-protein-coupled receptor allostery. Nature 2018; 559: 45–53. 10.1038/s41586-018-0259-z [DOI] [PubMed] [Google Scholar]
- 5.Wingler LM, Skiba MA, McMahon Cet al. Angiotensin and biased analogs induce structurally distinct active conformations within a GPCR. Science 2020; 367: 888–92. 10.1126/science.aay9813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suomivuori CM, Latorraca NR, Wingler LMet al. Molecular mechanism of biased signaling in a prototypical G protein-coupled receptor. Science 2020; 367: 881–7. 10.1126/science.aaz0326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wingler LM, Lefkowitz RJ. Conformational basis of G protein-coupled receptor signaling versatility. Trends Cell Biol 2020; 30: 736–47. 10.1016/j.tcb.2020.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wootten D, Christopoulos A, Marti-Solano Met al. Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat Rev Mol Cell Biol 2018; 19: 638–53. 10.1038/s41580-018-0049-3 [DOI] [PubMed] [Google Scholar]
- 9.Ye L, Van Eps N, Zimmer Met al. Activation of the A2A adenosine G-protein-coupled receptor by conformational selection. Nature 2016; 533: 265–8. 10.1038/nature17668 [DOI] [PubMed] [Google Scholar]
- 10.Gregorio GG, Masureel M, Hilger Det al. Single-molecule analysis of ligand efficacy in beta2AR-G-protein activation. Nature 2017; 547: 68–73. 10.1038/nature22354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Staus DP, Hu H, Robertson MJet al. Structure of the M2 muscarinic receptor-beta-arrestin complex in a lipid nanodisc. Nature 2020; 579: 297–302. 10.1038/s41586-020-1954-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kelly E, Bailey CP, Henderson G. Agonist-selective mechanisms of GPCR desensitization. Br J Pharmacol 2008; 153 Suppl 1: S379–88. 10.1038/sj.bjp.0707604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Komolov KE, Benovic JL. G protein-coupled receptor kinases: Past, present and future. Cell Signal 2018; 41: 17–24. 10.1016/j.cellsig.2017.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clark RB, Knoll BJ, Barber R. Partial agonists and G protein-coupled receptor desensitization. Trends Pharmacol Sci 1999; 20: 279–86. 10.1016/S0165-6147(99)01351-6 [DOI] [PubMed] [Google Scholar]
- 15.Gurevich VV, Gurevich EV. GPCR signaling regulation: the role of GRKs and arrestins. Front Pharmacol 2019; 10: 125. 10.3389/fphar.2019.00125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature 2009; 459: 356–63. 10.1038/nature08144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Warne T, Moukhametzianov R, Baker JGet al. The structural basis for agonist and partial agonist action on a beta(1)-adrenergic receptor. Nature 2011; 469: 241–4. 10.1038/nature09746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.January B, Seibold A, Whaley Bet al. Beta2-adrenergic receptor desensitization, internalization, and phosphorylation in response to full and partial agonists. J Biol Chem 1997; 272: 23871–9. 10.1074/jbc.272.38.23871 [DOI] [PubMed] [Google Scholar]
- 19.Baker JG. The selectivity of beta-adrenoceptor agonists at human beta1-, beta2- and beta3-adrenoceptors. Br J Pharmacol 2010; 160: 1048–61. 10.1111/j.1476-5381.2010.00754.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sears MR, Lotvall J. Past, present and future–beta2-adrenoceptor agonists in asthma management. Respir Med 2005; 99: 152–70. 10.1016/j.rmed.2004.07.003 [DOI] [PubMed] [Google Scholar]
- 21.van der Westhuizen ET, Breton B, Christopoulos Aet al. Quantification of ligand bias for clinically relevant beta2-adrenergic receptor ligands: implications for drug taxonomy. Mol Pharmacol 2014; 85: 492–509. 10.1124/mol.113.088880 [DOI] [PubMed] [Google Scholar]
- 22.Drake MT, Violin JD, Whalen EJet al. Beta-arrestin-biased agonism at the beta2-adrenergic receptor. J Biol Chem 2008; 283: 5669–76. 10.1074/jbc.M708118200 [DOI] [PubMed] [Google Scholar]
- 23.Rasmussen SG, DeVree BT, Zou Yet al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 2011; 477: 549–55. 10.1038/nature10361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cherezov V, Rosenbaum DM, Hanson MAet al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 2007; 318: 1258–65. 10.1126/science.1150577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swaminath G, Xiang Y, Lee TWet al. Sequential binding of agonists to the beta2 adrenoceptor. Kinetic evidence for intermediate conformational states. J Biol Chem 2004; 279: 686–91. 10.1074/jbc.M310888200 [DOI] [PubMed] [Google Scholar]
- 26.Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. J Neurosci Methods 1995; 25: 366–428. 10.1016/S1043-9471(05)80049-7 [DOI] [Google Scholar]
- 27.Masureel M, Zou Y, Picard LPet al. Structural insights into binding specificity, efficacy and bias of a beta2AR partial agonist. Nat Chem Biol 2018; 14: 1059–66. 10.1038/s41589-018-0145-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DeVree BT, Mahoney JP, Velez-Ruiz GAet al. Allosteric coupling from G protein to the agonist-binding pocket in GPCRs. Nature 2016; 535: 182–6. 10.1038/nature18324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Warne T, Edwards PC, Dore ASet al. Molecular basis for high-affinity agonist binding in GPCRs. Science 2019; 364: 775–8. 10.1126/science.aau5595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi L, Liapakis G, Xu Ret al. Beta2 adrenergic receptor activation. Modulation of the proline kink in transmembrane 6 by a rotamer toggle switch. J Biol Chem 2002; 277: 40989–96. 10.1074/jbc.M206801200 [DOI] [PubMed] [Google Scholar]
- 31.Yao X, Parnot C, Deupi Xet al. Coupling ligand structure to specific conformational switches in the beta2-adrenoceptor. Nat Chem Biol 2006; 2: 417–22. 10.1038/nchembio801 [DOI] [PubMed] [Google Scholar]
- 32.Rasmussen SG, Choi HJ, Fung JJet al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 2011; 469: 175–80. 10.1038/nature09648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manglik A, Kim TH, Masureel Met al. Structural insights into the dynamic process of beta2-adrenergic receptor signaling. Cell 2015; 161: 1101–11. 10.1016/j.cell.2015.04.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu J, Hu Y, Kaindl Jet al. Conformational complexity and dynamics in a muscarinic receptor revealed by NMR spectroscopy. Mol Cell 2019; 75: 53–65. 10.1016/j.molcel.2019.04.028 [DOI] [PubMed] [Google Scholar]
- 35.Huang W, Masureel M, Qu Qet al. Structure of the neurotensin receptor 1 in complex with beta-arrestin 1. Nature 2020; 579: 303–8. 10.1038/s41586-020-1953-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yin W, Li Z, Jin Met al. A complex structure of arrestin-2 bound to a G protein-coupled receptor. Cell Res 2019; 29: 971–83. 10.1038/s41422-019-0256-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanchez-Soto M, Verma RK, Willette BKAet al. A structural basis for how ligand binding site changes can allosterically regulate GPCR signaling and engender functional selectivity. Sci Signal 2020; 13: eaaw5885. 10.1126/scisignal.aaw5885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang Z, Yang F, Zhang Det al. Phosphorylation of G protein-coupled receptors: from the barcode hypothesis to the flute model. Mol Pharmacol 2017; 92: 201–10. 10.1124/mol.116.107839 [DOI] [PubMed] [Google Scholar]
- 39.Lee Y, Warne T, Nehmé Ret al. Molecular basis of β-arrestin coupling to formoterol-bound β1-adrenoceptor. Nature 2020; 583: 862–6. 10.1038/s41586-020-2419-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hanson MA, Cherezov V, Griffith MTet al. A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure 2008; 16: 897–905. 10.1016/j.str.2008.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu JJ, Horst R, Katritch Vet al. Biased signaling pathways in beta2-adrenergic receptor characterized by 19F-NMR. Science 2012; 335: 1106–10. 10.1126/science.1215802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mastronarde DN. SerialEM: a program for automated tilt series acquisition on Tecnai microscopes using prediction of specimen position. Microsc Microanal 2003; 9: 1182–3. 10.1017/S1431927603445911 [DOI] [Google Scholar]
- 43.Zheng SQ, Palovcak E, Armache J-Pet al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 2007; 14: 331–2. 10.1038/nmeth.4193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang K. Gctf: real-time CTF determination and correction. J Struct Biol 2016; 193: 1–12. 10.1016/j.jsb.2015.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zivanov J, Nakane T, Forsberg BOet al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 2018; 7: e42166. 10.7554/eLife.42166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kucukelbir A, Sigworth FJ, Tagare HD. Quantifying the local resolution of cryo-EM density maps. Nat Methods 2014; 11: 63–5. 10.1038/nmeth.2727 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.