

Abstract
Rebound bone loss following denosumab discontinuation is an important clinical challenge. Current treatment strategies to prevent this fail to suppress the rise and overshoot in osteoclast-mediated bone resorption. In this study, we use a murine model of denosumab treatment and discontinuation to show the temporal changes in osteoclast formation and activity during RANKL inhibition and withdrawal. We show that the cellular processes that drive the formation of osteoclasts and subsequent bone resorption following withdrawal of RANKL inhibition precede the rebound bone loss. Furthermore, a rise in serum TRAP and RANKL levels is detected before markers of bone turnover used in current clinical practice. These mechanistic advances may provide insight into a more defined window of opportunity to intervene with sequential therapy following denosumab discontinuation.
Keywords: osteoclast, RANKL, Denosumab, osteoporosis
Lay Summary
Stopping denosumab, a medication commonly used to improve bone mass by blocking formation of bone resorbing osteoclasts, leads to a rebound loss in the bone which was gained during treatment. Current strategies to prevent this bone loss fail in most cases as they are unable to prevent the rise and overshoot in bone resorption by osteoclasts. Thie stems from an incomplete understanding of how osteoclasts behave during denosumab treatment and after treatment is discontinued. We use a mouse model of this phenomenon to show how osteoclast formation and activity changes throughout this process. We show that increases in the processes that drive the formation of osteoclasts can be detected in the circulation before bone loss occurs. These findings could therefore provide insight into a targeted ‘window of opportunity’ to intervene and prevent the rebound bone loss following stopping denosumab in patients.
Graphical Abstract
Graphical Abstract.
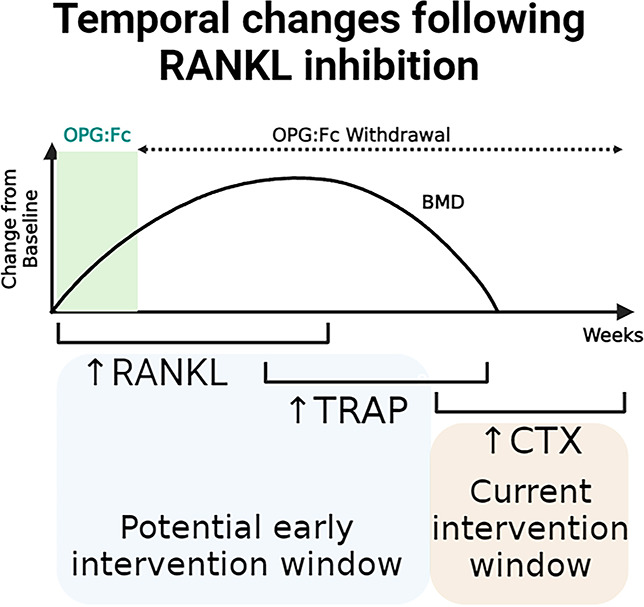
Introduction
Therapeutic agents inhibiting osteoclasts have revolutionized treatment of bone diseases including osteoporosis. Osteoclasts are highly specialized cells that differentiate from hematopoietic stem cells under local and systemic influences. The RANKL) its receptor RANK, and the decoy receptor osteoprotegerin (OPG) form the RANKL/RANK/OPG pathway, which plays a critical role in the differentiation and activation of osteoclasts, and therefore regulation of bone turnover.1 Inhibiting this interaction leads to increased bone mass and reduced bone resorption and has been the target of bone-directed therapies for conditions where suppression of osteoclast-mediated bone resorption is desired.
Denosumab is a fully human monoclonal antibody that binds to and inhibits RANKL, leading to inhibition of osteoclast differentiation and bone resorption. Denosumab has revolutionized the treatment of osteoporosis, and its continued use leads to continuous BMD gains with up to 10 yr of treatment.2
However, discontinuation of denosumab treatment leads to a rapid reversal of its therapeutic effects and a transient overshoot in bone turnover, which eventually returns to pre-treatment levels.3 This results in a net bone loss during this time and appears to be driven by osteoclast-mediated bone loss as measured by elevated markers of collagen breakdown, namely, CTX.3 However, why this rapid overshoot of bone turnover and bone loss following denosumab discontinuation occurs is unknown.
Clinical studies using sequential antiresorptive agents to inhibit this increase in bone resorption following denosumab discontinuation are predominantly observational or post hoc analyses and are therefore limited in providing a mechanism to explain this phenomenon nor a potential solution to manage these patients.4-7 Duration of denosumab treatment appears to impact outcomes following cessation, with patients receiving denosumab for a longer duration experiencing greater rates of bone loss and increased risk of vertebral fractures during the off-treatment period.8,9 Placebo-controlled randomized studies to examine the efficacy of interventions to prevent this bone loss are not ethically feasible due to the risk of bone loss and fractures in the control group.
Clinicians are faced with a dilemma, having to weigh risks associated with long-term anti-resorptive treatment, such as atypical femoral fractures, against the risks of bone loss and fractures if denosumab is discontinued. This problem stems from an incomplete understanding of osteoclast biology following withdrawal of RANKL inhibition, highlighting the need for pre-clinical investigations into the underlying mechanisms. Indeed, the discovery that osteoclasts undergo recycling via fission into osteomorphs, and re-fusion into active osteoclasts, and that during RANKL inhibition these fission products and osteoclast precursors may accumulate, provides new insight into this phenomenon.10
In the presence of RANKL inhibition, suppressed serum tartrate-resistant acid phosphatase (TRAP) levels reflected inhibition of osteoclast formation. Following cessation of RANKL inhibition, a rapid rise in serum TRAP preceded a dramatic loss in BMD in mice,10 confirming clinical findings that enhanced bone resorption follows treatment withdrawal. However, the cellular processes and changes in osteoclast biology that drive this rapid rise in serum TRAP are unclear, and the temporal relationship between serum TRAP and typical bone turnover markers CTX and P1NP has not yet been defined.
To address this, we aimed to define the key regulators of osteoclast formation and activity, and the longitudinal changes in bone turnover markers, to examine how these correlate with changes in BMD and bone microarchitecture during RANKL inhibition and following its cessation. We also examined these changes following longer periods of RANKL inhibition and compared the utility of serum TRAP, a marker of osteoclast activity, with markers of bone turnover utilized in clinical practice. We performed these studies using mice which respond to OPG:Fc with changes in serum bone turnover markers which are relevant to the human biological processes we aim to examine. Taken together, our preclinical investigations provide essential mechanistic insight to guide the timing of sequential therapy in patients discontinuing denosumab.
Materials and methods
Experimental mice
Animal experiments were performed in accordance with approved protocols from the Garvan Institute and St Vincent’s Hospital Animal Ethics Committee (ARA 18/03 and 21/17). All experiments were conducted in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes.
Female C57BL/KaLwRij (Harlan, Netherlands) or C57BL/6 J mice were obtained from the Australian BioResources. All mice were bred and maintained on a C57BL/KaLwRij background or C57BL/6J background under specific-pathogen free conditions. Animal experiments were performed at the Garvan Institute Biological Testing Facility. All animal holding areas in both facilities are maintained within a constant temperature of 21.4 °C with humidity range to avoid animal stress and to minimize experimental variability. Lighting mimics 12-h d/night cycles to stimulate circadian rhythms. Mice were acclimatized for 3 d upon arrival, and standard chow and water were provided ad libitum.
All mice entered their respective experiments aged 6–8 wk and group sizes were determined based on previous experiences with each model. This included studies of the effect of OPG:Fc in these models, in which power calculations were performed to estimate sample size. Using this, 8–10 mice were allocated to each group or otherwise as stated in the figure legends.
To compare the effects of OPG:Fc and anti-mouse RANKL antibody, C57BL/KaLwRij mice were randomly allocated to treatment groups. OPG:Fc or anti-mouse RANKL antibody was administered for 2 wk, and mice were culled at the end of the treatment phase at week 2 where tissue was harvested for their respective experiments outlined below.
To examine the effect of longer duration of RANKL inhibition, C57BL/KaLwRij mice were treated with OPG:Fc for 8 wk and were culled at the end of the study at week 23. Mice underwent DXA imaging and retro-orbital bleeds fortnightly or 3-weekly throughout the study.
To allow contemporaneous measurements of serum markers of osteoclast activity, C57BL6 mice were treated with OPG:Fc for 2 wk and were culled at week 2, 8, 11, and 13 to allow for collection of a large volume of serum. Throughout the study, mice underwent DXA imaging and retro-orbital bleed fortnightly. At each cull timepoint, a large volume of blood was collected through retro-orbital bleeding, and tissue was harvested for their respective experiments outlined below.
OPG:Fc and anti-mouse RANKL antibody treatment
OPG:Fc (Amgen Inc) was administered at a dose of 10 mg per kg i.p. 2-3 times weekly for 2 or 8 wk, a dose confirmed previously to abrogate osteoclasts.11 Anti-mouse RANKL antibody (BioXcell, BE0191) was administered at a dose of 5 mg per kg i.p. thrice weekly for 2 wk. Vehicle mice received saline.
DXA analysis of BMD
DXA (Faxitron Ultrafocus DXA, Hologic) was performed fortnightly or 3-weekly on anaesthetized mice under 3%-5% inhaled isoflurane. Analysis of the hind limb was performed using Vision DXA (Hologic) using a manually drawn region of interest encompassing the hindlimb to quantify the BMD.
Micro-computed tomography
Formalin-fixed right femora were imaged with the SkyScan 1772 micro-computed tomography (microCT) scanner (Bruker) at a resolution of 4.3 μm, 0.5 mm aluminum filter, 50 kv voltage, and 200 μA tube current. Images were captured every 0.4o through 360o and were reconstructed and analyzed using NRecon software (SkyScan). Bone structural parameters and nomenclature were utilized according to standardized guidelines.12 Three-dimensional reconstructed images of femora were generated using Drishti imaging software version 2.4 (ANU).
ROI selection and analyses were performed using CTAn software (Bruker). To compare the effect of OPG:Fc and mouse anti-RANKL antibody, trabecular and cortical bone parameters were calculated from scans performed at a voxel resolution of 5 μm in a 1 mm region of the trabecular compartment beginning 100 μm proximal to the distal femoral growth plate.
To examine the changes in bone parameters following treatment and during rebound bone loss following OPG:Fc withdrawal, trabecular bone parameters were calculated from scans performed at a voxel resolution of 5 μm in a 1 mm region beginning 200 μm proximal to the distal femoral growth plate to reduce the contribution of the primary spongiosa in the analysis. Cortical bone parameters were calculated from scans performed at a voxel resolution of 5 μm in a 0.5 mm region beginning 300 μm proximal to the distal femoral growth plate.
Measurement of TRAP5b, P1NP, CTX, and RANKL
Serum collected by retro-orbital bleeds, under isoflurane anesthesia, throughout animal phaseswas stored at −70° and then assessed for TRAP5b, P1NP, and CTX levels using ELISA kits (Immunodiagnostic Systems) or assessed for RANKL levels using ELISA kits (R&D Systems) following the manufacturer’s instructions.
Analysis of bone histomorphometry
Sectioning/TRAP staining of osteoclasts
Right femora samples were prepared for paraffin histomorphometric analysis by fixing in 4% paraformaldehyde for 24 h at 4 °C, then decalcifying samples in 0.34 M EDTA in PBS (pH 8.0). Samples were then processed for paraffin histology, and 3 μm sections were cut using a Leica Microtome Model RM2265. Paraffin femora sections were stained for TRAP to identify osteoclasts from other resident bone cells. The TRAP staining solution was prepared, comprising of 1000 μL of 0.20 g per mL sodium nitrite added to 1000 μL of the basic fuchsin solution until small bubbles appeared. The sodium nitrite/fuchsin solution was added to 0.35 g of tartaric acid dissolved in 350 mL 1 M sodium acetate buffer pH 5.4. Then, 0.20 g naphthol ASBI phosphate dissolved in 20 mL dimethylformamide was added until the solution turned bright pink. The mixed solution was filtered prior to immediate staining using a 0.22 μm vacuum filter unit (Corning).
Following dewaxing in xylene, sections were incubated in 1 M Tris–HCL buffer (pH 9.4) at 40 °C for 1 h. Throughout incubation, 350 mL of TRAP staining solution was incubated for 15 min at 37 °C. Sections were counterstained with hematoxylin then cover slipped with Eukitt.
Aperio image scanning
Right femora sections on Superfrost PLUS glass slides were scanned on the Aperio Scanscope CS2 model. An area of interest was indicated by a red rectangle placed on the Macro image. Utilizing an Olympus UPLXAPO objective lens at a 20× objective, high quality digital slides were created. Digital slides were modified on Aperio ImageScope (v12.3.2.8013) to show 3 ROI’s: 900 μm at 2.2× objective, 300 μm at 9.8× objective, and 300 μm at 7× objective.
Quantification of osteoclasts and osteoblasts
Quantification of osteoclast populations among the trabecular bone was performed with BioQuant Osteo (Version v21.5.60). Utilizing a Zeiss Axioplan Microscope (Zeiss, Germany) with a high resolution Jenoptik Camera at 10× objective, an ROI capturing a 3 mm region of trabecular bone, 1 mm from the top of the growth plate junction within the cortices was analyzed for each sample. TRAP-positive osteoclasts (bright pink cytoplasmic appearance) and cuboidal hematoxylin-stained osteoblasts were marked, and their cell surface-bone contact perimeters and trabecular bone surfaces were recorded. The total bone surface, no. of osteoclasts or osteoblasts per total bone surface, and osteoclast or osteoblast surface per total bone surface were determined. The structural and cellular parameters were calculated and expressed according to the ASBMR standardized nomenclature.12
RNA extraction
Harvested femora were frozen in liquid nitrogen and homogenized using the Polytron homogenizer probe (Kinematica) in TriReagent RNA isolation reagent (Sigma-Aldrich). RNA was sequentially precipitated from the homogenized sample by first separating the aqueous phase following the addition of chloroform and further precipitation with isopropanol and 3 M sodium acetate. The precipitated RNA was washed with 70% ethanol, resuspended in nuclease-free water, and stored at −70 °C.
Reverse transcription and quantitative polymerase chain reaction(qPCR)
Isolated RNA was used to generate cDNA using the Tetro cDNA synthesis kit (Meridian Life Science Inc) following the manufacturer’s instructions.
The following gene-specific TaqMan probes were used: mouse Rankl (mm00441906_m1), mouse Opg (mm00435454_m1), and mouse B2m (mm00437762_m1) for qPCR. The qPCR reaction was performed using TaqMan Gene Expression MasterMix (Thermo Fisher Scientific) and Life Technologies QuantStudio 7 instrument. For each sample, the threshold cycle (CT) values were processed according to the 2(-Delta Delta C(T)) method.13 Gene expression levels were normalized to the expression of the housekeeping gene B2m and presented relative to untreated vehicle controls.
Statistical methods
Results were analyzed using GraphPad Prism (Version 9, GraphPad Prism V9). One-way analysis of variance (ANOVA) and multiple comparisons were performed using Tukey’s correction, and unpaired t-tests were performed when comparing 2 populations. All data are expressed as mean with error bars representing standard deviation. For all statistical analyses, P-values < .05 were considered to be statistically significant.
Results
OPG:Fc models denosumab treatment and discontinuation in mice
Denosumab is a fully humanized monoclonal antibody against RANKL and does not bind murine RANKL, whereas human OPG is capable of binding directly to murine RANKL14 and able to reduce bone resorption and increase BMD.15,16 We have previously demonstrated RANKL inhibition and rebound bone loss following treatment withdrawal using OPG:Fc.10 To confirm this, we directly compared the efficacy of OPG:Fc and murine anti-RANKL antibody in wild-type mice in suppressing osteoclast activity and driving rebound bone loss upon discontinuation.
Treatment of mice with twice-weekly OPG:Fc (10 mg/kg) or thrice-weekly murine anti-RANKL antibody (5 mg/kg) (Figure 1A) for 2 wk significantly increased hindlimb BMD, which continued to increase for 6 wk following treatment. BMD peaked at week 8 in treated mice and the mean BMD was 12.7% and 15.2% higher than vehicle mean in OPG:Fc and murine anti-RANKL antibody-treated mice, respectively (P < 0.01 compared to vehicle). This was followed by a decrease in BMD to vehicle levels over the next 4 wk, (Figure 1Bi). Throughout the study, there was no significant difference in hindlimb BMD between mice treated with OPG:Fc and murine anti-RANKL antibody.
Figure 1.
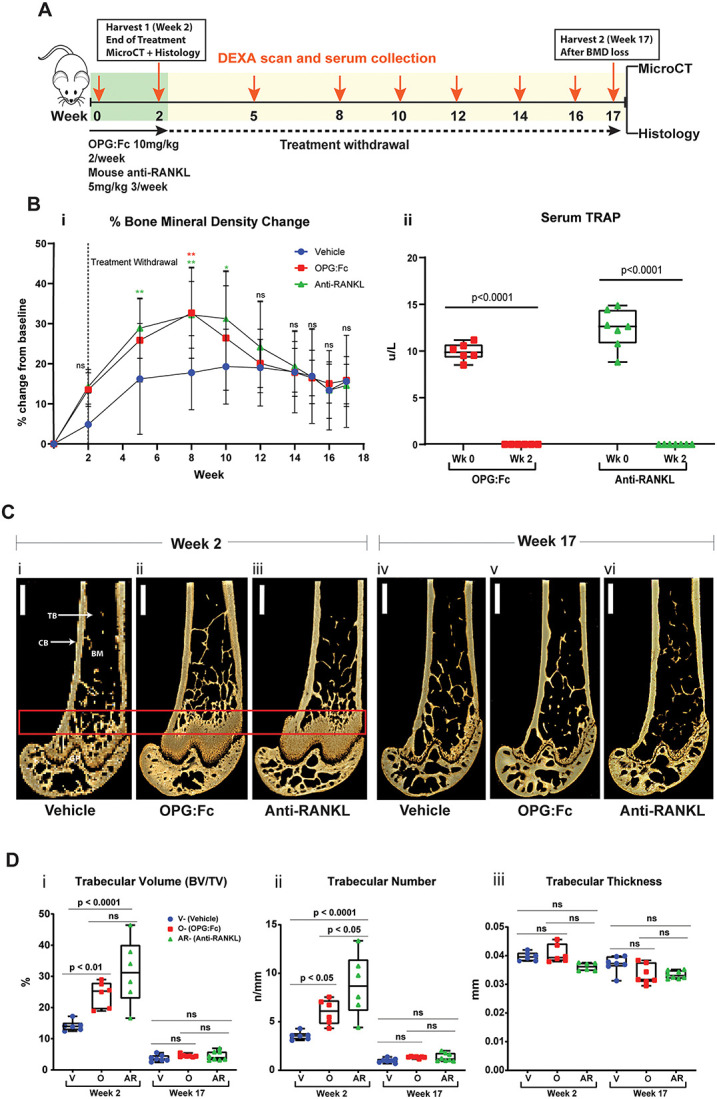
RANKL inhibition with OPG:Fc increases BMD, and treatment withdrawal leads to loss of bone mass and density. (A) Schematic of the experimental design to assess the effect of treatment with OPG:Fc or murine anti-RANKL antibody. (B) BMD and TRAP changes following treatment. (i) BMD shown as a percentage change from baseline levels following treatment with OPG:Fc (n = 9), murine anti-RANKL antibody (n = 9), or saline (vehicle, n = 9). Dotted line showing the end of treatment at week 2. Data represented as mean ± SD. Asterisks indicate P-values < .05 (*P < .05, **P < .01). (ii) Serum TRAP measured by ELISA at baseline and following 2 wk of treatment with OPG:Fc (n = 6) or murine anti-RANKL antibody (n = 7). Boxplots represent mean ± SD. (C) Representative 3D microCT reconstructed images of the right femur at the end of 2 wk of treatment (i)-(iii) and at the end of the study (iv)-(vi). Box highlighting the region of interest. CB, cortical bone; TB, trabecular bone; BM, bone marrow. (D) MicroCT analysis of the region of interest showing trabecular volume (i), number (ii), and thickness (iii) at the end of treatment (week 2, n = 6-7 per group) and at the end of the study (week 17, n = 9 per group). Boxplots represent mean ± SD. V, vehicle; O, OPG:Fc; AR, anti-RANKL.
Serum TRAP was completely suppressed following 2 wk of treatment with either OPG:Fc or murine anti-RANKL antibody (P < .0001, Figure 1Bii), confirming suppression of osteoclast activity with either treatment.
MicroCT analysis of the femur was performed at a 1 mm region of interest in the distal femur to compare the effects of treatment with OPG:FC or murine anti-RANKL antibody on the bone microarchitecture. This showed significantly increased trabecular bone structure at the end of 2 wk of treatment in both OPG:Fc and murine anti-RANKL antibody-treated mice compared to vehicle (Figure 1C, week 2). Trabecular volume (BV/TV) was 52.7% and 75.7% higher (P < 0.01 and P < 0.0001, respectively Figure 1Di), and trabecular number was 51.2% and 84.4% higher than vehicle levels in mice treated with OPG:Fc and murine anti-RANKL antibody, respectively (P < .05 and P < .0001, respectively, Figure 1Dii). The trabecular number was significantly higher in the murine anti-RANKL antibody group compared to the OPG:Fc group (P < .05, Figure 1Dii). There was no difference in trabecular thickness between all groups (Figure 1Diii). There was no significant difference between trabecular bone volume and thickness between mice treated with OPG:Fc or murine anti-RANKL antibody.
At week 17, following bone loss in treated mice, and when hindlimb BMD was equivalent in all groups, there was no difference in the trabecular microCT parameters between vehicle and mice treated with OPG:Fc or murine anti-RANKL antibody (Figure 1D, week 17). Cortical bone volume in the distal femur was increased in mice treated with OPG:Fc at the end of treatment compared to vehicle and murine anti-RANKL antibody-treated mice but was equivalent in all groups at week 17 (Supplementary Figure S1). This shows a similar response in trabecular bone structural parameters to both OPG:Fc and murine anti-RANKL treatment, with return to untreated vehicle levels following withdrawal.
Longer treatment with OPG:Fc increases the rate of bone loss following withdrawal
Increased duration of treatment with denosumab has been associated with increased risk of rebound bone loss and fractures.8,9 To examine this in our mouse models, we treated mice with OPG:Fc or saline for 8 wk instead of 2 wk (Figure 2A). In the longer treatment group, hindlimb BMD increased and continued to increase until week 16, where this reached levels 51.1% higher than vehicle levels from baseline (P < .001). This was followed by a rapid decline in BMD to vehicle levels with a decrease of 24.5% over a 4-wk period (weeks 16-20) (Figure 2B), which was greater than the rate of bone loss observed with 2 wk of treatment of 12.6% over a 4 wk period (Figure 1B). Interestingly, BMD continued to decline below vehicle levels between week 20 and 23, though there was no significant difference between the groups during this period.
Figure 2.

Longer duration of RANKL inhibition increases the rate of BMD loss and leads to sustained increases in serum TRAP following treatment withdrawal. (A) Schematic of the experimental design to assess the effect of longer duration of treatment with OPG:Fc on BMD and serum TRAP. N = 10 per group. (B) Longitudinal change in BMD presented as percentage change from baseline. Dotted line showing end of treatment at week 8. Data presented as mean ± SD. Asterisks indicate P-values < .05 (*P < .05, **P < .01, ***P < .001, ****P < .0001). (C) Longitudinal serum TRAP throughout the study. Dotted line showing end of treatment at week 8. Data presented as mean ± SD. Asterisks indicate P-values < .05 (**P < .01, ***P < .001).
Once again, we demonstrated that serum TRAP was suppressed with OPG:Fc treatment, remained markedly suppressed between week 8 and 14 (P < .01), and then showed a rapid overshoot in serum to 73.3% (P < .001) higher than vehicle just 2 wk later (Figure 2C). Serum TRAP remained significantly above vehicle levels between weeks 16-20, while BMD loss was ongoing, before dropping to vehicle levels by the end of the study at week 23. We again demonstrated that the rapid rise and overshoot above vehicle levels in serum TRAP (week 14-16) preceded the rebound loss in hindlimb BMD (from week 16).
Serum TRAP is elevated prior to P1NP and CTX following withdrawal of treatment with OPG:Fc
We have demonstrated that OPG:Fc treatment and withdrawal are representative of denosumab treatment and discontinuation in our model. Following our observation of serum TRAP rising prior to decline in hindlimb BMD, we sought to investigate how BMD and serum TRAP changes correlate with bone turnover markers used in clinical practice, namely CTX and P1NP, which are currently utilized to guide sequential therapy following denosumab discontinuation.
As shown previously, longitudinal analyses following 2 wk of OPG:Fc revealed suppressed serum TRAP to week 8 and a rise to control levels at week 10, prior to BMD loss between weeks 10 and 13 (Figure 3B and C). As expected, serum TRAP, P1NP, and CTX were all suppressed below control levels at the end of OPG:Fc treatment at week 2, and all markers remained suppressed at week 8 (Figures 3D and S2). At week 11, following the peak in BMD, serum TRAP was significantly elevated 34.8% above mean control levels, while serum P1NP and CTX had risen to reach control levels (Figures 3D and S2). By week 13, when BMD had returned to control levels in mice treated with OPG:Fc, serum TRAP remained significantly higher than control levels and it was only at this timepoint where serum P1NP and CTX were also significantly higher than vehicle levels (Figures 3D and S2).
Figure 3.
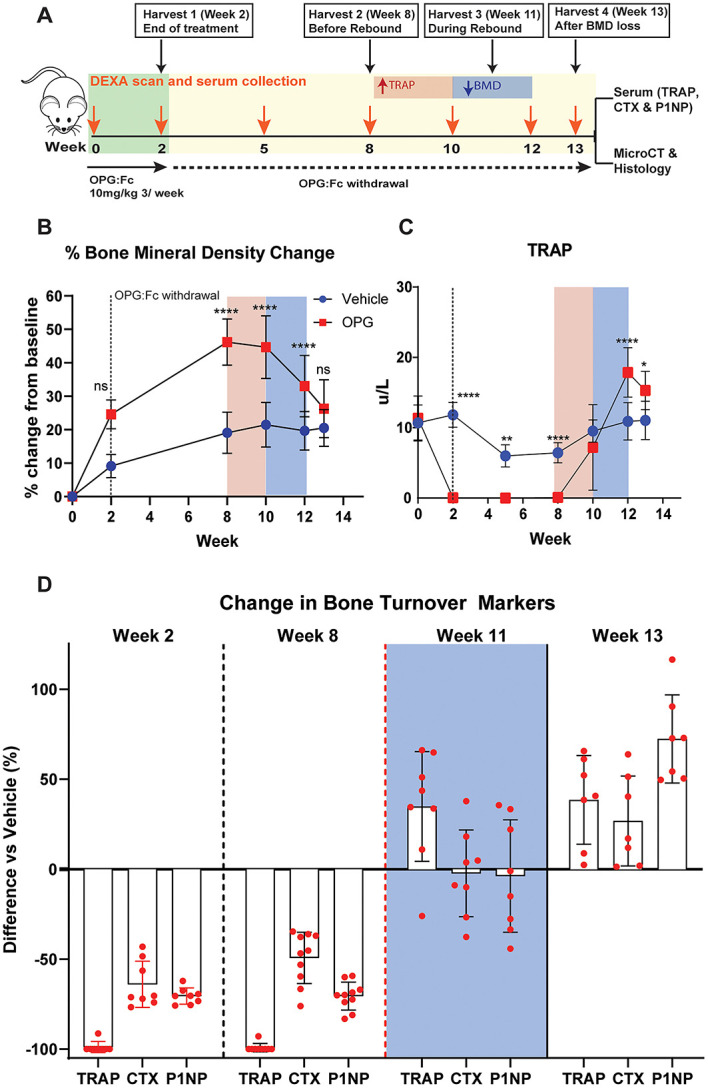
Serum TRAP rises above vehicle levels before P1NP and CTX following withdrawal of RANKL inhibition. (A) Schematic of the study design to harvest cohorts of mice at specific intervals following 2 wk of OPG:Fc treatment to allow for contemporaneous measurements of bone turnover markers, changes in bone microarchitecture, and histology. Pink box (left) highlights a time at which serum TRAP is rising from post treatment levels, while the blue box (right) highlights the time during which BMD loss is occurring. (B) Longitudinal changes in BMD. Data presented as mean ± SD. Asterisks indicate P-values < .05 (*P < .05, **P < .01, ***P < .001, ****P < .0001). (C) Longitudinal changes in serum TRAP. Data presented as mean ± SD. Asterisks indicate P-values < .05 (*P < .05, **P < .01, ***P < .001, ****P < .0001). (D) Changes in the bone turnover markers (TRAP, CTX, and P1NP) in mice treated with OPG:Fc compared to vehicle at each harvest timepoint, expressed as % change compared to vehicle mean. Week 11 highlighted in the blue box represents the time during which BMD loss is occurring.
Overall, this shows that an increase in osteoclast number and overshoot in osteoclast enzyme activity, as measured by serum TRAP, parallels bone loss more closely and was detected earlier than an overshoot in markers of bone turnover used in clinical practice, P1NP, and CTX.
Relative deterioration of bone microarchitecture is observed during rebound bone loss
To investigate the changes in bone microarchitecture during rebound bone loss following OPG:Fc treatment, harvested femora were examined with microCT (Figure 4A). Mice treated with OPG:Fc were harvested at time-points following treatment and during rebound bone loss (Figure 3A).
Figure 4.

Changes in bone microarchitecture during rebound bone loss following withdrawal of RANKL inhibition with OPG:Fc. (A) Representative 3D images of harvested femora showing differences in bone microarchitecture between mice treated with saline and OPG:Fc. Dashed red box denotes a region of interest examined at a 0.5 mm section located 3 mm above the growth plate (GP). Solid red box denotes region of interest examined at a 1 mm section located 2 mm above the growth plate. CB, cortical bone; TB, trabecular bone. (B) Differences in trabecular volume (i), number (ii), and thickness (iii) between mice treated with saline (vehicle) and OPG:Fc at the harvest timepoints. Boxplots represent mean ± SD. (C) Differences in cortical volume (i) and thickness (ii) between mice treated with saline (vehicle) and OPG:Fc at the harvest timepoints. Boxplots represent mean ± SD.
Trabecular bone volume, number, and thickness in the distal metaphyseal region were significantly higher in the femora of mice treated with OPG:Fc compared to their controls throughout the study (Figure 4B). Notably this difference was less evident by week 13 indicating a relative loss in trabecular volume during rebound BMD loss.
Cortical bone volume in the distal diaphysis was significantly higher in OPG:Fc-treated mice compared to vehicle at week 2 following treatment, at week 8 prior to the decline in BMD, and at week 11 while BMD loss was occurring (Figure 4Ci) but was equivalent to vehicle mice at week 13 following rebound bone loss. There was no difference in cortical thickness following treatment at week 2 but it was significantly lower in OPG:Fc-treated mice at each time-point thereafter (Figure 4Cii). The endosteal and periosteal perimeters were equivalent at the end of treatment but were higher in treated mice at each time-point thereafter (Figure S3). These changes in cortical parameters suggest that there is increased cortical volume initially, but there is increased bone resorption on the endosteal surface, leading to a thinner cortex and increased endosteal and periosteal perimeter.
Together, these results show that, following an initial improvement in bone microarchitecture following OPG:Fc treatment, loss in trabecular and cortical bone is observed while rebound BMD loss is occurring following the offset of RANKL inhibition.
Abundant osteoclasts are observed during rebound bone loss
To examine the changes in osteoclast number during OPG:Fc treatment and rebound bone loss, TRAP-positive cells were quantified in the distal femora at each harvest time point (Figure 5A).
Figure 5.
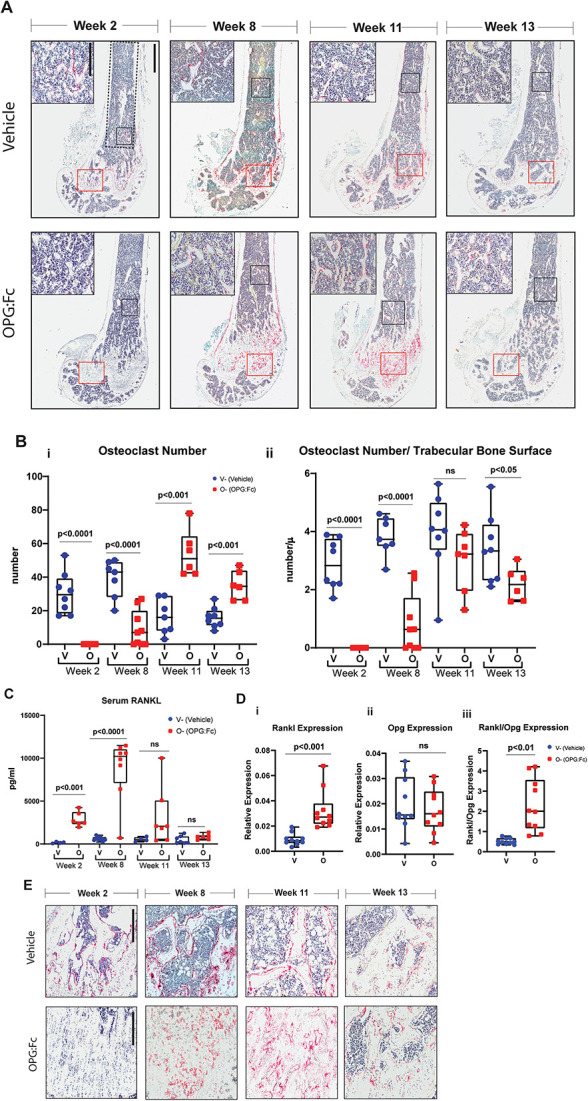
Changes in osteoclast number and activity during rebound bone loss following withdrawal of RANKL inhibition with OPG:Fc. (A) Representative histological images of femora stained with TRAP (red) harvested following treatment with saline or OPG:Fc. Analysis of osteoclast parameters on trabecular bone was performed within the ROI defined by the dotted line (scale bar 900 μm at 2.2× magnification). Representative high magnification images shown in top left corner and its corresponding ROI shown in the black box (scale bar 300 μm at 9.8× magnification). Red box (near bottom of image) denotes osteoclasts observed on trabecular bone surfaces as shown in magnified images in Figure 5E. (B) Quantification of number of osteoclasts (i) and osteoclast surface (ii) per trabecular bone surfaces at harvest timepoints week 2, 8, 11, and 13. Boxplots represent mean ± SD. (C) Quantification of serum RANKL following OPG:Fc treatment. Boxplots represent mean ± SD. (D) Quantification of mRNA expression of (i) Rankl, (ii) Opg, and (iii) Rankl:Opg in marrow depleted femora of mice treated with saline or OPG:Fc harvested at week 8. Boxplots represent mean ± SD. Representative images of osteoclasts observed on trabecular bone surfaces throughout the study in the ROI marked by the red box in Figure 5A (scale bar 300 μm at 7× magnification).
Following treatment with 2 wk of OPG:Fc, there was a complete absence of osteoclasts. Osteoclasts were detectable at week 8 but remained reduced in number compared to control. However, by week 11, osteoclast numbers were significantly higher than control levels, remaining elevated at week 13 (Figure 5Bi). When osteoclast numbers were normalized to trabecular bone surfaces, and the increase in trabecular bone volume with OPG:Fc treatment was accounted for (Figure 4B), osteoclasts numbers per trabecular bone surface were equivalent or lower in treated mice during rebound bone loss (Figure 5Bii). Osteoclast surfaces were also increased during rebound bone loss. But this was equivalent to vehicle levels when osteoclast surfaces were normalized to trabecular bone surfaces due to increased trabecular bone with OPG:Fc treatment at weeks 11 and 13, as was also demonstrated with MicroCT analysis (Figure S4).
Osteoblast number and surfaces were also quantified in this region of interest. Osteoblast numbers followed a similar pattern to osteoclasts during OPG:Fc treatment and rebound bone loss, with significantly lower numbers following treatment, starting to rise thereafter. When total osteoclast numbers were significantly higher than control at week 11, total osteoblast numbers remained equivalent to vehicle levels. By week 13, osteoblast numbers and surface were both significantly increased compared to control, but both number and surface were equivalent to control levels when normalized to trabecular bone surfaces (Figure S5).
In the most distal region of the metaphysis of the femur, quantification of osteoclasts was not possible owing to the difficulty in defining trabecular bone surfaces due to the retention of the primary spongiosa in mice treated with OPG:Fc. Nevertheless, images of this region show clearly that TRAP+ osteoclasts were absent at the end of treatment and abundant in numbers at weeks 8, 11, and 13 compared to vehicle (Figure 5E). On the endosteal surface, osteoclast parameters (number and surfaces) were not different between groups (data not shown).
Higher local and circulating RANKL levels provide a pro-osteoclastogenic environment prior to rebound bone loss
Serum RANKL measurement by ELISA demonstrated significantly higher levels of circulating RANKL following treatment with 2 wk of OPG:Fc, with levels 17.6-fold higher than vehicle levels at week 2 (P < .001, Figure 5C). Serum RANKL was maximal at week 8 with serum RANKL levels 14.7-fold higher compared to week 2 levels (P < .0001), indicating an abundance of circulating pro-osteoclastogenic factors just prior to BMD loss (Figure 5C). As rebound BMD loss was occurring, serum RANKL levels decreased. Serum RANKL levels were equivalent to vehicle levels once hindlimb BMD was equivalent.
We then performed quantitative PCR to measure mRNA expression of Rankl and Opg in marrow-depleted femora to examine the changes in osteoclastogenic factors in the bone microenvironment at week 8, when serum RANKL levels were highest. This showed significantly higher Rankl expression in OPG:Fc-treated bones, while Opg expression remained equivalent to control, leading to a significant shift in the Rankl:Opg expression, favoring osteoclastogenesis (Figure 5D).
Discussion
RANKL inhibition with denosumab has revolutionized the treatment of osteoporosis. However, the overshoot of bone resorption and rapid loss in BMD following its discontinuation presents an important clinical challenge in the long-term management of osteoporosis. This phenomenon remains incompletely understood. Combined with a lack of robust randomized control studies to define optimal management approaches, this has led to uncertainty around how to manage patients who have ceased or plan to cease denosumab therapy.
Animal models facilitate the direct examination of longitudinal changes in bone structure, bone cell activity, and local and serum markers of osteoclast activity during rebound bone loss following treatment withdrawal.
OPG:Fc treatment in mice models denosumab treatment and discontinuation
We used OPG:Fc to inhibit RANKL and model denosumab treatment and discontinuation. This approach models observations seen following denosumab discontinuation in clinical practice where gains in BMD on treatment are lost within 12 mo of discontinuation and a large overshoot in bone turnover markers PINP and CTX are detected.3,17 Importantly, we showed a similar pattern of response in hindlimb BMD, trabecular architecture, and serum TRAP in mice treated with OPG:Fc and murine anti-RANKL, confirming the relevance of OPG:Fc when modelling this scenario.
Longer duration of treatment with denosumab has been identified as a risk factor for increased bone loss and fractures.18 Our results align with this, with longer duration of RANKL inhibition with OPG:Fc leading to greater gains in hindlimb BMD and greater rates of BMD loss once treatment was discontinued: 24% (long) versus 12% (short) over 4 wk. Our model also showed a more sustained overshoot in bone resorption as measured by serum TRAP.
Patients discontinuing denosumab are at increased risk of fractures. Evaluation of iliac crest bone biopsy following discontinuation of denosumab showed a return to bone remodeling parameters similar to those observed in untreated postmenopausal women with osteoporosis.19 This was also observed in our study where femoral microCT parameters were equivalent between mice treated with OPG:Fc, murine anti-RANKL antibody, or saline at week 17 (Figure 1D). However, analysis of femora harvested while rebound bone loss was occurring showed a relative deterioration of BMD and trabecular bone microarchitecture (week 13), and a deficit in cortical thickness with widening of the cortical structure at weeks 11 and 13 (Figures 4C and S3).
Overshoot in serum TRAP occurs before a rise in serum P1NP and CTX during the rebound phenomenon
Bone turnover markers are used to predict fracture risk and monitor treatment response in the management of patients with osteoporosis. In 2010, the International Osteoporosis Foundation and International Federation of Clinical Chemistry and Laboratory Medicine recommended the use of the bone formation marker (P1NP) and bone resorption marker (CTX) as reference markers measured by standardized assays, and the use of these markers has guided clinical practice and trials since.20 In the setting of denosumab discontinuation, overshoot above control levels is seen in both P1NP and CTX,14 and a rise in CTX has been used to guide sequential therapy in randomized studies examining the use of zoledronate following denosumab to prevent bone loss.4
We demonstrated that serum TRAP, a marker of osteoclast enzymatic activity, rises prior to these bone turnover markers used in clinical practice. During bone resorption, type 1 collagen is digested by enzymes, and the degradation products of type 1 collagen (such as CTX) is released into circulation once bone resorption and breakdown of bone have already occurred. In our study, CTX and P1NP were only higher than vehicle levels once BMD loss had already occurred, and this may explain why studies intervening at a time of CTX rise failed to prevent BMD loss following denosumab discontinuation4,5,7 as the intervention was too late.
In comparison, TRAP is an enzyme produced by the osteoclast during osteoclast differentiation and bone resorption21,22 and is detectable earlier during osteoclastogenesis as well as during the process of bone resorption. This phenomenon was also seen in hRANKL mice receiving denosumab, where serum TRAP was suppressed below vehicle levels during treatment, then rose to vehicle levels before a significant overshoot in serum CTX was detected.23
TRAP is a useful marker to monitor response to antiresorptive treatment including denosumab, decreasing with treatment and correlating to changes in BMD and serum CTX.24-26 TRAP is also unaffected by food intake, whereas CTX and P1NP levels decrease after feeding, reducing diagnostic accuracy, and must be measured on a fasting sample.27 Therefore, a rise in serum TRAP from on-treatment levels may provide a more useful marker in the setting of denosumab discontinuation. Clinical investigations are required to determine the potential utility of this finding to inform timing of earlier intervention with sequential osteoclast-directed therapy in patients.
Elevated serum RANKL precedes an overshot in serum TRAP and BMD loss following withdrawal of RANKL inhibition
Following confirmation that a rapid formation of osteoclasts was driving the bone loss in our model, we aimed to define what was driving this response. Serum levels of RANKL were elevated 17-fold in mice following treatment with OPG:Fc and were significantly elevated before a rise in osteoclast number, serum TRAP, and BMD loss were detected. Our data align with cross-sectional clinical analyses of serum RANKL and TRAP at 6, 9, and 12 mo following the last dose of denosumab.26 In this clinical study, higher RANKL was observed 6 mo following the last denosumab dose, compared to levels measured at 9- and 12-mo, whereas serum TRAP was highest in the 9- and 12-mo groups,26 though the temporal relationship to changes in BMD was not examined.
This supports our observation that following withdrawal of RANKL inhibition with OPG:Fc, there is a rise in serum RANKL, which leads to a rise in serum TRAP and rebound bone resorption, releasing CTX into the circulation at the time of clinically detectable BMD loss.
However, this finding was not observed in another clinical study by Fassio and colleagues,28 who measured serum RANKL longitudinally following denosumab discontinuation and found a progressive increase in RANKL, which only reached statistical significance at 12 mo following denosumab discontinuation. Why a difference in the timing of elevated RANKL exists between these clinical studies is unclear. Sølling and colleagues hypothesized that this difference may be due to the difference in the number and mean age of participants and the RANKL assay utilized.26 TRAP was not measured in the study by Fassio and colleagues so the temporal relationship between RANKL and TRAP following denosumab discontinuation could not be examined in this study.
Notably, in our study, serum RANKL had returned to control levels by the end of our study once bone loss had completed and BMD also returned to control levels. Serial measurements of serum RANKL could be considered in future clinical studies examining denosumab discontinuation to further elucidate the pattern of change in osteoclastogenic factors. Nevertheless, our pre-clinical study provides a clear temporal pattern of accelerated osteoclast formation and activity in the lead up to BMD loss and elevated fracture risk.
Changes in Rankl expression promote osteoclast formation during rebound
Osteoclast precursors have been observed to accumulate during denosumab treatment and suggested to play a role in the rapid formation of osteoclasts following withdrawal.29 Our previous work using OPG:Fc showed osteoclasts undergoing fission to form daughter cells termed “osteomorphs,” which accumulated during OPG:Fc treatment.10 These RANK-positive cells were then capable of undergoing re-fusion to form mature osteoclasts following withdrawal of RANKL inhibition in a process called “osteoclast recycling.” The increase in RANKL following withdrawal of RANKL inhibition with OPG:Fc could therefore stimulate this pool of accumulated RANK-positive osteoclast precursors and osteomorphs to undergo differentiation, fusion, and activation to form a large number of osteoclasts and undergo bone resorption en masse.30 This is also supported by our previous work, where isolated bone marrow cells from OPG:Fc-treated mice were cultured in vitro with RANKL and M-CSF. The same number of bone marrow cells from OPG:Fc-treated mice produced more osteoclasts than cells from vehicle mice, and these cells were capable of forming resorption pits on dentine slices.10
To examine this mechanism further in our model and overcome limitations of not being able to assess protein levels of OPG, we examined local production of RANKL via quantification of RANKL and OPG mRNA. Indeed, a significant shift in the Rankl:Opg gene expression ratio in the marrow depleted bone prior to increased osteoclast formation and activity and BMD loss was demonstrated. A significant increase in Rankl expression was also observed in hRANKL mice receiving denosumab for 2 wk, though there was also a significant reduction in Opg expression, which was not observed in our study. Nevertheless, our local mRNA expression data indeed confirm out finding of elevated circulating sRANKL, providing an additional mechanism for the rapid formation of osteoclasts in our model.
In contrast to our previous findings, an increase in osteoclast progenitors, as determined by primary bone marrow cultures, was not detected in a study of hRANKL mice treated with denosumab.23 Single cell RNA sequencing of bone marrow harvested 2 wk following cessation of denosumab treatment in hRANKL mice revealed no change in myeloid lineage cells with denosumab. However, a trend toward more cells expressing osteomorph markers (such as Acp5, Axl, and Cadm1) was noted. The lack in significant increases in precursors and osteomorphs in this hRANKL mouse study may be due to technical isolation challenges, the timing of analysis post denosumab treatment, or may be influenced by the modified mouse model utilized.
A humanized RANKL mouse has been previously developed by Amgen, and denosumab was able to completely inhibit the chimeric RANKL, thereby increasing BMD.31 However, the baseline phenotype of this mouse was altered, with significantly lower serum TRAP levels and decreased osteoclast and osteoblast surfaces (despite equivalent trabecular volume) compared to wildtype.31 Although this novel hRANKL mouse demonstrated equivalent cortical thickness and trabecular volume to wild-type mice, bone cell activity and endogenous mouse RANKL/RANK/OPG signaling were not examined, therefore it is unclear if this would influence the differences observed in Rankl:Opg expression and osteoclast precursor populations following denosumab treatment.
Key resources table
| Reagent or resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Anti-mouse RANKL antibody | BioXcell | Cat#: BE0191 |
| Chemicals, peptides, and recombinant proteins | ||
| Ambion Nuclease-Free Water | Thermo Fisher Scientific | Cat#: AM9937 |
| Chloroform | Sigma-Aldrich | Cat#: C2432 |
| EDTA | Sigma-Aldrich | Cat#: E5134 |
| Ethyl alcohol, Pure | Sigma-Aldrich | Cat#: E7023 |
| Isopropanol (2-Propanol) | Sigma-Aldrich | Cat#: I9516 |
| Osteoprotegerin:Fc | Amgen Inc | N/A |
| Paraformaldehyde | Thermo Fisher Scientific | Cat#: ACR416785000 |
| PBS, pH 7.4 | GIBCO/Thermo Fisher Scientific | Cat#: 10010049 |
| TriReagent RNA Isolation Reagent | Sigma-Aldrich | Cat#: T9424 |
| Critical commercial assays | ||
| Mouse TRANCE/RANKL/TNFSF11 Quantikine ELISA Kit | R&D Systems | Cat#: MTR00 |
| Serum RatLaps (CTX-I) EIA | Abacus ALS Pty Ltd | Cat#: IDSAC06F1 |
| Serum P1NP ELISA -Mouse | Abacus ALS Pty Ltd | Cat#: IDSAC33F1 |
| Serum TRAP 5b ELISA – Mouse | Abacus ALS Pty Ltd | Cat#: IDSBTR103 |
| Taqman Gene Expression Assay | Thermo Fisher Scientific | Cat#: 4331182 |
| TaqMan Gene Expression MasterMix | Thermo Fisher Scientific | Cat#: 4369016 |
| Tetro cDNA Synthesis Kit | Meridian Life Science Inc | Cat#: BIO-65043 |
| Experimental models: Organisms/strains | ||
| C57BL/6J | Australian BioResources | ABR:000664 |
| C57BL/KaLwRij | Australian BioResources | ABR: 3076 |
| Software and algorithms | ||
| BioQuant Osteo Ver v21.5.60 | Bioquant Life Science | https://www.bioquant.com/ RRID:SCR_016423 |
| CTAn | Skyscan | https://www.bruker.com/ |
| Drishti-2.4 | 36 | https://github.com/nci/drishti; RRID:SCR_017999 |
| Vision DXA | Faxitron Bioptics/Hologic | https://www.faxitron.com/ |
| NRecon | Skyscan | https://www.bruker.com/ |
| Life Technologies QuantStudio 7 Real Time PCR System | Applied Biosystems | https://www.thermofisher.com/order/catalog/product/4485701#/4485701 RRID:SCR_020245 |
| Other | ||
| Polytron PT1200E Homogenizer | Kinematica AG | P/N: 9112017 |
Nevertheless, the conclusion that downregulation of OPG production by osteoblast and osteocytes in response to denosumab is sound and is supported by our observations showing a reduction in osteoblast parameters and the suppression of the osteoblast marker P1NP following OPG:Fc treatment, indicating decreased osteoblast number and activity in our model. These are complementary to our finding that elevated RANKL provides a local pro-osteoclastogenic environment prior to and during rebound bone loss. Additional studies examining changes in OPG in the bone microenvironment and osteocyte populations in our model using OPG:Fc to inhibit RANKL would be informative.
It is also important to note that other than bisphosphonates, which display a sustained effect on bone turnover following discontinuation, other bone-directed therapies also display a pattern of rise in bone resorption and loss of BMD following discontinuation. This has been observed following the discontinuation of hormone replacement therapy with estrogen,32 selective estrogen receptor modulators,33 odanacatib,34 and romosozumab.35 Although increases in fracture risk have not been associated with these reductions in BMD during off treatment periods, further pre-clinical studies employing a similar design to the present study and examining changes to RANKL/OPG signaling and serum TRAP would expand our understanding of this phenomenon across other treatments.
Our data provide the first model of denosumab discontinuation using OPG:Fc in mice with the aim to define the temporal cellular and molecular mechanisms driving the rebound phenomenon. Moreover, this is the first study to demonstrate that longer treatment with OPG:Fc drives a greater rebound stimulus of osteoclast activity and more rapid bone loss. It would be important to determine whether lower doses of OPG:Fc, which may be considered more physiologically relevant, would lead to similar outcomes; however, we did demonstrate a similar pattern of temporal bone gain and rebound loss with our OPG:Fc dosing protocol compared directly to a murine anti-RANKL antibody. In addition, we used intact growing mice in our studies. It would be important to explore our model in aged or mice following ovariectomy to truly mimic the post-menopausal bone microenvironment.
Importantly we also showed the potential for serum TRAP measures to be utilized to predict imminent BMD loss and guide timing of sequential therapy following denosumab discontinuation. At the cellular and molecular level, we revealed aberrant RANKL signaling and osteoclast formation in the weeks following OPG:Fc withdrawal, providing evidence to support our previous conclusion that osteoclast precursor accumulation and osteoclast recycling drive the rebound phenomenon following denosumab discontinuation.
Our studies were performed in growing and eugonadal mice, which is a limitation in the clinical translation of these findings as postmenopausal women represent the majority of patients receiving denosumab. Whether these changes in gene expression and osteoclast precursor and osteomorph populations are also seen in humans receiving denosumab is unclear and warrants further investigation.
Taken together, this study provides a novel framework for the biological mechanisms underpinning the rebound phenomenon and the potential use of alternative markers of bone resorption (serum RANKL and TRAP) to guide sequential therapy. Ultimately, this work will lead to an improved capacity to track rebound response to denosumab withdrawal and therefore our ability to initiate sequential therapy strategies to prevent bone loss and fractures in patients following denosumab discontinuation.
Supplementary Material
Acknowledgments
OPG:Fc used in this study was provided by Amgen Inc.
Contributor Information
Albert S Kim, Skeletal Diseases Program, Garvan Institute of Medical Research, Sydney, NSW, 2010, Australia; Faculty of Medicine, St Vincent’s Clinical School, UNSW Sydney, Sydney, NSW, 2010, Australia; Department of Diabetes and Endocrinology, Westmead Hospital, Sydney, NSW, 2145, Australia; Faculty of Health and Medicine, University of Sydney, Sydney, NSW, 2050, Australia.
Victoria E Taylor, Skeletal Diseases Program, Garvan Institute of Medical Research, Sydney, NSW, 2010, Australia.
Ariel Castro-Martinez, Skeletal Diseases Program, Garvan Institute of Medical Research, Sydney, NSW, 2010, Australia.
Suraj Dhakal, Skeletal Diseases Program, Garvan Institute of Medical Research, Sydney, NSW, 2010, Australia.
Amjad Zamerli, Skeletal Diseases Program, Garvan Institute of Medical Research, Sydney, NSW, 2010, Australia.
Sindhu Mohanty, Skeletal Diseases Program, Garvan Institute of Medical Research, Sydney, NSW, 2010, Australia.
Ya Xiao, Skeletal Diseases Program, Garvan Institute of Medical Research, Sydney, NSW, 2010, Australia.
Marija K Simic, Skeletal Diseases Program, Garvan Institute of Medical Research, Sydney, NSW, 2010, Australia; Department of Pathology, New York University Grossman School of Medicine, New York, NY, 10016, United States.
Jinchen Wen, Department of Psychology and Neuroscience, Duke University, Durham, NC, 27708, United States.
Ryan Chai, Skeletal Diseases Program, Garvan Institute of Medical Research, Sydney, NSW, 2010, Australia; Faculty of Medicine, St Vincent’s Clinical School, UNSW Sydney, Sydney, NSW, 2010, Australia.
Peter I Croucher, Skeletal Diseases Program, Garvan Institute of Medical Research, Sydney, NSW, 2010, Australia; Faculty of Medicine, St Vincent’s Clinical School, UNSW Sydney, Sydney, NSW, 2010, Australia.
Jacqueline R Center, Skeletal Diseases Program, Garvan Institute of Medical Research, Sydney, NSW, 2010, Australia; Faculty of Medicine, St Vincent’s Clinical School, UNSW Sydney, Sydney, NSW, 2010, Australia.
Christian M Girgis, Department of Diabetes and Endocrinology, Westmead Hospital, Sydney, NSW, 2145, Australia; Faculty of Health and Medicine, University of Sydney, Sydney, NSW, 2050, Australia.
Michelle M McDonald, Skeletal Diseases Program, Garvan Institute of Medical Research, Sydney, NSW, 2010, Australia; Faculty of Medicine, St Vincent’s Clinical School, UNSW Sydney, Sydney, NSW, 2010, Australia; Faculty of Health and Medicine, University of Sydney, Sydney, NSW, 2050, Australia.
Author contributions
Albert S. Kim (Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing—original draft, Writing—review & editing), Victoria E. Taylor (Data curation, Formal analysis), Ariel Castro-Martinez (Data curation, Methodology), Suraj Dhakal (Data curation, Formal analysis), Amjad Zamerli (Data curation), Sindhu Mohanty (Data curation), Ya Xiao (Data curation), Marija K. Simic (Data curation, Methodology), Jinchen Wen (Data curation), Ryan Chai (Supervision), Peter I. Croucher (Funding acquisition, Supervision, Writing—review & editing), Jacqueline R. Center (Conceptualization, Investigation, Supervision, Writing—review & editing), Christian M. Girgis (Conceptualization, Investigation, Writing—review & editing), and Michelle M. McDonald (Conceptualization, Formal analysis, Funding acquisition, Investigation, Supervision)
Funding
A.S.K. is supported by the UNSW Sydney University Postgraduate Award and the Australian Government Research Training Program Scholarship. This work is supported by the Mrs. Janice Gibson and the Ernest Heine Family Foundation and Cancer Council NSW project grant R20-05, American Society of Bone and Mineral Research Rising Star Award and Bone Health Foundation an Australia and New Zealand Bone and Mineral Society Grant in Aide.
Conflicts of interest
None declared.
Data availability
All raw data files can be provided on request.
References
- 1.Boyce BF, Xing L. The RANKL/RANK/OPG pathway. Curr Osteoporos Rep. 2007;5(3):98–104. 10.1007/s11914-007-0024-y. [DOI] [PubMed] [Google Scholar]
- 2.Bone HG, Wagman RB, Brandi ML, et al. 10 years of denosumab treatment in postmenopausal women with osteoporosis: results from the phase 3 randomised FREEDOM trial and open-label extension. Lancet Diabetes Endocrinol. 2017;5(7):513–523. 10.1016/S2213-8587(17)30138-9. [DOI] [PubMed] [Google Scholar]
- 3.Bone HG, Bolognese MA, Yuen CK, et al. Effects of denosumab treatment and discontinuation on bone mineral density and bone turnover markers in postmenopausal women with low bone mass. J Clin Endocrinol Metab. 2011;96(4):972–980. 10.1210/jc.2010-1502. [DOI] [PubMed] [Google Scholar]
- 4.Sølling AS, Harsløf T, Langdahl B. Treatment with Zoledronate Subsequent to Denosumab in Osteoporosis: a Randomized Trial. J Bone Miner Res. 2020;35(10):1858–1870. 10.1002/jbmr.4098. [DOI] [PubMed] [Google Scholar]
- 5.Anastasilakis AD, Papapoulos SE, Polyzos SA, Appelman-Dijkstra NM, Makras P. Zoledronate for the prevention of bone loss in women discontinuing denosumab treatment. a prospective 2-year clinical trial. J Bone Miner Res. 2019;34(12):2220–2228. 10.1002/jbmr.3853. [DOI] [PubMed] [Google Scholar]
- 6.Anastasilakis AD, Polyzos SA, Yavropoulou MP, et al. Comparative effect of zoledronate at 6 versus 18 months following denosumab discontinuation. Calcif Tissue Int. 2021;108(5):587–594. 10.1007/s00223-020-00785-1. [DOI] [PubMed] [Google Scholar]
- 7.Everts-Graber J, Reichenbach S, Ziswiler HR, Studer U, Lehmann T. A single infusion of zoledronate in postmenopausal women following denosumab discontinuation results in partial conservation of bone mass gains. J Bone Miner Res. 2020;35(7):1207–1215. 10.1002/jbmr.3962. [DOI] [PubMed] [Google Scholar]
- 8.Anastasilakis AD, Polyzos SA, Makras P, Aubry-Rozier B, Kaouri S, Lamy O. Clinical features of 24 patients with rebound-associated vertebral fractures after denosumab discontinuation: systematic review and additional cases. J Bone Miner Res. 2017;32(6):1291–1296. 10.1002/jbmr.3110. [DOI] [PubMed] [Google Scholar]
- 9.Burckhardt P, Faouzi M, Buclin T, Lamy O, The Swiss Denosumab Study G . Fractures after denosumab discontinuation: a retrospective study of 797 cases. J Bone Miner Res. 2021;36(9):1717–1728. 10.1002/jbmr.4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McDonald MM, Khoo WH, Ng PY, et al. Osteoclasts recycle via osteomorphs during RANKL-stimulated bone resorption. Cell. 2021;184(5):1330–47.e13. 10.1016/j.cell.2021.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McDonald MM, Morse A, Mikulec K, et al. Matrix metalloproteinase–driven endochondral fracture union proceeds independently of osteoclast activity. J Bone Miner Res. 2013;28(7):1550–1560. 10.1002/jbmr.1889. [DOI] [PubMed] [Google Scholar]
- 12.Dempster DW, Compston JE, Drezner MK, et al. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 2013;28(1):2–17. 10.1002/jbmr.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25(4):402–408. 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 14.Bossen C, Ingold K, Tardivel A, et al. Interactions of tumor necrosis factor (TNF) and TNF receptor family members in the mouse and human. J Biol Chem. 2006;281(20):13964–13971. 10.1074/jbc.M601553200. [DOI] [PubMed] [Google Scholar]
- 15.Morony S, Warmington K, Adamu S, et al. The inhibition of RANKL causes greater suppression of bone resorption and hypercalcemia compared with bisphosphonates in two models of humoral hypercalcemia of malignancy. Endocrinology. 2005;146(8):3235–3243. 10.1210/en.2004-1583. [DOI] [PubMed] [Google Scholar]
- 16.Samadfam R, Xia Q, Goltzman D. Co-treatment of PTH with osteoprotegerin or alendronate increases its anabolic effect on the skeleton of oophorectomized mice. J Bone Miner Res. 2007;22(1):55–63. 10.1359/jbmr.060915. [DOI] [PubMed] [Google Scholar]
- 17.McClung MR, Wagman RB, Miller PD, Wang A, Lewiecki EM. Observations following discontinuation of long-term denosumab therapy. Osteoporos Int. 2017;28(5):1723–1732. 10.1007/s00198-017-3919-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Everts-Graber J, Reichenbach S, Gahl B, Ziswiler HR, Studer U, Lehmann T. Risk factors for vertebral fractures and bone loss after denosumab discontinuation: A real-world observational study. Bone. 2021;144(1):115830. 10.1016/j.bone.2020.115830. [DOI] [PubMed] [Google Scholar]
- 19.Brown JP, Dempster DW, Ding B, et al. Bone remodeling in postmenopausal women who discontinued denosumab treatment: off-treatment biopsy study. J Bone Miner Res. 2011;26(11):2737–2744. 10.1002/jbmr.448. [DOI] [PubMed] [Google Scholar]
- 20.Vasikaran S, Eastell R, Bruyère O, et al. Markers of bone turnover for the prediction of fracture risk and monitoring of osteoporosis treatment: a need for international reference standards. Osteoporos Int. 2011;22(2):391–420. 10.1007/s00198-010-1501-1. [DOI] [PubMed] [Google Scholar]
- 21.Mira-Pascual L, Patlaka C, Desai S, et al. A novel sandwich ELISA for tartrate-resistant acid phosphatase 5a and 5b protein reveals that both isoforms are secreted by differentiating osteoclasts and correlate to the type I collagen degradation marker CTX-I in vivo and in vitro. Calcif Tissue Int. 2020;106(2):194–207. 10.1007/s00223-019-00618-w. [DOI] [PubMed] [Google Scholar]
- 22.Janckila AJ, Yam LT. Biology and clinical significance of tartrate-resistant acid phosphatases: new perspectives on an old enzyme. Calcif Tissue Int. 2009;85(6):465–483. 10.1007/s00223-009-9309-8. [DOI] [PubMed] [Google Scholar]
- 23.Fu Q, Bustamante-Gomez NC, Reyes-Pardo H, et al. Reduced OPG expression by osteocytes may contribute to rebound resorption after denosumab discontinuation. JCI insight. 2023;8(18). 10.1172/jci.insight.167790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nenonen A, Cheng S, Ivaska KK, et al. Serum TRACP 5b is a useful marker for monitoring alendronate treatment: comparison with other markers of bone turnover. J Bone Miner Res. 2005;20(10):1804–1812. 10.1359/JBMR.050403. [DOI] [PubMed] [Google Scholar]
- 25.Mori Y, Kasai H, Ose A, et al. Modeling and simulation of bone mineral density in Japanese osteoporosis patients treated with zoledronic acid using tartrate-resistant acid phosphatase 5b, a bone resorption marker. Osteoporos Int. 2018;29(5):1155–1163. 10.1007/s00198-018-4376-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sølling AS, Harsløf T, Jørgensen NR, Langdahl B. Changes in RANKL and TRAcP 5b after discontinuation of denosumab suggest RANKL mediated formation of osteoclasts results in the increased bone resorption. Osteoporos Int. 2023;34(3):599–605. 10.1007/s00198-022-06651-0. [DOI] [PubMed] [Google Scholar]
- 27.Gossiel F, Ugur A, Peel N, Walsh J, Eastell R. The clinical utility of TRACP-5b to monitor anti-resorptive treatments of osteoporosis. Osteoporos Int. 2022;33(6):1357–1363. 10.1007/s00198-022-06311-3. [DOI] [PubMed] [Google Scholar]
- 28.Fassio A, Adami G, Benini C, et al. Changes in Dkk-1, sclerostin, and RANKL serum levels following discontinuation of long-term denosumab treatment in postmenopausal women. Bone. 2019;123(1):191–195. 10.1016/j.bone.2019.03.019. [DOI] [PubMed] [Google Scholar]
- 29.Fontalis A, Gossiel F, Schini M, Walsh J, Eastell R. The effect of denosumab treatment on osteoclast precursor cells in postmenopausal osteoporosis. Bone Reports. 2020;13(Supplement):100457. 10.1016/j.bonr.2020.100457. [DOI] [PubMed] [Google Scholar]
- 30.Kim AS, Girgis CM, McDonald MM. Osteoclast recycling and the rebound phenomenon following denosumab discontinuation. Curr Osteoporos Rep. 2022;20(6):505–515. 10.1007/s11914-022-00756-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kostenuik PJ, Nguyen HQ, McCabe J, et al. Denosumab, a fully human monoclonal antibody to RANKL, inhibits bone resorption and increases BMD in knock-in mice that express chimeric (murine/human) RANKL. J Bone Miner Res. 2009;24(2):182–195. 10.1359/jbmr.081112. [DOI] [PubMed] [Google Scholar]
- 32.Tremollieres F, Pouilles J-M, Ribot C. Withdrawal of hormone replacement therapy is associated with significant vertebral bone loss in postmenopausal women. Osteoporos Int. 2001;12(5):385–390. 10.1007/s001980170107. [DOI] [PubMed] [Google Scholar]
- 33.Naylor K, Clowes J, Finigan J, Paggiosi M, Peel N, Eastell R. The effect of cessation of raloxifene treatment on bone turnover in postmenopausal women. Bone. 2010;46(3):592–597. 10.1016/j.bone.2009.10.043. [DOI] [PubMed] [Google Scholar]
- 34.Eisman JA, Bone HG, Hosking DJ, et al. Odanacatib in the treatment of postmenopausal women with low bone mineral density: three-year continued therapy and resolution of effect. J Bone Miner Res. 2011;26(2):242–251. 10.1002/jbmr.212. [DOI] [PubMed] [Google Scholar]
- 35.McClung MR, Brown JP, Diez-Perez A, et al. Effects of 24 months of treatment with romosozumab followed by 12 months of denosumab or placebo in postmenopausal women with low bone mineral density: a randomized, double-blind, phase 2, parallel group study. J Bone Miner Res. 2018;33(8):1397–1406. 10.1002/jbmr.3452. [DOI] [PubMed] [Google Scholar]
- 36.Limaye A. Drishti: a volume exploration and presentation tool. Developments in X-ray Tomography VIII. SPIE, 2012;8506:191–199. 10.1117/12.935640. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data files can be provided on request.