

Abstract
CLCs are dimeric chloride channels and anion/proton exchangers that regulate processes such as muscle contraction and endo-lysosome acidification. Common gating controls their activity; its closure simultaneously silences both protomers and its opening allows them to independently transport ions. Mutations affecting common gating in human CLCs cause dominant genetic disorders. The structural rearrangements underlying common gating are unknown. Here, using single-particle cryo-electron microscopy, we show that the prototypical Escherichia coli CLC-ec1 undergoes large-scale rearrangements in activating conditions. The slow, pH-dependent remodeling of the dimer interface leads to the concerted opening of the intracellular H+ pathways and is required for transport. The more frequent formation of short water wires in the open H+ pathway enables Cl− pore openings. Mutations at disease-causing sites favor CLC-ec1 activation and accelerate common gate opening in the human CLC-7 exchanger. We suggest that the pH activation mechanism of CLC-ec1 is related to the common gating of CLC-7.
Introduction
The CLC Cl− channels and anion/H+ exchangers play fundamental roles in human physiology. Mutations impairing their function lead to numerous disorders, including myotonia congenita, leukoencephalopathy, osteopetrosis, epilepsy, and lysosomal storage disorders 1,2. All CLCs are dimers, where each protomer forms an independent Cl− pore in the channels or mediates anion/H+ exchange in the transporters (Supplementary Fig. 1A-C) 2. The common gating process controls activation of CLC dimers: when this gate is closed, both protomers are inactive, and when it opens, they become activated and transport ions independently via single-pore gating 3–6. Common gating manifests as the characteristic double-barreled signature of the CLC single channels 3,7–10 and in double exponential relaxation kinetics of the macroscopic currents 11–13. Mammalian CLC transporters display voltage-dependent gating kinetics 14–19, attributed to common gating in CLC-7 16. Mutations causing dominant genetic disorders predominantly affect common gating in both channels and transporters (Supplementary Fig. 1D-E) 2,20–24, underscoring the importance of this process.
Within each protomer, the Cl− and H+ permeation pathways intersect at a highly conserved glutamate sidechain, Egate (E148 in CLC-ec1), and diverge at the intra- and extracellular ends 25,26 (Supplementary Fig. 1B). Single-pore gating occurs on the micro- to milli-second time scale and mostly entails Egate moving between the Cl− and H+ pathways to enable exchange 1,2. In contrast, common gating occurs over tens of milliseconds to seconds, and likely involves global rearrangements of the interprotomer interfaces in the transmembrane and cytosolic regions 3,9,12,13,15–19,27–29. Structural studies of the prototypical Escherichia coli exchanger CLC-ec1 revealed no large-scale structural changes, suggesting that, unlike its human counterparts, it is not gated, and its transport cycle entails only Egate movements 1,2. However, other reports suggested that CLC-ec1 undergoes Cl−- and pH-dependent conformational changes in regions distal to the Cl− pore, interpreted as steps in the transport cycle 30–33.
Here, we present cryogenic electron microscopy (cryoEM) structures of CLC-ec1 obtained at neutral and low pH with and without Cl−. The structures show that in conditions of maximal activity, CLC-ec1 adopts two conformations with drastically restructured and reduced transmembrane and soluble cytosolic dimer interfaces and increased solvent accessibility of the intracellular H+ pathways. Cysteine crosslinks preventing opening of the H+ pathway inhibit activity, showing it is required for function. Single-molecule fluorescence resonance energy transfer total internal reflection fluorescence (smFRET-TIRF) microscopy recordings show that interface remodeling occurs over seconds and, therefore, cannot reflect transport cycle steps. Molecular dynamics (MD) simulations show these rearrangements lead to increased transporter dynamics, formation of frequent short water wires connecting the intracellular milieu to Egate, and increased openings of the Cl− pores. Therefore, pH-dependent, slow, and concerted rearrangements of the dimer interface enable H+:Cl− exchange by each CLC-ec1 protomer. Mutations favoring these rearrangements in CLC-ec1 accelerate common gate opening in the mammalian CLC-7 transporter, suggesting the pH-dependent rearrangements of CLC-ec1 could be related to common gating of CLC-7.
Results
Structural basis of CLC-ec1 activation
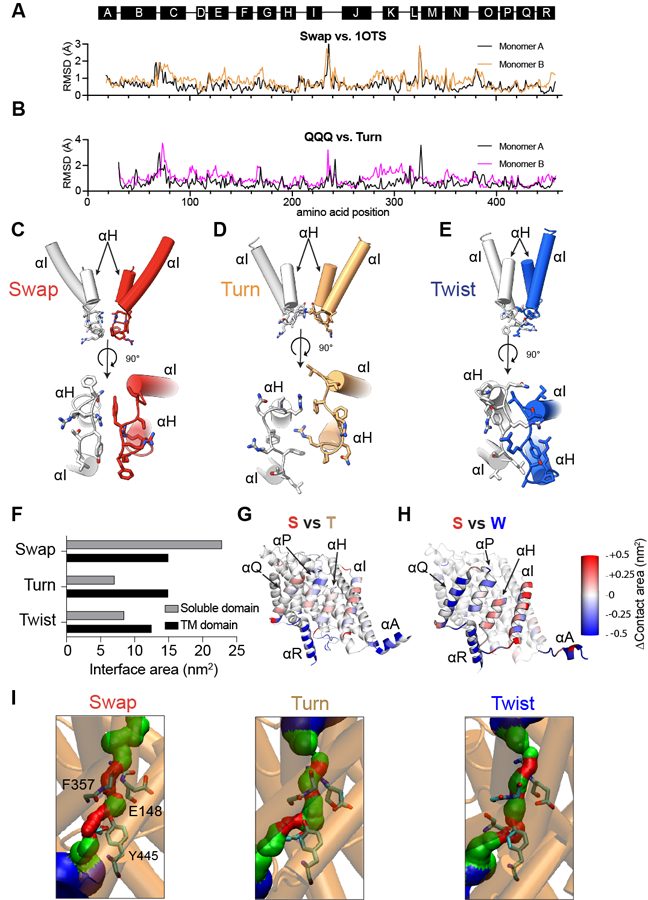
We used cryoEM to image detergent-solubilized CLC-ec1 in 0 or 100 mM Cl− at pH 7.5 and 4.5 to visualize pH- and Cl−-dependent rearrangements (Fig. 1A-J, Table 1). Data processing without symmetry constraints (Extended Data Fig. 1, 2, Table 1) resulted in nearly identical protomers within dimers under all conditions (Cα r.m.s.d.<1 Å). At pH 7.5 with and without Cl−, CLC-ec1 adopts a conformation similar to the crystallized CLC-ec1/FAB complex (Cα r.m.s.d. <1 Å; Fig. 1A-C,J; Extended Data Fig. 3A). In this “Swap” state the N-terminal α-helices (αA) are domain-swapped and interact with the C-terminal αR and the H-I loops of the opposite protomer, occluding the intracellular H+ pathway vestibules (Fig. 1A-C). The dimer interface (~3,780 Å2) consists of a transmembrane (TM) interface formed by the αH, αI, αP, and αQ helices (~1,500 Å2) and a soluble interface formed by αA and αR helices, the H-I loops (residues 201–214), and other minor interprotomer interactions (~2,300 Å2) (Extended Data Fig. 3C, F, G).
Figure 1: pH- and Cl−-dependent conformational rearrangements of CLC-ec1.
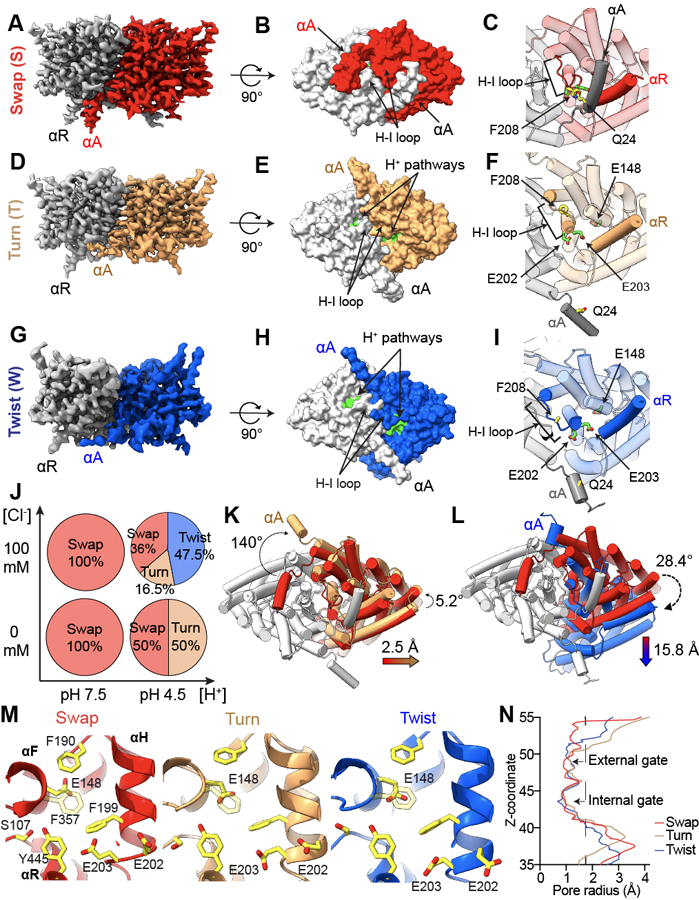
(A-I) Side views of the cryoEM density maps of CLC-ec1 in Swap (A), Turn (D), and Twist (G) imaged at pH 4.5 in 100 mM Cl−. Surface representation of CLC-ec1 in Swap (B), Turn (E), and Twist (H) viewed from the intracellular side. Residues lining the H+ pathway are colored in green (E113, E148, A182, L186, A189, F190, F199, E202, E203, M204, I402, T407 and Y445). Close-up view of gating of the H+ pathway by αA in Swap (C), Turn (F) and Twist (I). The dimers are shown in transparent cartoon representation apart from the H-I loops and helices αA and αR from opposing protomers. The H+ pathway residues E148, E202 and E203 are shown in green CPK stick representation. The interacting residues Q24 in αA and F208 in the H-I loop are shown in yellow CPK stick representation. In Twist the Coulomb density for Q24 and F208 side chains is insufficient for model building. In all panels one protomer is shown in gray and the other is colored (Swap in red, Turn in wheat, and Twist in blue). (J) A scheme showing pH and chloride conditions for cryoEM data collection, and conformational states observed in each condition: Swap (S), Turn (T), and Twist (W). The % of particles in each conformation are shown in the pie charts. (K-L) Cartoon representations of the bottom view of Turn (K) and Twist (L) aligned to Swap on protomers A. Color scheme is as in A-F. Dashed arrows denote protomer B or αA rotations, and colored arrows denote center of mass (COM) movements. (M) Close-up view of the Cl− and H+ pathways of CLC-ec1 in Swap (left), Turn (middle), and Twist (right). S107, Y445 and E148 are Cl− pathway residues, and H+ pathway residues are E148, F190, F199, E202, and E203. All residues are shown as yellow CPK sticks. (N) Pore radius profile along the z-axis of the Cl− pathway calculated using HOLE 78 for CLC-ec1 in Swap, Turn, and Twist.
Table 1:
CryoEM data acquisition, reconstruction, and model refinement statistics for WT CLC-ec1.
| Swap pH 4.5 100 mM Cl− (EMDB-24241) (PDB 7N8P) | Turn pH 4.5 100 mM Cl− (EMDB-24612) (PDB 7RP5) | Twist pH 4.5 100 mM Cl− (EMDB-24263) (PDB 7N9W) | Swap pH 4.5 0 mM Cl− (EMDB-24613) (PDB 7RP6) | Turn pH 4.5 0 mM Cl− (EMDB-24668) (PDB 7RSB) | Swap pH 7.5 100 mM Cl− (EMDB-24582) (PDB 7RNX) | Swap pH 7.5 0 mM Cl− (EMDB-24584) (PDB 7RO0) | |
|---|---|---|---|---|---|---|---|
| Data collection and processing | |||||||
| Magnification | 105,000 | 105,000 | 105,000 | 105,000 | 105,000 | 105,000 | 105,000 |
| Voltage (kV) | 300 | 300 | 300 | 300 | 300 | 300 | 300 |
| Electron exposure (e–/Å2) | 72.61 | 72.61 | 72.61 | 65.64 | 65.64 | 72.85 | 64.08 |
| Defocus range (μm) | −1.2 – −2.2 | −1.2 – −2.2 | −1.2 – −2.2 | −1.2 – −2.2 | −1.2 – −2.2 | −1.2 – −2.2 | −1.5 – −2.5 |
| Pixel size (Å) | 1.048 | 1.048 | 1.048 | 1.048 | 1.048 | 1.048 | 1.098 |
| Symmetry imposed | C1 | C1 | C1 | C1 | C1 | C1 | C1 |
| Initial particle images (no.) | 1,983,033 | 1,983,033 | 1,983,033 | 1,296,087 | 1,296,087 | 1,190,294 | 865,290 |
| Final particle images (no.) | 128,951 | 59.069 | 169,241 | 115,851 | 116,129 | 233,512 | 127,099 |
| Map resolution (Å) | 3.59 | 3.72 | 4.15 | 3.76 | 3.68 | 3.42 | 3.74 |
| FSC threshold | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 |
| Map resolution range (Å) | 2.4 – 6.9 | 2.6 – 7.1 | 2.9 – 7.4 | 2.6 – 7.1 | 2.6 – 7.1 | 2.6 – 7.1 | 2.4 – 6.9 |
| Refinement | |||||||
| Initial model used (PDB code) | 1OTS | 1OTS | 1OTS | 1OTS | 1OTS | 1OTS | 1OTS |
| Model resolution (Å) | 3.7 | 3.9 | 4.2 | 3.8 | 3.7 | 3.5 | 3.8 |
| FSC threshold | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| Map sharpening B factor (Å2) | −98 | −72 | −175 | −173 | −143 | −149 | −83 |
| Model composition | |||||||
| Non-hydrogen atoms | 6758 | 6578 | 6561 | 6753 | 6630 | 6758 | 6758 |
| Protein residues | 900 | 884 | 890 | 900 | 884 | 900 | 900 |
| Ligands | 2 | 2 | |||||
| B factors (Å2) | |||||||
| Protein | 57.66 | 63.50 | 86.57 | 43.59 | 57.96 | 52.55 | 63.61 |
| Ligand | 61.43 | 61.43 | |||||
| R.m.s. deviations | |||||||
| Bond lengths (Å) | 0.005 | 0.005 | 0.005 | 0.011 | 0.006 | 0.013 | 0.008 |
| Bond angles (°) | 0.883 | 1.003 | 1.074 | 1.122 | 1.064 | 1.063 | 1.021 |
| Validation | |||||||
| MolProbity score | 1.61 | 1.63 | 1.81 | 1.70 | 1.62 | 1.61 | 1.68 |
| Clashscore | 4.27 | 4.77 | 5.03 | 5.72 | 4.50 | 4.77 | 5.07 |
| Poor rotamers (%) | 0.29 | 0.46 | 1.4 | 0.49 | 0.75 | 0.44 | 0.74 |
| Ramachandran plot | |||||||
| Favored (%) | 94.08 | 94.31 | 92.23 | 94.31 | 94.10 | 94.75 | 93.74 |
| Allowed (%) | 5.92 | 5.69 | 6.66 | 5.69 | 5.90 | 5.25 | 6.23 |
| Disallowed (%) | 0.00 | 0.00 | 0.11 | 0.00 | 0.00 | 0.00 | 0.00 |
Ab initio reconstructions 34 of CLC-ec1 imaged at pH 4.5 without Cl−, show Swap and a second state, “Turn” (Fig. 1C,D,G; Extended Data Fig. 1, 2; Table 1), in similar amounts. In Turn, the αAs disengage from αRs and the H-I loops and turn away from the TM regions, while the H-I loops retract towards the TM region (Fig. 1E-F, Extended Data Fig. 3D). These movements expose the intracellular H+ pathway vestibules lined by the conserved acidic sidechains of E202 and E203 26,35–37 (Fig. 1D-F; Supplementary Fig. 1B,C). The TM dimer interface remains intact (~1,500 Å2), while the soluble interface decreases drastically (~700 Å2) (Extended Data Fig. 3D, F, G). Notably, the TM region of Turn resembles the crystal structure of the pH-insensitive CLC-ec1 E113Q/E148Q/E203Q (QQQ) mutant (Cα r.m.s.d. ~0.65 Å; Extended Data Fig. 3B) 31.
At pH 4.5 in 100 mM Cl−, we identified three major structural classes corresponding to Swap (36% of particles), Turn (~16.5%), and a “Twist” conformation (~47.5%). Twist features disengaged αAs and a large relative rotation and translation of the protomers (Fig. 1E,F,G,J; Extended Data Fig. 1, 2; Table 1). Further analysis (Methods) revealed several Twists differing slightly in subunit orientations, one of which we refined and modeled (Fig. 1G,H; Extended Data Fig. 1, 2). The TM interface in Twist is dramatically different from Swap and Turn; it is primarily formed by the tightly packed αH and αI helices (~1,200 Å2) (Fig. 1Extended Data Fig. 3F, H). The soluble interface slightly increases compared to Turn as the H-I loops relax towards the cytosol and interact (Extended Data Fig. 3D, F, H) (~800 Å2) (Extended Data Fig. 3D, F, G). In Twist, like in Turn, the H+ pathway is accessible to the intracellular solution (Fig. 1I).
Structural alignment of CLC-ec1 dimers on the A protomers highlights the large-scale inter-subunit movements between Swap, Turn, and Twist (Fig. 1K-L). In Turn and Twist, the protomers B rotate by ~5.2° and ~28.4o, and their centers of mass (COM) translate laterally by ~2.5 and ~15.8 Å compared to Swap. (Fig. 1K-L; Supplementary Movies 1,2). The Cl− pathways are similar in all states (Fig. 1M). Their external and internal gates (Fig. 1N, Extended Data Fig. 3I), respectively defined by the backbone atoms of R147, E148, I356, and F357 and by the side chains of S107 and Y445, remain constricted. CryoEM density for the E148 (Egate) sidechain is resolved in several structures (Fig. 1M, Extended Data Fig. 1G-J), showing it points towards the central Cl−-binding site or the intracellular H+ pathway. The resolution of these maps is insufficient to assign density to Cl− ions reliably. In all maps, we only observe dimers where both H+ pathways are closed (in Swap) or open (in Turn and Twist), suggesting that the rearrangements are cooperative (Fig. 1). In sum, in high activity conditions CLC-ec1 predominantly adopts the Turn and Twist conformations where inter- and intra-protomer rearrangements result in remodeled TM and soluble interfaces and opening of the intracellular H+ pathways.
Turn and Twist are activated conformations of CLC-ec1
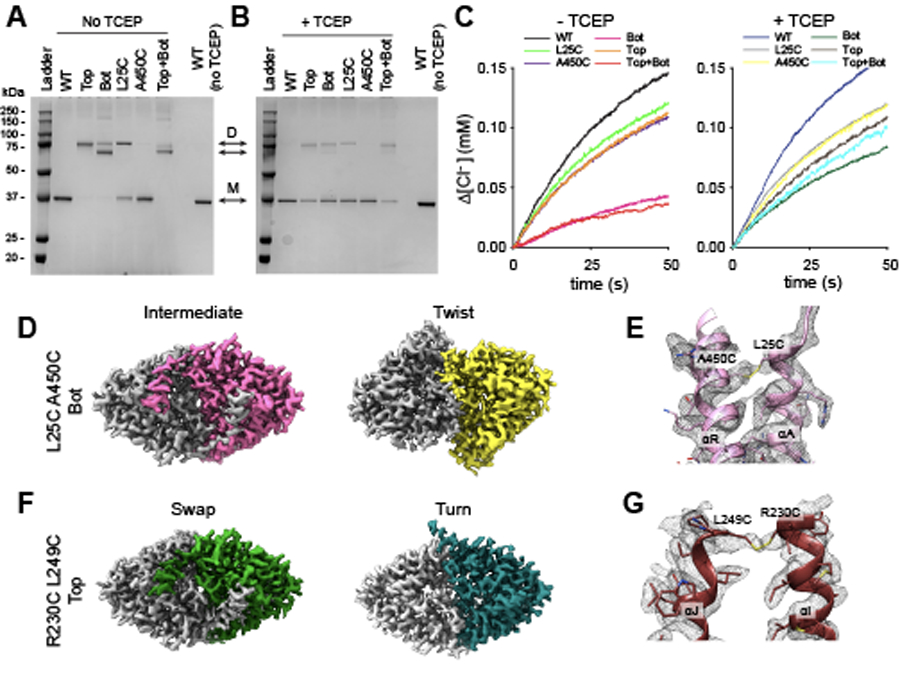
To test whether the opening of the H+ pathway in Turn and Twist, due to the disengagement of the αA and αR helices, is necessary for activity in CLC-ec1 we crosslinked these helices (Fig. 2A) by introducing cysteines at positions L25 and A450 in the background of Cys-lite CLC-ec1 38. A450C in αR migrates as a monomer on SDS-PAGE. In contrast, L25C in αA is a mixture of similar amounts of monomers and dimers, indicating spontaneous crosslinking between αAs and suggesting that these helices are dynamic (Extended Data Fig. 4A). Both single Cys substitutions transport Cl− at slightly reduced rates (Fig. 2C, Extended Data Fig. 4C, Supplementary Table 1). Thus, the αA-αA crosslinked L25C dimers are functional. The double L25C/A450C mutant (CLC-ec1 Bot) migrates as a dimer on SDS PAGE with a minor band (~20%) at a position corresponding to the αA-αA crosslink and a higher-mobility major band (~80%), corresponding to the double αA-αR/αA-αR crosslink (Extended Data Fig. 4A). TCEP reduces crosslinks in all constructs and results in predominantly monomeric bands (Extended Data Fig. 4B). Chloride efflux by CLC-ec1 Bot, is only ~20% of WT (Fig. 2B-C). The residual activity likely reflects transport by the ~20% of αA-αA crosslink (Extended Data Fig. 4A). Incubating CLC-ec1 Bot proteoliposomes with TCEP restores activity to ~50% of WT (Fig. 2B-C), consistent with the reduction of crosslinks only in dimers with cytosolic regions facing the outside of the liposomes. TCEP did not affect activity of WT or single Cys mutants (Fig. 2B-C).
Figure 2: Opening of the H+ pathway is required for transport by CLC-ec1.
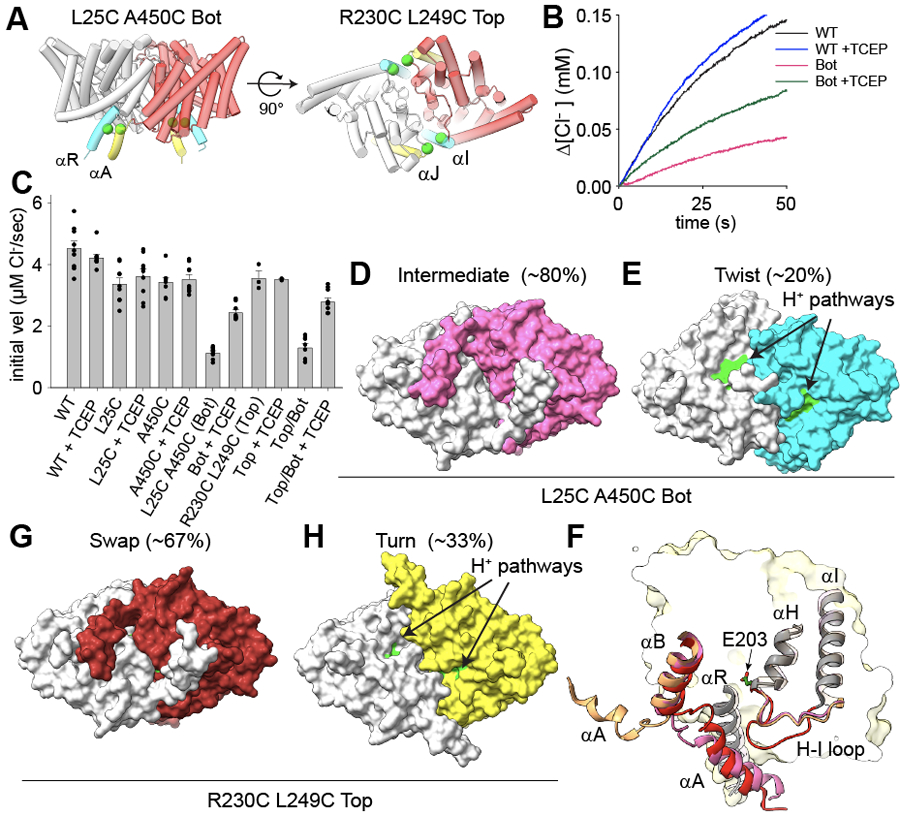
(A) Design of bottom (L25C/A450C, Bot) and top (R230C/L249C, Top) crosslinks. Side (top panel) and bottom (intracellular, bottom panel) views of CLC-ec1 (shown as cartoon with one protomer in gray and one in red) in Swap. Cα atoms of mutation sites are shown as green spheres. Crosslinked helices are colored in yellow and cyan. (B) Time courses of Cl− efflux mediated by CLC-ec1 WT without (black) and with TCEP (blue), and Bot without (pink) and with TCEP (green). (C) The initial velocity of Cl− efflux from proteo-liposomes containing WT or mutant CLC-ec1. All values are means ± S.D. Circles represent the values of individual experiments. Exact values, the number of repeats of independent experiments and preparations of proteo-liposome samples for all constructs are reported in Supplementary Table 1. (D-E) Surface representation of CLC-ec1 Bot in Intermediate (D) and Twist (E) viewed from the intracellular side. Residues lining the H+ pathway are colored as in Fig. 1. One protomer is shown in gray and the other is colored pink (Intermediate) or cyan (Twist). The percentages of particles in each conformation are indicated in brackets. (F) Close-up view of the H+ pathway vestibule in CLC-ec1 WT in Swap (red), Turn (wheat), and Bot Intermediate (pink). Helices αA, αB, αH, αI and connecting loops are shown in the cartoon representation; E203 is shown in stick representation. (G-H) Surface representation of CLC-ec1 Top crosslinked in Swap (G) and Turn (H) viewed from the intracellular side. Residues lining the H+ pathway are colored as in Fig. 1. One protomer is shown in gray and the other is colored in dark red (Top Swap) or limon (Top Turn).
CryoEM imaging of CLC-ec1 Bot at pH 4.5 and 100 mM Cl− shows it adopts two conformations: ~80% of particles feature interacting αA and αR helices and ~20% are in a Twist-like conformation with unresolved αA helices (Fig. 2C; Extended Data Fig. 4D, Extended Data Fig. 5A, Table 2). This latter conformation likely corresponds to the active αA-αA crosslinked exchangers. Coulomb density bridging L25C and A450C (Extended Data Fig. 4E) confirms crosslink formation in the major state. This state shows Swap-like engaged αA and αR, a Turn-like H-I loop, and an A-B loop intermediate between Swap- and Turn-like (Fig. 2F). Thus, H-I loop rearrangements under activating conditions can occur without αA and αR disengagement. When aligned to WT Swap on protomer A, the Bot protomer B shows a Turn-like COM translation of ~1.1 Å and ~5.0° rotation. Therefore, the inactive L25C/A450C crosslink is an intermediate between Swap and Turn, with engaged αA and αR. αAs partially occlude its H+ pathways, while E202 and E203 show increased solvent-accessible following H-I loop rearrangements (Fig. 2D). Thus, Swap-like states with interacting αA and αR helices are inactive or poorly active.
Table 2:
CryoEM data acquisition, reconstruction, and model refinement statistics for mutant CLC-ec1.
| Top Swap pH 4.5, 100 mM Cl− (EMDB-29884) (PDB 8GA1) | Top Turn pH 4.5, 100 mM Cl− (EMDB-29885) (PDB 8GA3) | Bot Interm. pH 4.5, 100 mM Cl− (EMDB-29890) (PDB 8GA5) | Bot Twist pH 4.5, 100 mM Cl− (EMDB-29899) (PDB 8GAH) | E202Y pH 4.5 100 mM Cl− (EMDB-29883) (PDB 8GA0) | |
|---|---|---|---|---|---|
| Data collection and processing | |||||
| Magnification | 105,000 | 105,000 | 105,000 | 105,000 | 105,000 |
| Voltage (kV) | 300 | 300 | 300 | 300 | 300 |
| Electron exposure (e–/Å2) | 56.11 | 56.11 | 58.65 | 58.65 | 51.79 |
| Defocus range (μm) | −1.2 – −2.2 | −1.2 – −2.2 | −0.9 – −1.9 | −0.9 – −1.9 | −1.2 – −2.2 |
| Pixel size (Å) | 0.825 | 0.825 | 0.825 | 0.825 | 1.048 |
| Symmetry imposed | C1 | C1 | C1 | C1 | C1 |
| Initial particle images (no.) | 3,356,123 | 3,356,123 | 4,019,373 | 4,019,373 | 932,285 |
| Final particle images (no.) | 722,867 | 361,089 | 1,603,825 | 429,018 | 71,031 |
| Map resolution (Å) | 2.66 | 3.21 | 2.69 | 2.99 | 3.31 |
| FSC threshold | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 |
| Map resolution range (Å) | 1.8 – 3.2 | 2.1 – 6.8 | 1.8 – 3.0 | 1.9 – 3.8 | 2.4 – 6.9 |
| Refinement | |||||
| Initial model used (PDB code) | 1OTS | 1OTS | 1OTS | 1OTS | 1OTS |
| Model resolution (Å) | 2.7 | 3.3 | 2.7 | 3.0 | 3.7 |
| FSC threshold | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| Map sharpening B factor (Å2) | −128 | −135 | −131 | −103 | −118 |
| Model composition | |||||
| Non-hydrogen atoms | 6781 | 6590 | 6789 | 6424 | 6598 |
| Protein residues | 900 | 886 | 900 | 864 | 882 |
| Ligands | 4 | 4 | 6 | 4 | |
| B factors (Å2) | |||||
| Protein | 15.27 | 33.09 | 35.07 | 29.73 | 32.28 |
| Ligand | 5.59 | 29.80 | 32.45 | 24.07 | |
| R.m.s. deviations | |||||
| Bond lengths (Å) | 0.005 | 0.004 | 0.005 | 0.005 | 0.004 |
| Bond angles (°) | 0.846 | 0.884 | 0.796 | 0.869 | 0.833 |
| Validation | |||||
| MolProbity score | 1.51 | 1.66 | 1.15 | 1.61 | 1.53 |
| Clashscore | 3.92 | 4.25 | 3.55 | 4.27 | 4.15 |
| Poor rotamers (%) | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Ramachandran plot | |||||
| Favored (%) | 95.20 | 92.97 | 98.10 | 94.07 | 95.22 |
| Allowed (%) | 4.80 | 7.03 | 1.90 | 5.93 | 4.78 |
| Disallowed (%) | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
To test whether Twist is required for function, we used the R230C/L249C mutant (CLC-ec1 Top), where a crosslink at the dimer interface should prevent inter-protomer movements 39 (Fig 2A). CLC-ec1 Top is dimeric on SDS-PAGE (Extended Data Fig. 4A) and nearly fully functional (Fig. 2C, Supplementary Table 1), as previously reported 39. CryoEM imaging at pH 4.5 and 100 mM Cl− showed only Swap and Turn (Fig. 2G-H; Extended Data Fig. 4F; Extended Data Fig. 5B; Table 1). Densities in the Coulomb maps confirm the formation of two crosslinks on the protomer interface (Extended Data Fig. 4G). Thus, the TM interface restructuring in Twist is not necessary for transport. However, Twist is likely functional because it is predominant in conditions of maximal activity (Fig. 1), it is observed in the partially active CLC-ec1 Bot (Fig. 2E), and monomeric CLC-ec1 mutants have no dimer interface and are functional 38,40. In sum, rearrangements opening of the H+ pathway are necessary for function. In contrast, the TM interface restructuring in Twist is unnecessary for activity, and its functional role remains unclear.
pH- and Cl−-dependent opening of the H+ pathway in membranes
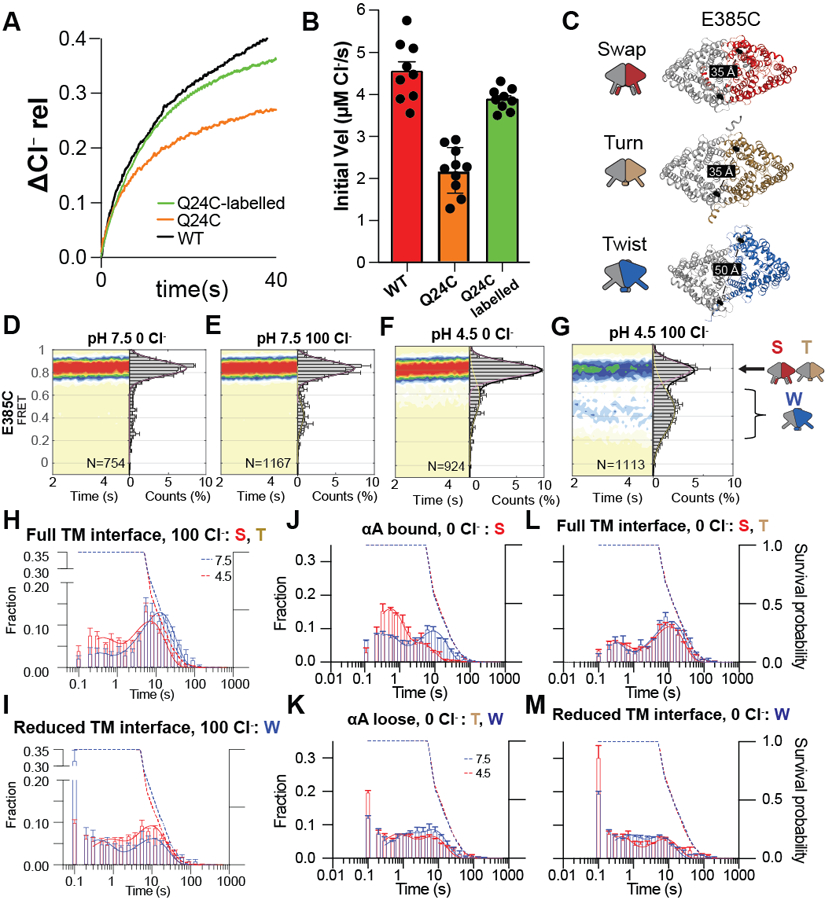
We used smFRET TIRF imaging 41 to determine the timing of the rearrangements leading to the H+ pathway opening. We introduced Q24C mutation in αA (Fig. 3A) within the Cys-lite CLC-ec1 background 38 and labelled it with a mixture of maleimide-activated donor LD555P and acceptor LD655 dyes 42,43. Unlabeled and labelled Q24C CLC-ec1 mediate pH-dependent Cl− flux at WT-like rates (Extended Data Fig. 6A, B, Supplementary Table 2). We reconstituted labelled proteins into liposomes, immobilized vesicles in the microscope chambers via biotin-phosphatidylethanolamine and monitored relative αA movements in identical internal and external ion concentrations. The distance between the Q24 residues of A and B protomers is short in Swap (αA bound), where the H+ pathway is closed. It increases when αAs disengage from αRs (αA loose) and the H+ pathways open in Turn and Twist (Fig. 3A). Labelled Q24C CLC-ec1 displays anticorrelated donor and acceptor fluorophore signals and discrete FRET efficiency states (Fig. 3B-C, Extended Data Fig. 7A-D). At pH 7.5, with and without Cl−, Q24C is predominantly in a high-FRET efficiency state (~0.85) consistent with engaged αA and αR and occluded H+ pathway (Fig. 3C,D). A small fraction is in a low-FRET state (~0.42), indicative of disengaged helices (Fig. 3A-D, Supplementary Table 2), consistent with minimal CLC-ec1 activity in these conditions 44,45. At pH 4.5 and 0 or 100 mM Cl−, Q24C populates a low-FRET (~0.56) state indicative of Turn and Twist (Fig. 3E,F, Supplementary Table 2). We do not interpret differences in the low-FRET states of Q24C at pH 4.5 and 7.5 structurally because disengaged αA helices are poorly resolved in cryoEM and likely dynamic (Fig. 1). Thus, in a membrane environment, CLC-ec1 is mostly in Swap at pH 7.5 and primarily in Turn and Twist at pH 4.5.
Figure 3: smFRET TIRF microscopy of inter-protomer dynamics in a membrane environment.
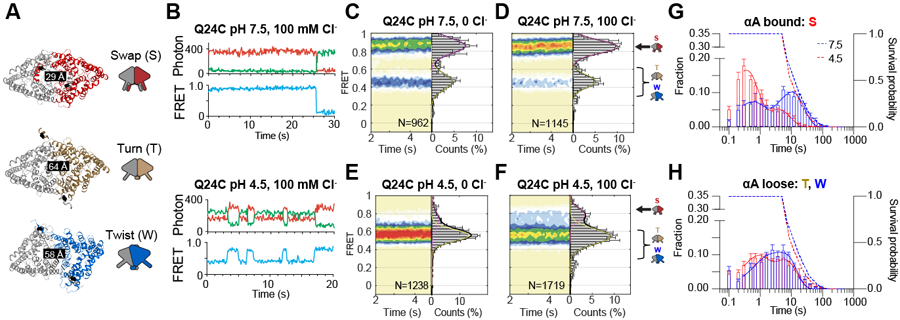
(A) The experimental design to monitor the opening and closing dynamics of the H+ pathway by tracking the movement of αA labelled with donor and acceptor fluorophores at position Q24C. CLC-ec1 dimer is shown as cartoon and colored as in Figure 1. The labeling sites are shown as black spheres, and inter-dye distances are shown on the structures. Schematic representations of Swap, Turn, and Twist are on the left. (B) Representative smFRET time traces in 100 mM Cl− for fluorescently labelled Q24C at pH 7.5 and pH 4.5. Shown are fluorescence emission traces (top) of LD555p (green) and LD655 (red) dyes and FRET efficiency (blue, bottom). (C-F) Population contour plots of labelled Q24C (bottom) at pH 7.5 in 0 mM Cl− (C), pH 7.5 in 100 mM Cl− (D), pH 4.5 in 0 mM Cl− (E), and pH 4.5 in 100 mM Cl− (F). N, the number of molecules used, is shown on each panel. Cumulative FRET state histograms are on the right of each panel plotted as means of three independent experiments ± SEM. Assignments of the FRET states to the protein conformations are on the right of panels D and F. (G-H) Dwell-time distributions at pH 4.5 (red) and 7.5 (blue) in 100 mM Cl− of FRET states corresponding to αA ‘bound’ (high FRET for Q24C) (G) or ‘loose’ (low FRET for Q24C) (H). Dashed lines are the dye survival plots, showing the fractions of the dyes that have not yet photo-bleached at pH 7.5 (blue) and pH 4.5 (red). Data are averages of 5 independent repeats, and error bars are SEM.
To monitor TM dimer interface rearrangements, we used the previously characterized E385C mutant 31 (Extended Data Fig. 6C). The distance between E385 residues increases from ~35 Å in Swap and Turn to ~50 Å in Twist. Labelled E385C CLC-ec1 predominantly shows a high-FRET efficiency at pH 7.5 with and without Cl− and at pH 4.5 without Cl−, consistently with cryoEM (Extended Data Fig. 6D-F). At pH 4.5 with Cl−, we observe a pronounced broad low-FRET peak (Extended Data Fig. 6G, Supplementary Table 2), consistent with Twist becoming a major conformation.
Single-molecule trajectories revealed discrete transitions between defined high- and low-FRET states in all conditions (Fig. 3B, Extended Data Fig. 7), indicating that the transitions are slower than the 0.1 s sampling rate. A single, averaged FRET state would be observed if transitions occurred on a faster time scale, like the ~0.5 ms Cl−:H+ exchange rate of CLC-ec1 44. The αA transitions between bound (high FRET, Swap) and loose conformations (low FRET, Turn and Twist) show bimodal dwell time distributions and means between ~0.3 and 20 seconds (Fig. 3G-H, Extended Data Fig. 6H-I, Extended Data Fig. 7A-D). These lifetimes are at the limits of our measurement (~0.3–20 s), determined by the sampling and the fluorophore photobleaching rates (Fig. 3G-H, Extended Data Fig. 6H-M). Therefore, the shortest and longest observed lifetimes might be over- and under-estimated, respectively. Nevertheless, the timing of αA movements, corresponding to the opening and closing of the H+ pathway, is at least 100–1000 fold slower than the exchange cycle. Therefore, these rearrangements might reflect slower regulatory events and not transport steps.
At pH 7.5, the dominant αA bound state has ~20 s lifetime (Figure 3G, blue), indicating that most molecules are non-dynamic (Fig. 3B, Extended Data Fig. 7B). A minor population of the αAs bound state is labile, transitioning to loose states with a ~0.6 s lifetime (Fig. 3G). The αA loose state is predominantly transient with lifetimes of ~2–5 seconds (Fig. 3H, blue). The acidification does not significantly affect the αA loose state (Fig. 3H, red), but αA bound at pH 4.5 shows mostly ~0.3 s dwells with non-dynamic periods shortened to ~4 s and less frequent (Fig. 3G, red), suggesting that protons facilitate αA disengagement from αR. Cl− does not affect the lifetimes of αA bound or loose states (Extended Data Fig. 6J-K). The full TM interface is long-lived (>1 s) in Swap and Turn under all conditions (Extended Data Fig. 6H,L). In contrast, Twist with the reduced interface is very short-lived under low activity conditions, and its lifetimes increase to >1s only in pH 4.5 and 100 mM Cl− (Extended Data Fig. 6I, M), consistent with increased Twist populations seen in cryoEM (Fig. 1) and the FRET population plots (Extended Data Fig. 6G).
In sum, the pH-dependent rearrangements of the soluble CLC-ec1 dimer interface, leading to the opening of the H+ pathway, are required for function, concerted and slow, suggesting they correspond to a form of activation gating.
H+ pathway opening favors Cl− pathway hydration and opening
We used molecular dynamics (MD) simulations to understand how opening of the H+ pathway regulates transport by CLC-ec1. We simulated CLC-ec1 dimers in Swap, Turn, Twist, and αA-αR cross-linked Bot Intermediate embedded in POPE/POPG membranes using amino acid protonation states mimicking pH 7.5 or 4.5 (Extended Data Fig. 7A) and with a Cl− ion in the central binding site (Supplementary Table 3). All systems were simulated in 10 independent 1 µs trajectories (Supplementary Table 3). In CLCs, H+ transport occurs via ordered water wires connecting Egate and the Cl− pathway to the intracellular H+ pathway vestibule with a free energy barrier lower when moving along shorter wires 25,26,31,35–37,46–49. αA and H-I loop restrict water access to the H+ pathway in Swap (Fig. 1B), keeping it mostly dehydrated. Ordered water wires form rarely (Extended Data Fig. 7B-F), are short-lived (Extended Data Fig. 7G), and long (7+ waters) (Fig. 4A, F). The Cl− pathway remains rigid during the simulations, and the external gate samples ‘open’ conformations (R > 1.8 Å), which are necessary to allow movement of a dehydrated Cl− ion, with a low ~0.57% probability (Fig. 4G-H). Like Swap, the crosslinked L25C/A450C Intermediate shows a poorly hydrated H+ pathway, rare and long water wires, and a low open probability (~0.67%) of the Cl− pathway (Fig. 4D, F-H). Thus, in states where αA interacts with αR, the H+ pathway is dehydrated and the Cl− pathway is closed preventing ion movement.
Figure 4: Substrate accessibilities of the H+ and Cl− pathways increase in Turn and Twist.
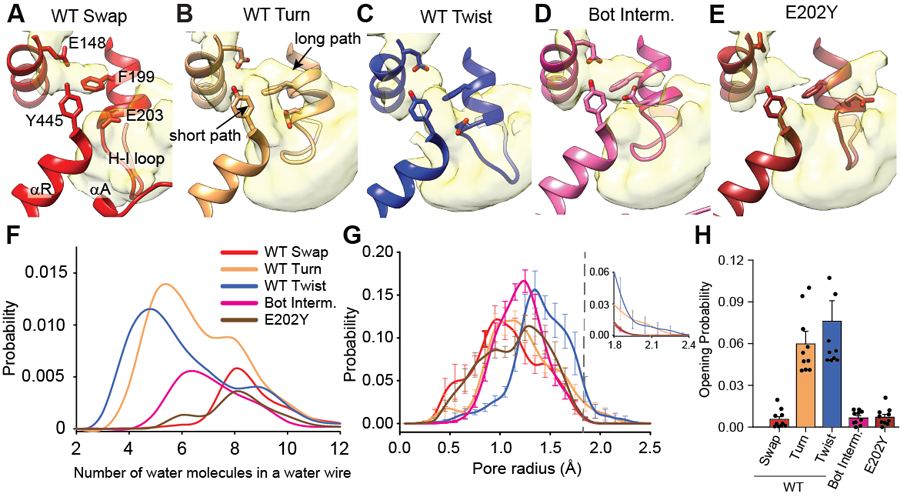
(A-E) Water accessibility of the intracellular H+ pathway vestibule CLC-ec1 WT in Swap (A), Turn (B), Twist (C), Bot Intermediate (D), and E202Y (E). αA, αR, and the H-I loop are shown in cartoon representation. E148, F199, E203, and Y445 are shown in stick representation. Water density is calculated from 10 independent 1 μs long MD trajectories for each system and is shown in surface representation using a threshold of 0.05. (F) Probability distribution of spontaneous formation of ordered water wires connecting E148 to the intracellular solution observed in Swap (red), Turn (wheat), Twist (blue), Bot Intermediate (pink), and E202Y (maroon), as a function of the number of water molecules in the wire. The probability is calculated over the 10 μs total duration of the simulations. (G) Probability distributions of the external gate radius sampled in all trajectories, using the same colors as in panel F. The external gate radius is calculated using the backbone atoms of residues 147, 148, 356, and 357. The top left panel shows the magnified probability distribution for R>1.8 Å. (H) Open probability of the external gate in Swap (red), Turn (wheat), Twist (blue), Bot Intermediate (pink), and E202Y (maroon) calculated as the integral of the area under the radius probability distribution curve for R>1.8 Å. Data in G-H are shown as means ± SEM of 10 independent 1 µs-long MD runs for each condition (Supplementary Table 4).
In Turn and Twist, the H+ pathway is open (Fig. 1) and hydrated (Fig. 4B,C), leading to the more frequent formation of short water wires (3–6 waters) between Egate and the intracellular solution (Fig. 4F). The Cl− pathway becomes dynamic in these states, resulting in a >10-fold increase in the open probability of the external gate to 6.0 and 8.4% in Turn and Twist (Fig. 4G,H, Extended Data Fig. 7H,I). These openings reflect movements of αM, displacing the backbone of I356 and F357 (Extended Data Fig. 7J). Thus, CLC-ec1 activation leads to increased protein dynamics, promoting H+ and Cl− transport by favoring formation of short water wires and opening of the Cl− permeation pathway.
pH-dependent stability of the CLC-ec1 dimer
Our structural and FRET data show that under high-transport conditions (pH 4.5, 100 mM Cl-), CLC-ec1 adopts three dimeric conformations: Swap, Turn, and Twist (Fig. 1, 3). In contrast, Swap is the predominant conformation at pH 7.5. Since the total dimer interface of Swap is larger than that of Turn or Twist (Extended Data Fig. 3F), we hypothesized that neutral pH should stabilize the dimeric state. We tested this prediction by measuring the proportion of fluorescently labelled CLC-ec1 monomers and dimers in membranes as a function of protein density and pH using a single-molecule photobleaching assay 38,40. Consistent with past work, CLC-ec1 exists in equilibrium between monomers and dimers with a macroscopic dissociation constant of (1.5 ± 0.6)·10−8 subunits/lipid at pH 4.5 38,40 (Fig. 5A, Extended Data Fig. 8A). In contrast, CLC-ec1 dimers do not dissociate significantly in the entire measurable surface protein density range at pH 7.5 (Fig. 5A, Extended Data Fig. 8A), indicating dimer stabilization. When we measured the dissociation constants of the functional αA deletion mutant ΔN29 CLC-ec1 40 we found they are (5.4 ± 2.8)·10−7 and (9.7 ± 2.8)·10−10 subunits/lipid at pH 4.5 and 7.5 (Fig. 5B, Extended Data Fig. 8A). Thus, deletion of αA weakens the dimer, consistent with a diminished total interface, but dissociation remains pH-dependent, indicating a role of other regions in this process. Thus, CLC-ec1 is predominantly dimeric in our experimental conditions, a state stabilized by the pH-dependent interactions of αA and other protein regions.
Figure 5. Mechanism of pH-dependent activation of CLC-ec1.
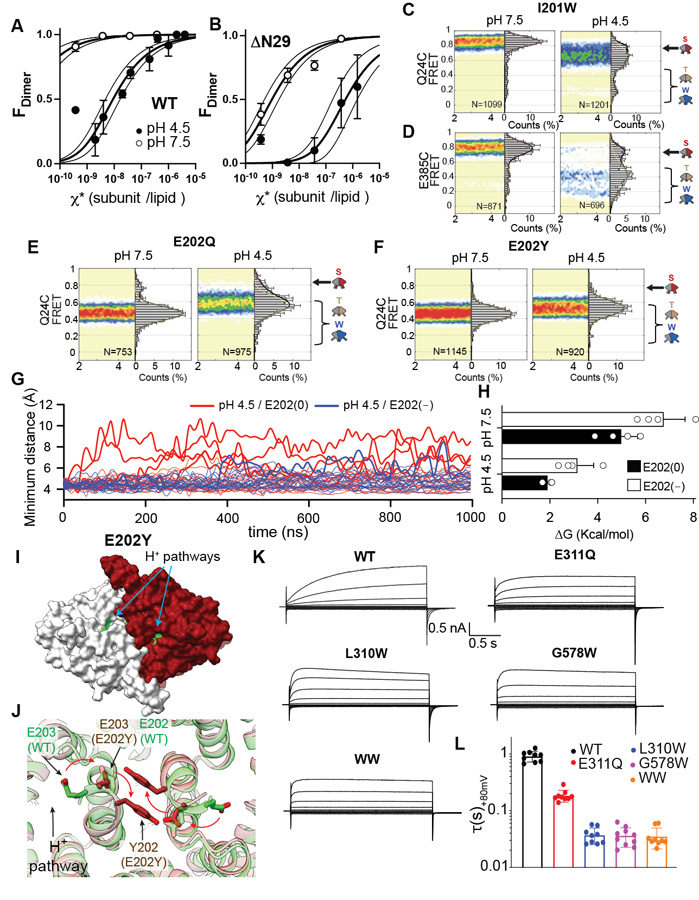
(A-B) CLC-ec1 dimerization isotherms in lipid bilayers at pH 7.5 and 4.5 for WT (A) and ΔN29 (B). Data are averages of at least 3 independent repeats, and error bars are SEM. (C-F) smFRET population contour plots of CLC-ec1 I201W labelled at Q24C (C) or E385C (D), E202Q (E), and E202Y (F) labelled at Q24C at pH 7.5 (left) and 4.5 (right) in 100 mM Cl−. Data are average of 3 independent repeats, and errors are SEM. (G) Time evolution of the minimum distance between the Cα atoms of residues in αA and αR of the sister protomer in 10 independent 1 μs long MD trajectories of Swap at pH 4.5 with E202 deprotonated (E202(−), blue) or protonated (E202(0), red). Thick lines indicate trajectories where the distance becomes > 9 Å and thin ones where it remains < 9 Å. (H) PMF calculations of the free energy costs (ΔG) of separating αA from αR in Swap with no Cl− in the central binding site when E202 is protonated (black) and ionized (white). ΔG is taken from the free energy profiles in Extended Data Fig. 9H. Individual data points and error bars (in SD) are estimated by calculating the PMF values in four consecutive blocks of the trajectories. (I) Surface representation of CLC-ec1 E202Y viewed from the intracellular side. One protomer is shown in gray and the other in maroon. Residues lining the H+ pathway are colored in as in Fig. 1. (J) Closeup view of the rearrangements at the dimer interface in E202Y (maroon) compared to WT Turn (green). E202 and E203 are shown in sticks and arrows denote their repositioning from WT to E202Y. Neighboring helices are shown in transparent cartoon representation. (K) Representative patch clamp currents of ClC-7pm (WT), E311Q, L310W, G578W, and L310W/G578W (WW) mutants. (L) Activation time constants, τ, at +80 mV of CLC-7pm WT, E311Q, L310W, G578W L310W/G578W. Shown are means of at least 3 independent repeats ± SEM. Data is reported in Supplementary Table 5.
Opening mechanism of the H+ pathway
The H-I loops (residues 201–214) participate in the soluble dimer interface of CLC-ec1, interact with the dynamic αAs (Extended Data Fig. 3F-H), and undergo significant rearrangements in Swap, Turn, and Twist (Extended Data Fig. 3C-E), suggesting they contribute to the pH-dependent activation of the CLC-ec1 dimer. We thus investigated their role in the opening of the H+ pathway. First, we introduced the I201W mutation, which destabilizes the CLC-ec1 dimer in detergent micelles 40 while retaining pH-dependent Cl− efflux activity (Extended Data Fig. 8B-E). Mutations at the corresponding residue in human CLCs are associated with disease 50–52 (Supplementary Table 4). At pH 7.5 and 100 mM Cl−, smFRET recordings yielded FRET efficiency distributions of I201W CLC-ec1 labelled at Q24C or E385C centered around high values, indicative of a closed H+ pathway and a full TM interface (Fig. 5C, D). At pH 4.5, both FRET efficiency distributions become broader and shift towards lower values, suggesting opening of the H+ pathway and dynamically rearranging interfaces (Fig. 5C, D). The lower FRET states comprise ~80% of the distribution, corresponding to a ~2-fold increase of Twist-like states in I201W relative to WT (Supplementary Table 2), supporting the notion that Twist is active.
The highly conserved neighboring E202 is the only titratable residue of the H+ pathway accessible to the intracellular solution in Swap (Fig. 1J), suggesting its protonation might play a role in pH-dependent gating. Furthermore, mutations at this position in human CLCs cause disease 52–59 (Supplementary Table 4). Therefore, we examined the effects of neutralizing mutations E202Q and E202Y 35,46 on αA disengagement. In striking contrast to WT and I201W, the smFRET efficiency distributions of labeled Q24C E202Q and Q24C E202Y CLC-ec1 become nearly pH-independent and centered around low FRET values (Fig. 5E-F). Thus, neutralizing E202 induces disengagement of αA. Consistently, in 4 of 10 MD trajectories of Cl−-free Swap at pH 4.5, where E202 is protonated, we observe αA spontaneously detaching from αR (Fig. 5G). In contrast, αA transiently disengaged only in one of 10 trajectories simulated under identical conditions but with deprotonated E202 (Fig. 5G). Potential of mean force (PMF) calculations show that E202 protonation lowers the energy barrier for disengagement of αA from αR by ~2.5 kcal mol−1 (Fig. 5H, Extended Data Fig. 8H-I, Supplementary Table 5). Thus, protonation of E202 promotes activation of CLC-ec1. Notably, the Turn-like QQQ mutant (Extended Data Fig. 3B), with a disengaged αA, was crystallized at high pH 31 where E202 is likely deprotonated. Thus, protonation of other residues in the H+ pathway can also facilitate Swap to Turn transition.
Hydration of the H+ pathway is essential for Cl− transport
Our smFRET experiments suggest that E202Q and E202Y are in Turn-like states. However, while E202Q shows WT-like transport, E202Y impairs the activity of dimeric CLC-ec1 (Extended Data Fig. 8B-C), but not the monomeric I201W/I422W mutant 35. Further, the smFRET efficiency distributions of E385C show that E202Q but not E202Y can adopt Twist-like states (Extended Data Fig. 8F-G). CryoEM imaging of E202Y at pH 4.5 and 100 mM Cl− shows it adopts a single Turn-like conformation with disengaged αAs (Fig. 5I; Extended Data Fig. 5C), as expected from smFRET (Fig. 5F, Extended Data Fig. 8G) and consistent with previous work 35. However, its H+ pathways and dimer interface are severely altered (Fig. 5J). The Y202 residues from two subunits form stacking interactions across the interface, distorting the C-terminal portion of αH and causing a 6 Å displacement of the E203 sidechain away from the H+ pathway (Fig. 5J). Thus, the H+ pathway vestibule becomes less electronegative and more hydrophobic. MD simulations show that in E202Y, the H+ pathway is open but poorly hydrated, so that E148 is connected to the intracellular solution only by rare and long and water wires (Fig. 4F). The Cl− pathway remains constricted with a low ~0.7% probability of the external gate opening (Fig. 4G,H), consistent with the low activity of E202Y. Therefore, hydration of the H+ pathway and dynamic openings of the Cl− pathway are correlated and essential for transport upon CLC-ec1 activation.
Effects of activating mutations on CLC-7
We probed how mutations promoting transitions from the inactive Swap to the transport-competent Turn and Twist in CLC-ec1 affect activation of the mammalian CLC-7 exchanger, which can adopt a Twist-like conformation with dissociated cytosolic domains, though its functional role is unknown 60. We generated E311Q (E202Q in CLC-ec1), L310W (I201W), and two additional mutants destabilizing the dimer interface, G578W (I422W) and L310W/G578W 40 within the plasma membrane-targeted CLC-7PM variant 61. Depolarizing potentials elicit the slow activation of the common gate in the WT CLC-7PM, which does not reach a steady state in 2 s 16,17 (Fig. 5K-L). All mutants expressed well and preserved the characteristic outward rectification and voltage dependence of WT CLC-7PM (Fig. 5E, Extended Data Fig. 8H). However, they displayed drastically accelerated kinetics of common gate activation, with currents reaching steady state with time constants of 30–200 ms (Fig. 5K-L, Supplementary Table 6), suggesting that conformations with opened and hydrated H+ pathways might correspond to states with an activated common gate.
Discussion
Our results unexpectedly show that the CLC-ec1 exchanger undergoes large-scale pH- and Cl−-dependent rearrangements, resulting in a profound remodeling of the TM and soluble dimer interfaces and leading to the opening of its H+ pathways (Fig. 1C,F,I; Extended Data Fig. 3C-E). Our structures show only dimer conformations with two H+ pathways simultaneously open or closed, suggesting these interprotomer rearrangements are cooperative. Opening of only one H+ pathway upon movements of one αA helix and one H-I loop is energetically unfavorable, perhaps because these regions form extensive interactions across the interface, which rearrange as the transporter transitions between the inactive Swap to the activated Turn and Twist states. The H+ pathway opening is required for exchange (Fig. 2) and occurs over time scales of seconds (Fig. 3H), suggesting it is a form of gating rather than a step in the exchange cycle.
Whereas mammalian CLC transporters are gated 16–18, CLC-ec1 was thought not to be gated, as only Swap had been seen in over 70 crystal structures 62,63 and engineered crosslinks preventing intersubunit movements in the TM region did not inhibit function 39. This led to the proposal that Swap is transport competent and that its pH dependence only reflected changes in the H+ concentration 45. However, our data suggest that Swap is inactive while Turn and Twist are active states of CLC-ec1. Swap is the predominant state in low activity conditions, its substrate permeation pathways are occluded and non-conductive, and crosslinks constraining Swap-like αA/αR interactions inhibit transport. In contrast, Turn and Twist predominate in transport conditions; their H+ pathways are open and hydrated, and their Cl− pathways visit conductive conformations more often. Finally, the E148Q mutant of CLC-ec1, which mediates H+-uncoupled Cl− transport 45, displays residual pH-dependent activity 31, suggestive of protonation-dependent rearrangements outside the transport cycle. Indeed, neutralization of two additional residues in the H+ pathway, E113 and E203, was necessary to completely ablate pH dependence of transport 31. Notably, the structure of the resulting CLC-ec1 QQQ 31 mutant (Extended Data Fig. 3B) is very similar to that of Turn. While the structure of QQQ was thought to capture an exchange cycle intermediate, our smFRET data show that transitions in and out of Turn are too slow for transport (Fig. 3), suggesting this mutant captured CLC-ec1 in an activated state. Twist is a dimeric CLC conformation not previously described at high resolution. Recent AFM measurements might have observed it at low pH in lipid membranes 33. Twist is an active state: it is the dominant conformation in membranes in transport conditions, its H+ pathways are open, and it displays the most frequent formation of short water wires and the highest open probability of the Cl− pore. Nevertheless, the Top crosslink, which cannot adopt Twist, has WT transport rates (Fig. 2) 39, suggesting Turn and Twist have comparable activities despite dramatically different TM interfaces (Fig. 1, Extended Data Fig. 3C-D). This is consistent with local rearrangements underlying the exchange cycle. Further work will define the role of Twist in CLC function.
Most CLC exchange mechanisms posit that the protein provides a rigid scaffold with transport entailing only rearrangements of sidechains 47,62–67. However, distal mutations can detrimentally affect the exchange. Our results provide a framework to interpret this long-standing conundrum. We show that Cl− and H+ pathways need to become dynamic to mediate ion exchange, indicating more extensive movements in CLCs than previously thought. Our findings that dimer interface rearrangements control these dynamics provide an initial framework to interpret how regions distal to ion pathways could regulate transport 32,68–72, and how mutations scattered throughout the TM and cytosolic regions can affect gating of human CLCs 2,20,21,73,74.
We propose that pH-dependent activation in CLC-ec1 entails the simultaneous opening of the H+ pathways in both protomers. Protonation of E202, (Fig. 6A,B) favors the retraction of the H-I loop toward the TM region and disengagement of αA from αR, relieving H+ pathway occlusion (Fig. 6C). The opening allows the intracellular H+ vestibules to become more hydrated and short water wires can connect Egate to the intracellular milieu to mediate H+ transport. The E202Y mutation renders the open H+ pathway hydrophobic and dry, abrogates Cl− pathway dynamics and ion transport. Therefore, hydration of the H+ pathway appears necessary to allow openings of the Cl− pathway and Cl−:H+ exchange (Fig. 6D). The proposed activation mechanism is an unusual form of gating as it does not remove a physical occlusion from the Cl− pathway but instead increases protein dynamics to allow transitions along the transport cycle. Further work will reveal whether this activation mechanism is conserved in human CLCs. However, most dominant disease-causing mutations localize at the dimer interfaces of human CLCs (Supplementary Fig. 1C-D) 2,21,24,73–75, and include positions equivalent to I201, E202, and I422W (Supplementary Table 4), suggesting they could alter the extent or mode of H+ pathway opening.
Figure 6. Activation mechanism of CLC-ec1.
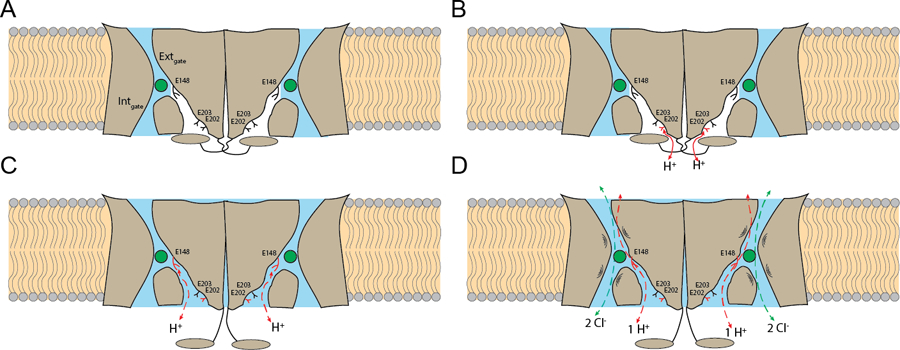
(A) The transporter is inactive. The H+ pathways are dehydrated (in white) and occluded by the cytosolic region (in gray), E148, E202 and E203 are deprotonated (in black), and the Cl− pathways (in blue) are constricted at the internal and external gates. (B) E202 is partially exposed to the intracellular solution and becomes protonated (in red). (C) The cytosolic regions disengage from the transmembrane regions and open the H+ pathways, that become hydrated (in blue) allowing formation of short water wires between E148 and the intracellular solution (red dashed arrows). (D) The Cl− pathways become dynamic, with openings of the internal or external gates to allow Cl−:H+ exchange to occur.
Several lines of evidence support the hypothesis that the rearrangements leading to the opening of the H+ pathway in CLC-ec1 are mechanistically related to the common gate activation characterized for many eukaryotic CLC channels and transporters 3,9,12,13,15–19,27–29. First, the rearrangements observed in CLC-ec1 are cooperative, pH-, and Cl−-dependent, as expected for common gating. Second, they are structurally conserved in CLC-7, which can adopt a Twist-like conformation with dissociated cytosolic regions and opened H+ pathways 60. Third, mutations that favor transitions into conformations with opened H+ pathways in CLC-ec1 accelerate common gate activation in hCLC-7 (Fig. 5K-L). Fourth, many mutations affecting common gating of human CLCs localize at the interface between the cytosolic and TM domains and the H-I loop (Supplementary Fig. 1C-D) 2,21,24,73–75. These considerations suggest that opening of the H+ pathway is likely a conserved feature of common gate activation. The mechanistic details will likely differ between homologues as common gate activation kinetics span nearly four orders of magnitude, and its voltage and pH dependencies differ drastically 2. Indeed, whereas the H+ pathway is gated by the the N-terminal cytosolic helix αA in CLC-ec1 (Fig. 1, Supplementary Fig. 2A,D), the large C-terminal CBS domains would play an analogous role in CLC-7 (Supplementary Fig. 2B-C). Thus, disruption of the CBS-TM interface via mutations or PIP2 depletion76,77 might accelerate common gate activation in CLC-7 by favoring dissociation of the cytosolic domains. In conclusion, we found that the CLC transporters are more dynamic than previously recognized and undergo large-scale subunit rearrangements that enable transport within each protomer. It will be fascinating to see whether and how these mechanisms are tuned to enable the complex regulation of these important proteins.
Materials and Methods
Protein expression and purification
Wild-type and mutant CLC-ec1 were expressed as described previously 45. Briefly, pASK90 plasmid containing CLC-ec1 gene was transformed into DH5α E. coli cells. Cells were grown to an OD of 1.2 – 1.8 at 37 °C with shaking at 250 rpm, induced with 0.2 μg/ml of anhydrotetracycline solubilized in dimethylformamide (Sigma-Aldrich, St. Louis, MO) and harvested 3 h post-induction. Cells were lysed by sonication and extracted with 2% n-decyl-β-D-maltopyranoside (DM) in breaking buffer (100 mM NaCl, 50 mM Tris pH 7.5) (w/v) at RT for 1–2h. Insoluble debris was pelleted, and the supernatant was loaded into a Talon column (Takara bio, Kusatsu, Japan). The column was washed with 30 ml of wash buffer (100 mM NaCl, 20 mM Tris pH 7.5, and 20 mM DM), and 15 ml of wash buffer with 30 mM imidazole and the protein eluted in wash buffer supplemented with 400 mM imidazole. The eluted protein was concentrated using a 50,000 MWCO centrifugal filter (Millipore-Sigma, Saint Louis, MO) and incubated for 1h with Lys-C (Sigma-Aldrich, St. Louis, MO) to remove His-tag. The protein was polished by size exclusion chromatography (SEC) using a Superdex 200 Increase column (GE Life Sciences, Little Chalfont, UK) in 100 mM NaCl, 20 mM Tris pH 7.5, and 5 mM DM (SEC buffer, pH 7.5). Mutations for crosslink constructs, R230C/L249C (Top), L25C/A450C (Bot), and L25C/ R230C/L249C/A450C (Top+Bot), were inserted in the background of Cys-lite CLC-ec1 C85A 38. In these constructs crosslink formed spontaneously during protein synthesis or purification, as in Top39. Crosslink formation was evaluated using SDS/PAGE gels (12%) with or without incubation with 30 mM Tris (2-carboxyethyl) phosphine (TCEP) (Sigma-Aldrich, St. Louis, MO) for 1h. TCEP stock solution was prepared in deionized water with 300 mM TCEP, then titrated to pH 7 with NaOH to prevent protein aggregation. Gel band intensity was analyzed with ImageJ 1.53K 79.
Liposome reconstitution and chloride flux recordings
Following established procedures80, E. coli polar lipids (Avanti, Alabaster, AL) were dried under N2, left overnight under vacuum, resuspended at 20 mg/ml in reconstitution buffer (RB): 300 mM KCl, 25 mM citric acid, 25 mM KH2PO4, pH 7.5 or pH 4.5 with H2SO4, 20 mM 3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS, Thermo Fisher Scientific, Grand Island, NY), and sonicated to clarity. After 1h the protein was added at 0.2 µg/mg protein/lipid (~3·10−6 subunits/lipid molar ratio). Detergent was removed using Bio-Beads SM-2 (Biorad). Liposomes were extruded through 0.4 µm Nucleopore polycarbonate filter (Cytiva, Emeryville, CA). Chloride efflux traces were acquired with DATAQ program, initiated with addition of Valynomicyn, and initial velocity was determined with a linear fit of the first 5 s of the trace44,80. To evaluate effects of TCEP, extruded vesicles were incubated for 1 hr in RB containing 30 mM TCEP. All initial velocities, number of repeats and of independent preparation are reported in Supplementary Table 1.
CryoEM sample preparation and data collection
Affinity-purified protein was polished by SEC on a Superdex 200 column (GE Healthcare) preequilibrated in 100 mM NaCl, 20 mM Tris, pH 7.5, and 3 mM DM immediately before freezing grids. For chloride-free conditions, NaCl was substituted with 100 mM NaGluconate. For pH 4.5 conditions, acetate was used and pH adjusted with NaOH. 3.5 μl of CLC-ec1 at ~1.6 mg/ml were applied to a glow-discharged UltrAuFoil R1.2–1.3 300-mesh gold grid or a homemade gold-coated copper grid (NYU Langone Health Cryo Electron Microscopy Laboratory)81 and incubated for 20 s at 100% humidity at 21°C. Grids were blotted for 1.5 s at +4 blot force and plunge frozen in liquid ethane with Vitrobot Mark IV (Thermo Fisher Scientific, Grand Island, NY). For all WT datasets, micrographs were acquired using a Titan Krios microscope (Thermo Fisher Scientific, Grand Island, NY) operated at 300 kV using a K2 Summit direct electron detector (Gatan), slit width of 20 eV on a GIF Quantum energy filter, the calibrated pixel size was 1.048 Å/pixel, exposure time 10 s, 65.64–72.85 e-/Å2 total dose, 2.7 mm spherical aberration, and −1.2 μm – 2.2 μm defocus range. For the pH 7.5 NaGluconate dataset, detector was equipped with a Cs corrector, calibrated pixel size was 1.098 Å/pixel, 64.08 e-/Å2 total dose, exposure time 8 s. For the Top and Bot crosslink datasets, micrographs were acquired with Titan Krios equipped with a Gatan K3 detector, 0.825 Å/ calibrated pixel size, 58.65–59.59 e-/Å2 doses, and −0.9 μm – 1.9 μm defocus range. For E202Y dataset, micrographs were acquired using Gatan K3 detector, 1.076 Å/pixel calibrated pixel size, 51.79 e-/Å2 total doses, and −1.2 μm – 2.0 μm defocus range. For each dataset ~6,000 -~12,000 micrographs were collected.
CryoEM data processing
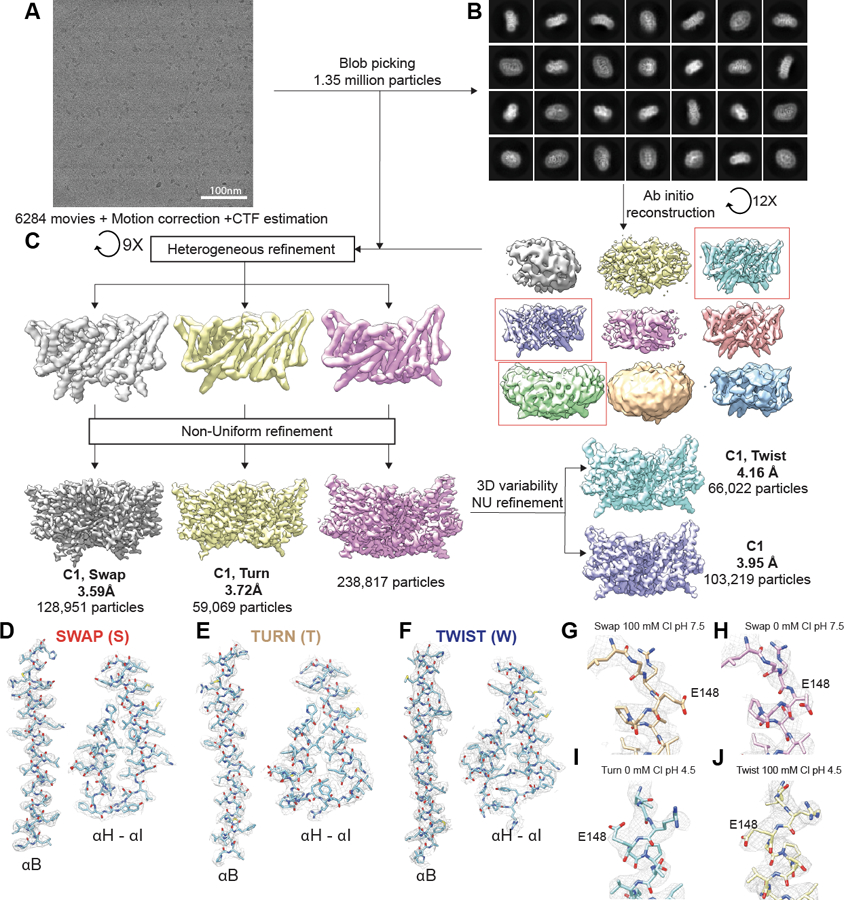
Datasets were processed using CryoSPARC v234. Briefly, movies were imported into CryoSPARC and frames were patch motion-corrected and subject to patch CTF estimation. Images with estimated CTF resolution of 10 Å and lower were removed. Blob picker was used to pick particles. Following 2D classification, the 10–20 best classes were used to pick particle again. Following 2 rounds of 2D classification, ab-initio reconstruction was performed 8–12 times gradually increasing the maximum resolution from 12 to 4 Å. The best 2–3 protein-like initial classes and 2–3 bad initial classes were respectively used as initial models or decoys in heterogeneous refinement where all picked particles were used as input. 3 additional rounds of heterogeneous refinements were performed for each protein-like class followed by several rounds of non-uniform refinement 82. Each conformation was subject to 3D variability analysis 83. For Twist in pH 4.5/NaCl buffer, several rounds of heterogeneous and non-uniform refinement were performed resulting in two maps that differ for small interprotomer rearrangements (Extended Data Fig. 1C). In one map a non-protein density, possibly a detergent molecule, was lodged within the H+ pathway. Thus, we built an atomic model only for the map showing an unobstructed H+ pathway. All datasets were processed using C1 symmetry. 3D variability analysis was also performed in early consensus refinements containing the particles from all the different conformations for all datasets to control for conformational heterogeneity.
Model building, refinement, validation, and availability
The crystal structure of CLC-ec1 (1OTS) was used as an initial model and docked into the density map in Chimera 1.16 84. Atomic model coordinates were refined against final density maps iteratively in Phenix 1.14 85, with secondary structure restraints and no NCS constraints, and manually in Coot 0.8.9.186. Models contain residues: Swap 12 – 461; Turn –19–461, Twist –17–461, Top/Swap –12–461, Top/Turn –19–461, Bot/Intermediate –12–461, Bot/Twist –30–461, and E202Y –22–461. The refined modified model was validated against the other half map (FSCfree). Structures belonging to the same conformational state are similar (r.m.s.d.<1 Å) regardless of pH and Cl− concentration. Therefore, structural comparisons used models determined at pH 4.5 100 mM Cl−. All accession codes are listed in Supplementary Table 7.
Protein labelling and reconstitution into liposomes for smFRET measurements
Q24C and E385C mutations were introduced into C85A CLC-ec1 38. Cell breaking and wash buffers were supplemented with 5 or 1 mM Tris(2-carboxyethyl)phosphine (TCEP) 38. CLC-ec1 mutants were labeled at pH 7.5 with maleimide activated LD555p-MAL and LD650-MAL fluorophores 42 using 1:2:2.5 molar ratios of CLC-ec1:LD555p:LD650 for Q24C and 1:2.5:2 for E385C for 15 min in the dark. Excess dye was removed by SEC. Labelling efficiency was determined using NanoDrop 2000c (Thermo Fisher Scientific, Grand Island, NY) using extinction coefficients (150,000 M−1 cm−1 for LD555p at 550 nm and 250,000 M−1 cm−1 for LD655 at 650 nm). Unspecific labelling efficiency, measured as described87, was 15–23% for LD555p and 10–15% for LD650. Labelling efficiency for CLC-ec1 Q24C and E385C was 70–95% and 55–65% for LD555p, and 65–85% and 90–105% for LD650. Labeled proteins were immediately reconstituted in liposomes as described above with the following differences: E. coli polar lipids were supplemented with 0.01% of 1,2-dioleoyl-sn-glycero-3-phophoethanolamine-N-(biotinyl) (Biotin PE, Avanti, Alabaster, AL). RB contained 100 mM KCl or KGluconate, 1.5 mM citrate, and 1.5 mM phosphate, buffered at pH 7.5 with H2SO4 (internal buffer). CLC-ec1 variants were added at a 5×10−5 subunits/lipid molar ratio and liposomes were extruded through 100 nm polycarbonate filters (Avanti, Alabaster, AL). Proteoliposomes were centrifuged at 21,100 g for 1 minute and the supernatant diluted at a 1:1000 ratio to a final lipid concentration of 20 μg/ml.
smFRET microscopy and data analysis
A home-built prism-based total internal reflection fluorescence (TIRF) microscope constructed around a Nikon Eclipse Ti41 was used for single-molecule FRET recordings. Briefly, samples were illuminated with a 532-nm laser (Laser Quantum), and fluorescence signals were separated with a T635lpxr dichroic filter (Chroma) mounted in a MultiCam apparatus (Cairn). Data was acquired using home-written acquisition software and scientific complementary metal-oxide-semiconductor (sCMOS) cameras (Hamamatsu). Instrument was controlled using custom software written in Labview (National Instruments). Quartz microscope slides and coverslips88 were coated with mPEG-SPA-1000 and NHS-PEG4-Biotin, and functionalized with 0.8 µM streptavidin 89,90. 10–15 µl of liposomes were injected into the microfluidic imaging chamber and immobilized via biotinylated lipids. Nonspecific binding was estimated by immobilizing the same amount of sample using BSA-passivated chambers before streptavidin functionalization. Unbound proteoliposomes were washed away with an external buffer (EB) containing 100 mM KCl (or KGluconate), 25 mM citrate, and 25 mM phosphate at pH 7.5 (or 4.5). EB was supplemented with an oxygen-scavenging system composed of 10–20 mM protocatechuic acid (PCA) and 150 nM protocatechuate-3,4-dioxygenase (PCD) 91, with 2 μM carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP, Sigma-Aldrich, St. Louis, MO) to maintain symmetric pH, and 1 μM valinomycin to collapse the voltage gradient. After injecting EB, proteoliposomes were equilibrated for 10–15 min before recording. Recordings were carried out with a 100 ms integration time using 120–160 mW laser power. Fluorescence intensities were extracted and analyzed using Spartan v3.7 (https://www.scottcblanchardlab.com/software)41 implemented in MATLAB (MathWorks). Fluorescence signals were corrected for spectral bleed-through and trajectories were extracted. Only trajectories with 50+ frames with a signal-to-noise ratio of >8 and ≤4 blinking events were analyzed further. Trajectories with multiple photobleaching events from LD555 or LD655 dyes were discarded. FRET efficiencies were calculated as . Population plots were fitted to 1, 2, or 3 Gaussians in Prism (GraphPad, San Diego, CA). Dwell-time distributions were obtained from trajectories idealized using a 3-state model corresponding to photobleached, helix A loose, and helix A bound for Q24C or photobleached, full interface, and reduced interface for E385C. Dwell-time histograms were fitted to a probability density function 92 in Prism. Each experiment was performed 3+ times using independent protein samples.
Cell culture and patch clamp electrophysiology
cDNAs were generously provided by Michael Pusch in a pcDNA3.1 vector containing both ClC-7PM (referred to as wild type) and OSTMI 93. 48 h before recording, cells were transfected with 200 ng of DNA using lipofectamine 3000 (Thermo Fisher Scientific, Grand Island, NY) according to the manufacturer’s instructions. Patch-clamp experiments were performed in whole-cell configuration. Pipettes were pulled from borosilicate glass capillaries (Kimble, DWK life sciences, Millville, NJ) with resistance of 2–3 MΩ. Intracellular solution: 130 mM CsCl, 2 mM EGTA, 2 mM MgSO4 and 10 mM HEPES at pH 7.4; extracellular solution: 140 mM NaCl, 2 mM MgSO4, 2 mM CaCl2 and 10 mM HEPES at pH 7.4. Voltage protocol: 25 ms at Vhold=−30 mV, 2 s Vpulse from −100 mV to +100 mV in 20 mV steps, 0.5 s Vtail=−60 mV. Data was acquired with Axopatch 200 amplifier (Molecular Devices, LLC, San Jose, CA, USA) at 50 kHz after filtering at 10 kHz with an eight-pole Bessel filter using pClamp (Molecular Devices, Union City, CA) and analyzed using the custom program ANA (available at http://users.ge.ibf.cnr.it/pusch/) and Prism (GraphPad, San Diego, CA). The step was fit to an exponential function to obtain activation kinetics:
Where is the current, and are the time-independent and time-dependent amplitudes, and is the time constant. For WT CLC-7 the whole trace was fit, for mutants with fast kinetics only the first 500 ms were fit. All CLC-7 constructs were recorded from 3+ independent transfections.
Free MD simulations
CryoEM coordinates of Swap (residues 12–461), Turn (residues 19–461), Twist (residues 17–461), Bot-Intermediate (residues 12–461), and E202Y (residues 22–461) determined at pH 4.5, 100 mM Cl− were used as initial conditions. Truncated sidechains of other residues were rebuilt using psfgen tool in VMD software version 1.9.394. For Bot-Intermediate, C25 and C450 were set to be covalently bonded. All simulation boxes were orthorhombic with dimensions of 120 Å × 120 Å × 100 Å with periodic boundary conditions applied along the x-y-z axes. The center of mass of CLC-ec1 was placed at the center of the box, with the longest dimer axis aligned to the diagonal of the x-y plane, to maximize distance between protein and its periodic image. Each system (~150,000 total atoms) was constructed using membrane builder tool in CHARMM-GUI website (http://www.charmm-gui.org/)95 as follows: the protein was embedded in a lipid membrane consisting of 270 POPE and 90 POPG molecules, solvated with ~31,000 waters, ~150 K+ and ~80 Cl− ions were added in the solvent space to neutralize the net charge of the system and mirror physiological ionic strength (100 mM). CHARMM36 force field96 was employed for the protein, lipids, and ions, and TIP3P model97 for waters. Simulations were performed with Gromacs v2020.2 98, interfaced with PLUMED v2.7 99 to apply bias. Long-range electrostatic interactions were treated with the Particle Mesh Ewald (PME) method100. A 12 Å cut-off distance was used for Lennard-Jones and real space Coulomb interaction. LINC algorithm101 was used to constrain all covalent bonds involving hydrogen atoms of the protein and water molecules. Initial energy minimization and equilibration steps were performed following the CHARMM-GUI setup. Ten (five) replicas were generated for each system by assigning initial velocities at 300 K using different random seed at start of equilibration. After equilibration, each replica ran for 20 ns with positional restraints on protein backbone atoms to relax the lipid structure around the protein, followed by production run for 1 µs with all positional restraints released. All simulations were performed in the semi-isotropic NPT ensemble at T=300 K and P=1 atm.
pKas of ionizable side chains (Asp, Glu, His, Lys and Arg) were calculated by PropKa v3.1 102,103 (Extended Data Fig. 8A). Residues were set to be protonated (doubly protonated for His) when calculated pKa ≥ 7.5 or 4.5, to mimic experimental conditions. In systems #1–6 (Supplementary Table 3), a Cl− ion was placed in Scen of each monomer. In systems #7–12 (Supplementary Table 3 and 5) all sites were empty and a half-sided harmonic restraint (Eq. 1), with force constant kcal/mol∙Å2 and reference distance r0= 30 Å, was applied to Cl− ions in solution104–106, to prevent binding
| (1) |
Analysis of external pore radius and formation of continuous water chains
The radius of the Cl− pathway was measured using HOLE78. The external gate was defined as the midpoint of the backbone atoms of R147, E148, I356, and F357. Formation of hydrogen bonded water chains between E148 and the intracellular solution in each MD frame was monitored as described31. The upper boundary of the intracellular solution space was defined as z = −12 Å, with the center of mass of protein set at z = 0 Å. The cutoff distance for hydrogen bonds was set at 2.8 Å for the O-O distances between waters, and water and E148 carboxyl oxygens. When more than one continuous water chains were found in the same MD frame, the shortest chain was considered. Probability distributions were normalized by the total number of MD frames.
PMF calculations
For forward potential of mean force (PMF) calculations the last snapshot of the equilibration run of free MD was used as initial conditions. The reaction coordinate (RC) of the PMF was defined as the minimum distance between positions in groups A (Cα atoms of Q12-K30 and the center of mass (COM) of Q12-R19 sidechains in protomer A) and B (Cα atoms of S446-L461, the sidechain COM of Q456-L461, and Cα atoms of Q207, F208, R403, Q437, K442, and P443 in protomer B). The minimum distance was calculated by Eq. 2 using Multicolvar tool implemented in PLUMED99, where was the distance between position in group A and position in group B, and Å.
| (2) |
105 umbrella sampling windows were generated with center positions, , between 3.5–13.9 Å at 0.1 Å intervals. A harmonic restraint, , was applied at every window with force constant kcal/mol∙Å2. Unfolding during pulling was prevented by applying alpha-helical restraints to and backbone dihedral angles of A13-L16 on αA and E457-Q460 on αR with kcal/mol∙rad2 at and . Initial configurations were equilibrated at for 10 ns. Final snapshot was taken as the initial configuration of adjacent windows (). The system was repeatedly pulled in this fashion until or 13.9 Å. Replica exchange umbrella sampling (REUS)107 simulation started from the last MD snapshot of the initial 10 ns run. The Metropolis Monte Carlo exchange criterion was applied every 20 ps to determine swapping of neighboring windows108,109. Average acceptance ratio for successful exchange of neighboring windows was ~15 % over all windows. Each window was sampled for 30 ns. The error bar of the resulting PMF was <1 kcal/mol. The RC values were recorded every 1 ps. The PMF was calculated using WHAM110. The tolerance parameter of the PMF convergence and the bin width in the WHAM calculation were 10−4 kcal/mol and 0.1 Å. ΔG of disengagement of αA was calculated as the PMF difference between the local energy minima at RC = 4 Å and > 9 Å. To determine hysteresis error, “reverse” PMFs were calculated for all systems following the same procedure as forward REUS run but with last MD snapshot of window of of the forward run as initial condition.
Photobleaching analysis of CLC-ec1 dimerization in membranes
WT or ΔN29 CLC-ec1 on a C85A/H234C background and with a hexahistidine tag on the C-terminus was expressed, purified, and labelled with either Cy5 Maleimide (i.e. Sulfo-Cyanine5 Maleimide, GE) or Cyanine5 Maleimide (Lumiprobe)38. Subunit labeling yields were calculated using the reported correction factors: and , resulting in subunit yields of (mean±sem, n = 6) for WT and (mean±sem, n = 6) for ΔN29. Protein was reconstituted into 2:1 POPE/POPG lipids (Avanti) in 300 mM KCl, 20 mM Citrate, 10 mM MES, 20 mM Hepes, pH to 4.5 or 7.5 at a defined subunit/lipid mole fraction, . After dialysis, liposomes were collected and freeze/thawed three times at −80°C/25°C. Samples were incubated at room temperature in the dark for 3–13 days and extruted 21 times through a 400 nm nucleopore membrane (Whatman) using a manual LiposoFast-basic extruder (Avestin) immediately prior to imaging using a single-molecule micro-mirror TIRF fluorescence microscope built on an RM21 platform (Mad City Labs Inc.) following the CoSMoS design111. Images were analyzed using custom MATLAB software112. Each spot in the image corresponded to a liposome containing 1+ subunits of labelled CLC-ec1. Discrete photobleaching events in liposomes were detected. Photobleaching probability distribution, (, , ) was determined for each sample. The dimer benchmark for (, , ) was determined by fitting WT pH 7.5 photobleaching data using a lab-built Poisson simulation in MATLAB38, assuming and lipid surface area 0.6 nm2. Best-fit corresponded to labeling yield of 0.85, with 0.10 background labeling, possibly as dark fluorophores reactivate on microscope. Expected monomer signal was simulated with these parameters, and dimer fraction, , was estimated by least-squares analysis of experimental vs. monomer-dimer photobleaching probability distributions. The corresponding vs. reactive mole fraction, , data were fit to equilibrium dimerization isotherms, assuming that the reaction persists in monomer-dimer equilibrium. Since the new structures indicate additional dimer states with linkage to protonation, the equilibrium data is reported as a macroscopic in subunits/lipid. All data is reported as mean±SEM, with n>3 independently prepared samples.
Extended Data
Extended Data Fig. 1. CryoEM image processing scheme for the dataset at pH 4.5 in 100 mM Cl−.
(A) Representative cryoEM micrographs of CLC-ec1. (B) Representative 2D classes from CryoSPARC. (C) Summary of the image processing procedure. Red frames indicate classes that were selected for further processing. (D-F) Density maps (mesh) for αB, αH, αI, and the H-I loop with the refined models of Swap (D), Turn (E), and Twist (F). (G-J) EM density (mesh) and atomic models of E148 for Swap at pH 7.5 and 100 mM Cl− (G), Swap at pH 7.5 and 0 mM Cl− (H), Turn at pH 4.5 and 0 mM Cl− (I), and Twist and pH 4.5 and 100 mM Cl− (J). Data acquisition and refinement parameters are reported in Table 1 and 2.
Extended Data Fig. 2. Validation of maps and models for WT CLC-ec1.
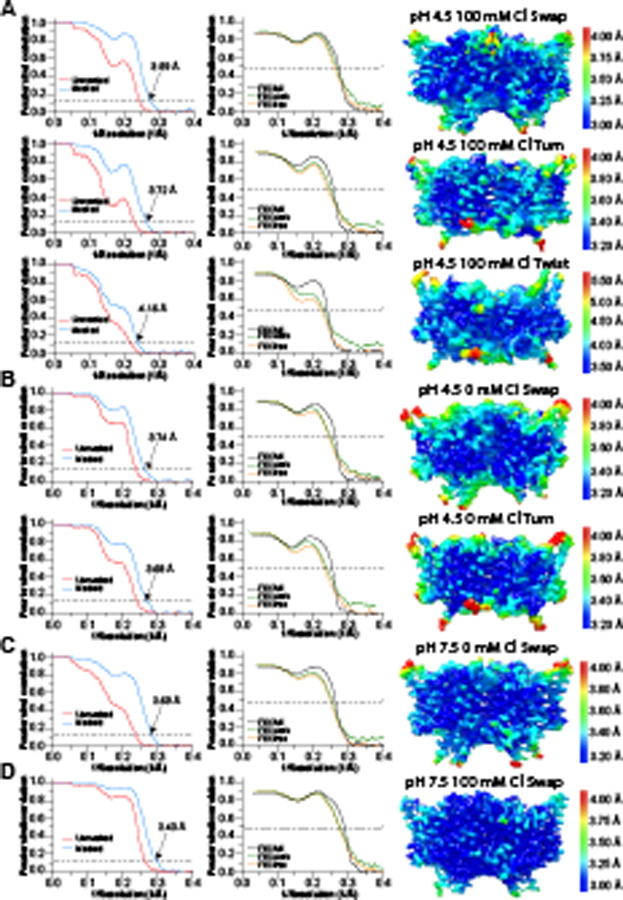
Fourier shell correlation (FSC) curves for the masked (tight mask from CryoSPARC) and unmasked density maps (left), FSC curves from model-map cross-validation (middle), and density maps colored by local resolution estimated in CryoSPARC (right) of maps obtained in the following conditions: pH 4.5 and 100 mM Cl− (A), pH 4.5 and 0 mM Cl− (B), pH 7.5 and 0 mM Cl− (C), pH 7.5 and 100 mM Cl− (D). Data acquisition and refinement parameters are reported in Table 1.
Extended Data Fig. 3. Structural comparisons between different CLC-ec1 conformations.
(A-B) Per residue Cα r.m.s.d. for CLC-ec1 in Swap at pH 7.5 and 100 mM Cl− relative to the crystal structure (PDB: 1OTS) (A) and for Turn at pH 4.5 and 100 mM Cl− relative to E113Q/E148Q/E203Q (QQQ, PDB: 6V2J) (B). (C-E) Close-up view of the rearrangements of the H-I loops in Swap (C), Turn (D), and Twist (E). The top panels are viewed from the plane of the membrane, and the bottom panels are viewed from the intracellular side. One protomer is colored gray, and the other is colored red (Swap), wheat (Turn), and Blue (Twist). Helices αH and αI are shown in cartoon representation, and residues Q207, F208, R209, Y210, T211, and L212 are shown in stick CPK representation. (F) Quantification of the surface area per protomer buried at the interface of the soluble (gray bars) and TM (black bars) domains in Swap, Turn, and Twist. (G-H) Cartoon representations of single protomers viewed from the dimer interface and colored by changes in per-residue contact areas in Turn (G) and Twist (H) relative to Swap. (I) Diameter of the Cl− permeation pathway visualized using HOLE 1 in cryoEM structures. The position of the external gate is defined as the midpoint of the backbone atoms of residues 147, 148, 356, and 357. Residues S107, E148, I356, and Y445 are shown as sticks. Blue indicates regions with R>2.3 Å, green 2.3 Å>R>1.15 Å, and red R< 1.15 Å.
Extended Data Fig. 4. Characterization of Top and Bot crosslinks.
(A-B) SDS PAGE showing the molecular mass of TCEP-treated (right) or untreated (left) CLC-ec1 WT, Top (R230C/L249C) and Bot (L25C/A450C) crosslinks, L25C and A450C single mutants, and the Top+Bot quadruple mutant. Arrows denoted by D and M indicate the position of the dimer and monomer, respectively. Note that L25C on helix αA spontaneously forms dimers, reflecting crosslinks between the two dynamic αA helices. This results in two dimer bands for the Bot and Top+Bot crosslinked constructs, where αA of one protomer is crosslinked either to αA or αR of the sister protomer. SDS PAGE results were consistent in three repeats from independently obtained proteo-liposome samples of WT and all mutants. (C) Time courses of Cl− efflux mediated by TCEP-treated (right) or untreated (left) proteoliposomes reconstituted with CLC-ec1 WT (black/blue), Bot (pink/dark green), L25C (light green/gray), Top (orange/brown), A450C (purple/yellow) and Top+Bot (red/cyan). Traces for WT and Bot ±TCEP are the same as those shown in Fig. 2. (D) Intracellular view of cryoEM density maps of CLC-ec1 Bot crosslink in Intermediate (left panel) and Twist (right panel) conformations imaged at pH 4.5 in 100 mM Cl−. One subunit is colored gray, and the other is pink (Intermediate) or yellow (Twist). (E) CryoEM density (mesh) contoured at 5.1 Å and atomic model of the crosslink between A450C and L25C in Bot Intermediate. (F) Intracellular view of cryoEM density maps of CLC-ec1 Top crosslink in Swap (left panel) and Turn (right panel) conformations imaged at pH 4.5 in 100 mM Cl−. One subunit is colored gray, and the other is green (Swap) or aquamarine (Turn). (G) CryoEM density (mesh) contoured at 6.0 σ and atomic model of the crosslink between A450C and L25C in Top Swap.
Extended Data Fig. 5. Validation of maps and models for CLC-ec1 mutants.
(A-C) FSC curves for the masked (tight mask from CryoSPARC) and unmasked density maps (left), FSC curves from model-map cross-validation (middle), and density maps colored by local resolution estimated in CryoSPARC (right) of maps obtained for CLC-ec1 (A) L25C/A450C Bot Intermediate and Twist, (B) R230C/L249C Top Swap and Turn, (C) E202Y. All data were collected at pH 4.5 and 100 mM Cl−. Data acquisition and refinement parameters are reported in Table 2.
Extended Data Fig. 6. Characterization of CLC-ec1 mutants used in smFRET experiments and the dynamics of the dimer interface.
(A) Representative normalized time course of chloride efflux from liposomes reconstituted at 0.2 μg protein/mg lipid with CLC-ec1 WT (black), Q24C (orange) and Q24C labelled with maleimide activated LD555p and LD650 fluorophores in the same molar ratio used in smFRET TIRF imaging (CLC-ec1:LD555p:LD650 = 1:2:2.5) (green). Traces were normalized to the total chloride content of the liposomes determined following the addition of detergent 2. (B) The initial velocity of Cl− efflux from proteo-liposomes containing WT CLC-ec1 (red), CLC-ec1 Q24C (orange), and Q24C-labelled (green) All values are means ± S.D. The number of repeats of independent experiments and preparations of proteo-liposome samples for all constructs are reported in Supplementary Table 1. (C) The experimental design to monitor dynamics of the TM dimer interface tracking the movement of E385C labelled with donor and acceptor fluorophores. CLC-ec1 dimer is shown as cartoon and colored as in Figure 1. The labeling site is shown as a black sphere, and inter-dye distances are shown on the structures. Schematic representations of Swap, Turn, and Twist are on the left. (D-G) Population contour plots of labelled E385C at pH 7.5 in 0 mM Cl− (D), pH 7.5 in 100 mM Cl− (E), pH 4.5 in 0 mM Cl− (F), and pH 4.5 in 100 mM Cl− (G). N, the number of molecules used to construct the plots, is shown on each panel. Cumulative FRET state histograms are on the right of each panel plotted as means of three independent experiments ± SEM. Assignments of the FRET states to the protein conformations are on the right of panel G. (H-M) Dwell-time distributions of FRET states corresponding to: ‘full TM interface’ (high FRET for E385C, Swap or Turn) (H) or ‘reduced TM interface’ (low FRET for E385C, Twist) (I) in 100 mM Cl− or 0 Cl− (L and M, respectively); αA bound (high FRET for Q24C, Swap) (J) or αA loose (low FRET for Q24C, Turn and Twist) (K) in 0 mM Cl-. Dashed lines are the fluorophore survival probability, which refers to the fraction of molecules where both LD555 and D655 survived longer than the time indicated in the x-axis at pH 7.5 (blue) and pH 4.5 (red). Data are averages of three independent repeats, and error bars are SEM.
Extended Data Fig. 7. Examples of smFRET recordings of CLC-ec1.
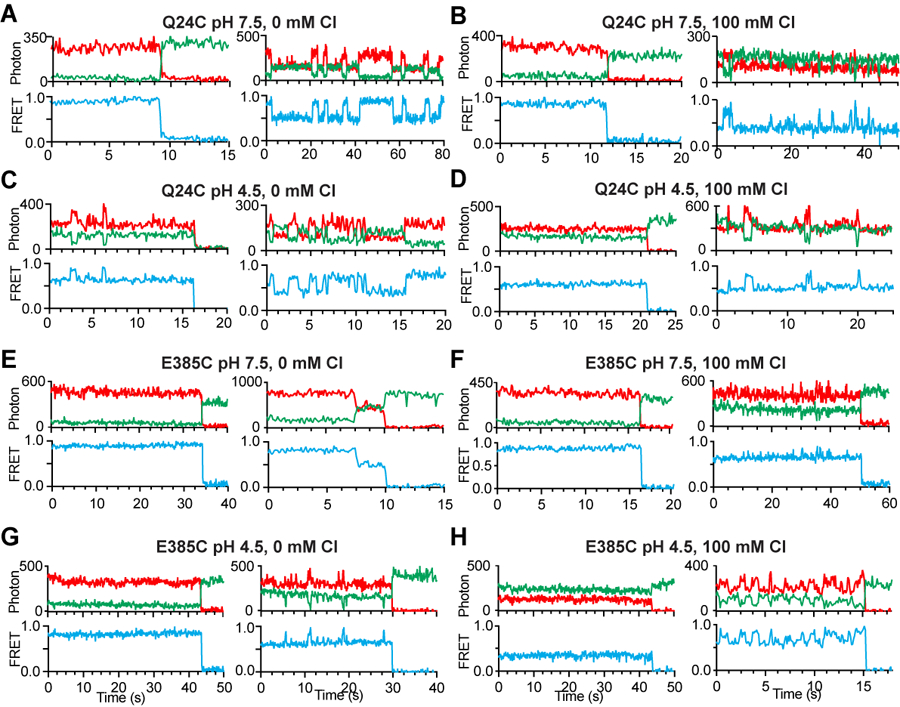
(A-H) Representative smFRET traces of CLC-ec1 labelled at E385C at pH 7.5 and 0 mM Cl− (A), pH 7.5 and 100 mM Cl− (B), pH 4.5 and 0 mM Cl− (C), pH 4.5 and 100 mM Cl− (D), or CLC-ec1 labelled at Q24C at pH 7.5 and 0 mM Cl− (E), pH 7.5 and 100 mM Cl− (F), pH 4.5 and 0 mM Cl− (G) and pH 4.5 and 100 mM Cl− (H). Fluorescence emissions from LD555p (green) and LD655 (red) dyes are shown in the top panels, and FRET efficiency (blue) is in the bottom panels. The traces were selected to illustrate the heterogeneity of the dynamic behaviors observed in all collected datasets. The quantitative analysis of the FRET-efficiency and dwell time distributions is reported in Figure 3 and Extended Data Fig. 6.
Extended Data Fig. 8. Water wire formation and Cl− pathway dynamics of CLC-ec1.
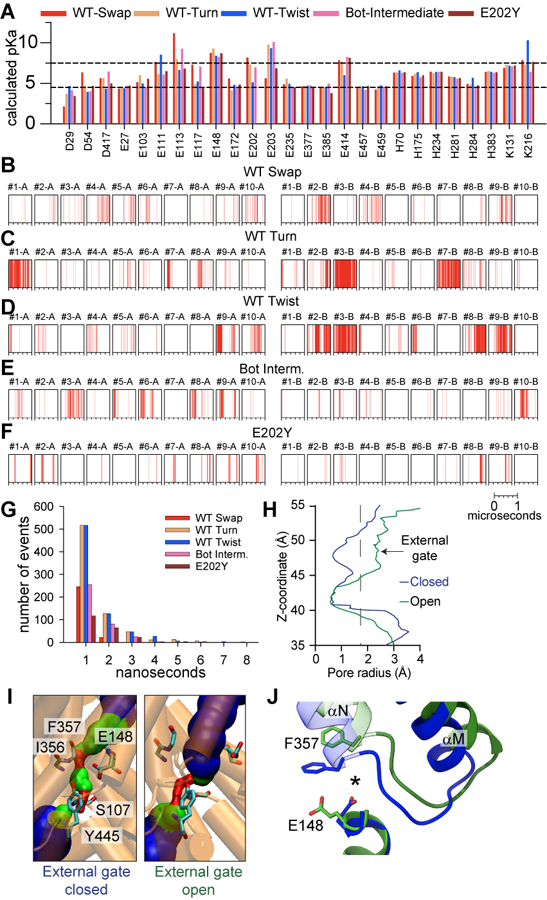
(A) The pKa values of the ionizable residues calculated using PropKa3,4. Black dotted lines correspond to pH 4.5 and 7.5. Data are not shown for residues with the calculated pKa values below 4.5 or above 7.5 in all conformations. (B-F) Spontaneous formation of water wires connecting Gluex and the intracellular bulk water within the H+ pathways of protomers A and B, observed in 10 independent 1 μs long simulations of CLC-ec1 WT in Swap (B), Turn (C), Twist (D), Bot Intermediate crosslinked (E), and E202Y (F). Each vertical line in the panels shows the occurrence of a water wire at that time point. The number of events of formation of water wires as a function of the lifetime of individual event. The lifetime of water wire is defined as the time duration for any hydrogen bond in the water wire to be disconnected. (H)Representative pore-radius profiles along the z-axis of the Cl− pathway, calculated using HOLE 1 for conformations with the open (green) and closed (blue) external gate seen in the MD simulations of Twist. (I) The diameter of the Cl− permeation pathway, visualized using HOLE 1 in MD snapshots of Twist with an open (right panel) and closed (left panel) extracellular gate. The external gate radius is calculated using the backbone atoms of residues 147, 148, 356, and 357. Residues S107, E148, I356, and Y445 are shown as sticks. Blue indicates regions with R>2.3 Å, green 2.3 Å>R>1.15 Å, and red R< 1.15 Å. (J) Representative snapshots of conformational rearrangements in the backbone of the Cl− pathway when the external gate is open (green) or closed (blue). * denotes the center of the Cl− permeation pathway.
Extended Data Fig. 9. Characterization of activating mutations in CLC-ec1 and CLC-7.
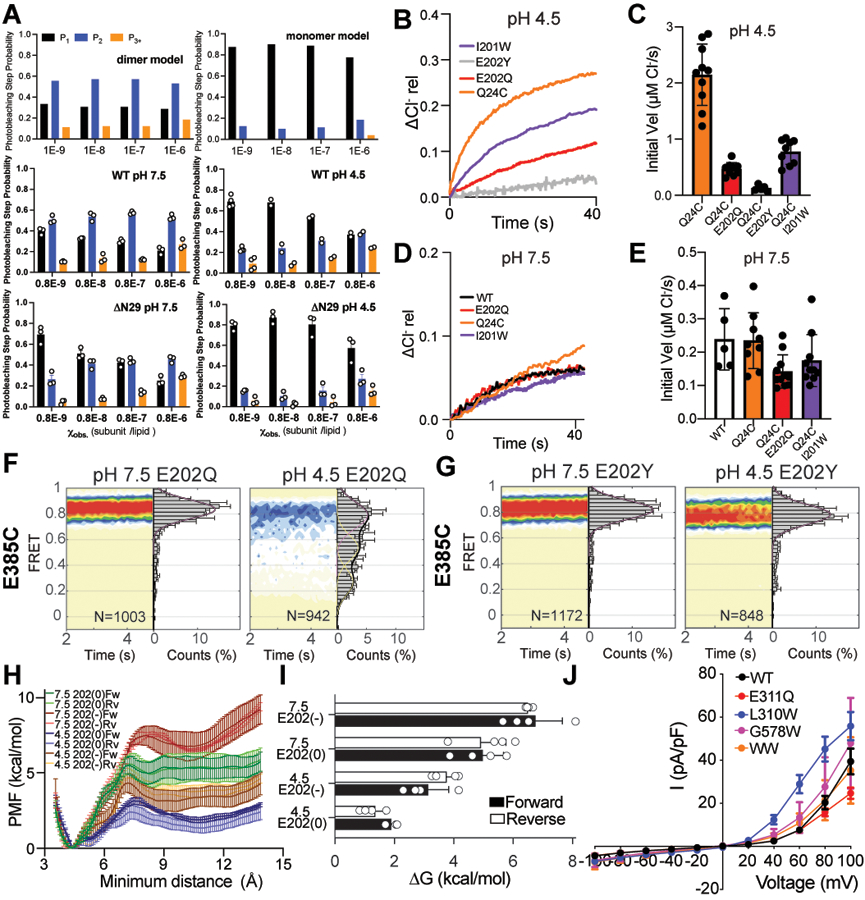
(A) Theoretical (top panels - dimer & monomer) and experimental (middle and bottom panels – WT & ΔN29 pH 7.5 & 4.5) photobleaching steps probability distributions (P1, P2, P3+) for CLC-ec1 in lipid bilayers as a function of the experimental subunit/lipid mole fraction, 𝜒obs. Data for WT and ΔN29 pH 7.5 & 4.5 are means ± SEM for n = 4 for WT pH 4.5 and n = 3 for all other systems, where n is the number of independently prepared samples. (B, D) Representative normalized time courses of chloride efflux from liposomes reconstituted at pH 4.5 (B) or 7.5 (D) with CLC-ec1 WT (black), Q24C (orange), I201W (purple), E202Q (red) and E202Y (grey) at 0.2 μg protein/mg lipid. Traces were normalized to the total chloride content of the liposomes determined following the addition of detergent 2. (C-E) Initial velocity of Cl− efflux at pH 4.5 (C) and 7.5 (E) of proteoliposomes reconstituted with CLC-ec1 WT (white), Q24C (orange), I201W (purple), E202Q (red) and E202Y (grey). All values are shown as means ± S.D. The number of repeats of independent experiments and preparations of proteo-liposome samples for all constructs are reported in Supplementary Table 1. (F-G) smFRET population contour plots of CLC-ec1 E202Q (F) and E202Y (G) labelled at E385C at pH 7.5 (left) and 4.5 (right) in 100 mM Cl−. Data are average of 3 independent repeats, and errors are SEM. (H) Forward and reverse potential of mean force (PMF) profile as a function of the minimum distance between αA of monomer A and αR of monomer B with E202 protonated, E202(0), or deprotonated, E202(−), at pH 4.5 or pH 7.5 with no Cl− in the central binding site. Molecular systems are listed in Supplementary Table 4. Error bars (in SD) are estimated by calculating the PMF values in four consecutive blocks of the trajectories. (I) Free energy difference between local energy minima for αA ‘bound’ to and ‘loose’ from αR with E202 protonated (E2020) or deprotonated (E202−) at pH 4.5 or pH 7.5 with no Cl− in the central binding site. ΔG is taken from the free energy profiles in panel H. Error bars are estimated in the same fashion as in panel H. (J) Current density-voltage curves for CLC-7PM WT (black), E311Q (red), L310W (blue), G578W (pink), and L310W/G578W (WW, orange). Data are averages of 10 independent repeats, and error bars are SEM.
Supplementary Material
Acknowledgements
The authors thank members of the Accardi and Boudker labs, Crina Nimigean and Simon Scheuring for helpful discussions. We thank Richard Hite for help with cryoEM data analysis. We thank Dr. Scott Blanchard for help establishing smFRET resources. The work was supported by National Institutes of Health (NIH) Grant GM128420 (to A.A.), R01GM120260 and R21GM126476 (to J.L.R.), and American Heart Association Grant 19PRE34380215 (to D.C.). The electron microcopy data were collected on the Tiran Krios at the Cryo-EM Core Facility at NYU Langone Health, with the assistance of William Rice and Bing Wang (RRID: SCR_019202). Some of this work was performed at the Simons Electron Microscopy Center and National Resource for Automated Molecular Microscopy located at the New York Structural Biology Center, supported by grants from the Simons Foundation (SF349247), NYSTAR, and the NIH National Institute of General Medical Sciences (GM103310). The computational resource of this research was provided in part by TACC Stampede2, SDSC Expanse, PSC Bridges2, and NCSA Delta through allocation number MCB200216 from the Extreme Science and Engineering Discovery Environment (XSEDE), which was supported by National Science Foundation grant number #1548562, and in part by HPC resources supported by the Scientific Computing Unit at Weill Cornell Medicine.
Footnotes
Competing Interests statement
The authors declare no competing interests.
Data Availability
All constructs are available on request to A.A. or O.B. All models and associated cryoEM maps have been deposited into the Electron Microscopy Data Bank (EMDB) and the Protein Data Bank (PDB) under the following accession codes CLC-ec1 pH 7.5 0 mM Cl− Swap (PDB: 7RNX; EMD-24582); CLC-ec1 pH 7.5 100 mM Cl− Swap (PDB: 7RO0; EMD-24584); CLC-ec1 pH 4.5 0 mM Cl− Swap (PDB: 7RP6; EMD-24668); CLC-ec1 pH 4.5 0 mM Cl− Turn (PDB:7RSB; EMD-24583); CLC-ec1 pH 4.5 100 mM Cl− Swap (PDB: 7N8P; EMD-24241); CLC-ec1 pH 4.5 100 mM Cl− Turn (PDB: 7RP5; EMD-24612); CLC-ec1pH 4.5 100 mM Cl− Twist (PDB: 7N0W; EMD-24263); R230C/L249C pH 4.5 100mM Cl− Swap (PDB: 8GA1; EMD-29884); R230C/L249C pH 4.5 100mM Cl− Turn (PDB: 8GA3; EMD-29885); L25C/A450C pH 4.5 100mM Cl− Intermediate (PDB:8GA5; EMD-29890); L25C/A450C pH 4.5 100mM Cl− Twist (PDB: 8GAH; EMD-29899); E202Y pH 4.5 100mM Cl− (PDB: 8GA0; EMD-29883) which are listed in Supplementary Table 7.The atomic coordinates of the free molecular dynamics simulation at t = 0 and 1 microseconds of systems 1–8 listed in Supplementary Table 3 are available at https://doi.org/10.6084/m9.figshare.24777321.v1.
References
- 1.Accardi A Structure and gating of CLC channels and exchangers. The Journal of Physiology 593, 4129–4138, doi: 10.1113/jp270575 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jentsch TJ & Pusch M CLC Chloride Channels and Transporters: Structure, Function, Physiology, and Disease. Physiological Reviews 98, 1493–1590, doi: 10.1152/physrev.00047.2017 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Miller C Open-state substructure of single chloride channels from Torpedo electroplax. Philos. Trans. R. Soc. Lond. B Biol. Sci 299, 401–411 (1982). [DOI] [PubMed] [Google Scholar]
- 4.Ludewig U, Pusch M & Jentsch TJ Independent gating of single pores in CLC-0 chloride channels. Biophys. J 73, 789–797 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weinreich F & Jentsch TJ Pores formed by single subunits in mixed dimers of different CLC chloride channels. J. Biol. Chem 276, 2347–2353 (2001). [DOI] [PubMed] [Google Scholar]
- 6.Middleton RE, Pheasant DJ & Miller C Homodimeric architecture of a ClC-type chloride ion channel. Nature 383, 337–340 (1996). [DOI] [PubMed] [Google Scholar]
- 7.Saviane C, Conti F & Pusch M The muscle chloride channel ClC-1 has a double-barreled appearance that is differentially affected in dominant and recessive myotonia. J. Gen. Physiol 113, 457–468 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jentsch TJ, Steinmeyer K & Schwarz G Primary structure of Torpedo marmorata chloride channel isolated by expression cloning in Xenopus oocytes. Nature 348, 510–514 (1990). [DOI] [PubMed] [Google Scholar]
- 9.Fischer M, Janssen AG & Fahlke C Barttin activates ClC-K channel function by modulating gating. J Am Soc Nephrol 21, 1281–1289, doi: 10.1681/ASN.2009121274 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stölting G, Fischer M & Fahlke C ClC-1 and ClC-2 form hetero-dimeric channels with novel protopore functions. Pflugers Arch 466, 2191–2204, doi: 10.1007/s00424-014-1490-6 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Pusch M, Ludewig U, Rehfeldt A & Jentsch TJ Gating of the voltage-dependent chloride channel CIC-0 by the permeant anion. Nature 373, 527–531 (1995). [DOI] [PubMed] [Google Scholar]
- 12.Accardi A & Pusch M Fast and slow gating relaxations in the muscle chloride channel CLC-1. J. Gen. Physiol 116, 433–444 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zúñiga L et al. The voltage-dependent ClC-2 chloride channel has a dual gating mechanism. J. Physiol 555, 671–682 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zifarelli G, Pusch M & Fong P Altered voltage-dependence of slowly activating chloride-proton antiport by late endosomal ClC-6 explains distinct neurological disorders. J Physiol 600, 2147–2164, doi: 10.1113/jp282737 (2022). [DOI] [PubMed] [Google Scholar]
- 15.Matsuda JJ et al. Overexpression of CLC-3 in HEK293T cells yields novel currents that are pH dependent. Am J Physiol Cell Physiol 294, 251–262 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Ludwig CF, Ullrich F, Leisle L, Stauber T & Jentsch TJ Common gating of both CLC transporter subunits underlies voltage-dependent activation of the 2Cl−/1H+ exchanger ClC-7/Ostm1. J Biol Chem 288, 28611–28619 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leisle L, Ludwig CF, Wagner FA, Jentsch TJ & Stauber T ClC-7 is a slowly voltage-gated 2Cl(−)/1H(+)-exchanger and requires Ostm1 for transport activity. EMBO J 30, 2140–2152 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Stefano S, Pusch M & Zifarelli G A single point mutation reveals gating of the human ClC-5 Cl−/H+ antiporter. J Physiol 591, 5879–5893, doi: 10.1113/jphysiol.2013.260240 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alekov AK & Fahlke C Channel-like slippage modes in the human anion/proton exchanger ClC-4. J Gen Physiol 133, 485–496 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suetterlin K et al. Translating genetic and functional data into clinical practice: a series of 223 families with myotonia. Brain : a journal of neurology 145, 607–620, doi: 10.1093/brain/awab344 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pusch M Myotonia caused by mutations in the muscle chloride channel gene CLCN1. Hum. Mutat 19, 423–434 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Dupré N et al. Clinical, electrophysiologic, and genetic study of non-dystrophic myotonia in French-Canadians. Neuromuscular disorders: NMD 19, 330–334, doi: 10.1016/j.nmd.2008.01.007 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Altamura C et al. The analysis of myotonia congenita mutations discloses functional clusters of amino acids within the CBS2 domain and the C-terminal peptide of the ClC-1 channel. Hum Mutat 39, 1273–1283, doi: 10.1002/humu.23581 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Zifarelli G The Role of the Lysosomal Cl(−)/H(+) Antiporter ClC-7 in Osteopetrosis and Neurodegeneration. Cells 11, doi: 10.3390/cells11030366 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leisle L et al. Divergent Cl− and H+ pathways underlie transport coupling and gating in CLC exchangers and channels. eLife 9, e51224, doi: 10.7554/eLife.51224 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Accardi A et al. Separate ion pathways in a Cl-/H+ exchanger. J. Gen. Physiol 126, 563–570. (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pusch M, Ludewig U & Jentsch TJ Temperature dependence of fast and slow gating relaxations of ClC-0 chloride channels. J. Gen. Physiol 109, 105–116 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bykova EA, Zhang XD, Chen TY & Zheng J Large movement in the C terminus of CLC-0 chloride channel during slow gating. Nat. Struct. Mol. Biol 13, 1115–1119 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Yu Y, Tsai MF, Yu WP & Chen TY Modulation of the slow/common gating of CLC channels by intracellular cadmium. J Gen Physiol 146, 495–508, doi: 10.1085/jgp.201511413 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abraham SJ et al. 13C NMR detects conformational change in the 100-kD membrane transporter ClC-ec1. Journal of biomolecular NMR 61, 209–226, doi: 10.1007/s10858-015-9898-7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chavan TS et al. A CLC-ec1 mutant reveals global conformational change and suggests a unifying mechanism for the CLC Cl–/H+ transport cycle. eLife 9, e53479, doi: 10.7554/eLife.53479 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khantwal CM et al. Revealing an outward-facing open conformational state in a CLC Cl(−)/H(+) exchange transporter. Elife 5, doi: 10.7554/eLife.11189 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heath GR et al. Localization atomic force microscopy. Nature 594, 385–390, doi: 10.1038/s41586-021-03551-x (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Punjani A, Rubinstein JL, Fleet DJ & Brubaker MA cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nature Methods 14, 290, doi: 10.1038/nmeth.4169 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Lim HH, Shane T & Miller C Intracellular Proton Access in a Cl−/H+ Antiporter. PLoS Biology 10, 10.1371/journal.pbio.1001441 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han W, Cheng RC, Maduke MC & Tajkhorshid E Water access points and hydration pathways in CLC H+/Cl− transporters. Proceedings of the National Academy of Sciences 111, 1819–1824, doi: 10.1073/pnas.1317890111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee S, Swanson JM & Voth GA Multiscale Simulations Reveal Key Aspects of the Proton Transport Mechanism in the ClC-ec1 Antiporter. Biophys J 110 1334–1345 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chadda R et al. The dimerization equilibrium of a ClC Cl−/H+ antiporter in lipid bilayers. eLife 5, e17438, doi: 10.7554/eLife.17438 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nguitragool W & Miller C CLC Cl− /H+ transporters constrained by covalent cross-linking. Proc. Natl. Acad. Sci. U. S. A 104, 20659–20665 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robertson JL, Kolmakova-Partensky L & Miller C Design, function and structure of a monomeric ClC transporter. Nature 468, 844–847 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Juette MF et al. Single-molecule imaging of non-equilibrium molecular ensembles on the millisecond timescale. Nat Methods 13, 341–344, doi: 10.1038/nmeth.3769 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Altman RB et al. Enhanced photostability of cyanine fluorophores across the visible spectrum. Nat Methods 9, 428–429, doi: 10.1038/nmeth.1988 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng Q, Jockusch S, Zhou Z & Blanchard SC The contribution of reactive oxygen species to the photobleaching of organic fluorophores. Photochemistry and photobiology 90, 448–454, doi: 10.1111/php.12204 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walden M et al. Uncoupling and turnover in a Cl-/H+ exchange transporter. J. Gen. Physiol 129, 317–329 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Accardi A & Miller C Secondary active transport mediated by a prokaryotic homologue of ClC Cl− channels. Nature 427, 803–807 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Lim HH & Miller C Intracellular proton-transfer mutants in a CLC Cl-/H+ exchanger. J Gen Physiol 133, 8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feng L, Campbell EB, Hsiung Y & MacKinnon R Structure of a Eukaryotic CLC Transporter Defines an Intermediate State in the Transport Cycle. Science 330, 635–641 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zdebik AA et al. Determinants of anion-proton coupling in mammalian endosomal CLC proteins. J. Biol. Chem 283, 4219–4227 (2008). [DOI] [PubMed] [Google Scholar]
- 49.Wang D & Voth GA Proton transport pathway in the ClC Cl−/H+ antiporter. Biophys. J 97, 121–131 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koty PP, Pegoraro E & Hoffman EP Linkage and mutation analysis of Thomsen and Becker myotonia families. American Journal of Human Genetics 55, Medium: X; Size: pp. A227.1323 (1994). [Google Scholar]
- 51.Lehmann-Horn F, Mailänder V, Heine R & George AL Myotonia levior is a chloride channel disorder. Human Molecular Genetics 4, 1397–1402, doi: 10.1093/hmg/4.8.1397 (1995). [DOI] [PubMed] [Google Scholar]
- 52.Palmer EE et al. Functional and clinical studies reveal pathophysiological complexity of CLCN4-related neurodevelopmental condition. Molecular Psychiatry 28, 668–697, doi: 10.1038/s41380-022-01852-9 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meyer-Kleine C, Steinmeyer K, Ricker K, Jentsch TJ & Koch MC Spectrum of mutations in the major human skeletal muscle chloride channel gene (CLCN1) leading to myotonia. Am. J. Hum. Genet 57, 1325–1334 (1995). [PMC free article] [PubMed] [Google Scholar]
- 54.Hoopes RR et al. Evidence for genetic heterogeneity in Dent’s disease. Kidney International 65, 1615–1620, doi: 10.1111/j.1523-1755.2004.00571.x (2004). [DOI] [PubMed] [Google Scholar]
- 55.Halbritter J et al. Fourteen Monogenic Genes Account for 15% of Nephrolithiasis/Nephrocalcinosis. Journal of the American Society of Nephrology 26, 543–551, doi: 10.1681/asn.2014040388 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Minamikawa S et al. Development of ultra-deep targeted RNA sequencing for analyzing X-chromosome inactivation in female Dent disease. Journal of Human Genetics 63, 589–595, doi: 10.1038/s10038-018-0415-1 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Tosetto E et al. Phenotypic and genetic heterogeneity in Dent’s disease--the results of an Italian collaborative study. Nephrol Dial Transplant 21, 2452–2463, doi: 10.1093/ndt/gfl274 (2006). [DOI] [PubMed] [Google Scholar]
- 58.Wang C et al. The virulence gene and clinical phenotypes of osteopetrosis in the Chinese population: six novel mutations of the CLCN7 gene in twelve osteopetrosis families. Journal of bone and mineral metabolism 30, 338–348, doi: 10.1007/s00774-011-0319-z (2012). [DOI] [PubMed] [Google Scholar]
- 59.Li L, Lv S-S, Wang C, Yue H & Zhang Z-L Novel CLCN7 mutations cause autosomal dominant osteopetrosis type II and intermediate autosomal recessive osteopetrosis. Mol Med Rep 19, 5030–5038, doi: 10.3892/mmr.2019.10123 (2019). [DOI] [PubMed] [Google Scholar]
- 60.Zhixuan Z, Long C, Jin H & Ji S Structure of the human CLC-7/Ostm1 complex reveals a novel state. JUSTC 53, 0306–0301–0306–0307, doi: 10.52396/JUSTC-2022-0172 (2023). [DOI] [Google Scholar]
- 61.Stauber T & Jentsch TJ Sorting motifs of the endosomal/lysosomal CLC chloride transporters. J Biol Chem 285, 34537–34548, doi: 10.1074/jbc.M110.162545 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dutzler R, Campbell EB, Cadene M, Chait BT & MacKinnon R X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature 415, 287–294 (2002). [DOI] [PubMed] [Google Scholar]
- 63.Dutzler R, Campbell EB & MacKinnon R Gating the selectivity filter in ClC chloride channels. Science 300, 108–112 (2003). [DOI] [PubMed] [Google Scholar]
- 64.Park E & MacKinnon R Structure of the CLC-1 chloride channel from Homo sapiens. eLife 7, e36629, doi: 10.7554/eLife.36629 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Park E, Campbell EB & MacKinnon R Structure of a CLC chloride ion channel by cryoelectron microscopy. Nature 541, 500–505, doi: 10.1038/nature20812 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Feng L, Campbell EB & MacKinnon R Molecular mechanism of proton transport in CLC Cl−/H+ exchange transporters. Proc Natl Acad Sci U S A 109, 11699–11704 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mayes HB, Lee S, White AD, Voth GA & Swanson JMJ Multiscale Kinetic Modeling Reveals an Ensemble of Cl−/H+ Exchange Pathways in ClC-ec1 Antiporter. Journal of the American Chemical Society 140, 1793–1804, doi: 10.1021/jacs.7b11463 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Basilio D, Noack K, Picollo A & Accardi A Conformational changes required for H(+)/Cl(−) exchange mediated by a CLC transporter. Nat Struct Mol Biol 21, 456–463 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Accardi A & Pusch M Conformational changes in the pore of CLC-0. J. Gen. Physiol 122, 277–293 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bell SP, Curran PK, Choi S & Mindell JA Site-directed fluorescence studies of a prokaryotic ClC antiporter. Biochemistry 45, 6773–6782 (2006). [DOI] [PubMed] [Google Scholar]
- 71.Osteen J & Mindell JA Zn2+ inhibition of CLC-4. Biophysical journal 95, 4668–4675 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Elvington S, Liu C & Maduke M Substrate-driven conformational changes in ClC-ec1 observed by fluorine NMR. EMBO J 28, 3090–3102 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pusch M Chloride channelopathies. Parmaceutical News 8, 45–51 (2001). [Google Scholar]
- 74.Kubisch C, Schmidt-Rose T, Fontaine B, Bretag AH & Jentsch TJ ClC-1 chloride channel mutations in myotonia congenita: variable penetrance of mutations shifting the voltage dependence. Hum. Mol. Genet 7, 1753–1760 (1998). [DOI] [PubMed] [Google Scholar]
- 75.Lourdel S et al. ClC-5 mutations associated with Dent’s disease: a major role of the dimer interface. Pflugers Arch 463, 247–256, doi: 10.1007/s00424-011-1052-0 (2012). [DOI] [PubMed] [Google Scholar]
- 76.Leray X et al. Tonic inhibition of the chloride/proton antiporter ClC-7 by PI(3,5)P2 is crucial for lysosomal pH maintenance. Elife 11, doi: 10.7554/eLife.74136 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang S et al. Molecular insights into the human CLC-7/Ostm1 transporter. Science Advances 6, eabb4747, doi: 10.1126/sciadv.abb4747 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Smart OS, Neduvelil JG, Wang X, Wallace BA & Sansom MS HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J Mol Graph 14, 354–360 (1996). [DOI] [PubMed] [Google Scholar]
- 79.Schneider CA, Rasband WS & Eliceiri KW NIH Image to ImageJ: 25 years of image analysis. Nature Methods 9, 671, doi: 10.1038/nmeth.2089 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods Only References
- 80.Fortea E & Accardi A A quantitative flux assay for the study of reconstituted Cl(−) channels and transporters. Methods Enzymol 652, 243–272 (2021). 10.1016/bs.mie.2021.01.026 [DOI] [PubMed] [Google Scholar]
- 81.Russo CJ & Passmore LA Electron microscopy: Ultrastable gold substrates for electron cryomicroscopy. Science (New York, N.Y.) 346, 1377–1380 (2014). 10.1126/science.1259530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Punjani A, Zhang H & Fleet DJ Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nature Methods 17, 1214–1221 (2020). 10.1038/s41592-020-00990-8 [DOI] [PubMed] [Google Scholar]
- 83.Punjani A & Fleet DJ 3D variability analysis: Resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM. Journal of Structural Biology 213, 107702 (2021). 10.1016/j.jsb.2021.107702 [DOI] [PubMed] [Google Scholar]
- 84.Pettersen EF et al. UCSF Chimera - A visualization system for exploratory research and analysis. Journal of Computational Chemistry 25, 1605–1612 (2004). 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 85.Liebschner D et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol 75, 861–877 (2019). 10.1107/s2059798319011471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta crystallographica. Section D, Biological crystallography 60, 2126–2132 (2004). 10.1107/s0907444904019158 [DOI] [PubMed] [Google Scholar]
- 87.Ciftci D et al. FRET-based Microscopy Assay to Measure Activity of Membrane Amino Acid Transporters with Single-transporter Resolution. Bio-protocol 11, e3970 (2021). 10.21769/BioProtoc.3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Joo C & Ha T Single-molecule FRET with total internal reflection microscopy. Cold Spring Harbor protocols 2012 (2012). 10.1101/pdb.top072058 [DOI] [PubMed] [Google Scholar]
- 89.Akyuz N et al. Transport domain unlocking sets the uptake rate of an aspartate transporter. Nature 518, 68–73 (2015). 10.1038/nature14158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Huysmans GHM, Ciftci D, Wang X, Blanchard SC & Boudker O The high-energy transition state of the glutamate transporter homologue GltPh. EMBO J, e105415 (2020). 10.15252/embj.2020105415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Aitken CE, Marshall RA & Puglisi JD An oxygen scavenging system for improvement of dye stability in single-molecule fluorescence experiments. Biophys J 94, 1826–1835 (2008). 10.1529/biophysj.107.117689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sigworth FJ & Sine SM Data transformations for improved display and fitting of single-channel dwell time histograms. Biophys. J 52, 1047–1054 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Di Zanni E et al. Pathobiologic Mechanisms of Neurodegeneration in Osteopetrosis Derived From Structural and Functional Analysis of 14 ClC-7 Mutants. Journal of Bone and Mineral Research 36, 531–545 (2021). 10.1002/jbmr.4200 [DOI] [PubMed] [Google Scholar]
- 94.Humphrey W, Dalke A & Schulten K VMD: visual molecular dynamics. J. Mol. Graph 14, 33–38 (1996). [DOI] [PubMed] [Google Scholar]
- 95.Wu EL et al. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. Journal of Computational Chemistry 35, 1997–2004 (2014). 10.1002/jcc.23702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Best RB et al. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput 8, 3257–3273 (2012). 10.1021/ct300400x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW & Klein ML Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 79, 926–935 (1983). 10.1063/1.445869 [DOI] [Google Scholar]
- 98.Abraham MJ et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015). 10.1016/j.softx.2015.06.001 [DOI] [Google Scholar]
- 99.Tribello GA, Bonomi M, Branduardi D, Camilloni C & Bussi G PLUMED 2: New feathers for an old bird. Computer Physics Communications 185, 604–613 (2014). 10.1016/j.cpc.2013.09.018 [DOI] [Google Scholar]
- 100.Darden T, York D & Pedersen L Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. The Journal of Chemical Physics 98, 10089–10092 (1993). 10.1063/1.464397 [DOI] [Google Scholar]
- 101.Berk H, Henk B, C. BHJ & M. FJGE LINCS: A linear constraint solver for molecular simulations. Journal of Computational Chemistry 18, 1463–1472 (1997). [DOI] [Google Scholar]
- 102.Søndergaard CR, Olsson MHM, Rostkowski M & Jensen JH Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. Journal of chemical theory and computation 7, 2284–2295 (2011). 10.1021/ct200133y [DOI] [PubMed] [Google Scholar]
- 103.Olsson MHM, Søndergaard CR, Rostkowski M & Jensen JH PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. Journal of chemical theory and computation 7, 525–537 (2011). 10.1021/ct100578z [DOI] [PubMed] [Google Scholar]
- 104.Im W & Roux B t. Ions and Counterions in a Biological Channel: A Molecular Dynamics Simulation of OmpF Porin from Escherichia coli in an Explicit Membrane with 1M KCl Aqueous Salt Solution. Journal of Molecular Biology 319, 1177–1197 (2002). 10.1016/S0022-2836(02)00380-7 [DOI] [PubMed] [Google Scholar]
- 105.Salari R, Joseph T, Lohia R, Hénin J & Brannigan G A Streamlined, General Approach for Computing Ligand Binding Free Energies and Its Application to GPCR-Bound Cholesterol. Journal of chemical theory and computation 14, 6560–6573 (2018). 10.1021/acs.jctc.8b00447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Alleva C et al. Na+-dependent gate dynamics and electrostatic attraction ensure substrate coupling in glutamate transporters. Science Advances 6, eaba9854 (2020). 10.1126/sciadv.aba9854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sugita Y, Kitao A & Okamoto Y Multidimensional replica-exchange method for free-energy calculations. The Journal of Chemical Physics 113, 6042–6051 (2000). 10.1063/1.1308516 [DOI] [Google Scholar]
- 108.Lee S, Liang R, Voth GA & Swanson JM Computationally Efficient Multiscale Reactive Molecular Dynamics to Describe Amino Acid Deprotonation in Proteins. Journal of chemical theory and computation 12, 879–891 (2016). 10.1021/acs.jctc.5b01109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Domański J, Sansom MSP, Stansfeld PJ & Best RB Atomistic mechanism of transmembrane helix association. PLOS Computational Biology 16, e1007919 (2020). 10.1371/journal.pcbi.1007919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kumar S, Rosenberg JM, Bouzida D, Swendsen RH & Kollman PA THE weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. Journal of Computational Chemistry 13, 1011–1021 (1992). 10.1002/jcc.540130812 [DOI] [Google Scholar]
- 111.Friedman LJ, Chung J & Gelles J Viewing dynamic assembly of molecular complexes by multi-wavelength single-molecule fluorescence. Biophys J 91, 1023–1031 (2006). 10.1529/biophysj.106.084004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Friedman LJ & Gelles J Multi-wavelength single-molecule fluorescence analysis of transcription mechanisms. Methods 86, 27–36 (2015). 10.1016/j.ymeth.2015.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All constructs are available on request to A.A. or O.B. All models and associated cryoEM maps have been deposited into the Electron Microscopy Data Bank (EMDB) and the Protein Data Bank (PDB) under the following accession codes CLC-ec1 pH 7.5 0 mM Cl− Swap (PDB: 7RNX; EMD-24582); CLC-ec1 pH 7.5 100 mM Cl− Swap (PDB: 7RO0; EMD-24584); CLC-ec1 pH 4.5 0 mM Cl− Swap (PDB: 7RP6; EMD-24668); CLC-ec1 pH 4.5 0 mM Cl− Turn (PDB:7RSB; EMD-24583); CLC-ec1 pH 4.5 100 mM Cl− Swap (PDB: 7N8P; EMD-24241); CLC-ec1 pH 4.5 100 mM Cl− Turn (PDB: 7RP5; EMD-24612); CLC-ec1pH 4.5 100 mM Cl− Twist (PDB: 7N0W; EMD-24263); R230C/L249C pH 4.5 100mM Cl− Swap (PDB: 8GA1; EMD-29884); R230C/L249C pH 4.5 100mM Cl− Turn (PDB: 8GA3; EMD-29885); L25C/A450C pH 4.5 100mM Cl− Intermediate (PDB:8GA5; EMD-29890); L25C/A450C pH 4.5 100mM Cl− Twist (PDB: 8GAH; EMD-29899); E202Y pH 4.5 100mM Cl− (PDB: 8GA0; EMD-29883) which are listed in Supplementary Table 7.The atomic coordinates of the free molecular dynamics simulation at t = 0 and 1 microseconds of systems 1–8 listed in Supplementary Table 3 are available at https://doi.org/10.6084/m9.figshare.24777321.v1.