

Abstract
Background
We aimed to correlate alterations in the rat sarcoma virus (RAS)/mitogen‐activated protein kinase pathway in vascular anomalies to the clinical phenotype for improved patient and treatment stratification.
Methods and Results
This retrospective multicenter cohort study included 29 patients with extracranial vascular anomalies containing mosaic pathogenic variants (PVs) in genes of the RAS/mitogen‐activated protein kinase pathway. Tissue samples were collected during invasive treatment or clinically indicated biopsies. PVs were detected by the targeted sequencing of panels of genes known to be associated with vascular anomalies, performed using DNA from affected tissue. Subgroup analyses were performed according to the affected genes with regard to phenotypic characteristics in a descriptive manner. Twenty‐five vascular malformations, 3 vascular tumors, and 1 patient with both a vascular malformation and vascular tumor presented the following distribution of PVs in genes: Kirsten rat sarcoma viral oncogene (n=10), neuroblastoma ras viral oncogene homolog (n=1), Harvey rat sarcoma viral oncogene homolog (n=5), V‐Raf murine sarcoma viral oncogene homolog B (n=8), and mitogen‐activated protein kinase kinase 1 (n=5). Patients with RAS PVs had advanced disease stages according to the Schobinger classification (stage 3–4: RAS, 9/13 versus non‐RAS, 3/11) and more frequent progression after treatment (RAS, 10/13 versus non‐RAS, 2/11). Lesions with Kirsten rat sarcoma viral oncogene PVs infiltrated more tissue layers compared with the other PVs including other RAS PVs (multiple tissue layers: Kirsten rat sarcoma viral oncogene, 8/10 versus other PVs, 6/19).
Conclusions
This comparison of patients with various PVs in genes of the RAS/MAPK pathway provides potential associations with certain morphological and clinical phenotypes. RAS variants were associated with more aggressive phenotypes, generating preliminary data and hypothesis for future larger studies.
Keywords: clinical characteristics, mosaicism, pathogenic variants, RAS/MAPK pathway, vascular anomalies
Subject Categories: Genetics, Genetics, Cell Signalling/Signal Transduction, Clinical Studies
Nonstandard Abbreviations and Acronyms
- BRAF
V‐Raf murine sarcoma viral oncogene homolog B
- CM
capillary malformation
- HRAS
Harvey rat sarcoma viral oncogene homolog
- KRAS
Kirsten rat sarcoma viral oncogene
- MAPK
mitogen‐activated protein kinase
- MAP2K1
mitogen‐activated protein kinase kinase 1
- NRAS
neuroblastoma ras viral oncogene homolog
- PI3K
phosphatidylinositol 3‐kinase
- PV
pathogenic variant
- RAS
rat sarcoma virus
- VM
venous malformation
Clinical Perspective.
What Is New?
This study reveals preliminary associations of rat sarcoma virus/V‐Raf murine sarcoma viral oncogene homolog B/mitogen‐activated protein kinase kinase 1 mosaicism with clinical phenotypes of extracranial vascular anomalies.
Lesions with Kirsten rat sarcoma viral oncogene pathogenic variants showed more aggressive infiltrative growth patterns across multiple tissue layers, whereas RAS variants were characterized by more advanced disease stages and more aggressive phenotypes.
What Are the Clinical Implications?
The potential benefit of combining clinical and genetic diagnostics may promote more individualized treatment regimens according to the underlying pathogenic variant, such as adding targeted therapeutics to multidisciplinary care.
Vascular anomalies comprise 2 large entities: vascular tumors and vascular malformations. They are currently categorized mainly on the basis of biological, histopathological, hemodynamic, and clinical findings, as provided by the International Society for the Study of Vascular Anomalies. 1 While vascular malformations are already present at birth and do not regress spontaneously, vascular tumors develop at different ages after birth and may regress on their own. Sporadic vascular malformations are congenital anomalies that are further classified into slow flow malformations (predominantly venous and lymphatic) as well as high‐flow arteriovenous malformations (AVMs). While isolated, nonsyndromic slow‐flow malformations in most cases follow a benign clinical course, AVMs are characterized by high morbidity, high progression rates, and thus challenging treatment, 2 , 3 , 4 as complete therapy is frequently not possible and the residual lesions continue to progress. The International Society for the Study of Vascular Anomalies classification was last updated in 2018 predominantly on the basis of novel descriptions of genes involved in the development of these rare lesions. More recently, rapidly increasing knowledge about the genetic and molecular basis of vascular malformations has transformed the understanding of the disease mechanisms, potentially allowing improved patient stratification with individualized treatment options. 5 It has been found that sporadic vascular malformations are often caused by somatic gain‐of‐function variants in genes encoding components of key cellular signaling pathways that regulate growth and differentiation. 6 The wide variability in the clinical phenotype is thought to depend on the cell type affected, the timing of the mutational event, and the degree and nature of pathway activation. In conditions caused by mutations affecting the phosphatidylinositol 3‐kinase/protein kinase B/mammalian target of rapamycin signaling pathway, genotype‐based stratification has led to early clinical trials of targeted therapies. 7 Somatic activating variants affecting the rat sarcoma virus (RAS)/mitogen‐activated protein kinase (MAPK) pathway were first described in intra‐ and extracranial, 8 , 9 , 10 and preliminary phenotypic correlations were reported. 11 , 12 , 13 The activation of certain signaling pathways not only occurs in vascular endothelial cells but similarly in adjacent soft tissue involving fibroblasts with altered extracellular matrix formation and increased extravascular inflammation. 14 While vascular malformations were formerly regarded as mere dysplastic diseases, the increasing evidence of activated signaling pathways, controlling growth and proliferation, suggests that vascular malformations instead are able to proliferate, which may in part explain the high progression rate reported after incomplete treatment. Therefore, it is currently under debate that malformations instead should be viewed as inborn malformative tumors, and that the dichotomous classification of vascular anomalies into tumors, on the one hand, and vascular malformations, on the other hand, is not accurate anymore. 15 Within this framework, more information is needed comparing the genotype of vascular anomalies with their associated phenotype. We present a cohort of vascular anomalies with mosaic pathogenic variants (PVs) in the V‐Raf murine sarcoma viral oncogene homolog B (BRAF), mitogen‐activated protein kinase kinase 1 (MAP2K1), and RAS genes detected by ultra‐deep next‐generation sequencing of affected tissue. Phenotypic and clinical associations including treatment response and recurrence are reported.
METHODS
The data sets used and analyzed during the current study are available from the corresponding author on reasonable request.
Primary Aim of the Study
This study aimed to correlate certain clinical phenotypes to genetic alterations in the RAS/MAPK pathway in a descriptive manner. Clinical features, 16 , 17 , 18 lesion extension, and progression rates are included as part of the phenotype.
Patient Cohort and Sample Collection
This retrospective multicenter cohort study was conducted by multidisciplinary vascular anomaly centers of 5 university hospitals in Germany, seeing a minimum of 150 new patients with vascular anomalies annually. Retrospective analyses were approved by the local ethics committee (University Hospital, LMU Munich), Project No. 23‐0337 (06/29/2023). All patients signed informed consent to participate. The Strengthening the Reporting of Genetic Association Studies 19 guidelines were used for appropriate reporting.
Core needle biopsies were collected between January 2019 to March 2023 under ultrasound guidance close to the center/nidus of the vascular lesion during invasive treatment or clinically indicated biopsies (before planned targeted therapy). Patients not receiving therapy do not routinely undergo biopsies for genetic testing at the participating institutions. Patients were eligible for this study, if a pathogenic variant in the RAS (Kirsten rat sarcoma viral oncogene [KRAS], Harvey rat sarcoma viral oncogene homolog [HRAS], neuroblastoma ras viral oncogene homolog [NRAS]), V‐Raf murine sarcoma viral oncogene homolog B (BRAF), or MAP2K1 genes was proven in fresh or formalin‐fixed paraffin‐embedded tissue biopsies from the vascular lesion. Exclusion criteria were clinical, pathological, or radiological doubts regarding the diagnosis of a vascular anomaly. Demographics, medical history, and clinical and radiological data, as well as treatment course and follow‐up assessments, were collected. Disease recurrence, when suspected clinically, was evaluated by magnetic resonance imaging. Increase or reoccurrence of clinical symptoms after treatment as well as increased lesion size or newly perfused vessels on magnetic resonance imaging compared with imaging findings after the last treatment was defined as progression.
DNA Extraction
Genomic DNA was extracted from native tissue samples and deparaffinated formalin‐fixed paraffin‐embedded samples using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) following the manufacturer's instructions. DNA concentration was measured using a Qubit 4 fluorometer (Invitrogen, Carlsbad, CA) and adjusted according to the requirements of subsequent experiments.
Mosaic Variant Screening
Methods for mosaic variant detection evolved during the course of this project. Initially, Sanger sequencing of KRAS, HRAS, NRAS, BRAF, and MAP2K1 hotspot exons was used, reaching a mosaic detection threshold of 10% to 15% variant allele frequency. To this end, mutation hotspot exons with flanking introns were amplified by polymerase chain reaction (PCR), and bidirectional Sanger sequencing was performed using a Big Dye Terminator Cycle Sequencing Kit on a 3500xl Genetic Analyzer (Applied Biosystems, Foster City, CA). Sequences were aligned using the Seqpilot analysis software (JSI Medical Systems, Kippenheim, Germany). Mosaic level for mutations was estimated by comparing the area under the curve of electropherograms for the wild type and mutant peaks in the forward and reverse sequencing directions.
The majority of samples were investigated by ultra‐deep sequencing of targeted multigene panels. This was carried out by paired‐end sequencing with 2×151 bp reads on a MiSeq system or NextSeq 550 system (Illumina, San Diego, CA) to obtain for each sample an average of 2.5M or 20M reads, respectively. Different target enrichment kits either with or without molecular barcoding by unique molecular identifiers were used during the course of this project: Illumina TruSeq Custom Amplicon Panel; Agilent SureSelect XT HS2 DNA Custom Panel with unique molecular identifiers (random 3‐bp duplex) (Agilent Technologies, Santa Clara, CA); Twist EF Custom Library Prep 2.0 of a Twist Custom Panel with Twist UMI Adapter (fixed 5‐bp or 6‐bp duplex) (Twist Bioscience, South San Francisco, CA). The target sequence comprised a panel of genes/gene hotspots that are known to be involved in vascular malformations or regional overgrowth. The gene content evolved over time (a complete list of genes/gene hotspots covered by the employed enrichment kit is available in Table S1). Library preparation was performed according to the respective manufacturer's instructions. Indexed sample libraries were equimolarly pooled for final multiplexed sequencing. Samples that underwent initial screening by Sanger sequencing and remained negative were not systematically reassessed by next‐generation sequencing later. Therefore, we may have missed cases with RAS/MAPK pathway mosaic variants, because variant allele frequencies for causative variants in mixed‐tissue DNA samples from vascular malformations are often <15%. However, as only mutation‐positive cases are reported and analyzed in this article, the results and conclusions are unlikely to be affected by false‐negative cases in the baseline cohort.
Genomic Data Analyses
Raw data (bcl format, binary base call sequence files) were uploaded to the varvis software package (Limbus Medical Technologies GmbH, Rostock, Germany) and processed (demultiplexing, read alignment, error correction) using the GRCh37 reference genome and the bioinformatics pipeline from varvis software version 1.23.3 with the manufacturer's standard settings. For unique molecular identifiers analysis (duplex sequencing data processing), duplex barcode sequences were extracted from the read sequence according to the manufacturer's user manual (Agilent SureSelect XT HS2 DNA Kits Protocol or Twist EF Library Preparation Kit, respectively) and reads were aligned to the reference genome. A minimum of 2 reads were required to define a strand‐specific consensus read. Strand‐specific consensus reads were then combined to create a final consensus read. Variant consensus reads were called down to a minimum variant allele frequency of 0.5% with a minimum of 2 aberrant consensus reads, thereby reaching a detection threshold for mosaic variants of at least 0.5% for most samples. The target regions typically had a mean sequencing depth of >2000× after demultiplexing, except for DNA samples from formalin‐fixed paraffin‐embedded tissue, which yielded variable but usually lower coverage.
Variant Confirmation By Digital PCR
Variants with an allele frequency close to the detection threshold were verified by digital PCR. Digital PCR was performed on a QIAcuity Four Digital PCR System (Qiagen, Hilden, Germany) on 26 k nanoplates using the QIAcuity Probe PCR Kit and digital PCR LNA Mutation Assays (QIAGEN GmbH, Hilden, Germany) containing specific probes for wild type as well as mutant alleles. Data analysis was performed using the QIAcuity Software Suite 2.1.8.23. The detection limit for the target variants was 0.1%.
Statistical Analysis
To analyze age and follow‐up time, the data were presented as median (range, minimum–maximum). Subgroup analyses were performed related to the affected genes. We used Fisher's exact test for categorial data and small sample sizes to assess the association of the different PV carriers with clinical phenotypes defined as lesion localization, lesion tissue involvement, Cho classification, 16 Schobinger classification, 17 extremity length discrepancy (yes/no), and associated segmental overgrowth (yes/no), as well as with therapy response defined as progression (yes/no). Analysis was conducted using SPSS version 26.0 (IBM Corp., Armonk, NY); all P values reported were 2‐tailed.
RESULTS
Patient Characteristics and Clinical Presentation
All patients included presented with clinically and radiologically confirmed extracranial vascular anomalies, had a somatic mutation in genes of the RAS pathway, and received multimodal treatment according to the anomaly type, clinical presentation, and patient preference (embolization, sclerotherapy, surgery, targeted medical therapy). Twenty‐nine patients were included in the study (Table 1) with a median age of 20 years (range, 5–55 years) at the time of genetic testing. The cohort consisted of 18 of 29 (62.1%) women and 11/29 (37.9%) men. All patients had extracranial vascular anomalies, 25 of 29 (86.2%) patients had vascular malformations, and 3 of 29 (10.3%) patients had benign vascular tumors, according to the International Society for the Study of Vascular Anomalies classification. 20 One patient (1/29, 3.4%) presented with both a vascular malformation (capillary malformation [CM]) as well as an additional vascular tumor (pyogenic granuloma), which was located on the CM. The mean age of the patients at clinical manifestation of the lesions was 2 years (range, 0–39 years). Vascular anomalies were located on the lower extremities in 11 of 29 patients (37.9%), followed by the head and neck region (5/29, 17.2%), the upper extremities (4/29, 13.8%), and the trunk (4/29; 13.8%). Five extensive lesions (5/29, 17.2%) involved the trunk as well as upper or lower extremities. Among all cases with vascular malformations, 24 of 26 (92.3%) patients presented simple or combined AVMs, while there was 1 of 26 (3.8%) simple venous malformation (VM) as well as 1 of 26 (3.8%) CM (Table 1). The distribution of AVMs according to the clinical Schobinger classification (n=24) was as follows: stage 1 (2/24, 8.3%), stage 2 (10/24, 41.7%), stage 3 (10/24, 41.7%), and stage 4 (2/24, 8.3%). AVMs were further classified angiographically according to the Cho classification (n=24): type I (1/24, 4.2%), type II (1/24, 4.2%), type IIIa (12/24, 50.0%), and type IIIb (10/24, 41.7%).
Table 1.
Patient Characteristics and Clinical Presentation of Study Cohort
| Patient no. | Age, y | Sex | Type of vascular anomaly | Subtype of vascular lesion | Tissue involvement | Lesion localization | Segmental overgrowth | Extremity length discrepancy | CNS occupancy | Angiographic classification (Cho*/Puig†) | Schobinger classification‡ | Treatment | Progression |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 5 | m | Vascular malformation | Combined (CVAVM) | (Sub)cutaneous | LE | Ipsilateral | IIIa* | 2 | Embolization | Yes | ||
| 2 | 34 | m | Vascular malformation | Arteriovenous | Multiple layers | LE | Ipsilateral | Yes | IIIa* | 3 | Embolization+resection | Yes | |
| 3 | 22 | w | Vascular malformation | Arteriovenous | Multiple layers | UE | IIIb* | 3 | Embolization | Yes | |||
| 4 | 54 | m | Vascular malformation | Arteriovenous | Multiple layers | LE | Ipsilateral | Yes | II* | 3 | Embolization+resection | Yes | |
| 5 | 17 | w | Vascular tumor | Benign | Multiple layers | Trunk+LE | … | … | Embolization | No | |||
| 6 | 26 |
w |
Vascular malformation |
Arteriovenous |
Multiple layers | HN | IIIb* | 3 | Embolization+targeted therapy (VEGF inhibitor+MEK inhibitor) | No | |||
| 7 | 17 | w | Vascular malformation | Syndromic (Parkes–Weber‐like phenotype) | Multiple layers | LE | Ipsilateral | Yes | IIIa* |
3 |
Sclerotherapy+embolization | Yes | |
| 8 | 21 | w | Vascular malformation | Arteriovenous (IFFVA) | Intramuscular | UE | Ipsilateral | IIIa* | 2 | None | NA | ||
| 9 | 55 | m | Vascular malformation | Arteriovenous | Multiple layers | Trunk+UE | Ipsilateral | IIIb* | 3 | Embolization+resection | Yes | ||
| 10 | 34 | w | Vascular malformation | Arteriovenous | Multiple layers | LE | Ipsilateral | IIIb* | 4 | Embolization | NA | ||
| 11 | 14 | w | Vascular malformation | Combined (CAVM) | Multiple layers | LE | Ipsilateral | Yes | IIIa* | 3 | Embolization | Yes | |
| 12 | 17 | m | Vascular malformation | Combined (CVAVM) | Multiple layers | LE | Ipsilateral | I* | 2 | Embolization | Yes | ||
| 13 | 20 | w | Vascular malformation | Arteriovenous | Intramuscular | UE | Yes | IIIb* | 3 | Embolization | Yes | ||
| 14 | 25 | m | Vascular tumor | Benign | Intramuscular | Trunk | Ipsilateral | … | … | Embolization+resection | Yes | ||
| 15 | 20 | m | Vascular malformation | Arteriovenous (IFFVA) | Intramuscular | Trunk | Ipsilateral | IIIb* | 2 | Resection | No | ||
| 16 | 14 | w | Vascular malformation (1)+vascular tumor (2) | CM (1) +benign (Pyogenic granuloma within CM; 2) | (Sub)cutaneous | Trunk | … | … | None | NA | |||
| 17 | 2 | w | Vascular malformation | Arteriovenous | Intramuscular | HN | IIIa* | 2 | Embolization | No | |||
| 18 | 17 | w | Vascular malformation | Arteriovenous | Intramuscular | LE | IIIa* | 3 | Embolization+resection | No | |||
| 19 | 4 | w | Vascular tumor | Benign | (Sub)cutaneous | Trunk | … | … | Embolization+resection | No | |||
| 20 | 10 | w | Vascular malformation | Arteriovenous | Multiple layers | LE | IIIa* | 3 | Targeted therapy (BRAF inhibitor) | No | |||
| 21 | 2 | m | Vascular malformation | Syndromic (Parkes–Weber‐like phenotype) | Multiple layers | Trunk+LE | Ipsilateral | Yes | IIIb* | 1 | None | NA | |
| 22 | 33 | w | Vascular malformation | Arteriovenous | Intramuscular | LE | Ipsilateral | Yes | IIIa* | 2 | Embolization | NA | |
| 23 | 50 | w | Vascular malformation | Arteriovenous | (Sub)cutaneous | Trunk+LE | IIIb* | 1 | Embolization | No | |||
| 24 | 35 | w | Vascular malformation | Venous | (Sub)cutaneous | UE | 1+ | … | Resection | Yes | |||
| 25 | 25 | m | Vascular malformation | Arteriovenous | Multiple layers | LE | IIIb* | 2 | Embolization | No | |||
| 26 | 25 | w | Vascular malformation | Arteriovenous | Multiple layers | Trunk+UE | Ipsilateral | Yes | IIIa* | 4 | Embolization+targeted therapy (MEK inhibitor) | No | |
| 27 | 20 | m | Vascular malformation | Arteriovenous | (Sub)cutaneous | HN | Ipsilateral | IIIb* | 2 | Embolization+resection | No | ||
| 28 | 21 | m | Vascular malformation | Arteriovenous | (Sub)cutaneous | HN | IIIa* | 2 | Embolization+resection | Yes | |||
| 29 | 16 | w | Vascular malformation | Arteriovenous | (Sub)cutaneous | HN | IIIa* | 2 | Embolization+resection | No |
CAVM indicates capillary arteriovenous malformation; CM, capillary malformation; CNS, central nervous system; CVAVM, capillary venous arteriovenous malformation; HN, head and neck; IFFVA, intramuscular fast‐flow vascular anomaly; LE, lower extremity; m, man; MEK, mitogen‐activated protein kinase kinase enzymes; NA, not available; UE, upper extremity; VEGF, vascular endothelial growth factor; and w, woman.
Cho classification according to Cho et al. 16
Puig classification according to Puig et al. 18
Schobinger classification according to Finn et al. 17
Treatment Course and Follow‐Up
In 23 of 29 (79.3%) cases, the vascular anomalies were treated by minimally invasive procedures (AVM embolization, VM sclerotherapy), in 11 of 29 (37.9%) cases a surgical resection was performed, and in 3 of 29 (10.3%) cases, patients received targeted medical therapies. The median follow‐up time was 23 months (range, 8–37 months). Overall, 12 of 24 (50.0%) patients experienced a progression in the clinical course after treatment. The individual treatment modalities and progression rates, as part of the phenotypic characterization, are described in detail in Table 1.
Mutational Spectrum in Extracranial Vascular Anomalies
The identified PVs involved RAS genes in 16 of 29 (55.2%) cases (see Figure 1): KRAS (10/29, 34.5%), HRAS (5/29, 17.2%), and NRAS (1/29, 3.4%). We also found PVs in BRAF (8/29, 27.6%; see Figure 2) and MAP2K1 (5/29, 17.2%; see Figure 3). Variant allele frequencies in DNA from lesional tissue samples ranged from 1% to 30% (Table 2). We identified 17 distinct PVs: 5 in KRAS, 5 in HRAS, 1 in NRAS, 2 in BRAF, and 4 in MAP2K1. Four PVs (c.1799T>A, p.Val600Glu in BRAF; c.35G>A, p.Gly12Asp in KRAS; c.183A>C, p.Gln61His in KRAS; c.171G>T, and p.Lys57Asn in MAP2K1) were recurrently observed in our cohort in 7/29 (24.1%), 5 of 29 (17.2), 2 of 29 (6.9%), and 2 of 29 (6.9%) patients, respectively. All other PVs were only identified once. None of the identified PVs was detected in leukocyte DNA or in control tissue samples, when available.
Figure 1. Patient 1: 20‐year‐old female patient with arteriovenous malformation (AVM) paravertebral and a HRAS pathogenic variant (PV).

A, Axial T1‐weighted turbo spin‐echo sequence image presenting an AVM (asterisk) located paravertebrally on the left side with intramuscular extension and involvement of the spinal canal and compression of the dural tube. Digital subtraction image before (B) and after (C) the first session of embolotherapy with ethylene‐vinyl‐alcohol copolymer (Squid‐18, BALT Germany GmbH). C, The image shows near complete embolization of the lesion (2020). D, Axial T1‐weighted turbo spin‐echo sequence image after 3 sessions of embolization presenting newly perfused vessels confirming clinically suspected progression, which was characterized by increasing pain and functional movement impairment (2023). Preprocedural (E) and periprocedural (F) digital subtraction images of the fourth embolotherapy session, both showing the newly embolized vascularized components of the progressive AVM (2023).
Figure 2. Patient 27: 20‐year‐old male patient with an infiltrative phenotype and a MAP2K1 pathogenic variant (PV).
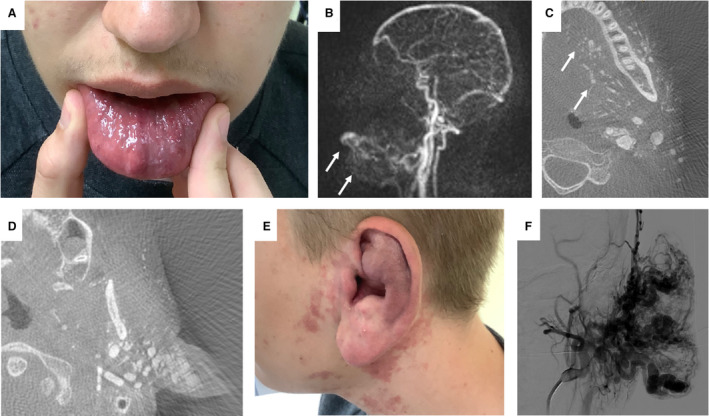
A, Clinical infiltration of the lip; notice the prominent blood vessels on the left side of the lower lip. B, Time‐resolved angiography with interleaved stochastic trajectories showing contrast enhancement of the lip and around the mandible. C and D, Contrast‐enhanced vessels in computed tomography reconstruction of the digital subtraction angiography. Notice the fine vessels infiltrating the tongue and the left side of the face, most prominent at the base of the ear. E, Correlating to the prominent vessels at the base of the ear is the enlarged ear, including the ear lobe. F, Digital subtraction angiography of the ear, demonstrating 3 large feeders into the outer ear.
Figure 3. Patient 20: 10‐year‐old female patient with an infiltrative arteriovenous malformation and a BRAF pathogenic variant (PV).
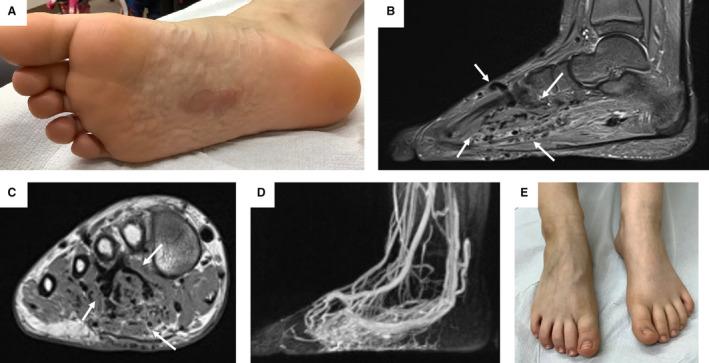
A, Clinical manifestation with capillary malformation (CM) of the sole of the right foot; also note the wormlike mass and dilated veins. B and C, Sagittal T2‐weighted turbo‐inversion recovery‐magnitude and coronal T1‐weighted turbo spin‐echo sequence images; note the flow voids in deeper tissues of the foot (labeled with white arrows). D, Time‐resolved angiography with interleaved stochastic trajectories angiography showing the marked hyperperfusion of the foot including cutaneous tissue. E, Correlating to the hyperperfusion, dilated veins can be seen on the back of the affected right foot of the patient.
Table 2.
Mutational Spectrum and Genotypic Characterization of Study Cohort
| Patient no. | Gene | cDNA; amino acid change | Source | Technology | Mutant allele fraction, % | Mutant allele count | Total allele count | dPCR confirmation, % |
|---|---|---|---|---|---|---|---|---|
| 1 | KRAS | c.35G>A; p.(Gly12Asp) | Native tissue (2) | Ultra‐deep NGS |
14.2 8.6 |
661 210 |
4661 2456 |
9.4 7.5 |
| 2 | KRAS | c.34G>T; p.(Gly12Cys) | Native tissue | Ultra‐deep NGS | 8.7 | 47 | 540 | 10.5 |
| 3 | KRAS | c.35G>A; p.(Gly12Asp) | Native tissue | Ultra‐deep NGS+UMI | 8.2 | 104 | 1273 | 5.4 |
| 4 | KRAS | c.183A>C; p.(Gln61His) | Native tissue | Ultra‐deep NGS+UMI | 6.7 | 135 | 2024 | 5.7 |
| 5 | KRAS | c.182A>G; p.(Gln61Arg) | Native tissue | Ultra‐deep NGS | 7.6 | 731 | 9610 | 7.6 |
| 6 | KRAS | c.183A>C; p.(Gln61His) | Native tissue | Ultra‐deep NGS+UMI | 9.4 | 106 | 1130 | 7.7 |
| 7 | KRAS | c.35G>A; p.(Gly12Asp) | Native tissue | Sanger NGS | 4.5 | 73 | 1619 | NA |
| 8 | KRAS | c.188_229dup; p.(Glu63_Glu76dup) | Native tissue | Ultra‐deep NGS+UMI | 4.7 | 122 | 2500 | NA |
| 9 | KRAS | c.35G>A; p.(Gly12Asp) | FFPE tissue | Ultra‐deep NGS+UMI | 4.5 | 76 | 1703 | 4.0 |
| 10 | KRAS | c.35G>A; p.(Gly12Asp) | Native tissue | Ultra‐deep NGS+UMI | 8.7 | 148 | 1698 | 11.4 |
| 11 | HRAS | c.172_177delinsGTCCTGGATGTT; p.(Thr58_Ala59delinsValLeuAspVal) | Native tissue | Ultra‐deep NGS | 6.7 | 37 | 535 | NA |
| 12 | HRAS | c.191_217dup; p.(Met72_Arg73insHisSerAlaMetArgAspGlnTyrMet) | Native tissue | Ultra‐deep NGS+UMI | 15.0 | 322 | 2146 | NA |
| 13 | HRAS | c.172_179delins; p.(Thr58_Gly60delinsValLeuAspValLeu) | FFPE tissue | Ultra‐deep NGS+UMI | 6.9 | 162 | 2331 | NA |
| 14 | HRAS | c.215_216insTTCCAGCGCCATGCGGGACCAGTACAT; p.(Tyr71_Met72insIleSerSerAlaMetArgAspGlnTyr) | Native tissue | Ultra‐deep NGS+UMI | 7.0 | 113 | 1614 | NA |
| 15 | HRAS | c.217_218ins27; p.(Met72_Arg73insProSerAlaMetArgAspGlnTyrMet) | FFPE tissue | Ultra‐deep NGS+UMI | 11.0 | 118 | 1077 | NA |
| 16 | NRAS | c.183A>T; p.(Gln61His) | Native tissue | Ultra‐deep NGS | 7.7 | 544 | 7031 | NA |
| 17 | BRAF | c.1517+2_1517+3insTACTCAGGT; p.? | Native tissue | Ultra‐deep NGS+UMI | 23.0 | 631 | 2744 | NA |
| 18 | BRAF | c.1799T>A; p.(Val600Glu) | Native tissue | Ultra‐deep NGS | 2.8 | 343 | 12 296 | 3.7 |
| 19 | BRAF | c.1799T>A; p.(Val600Glu) | FFPE tissue | Ultra‐deep NGS | 30.3 | 5118 | 16 913 | 37.0 |
| 20 | BRAF | c.1799T>A; p.(Val600Glu) | Native tissue | Sanger | 23 | … | … | 15.3 |
| 21 | BRAF | c.1799T>A; p.(Val600Glu) | Native tissue | Ultra‐deep NGS+UMI | 5.7 | 94 | 1656 | 5.3 |
| 22 | BRAF | c.1799T>A; p.(Val600Glu) | Native tissue | Ultra‐deep NGS+UMI | 10.4 | 191 | 1828 | 12.8 |
| 23 | BRAF | c.1799T>A; p.(Val600Glu) | Native tissue | Ultra‐deep NGS+UMI | 4.0 | 71 | 1754 | 7.2 |
| 24 | BRAF | c.1799T>A; p.(Val600Glu) | Native tissue | Ultra‐deep NGS | 9.5 | 218 | 2287 | NA |
| 25 | MAP2K1 | c.171G>T; p.(Lys57Asn) | Native tissue (2) | Sanger |
16 18 |
… | … | NA |
| 26 | MAP2K1 | c.169_170delinsCC; p.(Lys57Pro) | Native tissue (2) | Sanger |
16 20 |
… | … | NA |
| 27 | MAP2K1 | c.167A>C; p.(Gln56Pro) | Native tissue | Ultra‐deep NGS+UMI | 10.9 | 21 | 192 | NA |
| 28 | MAP2K1 | c.171G>T; p.(Lys57Asn) | Native tissue | Ultra‐deep NGS+UMI | 9.7 | 474 | 4886 | NA |
| 29 | MAP2K1 | c.171_185del; MAP2K1; p.(Gln58_Glu62del) | Native tissue | Ultra‐deep NGS+UMI | 0.9 | 15 | 1588 | NA |
dPCR indicates digital polymerase chain reaction; FFPE, formalin‐fixed paraffine‐embedded tissue; FT, fresh tissue; NA, not available; NGS, next‐generation sequencing; and UMI, unique molecular identifiers.
Genotype–Phenotype Associations
A comparison of clinical characteristics between patients with RAS, BRAF, and MAP2K1 PVs is shown in Table 3. Patients with RAS PVs had a higher score according to Schobinger classification (3.0 [range, 2–4] versus 2.0 [range, 1–3] versus 2.0 [range, 2–4]; P=0.037) and progression after treatment was more frequently observed (76.9% versus 16.7% versus 20.0%; P=0.024). There were no remarkable differences in the prevalence of clinical manifestations such as leg length discrepancy, associated segmental overgrowth or in the angiographic classification according to Cho (see Table 3). In addition, there was no evident association between lesion localization and the different genotypes (see Table 3).
Table 3.
Comparison of Clinical Characteristics Among the RAS, BRAF, and MAP2K1 PV Carriers
| Clinical characteristics | RAS | BRAF (n) | MAP2K1 | P value‡ |
|---|---|---|---|---|
| Age, y | n=16 | n=8 | n=5 | |
| Median (range) | 20 (5–55) | 14 (2–50) | 21 (16–24) | |
| Lesion localization, n (%) | n=16 | n=8 | n=5 | 0.313 |
| Lower extremity | 7 (43.8) | 3 (37.5) | 1 (20.0) | |
| Upper extremity | 3 (18.8) | 1 (12.5) | 0 (0.0) | |
| Trunk | 3 (18.8) | 1 (12.5) | 0 (0.0) | |
| Trunk+lower extremity | 1 (6.3) | 2 (25.0) | 0 (0.0) | |
| Trunk+upper extremity | 1 (6.3) | 0 (0.0) | 1 (20.0) | |
| Head and neck | 1 (6.3) | 1 (12.5) | 3 (60.0) | |
| Cho classification for AVMs*, n (%) | 0.960 | |||
| I | 1 (7.7) | 0 (0.0) | 0 (0.0) | |
| II | 1 (7.7) | 0 (0.0) | 0 (0.0) | |
| IIIa | 5 (38.5) | 4 (66.7) | 3 (60.0) | |
| IIIb | 6 (46.2) | 2 (33.3) | 2 (40.0) | |
| Schobinger classification for AVMs,† n (%) | n=13 | n=6 | n=5 | 0.037 |
| 1 | 0 (0.0) | 2 (33.3) | 0 (0.0) | |
| 2 | 4 (30.8) | 2 (33.3%) | 4 (80.0) | |
| 3 | 8 (61.5) | 2 (33.3) | 0 (0.0) | |
| 4 | 1 (7.7) | 0 (0.0) | 1 (20.0) | |
| Extremity length discrepancy, n (%) | n=16 | n=8 | n=5 | 1.000 |
| Yes | 4 (25.0) | 2 (25.0) | 1 (20.0) | |
| No | 12 (75.0) | |||
| Associated segmental overgrowth, n (%) | n=16 | n=8 | n=5 | 0.117 |
| Yes | 11 (68.8) | 2 (25.0) | 2 (40.0) | |
| No | 5 (31.3) | 6 (75) | 3 (60.0) | |
| Progression, n (%) | n=13 | n=6 | n=5 | 0.024 |
| Yes | 10 (76.9) | 1 (16.7) | 1 (20.0) | |
| No | 3 (23.1) | 5 (83.3) | 4 (80.0) |
AVM indicates arteriovenous malformation; BRAF, V‐Raf murine sarcoma viral oncogene homolog B; MAP2K1, mitogen‐activated protein kinase kinase 1; PV, pathogenic variant; and RAS, rat sarcoma virus.
Cho classification according to Cho et al. 16
Schobinger classification according to Finn et al. 17
Fisher's exact test for categorial data.
Lesions with KRAS PVs infiltrated multiple tissue layers more frequently than subcutaneous or muscular tissue only compared with the other PVs (KRAS, 80.0% versus HRAS, 40.0% versus NRAS, 0.0% versus BRAF, 25.0% versus MAP2K1, 40.0%; P=0.036; see Table 4).
Table 4.
Comparison of Tissue Involvement Among the KRAS, HRAS, NRAS, BRAF, and MAP2K1 PV Carriers
| Clinical characteristics | KRAS | HRAS | NRAS | BRAF | MAP2K1 | P value† |
|---|---|---|---|---|---|---|
| Age, y | n=10 | n=5 | n=1 | n=8 | n=5 | |
| Median (range) | 24 (5–55) | 17 (14–20) | 14 (2–50) | 21 (16–24) | ||
| Lesion tissue involvement*, n (%) | n=10 | n=5 | n=1 | n=8 | n=5 | 0.036 |
| (Sub)cutaneous | 1 (10.0) | 0 (0.0) | 1 (100.0) | 3 (37.5) | 3 (60.0) | |
| Intramuscular | 1 (10.0) | 3 (60.0) | 0 (0.0) | 3 (37.5) | 0 (0.0) | |
| Multiple layers 1 | 8 (80.0) | 2 (40.0) | 0 (0.0) | 2 (25.0) | 2 (40.0) |
BRAF, V‐Raf murine sarcoma viral oncogene homolog B; HRAS, Harvey rat sarcoma viral oncogene homolog; MAP2K1, mitogen‐activated protein kinase kinase 1; NRAS, neuroblastoma RAS viral oncogene homolog; and PV, pathogenic variant.
Multiple tissue layers include (sub)cutaneous, muscular, and osseous involvement of the vascular anomaly.
Fisher's exact test for categorial data.
DISCUSSION
This retrospective multicenter cohort study describes potential correlations between mosaic‐activating PVs in 5 genes of the RAS/MAPK pathway (KRAS, HRAS, NRAS, BRAF, and MAP2K1) and phenotypic characteristics. Lesions with KRAS PVs showed a more aggressive infiltrative growth pattern across multiple tissue layers. Furthermore, RAS variants were characterized by more advanced disease stages and higher progression rates. No differences were observed in the angiographic classification of lesions or in the prevalence of associated disease manifestations such as leg length discrepancy and segmental overgrowth.
Initially, KRAS PVs in vascular anomalies were mainly described in intracranial vascular malformations 9 , 21 , 22 but more recent studies have increasingly reported them in extracranial lesions. 8 , 11 , 23 Al‐Olabi et al 8 found KRAS PVs in 4 of 135 patients with slow‐flow malformations and in 3 of 25 patients with extracranial AVMs. In intracranial AVMs, KRAS PVs are mostly found at the hotspot in codon 12 (G12V and G12D), which is also frequently mutated in cancer. In accordance with this, 50% of KRAS PVs in our study were c.35G>A, p.Gly12Asp (G12D). In 2 cases, we found the KRAS PV c.183A>T, p.Gln61His (Q61H). This PV was reported by El Sissy et al 11 as the most frequent distinct variant (67%) in their cohort consisting of 6 extracranial AVMs with KRAS PVs. Various other KRAS PVs have been reported in vascular anomalies. Ten Broek et al 13 presented 8 different variants in a cohort with 9 KRAS PVs, which were found in various types of vascular malformations. In addition to the most prevalent KRAS variants, we identified 5 HRAS PVs and 1 NRAS PV that were nonrecurrent in our cohort. Notably, all observed HRAS PVs represented in‐frame deletions/insertions affecting the switch‐II region (amino acids 58–76) of the HRAS protein. Eijkelenboom et al 24 presented five HRAS in frame insertions in that region in patients with vascular malformations/overgrowth syndromes and provided functional data that showed the inability of guanine nucleotide exchange factors to induce GTP loading and reduced intrinsic and GTPase‐activating protein–stimulated GTP hydrolysis of mutant HRAS proteins. These opposing effects led to a net increase in MAPK activation in a cellular model system and were supposed to cause decoupling from activating upstream cellular signals. Our observations provide further evidence that variants of that particular type are characteristic of HRAS PVs associated with vascular anomalies, which often present as atypical vascular malformation/overgrowth syndromes. The HRAS PV c.172_177delinsGTCCTGGATGTT, p.Thr58_Ala59delinsValLeuAspVal, which was found in 1 of our patients, has previously been reported by Konczyk et al 25 in a patient with facial AVM and associated adipose tissue overgrowth of the right cheek. Our patient with this distinct variant presented with an extensive AVM of the lower extremity, associated capillary lesions, leg length discrepancy, and segmental overgrowth, as well as recurrent pain (patient 11). There was no difference observed between the patients with HRAS versus KRAS PVs in our cohort regarding the occurrence of clinical characteristics, such as segmental overgrowth. Consistent with our findings, NRAS PVs tend to be in the minority in most cohorts reporting vascular anomalies and RAS variants. 8 , 26 In the literature, 1 distinct NRAS PV, c.182A>G, p.Gln61Arg (Q61R), was recurrently identified in complex lymphatic anomalies, such as generalized lymphatic anomaly or kaposiform lymphangiomatosis. 27 , 28 , 29 , 30 However, the NRAS PV affecting the same codon, c.183A>T, p.Gln61His, was confirmed in 1 patient presenting with a CM on the costal arch and a vascular tumor (pyogenic granuloma) located on this CM. Consistent with this, Ten Broek et al 13 described 1 case with both multiple eruptive pyogenic granulomas and a CM with the same NRAS PV. In addition, a cohort of patients with pyogenic granuloma was reported, in which 1 NRAS PV was found, while most of pyogenic granulomas contained GNAQ PVs. 31 Interestingly, NRAS PVs have not been reported in AVMs so far, either in our series or in the literature. Concerning all 16 cases with RAS PVs in our cohort, we noticed that these anomalies presented mainly with complex phenotypes, such as extensive combined vascular malformations or associated segmental overgrowth (68%). Additionally, among these 16 cases, there were 2 intramuscular hemangioma‐like anomalies that may have overlapping clinical features with AVMs (intramuscular fast‐flow vascular anomaly; patients 12 and 15), similar to patients described by Goss et al 32 and Sudduth et al. 33
In general, our results suggest that RAS variants are characterized by more advanced disease stage according to Schobinger and higher progression rates (69%), which may pave the way for individualized and intensified multimodal treatment regimens according to the underlying PVs. In a cohort of 18 extracranial AVMs, El Sissy et al 11 also found a higher progression rate of the 6 patients with KRAS PVs compared with 7 patients with MAP2K1 PVs. This finding is therefore now being replicated in a second cohort. Further, we found, that lesions with KRAS PVs tend to present a more aggressive infiltrative growth pattern across multiple tissue layers, which has not yet been established in the literature. However, these preliminary findings should be interpreted with caution due to the small sample sizes. If confirmed in further studies, this may also have an impact on patient stratification and may further shift the choice of therapy options.
BRAF PVs have previously been detected in tissue samples from various intra‐ and extracranial vascular anomalies including fast‐flow malformations, isolated slow‐flow malformations, and vascular tumors such as pyogenic granulomas. 8 , 11 , 34 , 35 In these cohorts, all reported cases presented the BRAF PV, c.1799T>A, p.Val600Glu, which was also the predominating BRAF PV in our study. Additionally, in 1 case (patient 17), we identified a novel mosaic BRAF variant, c.1517+2_1517+3insTACTCAGGT, that predicts the insertion of 3 amino acids in the protein p.Arg506_Lys507insLeuLeuArg. Comparing the clinical phenotype associated with the few cases where BRAF PVs have been previously described in the literature, 8 , 11 , 34 , 35 8 patients with BRAF PVs in our cohort showed similarly variable clinical phenotypes including AVMs of limited extension (patients 17, 18, 22, and 23), a more extensive AVM with multiple‐layer tissue involvement (patient 20), a complex combined Parkes–Weber‐like phenotype (patient 21), a subungual VM (patient 24), and a diffuse infantile fibromatosis (patient 19). Associated segmental overgrowth and multiple tissue layer involvement were observed in 25% each. The latter was less prevalent compared with KRAS PVs, which may indicate better and less challenging conditions for surgical or interventional treatment approaches. While the nature of vascular anomalies varied, patients had less severe symptoms and a lower progression rate compared with those with RAS PVs.
MAP2K1 PVs were found in 5 cases (17%) of our cohort with the recurrent MAP2K1 PV, c.171G>T, p.Lys57Asn, found twice, while 3 other MAP2K1 PVs were identified in single cases. These PVs were all missense or small in‐frame deletions affecting amino acid residues adjacent to or within the protein's negative regulatory domain and have been previously reported in an AVM cohort by Couto et al. 12 Several of these variants have been found in cancers and shown to increase MAP2K1 activity. 36 , 37 In our cohort, patients with MAP2K1 PVs all had AVMs, which is in contrast to the RAS and BRAF PVs including a broader clinical spectrum of vascular anomalies. A certain selection bias for this observation cannot be excluded, and this observation needs yet to be evaluated in other cohorts. Lesions with MAP2K1 PVs were classified as Schobinger stage II in 80% and were accompanied by segmental or local overgrowth in 20%. Similar to the BRAV PVs in our cohort, MAP2K1 PVs presented lower progression rates (20%) compared with KRAS PVs. El Sissy et al 11 reported comparable results regarding differences in Schobinger classification and progression rates after treatment (29% versus 100%) among 7 patients with MAP2K1 and 6 patients with KRAS PVs in facial AVMs. At this preliminary state of knowledge, these results should be interpreted with the limitation that small group sizes were analyzed in our cohort as well as the cohorts reported previously.
RAS, BRAF, or MAP2K1 have long been implicated in various malignancies, in which they play a driving role in tumor growth. This knowledge has led to the development of targeted therapies using small molecule inhibitors of the RAS/MAPK pathway, which are also considered as potential new therapies for vascular anomalies driven by overactivation of this pathway. Mitogen‐activated protein kinase kinase inhibition with trametinib was used off‐label in 2 patients in this cohort; both patients subsequently showed a reduction of symptoms and no progression. Lekwuttikarn et al 38 reported an 11‐year‐old female patient presenting with an extracranial MAP2K1‐mutated AVM treated by trametinib. One month after treatment initiation, the vascular malformation presented with clinically decreased perfusion and discoloration, and magnetic resonance imaging after 6 months confirmed an objective decrease in size. Al‐Olabi et al 8 reported the treatment of AVM‐BRAF mutant zebrafish with the BRAF inhibitor (vemurafinib), which resulted in hemodynamic improvement through less distorted vasculature. One patient in our cohort (patient 20) with an AVM of the foot due to a BRAF mutation was treated with the BRAF inhibitor dabrafenib due to pain and lack of promising surgical or interventional treatment approaches. Under this therapy, the pain quickly abated, and she had a substantial functional improvement, while no severe adverse events occurred during this therapy. A specific KRAS G12C inhibitor (sotorasib) was recently developed for the treatment of KRAS G12C–mutated non‐small‐cell lung cancer. 39 The latter may be suggested specifically for patients with AVM with the KRAS G12C PVs that made up an important part of all KRAS PVs in our cohort. Sotorasib would represent an even more specific targeted treatment compared with unselective mitogen‐activated protein kinase kinase inhibition, for example, with trametinib. This approach may harbor potential benefits, especially in light of the high progression rates in patients with KRAS AVMs but will need further study in clinical trials.
Generally, the growing body of literature as well as this study support the notion that the clinical spectrum of vascular anomalies driven by PVs in components of the canonical RAS/MAPK pathway is becoming broader than anticipated after the first published findings in selected patient cohorts, and together with their broadening the clinical spectra associated with individual genes also tend to merge. There is still a predominance of fast‐flow anomalies, but mixed and low‐flow malformations such as VMs also occur. Notably, MAP2K1 variants have exclusively been observed in AVMs so far. The validation and further resolution of genotype–phenotype correlations in vascular anomalies deserve continued efforts and studies in larger cohorts, before they become a routine basis of clinical decision making. As such, this works in a hypothesis‐generating manner to set up larger multicenter cohorts, potentially on the basis of registries, as large randomized controlled trials on vascular anomalies are unlikely to being realized in the near future. 40
This study reveals preliminary associations of RAS/BRAF/MAP2K1 mosaicism with clinical phenotypes of extracranial vascular anomalies. Lesions with KRAS PVs tended to show more infiltrative growth patterns across multiple tissue layers. Furthermore, RAS variants were characterized by more advanced disease stages and potentially higher progression rates, reflecting a more aggressive phenotype. The benefit of combining clinical and genetic diagnostics may promote more individualized treatment regimens according to the underlying PV such as by adding targeted therapeutics to multidisciplinary care.
Sources of Funding
Dr Schmidt received intramural funding (Munich Medical & Clinician Scientist Program, LMU Munich; https://www.med.lmu.de/karriere/mcsp/index.html). The funder played no role in study design, data collection, analysis and interpretation of data, or the writing of this manuscript. We do not report any external funding for this study.
Disclosures
Dr Kapp received consultation fees from Novartis. The remaining authors have no disclosures to report.
Supporting information
Table S1
Acknowledgments
Some of the authors are members of the German Reference Network for Vascular Anomalies. Conceptualization was contributed by Drs Schmidt, Kapp, Zenker, and Wildgruber. Dr Schmidt appears first in the author list because of her involvement in the initiation of the project due to project‐related intramural funding. Vascular anomalies were clinically and radiologically diagnosed and treated by Drs Schmidt, Kapp, Brill, Vielsmeier, Michel, Seidensticker, Uller, Wohlgemuth, and Wildgruber. Targeted sequencing of panels of genes was performed by Schanze and Zenker. Drs Schmidt, Kapp, Kimm, and Wildgruber contributed to the investigation and writing of the original draft. All authors edited and approved the final version of the manuscript.
A complete list of the APOLLON Investigators can be found in the Supplemental Material.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.123.033287
For Sources of Funding and Disclosures, see page 13.
References
- 1. Wassef M, Borsik M, Cerceau P, Faucon B, Laurian C, Le Clerc N, Lemarchand‐Venencie F, Massoni C, Salvan D, Bisdorff‐Bresson A. Classification des tumeurs et malformations vasculaires. Apport de la classification ISSVA 2014/2018. Article in French. Ann Pathol. 2021;41:58–70. doi: 10.1016/j.annpat.2020.11.004 [DOI] [PubMed] [Google Scholar]
- 2. Schmidt VF, Olivieri M, Häberle B, Masthoff M, Deniz S, Sporns PB, Wohlgemuth WA, Wildgruber M. Interventional treatment options in children with extracranial vascular malformations. Hamostaseologie. 2022;42:131–141. doi: 10.1055/a-1728-5686 [DOI] [PubMed] [Google Scholar]
- 3. Schmidt VF, Masthoff M, Goldann C, Deniz S, Öcal O, Häberle B, Köhler M, Seidensticker M, Ricke J, Wohlgemuth WA, et al. Percutaneous sclerotherapy of venous malformations of the hand: a multicenter analysis. Cardiovasc Intervent Radiol. 2021;44:1543–1550. doi: 10.1007/s00270-021-02926-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schmidt VF, Masthoff M, Goldann C, Ehrl D, Deniz S, Öcal O, Seidensticker M, Ricke J, Köhler M, Brill R, et al. Image‐guided embolization of arteriovenous malformations of the hand using ethylene‐vinyl alcohol copolymer. Diagn Interv Radiol. 2022;28:486–494. doi: 10.5152/dir.2022.21644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schmidt VF, Masthoff M, Czihal M, Cucuruz B, Häberle B, Brill R, Wohlgemuth WA, Wildgruber M. Imaging of peripheral vascular malformations—current concepts and future perspectives. Mol Cell Pediatr. 2021;8:19. doi: 10.1186/s40348-021-00132-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Queisser A, Seront E, Boon LM, Vikkula M. Genetic basis and therapies for vascular anomalies. Circ Res. 2021;129:155–173. doi: 10.1161/CIRCRESAHA.121.318145 [DOI] [PubMed] [Google Scholar]
- 7. Nathan N, Keppler‐Noreuil KM, Biesecker LG, Moss J, Darling TN. Mosaic disorders of the PI3K/PTEN/AKT/TSC/MTORC1 signaling pathway. Dermatol Clin. 2017;35:51–60. doi: 10.1016/j.det.2016.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Al‐Olabi L, Polubothu S, Dowsett K, Andrews KA, Stadnik P, Joseph AP, Knox R, Pittman A, Clark G, Baird W, et al. Mosaic RAS/MAPK variants cause sporadic vascular malformations which respond to targeted therapy. J Clin Invest. 2018;128:1496–1508. doi: 10.1172/JCI98589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nikolaev SI, Vetiska S, Bonilla X, Boudreau E, Jauhiainen S, Rezai Jahromi B, Khyzha N, DiStefano PV, Suutarinen S, Kiehl TR, et al. Somatic activating KRAS mutations in arteriovenous malformations of the brain. N Engl J Med. 2018;378:250–261. doi: 10.1056/NEJMoa1709449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schmidt VF, Wieland I, Wohlgemuth WA, Ricke J, Wildgruber M, Zenker M. Mosaic rasopathy due to KRAS variant G12D with segmental overgrowth and associated peripheral vascular malformations. Am J Med Genet A. 2021;185:3122–3128. doi: 10.1002/ajmg.a.62386 [DOI] [PubMed] [Google Scholar]
- 11. El Sissy FN, Wassef M, Faucon B, Salvan D, Nadaud S, Coulet F, Adle‐Biassette H, Soubrier F, Bisdorff A, Eyries M. Somatic mutational landscape of extracranial arteriovenous malformations and phenotypic correlations. J Eur Acad Dermatol Venereol. 2022;36:905–912. doi: 10.1111/jdv.18046 [DOI] [PubMed] [Google Scholar]
- 12. Couto JA, Huang AY, Konczyk DJ, Goss JA, Fishman SJ, Mulliken JB, Warman ML, Greene AK. Somatic MAP2K1 mutations are associated with extracranial arteriovenous malformation. Am J Hum Genet. 2017;100:546–554. doi: 10.1016/j.ajhg.2017.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ten Broek RW, Eijkelenboom A, van der Vleuten CJM, Kamping EJ, Kets M, Verhoeven BH, Grünberg K, Schultze Kool LJ, Tops BBJ, Ligtenberg MJL, et al. Comprehensive molecular and clinicopathological analysis of vascular malformations: a study of 319 cases. Genes Chromosomes Cancer. 2019;58:541–550. doi: 10.1002/gcc.22739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wei T, Shalin S, Draper E, Miller E, Zhang H, Sun R, Lee M, Albert G, Richter GT. Abnormal elastin and collagen deposition is present in extracranial arteriovenous malformations: a comparison to intracranial disease. Histol Histopathol. 2019;34:1355–1363. doi: 10.14670/HH-18-129 [DOI] [PubMed] [Google Scholar]
- 15. Greene AK, Liu AS, Mulliken JB, Chalache K, Fishman SJ. Vascular anomalies in 5,621 patients: guidelines for referral. J Pediatr Surg. 2011;46:1784–1789. doi: 10.1016/j.jpedsurg.2011.05.006 [DOI] [PubMed] [Google Scholar]
- 16. Cho SK, Do YS, Shin SW, Kim DI, Kim YW, Park KB, Kim EJ, Ahn HJ, Choo SW, Choo IW. Arteriovenous malformations of the body and extremities: analysis of therapeutic outcomes and approaches according to a modified angiographic classification. J Endovasc Ther. 2006;13:527–538. doi: 10.1583/05-1769.1 [DOI] [PubMed] [Google Scholar]
- 17. Finn MC, Glowacki J, Mulliken JB. Congenital vascular lesions: clinical application of a new classification. J Pediatr Surg. 1983;18:894–900. doi: 10.1016/S0022-3468(83)80043-8 [DOI] [PubMed] [Google Scholar]
- 18. Puig S, Aref H, Chigot V, Bonin B, Brunelle F. Classification of venous malformations in children and implications for sclerotherapy. Pediatr Radiol. 2003;33:99–103. doi: 10.1007/s00247-002-0838-9 [DOI] [PubMed] [Google Scholar]
- 19. Little J, Higgins JP, Ioannidis JP, Moher D, Gagnon F, von Elm E, Khoury MJ, Cohen B, Davey‐Smith G, Grimshaw J, et al. STrengthening the REporting of Genetic Association studies (STREGA)—an extension of the STROBE statement. Genet Epidemiol. 2009;33:581–598. doi: 10.1002/gepi.20410 [DOI] [PubMed] [Google Scholar]
- 20. ISSVA classification of vascular anomalies ©2018 International Society for the Study of Vascular Anomalies . Published April 2014. Date updated May 2018. Assessed May 25, 2023. issva.org/classification; https://www.issva.org/UserFiles/file/ISSVA‐Classification‐2018.pdf
- 21. Oka M, Kushamae M, Aoki T, Yamaguchi T, Kitazato K, Abekura Y, Kawamata T, Mizutani T, Miyamoto S, Takagi Y. KRAS G12D or G12V mutation in human brain arteriovenous malformations. World Neurosurg. 2019;126:e1365–e1373. doi: 10.1016/j.wneu.2019.03.105 [DOI] [PubMed] [Google Scholar]
- 22. Goss JA, Huang AY, Smith E, Konczyk DJ, Smits PJ, Sudduth CL, Stapleton C, Patel A, Alexandrescu S, Warman ML, et al. Somatic mutations in intracranial arteriovenous malformations. PLoS One. 2019;14:e0226852. doi: 10.1371/journal.pone.0226852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nozawa A, Ozeki M, Niihori T, Suzui N, Miyazaki T, Aoki Y. A somatic activating KRAS variant identified in an affected lesion of a patient with gorham‐stout disease. J Hum Genet. 2020;65:995–1001. doi: 10.1038/s10038-020-0794-y [DOI] [PubMed] [Google Scholar]
- 24. Eijkelenboom A, van Schaik FMA, van Es RM, Ten Broek RW, Rinne T, van der Vleuten C, Flucke U, Ligtenberg MJL, Rehmann H. Functional characterisation of a novel class of in‐frame insertion variants of KRAS and HRAS. Sci Rep. 2019;9:8239. doi: 10.1038/s41598-019-44584-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Konczyk DJ, Goss JA, Smits PJ, Huang AY, Al‐Ibraheemi A, Sudduth CL, Warman ML, Greene AK. Arteriovenous malformation associated with a HRAS mutation. Hum Genet. 2019;138:1419–1421. doi: 10.1007/s00439-019-02072-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Siegel DH, Cottrell CE, Streicher JL, Schilter KF, Basel DG, Baselga E, Burrows PE, Ciliberto HM, Vigh‐Conrad KA, Eichenfield LF, et al. Analyzing the genetic spectrum of vascular anomalies with overgrowth via cancer genomics. J Invest Dermatol. 2018;138:957–967. doi: 10.1016/j.jid.2017.10.033 [DOI] [PubMed] [Google Scholar]
- 27. Ozeki M, Aoki Y, Nozawa A, Yasue S, Endo S, Hori Y, Matsuoka K, Niihori T, Funayama R, Shirota M, et al. Detection of NRAS mutation in cell‐free DNA biological fluids from patients with kaposiform lymphangiomatosis. Orphanet J Rare Dis. 2019;14:215. doi: 10.1186/s13023-019-1191-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Barclay SF, Inman KW, Luks VL, McIntyre JB, Al‐Ibraheemi A, Church AJ, Perez‐Atayde AR, Mangray S, Jeng M, Kreimer SR, et al. A somatic activating NRAS variant associated with kaposiform lymphangiomatosis. Genet Med. 2019;21:1517–1524. doi: 10.1038/s41436-018-0390-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Manevitz‐Mendelson E, Leichner GS, Barel O, Davidi‐Avrahami I, Ziv‐Strasser L, Eyal E, Pessach I, Rimon U, Barzilai A, Hirshberg A, et al. Somatic NRAS mutation in patient with generalized lymphatic anomaly. Angiogenesis. 2018;21:287–298. doi: 10.1007/s10456-018-9595-8 [DOI] [PubMed] [Google Scholar]
- 30. Chowers G, Abebe‐Campino G, Golan H, Vivante A, Greenberger S, Soudack M, Barkai G, Fox‐Fisher I, Li D, March M, et al. Treatment of severe kaposiform lymphangiomatosis positive for NRAS mutation by MEK inhibition. Pediatr Res. 2023;94:1911–1915. doi: 10.1038/s41390-022-01986-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Groesser L, Peterhof E, Evert M, Landthaler M, Berneburg M, Hafner C. BRAF and RAS mutations in sporadic and secondary pyogenic granuloma. J Invest Dermatol. 2016;136:481–486. doi: 10.1038/JID.2015.376 [DOI] [PubMed] [Google Scholar]
- 32. Goss JA, Konczyk DJ, Smits PJ, Kozakewich HPW, Alomari AI, Al‐Ibraheemi A, Taghinia AH, Dickie BH, Adams DM, Fishman SJ, et al. Intramuscular fast‐flow vascular anomaly contains somatic MAP2K1 and KRAS mutations. Angiogenesis. 2019;22:547–552. doi: 10.1007/s10456-019-09678-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sudduth CL, McGuire AM, Smits PJ, Konczyk DJ, Al‐Ibraheemi A, Fishman SJ, Greene AK. Arteriovenous malformation phenotype resembling congenital hemangioma contains KRAS mutations. Clin Genet. 2020;98:595–597. doi: 10.1111/cge.13833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zenner K, Jensen DM, Dmyterko V, Shivaram GM, Myers CT, Paschal CR, Rudzinski ER, Pham MM, Cheng VC, Manning SC, et al. Somatic activating BRAF variants cause isolated lymphatic malformations. HGG Adv. 2022;3:100101. doi: 10.1016/j.xhgg.2022.100101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Strobel K, Maurus K, Hamm H, Roth S, Goebeler M, Rosenwald A, Wobser M. Recurrent alterations in the MAPK pathway in sporadic pyogenic granuloma of childhood. Acta Derm Venereol. 2022;102:adv00715. doi: 10.2340/actadv.v102.1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nikolaev SI, Rimoldi D, Iseli C, Valsesia A, Robyr D, Gehrig C, Harshman K, Guipponi M, Bukach O, Zoete V, et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat Genet. 2012;44:133–139. doi: 10.1038/ng.1026 [DOI] [PubMed] [Google Scholar]
- 37. Choi YL, Soda M, Ueno T, Hamada T, Haruta H, Yamato A, Fukumura K, Ando M, Kawazu M, Yamashita Y, et al. Oncogenic MAP2K1 mutations in human epithelial tumors. Carcinogenesis. 2012;33:956–961. doi: 10.1093/carcin/bgs099 [DOI] [PubMed] [Google Scholar]
- 38. Lekwuttikarn R, Lim YH, Admani S, Choate KA, Teng JMC. Genotype‐guided medical treatment of an arteriovenous malformation in a child. JAMA Dermatol. 2019;155:256–257. doi: 10.1001/jamadermatol.2018.4653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, Italiano A, Schuler M, Borghaei H, Barlesi F, et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N Engl J Med. 2021;384:2371–2381. doi: 10.1056/NEJMoa2103695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schmidt VF, Masthoff M, Vielsmeier V, Seebauer CT, Cangir Ö, Meyer L, Mükke A, Lang W, Schmid A, Sporns PB, et al. Clinical outcome and quality of life of multimodal treatment of extracranial arteriovenous malformations: the apollon study protocol. Cardiovasc Intervent Radiol. 2023;46:142–151. doi: 10.1007/s00270-022-03296-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1