

Abstract
The rational design of materials with cell-selective membrane activity is an effective strategy for the development of targeted molecular imaging and therapy. Here we report a new class of cationic multidomain peptides (MDPs) that can undergo enzyme-mediated molecular transformation followed by supramolecular assembly to form nanofibers in which cationic clusters are presented on a rigid β-sheet backbone. This structural transformation, which is induced by cells overexpressing the specific enzymes, led to a shift in the membrane perturbation potential of the MDPs, and consequently enhanced cell uptake and drug delivery efficacy. We envision the directed self-assembly based on modularly designed MDPs is a highly promising approach to generate dynamic supramolecular nanomaterials with emerging membrane activity for a range of disease targeted molecular imaging and therapy applications.
Keywords: self-assembly, membrane activity, molecular imaging, cell selectivity, drug delivery
Graphical Abstract
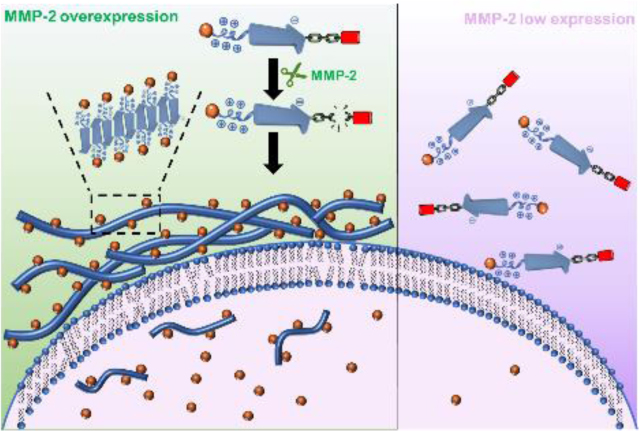
Molecularly designed cationic peptides having low membrane activity undergo cell-specific enzyme triggered chemical transformation and self-assembly to form supramolecular cationic clusters with enhanced membrane activity. The cell-selective mebrane activity by in situ formed peptide assembly is a highly desirable feature for targeted molecular imaging and therapy.
Membrane-active peptides are an important class of materials that function by interacting with the cell membrane to cause membrane disruption or direct translocation for cargo delivery.[1] The majority of these peptides adopt a global amphiphilic conformation upon membrane binding in which cationic and hydrophobic amino acids are segregated into clusters on the opposite faces of a peptide secondary structure, commonly an α-helix or β-sheet. While global amphiphiles are a common motif for the design of membrane-active molecules and polymers, recent efforts suggested preorganization of locally clustered facial or linear amphiphiles on a macromolecular backbone prior to membrane binding can be an effective approach to further increase membrane activity.[2] This is largely due to the reduced entropic loss associated with a membrane-induced conformational change of pre-assembled amphiphilic clusters. It was shown that a rigid backbone, such as a long α-helix forming polypeptide, is more favorable for inducing an ordered global arrangement of the amphiphilic clusters while maintaining sufficient local conformational flexibility needed for effective membrane interactions. Using the approach of supramolecular peptide assembly, our group reported a library of membrane-active nanofibers in which local amphiphilic clusters are presented on a rigid self-assembled β-sheet peptide nanofibers to induce potent membrane activity.[3]
A general strategy for the construction of membrane-active nanofibers involves self-assembly of a de novo designed cationic multidomain peptide (MDP) which has the tendency to form supramolecular β-sheet nanofibers. As demonstrated in our previous studies, the emerging membrane activity is largely attributed to the formation of supramolecular nanofibers as constitutional peptide isomers which have the same secondary structures but exist as monomers or small oligomers had much lower membrane activity.[3a, 3e] Upon self-assembly, supramolecular ionic clusters are displayed on the rigid nanofiber backbone and their conformational flexibility can be further tuned by changing the numbers of cationic and anionic amino acids on MDPs to enhance the membrane activity.
Inspired by the work on proteolytically activated cell penetrating peptides and recent advances in trigger-responsive peptide self-assembly,[4] in this report we aim to explore a trigger-responsive mechanism to fabricate nanofibers with control over membrane activity in a cell-selective manner. The ability of these nanofibers to perturb cell membranes is achieved through enzymatic-mediated molecular transformation and supramolecular assembly of modularly designed MDPs to form membrane-active supramolecular nanofibers. It is worth mentioning that our approach to achieving cell selective membrane activity is different from that of conjugation of a shielded natural cell penetrating peptide on a nanoparticle surface which is re-activated upon reaching the target site.[5] Rather, we used a modular self-assembly approach in which monomeric peptides with low membrane activity undergo enzyme-triggered chemical transformation followed by self-assembly to form supramolecular nanofibers with enhanced membrane activity.
Fig. 1 shows the chemical structure of a modularly designed MDP which can undergo a selective chemical transformation and supramolecular assembly to form membrane-active nanofibers. The MDP is designed to have three modules. A key module is the membrane-active self-assembling (SA) module, the sequence of which was optimized through our previous screening studies.[3e] In this work, we selected K10(QW)6E3 as the membrane-active SA module in which the subscript represents the numbers of repeating units for lysine (K), glutamic acid (E) and the alternating glutamine (Q) and tryptophan (W). A cationic capping (CC) module consisting of oligolysines is attached to the C-terminus of the SA module through a labile linker (LL) module. The hypothesis is in the presence of the cationic CC domain, due to the abundance of the cationic charges and increased electrostatic repulsion, MDPs do not self-assemble and therefore have weak membrane activity. When the external stimulus is applied under specific cellular condition, the LL domain is cleaved to release the CC domain. Consequently, the ability of MDPs to self-assemble is restored to form nanofibers with improved membrane activity. As a proof-of-concept study, we chose matrix metalloproteinase 2, MMP-2 as our initial cellular target due to its overexpression by cancer cells.[6] MMPs have also been used as a model enzyme for the development of various targeted biomaterials.[7] We synthesized an MDP consisting of an MMP-2 responsive substrate (PLGLAG) as the linker, termed as CS-MDP and a control MDP with a scrambled linker sequence of LALGPG but otherwise identical, termed as NS-MDP. Both peptides were investigated for their enzymatic-dependent self-assembly in aqueous buffer solution and membrane activity in cancer cell lines with high and low levels of MMP-2.
Figure 1.

Color-coded chemical structure (A) and self-assembly (B) of a modularly designed CS-MDP to form supramolecular cationic nanofibers. Blue: self-assembling (SA) module, K10(QW)6E3 which has the intrinsic membrane activity upon self-assembly. Green: MMP-2 labile linker, PLGLAG. Red: Cationic capping (CC) module consisting of five lysine residues. Black: Three glycines were included between the SA module and MMP-2 linker. The hypothesis is in the presence of the CC domain, due to the abundance of the cationic charges and electrostatic repulsion, CS-MDPs do not self-assemble and therefore have weak membrane activity. When the external stimulus is applied to remove the CC domain, the self-assembling capability of K10(QW)6E3 is restored to form supramolecular nanofibers with enhanced membrane activity.
As expected, the CS-MDP is susceptible to MMP-2 cleavage and the mass of the peptide fragments confirms the cleavage reaction occurs between glycine and leucine in the LL domain (Fig. S2). Using high performance liquid chromatography, the MMP-2 cleavage efficiency was determined to be 75% after 4 h of enzymatic treatment, which further increased to 91% upon 24 h of incubation (Fig. S3). Circular dichroism (CD) spectroscopy shows a secondary structural transition from a random coil to a predominant β-sheet conformation upon MMP-2 treatment (Fig. 2A). The minimum absorption at 215 nm became more intense and narrower with the incubation time, suggesting a time-dependent structural transition process. In comparison, peptides with a scrambled MMP-2 linker, NS-MDPs adopted a random coil regardless of MMP-2 treatment (Fig. S4). The change of the molecular structure was accompanied by the formation of larger macromolecular species, which were identified through analytical ultracentrifugation (AUC) experiments. Specifically, we performed a sedimentation velocity (SV) experiment to estimate the apparent molecular weight and weight distribution for both CS-MDPs and NS-MDPs in the presence and absence of MMP-2 (Fig. 2B and Fig. S5). Without MMP-2, both peptides exist as monomers with an MW at ~5.1 kDa. This result validates our hypothesis that the electrostatic repulsion among the CC domain is sufficiently strong to prevent peptides from self-assembling. Upon MMP-2 treatment, while NS-MDP still adopted monomers, new species with larger molecular weights were observed for CS-MDPs at ~ 3.4 MDa and ~ 4.9 MDa after 4 h of incubation with MMP-2. The molecular weight further increases upon extended incubation showing the major population at ~ 4.9 MDa and ~ 5.6 MDa after 24 h of enzymatic treatment. Based on our design principle, detaching the CC domain from the SA domain causes a reduction of the repulsive forces and therefore shifts the equilibrium toward self-assembly.
Figure 2.

(A) CD spectra of CS-MDPs with and without MMP-2 showing enzyme-induced structural transition to β-sheets. (B) Continuous sedimentation coefficient distribution, c(s) curve showing the formation of larger species upon MMP-2 treatment. Peptides were prepared in Tris buffer (20 mM, pH=7.4) containing 5 mM CaCl2 and 20 mM ZnCl2 with a final peptide concentration at 20 μM.
Negatively stained transmission electron microscopy (TEM) was further used to examine the self-assembled nanostructures of CS-MDP upon MMP-2 treatment. TEM shows spherical aggregates for both NS-MDP and CS-MDP without MMP-2 (Fig. 3A and Fig. S6), which is likely due to non-specific aggregation of the monomeric peptides (as confirmed by the solution-state AUC analysis) under the drying condition used to prepare negatively stained TEM samples. Upon MMP-2 treatment, while NS-MDPs still adopted spherical aggregates (Fig. S6), nanofibers were observed for CS-MDP (Fig. 3B and 3C). Extended incubation of CS-MDP with the enzyme led to an increase of fiber length from an average of 122 nm at 4 h to 268 nm at 24 h (Fig. 3D), indicating a time-dependent self-assembly process, which is also consistent with the trend of secondary structure change observed by CD spectroscopy. These long nanofibers are composed of cleaved fragments of the cationic SA domain. They are arranged perpendicular to the long fiber axis to form pre-organized supramolecular cationic clusters to interact with the negatively charged cell membrane. Based on the established structural repeats of cross-β sheets at 4.7 Å along the fiber axis,[8] we can then calculate the number of peptide chains in a single nanofiber using the formula of “2 x (average length of a fiber (nm) / 0.47 nm)” where the factor of 2 reflects the bilayer, sandwich-like cross-β sheets packing. Given the average length of the fiber at 268 nm, approximately 1140 peptide chains were estimated in a single nanofiber. Meanwhile, AUC results showed the average MW of peptide nanofiber upon MMP-2 treatment for 24 h is at 5.3 MDa. Considering the MW of a single peptide chain at 4935 Da, the number of peptide chains within a nanofiber would be 5.3 × 106 Da/4935 Da = 1070, which is in good agreement with the TEM analysis.
Figure 3.

TEM images of (A) CS-MDPs in the absence of MMP-2; (B) CS-MDPs treated with MMP-2 for 4 h; (C) CS-MDPs treated with MMP-2 for 24 h showing MMP-2 triggered self-assembly to form nanofibers; (D) Quantification of the average fiber length by Gaussian fitting. The measurements were based on a total number of 100 fibers formed by CS-MDPs upon treatment with MMP-2 for 4 h and 24 h. Peptides were prepared in Tris buffer (20 mM, pH=7.4) containing 5 mM CaCl2 and 20 mM ZnCl2 with a final peptide concentration at 100 μM.
The formation of enzymatic-triggered supramolecular cationic clusters was further harnessed to design nanofibers with cell-selective membrane activity. Cell selectivity is largely controlled by the endogenous enzyme secreted by cells. Three cancer cell lines, i.e. esophageal cancer cells (KYSE-30), lung cancer cells (A549) and cervical cancer cells (HeLa), which are known to have different levels of endogenous MMP-2, were used to investigate cell-selective membrane activity. The MMP-2 enzymatic activity was quantified through the SensoLyte MMP-2 assay (Fig. S7) and the results are consistent with the relative activity trend reported in the literature,[9] namely KYSE-30 and A549 cells have higher levels of MMP-2 than HeLa cells. It should be noted that the MMP-2 concentration determined in the KYSE-30 and A549 cell culture was ~ 10-fold lower than what is reported in vivo at the diseased issues.[10] However, despite the reduced MMP-2 activity in vitro, induced membrane activity of CS-MDPs was observed. We used confocal laser scanning microscopy (CLSM) to monitor cell uptake of fluorescently labelled CS-MDPs and NS-MDPs in which an NBD fluorescence dye was appended at the N-terminus of the SA domain, denoted as NBD-CS-MDP and NBD-NS-MDP (Table S1, Fig. S1(3) and Fig. S1(4)). Peptides were incubated with cells for 24 h followed by thorough washing before CLSM imaging. As shown in Fig. 4, upon incubation with CS-MDPs, MMP-2 overexpressed KYSE-10 and A549 cells show much higher fluorescence intensity than HeLa cells which have a low level of endogenous MMP-2. The addition of exogenous MMP-2 to HeLa cell culture dramatically enhanced the fluorescence intensity, further confirming the important role of MMP-2 in activating the membrane activity (Fig. S8). As shown in Fig. 4A, much stronger fluorescence is observed for KYSE-10 and A549 cells treated with CS-MDP than those treated with NS-MDP. In contrast, fluorescence intensity is comparable between CS-MDP and NS-MDP treated HeLa cells with a low level of endogenous MMP-2. CS-MDP and NS-MDP have the same amino acid composition but differ in their MMP-2 susceptibility. The enhanced membrane activity of CS-MDP toward KYSE-10 and A549 cells is mostly attributed to MMP-2 mediated cleavage and self-assembly to form supramolecular cationic clusters. NS-MDP do not respond to enzymes and therefore remain intact as monomers with low membrane activity. Flow cytometry results show the same trend of fluorescence change, further supporting the role of MMP-2 in the molecular transformation and supramolecular cationic clusters with induced membrane activity (Fig. 4B). It is notable that although CS-MDP seem to be mostly localized in the pericellular region of KYSE-30 or A549 cells, they are effective to perturb cell membranes for enhanced drug delivery. To further correlate the membrane activity with β-sheet nanofiber formation, we performed a Congo Red (CR) staining assay in which CR is used to selectively stain cross β-sheet nanofibers.[11] As shown in Fig. S9, increased CR fluorescence is observed for NBD-CS-MDP treated A549 than NBD-NS-MDP treated cells, suggesting the formation of nanofiber by CS-MDP and the correlation between supramolecular nanofibers and their membrane activity.
Figure 4.

(A) CLSM images of cells upon incubation with NBD labeled CS-MDP and NS-MDP showing induced membrane activity of CS-MDP toward MMP-2 overexpressed KYSE-30 and A549 cells in comparison to HeLa cells with a low endogenous level of MMP-2. (B) Flow cytometry measurement of different cells treated with NBD-labeled CS-MDP and NS-MDP. Incubation time: 24 h. The final peptide concentration in the culture medium is 20 μM. Scale bar: 50 μm. Statistic significant difference is indicated by ***p < 0.001.
A preliminary study of in vitro drug efficacy was performed using doxorubicin (DOX) as a model anticancer drug molecule. A disulfide linker was introduced between DOX and the SA domain in order to release drugs in the reducing cellular microenvironment upon cell uptake.[3d, 11d, 12] DOX conjugated CS-MDP and NS-MDP termed as DOX-CS-MDP and DOX-NS-MDP (Table S1) were synthesized through an established procedure using a thiol-disulfide exchange reaction (Fig. S10).[13] The disulfide DOX derivative compound used for peptide conjugation was confirmed by one- and two-dimensional 1H-NMR and electrospray ionization mass spectrometry (ESI-MS) (Fig. S11). DOX peptide conjugates were confirmed by ESI-MS (Fig. S1(5) and Fig. S1(6)). Intracellular DOX cleavage and release were confirmed by CLSM showing minimum co-localization of green and red fluorescence in KYSE-30 and A549 cells treated with dual NBD and DOX labeled CS-MDPs, named as DOX-(NBD)CS-MDP (Table S1 and Fig. S1(7)) in which NBD was covalently linked at the N-terminus of CS-MDP (Fig. S12). Cytotoxicity of free DOX, DOX-CS-MDP and DOX-NS-MDP were evaluated against all three cell lines. As shown in Fig. S13A, while DOX alone elicits moderate cytotoxicity toward three cell lines, DOX-CS-MDP had much higher toxicity toward MMP-2 overexpressed cell lines than low-expression cells, showing cell viability at 23% toward A549, 20% toward KYSE-30 but 90% toward HeLa cells, respectively. In contrast, no prominent selectivity was achieved for DOX-NS-MDP. Notably, CS-MDP and NS-MDP alone show comparable cell viability against all three cells (Fig. S13B) at ~70%−80%. The cell-selective drug efficacy/toxicity is largely attributed to the enhanced membrane activity of CS-MDPs upon enzyme-mediated supramolecular assembly that can facilitate the transport and release of DOX to the cytoplasm as confirmed by flow cytometry showing much higher intracellular DOX fluorescence for cells treated with DOX-CS-MDP than those treated with DOX-NS-MDP (Fig. S14).
We demonstrated a modular design approach to the fabrication of supramolecular peptide assembly with cell-selective membrane activity. The approach is driven by our recent discovery that supramolecular ionic clusters on a β-sheet nanofiber scaffold can effectively perturb the cell membrane compared to their monomeric counterparts. In this work, cell selectivity is achieved through the rational design of MDPs that can undergo cell-specific enzyme-mediated molecular and supramolecular transformation converting a monomeric peptide with low membrane activity to nanofibers with enhanced membrane activity. Because the design of MDP building blocks is highly modular, we can readily change the labile linker domain in the linear peptide sequence to respond to different disease-specific physiological conditions.
Supplementary Material
Acknowledgements
This study was supported by the National Science Foundation (DMR1824614), start-up fund from the University of Texas at Arlington and U.S. National Institutes of Health (NIH) Grants R01 CA185055 and S10OD025230 (to Z.P.). S.H. was supported by the Welch Foundation (Y-2031). NMR facilities were supported by the National Science Foundation CHE-0840509 (CRIF: MU).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- [1].a) Frankel AD, Pabo CO, Cell 1988, 55, 1189–1193; [DOI] [PubMed] [Google Scholar]; b) Green M, Loewenstein PM, Cell 1988, 55, 1179–1188; [DOI] [PubMed] [Google Scholar]; c) Zorko M, Langel U, Adv. Drug Deliv. Rev. 2005, 57, 529–545; [DOI] [PubMed] [Google Scholar]; d) McErlean EM, Ziminska M, McCrudden CM, McBride JW, Loughran SP, Cole G, Mulholland EJ, Kett V, Buckley NE, Robson T, Dunne NJ, McCarthy HO, Control J. Release 2021, 330, 1288–1299. [DOI] [PubMed] [Google Scholar]
- [2].a) Jiang Y, Han M, Bo Y, Feng Y, Li W, Wu JR, Song Z, Zhao Z, Tan Z, Chen Y, Xue T, Fu Z, Kuo SH, Lau GW, Luijten E, Cheng J, ACS Cent. Sci. 2020, 6, 2267–2276; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lu H, Wang J, Bai Y, Lang JW, Liu S, Lin Y, Cheng J, Nat. Commun. 2011, 2, 206; [DOI] [PubMed] [Google Scholar]; c) Rahman MA, Jui MS, Bam M, Cha Y, Luat E, Alabresm A, Nagarkatti M, Decho AW, Tang C, ACS Appl. Mater. Interfaces 2020, 12, 21221–21230; [DOI] [PubMed] [Google Scholar]; d) Rahman MA, Bam M, Luat E, Jui MS, Ganewatta MS, Shokfai T, Nagarkatti M, Decho AW, Tang C, Nat. Commun. 2018, 9, 5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Xu D, Dustin D, Jiang L, Samways DS, Dong H, Chem. Commun. 2015, 51, 11757–11760; [DOI] [PubMed] [Google Scholar]; b) Xu D, Jiang L, DeRidder L, Elmore B, Bukhari M, Wei Q, Samways DSK, Dong H, Mol. Biosyst. 2016, 12, 2695–2699; [DOI] [PubMed] [Google Scholar]; c) Xu D, Samways DSK, Dong H, Bioact. Mater. 2017, 2, 260–268; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Yang S, Xu D, Dong H, J. Mater. Chem. B 2018, 6, 7179–7184; [DOI] [PubMed] [Google Scholar]; e) Yang S, Dong H, RSC Adv. 2020, 10, 29469–29474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Jiang T, Olson ES, Nguyen QT, Roy M, Jennings PA, Tsien RY, Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 17867–17872; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shi J, Schneider JP, Angew Chem. Int. Ed. Engl. 2019, 58, 13706–13710; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kalafatovic D, Nobis M, Son J, Anderson KI, Ulijn RV, Biomaterials 2016, 98, 192–202; [DOI] [PubMed] [Google Scholar]; d) Wang Y, Zhan J, Chen Y, Ai S, Li L, Wang L, Shi Y, Zheng J, Yang Z, Nanoscale 2019, 11, 13714–13719; [DOI] [PubMed] [Google Scholar]; e) Ji W, Yuan C, Chakraborty P, Makam P, Bera S, Rencus-Lazar S, Li J, Yan X, Gazit E, ACS Nano 2020, 14, 7181–7190; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Lin YA, Ou YC, Cheetham AG, Cui H, Biomacromolecules 2014, 15, 1419–1427; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Webber MJ, Newcomb CJ, Bitton R, Stupp SI, Soft Matter. 2011, 7, 9665–9672; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Lin BF, Megley KA, Viswanathan N, Krogstad DV, Drews LB, Kade MJ, Qian Y, Tirrell MV, J. Mater. Chem. 2012, 22; [Google Scholar]; i) Tanaka A, Fukuoka Y, Morimoto Y, Honjo T, Koda D, Goto M, Maruyama T, J. Am. Chem. Soc. 2015, 137, 770–775; [DOI] [PubMed] [Google Scholar]; j) Wang H, Feng Z, Wang Y, Zhou R, Yang Z, Xu B, J. Am. Chem. Soc. 2016, 138, 16046–16055; [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Bellomo EG, Wyrsta MD, Pakstis L, Pochan DJ, Deming TJ, Nat. Mater. 2004, 3, 244–248; [DOI] [PubMed] [Google Scholar]; l) Collier JH, Hu BH, Ruberti JW, Zhang J, Shum P, Thompson DH, Messersmith PB, J. Am. Chem. Soc. 2001, 123, 9463–9464; [DOI] [PubMed] [Google Scholar]; m) Bowerman CJ, Nilsson BL, J. Am. Chem. Soc. 2010, 132, 9526–9527. [DOI] [PubMed] [Google Scholar]
- [5].a) Jiang T, Zhang Z, Zhang Y, Lv H, Zhou J, Li C, Hou L, Zhang Q, Biomaterials 2012, 33, 9246–9258; [DOI] [PubMed] [Google Scholar]; b) Li J, Liu F, Shao Q, Min Y, Costa M, Yeow EK, Xing B, Adv. Healthc. Mater. 2014, 3, 1230–1239. [DOI] [PubMed] [Google Scholar]
- [6].a) Kessenbrock K, Plaks V, Werb Z, Cell 2010, 141, 52–67; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Noel A, Jost M, Maquoi E, Semin. Cell Dev. Biol. 2008, 19, 52–60. [DOI] [PubMed] [Google Scholar]
- [7].a) Sun Z, Li R, Sun J, Peng Y, Xiao L, Zhang X, Xu Y, Wang M, ACS Appl. Mater. Interfaces 2017, 9, 40614–40627; [DOI] [PubMed] [Google Scholar]; b) Son J, Kalafatovic D, Kumar M, Yoo B, Cornejo MA, Contel M, Ulijn RV, ACS Nano 2019, 13, 1555–1562; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Yang Z, Ma M, Xu B, Soft Matter 2009, 5, 2546–2548. [Google Scholar]
- [8].Kirschner DA, Abraham C, Selkoe DJ, Proc. Natl. Acad. Sci. U.S.A. 1986, 83, 503–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Mohammadi F, Javid H, Afshari AR, Mashkani B, Hashemy SI, Mol. Biol. Rep. 2020, 47, 4263–4272; [DOI] [PubMed] [Google Scholar]; b) Wang Y, Lin T, Zhang W, Jiang Y, Jin H, He H, Yang VC, Chen Y, Huang Y, Theranostics 2015, 5, 787–795; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Han M, Huang-Fu M-Y, Guo W-W, Guo N-N, Chen J, Liu H-N, Xie Z-Q, Lin M-T, Wei Q-C, Gao J-Q, ACS Appl. Mater. Interfaces 2017, 9, 42459–42470. [DOI] [PubMed] [Google Scholar]
- [10].a) Morgia G, Falsaperla M, Malaponte G, Madonia M, Indelicato M, Travali S, Mazzarino MC, Urol. Res. 2005, 33, 44–50; [DOI] [PubMed] [Google Scholar]; b) Groblewska M, Mroczko B, Gryko M, Pryczynicz A, Guzinska-Ustymowicz K, Kedra B, Kemona A, Szmitkowski M, Tumour Biol. 2014, 35, 3793–3802; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Isaacson KJ, Martin Jensen M, Subrahmanyam NB, Ghandehari H, Control J. Release 2017, 259, 62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Williams RJ, Smith AM, Collins R, Hodson N, Das AK, Ulijn RV, Nat. Nanotechnol. 2009, 4, 19–24; [DOI] [PubMed] [Google Scholar]; b) Espargaro A, Llabres S, Saupe SJ, Curutchet C, Luque FJ, Sabate R, Angew Chem. Int. Ed. Engl. 2020, 59, 8104–8107; [DOI] [PubMed] [Google Scholar]; c) Khurana R, Uversky VN, Nielsen L, Fink AL, J. Biol. Chem. 2001, 276, 22715–22721; [DOI] [PubMed] [Google Scholar]; d) Chen W, Li S, Lang JC, Chang Y, Pan Z, Kroll P, Sun X, Tang L, Dong H, Small 2020, 16, e2002780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Guo WW, Zhang ZT, Wei Q, Zhou Y, Lin MT, Chen JJ, Wang TT, Guo NN, Zhong XC, Lu YY, Yang QY, Han M, Gao J, Biomacromolecules 2020, 21, 444–453. [DOI] [PubMed] [Google Scholar]
- [13].Song Q, Chuan X, Chen B, He B, Zhang H, Dai W, Wang X, Zhang Q, Drug Deliv. 2016, 23, 1734–1746. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.