

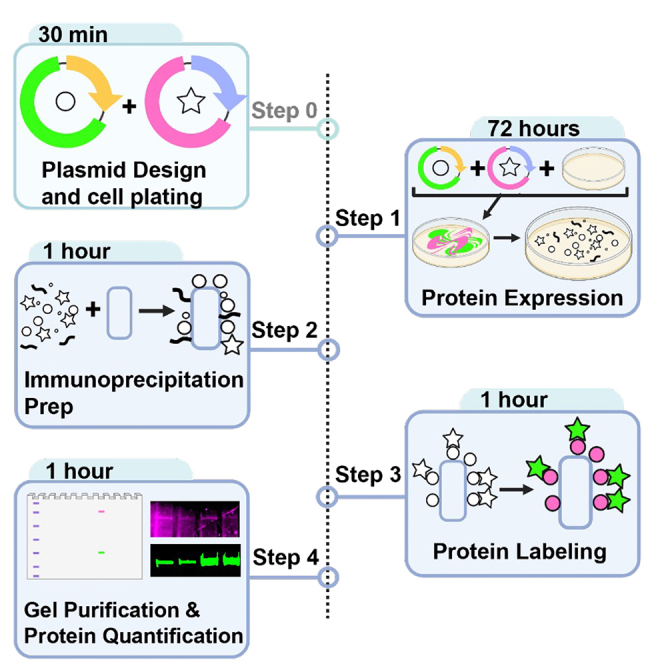
Summary
Here, we present a protocol to quantify interactions among difficult-to-express proteins from Drosophila cells using the select western blot-free tagged-protein interaction (SWFTI) assay. We describe steps for plasmid design, cell plating, protein expression, and immunoprecipitation preparation. We then detail procedures for protein labeling, gel purification, and protein quantification. This protocol offers a fluorescence-based technique for rapid quantification of ectopically expressed proteins that are fused to SNAP and CLIP tags without the need for membrane transfer.
For complete details on the use and execution of this protocol, please refer to Lin et al.1
Subject areas: Molecular Biology, Molecular/Chemical Probes, Protein Biochemistry, Protein expression and purification
Graphical abstract
Highlights
-
•
Steps for transiently transfecting insect cells with two plasmids
-
•
Pull-down procedure for capturing SNAP/CLIP-tagged interacting proteins
-
•
How to resolve and image SNAP/CLIP-tagged proteins
-
•
Protocol for quantifying interactions by use of a doubly labeled reference protein
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Here, we present a protocol to quantify interactions among difficult-to-express proteins from Drosophila cells using the select western blot-free tagged-protein interaction (SWFTI) assay. We describe steps for plasmid design, cell plating, protein expression, and immunoprecipitation preparation. We then detail procedures for protein labeling, gel purification, and protein quantification. This protocol offers a fluorescence-based technique for rapid quantification of ectopically expressed proteins that are fused to SNAP and CLIP tags without the need for membrane transfer.
Before you begin
The protocol below describes the steps taken to characterize the interaction between the Drosophila melanogaster circadian clock proteins Cryptochrome (CRY) and Timeless (TIM) in S2 cells. This protocol may be used for any protein of interest (POI) and cell type, provided the targets can be recombinantly expressed as the SNAP/CLIP fusion proteins in the cells of interest. SNAP and CLIP protein tags (182 residues, 19.4 kDa, respectively) are available from Proteintech and New England Biolabs2 and are based on engineered versions of O6-alkylguanine-DNA-alkyltransferase (hATG) that react with fluorescent dyes to form covalent adducts.3,4 The SNAP-tag protein reacts with O6-benzylguanine (BG) and the CLIP-tag incorporates benzylcytosine with orthogonal reactivity to the SNAP-tag. Of note, users should have access to a sterile environment for cell culturing, an appropriate incubator to support cell growth, and a centrifuge that can operate between 210 and 15,000 × g. It is advised to test the performance of the SNAP/CLIP dyes, such as excitability by the fluorescence microscope or imaging instrument intended for SWFTI assay use before experimentation.
The protocol below describes the specific steps for examining the interaction between circadian clock proteins in S2 cells under comparative conditions of dark and light exposure. However, in principle this protocol is general for any protein type under a variety of comparative conditions.
Plasmid design
Timing: 20 min, designed prior to transfection
This section accomplishes plasmid design for proteins of interest.
-
1.
Design plasmids for two different POI so that one plasmid contains the protein sequence fused to a sequence coding for a SNAP tag and the other contains a sequence coding for a CLIP tag.5
Note: The tags may be placed at either termini of the POI, or even internal to the coding sequence if they do not disrupt folding or function. For protein expression in S2 cells, pAc plasmid using an Ac5 promoter is advisable. Each protein can be in its own plasmid, as the two plasmids will be transfected together. To facilitate immunoprecipitation myc or HA epitope tags, for example, can be appended to each cloned protein.
CRITICAL: When designing plasmids consider the linker type and length between protein tags. It is recommended that users optimize the linker lengths and composition on a protein-specific basis. For guidance on choosing linkers, one can consult Grawe et al.6
For example, for CRY to bind TIM, tags should not be located at the C-terminus of CRY and N-terminus of TIM as those are the locations of the CRY:TIM binding interface. Taking into consideration that the binding event may require changes in protein conformation, a semi-flexible linker was utilized.
Preparation of cells
This section accomplishes cell preparation for protein expression.
Note: The labeling protocol described here has been optimized for adherent S2 cells. The procedure can also be adapted for other adherent cell types. This procedure has not been tested on non-adherent cell types but seeding density and differences in lysis isolation are factors that should be considered if adapting the protocol to non-adherent cells.7
-
2.
Plate cells in a 100 mm tissue culture plate, such that they will be ∼80% confluent in 72 h. For S2 cells, 1.0 × 107 cells are plated in a 100 mm plate containing growth media (medium supplemented with 10% fetal bovine serum; FBS) so that the total volume of cells and growth media is 10 mL in each plate.
Note: Expression plasmids can be introduced into the cells via transfection 24 h after plating the cells.
A plate of healthy S2 cells at ∼80% confluency viewed through an optical microscope at 10x magnification should look similar to Figure 1A . S2 cells that appear linear or shriveled, require extended doubling time, or otherwise look unhealthy may not be appropriate for efficient transfection (Figure 1B).
Figure 1.

Assessment of S2 cell health for transfection
(A) S2 cells at ∼80% confluency appearhomogeneously round.
(B) S2 cells that are overconfluent (80%<), do not appear primarily round, and/or are kept in cloudy media indicating excessive cell death or contamination. The latter may negatively affect transfection efficacy.
Preparation of reagents
This section accomplishes reagent preparation.
-
3.
Filter sterile 1X phosphate buffered saline (PBS) with membrane filter unit, 200 mL, store at 4°C for up to 6 months.
-
4.
Lysis buffer, 200 mL, store at 4°C for up to 6 months before addition of protease inhibitor. Once protease inhibitor has been added, store at 4°C for up to 24 h.
Note: Protease inhibitor should be added fresh to the lysis buffer. The concentration of protease inhibitor may need to be adapted for different cell types or protein of interest to be isolated. For example, to make 3 mL of complete lysis buffer, combine 2.7 mL of lysis buffer and 300 μL of 10X protease inhibitor.
-
5.
Prepare Immunoprecipitation buffer, 200 mL, store at 4°C for up to 6 months. Prepare TBST buffer, 2 L, also store at 4°C.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Dithiothreitol (DTT) | Oakwood Chemical | 3483123 |
| Fetal bovine serum | Sigma | F4135 |
| SNAP-Cell 647-SiR | New England Biolabs | S9102S |
| CLIP-Cell 505 | New England Biolabs | S9217S |
| MG-132 proteosome inhibitor | Cayman Chemical | 10012628 |
| Magnetic Myc resin | Thermo Fisher Scientific | 88842 |
| Magnetic HA resin | Thermo Fisher Scientific | 88836 |
| Mini-PROTEAN TGX Stain-Free gels (4%–20%, 12-well comb, 20 μL) | Bio-Rad | 4568095 |
| TransIT-insect transfection reagent | Mirus Bio | 10766-896 |
| Experimental models: Cell lines | ||
| Drosophila S2 cells | ATTC | CRL1963 |
| Other | ||
| 0.45 μM PVDF membrane filter | Milipore Sigma | S2HVU05RE |
| 16-tube SureBeads magnetic rack | Bio-Rad | 1614916 |
Materials and equipment
Lysis Buffer
| Reagent | Final concentration | Amount from stock | Reagent stock |
|---|---|---|---|
| Tris pH 8 | 50 mM | 15 mL | 1 M |
| NaCl | 150 mM | 45 mL | 3 M |
| IGEPAL CA-630 | 1% | 3 mL | 100% |
| Glycerol | 10% | 30 mL | 100% |
| MG-132 Protease Inhibitor | N/A | N/A | 100% |
| ddH2O | N/A | 207 mL | 100% |
| Total | N/A | 300 mL | N/A |
Tris-buffered saline and tween 20 (TBST) Reagent
| Reagent | Final concentration | Amount from stock | Reagent stock |
|---|---|---|---|
| Tris pH 7.6 | 20 mM | 40 mL | 1 M |
| NaCl | 150 mM | 300 mL | 3 M |
| tween 20 | 0.01% | 1 mL | 100% |
| ddH2O | N/A | 1659 mL | 100% |
| Total | N/A | 2000 mL | N/A |
Immunoprecipitation Buffer
| Reagent | Final concentration | Amount from stock | Reagent stock |
|---|---|---|---|
| Tris pH 8 | 50 mM | 15 mL | 1 M |
| NaCl | 150 mM | 45 mL | 3 M |
| Glycerol | 10% | 30 mL | 100% |
| ddH2O | N/A | 210 mL | 100% |
| Total | N/A | 300 mL | N/A |
Step-by-step method details
Transiently co-transfect cells with two plasmids: Day 1
This section accomplishes cell transfection for protein production.
-
1.
Follow Mirus Optimization Protocol for Plasmid DNA Delivery.8
Note: TransIT-Insect Transfection Reagent is advised for S2 cells.
-
2.
Incubate cells for 72 h.
Harvest cells: Day 4
This section accomplishes cell harvesting for assay use.
-
3.
To limit proteolysis, add 100x MG-132 (optimal working concentration range 5–50 μM depending on cell type) dropwise to each 100 mm plate.
Note: To compare two conditions (e.g., light vs dark), split cells from the 100 mm plate evenly between two 60 mm plates.
-
4.
Allow 2 h for cells to re-adhere to the 60 mm plates in a 27°C incubator.
-
5.
Subject each of the two 60 mm plates to different experimental conditions being tested (ex. light-dependent interactions, temperature, drug screening, genetic or RNAi screening, etc.), usually for the same amount of time, unless time is a variable being explored.
Note: For example, to test the effect of blue light on CRY:TIM binding, place 60 mm plates in a completely dark temperature-controlled environment, which serves as the control. Place the other 60 mm plate in an exclusively blue light illuminated temperature-controlled environment, which serves as the independent variable. Both plates should be in their respective environments for 1 h.
-
6.
Gently lift cells from the plate using a cell scraper and collect cells into a 5 mL tube for each 60 mm plate.
-
7.
Spin cells at 210 × g for 5 min. Discard supernatant and resuspend cells in 3 mL PBS.
-
8.
Repeat step 7 two times. Then spin down cells again at 210 × g for 5 min, discard supernatant, and store at −80°C for at least 12 h.
Immunoprecipitation and lysate sample preparation: Day 5
This section accomplishes first half of sample preparation for SDS-PAGE gel loading.
-
9.Thaw cell pellets on ice.
-
a.Resuspend the pellet in 400 μL lysis buffer with 1x protease inhibitor.
-
b.Allow the lysate to rest on ice for 30 min.
-
c.While lysate is on ice, put 10 μL magnetic myc resin in two 1.7 mL Eppendorf tubes, and 10 μL magnetic HA resin in two other 1.7 mL Eppendorf tubes.
-
a.
-
10.
Use a magnetic rack, such as 16-Tube SureBeads Magnetic Rack, to separate each resin from the storing solution, and remove all the storing solution, leaving only the resin behind.
-
11.
Using the magnetic rack, wash resin with 500 μL cold TBST, wait until all resin separates from TBST, and remove TBST.
Note: The detergent in TBST prevents resin aggregation.
-
12.
Repeat step 11 two times.
-
13.
Spin down lysate at 15,000 × g for 15 min at 4°C. Move supernatant into a new tube.
-
14.Wash resin once more with cold TBST.
-
a.Remove TBST.
-
b.300 μL supernatant to each tube.
-
c.Reserve remaining supernatant.
-
a.
-
15.If the condition being tested is light/dark, (such as for the CRY:TIM interactions) wrap the Eppendorf tubes containing cells that were left in the dark environment during Day 5 in aluminum foil.
-
a.Rotate all four Eppendorf tubes with intermittent resuspensions overnight at 4°C.Note: If the dark samples are extremely light sensitive, it may be necessary to handle dark samples exclusively under red light to visualize light/dark discrimination in step 26.
-
b.Put all 4 Eppendorf tubes rotating with manual intermittent jolts overnight at 4°C.CRITICAL: The intermittent resuspensions are necessary to prevent resin from settling and compacting in the tube. Flicking the tube is usually sufficient.
-
a.
-
16.
Dilute reserved supernatant so that it is 1 part supernatant, 2 parts IP buffer, and add additional protease inhibitor.
Note: Moving forward the supernatant and IP buffer mixture without resin will be referred to as the lysate samples.
-
17.
Flash freeze lysate samples and store at −80°C overnight.
Note: freezing lysate is not necessary if next step is done immediately following the 6–8 h incubation.
Protein interaction quantification: Day 6
This section accomplishes sample preparation for SDS-Page gel loading.
-
18.
Remove Eppendorf tubes from rotator.
-
19.Use the magnetic rack to separate the resin from the supernatant.
-
a.Remove the supernatant and wash resin with TBST.
-
b.Repeat this step 3 times.
-
a.
-
20.
Remove TBST from resin and elute protein off the resin with 27 μL immunoprecipitation buffer.
-
21.Remove lysate from −80°C and move 27 μL to a separate microcentrifuge tube.
-
a.Add 1 μL of 0.1 mM SNAP-Cell 647-SiR dye and 1 μL of 0.1 mM CLIP-Cell 505 dye to both lysate and immunoprecipitation samples.
-
b.Rotate the lysate and immunoprecipitation samples in the dark for 1 h at room temperature for proper tag labeling.
-
a.
-
22.
Remove all samples from the rotator. Add 1 μL DTT, 0.5 μL β-mercaptoethanol, and 10 μL laemmli sample buffer (Bio Rad) to each sample and boil at 95°C for 10 min.
-
23.
Load 20 μL of lysate and 20 μL immunoprecipitated protein in separate lanes of an SDS-Page gel, such as Mini-PROTEAN TGX Stain-Free Gels (4%–20%, 12-well comb, 20 μL). Load a control to normalize the signals of the samples.
Note: Stain-free gels are advised so that there is no dye interference with the fluorescence signals. Stain-free gels provide for facile evaluation of total protein loaded in each lane without use of dyes.
-
24.
Electrophorese the gel to the degree that the POIs are suitably resolved.
Note: The gel should be run in the dark so as not to photo-bleach the SNAP and CLIP tags.
-
25.
Immediately excite each gel-entrapped fluorophore at different time exposures for later protein quantification using software such as Image J.
Note: Proteins containing the SNAP tag with SNAP-Cell 647-SiR dye can be visualized by excitation at 647 nm; proteins containing the CLIP tag with CLIP-Cell 505 dye can be visualized by excitation at 505 nm. It is advised to use instrumentation suitable for visible light and far red/near infrared fluorescence detection for a stain-free gel, such as a Bio-Rad ChemiDoc.
DO NOT stain the gel for typical protein visualization (e.g., Coomassie stain) prior to fluorophore excitation as that may limit the sensitivity of the assay. Once protein detection by fluorescence is complete it is possible to stain the gel.
For a loading control, total protein can be visualized and quantified by UV light activation of a stain-free gel. But, be sure to analyze by UV after imaging CLIP/SNAP fluorescent dyes.
Note: TBST may be substituted with PBS for resin washing if detergent use is a concern. This substitution may however lead to increased non-specific binding and/or protein aggregation on the resin.
Binding affinity quantification: Day 7
This section accomplishes quantification of interactions between proteins of interest.
-
26.
Fluorescence intensities of both the CLIP tagged protein (ex. CRY or ]) and SNAP tagged protein (ex. TIM or [) are normalized relative to a known amount of a standard pure protein containing both CLIP and SNAP tags (ex. CLIP-CRY-SNAP). Normalization is necessary to deduce relative amounts of each protein bound to the resin.
Note: Apparent binding constants ( are calculated assuming the resin largely captures the equilibrium binding distribution prior to immunoprecipitation and the dissociation time constants are not much faster than seconds.1
The following calculations are based on a 1:1 binding stoichiometry between TIM [B] and CRYΔ ( CRY residues 1–520, lacking the C-terminal tail) [A].
-
27.
Calculate the apparent binding protein constants as follows:
| (Equation 1) |
| (Equation 2) |
| (Equation 3) |
The fraction of B bound to A (F) is derived by calculating the amount of B in the pull-down compared to the amount of A in the pull-down and standardizing the amounts by the fluorescence signal from the known amount of the standard (CLIP-CRY-SNAP), which must be run and imaged on the same gel. To estimate the concentration of free A [A], high expression levels of A are assumed, i.e., [A] >> [AB]; therefore [A] ∼ [A] + [AB] = Atotal , where Atotal is the sum of bound and unbound A in the cell lysate sample. With [A] and F in hand, the apparent KD is calculated from Equation 3 above. An excel spreadsheet calculator is provided to aid analysis (File S1: Apparent KD Value Calculator.xlsx).
Expected outcomes
If the procedure was successful, then exciting the tags with light of the appropriate wavelengths should result in a fluorescent band on the gel at the molecular weight corresponding to the size of the tagged protein(s) as shown in Figure 2. The SWFTI protocol provides a reliably reproducible and efficient method for protein expression analysis that can be applied to a wide range of biological samples and research questions.
Figure 2.

Example of expected outcome for the SWFTI assay
A TIM tagged with an HA tag and SNAP tag was used as the bait to pull down a myc- and CLIP-tagged CRY in a bait-and-prey style immunoprecipitation. Both CRY and TIM were incubated together with HA resin with TIM as the bait. SNAP-Cell 647-SiR dye was utilized for SNAP-tagged TIM detection. The SNAP tag is excited at 647 nm and fluorescence is detected at a gel position corresponding near 120 kD, matching the expected molecular weight for the SNAP-tagged TIM protein being probed (pink). The CLIP-tagged CRY was visualized by exciting the CLIP-Cell 505 dye at 495 nm. Detection at expected excitation wavelength indicates positive protein detection from a stain-free gel. The data indicates that more CRY is bound to TIM in the light (L) compared to in the dark or absence of irradiation (D). In the assay shown, PBS was used to wash the resin instead of the suggested TBST resulting in less TIM visualized in the pull-down relative to the lysate due to protein aggregation on the resin.
The SWFTI protocol was initially designed to detect and quantify the protein expression levels of D. melanogaster proteins Timeless and Cryptochrome, which are known to play key roles in regulating the circadian clock.1,9 The expected outcomes of this analysis are to gain a better understanding of how these proteins are expressed over time, as well as to identify any potential changes in their expression patterns under different experimental conditions. By characterizing the expression profiles of Timeless and Cryptochrome, we may gain insights into their role in the circadian clock and how disruptions in their protein expression may contribute to circadian rhythm disorders.10
Limitations
A possible limitation of this protocol is in investigating the interaction between multiple proteins at one time, where more than one protein containing the same fluorophore tag has similar molecular weights, leading to bleed over of the fluorescence signal; this complication could be compensated by another dimension of separation (e.g., isoelectric point).
Photobleaching of the fluorophore by labeling the proteins in a light environment may cause problems in detection. Low transfection efficiency may limit the ability of the SWFTI assay to quantify protein expression accurately. In addition, binding affinities measured by SWFTI are only estimates as the pull-down is not under equilibrium conditions; however, comparisons to derive relative affinities are valid.
Apparent KDs won’t be possible unless [A] is sufficiently overexpressed such that Atotal ≃ [A].
The tag might interfere with the binding interaction or even the expression, non-specific reactivity of the dyes is generally not a problem but does occur to some extent – (as exemplified by the fluorescence of the molecular weight markers) ; one could imagine a case where a protein reacts especially strongly – but this can be tested in the absence of the tags.
Troubleshooting
Problem 1
Low protein yield
Potential solution
Ensure the DNA plasmid is of high quality and free of contaminants. Cell contamination may also significantly reduce protein expression; if you suspect your expression system is contaminated discard cells and re-express in new cells. Please refer to the corresponding protocol step 3 of “before you begin”.
Problem 2
Variability in results
Potential solution
Ensure the experimental conditions are consistent and that the plasmid DNA is of consistent quality. Additionally, consider use of sufficient replicate samples to reduce the impact of variability and apply appropriate statistical analysis to assess significance. Testing different plasmid ratios may be necessary for optimizing protein expression. Please refer to the corresponding protocol step 1 of “step-by-step method details”.
Problem 3
Proteolysis and degradation
Potential solution
Increasing proteolytic inhibitor concentration may be necessary to reduce protein degradation. Check that the protease inhibitor is preventing proteolysis in a negative control experiment where it is removed. Please refer to the corresponding protocol step 3 of “step-by-step method details”.
Problem 4
No signal
If a protein band does not fluoresce on the gel at the expected excitation for emission, it is possible that the protein tag has become photobleached from light exposure during a previous step. Alternatively, it is also possible that the protein was degraded, resulting in the SNAP or CLIP tag running with the gel front.
Potential solution
After an unsuccessful imaging attempt it is advised to stain the gel with Silver Stain or Coomassie to check if the protein is present at the expected molecular weight. For label-free imaging, repeat the SWFTI assay being conscientious to limit any ultraviolet light prior to fluorescent detection. Please refer to the corresponding protocol step 25 of “step-by-step method details”.
Problem 5
Band shift
If a band fluoresces at an unexpected molecular weight, this could indicate protein degradation. Alternatively, if the protein construct was designed to have multiple tags, it is also possible that one of the other tags was cleaved in a previous step resulting in a protein band shift.
Potential solution
Check that the protease inhibitor is functional on a fusion-protein control. One can also transfer the protein in the gel onto a PVDF membrane and probe for epitope tags included in the protein construct and the proteins themselves by more conventional methods. Please refer to the corresponding protocol step 24 of “step-by-step method details".
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Brian Crane (bc69@cornell.edu).
Technical contact
Further information and technical questions should be directed to and will be fulfilled by the technical contact, Dr. Changfan Lin (cl2275@cornell.edu).
Materials availability
This study did not generate new unique materials or reagents.
Data and code availability
This study did not generate new databases. Attached is an Excel calculator, where users can input band intensities, and calculate KD values.
Acknowledgments
The study was supported by the NIH grant R35GM122535.
Author contributions
C.D. and C.L. performed the experiments. C.D., C.L., and B.R.C. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2024.103171.
Contributor Information
Changfan Lin, Email: cl2275@cornell.edu.
Brian R. Crane, Email: bc69@cornell.edu.
Supplemental information
References
- 1.Lin C., Schneps C.M., Chandrasekaran S., Ganguly A., Crane B.R. Mechanistic insight into light-dependent recognition of Timeless by Drosophila Cryptochrome. Structure. 2022;30:851–861.e5. doi: 10.1016/j.str.2022.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kai J. SNAP-tag® Technologies: Novel Tools to Study Protein Function. NEB Expressions 2008;3. New England Biolabs. https://www.neb.com/en-us/tools-and-resources/feature-articles/snap-tag-technologies-novel-tools-to-study-protein-function.
- 3.Erdmann R.S., Baguley S.W., Richens J.H., Wissner R.F., Xi Z., Allgeyer E.S., Zhong S., Thompson A.D., Lowe N., Butler R., et al. Labeling Strategies Matter for Super-Resolution Microscopy: A Comparison between HaloTags and SNAP-tags. Cell Chem. Biol. 2019;26:584–592.e6. doi: 10.1016/j.chembiol.2019.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keppler A., Gendreizig S., Gronemeyer T., Pick H., Vogel H., Johnsson K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol. 2003;21:86–89. doi: 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- 5.Gautier A., Juillerat A., Heinis C., Corrêa I.R., Jr., Kindermann M., Beaufils F., Johnsson K. An engineered protein tag for multiprotein labeling in living cells. Chem. Biol. 2008;15:128–136. doi: 10.1016/j.chembiol.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 6.Gräwe A., Stein V. Linker Engineering in the Context of Synthetic Protein Switches and Sensors. Trends Biotechnol. 2021;39:731–744. doi: 10.1016/j.tibtech.2020.11.007. [DOI] [PubMed] [Google Scholar]
- 7.Gibco . Thermo Fisher Scientific; 2020. Cell Culture Basics Handbook; pp. 1–132. [Google Scholar]
- 8.Mirus . Mirus Bio LLC; 2014. TransIT®-Insect Transfection Reagent Protocol; pp. 1–6. [Google Scholar]
- 9.Lin C., Feng S., DeOliveira C.C., Crane B.R. Cryptochrome-Timeless structure reveals circadian clock timing mechanisms. Nature. 2023;617:194–199. doi: 10.1038/s41586-023-06009-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Allada R., Chung B.Y. Circadian organization of behavior and physiology in Drosophila. Annu. Rev. Physiol. 2010;72:605–624. doi: 10.1146/annurev-physiol-021909-135815. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate new databases. Attached is an Excel calculator, where users can input band intensities, and calculate KD values.