

Key Points
Question
Among patients with moderately to severely active ulcerative colitis, does risankizumab improve clinical remission rates compared with placebo when risankizumab is administered as an induction and a maintenance therapy?
Findings
Among the 975 patients analyzed in the induction trial, 1200 mg of risankizumab significantly increased the rates of clinical remission at 12-week follow-up compared with placebo (20.3% vs 6.2%, respectively). Among 548 patients included in the primary efficacy analysis for the maintenance trial, 180 mg of risankizumab and 360 mg of risankizumab significantly increased the rates of clinical remission (40.2% and 37.6%, respectively) compared with placebo (25.1%).
Meaning
Risankizumab improved the rates of clinical remission when used as an induction and a maintenance therapy for patients with moderately to severely active ulcerative colitis.
Abstract
Importance
The clinical effects of risankizumab (a monoclonal antibody that selectively targets the p19 subunit of IL-23) for the treatment of ulcerative colitis are unknown.
Objective
To evaluate the efficacy and safety of risankizumab when administered as an induction and a maintenance therapy for patients with ulcerative colitis.
Design, Setting, and Participants
Two phase 3 randomized clinical trials were conducted. The induction trial was conducted at 261 clinical centers (in 41 countries) and enrolled 977 patients from November 5, 2020, to August 4, 2022 (final follow-up on May 16, 2023). The maintenance trial was conducted at 238 clinical centers (in 37 countries) and enrolled 754 patients from August 28, 2018, to March 30, 2022 (final follow-up on April 11, 2023). Eligible patients had moderately to severely active ulcerative colitis; a history of intolerance or inadequate response to 1 or more conventional therapies, advanced therapies, or both types of therapies; and no prior exposure to risankizumab.
Interventions
For the induction trial, patients were randomized 2:1 to receive 1200 mg of risankizumab or placebo administered intravenously at weeks 0, 4, and 8. For the maintenance trial, patients with a clinical response (determined using the adapted Mayo score) after intravenous treatment with risankizumab were randomized 1:1:1 to receive subcutaneous treatment with 180 mg or 360 mg of risankizumab or placebo (no longer receiving risankizumab) every 8 weeks for 52 weeks.
Main Outcomes and Measures
The primary outcome was clinical remission (stool frequency score ≤1 and not greater than baseline, rectal bleeding score of 0, and endoscopic subscore ≤1 without friability) at week 12 for the induction trial and at week 52 for the maintenance trial.
Results
Among the 975 patients analyzed in the induction trial (aged 42.1 [SD, 13.8] years; 586/973 [60.1%] were male; and 677 [69.6%] were White), the clinical remission rates at week 12 were 132/650 (20.3%) for 1200 mg of risankizumab and 20/325 (6.2%) for placebo (adjusted between-group difference, 14.0% [95% CI, 10.0%-18.0%], P < .001). Among the 548 patients analyzed in the maintenance trial (aged 40.9 [SD, 14.0] years; 313 [57.1%] were male; and 407 [74.3%] were White), the clinical remission rates at week 52 were 72/179 (40.2%) for 180 mg of risankizumab, 70/186 (37.6%) for 360 mg of risankizumab, and 46/183 (25.1%) for placebo (adjusted between-group difference for 180 mg of risankizumab vs placebo, 16.3% [97.5% CI, 6.1%-26.6%], P < .001; adjusted between-group difference for 360 mg of risankizumab vs placebo, 14.2% [97.5% CI, 4.0%-24.5%], P = .002). No new safety risks were detected in the treatment groups.
Conclusion and Relevance
Compared with placebo, risankizumab improved clinical remission rates in an induction trial and in a maintenance trial for patients with moderately to severely active ulcerative colitis. Further study is needed to identify benefits beyond the 52-week follow-up.
Trial Registration
ClinicalTrials.gov Identifiers: NCT03398148 and NCT03398135
These 2 randomized clinical trials compare the efficacy and safety of risankizumab vs placebo during an induction trial and a maintenance trial in patients with ulcerative colitis.
Introduction
Ulcerative colitis is a chronic, immune-mediated inflammatory bowel disease associated with diarrhea, rectal bleeding, and bowel urgency.1 In North America, the prevalence of ulcerative colitis is 0.4% and it affects approximately 1.5 million people.1 Ulcerative colitis symptoms are associated with reduced quality of life, impaired social and psychological function, and increased health care costs.1,2,3 Current therapies, including corticosteroids and immunomodulators, are limited by lack of initial response, loss of response, and potential adverse events (such as increased risk of infections or malignancy). Tumor necrosis factor inhibitors, such as infliximab and adalimumab, are a first-line therapy for ulcerative colitis; however, approximately one-third of patients will not respond to initial treatment with a tumor necrosis factor inhibitor.4
The cytokine IL-23 is implicated in intestinal inflammation and ulcerative colitis pathogenesis,5 and it stimulates the proliferation of inflammatory cell populations and supports the activation of other cytokines, including IL-17 and IL-22. Ustekinumab, which targets the p40 subunit shared between IL-12 and IL-23, and mirikizumab, which targets the p19 subunit specific to IL-23, have previously demonstrated the therapeutic potential of this pathway in ulcerative colitis.6,7 Risankizumab is a monoclonal IgG-1 antibody that selectively targets the IL-23 p19 subunit, blocking signaling through the IL-23 receptor. Risankizumab has been approved by the US Food and Drug Administration and the European Medicines Agency for the treatment of Crohn disease, plaque psoriasis, and psoriatic arthritis; has been approved by the Pharmaceuticals and Medical Devices Agency for the treatment of palmoplantar pustulosis; and is being investigated for the treatment of ulcerative colitis.8,9,10
This report describes the results of 2 phase 3 trials that evaluated the efficacy and safety of risankizumab compared with placebo as an induction and a maintenance therapy in patients with moderately to severely active ulcerative colitis.
Methods
Trial Design
Two phase 3, multicenter, double-blind, placebo-controlled randomized clinical trials were conducted. The induction trial was called the INSPIRE substudy 2 and the maintenance trial was called the COMMAND substudy 1 (additional information appears in eMethods 1 and eFigure 1 in Supplement 1). An independent ethics committee or institutional review board at each study site approved the trial protocol (Supplement 2), the informed consent forms, and the recruitment materials before patient enrollment. The studies were conducted in accordance with the International Conference for Harmonization guidelines, applicable regulations, and the Declaration of Helsinki. All patients provided written informed consent.
Enrollment for the induction trial was conducted at 261 clinical centers in 41 countries from November 5, 2020, to August 4, 2022, with final follow-up on May 16, 2023 (Figure 1). Enrollment for the maintenance trial was conducted at 238 clinical centers in 37 countries from August 28, 2018, to March 30, 2022, with the final follow-up on April 11, 2023 (Figure 2).
Figure 1. Flow of Patients in the Induction Trial.

aDetermination of eligibility was made at the clinical sites at the time of enrollment. The specific reasons for not meeting screening criteria were not collected.
bThe 2:1 randomization was stratified by the number of prior biological drugs (0 or 1 vs >1) each patient received that produced an inadequate response, current use of steroids (yes vs no), and adapted Mayo score (≤7 vs >7).
cThe patients included in the primary efficacy analysis received at least 1 dose of study drug during the 12-week, double-blind induction period.
Figure 2. Flow of Patients in the Maintenance Trial.
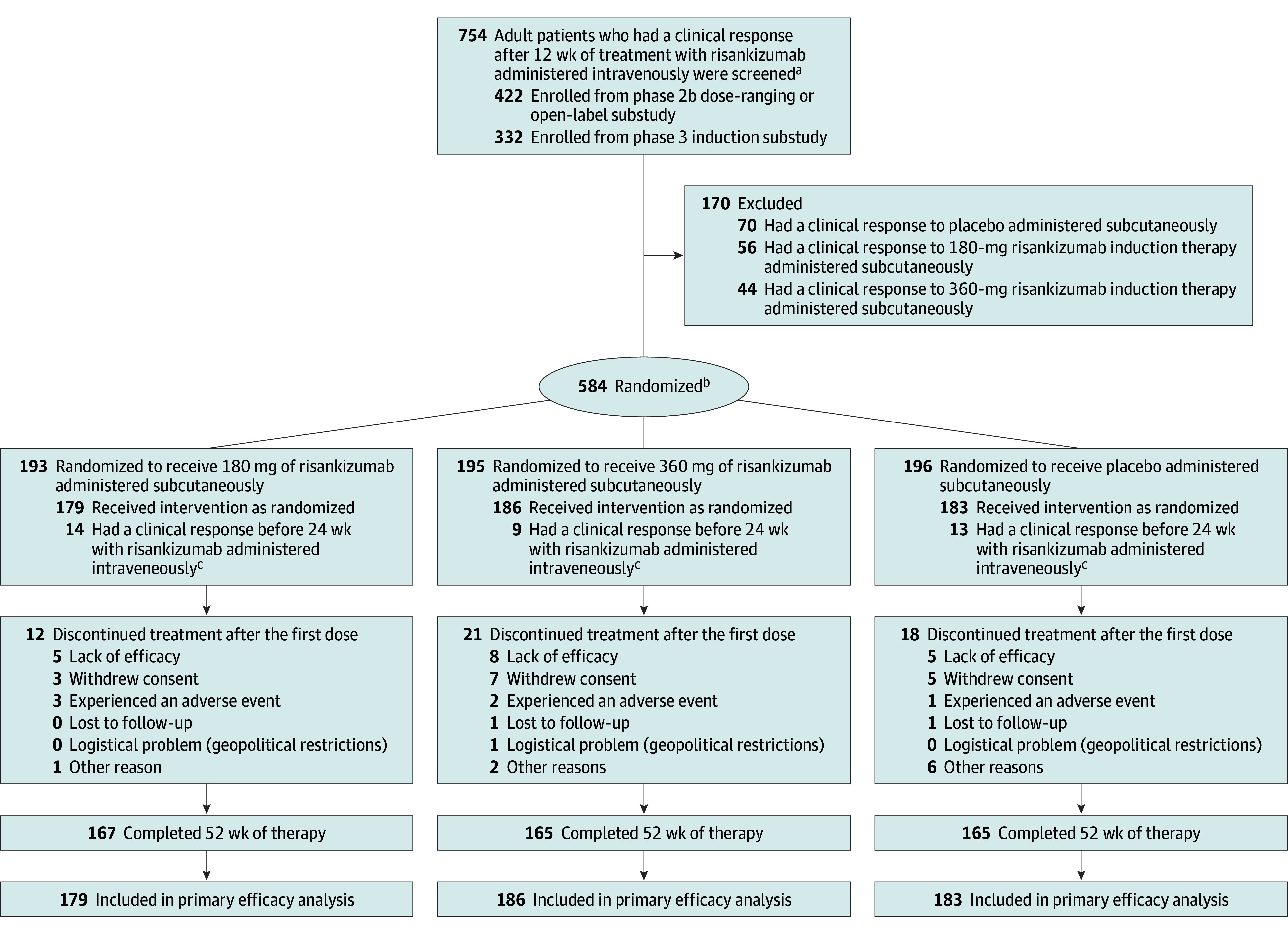
aThe patients with an inadequate response to risankizumab could receive an additional 12 weeks of therapy with risankizumab (during an extended period of induction therapy in the phase 3 substudy; data not shown).
bIncluded in the safety analysis.
cReceived 1800-mg intravenous dose in phase 2b study or 1200-mg intravenous dose in phase 3 study. These individuals were included in the safety analysis only (not included in the primary efficacy analysis).
Inclusion and Exclusion Criteria
Patients eligible for the induction trial (1) were 18 years to 80 years of age with a diagnosis of ulcerative colitis for at least 3 months, (2) had an adapted Mayo score of 5 to 9 points (range, 0-9) (the adapted Mayo score is composed of the categorical subscores for stool frequency [range, 0-3; a score of 0 for a “normal” number of stools and a score of 3 for ≥5 stools than “normal”], rectal bleeding [range, 0-3; a score of 0 for no blood seen and a score of 3 for blood passed alone], and an endoscopic score [range, 0-3; a score of 0 for “normal” appearance of mucosa and a score of 3 for severe disease, including spontaneous bleeding and ulcerations]), (3) had an endoscopic subscore of 2 to 3 that was confirmed by central review, and (4) had a history of intolerance or inadequate response to conventional therapy alone, or to 1 or more advanced therapies (eg, infliximab, adalimumab, golimumab, vedolizumab, tofacitinib, filgotinib, upadacitinib, ozanimod), or to a combination of conventional and advanced therapies. Exclusion criteria included prior exposure to p40 inhibitors (eg, ustekinumab) or p19 inhibitors (eg, risankizumab or mirikizumab). The full eligibility criteria appear in the eMethods 2 in Supplement 1.
Patients eligible for the maintenance trial (1) had been enrolled in an induction trial and (2) had an adequate response to risankizumab at 12- or 24-week follow-up (an adequate response was defined as a decrease of ≥30% from baseline, a reduction of 2 points on the adapted Mayo score, and a decrease of ≥1 in rectal bleeding score or an absolute rectal bleeding score ≤1). The induction trial consisted of the phase 3 induction substudy and a phase 2b induction substudy, which included a dose-ranging study followed by an open-label substudy (eMethods 3 and eFigure 1 in Supplement 1). The phase 2b dose-finding substudy evaluated the efficacy and safety of risankizumab as an induction treatment to identify the appropriate induction dose of risankizumab for further evaluation in a phase 3 trial.
Randomization and Blinding
For the induction trial, patients were randomized 2:1 to receive 1200 mg of risankizumab or placebo administered intravenously at weeks 0, 4, and 8 (Figure 1). The dose of risankizumab was based on the results of a phase 2b substudy (eFigures 2-3 and eTables 1-2 in Supplement 1). Randomization was stratified by the presence of baseline corticosteroid use (yes, no), baseline adapted Mayo score (≤7, >7), and a history of intolerance or inadequate response to advanced therapies (0, 1, >1 treatments).
Patients from the induction trial with an adequate clinical response (determined using the adapted Mayo score) after 12 or 24 weeks of risankizumab therapy administered intravenously were randomized in the maintenance trial 1:1:1 to 180 mg or 360 mg of risankizumab or placebo (no longer receiving risankizumab) administered subcutaneously every 8 weeks for 52 weeks (Figure 2 and eFigure 1 in Supplement 1). Randomization was stratified by history of inadequate response to advanced therapy (yes, no), last risankizumab induction dose (600 mg, 1200 mg, 1800 mg administered intravenously), and clinical remission status (per local evaluation) at the last visit of the induction trial (yes, no). Although patients with an adequate clinical response to treatment with risankizumab at either weeks 12 or 24 were included in the safety outcomes analysis, only randomized patients who (1) received at least 1 dose of the study drug and (2) had an adequate clinical response to risankizumab administered intravenously after 12 weeks of treatment were included in the primary efficacy analysis for the maintenance trial.
Randomization was performed using web-based interactive response technology (Endpoint Clinical, version 3.0) with block randomization methods. The block randomization schedules were generated by randomization specialists at AbbVie (sponsor of the 2 trials) and distributed to the interactive response technology vendor for randomization. The interactive response technology was used to provide the appropriate medication kit numbers that were dispensed to each patient according to their randomized treatment group. Patients, investigators, and study personnel involved in the conduct of the trial or in the analyses were blinded to treatment assignments until data analysis.
Administration of Risankizumab and Placebo
An unblinded pharmacist (or qualified designee) prepared the intravenous and subcutaneous solutions consisting of saline with an equal volume of the study drug or placebo according to group assignment. The study drug (risankizumab) or placebo was administered with covered syringes. The risankizumab and placebo subcutaneous kits were identical in appearance.
In the induction trial, patients taking stable doses of corticosteroids, aminosalicylates, or immunomodulators at baseline continued these treatments throughout the trial. In the maintenance trial, patients undergoing corticosteroid therapy were required to taper by week 8. Starting at week 16 of the maintenance trial, patients in any treatment group could receive open-label risankizumab therapy (ie, 1 single dose of risankizumab administered intravenously followed by 360 mg administered subcutaneously every 8 weeks) in the event of loss of adequate clinical response (defined as a rectal bleeding score of ≥1 point than the week 0 value or an endoscopic subscore of 2 or 3).
Primary Outcome in the Induction and Maintenance Trials
In the induction trial, the primary outcome was clinical remission, which was determined using the adapted Mayo score (stool frequency score ≤1 and not greater than baseline, rectal bleeding score of 0, and endoscopic subscore ≤1 without friability) at 12-week follow-up, which is consistent with regulatory guidance.11,12,13 In the maintenance trial, the primary outcome was clinical remission, which was determined using the adapted Mayo score (the components of the score were defined similarly to the primary outcome in the induction trial), at 52-week follow-up.
Secondary Outcomes in the Induction and Maintenance Trials
At 12-week follow-up in the induction trial and at 52-week follow-up in the maintenance trial, the prespecified secondary outcomes were (1) clinical response, which was determined using the adapted Mayo score (a decrease of ≥30% and ≥2 points from baseline and a decrease in rectal bleeding score of ≥1 or an absolute rectal bleeding score of ≤1), (2) clinical response determined using the partial adapted Mayo score (in the induction trial only at week 4; decrease of ≥30% and 1 point from baseline and a decrease in rectal bleeding score ≥1 or an absolute rectal bleeding score ≤1), (3) endoscopic improvement (endoscopic subscore ≤1 without friability), (4) endoscopic remission (endoscopic subscore of 0), (5) histological, endoscopic, and mucosal improvement (endoscopic subscore of 0 or 1 without friability and Geboes score ≤3.1), (6) histological, endoscopic, and mucosal remission (endoscopic subscore of 0 and Geboes score <2.0), (7) absence of bowel urgency, (8) absence of abdominal pain, (9) absence of nocturnal bowel movements, (10) absence of tenesmus, and (11) change from baseline for fecal incontinence, sleep interruption due to ulcerative colitis symptoms, fatigue score (determined using the Functional Assessment of Chronic Illness Therapy-Fatigue14 [FACIT-F; score range, 0-52 points; a higher score indicates less fatigue; a meaningful improvement was a change of 4-9 points [“minimally improved” or “much improved”] according to the Patient Global Impression of Change), score on the Inflammatory Bowel Disease Questionnaire (IBDQ; score range, 32-224 points; a higher score indicates better quality of life; a meaningful improvement was a change of 15 points), and in ulcerative colitis–related hospitalizations (eMethods 3 in Supplement 1).15
The secondary outcomes of histological, endoscopic, and mucosal improvement, absence of bowel urgency, absence of abdominal pain, absence of nocturnal bowel movements, absence of tenesmus, fecal incontinence, and sleep interruption were added to the induction trial on December 16, 2022, to align with the update from the Selecting Therapeutic Targets in Inflammatory Bowel Disease initiative.16 These secondary outcomes were added before any results were analyzed.
The maintenance trial evaluated the following outcomes at 52-week follow-up: maintenance of clinical remission (determined using the adapted Mayo score) at week 0 and at week 52, corticosteroid-free clinical remission, and maintenance of endoscopic improvement at week 0 and at week 52.
Additional Prespecified Analyses
For the induction and maintenance trials, additional efficacy analyses were performed including (1) the patients who had documented treatment intolerance or had an inadequate clinical response to 1 or more advanced therapies (infliximab, adalimumab, golimumab, vedolizumab, tofacitinib, filgotinib, upadacitinib, ozanimod) and (2) the patients who were tolerant of treatment or had an adequate response to at least 1 type of advanced therapy. Type I error was not controlled for this analysis.
The inflammatory biomarkers of high-sensitivity C-reactive protein (CRP) and fecal calprotectin were exploratory outcomes and were included to characterize disease activity.16 Change from baseline in high-sensitivity CRP was evaluated at weeks 0, 4, 8, and 12 in the induction trial and at weeks 0, 24, and 52 in the maintenance trial. Fecal calprotectin was assessed at weeks 0, 4, and 12 in the induction trial and at weeks 0, 24, and 52 in the maintenance trial.
Post Hoc Analysis
Serum concentrations of IL-22 were measured at weeks 0, 4, 12, and 52 in the maintenance trial using the SMC high-sensitivity immunoassay (EMD Millipore) via a quantitative fluorescent sandwich immunoassay technique.
Statistical Analysis
For the induction trial, a sample size of 966 patients provided 90% power or greater (based on an assumption of 6% for clinical remission [determined using the adapted Mayo score] in the placebo group and 16% in the risankizumab group at week 12) to detect a treatment difference of 10% for the primary outcome of clinical remission using the 2-sided Miettinen and Nurminen test at a significance level of .05.6 The statistical analysis plan appears in Supplement 2.
For the maintenance trial, a sample size of 573 patients provided 90% power or greater (based on an assumption of 22% for clinical remission [determined using the adapted Mayo score] in the placebo group and 42% in the 180-mg and 360-mg risankizumab groups at week 52) to detect a treatment difference of 20% for the primary outcome of clinical remission (between each risankizumab dose group and the placebo group) using the 2-sided Miettinen and Nurminen test at a significance level of .025, adjusting the .05-significance level for the 2 comparisons.6
Induction Trial
The primary efficacy analysis for the induction trial included randomized patients who received at least 1 dose of study drug. The demographics and baseline characteristics for each treatment group were summarized using descriptive statistics. To control for the family-wise type I error rate at a 2-sided significance level of .05, the primary outcome was tested at a prespecified 2-sided significance level of .05 for risankizumab compared with placebo. If the primary outcome achieved significance, continued testing of the secondary outcomes followed a prespecified weight of allocation as indicated in the graphical multiple testing procedure (eFigure 4 in Supplement 1). No type I error control was applied to the exploratory outcomes.
The categorical efficacy outcomes (eg, percentage of patients achieving clinical or endoscopic remission) were analyzed using the Cochran-Mantel-Haenszel test for common risk difference. Continuous efficacy outcomes (eg, change from baseline in FACIT-F or IBDQ total score) were analyzed using a mixed-effect model and a repeated-measures method or analysis of covariance that included the categorical fixed effects of treatment, stratification factors, and the continuous fixed covariates of baseline measurement. Safety was assessed through week 12 of follow-up according to adverse events reported among all the patients who were randomized and received at least 1 dose of risankizumab or placebo (eResults 1-3 in Supplement 1). The safety data were summarized descriptively.
Maintenance Trial
To control for the family-wise type I error rate at a 2-sided significance level of .05, the primary outcome was tested at the prespecified 2-sided significance level of .025 for each risankizumab dose compared with placebo. If the primary outcome achieved significance, continued testing of the secondary outcomes followed a prespecified weight of allocation (eFigure 5 in Supplement 1). The secondary outcomes (prespecified significance level of .025) were maintenance of clinical remission, corticosteroid-free clinical remission, and maintenance of endoscopic improvement.
No control for the type I error rate was applied to the exploratory outcomes or the subgroup analyses. The same assessment methods were used for the categorical and continuous efficacy outcomes for both the induction and maintenance trials. Safety was assessed among patients with an adequate clinical response to risankizumab (administered intravenously) at 12 weeks or 24 weeks. These patients received at least 1 dose of risankizumab during the maintenance trial. The safety data were summarized descriptively.
Missing Data
The primary approach for handling missing data was nonresponder imputation while incorporating multiple imputation to handle missing data due to logistical restrictions because of the COVID-19 pandemic or geopolitical restrictions for the categorical variables. Multiple imputations were used that incorporated the return to baseline values for the continuous variables. No missing data were imputed for the safety analyses. All statistical analyses were completed using SAS version 9.4 or newer (SAS Institute Inc).
Results
Of the 1430 patients screened for the induction trial, 977 were randomized and 975 received 1 or more doses of risankizumab (n = 650) or placebo (n = 325) administered intravenously (Figure 1). Among these 975 patients (aged 42.1 [SD, 13.8] years; 586/973 [60.1%] were male; and 677 [69.6%] were White), 637/650 (98%) in the risankizumab group completed 12-week follow-up compared with 298/325 (92%) in the placebo group.
Of the 584 patients randomized in the maintenance trial (all treatments administered subcutaneously), 548 had an adequate clinical response to risankizumab at 12 weeks during the induction trial and these patients were included in the primary efficacy population (179 in the 180 mg of risankizumab group, 186 in the 360 mg of risankizumab group, and 183 in the placebo group [no longer receiving risankizumab]) (Figure 2). Among these 548 patients (aged 40.9 [SD, 14.0] years; 313 [57.1%] were male; and 407 [74.3%] were White) with an adequate clinical response to risankizumab, 167/179 (93%) in the 180 mg of risankizumab group, 165/186 (89%) in the 360 mg of risankizumab group, and 165/183 (90%) in the placebo group (no longer receiving risankizumab) completed 52-week follow-up.
Of those included in the primary efficacy population, 503/975 patients (52%) in the induction trial and 411/548 patients (75%) in the maintenance trial had a history of intolerance or inadequate response to advanced therapies (Table 1). At week 0 in the maintenance trial, 44/179 patients (25%) in the 180 mg of risankizumab group, 40/186 patients (22%) in the 360 mg of risankizumab group, and 53/183 patients (29%) in the placebo group (no longer receiving risankizumab) were in clinical remission (eTable 3 in Supplement 1).
Table 1. Baseline Characteristics of Patients Participating in the Induction and Maintenance Trials.
| Induction trial (12 wk) | Maintenance trial (52 wk)a | ||||
|---|---|---|---|---|---|
| 1200 mg of risankizumab administered intravenously (n = 650) |
Placebo administered intravenously (n = 325) |
Risankizumab administered subcutaneously | Placebo administered subcutaneously (n = 183) |
||
| 180 mg (n = 179) | 360 mg (n = 186) | ||||
| Sex, No. (%) | |||||
| Female | 265 (40.8) | 124 (38.2) | 74 (41.3) | 79 (42.5) | 82 (44.8) |
| Male | 385 (59.2) | 201 (61.8) | 105 (58.7) | 107 (57.5) | 101 (55.2) |
| Age, mean (SD), y | 41.8 (13.5) | 42.8 (14.3) | 40.9 (14.7) | 42.5 (12.9) | 39.2 (14.2) |
| Body mass index | |||||
| No. of patients | 647 | 325 | 177 | 186 | 183 |
| Mean (SD)b | 24.7 (5.3) | 24.9 (5.2) | 24.9 (5.4) | 24.2 (4.9) | 24.2 (5.3) |
| Race, No. (%)c | |||||
| No. of patients | 648 | 325 | 179 | 186 | 183 |
| American Indian or Alaska Native | 1 (0.2) | 0 | 0 | 0 | 0 |
| Asian | 171 (26.4) | 96 (29.5) | 36 (20.1) | 51 (27.4) | 46 (25.1) |
| Black or African American | 12 (1.9) | 7 (2.2) | 4 (2.2) | 1 (0.5) | 2 (1.1) |
| Native Hawaiian or Other Pacific Islander | 0 | 0 | 0 | 0 | 0 |
| Multipled | 5 (0.8) | 4 (1.2) | 0 | 1 (0.5) | 0 |
| White | 459 (70.8) | 218 (67.1) | 139 (77.7) | 133 (71.5) | 135 (73.8) |
| Ethnicity, No. (%) | |||||
| Hispanic or Latino | 44 (6.8) | 20 (6.2) | 19 (10.6) | 11 (5.9) | 5 (2.7) |
| Non-Hispanic or non-Latino | 604 (93.2) | 305 (93.8) | 160 (89.4) | 175 (94.1) | 178 (97.3) |
| Disease duration, mean (SD), y | 7.7 (6.9) | 8.1 (7.0) | 8.5 (7.4) | 9.3 (7.1) | 8.2 (7.2) |
| Location and extent of disease, No. (%) | |||||
| Left side | 313 (48.2) | 150 (46.2) | 84 (46.9) | 92 (49.5) | 85 (46.4) |
| Extensive or pancolitis | 334 (51.4) | 174 (53.5) | 94 (52.5) | 94 (50.5) | 98 (53.6) |
| Limited to rectum | 3 (0.5) | 1 (0.3) | 1 (0.6) | 0 | 0 |
| Adapted Mayo scoree | |||||
| No. of patients | 649 | 325 | 179 | 186 | 183 |
| Score, mean (SD) | 7.1 (1.2) | 7.1 (1.3) | 7.2 (1.2) | 7.0 (1.3) | 7.2 (1.2) |
| Score category, No. (%) | |||||
| ≤7 | 376 (57.9) | 190 (58.5) | 102 (57.0) | 109 (58.6) | 92 (50.3) |
| >7 | 273 (42.1) | 135 (41.5) | 77 (43.0) | 77 (41.4) | 91 (49.7) |
| Endoscopic subscore, mean (SD)f | 2.7 (0.5) | 2.7 (0.5) | 2.7 (0.4) | 2.7 (0.5) | 2.7 (0.5) |
| Score category, No. (%) | |||||
| 2 | 208 (32.0) | 94 (28.9) | 47 (26.3) | 63 (33.9) | 52 (28.4) |
| 3 | 442 (68.0) | 231 (71.1) | 132 (73.7) | 123 (66.1) | 131 (71.6) |
| High-sensitivity C-reactive protein | |||||
| No. of patients | 638 | 318 | 177 | 184 | 179 |
| Median (IQR), mg/Lg | 3.4 (1.2-8.6) | 4.0 (1.2-9.5) | 4.7 (1.5-13.6) | 2.8 (1.0-7.7) | 4.2 (1.5-12.0) |
| Fecal calprotectin | |||||
| No. of patients | 602 | 302 | 151 | 166 | 162 |
| Median (IQR), μg/gh | 1530.0 (592.0-3196.0) | 1624.0 (601.0-3493.0) | 1605.0 (611.0-3180.0) | 1568.0 (499.0-3884.0) | 1514.5 (729.0-2910.0) |
| Medication use, No. (%) | |||||
| Immunosuppressants | 108 (16.6) | 53 (16.3) | 35 (19.6) | 32 (17.2) | 37 (20.2) |
| Aminosalicylates | 475 (73.1) | 238 (73.2) | 119 (66.5) | 135 (72.6) | 117 (63.9) |
| Corticosteroids | 236 (36.3) | 112 (34.5) | 74 (41.3) | 59 (31.7) | 68 (37.2) |
| Treatment response history, No. (%) | |||||
| Inadequate to advanced therapyi,j | 333 (51.2) | 170 (52.3) | 134 (74.9) | 139 (74.7) | 138 (75.4) |
| Inadequate to nonadvanced therapyj | 317 (48.8) | 155 (47.7) | 45 (25.1) | 47 (25.3) | 45 (24.6) |
| No. of times patient had an inadequate response to advanced therapy, No. (%)j | |||||
| 0 | 317 (48.8) | 155 (47.7) | 45 (25.1) | 47 (25.3) | 45 (24.6) |
| 1 | 153 (23.5) | 80 (24.6) | 52 (29.1) | 55 (29.6) | 62 (33.9) |
| 2 | 112 (17.2) | 55 (16.9) | 44 (24.6) | 37 (19.9) | 40 (21.9) |
| >2 | 68 (10.5) | 35 (10.8) | 38 (21.2) | 47 (25.3) | 36 (19.7) |
| No. of times with inadequate response to anti–tumor necrosis factor therapy, No. (%)j | |||||
| No. of patients | 333 | 170 | 134 | 139 | 138 |
| 0 | 48 (14.4) | 28 (16.5) | 11 (8.2) | 11 (7.9) | 7 (5.1) |
| 1 | 209 (62.8) | 112 (65.9) | 71 (53.0) | 85 (61.2) | 92 (66.7) |
| 2 | 68 (20.4) | 22 (12.9) | 50 (37.3) | 39 (28.1) | 35 (25.4) |
| >2 | 8 (2.4) | 8 (4.7) | 2 (1.5) | 4 (2.9) | 4 (2.9) |
The patients in the maintenance trial (and included in the primary efficacy analysis) received at least 1 dose of risankizumab either during the 12-week induction trial or during the 52-week maintenance trial. Additional information appears in eTable 3 in Supplement 1.
Calculated as weight in kilograms divided by height in meters squared.
Patients were asked to respond to closed category questions with the option of selecting multiple race categories.
Selected 2 or more race categories.
Measures disease activity in patients with ulcerative colitis. There are 3 components of the score: the stool frequency score, the rectal bleeding score, and an endoscopic subscore. Each score is measured on a scale from 0 to 3 and then the scores are combined. The combined score range is 0 to 9. A score of 5 to 9 points indicates moderate to severe active disease; a score of 0 indicates inactive disease.
Evaluated for each observed segment of the colon (rectum, sigmoid, descending colon, transverse colon, and ascending colon or cecum) using a 4-point scale. A higher score indicates more severe disease.
The reference range is 0 mg/L to 10 mg/L.
The reference value is less than 50 μg/g.
Advanced therapies included infliximab, adalimumab, golimumab, vedolizumab, tofacitinib, filgotinib, upadacitinib, and ozanimod.
An inadequate response was defined as lack of response to treatment or the patient experienced unacceptable therapy-related adverse effects.
At week 12 of the induction trial, there were missing data for the primary outcome of clinical remission in 15/650 patients (2.3%) in the 1200 mg of risankizumab group and in 18/325 patients (5.5%) in the placebo group. At week 52 of the maintenance trial, there were missing data for the primary outcome of clinical remission in 18/179 patients (10.1%) in the 180 mg of risankizumab group, in 25/186 patients (13.4%) in the 360 mg of risankizumab group, and in 23/183 patients (12.6%) in the placebo group (no longer receiving risankizumab).
Induction Trial
Primary Outcome
At week 12, risankizumab significantly improved clinical remission rates compared with placebo (20.3% vs 6.2%, respectively; adjusted between-group difference, 14.0% [95% CI, 10.0%-18.0%], P < .001) (Table 2 and eFigure 6 and eTable 4 in Supplement 1).
Table 2. Primary and Key Secondary Outcomes in the Induction Trial.
| No. (%) [95% CI]a | Adjusted between-group difference, % (95% CI)b | P valuec | ||
|---|---|---|---|---|
| 1200 mg of risankizumab administered intravenously (n = 650) |
Placebo administered intravenously (n = 325) |
|||
| Primary outcome | ||||
| Clinical remission (determined using the adapted Mayo score)d,e | 132 (20.3) [17.2 to 23.4] | 20 (6.2) [3.6 to 8.9] | 14.0 (10.0 to 18.0) | <.001 |
| Secondary outcomes | ||||
| Clinical responsef | ||||
| Determined using the adapted Mayo scored | 418 (64.3) [60.6 to 67.9] | 116 (35.7) [30.5 to 40.9] | 28.6 (22.3 to 34.8) | <.001 |
| Determined using the partial adapted Mayo score at wk 4g | 339 (52.2) [48.3 to 56.0] | 99 (30.5) [25.5 to 35.5] | 21.8 (15.6 to 28.1) | <.001 |
| Improvement | ||||
| Endoscopich | 237 (36.5) [32.8 to 40.2] | 39 (12.1) [8.5 to 15.6] | 24.3 (19.3 to 29.4) | <.001 |
| Histological, endoscopic, and mucosali | 159 (24.5) [21.2 to 27.8] | 25 (7.7) [4.8 to 10.6] | 16.6 (12.3 to 21.0) | <.001 |
| Endoscopic remissionj | 69 (10.6) [8.2 to 13.0] | 11 (3.4) [1.4 to 5.4] | 7.2 (4.2 to 10.2) | <.001 |
| No bowel urgencyk | 287 (44.1) [40.3 to 47.9] | 90 (27.7) [22.8 to 32.6] | 16.3 (10.3 to 22.4) | <.001 |
| No abdominal paink | 232 (35.8) [32.1 to 39.4] | 86 (26.5) [21.7 to 31.3] | 9.3 (3.4 to 15.3) | .002 |
| Histological, endoscopic, and mucosal remissionl | 41 (6.3) [4.4 to 8.2] | 2 (0.6) [0 to 1.5] | 5.6 (3.5 to 7.7) | <.001 |
| Mean change (95% CI) | ||||
| 13-Item Functional Assessment of Chronic Illness Therapy-Fatigue scale scorem | 7.9 (7.0 to 8.7) | 3.3 (2.1 to 4.5) | 4.5 (3.1 to 6.0) | <.001 |
| 32-Question Inflammatory Bowel Disease Questionnaire scoren | 42.6 (39.7 to 45.6) | 24.3 (20.2 to 28.5) | 18.3 (13.4 to 23.3) | <.001 |
| ≥1 Ulcerative colitis–related hospitalization | 5 (0.8) [0.1 to 1.4] | 18 (5.5) [3.1 to 8.0] | −4.8 (−7.3 to −2.2) | <.001 |
| No nocturnal bowel movementsk | 437 (67.3) [63.7 to 70.9] | 140 (43.1) [37.7 to 48.5] | 24.2 (17.9 to 30.5) | <.001 |
| No tenesmusk | 317 (48.7) [44.9 to 52.6] | 98 (30.2) [25.2 to 35.1] | 18.6 (12.4 to 24.8) | <.001 |
| Fecal incontinence, mean change (95% CI), episodes/wko | −3.8 (−4.3 to −3.4) | −2.2 (−2.9 to −1.6) | −1.6 (−2.4 to −0.9) | <.001 |
| Sleep interrupted due to ulcerative colitis symptoms, mean change (95% CI), d/wkp | −2.5 (−2.7 to −2.3) | −1.5 (−1.8 to −1.2) | −1.0 (−1.3 to −0.6) | <.001 |
Unless otherwise indicated. All outcomes were controlled for multiplicity. The results for the categorical outcomes are based on nonresponder imputation while incorporating multiple imputation to handle missing data due to logistical restrictions because of the COVID-19 pandemic or geopolitical restrictions. The results for the continuous outcomes are based on return to baseline multiple imputation.
Calculated using the Mantel-Haenszel common rate difference with nonresponder imputation while incorporating multiple imputation to handle missing data due to logistical restrictions because of the COVID-19 pandemic or geopolitical restrictions for the categorical outcomes and using analysis of covariance or mixed-effect model and a repeated-measures method with return to baseline multiple imputation for the continuous outcomes.
All comparisons were statistically significant according to hierarchical testing.
The adapted Mayo score measures disease activity in patients with ulcerative colitis. There are 3 components of the score: the stool frequency score, the rectal bleeding score, and an endoscopic subscore. Each score is measured on a scale from 0 to 3 and then the scores are combined. The combined score range is 0 to 9. A score of 5 to 9 points indicates moderate to severe active disease; a score of 0 indicates inactive disease.
Clinical remission was defined as a stool frequency score of 1 or less and not greater than the score at baseline. A score of 0 was given for patients with rectal bleeding. An endoscopic subscore was used to evaluate each segment of the colon (rectum, sigmoid, descending colon, transverse colon, and ascending colon or cecum) using a 4-point scale. A higher score indicates more severe disease. An endoscopic subscore of 1 or less was given for patients without friability.
Clinical response was defined as a decrease of at least 30% and a decrease of at least 2 points from baseline. At week 4, clinical response was defined as a decrease of at least 30% and a decrease of 1 point from baseline. At all weeks, clinical response was also determined by a decrease in rectal bleeding score of 1 or greater or an absolute rectal bleeding score of 1 or less.
The partial adapted Mayo score is a noninvasive assessment of disease severity in patients with ulcerative colitis. There are 2 components of the score: stool frequency subscore and rectal bleeding subscore. Each score is measured on a scale of 0 to 3 and then the scores are combined. The combined score range is 0 to 6. A score of 6 indicates active disease and spontaneous bleeding; a score of 0 indicates inactive disease.
Defined by an endoscopic subscore of 1 or less without friability.
Defined by an endoscopic subscore of 0 or 1 without friability and a Geboes score of 3.1 or less.
Defined by an endoscopic subscore of 0.
A 4-point scale was used to evaluate symptom frequency and severity over time. A higher score signified more severe pain. The mean of the scores from the most recent 3 days and up to 10 days prior to each study visit was calculated.
Defined by an endoscopic subscore of 0 and a Geboes score of less than 2.0.
Assesses how fatigue is associated with disease and how it has an effect on daily activities and function, including tiredness, weakness, listlessness, and lack of energy. The score range is 0 to 52 points. A higher score reflects less fatigue (expressed as a least-squares mean change from baseline). An increase in the score from baseline reflects improvement in fatigue. There were 614 patients in the 1200-mg group; placebo, 308.
Evaluates quality of life. The 32 questions are divided into 4 dimensions: bowel symptoms, systemic symptoms, emotional function, and social function. The total score range is 32 to 224. A higher score reflects better quality of life. An increase in the score from baseline reflects improvement in quality of life. There were 619 patients in the 1200-mg group; placebo, 310.
Quantified the number of weekly episodes of accidental bowel leakage prior to each study visit. A reduction from baseline score indicated improvement. There were 602 patients in the 1200-mg group; placebo, 288.
Quantified the number of nights with sleep interruption due to ulcerative colitis symptoms in the most recent 7 days prior to each study visit. A reduction from baseline score indicated improvement. There were 602 patients in the 1200-mg group; placebo, 288.
Secondary Outcomes
Risankizumab significantly improved clinical response (determined using the adapted Mayo score) compared with placebo (64.3% vs 35.7%, respectively; between-group difference, 28.6% [95% CI, 22.3%-34.8%], P < .001), clinical response determined using the partial adapted Mayo score at week 4 (52.2% vs 30.5%; between-group difference, 21.8% [95% CI, 15.6%-28.1%], P < .001), endoscopic improvement (36.5% vs 12.1%; between-group difference, 24.3% [95% CI, 19.3%-29.4%], P < .001), endoscopic remission (10.6% vs 3.4%; between-group difference, 7.2% [95% CI, 4.2%-10.2%], P < .001), histological, endoscopic, and mucosal improvement (24.5% vs 7.7%; between-group difference, 16.6% [95% CI, 12.3%-21.0%], P < .001), and histological, endoscopic, and mucosal remission (6.3% vs 0.6%; between-group difference, 5.6% [95% CI, 3.5%-7.7%], P < .001) (Table 2 and eFigures 7-8 in Supplement 1).
Risankizumab significantly improved the patient-reported outcome of absence of bowel urgency compared with placebo (44.1% vs 27.7%, respectively; between-group difference, 16.3% [95% CI, 10.3% to 22.4%], P < .001), absence of abdominal pain (35.8% vs 26.5%; between-group difference, 9.3% [95% CI, 3.4% to 15.3%], P = .002), absence of nocturnal bowel movements (67.3% vs 43.1%; between-group difference, 24.2% [95% CI, 17.9% to 30.5%], P < .001), absence of tenesmus (48.7% vs 30.2%; between-group difference, 18.6% [95% CI, 12.4% to 24.8%], P < .001), fecal incontinence (mean change from baseline, −3.8 vs −2.2 episodes/week; between-group difference, −1.6 [95% CI, −2.4 to −0.9] episodes/week, P < .001), and sleep interruption (mean change from baseline, −2.5 vs −1.5 days/week; between-group difference, −1.0 [95% CI, −1.3 to −0.6] days/week, P < .001) (Table 2).
Risankizumab significantly improved fatigue compared with placebo (change in mean FACIT-F score from baseline, 7.9 vs 3.3, respectively; between-group difference, 4.5 [95% CI, 3.1 to 6.0], P < .001) and health-related quality of life (change in mean IBDQ score from baseline, 42.6 vs 24.3; between-group difference, 18.3 [95% CI, 13.4 to 23.3], P < .001) (Table 2 and eFigure 9A in Supplement 1). The occurrence of ulcerative colitis–related hospitalizations was significantly reduced in the risankizumab group compared with the placebo group through week 12 (0.8% vs 5.5%, respectively; between-group difference, −4.8% [95% CI, −7.3% to −2.2%], P < .001) (Table 2).
Maintenance Trial
Primary Outcome
At week 52, each dose of risankizumab significantly improved clinical remission rates compared with placebo (no longer receiving risankizumab) (40.2% for 180 mg of risankizumab and 37.6% for 360 mg of risankizumab vs 25.1% for placebo (between-group difference for 180 mg of risankizumab vs placebo, 16.3% [97.5% CI, 6.1%-26.6%], P < .001; between-group difference for 360 mg of risankizumab vs placebo, 14.2% [97.5% CI, 4.0%-24.5%], P = .002) (Table 3 and eTable 5 in Supplement 1).
Table 3. Primary and Key Secondary Outcomes in the Maintenance Trial.
| No. (%) [97.5% CI]a | Risankizumab (180 mg) vs placebo | Risankizumab (360 mg) vs placebo | |||||
|---|---|---|---|---|---|---|---|
| Risankizumab administered subcutaneously | Placebo administered subcutaneously (n = 183) | ||||||
| 180 mg (n = 179) | 360 mg (n = 186) | Adjusted between-group difference, % (97.5% CI)b | P valuec | Adjusted between-group difference, % (97.5% CI)b | P valuec | ||
| Primary outcome | |||||||
| Clinical remission (determined using the adapted Mayo score)d,e | 72 (40.2) [31.9 to 48.4] | 70 (37.6) [29.7 to 45.6] | 46 (25.1) [17.9 to 32.3] | 16.3 (6.1 to 26.6) | <.001 | 14.2 (4.0 to 24.5) | .002 |
| Secondary outcomes | |||||||
| Clinical response (determined using the adapted Mayo score)d,f | 122 (68.2) [60.4 to 76.0] | 116 (62.3) [54.4 to 70.3] | 95 (51.9) [43.6 to 60.2] | 17.1 (6.2 to 28.0) | <.001 | 11.5 (0.3 to 22.6) | .02 |
| Improvement | |||||||
| Endoscopicg | 91 (50.8) [42.4 to 59.2] | 90 (48.3) [40.1 to 56.5] | 58 (31.7) [24.0 to 39.4] | 20.1 (9.2 to 30.9) | <.001 | 17.4 (6.6 to 28.3) | <.001 |
| Histological, endoscopic, and mucosalh | 77 (42.8) [34.5 to 51.1] | 79 (42.2) [34.1 to 50.4] | 43 (23.5) [16.5 to 30.5] | 20.2 (9.9 to 30.5) | <.001 | 19.8 (9.5 to 30.0) | <.001 |
| Endoscopic remissioni | 41 (23.2) [16.1 to 30.3] | 45 (24.3) [17.3 to 31.4] | 27 (14.8) [8.9 to 20.6] | 9.5 (0.8 to 18.2) | .01 | 9.6 (0.9 to 18.2) | .01 |
| No bowel urgencyj | 96 (53.6) [45.3 to 62.0] | 92 (49.4) [41.2 to 57.6] | 57 (31.1) [23.5 to 38.8] | 22.6 (11.8 to 33.5) | <.001 | 18.4 (7.4 to 29.3) | <.001 |
| No abdominal painj | 84 (46.9) [38.6 to 55.3] | 70 (37.8) [29.8 to 45.8] | 54 (29.5) [22.0 to 37.1] | 17.0 (6.0 to 28.0) | <.001 | 8.2 (−2.6 to 19.0) | .09 |
| Histological, endoscopic, and mucosal remissionk | 23 (12.9) [7.3 to 18.6] | 29 (15.6) [9.7 to 21.6] | 18 (9.8) [4.9 to 14.8] | 4.0 (−3.1 to 11.2) | .21 | 6.1 (−1.2 to 13.4) | .06 |
| Mean change from induction baseline (95% CI) | |||||||
| 13-Item Functional Assessment of Chronic Illness Therapy-Fatigue scale scorel | 10.9 (8.5 to 13.4) | 10.3 (7.8 to 12.8) | 7.0 (4.6 to 9.5) | 3.9 (0.8 to 7.0) | .005 | 3.3 (0.2 to 6.3) | .02 |
| 32-Question Inflammatory Bowel Disease Questionnaire scorem | 52.6 (43.8 to 61.3) | 50.3 (41.0 to 59.5) | 35.0 (26.0 to 44.1) | 17.5 (6.6 to 28.4) | <.001 | 15.2 (3.7 to 26.8) | .003 |
| ≥1 Ulcerative colitis–related hospitalization, patients/100 person-years (97.5% CI) | 0.6 (0 to 1.9) | 1.2 (0 to 3.1) | 3.1 (0 to 6.1) | −2.5 (−5.8 to 0.8) | .09 | −1.8 (−5.5 to 1.8) | .25 |
| No nocturnal bowel movementj | 75 (41.9) [33.6 to 50.2] | 81 (43.5) [35.3 to 51.6] | 55 (30.1) [22.5 to 37.7] | 12.0 (2.0 to 21.9) | .007 | 14.8 (4.8 to 24.7) | <.001 |
| No tenesmusj | 66 (36.9) [28.8 to 45.0] | 68 (36.8) [28.8 to 44.8] | 43 (23.5) [16.5 to 30.5] | 13.1 (3.4 to 22.9) | .003 | 14.4 (4.5 to 24.3) | .001 |
| Mean change from induction baseline (97.5% CI) | |||||||
| Fecal incontinence, episodes/wkn | −3.4 (−4.9 to −2.0) | −2.9 (−4.5 to −1.3) | −2.8 (−4.4 to −1.1) | −0.7 (−2.8 to 1.5) | .48 | −0.1 (−2.4 to 2.2) | .92 |
| Sleep interrupted due to ulcerative colitis symptoms, d/wko | −2.6 (−3.2 to −1.9) | −2.5 (−3.1 to −1.9) | −1.8 (−2.4 to −1.2) | −0.8 (−1.7 to 0.07) | .04 | −0.7 (−1.5 to 0.1) | .06 |
| Clinical remission | |||||||
| Maintenance, No./total (%) [97.5% CI]p | 31/44 (70.2) [54.7 to 85.8] | 20/40 (50.0) [32.3 to 67.7] | 21/53 (39.6) [24.6 to 54.7] | 29.2 (7.4 to 51.0) | .003 | 12.5 (−10.5 to 35.6) | .22 |
| Corticosteroid-freeq | 71 (39.6) [31.4 to 47.8] | 69 (37.1) [29.2 to 45.0] | 46 (25.1) [17.9 to 32.3] | 15.8 (5.6 to 26.0) | <.001 | 13.7 (3.5 to 24.0) | .003 |
| Maintenance of endoscopic improvement, No./total (%) [97.5% CI]r | 45/61 (73.6) [60.9 to 86.3] | 37/68 (54.1) [40.5 to 67.7] | 37/78 (47.4) [34.8 to 60.1] | 23.9 (6.4 to 41.4) | .002 | 4.8 (−13.7 to 23.2) | .56 |
Unless otherwise indicated. All outcomes were controlled for multiplicity. The results for the categorical outcomes are based on nonresponder multiple imputation to handle missing data due to logistical restrictions because of the COVID-19 pandemic or geopolitical restrictions. The results for the continuous outcomes are based on return to baseline multiple imputation. The patients in the maintenance trial (and included in the primary efficacy analysis) received at least 1 dose of risankizumab either during the 12-week induction trial or during the 52-week maintenance trial.
Calculated using the Mantel-Haenszel common rate difference with nonresponder imputation while incorporating multiple imputation to handle missing data due to logistical restrictions because of the COVID-19 pandemic or geopolitical restrictions for categorical outcomes and using analysis of covariance or mixed-effect model and a repeated-measures method with return to baseline multiple imputation for continuous outcomes.
Hierarchical testing was used to calculate the P values.
The adapted Mayo score measures disease activity in patients with ulcerative colitis. There are 3 components of the score: the stool frequency score, the rectal bleeding score, and an endoscopic subscore. Each score is measured on a scale from 0 to 3 and then the scores are combined. The combined score range is 0 to 9. A score of 5 to 9 points indicates moderate to severe active disease; a score of 0 indicates inactive disease.
Clinical remission was defined as a stool frequency score of 1 or less and not greater than the score at baseline. A score of 0 was given for patients with rectal bleeding. An endoscopic subscore was used to evaluate each segment of the colon (rectum, sigmoid, descending colon, transverse colon, and ascending colon or cecum) using a 4-point scale. A higher score indicates more severe disease. An endoscopic subscore of 1 or less was given for patients without friability.
Clinical response was defined as a decrease of at least 30% and a decrease of at least 2 points from baseline and was determined by a decrease in rectal bleeding score of 1 or greater or an absolute rectal bleeding score of 1 or less.
Defined by an endoscopic subscore of 1 or less without friability.
Defined by an endoscopic subscore of 0 or 1 without friability and a Geboes score of 3.1 or less.
Defined by an endoscopic subscore of 0.
The patients were assessed using a 4-point scale to evaluate symptom frequency and severity over time. A higher score signified more severe pain. The mean of the scores from the most recent 3 days and up to 10 days prior to each study visit was calculated.
Defined by an endoscopic subscore of 0 and a Geboes score of less than 2.0.
Assesses how fatigue is associated with disease and how it has an effect on daily activities and function, including tiredness, weakness, listlessness, and lack of energy. The score range is 0 to 52 points. A higher score reflects less fatigue (expressed as a least-squares mean change from baseline). An increase in the score from baseline reflects improvement in fatigue. For the assessment of the FACIT-F, there were 166 patients in the 180 mg of risankizumab group; 163 in the 360 mg of risankizumab group; and 171 in the placebo group.
Evaluates quality of life. The 32 questions are divided into 4 dimensions: bowel symptoms, systemic symptoms, emotional function, and social function. The total score range is 32 to 224. A higher score reflects better quality of life. An increase in the score from baseline reflects improvement in quality of life. For the evaluation of the Inflammatory Bowel Disease Questionnaire, there were 168 patients in the 180 mg of risankizumab group; 168 in the 360 mg of risankizumab group; and 172 in the placebo group.
Quantified the number of weekly episodes of accidental bowel leakage prior to each study visit. A reduction from baseline induction score indicated improvement. For the evaluation of fecal incontinence, there were 66 patients in the 180 mg of risankizumab group; 70 in the 360 mg of risankizumab group; and 69 in the placebo group.
Quantified the number of nights with sleep interruption due to ulcerative colitis symptoms in the most recent 7 days prior to each study visit. A reduction from baseline induction score indicated improvement. For the evaluation of sleep disruption, there were 66 patients in the 180 mg of risankizumab group; 70 in the 360 mg of risankizumab group; and 69 in the placebo group.
Defined as clinical remission at week 52 among patients who were in remission at week 0 of maintenance.
Defined as clinical remission at week 52 among patients who abstained from corticosteroid use for 90 days or longer prior to the assessment at week 52.
Defined as endoscopic improvement at week 52 among patients who were in remission at week 0 of maintenance.
Secondary Outcomes
Each dose of risankizumab significantly improved clinical response (determined using the adapted Mayo score) compared with placebo (68.2% for 180 mg of risankizumab and 62.3% for 360 mg of risankizumab vs 51.9% for placebo; between-group difference for 180 mg of risankizumab vs placebo, 17.1% [97.5% CI, 6.2%-28.0%], P < .001; between-group difference for 360 mg of risankizumab vs placebo, 11.5% [97.5% CI, 0.3%-22.6%], P = .02) (Table 3).
Each dose of risankizumab significantly increased (1) endoscopic improvement compared with placebo (50.8% for 180 mg of risankizumab and 48.3% for 360 mg of risankizumab vs 31.7% for placebo; between-group difference for 180 mg of risankizumab vs placebo, 20.1% [97.5% CI, 9.2%-30.9%], P < .001; between-group difference for 360 mg of risankizumab vs placebo, 17.4% [97.5% CI, 6.6%-28.3%], P < .001), (2) endoscopic remission (23.2% for 180 mg of risankizumab and 24.3% for 360 mg of risankizumab vs 14.8% for placebo; between-group difference for 180 mg of risankizumab vs placebo, 9.5% [97.5% CI, 0.8%-18.2%], P = .01; between-group difference for 360 mg of risankizumab vs placebo, 9.6% [97.5% CI, 0.9%-18.2%], P = .01), and (3) histological, endoscopic, and mucosal improvement (42.8% for 180 mg of risankizumab and 42.2% for 360 mg of risankizumab vs 23.5% for placebo; between-group difference for 180 mg of risankizumab vs placebo, 20.2% [97.5% CI, 9.9%-30.5%], P < .001; between-group difference for 360 mg of risankizumab vs placebo, 19.8% [97.5% CI, 9.5%-30.0%], P < .001).
Modest differences between each dose of risankizumab and placebo were observed for histological, endoscopic, and mucosal remission (12.9% for 180 mg of risankizumab and 15.6% for 360 mg of risankizumab vs 9.8% for placebo; between-group difference for 180 mg of risankizumab vs placebo, 4.0% [97.5% CI, −3.1% to 11.2%], P = .21; between-group difference for 360 mg of risankizumab vs placebo, 6.1% [97.5% CI, −1.2% to 13.4%], P = .06).
Numerical differences were observed in the following patient-reported outcomes: (1) absence of bowel urgency (53.6% for 180 mg of risankizumab and 49.4% for 360 mg of risankizumab vs 31.1% for placebo; between-group difference for 180 mg of risankizumab vs placebo, 22.6% [97.5% CI, 11.8% to 33.5%], P < .001; between-group difference for 360 mg of risankizumab vs placebo, 18.4% [97.5% CI, 7.4% to 29.3%], P < .001), (2) absence of abdominal pain (46.9% for 180 mg of risankizumab and 37.8% for 360 mg of risankizumab vs 29.5% for placebo; between-group difference for 180 mg of risankizumab vs placebo, 17.0% [97.5% CI, 6.0% to 28.0%], P < .001; between-group difference for 360 mg of risankizumab vs placebo, 8.2% [97.5% CI, −2.6% to 19.0%], P = .09), (3) absence of nocturnal bowel movements (41.9% for 180 mg of risankizumab and 43.5% for 360 mg of risankizumab vs 30.1% for placebo; between-group difference for 180 mg of risankizumab vs placebo, 12.0% [97.5% CI, 2.0% to 21.9%], P = .007; between-group difference for 360 mg of risankizumab vs placebo, 14.8% [97.5% CI, 4.8% to 24.7%], P < .001), and (4) absence of tenesmus (36.9% for 180 mg of risankizumab and 36.8% for 360 mg of risankizumab vs 23.5% for placebo; between-group difference for 180 mg of risankizumab vs placebo, 13.1% [97.5% CI, 3.4% to 22.9%], P = .003; between-group difference for 360 mg of risankizumab vs placebo, 14.4% [97.5% CI, 4.5% to 24.3%], P = .001).
Numerical differences were observed for fecal incontinence (mean change from baseline, −3.4 episodes/week for 180 mg of risankizumab and −2.9 episodes/week for 360 mg of risankizumab vs −2.8 episodes/week for placebo; between-group difference for 180 mg of risankizumab vs placebo, −0.7 [97.5% CI, −2.8 to 1.5] episodes/week, P = .48; between-group difference for 360 mg of risankizumab vs placebo, −0.1 [97.5% CI, −2.4 to 2.2] episodes/week, P = .92) and for sleep interruption (mean change from baseline, −2.6 days/week for 180 mg of risankizumab and −2.5 days/week for 360 mg of risankizumab vs −1.8 days/week for placebo; between-group difference for 180 mg of risankizumab vs placebo, −0.8 [97.5% CI, −1.7 to 0.07] days/week, P = .04; between-group difference for 360 mg of risankizumab vs placebo, −0.7 [97.5% CI, −1.5 to 0.1] days/week, P = .06).
Numerical differences were also observed for fatigue (change in mean FACIT-F score from baseline, 10.9 for 180 mg of risankizumab and 10.3 for 360 mg of risankizumab vs 7.0 for placebo; between-group difference for 180 mg of risankizumab vs placebo, 3.9 [97.5% CI, 0.8 to 7.0], P = .005; between-group difference for 360 mg of risankizumab vs placebo, 3.3 [97.5% CI, 0.2 to 6.3], P = .02) and for quality of life (change in mean IBDQ score from baseline, 52.6 for 180 mg of risankizumab and 50.3 for 360 mg of risankizumab vs 35.0 for placebo; between-group difference for 180 mg of risankizumab vs placebo, 17.5 [97.5% CI, 6.6 to 28.4], P < .001; between-group difference for 360 mg of risankizumab vs placebo, 15.2 [97.5% CI, 3.7 to 26.8], P = .003). The occurrence of ulcerative colitis–related hospitalizations decreased for patients treated with risankizumab compared with placebo (0.6/100 person-years for 180 mg of risankizumab and 1.2/100 person-years for 360 mg of risankizumab vs 3.1/100 person-years for placebo; between-group difference for 180 mg of risankizumab vs placebo, −2.5/100 person-years [97.5% CI, −5.8 to 0.8/100 person-years], P = .09; between-group difference for 360 mg of risankizumab vs placebo, −1.8/100 person-years [97.5% CI, −5.5 to 1.8/100 person-years], P = .25).
Higher proportions of patients treated with 180 mg of risankizumab were able to sustain clinical remission from maintenance week 0 to week 52 compared with placebo (70.2% for 180 mg of risankizumab and 50.0% for 360 mg of risankizumab vs 39.6% for placebo; between-group difference for 180 mg of risankizumab vs placebo, 29.2% [97.5% CI, 7.4% to 51.0%], P = .003; between-group difference for 360 mg of risankizumab vs placebo, 12.5% [97.5% CI, −10.5% to 35.6%], P = .22).
Among patients who continued with any dose of risankizumab, there were numerically improved rates for corticosteroid-free clinical remission compared with placebo (39.6% for 180 mg of risankizumab and 37.1% for 360 mg of risankizumab vs 25.1% for placebo; between-group difference for 180 mg of risankizumab vs placebo, 15.8% [97.5% CI, 5.6%-26.0%], P < .001; between-group difference for 360 mg of risankizumab vs placebo, 13.7% [97.5% CI, 3.5%-24.0%], P = .003) and for overall corticosteroid discontinuation (64.9% for 180 mg of risankizumab and 54.2% for 360 mg of risankizumab vs 36.8% for placebo; between-group difference for 180 mg of risankizumab vs placebo, 28.4% [97.5% CI, 14.0%-42.8%], P < .001; between-group difference for 360 mg of risankizumab vs placebo, 20.7% [97.5% CI, 4.9%-36.6%], P = .01) (Table 3 and eFigure 10 in Supplement 1).
Among the 72 patients treated with risankizumab and who had clinical remission at week 52, there were 71 (98.6%) not taking corticosteroids. Higher proportions of patients treated with 180 mg of risankizumab with endoscopic improvement at week 0 of maintenance were able to sustain their status to week 52 compared with placebo (73.6% for 180 mg of risankizumab and 54.1% for 360 mg of risankizumab vs 47.4% for placebo; between-group difference for 180 mg of risankizumab vs placebo, 23.9% [97.5% CI, 6.4% to 41.4%], P = .002; between-group difference for 360 mg of risankizumab vs placebo, 4.8% [97.5% CI, −13.7% to 23.2%], P = .56).
Additional Prespecified Subgroup Analyses in the Induction and Maintenance Trials
In the induction trial, the clinical remission rates were numerically improved for patients without a history of intolerance or inadequate response to advanced therapy and treated with risankizumab compared with patients in the placebo group (29.7% vs 8.4%, respectively; between-group difference, 21.3% [95% CI, 14.6% to 27.9%]), and also for patients with a history of inadequate response to advanced therapy (11.4% vs 4.3%; between-group difference, 7.2% [95% CI, 2.6%-11.8%]) (eTable 6 in Supplement 1).
In the maintenance trial, the patients without a history of inadequate response to advanced therapy and treated with either dose of risankizumab (180 mg or 360 mg) had numerically improved clinical remission rates compared with placebo (50.9% for 180 mg of risankizumab and 61.7% for 360 mg of risankizumab vs 31.1% for placebo; between-group difference for 180 mg of risankizumab vs placebo, 19.8% [95% CI, −0.2% to 39.7%]; between-group difference for 360 mg of risankizumab vs placebo, 30.6% [95% CI, 11.2% to 50.0%]). Patients with a history of inadequate response to advanced therapy and treated with either dose of risankizumab also had numerically improved rates of clinical remission compared with placebo during maintenance (36.6% for 180 mg of risankizumab and 29.5% for 360 mg of risankizumab vs 23.2% for placebo; between-group difference for 180 mg of risankizumab vs placebo, 13.4% [95% CI, 2.6% to 24.2%]; between-group difference for 360 mg of risankizumab vs placebo, 6.3% [95% CI, −4.0% to 16.7%]).
A numerically greater improvement was observed in the induction trial for endoscopic improvement in patients treated with risankizumab (47.6% in those without a history of inadequate response to advanced therapy vs 25.9% in those with a history of inadequate response to prior advanced therapy), endoscopic remission (16.7% vs 4.8%, respectively), histological, endoscopic, and mucosal improvement (33.4% vs 16.0%), and histological, endoscopic, and mucosal remission (10.7% vs 2.1%). Similar results were observed for maintenance in patients treated with 180 mg of risankizumab; there were numerically greater increases in the secondary clinical and endoscopic outcomes among those without a history of intolerance or inadequate response to advanced therapy vs those with a history of inadequate response to advanced therapy, including endoscopic improvement (59.8% vs 47.8%, respectively), endoscopic remission (36.6% vs 18.7%), histological, endoscopic, and mucosal improvement (54.8% vs 38.8%), and histological, endoscopic, and mucosal remission (27.0% vs 8.2%).
Numerically greater differences were also observed for maintenance in secondary clinical and endoscopic outcomes in patients treated with 360 mg of risankizumab without a history of intolerance or inadequate response to advanced therapy vs those with a history of inadequate response to advanced therapy, including endoscopic improvement (76.2% vs 38.8%, respectively), endoscopic remission (51.6% vs 15.1%), histological, endoscopic, and mucosal improvement (69.3% vs 33.1%), and histological, endoscopic, and mucosal remission (32.1% vs 10.1%).
For the induction trial, patients in the 1200 mg of risankizumab group had numerically larger reductions in high-sensitivity CRP from baseline to week 4 (−4.2 mg/L vs −1.4 mg/L for placebo) and from baseline to week 12 (−4.4 mg/L vs −0.3 mg/L, respectively) (between-group difference in mean change from baseline to week 4, −2.8 mg/L [95% CI, −4.0 to −1.5 mg/L], P < .001; between-group difference in mean change from baseline to week 12, −4.1 mg/L [95% CI, −5.5 to −2.7 mg/L], P < .001) (eFigure 11A-B in Supplement 1). Similar results were observed for fecal calprotectin from baseline to week 4 (−1106 mg/kg for risankizumab vs 306 mg/kg for placebo) and from baseline to week 12 (−1804 mg/kg vs −928 mg/kg, respectively); between-group difference in mean change from baseline to week 4, −1457 mg/kg [95% CI, −2036 to −879 mg/kg], P < .001; between-group difference in mean change from baseline to week 12, −876 mg/kg [95% CI, −1300 to −453 mg/kg], P < .001) (eFigure 11C-D in Supplement 1).
For the maintenance trial, a numeric reduction was observed in high-sensitivity CRP at week 52 (0.2 mg/L for 180 mg of risankizumab and −0.2 mg/L for 360 mg of risankizumab vs 2.1 mg/L for placebo; between-group difference for 180 mg of risankizumab vs placebo, −1.8 mg/L [97.5% CI, −3.4 to −0.3 mg/L], P = .02; between-group difference for 360 mg of risankizumab vs placebo, −2.2 mg/L [97.5% CI, −3.8 to −0.6 mg/L], P = .006) and in fecal calprotectin (−358 mg/kg for 180 mg of risankizumab and −120 mg/kg for 360 mg of risankizumab vs 500 mg/kg for placebo; between-group difference for 180 mg of risankizumab vs placebo, −858.6 mg/kg [97.5% CI, −1502.8 to −214.4 mg/kg], P = .009; between-group difference for 360 mg of risankizumab vs placebo, −620.5 mg/kg [97.5% CI, −1260.4 to 19.4 mg/kg], P = .06) (eFigure 11E-H in Supplement 1).
Post Hoc Analysis
Serum IL-22 concentrations were evaluated to confirm risankizumab IL-23 pathway engagement. Among patients receiving placebo throughout both the induction trial (administered intravenously) and the maintenance trial (administered subcutaneously), no difference was observed in the mean change in IL-22 from baseline to week 4 (−29.7%) and from baseline to week 52 (−31.9%) (P = .27 for week 4 and P = .18 for week 52; eFigure 11I in Supplement 1). In contrast, patients receiving risankizumab throughout both the induction trial and the maintenance trial showed numeric suppression of IL-22 levels from baseline to week 4 (−47.5% for 180 mg, P < .001; −30.2% for 360 mg, P = .02) and from baseline to week 52 (−66.3% for 180 mg and −57.0% for 360 mg, P < .001 for both outcomes) (eFigure 11I in Supplement 1). Patients who received risankizumab in the induction trial demonstrated suppression at week 4 (−51.7%) continuing to week 52 (−62.1% in placebo group), suggesting a durable effect of risankizumab on the IL-23 pathway after 12 weeks of induction therapy (P < .001 for both outcomes).
Safety Outcomes for the Induction and Maintenance Trials
In the induction trial, the most frequently reported adverse events (using the Medical Dictionary for Regulatory Activities preferred terms) were COVID-19 (4.8%) and anemia (3.4%) in the risankizumab group and colitis ulcerative (10.2%) and anemia (6.5%) in the placebo group (Table 4). The rate of serious adverse events was 2.3% for risankizumab compared with 10.2% for placebo. One treatment-emergent death occurred in the risankizumab group and was due to respiratory failure caused by COVID-19 pneumonia.
Table 4. Treatment-Emergent Adverse Events Through Week 12 of the Induction Trial and Through Week 52 of the Maintenance Trial.
| Adverse events, No. (%) | |||||
|---|---|---|---|---|---|
| Induction trial (12 wk)a | Maintenance trial (52 wk) | ||||
| 1200 mg of risankizumab administered intravenously (n = 651) |
Placebo administered intravenously (n = 324) |
Risankizumab administered subcutaneously | Placebo administered subcutaneously (n = 196) |
||
| 180 mg (n = 193) | 360 mg (n = 195) | ||||
| Adverse events | |||||
| Any | 274 (42.1) | 161 (49.7) | 140 (72.5) | 138 (70.8) | 150 (76.5) |
| Possibly related to the study drugb | 61 (9.4) | 26 (8.0) | 36 (18.7) | 34 (17.4) | 41 (20.9) |
| Leading to discontinuation of study drug | 4 (0.6) | 12 (3.7) | 3 (1.6) | 5 (2.6) | 3 (1.5) |
| Related to COVID-19 | 35 (5.4) | 19 (5.9) | 20 (10.4) | 26 (13.3) | 27 (13.8) |
| Seriousc | 15 (2.3) | 33 (10.2) | 10 (5.2) | 10 (5.1) | 16 (8.2) |
| Severed | 16 (2.5) | 33 (10.2) | 3 (1.6) | 6 (3.1) | 10 (5.1) |
| Died | 1 (0.2)e | 0 | 0 | 1 (0.5)f | 0 |
| Most frequent adverse events (≥5% in any treatment group) g | |||||
| COVID-19 | 31 (4.8) | 19 (5.9) | 17 (8.8) | 26 (13.3) | 23 (11.7) |
| Anemia | 22 (3.4) | 21 (6.5) | 1 (0.5) | 1 (0.5) | 2 (1.0) |
| Arthralgia | 20 (3.1) | 5 (1.5) | 11 (5.7) | 18 (9.2) | 9 (4.6) |
| Headache | 19 (2.9) | 7 (2.2) | 9 (4.7) | 8 (4.1) | 15 (7.7) |
| Nasopharyngitis | 18 (2.8) | 8 (2.5) | 18 (9.3) | 12 (6.2) | 16 (8.2) |
| Colitis ulcerativeh | 11 (1.7) | 33 (10.2) | 25 (13.0) | 27 (13.8) | 29 (14.8) |
| Treatment-emergent adverse events of special interest g , i | |||||
| Hypersensitivityj | 24 (3.7) | 6 (1.9) | 20 (10.4) | 10 (5.1) | 10 (5.1) |
| Hepatic eventsk | 10 (1.5) | 14 (4.3) | 3 (1.6) | 13 (6.7) | 1 (0.5) |
| Serious infections | 4 (0.6) | 4 (1.2) | 2 (1.0) | 1 (0.5) | 4 (2.0) |
| Injection site reactions | 4 (0.6) | 4 (1.2) | 7 (3.6) | 5 (2.6) | 2 (1.0) |
| Herpes zoster | 2 (0.3) | 0 | 2 (1.0) | 1 (0.5) | 3 (1.5) |
| Opportunistic infectionl | 0 | 0 | 0 | 1 (0.5) | 0 |
| Active tuberculosis | 0 | 0 | 0 | 0 | 0 |
| Serious hypersensitivity | 0 | 0 | 0 | 0 | 0 |
| Adjudicated anaphylactic reactions | 0 | 0 | 0 | 0 | 0 |
| Adjudicated major cardiovascular events | 0 | 0 | 0 | 0 | 0 |
| Malignancies (all types) | 0 | 2 (0.6) | 0 | 2 (1.0) | 1 (0.5) |
| Non–melanoma skin cancer | 0 | 0 | 0 | 0 | 1 (0.5) |
One patient randomized to 1200 mg of risankizumab was treated with placebo.
Assessed by study investigators.
Met any of the following criteria: death, life-threatening, hospitalization or prolongation of hospitalization, congenital anomaly, persistent or significant disability or incapacity, important medical event requiring a medical or a surgical intervention to prevent serious outcome.
Classified as grade 3 or above using version 4.03 of the Common Terminology Criteria for Adverse Events.
Due to respiratory failure caused by COVID-19 pneumonia.
Due to the colon adenocarcinoma and not related to study drug.
Ordered by decreasing frequency in the 1200 mg of risankizumab group.
Medical Dictionary for Regulatory Activities (MedDRA) preferred term. Includes, but is not limited to, worsening of the underlying disease, which was defined at the investigator’s discretion.
Treatment-emergent adverse events began either on or after the first dose of the study drug, during the induction period, before the first dose of study drug during the extended treatment period, or within 140 days after the last dose of the study drug in the maintenance trial.
Includes both nonserious and serious hypersensitivity reaction events. The events were identified with standardized MedDRA queries. The broader medical concept terms related to the injection and infusion site (ie, injection site rash) overlap with customized MedDRA queries for injection site reaction.
Identified with search criteria covering the standardized MedDRA queries of hepatic failure, fibrosis, and cirrhosis and other liver damage–related conditions, hepatitis, noninfectious, cholestasis and jaundice of hepatic origin, liver-related investigations, signs and symptoms, and liver-related coagulation and bleeding disturbances.
Excludes tuberculosis and herpes zoster.
In the maintenance trial, the most frequently reported adverse events among all treatment groups were colitis ulcerative (13.0% in the 180 mg of risankizumab group and 13.8% in the 360 mg of risankizumab group vs 14.8% in the placebo group) and COVID-19 (8.8% in the 180 mg group and 13.3% in the 360 mg group vs 11.7% for placebo). Colitis ulcerative refers to the worsening of the underlying disease, which was defined at the investigator’s discretion. Serious adverse events were reported in 5.2% in the 180 mg group and 5.1% in the 360 mg group vs 8.2% in the placebo group.
One non–treatment-emergent death in the 360 mg of risankizumab group due to colon adenocarcinoma was reported, which existed prior to administration of the first dose of the study drug. Malignancies were reported in 2 patients undergoing treatment with risankizumab (1 invasive ductal breast carcinoma in a patient with a history of a breast lump and 1 non–treatment-emergent colon adenocarcinoma, which was fatal). Both events were determined to be unrelated to the study drug. Exposure-adjusted event rates for the induction and maintenance trials appear in eTables 7-10 in Supplement 1.
Discussion
In the phase 3 induction and maintenance trials, treatment with risankizumab improved rates of clinical remission compared with placebo in patients with moderately to severely active ulcerative colitis. In the induction trial, treatment with risankizumab improved endoscopic and histological secondary outcomes characterized by endoscopic improvement; endoscopic remission; and histological, endoscopic, and mucosal improvement. These improvements were also observed in the maintenance trial.
Endoscopic and histological inflammation in patients with ulcerative colitis have been identified as important indicators of disease activity.16 Adequate healing (as measured by histological, endoscopic, and mucosal improvement) has been associated with improved long-term outcomes in patients with ulcerative colitis, including decreased steroid use, hospitalization, and colectomy avoidance.17 After 1 year of treatment with either maintenance dose of risankizumab (180 mg or 360 mg administered subcutaneously), more than 40% of patients had histological and endoscopic improvement. Further research is needed to define the optimal assessment of histological healing as a measure of disease remission and its long-term effect.
In the maintenance trial, 75% of the patient population had a history of inadequate response to advanced therapies. Safety outcomes in this trial were consistent with previously reported safety outcomes in clinical trials of risankizumab for Crohn disease.18,19 Many patients with inflammatory bowel disease have a history of intolerance or an inadequate response to advanced therapies (including infliximab, adalimumab, golimumab, and vedolizumab; the Janus kinase inhibitors tofacitinib, filgotinib, and upadacitinib; and ozanimod, which is a sphingosine-1-phosphate receptor modulator) that inhibit inflammatory pathways of inflammatory bowel disease.20,21
Evidence from other trials indicates that response rates are typically higher for patients without a history of inadequate response to advanced therapy than the rates for patients with a history of inadequate response to advanced therapy.6,7,22,23 This finding is consistent with the results reported here from the induction and maintenance trials. Numerically higher clinical remission and endoscopic outcomes were observed in patients without a history of inadequate response to advanced therapy during the maintenance trial for the 360-mg dose of risankizumab compared with 180-mg dose, but not in patients with a history of inadequate response to advanced therapy. Although the reason for the attenuated efficacy of anti-inflammatory agents in patients with ulcerative colitis and a history of inadequate response to advanced therapy is not fully understood, there is accumulating evidence that uncontrolled inflammation may be associated with immunological, structural, and neuromuscular changes in the diseased colon that may be less responsive to treatment.24,25
Limitations
The 2 randomized clinical trials had several limitations. First, in the maintenance trial, drug levels of risankizumab were detectable until week 16 in the placebo group (no longer receiving risankizumab but received it during the induction trial), demonstrating continued risankizumab exposure from the induction trial; this may potentially inflate the response rates for outcomes in the placebo group during the maintenance trial.19 Persistent suppression of serum concentrations of IL-22 (an effector cytokine and biomarker of anti–IL-23 therapy) was observed from week 4 to week 52.
Second, patients with prior exposure to ustekinumab were excluded from the trial. The efficacy of risankizumab in ulcerative colitis for patients with prior exposure to emerging therapies in the evolving inflammatory bowel disease treatment landscape requires further study. Third, follow-up beyond 52 weeks is not available.
Conclusions
Compared with placebo, risankizumab improved clinical remission rates in an induction trial and in a maintenance trial for patients with moderately to severely active ulcerative colitis. Further study is needed to identify benefits beyond the 52-week follow-up.
eMethods 1. Methods and Statistical Analysis for Phase 2b Induction Substudy
eMethods 2. Full Patient Eligibility Criteria in the Final Protocol Amendment of the Phase 2b and Phase 3 INSPIRE Induction Study
eMethods 3. Supplementary Methods for the Phase 3 Induction and Maintenance Studies
eFigure 1. Key Study Design Features and Trial Profile of Induction and Maintenance Studies
eFigure 2. Trial Profile of the Dose-Ranging Phase 2b Substudy
eFigure 3. Key Clinical and Endoscopic Endpoints and Inflammatory Biomarkers for the Dose-Ranging Phase 2b Substudy
eFigure 4. Graphical Multiple-Testing Procedure for the Phase 3 Induction Study
eFigure 5. Graphical Multiple-Testing Procedure For the Phase 3 Maintenance Study
eFigure 6. Clinical Remission per Adapted Mayo Score by Subgroup Analysis at Week 12 of Induction and Week 52 of Maintenance
eFigure 7. Percent of Patients Who Achieved Histologic Remission at Week 12 of Induction and Week 52 of Maintenance
eFigure 8. Percent of Patients that Achieved Clinical Response per Partial Adapted Mayo Score Through Week 12 of INSPIRE
eFigure 9. Patient-Reported Outcomes Among Patients Who Reported Having Symptoms at Baseline of Phase 3 Induction
eFigure 10. Discontinuation of Corticosteroids at Week 52 of Maintenance Among Patients Taking Corticosteroids at Baseline of Induction
eFigure 11. Change From Baseline in Biomarkers During the Phase 3 Induction and Maintenance Studies
eTable 1. Baseline Characteristics for the Dose-Ranging Phase 2b Induction Substudy
eTable 2. Treatment-Emergent Adverse Events From the Dose-Ranging Phase 2b Induction Substudy
eTable 3. Additional Baseline Characteristics and Demographics
eTable 4. Sensitivity Analysis for the Primary Endpoint Clinical Remission per Adapted Mayo Score for the Phase 3 Induction and Maintenance Studies
eTable 5. Primary and Secondary Endpoints at Week 52 of Maintenance for All Patients Who Responded to 12-Week Risankizumab 1200 mg Induction Therapy and Were Randomized to Maintenance
eTable 6. Prespecified Primary and Secondary Endpoints in Non-Advanced and Advanced Therapy-Inadequate Response Patients in the Phase 3 Induction and Maintenance Studies
eTable 7. Exposure-Adjusted Event Rates for Treatment-Emergent Adverse Events Through Week 12 of Induction and Week 52 of Maintenance
eTable 8. Patients Meeting Criteria for Liver-Related Elevations During Induction and Maintenance
eTable 9. Summary of Mean Change From Baseline in Key Chemistry Values During Induction and Maintenance
eTable 10. Summary of Treatment-Emergent Adverse Events Through Week 52 of Maintenance for All Patients Who Responded to 12-Week Risankizumab 1200 mg Intravenous Induction Therapy and Were Randomized to Maintenance
eResults 1. Results for the Dose-Ranging Phase 2b Induction Substudy
eResults 2. Additional Safety and Efficacy Outcomes for the Phase 3 Induction and Maintenance Study
eResults 3. Pharmacokinetics and Immunogenicity Results From Induction and Maintenance Studies
eReferences
Trial protocol and statistical analysis plan
Nonauthor collaborators
Data sharing statement
References
- 1.Gros B, Kaplan GG. Ulcerative colitis in adults: a review. JAMA. 2023;330(10):951-965. doi: 10.1001/jama.2023.15389 [DOI] [PubMed] [Google Scholar]
- 2.Feig JL, Gribetz ME, Lebwohl MG. Chronic lichen sclerosus successfully treated with intralesional adalimumab. Br J Dermatol. 2016;174(3):687-689. doi: 10.1111/bjd.14212 [DOI] [PubMed] [Google Scholar]
- 3.Han SW, McColl E, Barton JR, James P, Steen IN, Welfare MR. Predictors of quality of life in ulcerative colitis: the importance of symptoms and illness representations. Inflamm Bowel Dis. 2005;11(1):24-34. doi: 10.1097/00054725-200501000-00004 [DOI] [PubMed] [Google Scholar]
- 4.Gisbert JP, Chaparro M. Primary failure to an anti-TNF agent in inflammatory bowel disease: switch (to a second anti-TNF agent) or swap (for another mechanism of action)? J Clin Med. 2021;10(22):5318. doi: 10.3390/jcm10225318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verstockt B, Salas A, Sands BE, et al. ; Alimentiv Translational Research Consortium (ATRC) . IL-12 and IL-23 pathway inhibition in inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2023;20(7):433-446. doi: 10.1038/s41575-023-00768-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sands BE, Sandborn WJ, Panaccione R, et al. ; UNIFI Study Group . Ustekinumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2019;381(13):1201-1214. doi: 10.1056/NEJMoa1900750 [DOI] [PubMed] [Google Scholar]
- 7.D’Haens G, Dubinsky M, Kobayashi T, et al. ; LUCENT Study Group . Mirikizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2023;388(26):2444-2455. doi: 10.1056/NEJMoa2207940 [DOI] [PubMed] [Google Scholar]
- 8.Skyrizi (risankizumab): highlights of prescribing information. Package insert. AbbVie; 2022. Accessed July 20, 2023. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/761262s000lbl.pdf
- 9.Skyrizi (risankizumab): annex I summary of product characteristics. Package insert. AbbVie. Accessed August 5, 2023. https://www.ema.europa.eu/en/documents/product-information/skyrizi-epar-product-information_en.pdf
- 10.Skyrizi (risankizumab): prescribing information: 4th edition. AbbVie. Accessed July 12, 2023. https://www.pmda.go.jp/english/review-services/reviews/approved-information/drugs/0001.html#select19
- 11.Ulcerative colitis: clinical trial endpoints: guidance for industry. News release. US Food and Drug Administration; August 2016. Accessed June 12, 2024. https://www.fda.gov/media/99526/download
- 12.Ulcerative colitis: developing drugs for treatment guidance for industry. News release. US Food and Drug Administration; April 2022. Accessed June 12, 2024. https://www.fda.gov/media/158016/download
- 13.Australian Therapeutic Goods Administration . International scientific guideline: guideline on the development of new medicinal products for the treatment of ulcerative colitis. Accessed June 12, 2024. https://www.tga.gov.au/resources/resource/international-scientific-guidelines/international-scientific-guideline-guideline-development-new-medicinal-products-treatment-ulcerative-colitis
- 14.Loftus EV Jr, Ananthakrishnan AN, Lee WJ, et al. Content validity and psychometric evaluation of the Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-Fatigue) in patients with Crohn’s disease and ulcerative colitis. Pharmacoecon Open. 2023;7(5):823-840. doi: 10.1007/s41669-023-00419-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feagan BG, Rochon J, Fedorak RN, et al. ; North American Crohn’s Study Group Investigators . Methotrexate for the treatment of Crohn’s disease. N Engl J Med. 1995;332(5):292-297. doi: 10.1056/NEJM199502023320503 [DOI] [PubMed] [Google Scholar]
- 16.Turner D, Ricciuto A, Lewis A, et al. ; International Organization for the Study of IBD . STRIDE-II: an update on the Selecting Therapeutic Targets in Inflammatory Bowel Disease (STRIDE) initiative of the International Organization for the Study of IBD (IOIBD): determining therapeutic goals for treat-to-target strategies in IBD. Gastroenterology. 2021;160(5):1570-1583. doi: 10.1053/j.gastro.2020.12.031 [DOI] [PubMed] [Google Scholar]
- 17.Parkes G, Ungaro RC, Danese S, et al. Correlation of mucosal healing endpoints with long-term clinical and patient-reported outcomes in ulcerative colitis. J Gastroenterol. 2023;58(10):990-1002. doi: 10.1007/s00535-023-02013-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.D’Haens G, Panaccione R, Baert F, et al. Risankizumab as induction therapy for Crohn’s disease: results from the phase 3 ADVANCE and MOTIVATE induction trials. Lancet. 2022;399(10340):2015-2030. doi: 10.1016/S0140-6736(22)00467-6 [DOI] [PubMed] [Google Scholar]
- 19.Ferrante M, Panaccione R, Baert F, et al. Risankizumab as maintenance therapy for moderately to severely active Crohn’s disease: results from the multicentre, randomised, double-blind, placebo-controlled, withdrawal phase 3 FORTIFY maintenance trial. Lancet. 2022;399(10340):2031-2046. doi: 10.1016/S0140-6736(22)00466-4 [DOI] [PubMed] [Google Scholar]
- 20.Ben-Horin S, Chowers Y. Tailoring anti-TNF therapy in IBD: drug levels and disease activity. Nat Rev Gastroenterol Hepatol. 2014;11(4):243-255. doi: 10.1038/nrgastro.2013.253 [DOI] [PubMed] [Google Scholar]
- 21.Singh S, Murad MH, Fumery M, Dulai PS, Sandborn WJ. First- and second-line pharmacotherapies for patients with moderate to severely active ulcerative colitis: an updated network meta-analysis. Clin Gastroenterol Hepatol. 2020;18(10):2179-2191.e6. doi: 10.1016/j.cgh.2020.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feagan BG, Rubin DT, Danese S, et al. Efficacy of vedolizumab induction and maintenance therapy in patients with ulcerative colitis, regardless of prior exposure to tumor necrosis factor antagonists. Clin Gastroenterol Hepatol. 2017;15(2):229-239.e5. doi: 10.1016/j.cgh.2016.08.044 [DOI] [PubMed] [Google Scholar]
- 23.Sandborn WJ, Vermeire S, Peyrin-Biroulet L, et al. Etrasimod as induction and maintenance therapy for ulcerative colitis (ELEVATE): two randomised, double-blind, placebo-controlled, phase 3 studies. Lancet. 2023;401(10383):1159-1171. doi: 10.1016/S0140-6736(23)00061-2 [DOI] [PubMed] [Google Scholar]
- 24.Friedrich M, Pohin M, Jackson MA, et al. ; Oxford IBD Cohort Investigators; Roche Fibroblast Network Consortium . IL-1-driven stromal-neutrophil interactions define a subset of patients with inflammatory bowel disease that does not respond to therapies. Nat Med. 2021;27(11):1970-1981. doi: 10.1038/s41591-021-01520-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Torres J, Billioud V, Sachar DB, Peyrin-Biroulet L, Colombel JF. Ulcerative colitis as a progressive disease: the forgotten evidence. Inflamm Bowel Dis. 2012;18(7):1356-1363. doi: 10.1002/ibd.22839 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eMethods 1. Methods and Statistical Analysis for Phase 2b Induction Substudy
eMethods 2. Full Patient Eligibility Criteria in the Final Protocol Amendment of the Phase 2b and Phase 3 INSPIRE Induction Study
eMethods 3. Supplementary Methods for the Phase 3 Induction and Maintenance Studies
eFigure 1. Key Study Design Features and Trial Profile of Induction and Maintenance Studies
eFigure 2. Trial Profile of the Dose-Ranging Phase 2b Substudy
eFigure 3. Key Clinical and Endoscopic Endpoints and Inflammatory Biomarkers for the Dose-Ranging Phase 2b Substudy
eFigure 4. Graphical Multiple-Testing Procedure for the Phase 3 Induction Study
eFigure 5. Graphical Multiple-Testing Procedure For the Phase 3 Maintenance Study
eFigure 6. Clinical Remission per Adapted Mayo Score by Subgroup Analysis at Week 12 of Induction and Week 52 of Maintenance
eFigure 7. Percent of Patients Who Achieved Histologic Remission at Week 12 of Induction and Week 52 of Maintenance
eFigure 8. Percent of Patients that Achieved Clinical Response per Partial Adapted Mayo Score Through Week 12 of INSPIRE
eFigure 9. Patient-Reported Outcomes Among Patients Who Reported Having Symptoms at Baseline of Phase 3 Induction
eFigure 10. Discontinuation of Corticosteroids at Week 52 of Maintenance Among Patients Taking Corticosteroids at Baseline of Induction
eFigure 11. Change From Baseline in Biomarkers During the Phase 3 Induction and Maintenance Studies
eTable 1. Baseline Characteristics for the Dose-Ranging Phase 2b Induction Substudy
eTable 2. Treatment-Emergent Adverse Events From the Dose-Ranging Phase 2b Induction Substudy
eTable 3. Additional Baseline Characteristics and Demographics
eTable 4. Sensitivity Analysis for the Primary Endpoint Clinical Remission per Adapted Mayo Score for the Phase 3 Induction and Maintenance Studies
eTable 5. Primary and Secondary Endpoints at Week 52 of Maintenance for All Patients Who Responded to 12-Week Risankizumab 1200 mg Induction Therapy and Were Randomized to Maintenance
eTable 6. Prespecified Primary and Secondary Endpoints in Non-Advanced and Advanced Therapy-Inadequate Response Patients in the Phase 3 Induction and Maintenance Studies
eTable 7. Exposure-Adjusted Event Rates for Treatment-Emergent Adverse Events Through Week 12 of Induction and Week 52 of Maintenance
eTable 8. Patients Meeting Criteria for Liver-Related Elevations During Induction and Maintenance
eTable 9. Summary of Mean Change From Baseline in Key Chemistry Values During Induction and Maintenance
eTable 10. Summary of Treatment-Emergent Adverse Events Through Week 52 of Maintenance for All Patients Who Responded to 12-Week Risankizumab 1200 mg Intravenous Induction Therapy and Were Randomized to Maintenance
eResults 1. Results for the Dose-Ranging Phase 2b Induction Substudy
eResults 2. Additional Safety and Efficacy Outcomes for the Phase 3 Induction and Maintenance Study
eResults 3. Pharmacokinetics and Immunogenicity Results From Induction and Maintenance Studies
eReferences
Trial protocol and statistical analysis plan
Nonauthor collaborators
Data sharing statement