

Abstract

The air–water interface (AWI) is a ubiquitous reaction field different from the bulk phase where unexpected reactions and physical processes often occur. The AWI is a region where air contacts cloud droplets, aerosol particles, the ocean surface, and biological surfaces such as fluids that line human epithelia. In Earth’s atmosphere, short-lived intermediates are expected to be generated at the AWI during multiphase reactions. Recent experimental developments have enabled the direct detection of atmospherically relevant, short-lived intermediates at the AWI. For example, spray ionization mass spectrometric analysis of water microjets exposed to a gaseous mixture of ozone and water vapor combined with a 266 nm laser flash photolysis system (LFP-SIMS) has been used to directly probe organic peroxyl radicals (RO2·) produced by interfacial hydroxyl radicals (OH·) + organic compound reactions. OH· emitted immediately after the laser flash photolysis of carboxylic acid at the gas–liquid interface have been directly detected by time-resolved, laser-induced florescence techniques that can be used to study atmospheric multiphase photoreactions. In this Featured Article, we show some recent experimental advances in the detection of atmospherically important intermediates at the AWI and the associated reaction mechanisms. We also discuss current challenges and future prospects for atmospheric multiphase chemistry.
1. Introduction
Earth’s atmosphere is composed of gas, liquid, and solid phases and their interfaces.1 The interfaces are sometimes regarded as a fourth phase that can be distinguished from the other phases by its inhomogeneous nature. The air–water interface (AWI), arguably the most common interface, is a widespread reaction field in the atmosphere where unique reactions and physical processes often occur.2 The AWI includes regions where air is in contact with cloud droplets, aerosol particles, the surface of the ocean, biological surfaces such as the wet film on a plant, human skin, and epithelial-lining fluids (ELFs). The following are among the key distinguishing features of AWIs. They are associated with a sharp decrease of water density from 1 g cm–3 to zero within just a few angstrom, and the dielectric constant, hydrogen-bonding network, ion distribution, and the associated properties dramatically change as a function of depth.2 The fact that a “pure” AWI is negatively charged has been known for a century.3−6 Although the cause of the negative charge is still in debate,7−9 it is generally accepted that the accumulation of OH– at the AWI if the bulk pH exceeds ∼4 leads to the negative charge.10,11 Experiments have shown that adding salt to water changes the distribution of both OH– and H+ at the AWI.10 It has been demonstrated that (H3O+)(H2O)≤4 is available to the topmost layers of the AWI if the bulk pH is less than ∼4,10,12 and this species acts as a superacid.13 Because some ions at the AWI are less hydrated or distortedly hydrated,13,14 there is a huge acceleration of reaction rates compared with the bulk liquid phase, e.g., Fe2+ + H2O2.15 Because the average hydration number for a water molecule at the AWI is 2.7 (cf., 3.9 in the bulk solution), some chemical species are preferentially adsorbed therein.16,17
In the atmosphere, a variety of intermediates are expected to be generated at the AWI during multiphase reactions. Multiphase processes can alter the composition of gas-phase species and contribute to the aging of atmospheric condensed phases, thereby change a variety of properties (hydrophilicity, size, phase, etc.) that impact the radiation balance of Earth. The aging process that occurs at the AWI can also change the oxidation–reduction potential and toxicity of aerosol particles. The uptake of gaseous hydroxyl radicals (OH·), an atmospheric detergent, on the AWI of atmospheric condensed phases such as cloud droplets/aerosols would cause a rapid oxidation of surface-active organic compounds therein. An OH· can abstract a hydrogen (H) atom from an alkyl group or add to a carbon–carbon double bond (C=C). Both reaction pathways yield peroxyl radical (RO2·) intermediates under the high-O2 conditions in Earth’s atmosphere.18 The RO2· undergo further reactions that lead to the production of closed-shell products, some of which are emitted to the gas phase or diffuse into the bulk phase. It is worth mentioning that the self-reaction and cross-reaction of RO2· (i.e., RO2· + RO2·, RO2· + R′O2·) are generally much faster in the aqueous phase and at the AWI than in the gas phase because of the stabilization of ROOOOR(R′)* by solvent.19,20 The rate constants are often on the order of 109 M–1 s–1 in the aqueous phase.19 The OH· can also react with reactive thiol/thiolate groups in bioaerosol particles, which are mixtures of diverse molecules originating from living systems, to form short-lived, S-containing intermediates.21 Some biological molecules such as glutathione (GSH) can trap OH· and “steer” it to react with the thiol/thiolate group without causing H-abstraction from other moieties (e.g., alkyl groups).22 Such a “radical recognition process” could occur at the AWI of bioaerosols and ELFs.
Recent experimental, theoretical, and modeling studies have greatly advanced our knowledge of atmospheric intermediates at the AWI and the related chemistry.2,23,24 In this Featured Article, we report experimental results derived from a spray ionization mass spectrometric study of liquid microjets exposed to reactive gases combined with a 266 nm laser flash photolysis system (LFP-SIMS).20 We were able to use LFP-SIMS to directly probe atmospherically relevant, short-lived intermediates, including radicals. We focused on the detection of atmospherically important radicals (RO2·, OH·, and Criegee intermediates) and closed-shell sulfenic acid at the AWI. Although other intermediates, including oxo-ferryl [FeIV=O],15 I2–/IBr– radical anions,25 and carbocations,26 were also detected at the AWI by mass spectrometry, we do not discuss those intermediates here. We also report a recently developed technique that combines LFP and laser-induced fluorescence (LIF) for analysis of multiphase photoreactions (m-LFP-LIF).27 This method enabled us to successfully detect the OH· emitted to the gas phase during the photolysis of a liquid, nonanoic acid at the gas–liquid interface. Both new methods hold promise for elucidating the detailed mechanisms of atmospheric multiphase reactions.
2. Experimental Section
2.1. Laser Flash Photolysis + Spray Ionization Mass Spectrometry (LFP-SIMS)
The experiments involved the injection of water microjets containing 10–5 to 10–1 M organic reactant into the spraying chamber of a mass spectrometer at atmospheric pressure and ambient temperature (Figure 1A). Water microjets, a mixture of cylindrical jets and microdroplets, were exposed to nearly orthogonal gas-phase OH· beams generated by 266 nm photolysis of a gaseous mixture of O3/O2/H2O/N2 for the study of oxidations of organic molecules initiated by OH· at the AWI.20 The species produced on the surface of the microjets were analyzed in situ via an online quadrupole mass spectrometer (Agilent 6130 Quadrupole LC/MS Electrospray System, mass range m/z ≥ 50). Using this technique (laser flash photolysis + spray ionization mass spectrometry, LFP-SIMS), we monitored the prompt (within the ∼10-μs lifetime of the intact microjets) formation of anionic products at the AWI of the microjets from the reaction of gaseous OH· with the aqueous reactant.20 Aqueous solutions containing organic reactant were pumped (100 μL min–1) into the spraying chamber of the mass spectrometer through a grounded stainless steel needle (100-μm bore) coaxial with a sheath (internal diameter n = 200 μm, external diameter = 300 μm) that issued nebulizer N2(g) at a high gas velocity (vg ∼ 160 m s–1).28 A key feature of the instrument was that the microjets that issued from the nozzle source were orthogonal to the polarized inlet to the mass spectrometer. In such a configuration, a large part of the core components of the microdroplets went to sink, while surface-active ions that were selectively emitted from submicron droplets that originated from the AWI of the initial microdroplets to the gas phase by rapid evaporation of the water solvent were collected by applying an electric bias to the inlet of mass spectrometer (see below). Reaction intermediates/products were detected without any manipulation by mass spectrometry in less than 1 ms.
Figure 1.

A) Schematic diagrams of LFP-SIMS and B) the bag-breakup mechanism of droplet. This experimental method allows for the in situ monitoring of reactions of OH· with a sample reactant on the surface of aqueous microdroplets. Adapted with permission from refs (20) and (29). Copyright 2014/2016 American Chemical Society.
The surface specificity of the present method has been demonstrated in several experiments.10,12,15,28,30,31 For example, the mass spectral signal intensities obtained from equimolar mixtures of solutions of carboxylic acids Rn-COOH (n = 1–7) have been found to increase as a function of n (i.e., hydrophobicity).29 Because all Rn-COOH (n = 1–7) have similar pKa values of 4.8 ± 0.1, the observed tendency is attributable to the relative ion populations at the AWI. In addition, the mass spectral signal intensity of iodide I– detected at m/z = 127 has been found to be 3.04 ± 0.24 times that of bromide Br– at m/z = 79 and 81 from equimolar aqueous solutions of NaI and NaBr.31 The mass spectral signal intensity has been found to follow the order ClO4– > I– > Br– ∼ NO3–,31 in excellent agreement with the order of ion affinity at the AWI determined by other interface-sensitive techniques such as sum frequency generation spectroscopy (SFG), X-ray photoelectron spectroscopy (XPS), and theoretical calculations.32−34 Notably, the relative signal intensity of I– among that of total anions from an equimolar mixture of NaI + NaBr (i.e., S(I–)/ [S(I–) + S(Br–)]) increased as a function of the nebulizer N2 gas velocity (vg) and, as expected, extrapolated to 1 as vg → ∞ because only I– was detected at the topmost layers of the AWI.28 In contrast, the fact that it extrapolated to 0.5 as vg → 0 indicated that the bulk solution contained equimolar amounts of I– and Br–.28 This result is explained by the well-known bag-breakup mechanism associated with a liquid film whose thickness and composition are controlled aerodynamically by gas flow (Figure 1B).6,28−30 In our experimental configuration, after a short (≤10 μs) period of exposure to OH·(g), the microdroplets (>10 μm) were transformed into a film and rims, in which surface-active and nonsurface-active species accumulated, respectively. The film decomposed into submicron droplets, whereas the rims remained as large droplets. Ions were emitted from the submicron droplets to the gas phase by evaporation of water and were detected by a quadrupole mass spectrometer. The mass spectra therefore reflected the compositions of the ions present at the AWI of the initial microjets.28−30,35
The 266 nm pulse beam shot by the Nd3+:YAG laser setup was used to generate OH·(g) in situ (Figure 1).20,36−38 The photodissociation of O3(g) by 266 nm photons produced O(1D), followed by the reaction with H2O(g) within ∼6 ns (rate constant k = 2.2 × 10–10 cm3 molecule–1 s–1), in competition with its deactivation by N2(g)/O2(g) into O(3P). [OH·(g)]0 values were evaluated from the number of photons per unit area, [O3(g)], and gas-phase kinetics.20 [OH·] at the surface of microjets would be lower than the [OH(g)]0 estimated at the laser photolysis spot because of partial losses via gas-phase OH· recombination into nonreactive H2O2. Thus, [OH·(g)]0 values were regarded as the upper limits to [OH·] on the surface of microjets. Kinetic analysis showed that the reaction of gaseous O(1D) with reactant (aq) was too slow to compete with the gas-phase reaction with water vapor under the experimental conditions.20 Also, O(3P) was converted mainly into O3 by reaction with excess O2(g). We confirmed that depletion of the reactant signal and formation of the product signal required both the participation of 266 nm photons and O3(g). Therefore, no photolysis occurred on/in water microjets of dissolved reactants. Because the microjets broke up within 10–5 s after being ejected from the nozzle, whereas the laser pulsed every 10–1 s (i.e., 10 Hz), we assumed that the reactions always took place at the AWI of fresh solutions.
2.2. Laser Flash Photolysis + Laser-Induced Fluorescence for Multiphase Photoreactions (m-LFP+LIF)
We developed a new experimental method to directly detect gaseous OH· emitted from the gas–liquid interface. The ultraviolet LFP of liquid nonanoic acid (NA) at the gas–liquid interface was combined with the detection of OH·(g) by LIF to investigate multiphase photoreactions (m-LFP-LIF).27Figure 2 shows a schematic diagram of the m-LFP-LIF.
Figure 2.

Schematics of 213 nm photolysis of liquid- and gas-phase organic molecules and OH· detection by LIF. (a) Liquid phase photolysis: the photolysis laser light was incident on the sample at 45° to the normal from the surface. (b) Gas-phase photolysis: two lasers were crossed in a gas-filled chamber. In both experiments, nitrogen buffer gas was introduced into the chamber up to about 80 Pa. Adapted with permission from ref (27). Copyright 2022 American Chemical Society.
The m-LFP-LIF experiments were carried out in a
stainless-steel
chamber. A silicon (111) substrate was deposited on a nickel-plated
copper stage in the chamber. The temperature of the stage was controllable
by a thermostatic water bath (Thomas, TRL-11LP) and monitored with
a type-K thermocouple. About 2 mL of neat liquid NA (purity: >98.0%,
Tokyo Chemical Industry) was placed on the substrate, and then the
chamber was evacuated to a background pressure of 1.1 Pa by two dry
pumps (Kashiyama, NeoDry15E). The liquid NA had a low vapor pressure
of about 0.2 Pa at room temperature, and it could be introduced directly
into the medium–vacuum chamber without boiling. The liquid
NA was kept at a temperature of ∼295 K to keep its vapor pressure
constant during the experiments. The fifth harmonic (213 nm) generated
by a pulsed Nd:YAG laser (Lotis TII, LS-2137U–N and VM-TIM,
YHG-5) was used for photolysis. This laser beam was incident on the
liquid NA sample at an angle of 45° to the normal from its surface.
The pulse energy and diameter of the beam—500 μJ and
4 mm, respectively—allowed only single-photon absorption. The
typical thickness of liquid NA placed on the substrate was about 810
± 70 μm, thick enough to ignore possible photoreactions
at the interface between the liquid and the substrate. The OH·
desorbed from the liquid NA were detected by 315 nm LIF via the A2Σ (v = 1) → X2Π
(v = 1) transition. The excited A2Σ
(v = 1) state was produced by pulsed 282 nm radiation
from a frequency-doubled dye laser (Lambda Physik, SCANmate 1 with
BBO crystal) pumped by the second harmonic (532 nm) generated by a
Nd:YAG laser (VM-TIM, VM-45TF5). This probe laser beam was injected
into the chamber after a specified delay from the photolysis laser.
The pulse energy and diameter of the beam were approximately 50 μJ
and 2 mm, respectively. We used z to represent the
distance between the liquid surface and the center of the probe laser,
and the shortest distance between the centers of the two lasers (d) could be written as 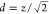 . The OH· LIF was
transmitted outside
the chamber by a liquid light guide (Lumatec, Series 300) and then
detected with a photomultiplier tube (PMT, Hamamatsu, R1104). The
distance between the guide and the center of the probe laser was fixed
at 5 mm to keep constant the solid angle for detection of the OH·
LIF. Time-resolved OH· LIF intensities were recorded by a digital
oscilloscope (Iwatsu, DS-5624A). The measurements were typically performed
at a repetition rate of 10 Hz using a digital delay-pulse generator
(Quantum Composers, 9428). See ref27 for
more experimental information.
. The OH· LIF was
transmitted outside
the chamber by a liquid light guide (Lumatec, Series 300) and then
detected with a photomultiplier tube (PMT, Hamamatsu, R1104). The
distance between the guide and the center of the probe laser was fixed
at 5 mm to keep constant the solid angle for detection of the OH·
LIF. Time-resolved OH· LIF intensities were recorded by a digital
oscilloscope (Iwatsu, DS-5624A). The measurements were typically performed
at a repetition rate of 10 Hz using a digital delay-pulse generator
(Quantum Composers, 9428). See ref27 for
more experimental information.
3. Results and Discussion
3.1. Peroxyl Radical (RO2·)
RO2 can be formed by H-abstraction from an alkyl group in the presence of O2. In typical atmospheric environments, H-abstraction is predominantly caused by OH·, and partly by NO3· (nighttime) and Cl· atoms (marine environment). It is noteworthy that in the atmosphere the concentration of O2 is always quite large, i.e., ∼5.2 × 1018 molecules cm–3 in the air and ∼0.26 mM in water at 1 atm, 298 K. It is therefore unlikely that alkyl radicals (R·) would not react with O2 in most media. The reaction R· + O2 usually yields RO2· under ambient conditions. Theoretical calculations predict that the concentration of O2 at the AWI would be larger than the value in bulk water estimated from the Henry’s law constant.39
To the best of our knowledge, the first report of the direct detection of RO2· at the AWI was published in 2014.20 Water microjets containing 0.1 mM acetic/hexanoic/octanoic acid were exposed to gas-phase OH· beams generated by 266 nm photolysis of a gaseous mixture of O3/O2/H2O/N2.20Figure 3A and B show the mass spectra of n-octanoic acid microjets exposed to O3(g)/O2(g)/H2O(g)/N2(g) mixtures with the 266 nm laser on and off. In the presence O3(g) but without a laser, the failure of any new product signals to appear (blue traces in Figure 3B) confirmed that the observed intermediates/products clearly came from reactions that involved radicals. In Figure 3B, the m/z = 174 = 143 (octanoate anion, OA) – 1 (H atom) + 32 (O2) corresponds to octanoic peroxyl radicals, RO2·. It appeared that OH· initially abstracted an H atom of OA, followed by the addition of O2 to the alkyl radical. Note that although we could not distinguish which H atom was abstracted in the case of the OH· reaction with OA, interface-specific H-abstraction occurred for long-chain dicarboxylic acids (see below).37
Figure 3.

Negative ion mass spectrum of aqueous 0.1 mM octanoic acid microjets with O3(g) ([O3(g)] = 1.6 × 1016 molecules cm–3) obtained by LFP-SIMS in the absence and presence of OH·(g) (A). B is an enlargement of the mass spectrum of A. C is the mass spectrum obtained from 0.1 mM octanoic acid in 99.9% D2O microjets. Blue: with O3/H2O(g), laser off; Red: with O3/H2O(g), 266 nm laser on (∼8 ns, 40 mJ, 10 Hz). Note that the mass spectral signal at m/z 174 for RO2 did not shift by replacement of H2O by D2O. Adapted with permission from ref (20). Copyright 2014 American Chemical Society.
The product signals originating from the reactions associated with RO2· intermediates were observed at m/z = 157 = 174 (RO2·) – 16 (O) – 1 (H) for carbonyls (RC=O), m/z = 159 = 174 (RO2·) – 16 (O) + 1(H) for alcohols (ROH), and m/z = 175 = 143 (OA) – 1 (H) + 32 (O2) + 1 (H) for hydroperoxides (ROOH). The RC=O and ROH products came from the well-known self-reaction RO2· + RO2· via the Bennett–Summers and the Russell mechanism, respectively.19 The ROOH was expected to come from the reaction of RO2· with HO2· at the AWI.40
Figure 3C shows the mass spectra of n-octanoic acid in 99.9% D2O microjets exposed to O3(g)/O2(g)/H2O(g)/N2(g) mixtures with the 266 nm laser on and off. It has been shown that in experiments with D2O as a solvent, exchanges of hydrogen atoms causes −OH and −OOH groups to become −OD and −OOD, respectively, and the product signals shift by +1 Da. The experiment thus provided evidence that the nonshifted m/z 174 signal was attributable to RO2· that lacked exchangeable H-D atoms in its structure, in contrast with other products that possessed −OH and −OOH groups. The carbonyl products were also not shifted by replacement of H2O by the D2O solvent because these compounds do not possess any exchangeable H(D)-atoms. Additional experiments in H216O:H218O (v:v = 1:1) solvents revealed that these products did not incorporate O atoms from water. This conclusion was consistent with the proposed mechanism of formation of RO2· and associated closed-shell products.
Figure 4 shows the decay of OA and the increases of the RC=O, ROH, and RO2· signals as a function of 266 nm laser energy per pulse. The laser energies in the figure can be converted into [OH·], e.g., 1, 5, 10, 20, 30, and 40 mJ pulse–1 correspond to [OH·(g)] ≈ 4.9 × 1013, 2.2 × 1014, 4.4 × 1014, 8.1 × 1014, 1.1 × 1015, and 1.4 × 1015 radicals cm–3, respectively (see Experimental Section). These values were the upper limits of actual [OH·]0 collisions in the microjets. We note that reactant conversions during the 10-μs reaction times of our experiments were comparable to those in which aerosol droplets exposed to typical tropospheric [OH·] = 106 radicals cm–3 concentrations for 8–233 min. It is apparent that reactant losses and concomitant formation of products display biexponential rather than single-exponential behavior; sharp initial effects are followed by attenuated responses. This evidence suggests that OH· reacts rapidly with alkyl chains and therefore depletes them in the outermost interfacial layer of the AWI, but above a certain threshold dose, the self-reaction of OH· starts to dominate and leads to formation of H2O2. The milder biexponential behavior observed in larger [OA] (Figure 4C) is consistent with a mechanism where OH· begins to preferentially react with alkyl chains rather than with OH· itself. A replenishment of OA from the bulk to the surface by diffusion may also contribute to the observed behavior. By adapting the reported adsorption isotherms of OA at the AWI,41 we estimated that 0.05 mM and 0.5 mM OA covered ∼3% and ∼70% of the water surface, respectively. Because the RO2· signals reached a plateau, the production and consumption of closed-shell products such as RC=O and ROH became balanced at a certain OH· exposure.
Figure 4.

Mass spectral signal intensities from aqueous 0.1 mM (A, B) or 1.0 mM (C, D) octanoic acid microjets exposed to O3(g)/O2(g)/H2O(g)/N2(g) mixtures, [O3(g)] ∼ 1.5 × 1016 molecules cm–3, irradiated with 266 nm laser beams as functions of laser energy (in mJ pulse–1). Adapted with permission from ref (20). Copyright 2014 American Chemical Society.
Notably, the same product signals at m/z = M (reactant anion) – 1 (H) + 32 (O2) were observed in the experiments that involved OH·(g) exposure to microjets with acetic acid, hexanoic acid, C4–C8 dicarboxylic acids, cis-pinonic acid, and benzoic acid.20,37,42,43 The fact that none of these product signals were shifted by replacement of H2O by D2O as the solvent supported the assignment for RO2·. In remarkable contrast, no RO2 signals were observed for the reaction of OH·(g) with aqueous S atom-containing compounds: dimethyl sulfoxide (aq), glutathione (aq), glutathione disulfide (aq), and a model peptide of surfactant protein B (aq).36,38,44 The implication is that S atom(s) would react selectively with OH· and thereby bypass H-abstraction from alkyl groups (see sulfenic acid section).
The RO2· signal was observed not only from OH-oxidation of alkyl groups but also from OH-oxidation of benzene rings. Figure 5 shows the mass spectra of benzoic acid in 99.9% D2O microjets exposed to O3(g)/O2(g)/H2O(g)/N2(g) mixtures with the 266 nm laser on (red) and off (gray). The signal of RO2·, formed by H-abstraction from the benzene ring followed by O2 addition, appeared at m/z = 152 = 121 (benzoate anion, BA) – 1 (H) + 32 (O2) and did not shift in H2O vs D2O as the solvent, in contrast with other signals of closed-shell products possessing −OH/–OOH (see inset of Figure 5).43 The product signals observed at m/z 137 and 171 when H2O was the solvent were shifted to m/z 138 and 173 when D2O was the solvent. Those signals were attributed to hydroxyl-benzoates (BA–OH) and hydroxyl-hydroperoxyl-benzoate (BA(−OH)(−OOH)), respectively. We also performed experiments using C6D5COOH in H2O vs D2O. The results clearly showed that a single H(D)-atom was abstracted from the benzene ring followed by O2 addition to form RO2·. Because H-abstraction from a benzene ring by OH· is a very minor process in the gas-phase or the aqueous phase under ambient temperatures,45,46 we inferred that this reaction proceeded via an interface-specific mechanism fueled by partially hydrated (OH·)n at the AWI. Theoretical calculations suggested that the transition states of the reaction of OH· with benzene was sensitively modulated by the presence of (H2O)n.47 The implication was that the rate constant and reaction pathways for the OH· + benzoic acid reaction differed between the AWI and the gas-phase or bulk reaction.
Figure 5.

Negative ion mass spectra of 0.5 mM benzoic acid in D2O microjets in the presence of 6.9 × 1015 molecules cm–3 O3(g) in O2(g)/H2O(g)/N2(g) mixtures with 266 nm laser pulses (40 mJ, ∼ 8 ns, 10 Hz) off (gray) or on (red). Reproduced from ref (43). Available under a CC-BY-NC 3.0 license. Copyright 2016 Royal Society of Chemistry.
Intriguingly, a non-negligible mass spectrometric signal for a putative benzoic hydrotrioxide (ROOOH) that was observed at m/z 169 in H2O was shifted to 170 when the solvent was D2O.43 The species might be formed by the reaction between RO2· (m/z 152) and OH· (17 Da) at the AWI. Results obtained from experiments using C6D5COOH in H2O vs D2O supported the assignment for ROOOH. Kinetic studies of the gas-phase reactions of RO2· with OH· have shown an inverse relationship between temperature and the rate constant that implies the existence of ROOOH.48 In organic chemistry, ROOOH formed during low-temperature ozonolysis of saturated organic compounds in organic solvents has been used as a source of singlet oxygen (1O2).49 Gaseous ROOOH formed in the OH-oxidation of organic compounds (trimethyl amine, isoprene, etc.) has recently been detected as iodide adducts by mass spectrometry.50 We suggest that ROOOH can be formed not only in the gas phase or bulk organic phase but also at the AWI. The surface accumulation of RO2· and OH· as well as the presence of water molecules as a quencher of excited trioxide (ROOOH*) may enhance the production of ROOOH at the AWI.
3.2. Hydroxyl Radical (OH·)
Arguably, OH· is the most important radical in the atmosphere. OH· plays central roles in the HOx cycle, the degradation of volatile organic compounds (VOCs) in the gas phase, and the photochemical aging of atmospheric condensed phases. OH· is formed predominantly by the reaction of O(1D) with water vapor and the photolysis of HONO in most atmospheric environments.51 OH· is also one of the most reactive oxygen species (ROS) in vivo, where it initiates the oxidation/degradation of biomolecules. Despite its exceptional importance, however, OH· has not yet been directly detected at the AWI. Theoretical calculations predict that OH· would preferentially accumulate at the AWI.52 Experimental studies have demonstrated that heterogeneous reactions of gaseous OH· with aqueous species proceed via the Langmuir–Hinshelwood (LH) mechanism wherein initial adsorption of the radical at the AWI is followed by a reaction with a surface-bound species. This mechanism supports the affinity of OH· at the AWI.20,53
An LFP-SIMS study of the reaction of OH·(g) with dicarboxylic acid(aq), HOOC-Rn-COOH (n = 2–6), has supported the LH mechanism.37 It has been found that during the exposure of OH·(g) to water microjets containing sub-mM HOOC-Rn-COOH, the reaction products always include HOOC-Rn-1-COOH, which are formed from the β-scission of the alkoxyl radicals HOOC-Rn(O·)-COOH by splitting HOCO· radicals. Instead, the astonishing result is the absence of any production of HOOC-R≤n-2-COOH. For example, the heterogeneous OH·(g)-oxidation of HOOC-R6-COOH(aq) (suberic acid) produced a signal of HOOC-R5-COO– but not of HOOC-R4-COO– or other shorter-chain products. This result is in sharp contrast with the results obtained from the study of homogeneous OH-oxidations of HOOC-Rn-COOH in the bulk water phase, which have shown the production of multiple shorter-chain HOOC-R≤n-1-COOH.54−56 Clearly, the initial attack of OH· exclusively occurred on the H atom of α-CH2 groups of undissociated terminal HOOC moieties in the topmost layers of the AWI (Figure 6). This scenario is consistent with the interface-specific orientation of HOOC-Rn-COOH at the AWI revealed by vibrational sum frequency spectroscopy.57 That study has shown that dicarboxylic acids (HOOC-Rn-COOH, n = 3, 4) that reside at the AWI act like a bridge by protruding their backbone alkyl group parallel to the interface and pointing their hydrophilic COOH head groups into the aqueous phase. This interface-specific mechanism can occur only if the reaction proceeds via the LH mechanism, whereby OH· adsorbs onto the AWI and has enough time to selectively abstract an H atom from the surface-available HOOC-Rn-COOH before diffusing to the bulk phase. Indeed, hydrophilic oxalic acid and malonic acid (n = 0, 1) are not oxidized by OH·(g) under the same experimental conditions.37 Note that if the Eley–Rideal (ER) mechanism had prevailed, multiple shorter chain HOOC-R≤n–1-COOH products should have been observed. These results indicated that OH· preferentially resided at the AWI, and its reaction proceeded via the LH mechanism.
Figure 6.

A schematic illustration of OH·(g) uptake and subsequent reactions at the air–water interface. Interface-specific H-abstraction from the α-CH2 group of suberic acid occurs. OH· is consumed by the reaction with surface-available suberic acid or with OH· itself at the topmost layers via the Langmuir–Hinshelwood mechanism before diffusing into the deeper layers, where hydrophilic oxalic acid largely resides.
In 2016, Rossignol et al. reported that a monolayer of nonanoic acid (NA) at the AWI absorbs UV radiation at wavelengths present in sunlight (280–330 nm) and then produces gas-phase organic compounds such as aldehydes and ketones (≤C9) that eventually serve as aerosol precursors.58 As one of the primary photochemical reactions of NA molecules at the AWI, they also proposed that a weak absorption by a singlet–triplet transition centered around 270 nm induces direct photodissociation (C–O bond fission) and formation of several radicals, especially gaseous OH·.58
Although we could not directly detect OH· at the AWI, we succeeded in detecting gaseous OH· emitted from photodissociation at the surface of a neat liquid NA by our newly developed m-LFP-LIF.27
Figure 7 shows normalized OH-LIF intensities as a function of delay time between the photolysis and probe lasers obtained in the 213 nm photolysis of liquid- and gas-phase NAs. The intensities from the liquid-phase photolysis (red circles in Figure 7) were measured by changing the distance between the liquid surface and the center of the dye laser from 3 mm to 7 mm (z in Figure 2a). The peak delay time for the OH-LIF intensity increased as z increased from 3 to 7 mm. This observation suggested that the measured OH· came from the liquid surface. Nevertheless, gaseous NA molecules were also present in the chamber because the vapor pressure of liquid NA is ∼0.2 Pa at room temperature.59 We therefore carried out the 213 nm photolysis of gas-phase NA separately to evaluate the contribution of the photolysis of gas-phase NA to the OH-LIF signals in the liquid experiment. The OH-LIF intensities from the gas-phase photolysis (black vertical bars in Figure 7) were also measured by changing the distance between the photolysis and probe lasers from 2 mm to 5 mm (d′ in Figure 2b). The distance was set to be approximately the same as that in the liquid-phase experiment (i.e., d ≈ d′ in Figures 2a and b). When the two lasers were close to each other (d′ = 2 mm), an OH-LIF signal due to gas-phase NA photolysis was observed. However, the OH· became barely detectable when d′ was increased to 5 mm. The difference between the OH-LIF intensities measured in the liquid and gas phases should correspond to the amount of OH· desorbed from the liquid NA, which was much larger than the amount formed in the gas-phase NA photolysis. These results confirmed that the dominant part of the OH-LIF signals during the liquid-phase experiment originated from OH· desorbed from the liquid NA surface.
Figure 7.

Normalized
OH-LIF intensities as a function of the delay time between
the photolysis and probe lasers. Red circles and black vertical bars
indicate the intensities measured in the liquid- and gas-phase NA
experiments, respectively. Each data point represents the average
of 160 measurement cycles. The error bars indicate ± one standard
deviation. For comparison, the OH-LIF intensities are normalized by
the photon fluences of the photolysis laser. Both experiments were
conducted with similar distances between the centers of the two lasers
(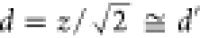 ). Adapted with permission
from ref (27). Copyright
2022 American
Chemical Society.
). Adapted with permission
from ref (27). Copyright
2022 American
Chemical Society.
The results in Figure 7 clearly indicated that OH· desorbed after UV irradiation to liquid-phase NA. To clarify whether the OH· were formed directly from liquid NA molecules or from secondary photoproducts that accumulated on the liquid surface, we conducted single-shot experiments. Figure 8 shows the shot-to-shot evolution of the OH-LIF intensity as a function of the number of measurement cycles in the photolysis of liquid NA. The intensities were recorded at the on- and off-resonance wavelengths for the excitation of OH·. During the single-shot experiments, the pulsed probe laser was set to be injected into the chamber. At the on-resonance wavelength (black lines in Figure 8), the intensities rose steeply just after the 213 nm irradiation. They remained almost constant during the subsequent 400 shots of irradiation in all measurements at repetition rates of 1, 2, 5, and 10 Hz from the two lasers. The uncertainties of the OH-LIF signal intensities were derived from the energy fluctuations of the two lasers. At the off-resonance wavelength, no OH-LIF signals appeared after 213 nm irradiation (gray lines in Figure 8). Instead, the signal intensities corresponded to the background signal intensities derived from stray light and the scattering of the probe laser. These results meant that the photolysis of NA molecules formed gaseous OH· as a primary photoproduct, and the OH· desorbed by the photolysis of secondary photoproducts that accumulated on the liquid surface was negligible at time scales of 0.1–1 s.
Figure 8.

Shot-to-shot evolution of the OH-LIF intensity as a function of the number of measurement cycles in the photolysis of liquid NA. The data were obtained by single-shot data acquisition at laser repetition rates of 1, 2, 5, and 10 Hz. Black and gray lines indicate results measured at the on- and off-resonance wavelengths of OH· excitation, respectively. The 213 nm photolysis laser irradiation of the liquid NA surface commenced at n = 0. Adapted with permission from ref (27). Copyright 2022 American Chemical Society.
The results shown in Figures 7 and 8 demonstrate the desorption of gaseous OH· following the 213 nm photolysis of liquid NA. We succeeded in quantifying this process of OH· formation by comparing the results with the 213 nm photolysis of gaseous acetic acid (see ref (27) for details).
3.3. Criegee Intermediates (RR′COO)
Carbonyl oxide diradicals/zwitterions (RR′C·OO·/RR′C=O+−O–), known as Criegee intermediates (CIs), are a key atmospheric species predominantly formed by the ozonolysis of organic compounds possessing C=C bond(s) such as terpenoids.60 Theoretical calculations have suggested that the reaction mechanisms involving CIs at AWIs differ from those in the gas phase or liquid phase.61−63 Because most CIs can react with water molecules, it is generally difficult to detect CIs at the AWI. To the best of our knowledge, direct detection of CIs at the AWI or gas–liquid interface has not been achieved. Instead, organic hydroperoxides (ROOHs) formed from the reactions of CIs with compounds possessing −OH moieties have been observed as the chloride ion (Cl–)-adducts by mass spectrometry and used for studies of the kinetics of CI production/consumption at the AWI.64−66 O3(g) exposure to water microjets containing atmospherically important sesquiterpenes (β-caryophyllene and α-humulene) and acetonitrile (for dissolving the hydrophobic sesquiterpenes) produce α-hydroxy-hydroperoxides (α-HHs) at the AWI that have been detected by SIMS as α-HH-Cl– signals in the presence of submillimolar NaCl.67 The signals at m/z 305;307 = 204 (sesquiterpene) + 48 (O3) + 18 (H2O) + 35;37 (chloride ion) indicate the rapid formation of α-HHs from the reaction of the CIs of hydrophobic sesquiterpenes with water molecules (H2O)n at the AWI. Both α-HHs and other ROOHs from the reactions of CIs with carboxylic acids and alcohols at the AWI have been successfully detected as their Cl–-adducts.67,68 Interestingly, the ROOH-Cl– signals from the reaction of the CIs of sesquiterpene with less surface-active species such as acetic acid are negligible. The implication is that the observed reactions proceed largely at the AWI.67,68 Note that the gas-phase rate constant for the reaction of CIs with acetic acid is extremely large, ∼ 10–10 cm3 molecule–1 s–1.60,69 In fact, the most prominent ROOH-Cl– signals have been observed from the reaction of the CIs of sesquiterpene with cis-pinonic acid,70 which is known as an exceptionally surface-active C10 carboxylic acid produced from the ozonolysis of α-pinene.42,71 The mass spectral signals of α-HHs derived from the reaction of the CIs of α-terpineol with (H2O)n at the AWI is just mildly influenced by the bulk pH of water microjets between 1 and 11.72 This result implies that reactions involving the CIs of α-terpineol are not affected by the excess presence of H+/OH– at the AWI. It should be noted that α-HHs are also reactive intermediates in aqueous solution because they rapidly decompose in the presence of water.73 The H+-catalyzed decomposition has been found to be so fast that α-HHs cannot survive for more than a few seconds in aqueous solution at pH = 1–2 (typical pH of aerosols).74 It will thus be very challenging to detect such α-HHs from atmospheric condensed phases collected by field measurements.64
Notably, it has been found that the CIs of sesquiterpenes react with hydrophilic levoglucosan (Levo), a key tracer of biomass burning, to form α-alkoxy-hydroperoxides (α-AHs) at the AWI (Figure 9).75 The mass spectral signals at m/z 449;451 = 204 (β-caryophyllene) + 48 (O3) + 162 (Levo) + 35;37 (chloride ion) clearly indicate the incorporation of the CIs of β-caryophyllene into Levo, and the formation of the corresponding α-AHs have been detected as their Cl– adducts. The mass signals attributed to the chloride adducts of Levo and α-HHs have been observed along with the corresponding carboxylate derived from the isomerization of the CIs. Experiments using D2O and H218O as solvents have supported the assignments of products. The reactivity of CIs toward OH-species generally increases as the gas-phase acidity of the OH-group increases,76 and Levo actually possesses much more acidic OH-groups than normal alkyl alcohols.77 Levo can therefore react with surface-available CIs at the AWI.75 We also found that not only Levo but also glucose, arabitol and mannitol can react with CIs at the AWI. Given the proposed structure shown in Figure 9, the α-AHs from Levo + CIs are expected to diffuse to the bulk phase, followed by decomposition to alcohols and other products in atmospheric condensed phases.78 In this process, the −OOH moiety would be converted to −OH without producing radicals.79 The products possessing multiple −OH groups would be further degraded by OH· reactions in the bulk.80 Those reactions could potentially lead to the formation of extremely low volatility organic compounds (ELVOCs) or highly oxygenated organic molecules (HOMs).
Figure 9.

Negative ion mass spectra of 1 mM β-caryophyllene + 0.2 mM NaCl + 100 mM levoglucosan in microjets of an acetonitrile:water (4:1 = vol:vol) solution (gray) or exposed to O3(g) (red, E = 2.4 × 1011 molecules cm–3 s) at 1 atm and 298 K. The m/z 449;451 signals correspond to chloride-adducts of α-alkoxy-hydroperoxide (α-AH) from the reaction between the CIs of β-caryophyllene and levoglucosan at the AWI. Adapted with permission from ref (75). Copyright 2017 American Chemical Society.
3.4. Sulfenic Acid (RSOH)
Sulfenic acid (RSOH) is a key intermediate in the reaction of biological cysteine-containing species such as GSH and proteins.81 Compounds possessing the thiol (−SH) group are easily oxidized into RSOH by ROS, and the RSOHs are further oxidized to sulfinic acid (RSO2H) followed by the formation of sulfonic acid (RSO3H) or reacted with compounds possessing −SH to form a disulfide (RSSR(R′)). The fate of RSOH is delicately controlled by circumstances, e.g., pH, the concentration of ROS, the distance between thiol groups, and the molecular structure.82 The GSOH and the associated products may act as signaling molecules that control the redox balance and signal transduction in ELFs.
Bioaerosols are composed of a variety of biological molecules, including free amino acids, lipids, proteins, DNA, and their fractured molecules.21,83 Although accurate rates of emission of bioaerosols are still unknown, they may be a major fraction of atmospheric aerosols.84 Bioaerosols are expected to react with gaseous reactants, as are other aerosol particles, during the time from their emission to their deposition. Given that −SH (and thiolate −S–) is one of the most reactive functional groups among all the components of bioaerosols, RSOH and its associated products could be formed at the AWI during the heterogeneous reactions of bioaerosols with gaseous reactants such as OH·. However, it has been challenging to directly detect RSOH because of its high reactivity and short lifetime.81
Glutathione sulfenic acid (GSOH) was first detected at the AWI by LFP-SIMS in 2015.36 Water microjets containing 0.1 mM GSH (typical [GSH] in ELFs)85 were exposed to gas-phase OH· beams generated by 266 nm photolysis of a gaseous mixture of O3/O2/H2O/N2. The oxidation of aqueous GSH by gaseous OH· was found to produce GSOH, which was further oxidized to glutathione sulfinic acid (GSO2H) and glutathione sulfonic acid (GSO3H) (Figure 10). The proposed mechanism of GSOH formation is the addition of OH· to the S atom of GSH to produce a GSH–OH adduct, followed by reaction with O2 to produce GSOH and HO2 or with OH· to produce GSOH and H2O at the AWI.36 Because the rate of production of GSOH does not depend on the square of [OH·] (i.e., laser energy), the former production pathway was more plausible. These results have been consistent with a mechanism in which OH· is captured by the GSH carboxylate groups and steered toward the reactive – SH group by a concerted process involving multiple H-bonded interactions (see below).22
Figure 10.

GSH (m/z 306) (A), and GSOH (m/z 322), GSO2H (m/z 338), GSO3H (m/z 354) (B) mass spectral signal intensities from aqueous 0.1 mM GSH (pH 4.4) microjets exposed to O3/O2/H2O/N2(g) mixtures at [O3(g)] ∼ 4.2 × 1015 molecules cm–3, irradiated with 266 nm laser beams as functions of laser energy (in mJ pulse–1). (C) Closeup of B data at lower laser energies. Adapted with permission from ref (36). Copyright 2015 American Chemical Society.
The same GSOnH (n = 1–3) products, along with oxo-ferryl (FeIV=O) intermediates,15 have been observed in experiments involving gaseous ozone exposure to microjets containing GSH and FeCl2.36 The implication is that an interfacial Fenton-like reaction could also add O atom(s) to GSH. Note that GSH has been found to be unreactive toward O3 in the absence of Fe2+ when the pH is <4. This result is consistent with the pKa of the thiol group (pKa = 7–8),86 and the thiol is much less reactive toward O3 than the thiolate.87
We performed additional experiments using water microjets containing 0.1 mM GSSG exposed to gas-phase OH· beams. Interestingly, GSSG was also oxidized by OH· at the AWI to form GSO1–3H.36 We also found evidence that SP-B1–25, a 25-residue polypeptide surrogate of human lung surfactant protein B possessing two cysteine groups, was oxidized by OH· and O3 at the AWI to form primary On-containing products/intermediates (n ≤ 5).44 Theoretical calculations have indicated that cysteines Cys8 and Cys11 as well as tryptophan Trp9 in the hydrophobic section of SP-B1–25 are present in the topmost layers of the AWI,88 and these groups are exceptionally reactive toward OH·/O3.89 We concluded that Trp9 accepted three O atoms and that Cys8 and Cys11 accepted one O atom each during the reactions of OH·/O3 at the AWI. The absence of mass spectral signals for On≥6-containing products implied that further oxidation of the −SOH groups did not occur because of steric hindrance in the SP-B1–25 structure at the AWI.
These results indicated that RSOH could be formed from compounds possessing thiol groups because of the presence of OH· at the AWI of bioaerosols or ELFs. Subsequent OH-oxidations of GSOH lead to the production of GSO3H rather than to the generally accepted GSSG. We infer that OH· that naturally accumulates at the AWI selectively reacts with GSOH.52 The orientations of GSOH at the AWI may also play a key role. If the reactive −SOH group is located in the topmost layers, it will more easily react with OH· than with bulky GSH. This interface-specific selectivity would explain the observed GSO2H/GSO3H formation over GSSG at the AWI in the wide range of [GSH], which is 0.01–100 mM.36 Future theoretical calculations will shed light on this interface-specific mechanism for the GSH + OH· reaction. The fates and roles of “end-product” GSO3H in bioaerosols and ELFs have yet to be identified.
4. Summary and Future Perspectives
In this Featured Article, we reported some recent advances in the detection of atmospherically relevant intermediates and the associated reaction mechanisms at the AWI. We showed that LFP-SIMS was a powerful method to prove that short-lived (<1 ms) intermediates at the AWI formed during the heterogeneous reaction of OH·(g) with organic species (aq). Use of m-LFP-LIF enabled us to directly monitor OH· emitted from the surface of liquid samples irradiated by UV laser beams.
Organic peroxyl radicals (RO2·) produced from the heterogeneous reactions of gaseous hydroxyl radicals (OH·) with aqueous carboxylic acids were successfully detected by LFP-SIMS. The time scale of the LFP-SIMS, less than one millisecond, enabled us to detect short-lived reactive intermediates. The RO2· signals, which appeared at m/z = M (reactant) – 1 (H atom) + 32 (O2), were not changed by switching the solvent from H2O to D2O or H218O. Investigation of the H-D exchange was found to be a powerful way to precisely assign products. The RO2-associated products, including carbonyls, alcohols, and hydroperoxides (ROOH), were also detected by LFP-SIMS. In the case of benzoic acid, we also observed a tentatively assigned hydrotrioxide (ROOOH) that might be generated from the reaction of the RO2· with OH· at the AWI.
Although theoretical calculations predict that the concentration of OH· is higher in the AWI than the bulk phase, its direct detection at the AWI has not been achieved. The LFP-SIMS studies, however, indirectly supported the interfacial affinity of OH· by showing that the heterogeneous reaction of OH·(g) with aqueous dicarboxylic acids proceeds via the Langmuir–Hinshelwood (LH) mechanism.37 The observed interface-specific H-abstraction from the α-CH2 of long-chain dicarboxylic acids by OH· indicates that the lifetime of OH· is long enough for the reaction to occur at the AWI before it diffuses into the aqueous bulk phase. An m-LFP-LIF study has reported the direct detection of OH·(g) emitted from photoirradiation of liquid nonanoic acid (NA), a key carboxylic acid that covers the AWI of marine aerosols and the ocean surface, by a UV laser shot.27 That study has revealed that the yield of OH· is just a few percent compared to the yield from the photolysis of gaseous acetic acid. The implication is that interface-specific photodissociation of NA occurred. Indeed, vibrational sum frequency generation spectroscopy has revealed that NA forms a dimer at the gas–liquid interface, and formation of that dimer may preclude the OH· split from the −C(=O)OH moiety.27 The implication is that the yield of OH· emitted from the AWI of marine aerosols/ocean and human skin90 is controlled by interface-specific photoreactions.
Criegee intermediates (CIs) are key atmospheric intermediates formed from ozonolysis of organic compounds possessing C=C bond(s) and are successfully trapped at the AWI by water molecules, carboxylic acids, and alcohols. We used SIMS to detect CIs as ROOH-Cl– adducts. For example, levoglucosan, a representative atmospheric saccharide formed by biomass burning, can react with the CIs of β-caryophyllene to form α-alkoxy-hydroperoxides at the AWI that we detected at m/z 449;451 = 204 (β-caryophyllene) + 48 (O3) + 162 (Levo) + 35;37 (chloride ion). We thus found that the reactions of CIs with OH-species preferentially proceeded at the AWI, and the reactivity was controlled by the surface-availability of OH-species and the acidity of the gas phase.66 Because such α-alkoxy-hydroperoxides and other observed ROOHs are hydrophilic, they diffuse into the bulk phase and are then decomposed by H+/water catalyzed reactions.64,91 During this process, the −OOH moiety would be converted to the −OH moiety by production of H2O2 but not OH·. The products possessing multiple −OH groups would be further degraded by reaction with OH· in the bulk.80 These reactions would lead to the production of the ELVOCs or HOMs found in atmospheric condensed phases.
An LFP-SIMS study has demonstrated that aqueous glutathione (GSH), a key antioxidant tripeptide in vivo, reacts with OH·(g) to produce glutathione sulfenic acid, GSOH, rather than GSSG at the AWI. The GSOH has been directly detected by mass spectrometry.36 The further oxidation of GSOH by OH· leads to the formation of a glutathione sulfinic acid GSO2H intermediate and glutathione sulfonic acid GSO3H product. GSSG and SP-B1–25, a 25-residue polypeptide surrogate of human lung surfactant protein B, have also been found to produce the corresponding sulfenic acids at the AWI.36,44
Although the direct detection of atmospherically relevant intermediates at the AWI has in some cases been achieved, there remain numerous challenges in this field. For example, careful consideration should be given to how the experimental results obtained from LFP-SIMS can be translated to heterogeneous processes that occur at the surface of ambient atmospheric droplets.92 Furthermore, it has been shown that traces of impurities (0.1% at most) intrinsically contained in reagents could cause unwanted photoreactions.93 Development of new experimental techniques and the associated theoretical calculations are clearly needed. We summarize future perspectives below.
4.1. Detection of OH· at the AWI
In situ detection of OH· at the AWI or in condensed phases is a highly challenging task because of its short lifetime. LIF is a powerful method to probe OH· because of its high selectivity and sensitivity, and it may be used to directly detect OH· at the AWI. Given that LIF requires vacuum conditions, however, using water as a solvent is challenging. A suitable initial trial would be to detect OH· at the gas-organic solvent interface. It may be possible to detect OH· at the interface between the gas and organic solvent when the thin liquid organic film is photoirradiated under medium-vacuum conditions. In situ detection of OH· at the interface will provide valuable information about the fate of OH· in the medium, e.g., how vibrationally excited OH· can be quenched by interfacial molecules.
4.2. Detection of O3 at the AWI
Both experimental and theoretical studies have suggested that O3 has a high affinity at the AWI.94−96 Theoretical calculations have predicted that photodissociation of O3 differs at the AWI and in the gas-phase.16,97 The simulated adsorption of O3 at the AWI is thermodynamically spontaneous, and the adsorbed O3 is more stable at the AWI than in the gas-phase by 1.3 kcal/mol and in bulk water by 3.0 kcal/mol.97 The calculated absorption cross sections of O3 at wavelengths of 200–700 nm are generally larger at the AWI than in the gas phase. Calculations predict that the photodissociation of O3 at the AWI may lead to an enhancement of the rate of OH· formation by 4 orders of magnitude. If that prediction is correct, more production of OH· would occur during the sunlight-photolysis of O3 at the AWI of cloud droplets and aerosol particles than previously assumed. Because this scenario potentially changes the current understanding of HOx cycles in the atmosphere, detecting and revealing the associated photoreactions of O3 at the AWI is a pivotal challenge. A recent study has shown that trapping of O3 by Br– at the gas–water interface leads to formation of an interface-specific Br−OOO– complex that was identified by XPS.98 LFP-SIMS may be used to investigate the photoreaction of O3 at the AWI by changing the currently pulsed laser to a continuous-wave laser system. That transition is underway in our laboratory.
4.3. Detection of Criegee Intermediates at the AWI
Although short-chain CIs have been directly detected in the gas phase,60,99 detecting CIs at the AWI is intrinsically difficult because of their reactivity toward water molecules. CIs can probably be detected at the air-organic solvent interface. Another issue, however, lies in the fact that CIs can generally isomerize into a vinyl hydroperoxide or other compounds. Mass spectrometric detection of CIs at the air-organic solvent interface will therefore be needed to distinguish between these isomers. Direct detection of CIs will shed light on the reaction rates of CIs at the interface, which are predicted to be much different from those in the gas phase or liquid phase.61,62 Elucidation of the mechanism responsible for ozonolysis of organic compounds possessing C=C bonds at the gas–liquid interface will also be helpful. Those mechanisms are highly relevant to the issue of interaction between air pollutants and biological surfaces such as human skin.100−102
4.4. Detection of ROOOH at the AWI
As shown in the LFP-SIMS study of the reaction between OH·(g) and benzoic acid(aq) at the AWI,43 direct detection by mass spectrometry of the hydrotrioxide formed from RO2· and OH· will be revealing. A recent experimental study has indeed shown that ROOOH(g) can be detected by mass spectrometry.50 ROOOH at the AWI may be detected by LFP-SIMS under certain conditions because ROOOH can be stabilized by the presence of interfacial water molecules. We have detected some organic hydroperoxides (ROOH) as their Cl–-adducts by SIMS in the presence of submillimolar amounts of NaCl. Therefore, ROOOHs at the AWI may also be detected as their ROOOH-Cl– adducts by SIMS. The direct detection of ROOOHs at the AWI may reveal hitherto unknown roles of ROOOHs in atmospheric multiphase chemistry.48
4.5. Radical Recognition Process at the AWI
No evidence for H-abstraction from GSH (aq), GSSG (aq), or SP-B1–25 (aq) by OH·(g) has been found in LFP-SIMS experiments, despite the fact that these molecules actually possess multiple functionalities (−CH3, −CH2–, −NH2, etc.) that would be expected to donate an H atom to OH·.36,38,44 In contrast, the production of the corresponding sulfenic acids by all these reactions indicates that OH· is exclusively bonded to the sulfur atom of the thiol/thiolate group. This peculiar observation may be related to a “radical recognition process” that occurs specifically at the AWI. The key features of the radical recognition process at the AWI originate from (a) the flexibility,103,104 (b) multiple hydrogen-bonding ability,22 and (c) interface-specific orientation88 of the biomolecule. Theoretical calculations have shown that the interaction between GSH and OH· involves two distinct steps: catching and steering.22 Because the catching mechanism occurs via complex formation between carboxylate anions and OH·, both terminal residues of GSH serve as “recognition sites”. The relative enthalpy changes of formation of these GSH–OH complexes are in the range of −42.4 to −27.8 kJ mol–1.22 The flexibility of the glycine residue enables the formation of further interactions of other parts of glutathione with the lone electron pair of OH·. As a result, OH· is steered toward the thiol group of the cysteine residue. This catch and steering process results in the successful trap of OH· by the S atoms of GSH. Furthermore, the interface-specific orientation of biomolecules at the AWI would cause the recognition process to differ between the AWI and bulk phase. Theoretical calculations have shown some residues of SP-B1–25, such as cysteines, are located at the topmost layers and are less hydrated, whereas others, such as a methionine, reside in a deeper aqueous phase.88 Indeed, our experimental results from the SP-P1–25 + OH· reaction at the AWI implied that the surface-available cysteine residues were preferentially oxidized by OH· coming from the gas phase, whereas the buried methionine was not.44 Future theoretical calculations will shed light on the interface-specific radical recognition process, that will be pivotal for understanding the multiphase chemistry of bioaerosols and ELFs.
4.6. Photochemistry of “Pure” Organic Compounds at the AWI
As described in Section 3.2, previous laboratory studies have proposed that irradiating saturated fatty acids such as NA at the AWI with UV wavelengths longer than 295 nm can produce OH· and organic radicals in the troposphere.58,105 However, our understanding of photochemical reactions of liquid organic compounds is still at an early stage, and many questions have yet to be addressed even in the neat liquid phase, let alone at the AWI. Considering that the mass fraction of organic matter in sea spray aerosol particles (d = 150 nm) is ∼40% and the majority of organic carbon is insoluble in water,106 quantitative information about UV absorption cross sections of neat liquid organic compounds are desirable as a first step in understanding the photochemistry that occurs on/in sea spray aerosols. Despite its importance, there is currently no database for UV absorption spectra of liquid organic compounds relevant to atmospheric chemistry. This situation is in sharp contrast to the study of gas-phase molecules, for which databases now contain UV absorption cross sections of many gaseous molecules and even radicals.107−109
An intrinsic problem in obtaining accurate UV absorption spectra for neat liquid organic compounds is the contribution of impurities. Commercially available reagents are implicitly assumed to be sufficiently pure for most laboratory experiments, and only a few previous atmospheric chemistry studies have involved purification of reagents.110 In this context, we recently revealed that the previously reported absorption of wavelengths longer than 250 nm by neat liquid NA originates from traces of impurities (0.1% at most) intrinsically contained in NA reagents (Figure 11).93 Absorption cross sections of liquid NA have been obtained for a wavelength range of 190–310 nm, which gives an upper limit of 1.0 × 10–9 s–1 for the rate of photolysis (J-value) of neat liquid NA in the troposphere. Our study strongly suggests that pure NA is photochemically inert in the troposphere, in contrast to previous predictions.58 We suggest that photochemical studies regarding liquid phase and gas–liquid interfaces require careful evaluation of trace impurities and contaminants (see also ref93 for details).
Figure 11.

UV absorption spectra and absorption cross sections of liquid nonanoic acid at room temperature. (A) UV absorption spectra of nonanoic acid: (a) reported by Rossignol et al.58 for a commercial sample (purity, 97%) and measured here for a commercial sample (purity, > 98.0%) (b) before and (c) after purification by recrystallization. Optical path lengths were 10 mm. (B) The corresponding calculated absorption cross sections. Reproduced from ref (93). Available under a CC-BY 4.0 license. Copyright 2023 American Association for the Advancement of Science.
Experimental measurements of UV spectra of atmospherically relevant molecules and radicals at the AWI are challenging. Nevertheless, recent progress in linear and nonlinear spectroscopic methods have led to successful acquisition of electronic spectra of organic compounds at the AWI and liquid/liquid interfaces in the UV- and visible wavelength regions.111−113 Application of these spectroscopic techniques to atmospheric multiphase chemistry is a promising approach for improving the understanding of photochemical reactions at the AWI. Another approach we are currently planning involves a new apparatus for direct detection of gaseous radical products (e.g., OH·) formed following photochemistry at the AWI. The method involves laser flash photolysis under atmospheric pressure and radical probing by LIF in a vacuum. This apparatus can allow us to investigate the yields of radicals in photochemical reactions at the AWI. The results can be used for theoretical and modeling studies in climate science and give insights into the interface-specific reaction mechanisms.
Acknowledgments
The authors would like to thank the many collaborators who have contributed to the works described in this Featured Article. Especially, SE is grateful to Prof. Michael R. Hoffmann and Dr. A. J. Colussi (Caltech) for their long-time collaborations. This work was partly supported by KAKENHI grants from the Japan Society for the Promotion of Science (nos. 23H00525, 23K18526, 24H02113) and the Morino Foundation for Molecular Science.
Biographies

Shinichi Enami is a full Professor at the University of Tsukuba. He received a B.S. (2002), a M.S. (2004), and a Ph.D. (2006) from Kyoto University (Mentor: Prof. Masahiro Kawasaki). After working for four-and-a-half years (2006/10–2011/3) as a Postdoctoral Researcher at the California Institute of Technology (with Prof. Michael R. Hoffmann and Dr. A. J. Colussi), he joined Kyoto University as an Associate Professor (2011–2016, the Hakubi Project). After Kyoto University, he worked at the National Institute for Environmental Studies (2016–2021 as a Senior Researcher, 2021–2023 as a Chief Senior Researcher), and then moved to his current position. His recent research includes atmospheric multiphase chemistry, biosurface chemistry, and molecular-level inhomogeneity on/in liquid phases.

Naoki Numadate is an Assistant Professor at the University of Tsukuba. He received a B.S. (2013) and a M.S. (2015) from Sophia University, and a Ph.D. (2019) from Tokyo Metropolitan University (Mentor: Prof. Hajime Tanuma). After working for a year as a Postdoctoral Researcher at the University of Electro-Communications, he then worked for three-and-a-half years as a Project Assistant Professor at the University of Tokyo. After the University of Tokyo, he moved to his current position. His recent research includes dynamics and kinetics studies of gas–liquid interfacial reactions.

Tetsuya Hama is as an Associate Professor at the University of Tokyo. He received his B.Eng. (2007), M.Eng. (2009), and Ph.D. (2010) in engineering from Kyoto University for his study of the photodissociation dynamics of water ice. He joined the Institute of Low Temperature Science at Hokkaido University as an Assistant Professor in 2010. Since 2020, he has worked at the Komaba Institute for Science at the University of Tokyo. His research focuses on surface chemistry and photochemistry relevant to atmospheric chemistry and astrochemistry, with an emphasis on liquid and solid water.
The authors declare no competing financial interest.
References
- Finlayson-Pitts B. J. Reactions at Surfaces in the Atmosphere: Integration of Experiments and Theory as Necessary (but Not Necessarily Sufficient) for Predicting the Physical Chemistry of Aerosols. Phys. Chem. Chem. Phys. 2009, 11, 7760–7779. 10.1039/b906540g. [DOI] [PubMed] [Google Scholar]
- Ruiz-Lopez M. F.; Francisco J. S.; Martins-Costa M. T. C.; Anglada J. M. Molecular Reactions at Aqueous Interfaces. Nat. Rev. Chem. 2020, 4, 459–475. 10.1038/s41570-020-0203-2. [DOI] [PubMed] [Google Scholar]
- Gray-Weale A.; Beattie J. K. An Explanation for the Charge on Water’s Surface. Phys. Chem. Chem. Phys. 2009, 11, 10994–11005. 10.1039/b901806a. [DOI] [PubMed] [Google Scholar]
- Beattie J. K.; Djerdjev A. N.; Warr G. G. The Surface of Neat Water Is Basic. Faraday Discuss. 2009, 141, 31–39. 10.1039/B805266B. [DOI] [PubMed] [Google Scholar]
- Bai C.; Herzfeld J. Surface Propensities of the Self-Ions of Water. ACS Cent. Sci. 2016, 2, 225–231. 10.1021/acscentsci.6b00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilch L. W.; Maze J. T.; Smith J. W.; Ewing G. E.; Jarrold M. F. Charge Separation in the Aerodynamic Breakup of Micrometer-Sized Water Droplets. J. Phys. Chem. A 2008, 112, 13352–13363. 10.1021/jp806995h. [DOI] [PubMed] [Google Scholar]
- Devlin S. W.; Bernal F.; Riffe E. J.; Wilson K. R.; Saykally R. J. Spiers Memorial Lecture: Water at Interfaces. Faraday Discuss. 2024, 249, 9–37. 10.1039/D3FD00147D. [DOI] [PubMed] [Google Scholar]
- Herzfeld J. Art, Fact and Artifact: Reflections on the Cross-Talk between Theory and Experiment. Phys. Chem. Chem. Phys. 2024, 26, 9848–9855. 10.1039/D4CP00005F. [DOI] [PubMed] [Google Scholar]
- Zhang P.; Feng M.; Xu X. Double-Layer Distribution of Hydronium and Hydroxide Ions in the Air-Water Interface. ACS Phys. Chem. Au 2024, 10.1021/acsphyschemau.3c00076. [DOI] [Google Scholar]
- Enami S.; Hoffmann M. R.; Colussi A. J. Proton Availability at the Air/Water Interface. J. Phys. Chem. Lett. 2010, 1, 1599–1604. 10.1021/jz100322w. [DOI] [Google Scholar]
- Mishra H.; Enami S.; Nielsen R. J.; Stewart L. A.; Hoffmann M. R.; Goddard W. A.; Colussi A. J. Bronsted Basicity of the Air-Water Interface. Proc. Nat. Acad. Sci. U. S. A. 2012, 109, 18679–18683. 10.1073/pnas.1209307109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enami S.; Hoffmann M. R.; Colussi A. J. Molecular Control of Reactive Gas Uptake ″on Water″. J. Phys. Chem. A 2010, 114, 5817–5822. 10.1021/jp1019729. [DOI] [PubMed] [Google Scholar]
- Enami S.; Stewart L. A.; Hoffmann M. R.; Colussi A. J. Superacid Chemistry on Mildly Acidic Water. J. Phys. Chem. Lett. 2010, 1, 3488–3493. 10.1021/jz101402y. [DOI] [Google Scholar]
- Gladich I.; Chen S.; Yang H.; Boucly A.; Winter B.; van Bokhoven J. A.; Ammann M.; Artiglia L. Liquid-Gas Interface of Iron Aqueous Solutions and Fenton Reagents. J. Phys. Chem. Lett. 2022, 13, 2994–3001. 10.1021/acs.jpclett.2c00380. [DOI] [PubMed] [Google Scholar]
- Enami S.; Sakamoto Y.; Colussi A. J. Fenton Chemistry at Aqueous Interfaces. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 623–628. 10.1073/pnas.1314885111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J.; Kumar M.; Anglada J. M.; Martins-Costa M. T. C.; Ruiz-Lopez M. F.; Zeng X. C.; Francisco J. S. Atmospheric Spectroscopy and Photochemistry at Environmental Water Interfaces. Annu. Rev. Phys. Chem. 2019, 70, 45–69. 10.1146/annurev-physchem-042018-052311. [DOI] [PubMed] [Google Scholar]
- Limmer D. T.; Götz A. W.; Bertram T. H.; Nathanson G. M. Molecular Insights into Chemical Reactions at Aqueous Aerosol Interfaces. Annu. Rev. Phys. Chem. 2024, 10.1146/annurev-physchem-083122-121620. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Barraza K. M.; Upton K. T.; Beauchamp J. L. Time Resolved Study of Hydroxyl Radical Oxidation of Oleic Acid at the Air-Water Interface. Chem. Phys. Lett. 2017, 683, 76–82. 10.1016/j.cplett.2017.05.051. [DOI] [Google Scholar]
- von Sonntag C.; Schuchmann H. P. The Elucidation of Peroxyl Radical Reactions in Aqueous-Solution with the Help of Radiation-Chemical Methods. Angew. Chem. Int. Edt. 1991, 30, 1229–1253. 10.1002/anie.199112291. [DOI] [Google Scholar]
- Enami S.; Hoffmann M. R.; Colussi A. J. In Situ Mass Spectrometric Detection of Interfacial Intermediates in the Oxidation of RCOOH(Aq) by Gas-Phase OH-Radicals. J. Phys. Chem. A 2014, 118, 4130–4137. 10.1021/jp503387e. [DOI] [PubMed] [Google Scholar]
- Estillore A. D.; Trueblood J. V.; Grassian V. H. Atmospheric Chemistry of Bioaerosols: Heterogeneous and Multiphase Reactions with Atmospheric Oxidants and Other Trace Gases. Chem. Sci. 2016, 7, 6604–6616. 10.1039/C6SC02353C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiser B.; Jójárt B.; Csizmadia I. G.; Viskolcz B. Glutathione - Hydroxyl Radical Interaction: A Theoretical Study on Radical Recognition Process. PLoS One 2013, 8, e73652 10.1371/journal.pone.0073652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J.; Kumar M.; Francisco J. S.; Zeng X. C. Insight into Chemistry on Cloud/Aerosol Water Surfaces. Acc. Chem. Res. 2018, 51, 1229–1237. 10.1021/acs.accounts.8b00051. [DOI] [PubMed] [Google Scholar]
- Poschl U.; Shiraiwa M. Multiphase Chemistry at the Atmosphere-Biosphere Interface Influencing Climate and Public Health in the Anthropocene. Chem. Rev. 2015, 115, 4440–4475. 10.1021/cr500487s. [DOI] [PubMed] [Google Scholar]
- Enami S.; Hoffmann M. R.; Colussi A. J. Halogen Radical Chemistry at Aqueous Interfaces. J. Phys. Chem. A 2016, 120, 6242–6248. 10.1021/acs.jpca.6b04219. [DOI] [PubMed] [Google Scholar]
- Ishizuka S.; Matsugi A.; Hama T.; Enami S. Interfacial Water Mediates Oligomerization Pathways of Monoterpene Carbocations. J. Phys. Chem. Lett. 2020, 11, 67–74. 10.1021/acs.jpclett.9b03110. [DOI] [PubMed] [Google Scholar]
- Numadate N.; Saito S.; Nojima Y.; Ishibashi T.; Enami S.; Hama T. Direct Observation and Quantitative Measurement of OH Radical Desorption During the Ultraviolet Photolysis of Liquid Nonanoic Acid. J. Phys. Chem. Lett. 2022, 13, 8290–8297. 10.1021/acs.jpclett.2c02199. [DOI] [PubMed] [Google Scholar]
- Enami S.; Colussi A. J. Long-Range Specific Ion-Ion Interactions in Hydrogen-Bonded Liquid Films. J. Chem. Phys. 2013, 138, 184706. 10.1063/1.4803652. [DOI] [PubMed] [Google Scholar]
- Enami S.; Fujii T.; Sakamoto Y.; Hama T.; Kajii Y. Carboxylate Ion Availability at the Air-Water Interface. J. Phys. Chem. A 2016, 120, 9224–9234. 10.1021/acs.jpca.6b08868. [DOI] [PubMed] [Google Scholar]
- Enami S.; Colussi A. J. Long-Range Hofmeister Effects of Anionic and Cationic Amphiphiles. J. Phys. Chem. B 2013, 117, 6276–6281. 10.1021/jp401285f. [DOI] [PubMed] [Google Scholar]
- Enami S.; Mishra H.; Hoffmann M. R.; Colussi A. J. Hofmeister Effects in Micromolar Electrolyte Solutions. J. Chem. Phys. 2012, 136, 154707. 10.1063/1.4704752. [DOI] [PubMed] [Google Scholar]
- Tian C. S.; Byrnes S. J.; Han H. L.; Shen Y. R. Surface Propensities of Atmospherically Relevant Ions in Salt Solutions Revealed by Phase-Sensitive Sum Frequency Vibrational Spectroscopy. J. Phys. Chem. Lett. 2011, 2, 1946–1949. 10.1021/jz200791c. [DOI] [Google Scholar]
- Ghosal S.; Hemminger J. C.; Bluhm H.; Mun B. S.; Hebenstreit E. L. D.; Ketteler G.; Ogletree D. F.; Requejo F. G.; Salmeron M. Electron Spectroscopy of Aqueous Solution Interfaces Reveals Surface Enhancement of Halides. Science (New York, N.Y.) 2005, 307, 563–566. 10.1126/science.1106525. [DOI] [PubMed] [Google Scholar]
- Jungwirth P.; Tobias D. J. Specific Ion Effects at the Air/Water Interface. Chem. Rev. 2006, 106, 1259–1281. 10.1021/cr0403741. [DOI] [PubMed] [Google Scholar]
- Enami S.; Ishizuka S.; Colussi A. J. Chemical Signatures of Surface Microheterogeneity on Liquid Mixtures. J. Chem. Phys. 2019, 150, 024702. 10.1063/1.5055684. [DOI] [PubMed] [Google Scholar]
- Enami S.; Hoffmann M. R.; Colussi A. J. OH-Radical Specific Addition to Glutathione S-Atom at the Air-Water Interface: Relevance to the Redox Balance of the Lung Epithelial Lining Fluid. J. Phys. Chem. Lett. 2015, 6, 3935–3943. 10.1021/acs.jpclett.5b01819. [DOI] [PubMed] [Google Scholar]
- Enami S.; Hoffmann M. R.; Colussi A. J. Stepwise Oxidation of Aqueous Dicarboxylic Acids by Gas-Phase OH Radicals. J. Phys. Chem. Lett. 2015, 6, 527–534. 10.1021/jz502432j. [DOI] [PubMed] [Google Scholar]
- Enami S.; Sakamoto Y.; Hara K.; Osada K.; Hoffmann M. R.; Colussi A. J. Sizing” Heterogeneous Chemistry in the Conversion of Gaseous Dimethyl Sulfide to Atmospheric Particles. Environ. Sci. Technol. 2016, 50, 1834–1843. 10.1021/acs.est.5b05337. [DOI] [PubMed] [Google Scholar]
- Vacha R.; Slavicek P.; Mucha M.; Finlayson-Pitts B. J.; Jungwirth P. Adsorption of Atmospherically Relevant Gases at the Air/Water Interface: Free Energy Profiles of Aqueous Solvation of N2, O2, O3, OH, H2O, HO2, and H2O2. J. Phys. Chem. A 2004, 108, 11573. 10.1021/jp046268k. [DOI] [Google Scholar]
- Stone D.; Whalley L. K.; Heard D. E. Tropospheric OH and HO2 Radicals: Field Measurements and Model Comparisons. Chem. Soc. Rev. 2012, 41, 6348–6404. 10.1039/c2cs35140d. [DOI] [PubMed] [Google Scholar]
- Lunkenheimer K.; Barzyk W.; Hirte R.; Rudert R. Adsorption Properties of Soluble, Surface-Chemically Pure N-Alkanoic Acids at the Air/Water Interface and the Relationship to Insoluble Monolayer and Crystal Structure Properties. Langmuir 2003, 19, 6140–6150. 10.1021/la034379p. [DOI] [Google Scholar]
- Enami S.; Sakamoto Y. OH-Radical Oxidation of Surface-Active cis-Pinonic Acid at the Air-Water Interface. J. Phys. Chem. A 2016, 120, 3578–3587. 10.1021/acs.jpca.6b01261. [DOI] [PubMed] [Google Scholar]
- Enami S.; Hoffmann M. R.; Colussi A. J. Extensive H-Atom Abstraction from Benzoate by OH-Radicals at the Air-Water Interface. Phys. Chem. Chem. Phys. 2016, 18, 31505–31512. 10.1039/C6CP06652F. [DOI] [PubMed] [Google Scholar]
- Enami S.; Colussi A. J. OH-Radical Oxidation of Lung Surfactant Protein B on Aqueous Surfaces. Mass Spectrom. 2018, 7, 1–7. 10.5702/massspectrometry.S0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X. M.; Schuchmann M. N.; Vonsonntag C. Oxidation of Benzene by the OH Radical - A Product and Pulse-Radiolysis Study in Oxygenated Aqueous-Solution. J. Chem. Soc. Perkin Trans. 2 1993, 289–297. 10.1039/p29930000289. [DOI] [Google Scholar]
- Bohn B.; Zetzsch C. Gas-Phase Reaction of the OH-Benzene Adduct with O2: Reversibility and Secondary Formation of HO2. Phys. Chem. Chem. Phys. 1999, 1, 5097–5107. 10.1039/a904887a. [DOI] [Google Scholar]
- DeMatteo M. P.; Poole J. S.; Shi X. F.; Sachdeva R.; Hatcher P. G.; Hadad C. M.; Platz M. S. On the Electrophilicity of Hydroxyl Radical: A Laser Flash Photolysis and Computational Study. J. Am. Chem. Soc. 2005, 127, 7094–7109. 10.1021/ja043692q. [DOI] [PubMed] [Google Scholar]
- Fittschen C.; Al Ajami M.; Batut S.; Ferracci V.; Archer-Nicholls S.; Archibald A. T.; Schoemaecker C. ROOOH: A Missing Piece of the Puzzle for OH Measurements in Low-No Environments?. Atmos. Chem. Phys. 2019, 19, 349–362. 10.5194/acp-19-349-2019. [DOI] [Google Scholar]
- Stary F. E.; Emge D. E.; Murray R. W. Ozonization of Organic Substrates. Hydrotrioxide Formation and Decomposition to Give Singlet Oxygen. J. Am. Chem. Soc. 1976, 98, 1880–1884. 10.1021/ja00423a039. [DOI] [Google Scholar]
- Berndt T.; Chen J.; Kjærgaard E. R.; Møller K. H.; Tilgner A.; Hoffmann E. H.; Herrmann H.; Crounse J. D.; Wennberg P. O.; Kjaergaard H. G. Hydrotrioxide (ROOOH) Formation in the Atmosphere. Science (New York, N.Y.) 2022, 376, 979–982. 10.1126/science.abn6012. [DOI] [PubMed] [Google Scholar]
- Akimoto H.; Hirokawa J.. Historical Background of Atmospheric Secondary Aerosol Research. In Atmospheric Multiphase Chemistry; Wiley: 2020; pp 1–11. [Google Scholar]
- Roeselova M.; Vieceli J.; Dang L. X.; Garrett B. C.; Tobias D. J. Hydroxyl Radical at the Air-Water Interface. J. Am. Chem. Soc. 2004, 126, 16308–16309. 10.1021/ja045552m. [DOI] [PubMed] [Google Scholar]
- Bagot P. A. J.; Waring C.; Costen M. L.; McKendrick K. G. Dynamics of Inelastic Scattering of OH Radicals from Reactive and Inert Liquid Surfaces. J. Phys. Chem. C 2008, 112, 10868–10877. 10.1021/jp8024683. [DOI] [Google Scholar]
- Yang L. M.; Ray M. B.; Yu L. E. Photooxidation of Dicarboxylic Acids- Part II: Kinetics, Intermediates and Field Observations. Atmos. Environ. 2008, 42, 868–880. 10.1016/j.atmosenv.2007.10.030. [DOI] [Google Scholar]
- Yang L. M.; Ray M. B.; Yu L. E. Photooxidation of Dicarboxylic Acids- Part 1: Effects of Inorganic Ions on Degradation of Azelaic Acid. Atmos. Environ. 2008, 42, 856–867. 10.1016/j.atmosenv.2007.10.029. [DOI] [Google Scholar]
- Charbouillot T.; Gorini S.; Voyard G.; Parazols M.; Brigante M.; Deguillaume L.; Delort A. M.; Mailhot G. Mechanism of Carboxylic Acid Photooxidation in Atmospheric Aqueous Phase: Formation, Fate and Reactivity. Atmos. Environ. 2012, 56, 1–8. 10.1016/j.atmosenv.2012.03.079. [DOI] [Google Scholar]
- Valley N. A.; Blower P. G.; Wood S. R.; Plath K. L.; McWilliams L. E.; Richmond G. L. Doubling Down: Delving into the Details of Diacid Adsorption at Aqueous Surfaces. J. Phys. Chem. A 2014, 118, 4778–4789. 10.1021/jp501498h. [DOI] [PubMed] [Google Scholar]
- Rossignol S.; Tinel L.; Bianco A.; Passananti M.; Brigante M.; Donaldson D. J.; George C. Atmospheric Photochemistry at a Fatty Acid-Coated Air-Water Interface. Science (New York, N.Y.) 2016, 353, 699–702. 10.1126/science.aaf3617. [DOI] [PubMed] [Google Scholar]
- Ambrose D.; Ghiassee N. B. Vapour Pressures and Critical Temperatures and Critical Pressures of Some Alkanoic Acids: C1 to C10. J. Chem. Thermodyn. 1987, 19, 505–519. 10.1016/0021-9614(87)90147-9. [DOI] [Google Scholar]
- Chhantyal-Pun R.; Khan M. A. H.; Taatjes C. A.; Percival C. J.; Orr-Ewing A. J.; Shallcross D. E. Criegee Intermediates: Production, Detection and Reactivity. Int. Rev. Phys. Chem. 2020, 39, 385–422. 10.1080/0144235X.2020.1792104. [DOI] [Google Scholar]
- Zhong J.; Kumar M.; Zhu C. Q.; Francisco J. S.; Zeng X. C. Surprising Stability of Larger Criegee Intermediates on Aqueous Interfaces. Angew. Chem. Int. Edt. 2017, 56, 7740–7744. 10.1002/anie.201702722. [DOI] [PubMed] [Google Scholar]
- Zhu C. Q.; Kumar M.; Zhong J.; Li L.; Francisco J. S.; Zeng X. C. New Mechanistic Pathways for Criegee-Water Chemistry at the Air/Water Interface. J. Am. Chem. Soc. 2016, 138, 11164–11169. 10.1021/jacs.6b04338. [DOI] [PubMed] [Google Scholar]
- Xiao P.; Yang J.-J.; Fang W.-H.; Cui G. QM/MM Studies on Ozonolysis of α-Humulene and Criegee Reactions with Acids and Water at Air-Water/Acetonitrile Interfaces. Phys. Chem. Chem. Phys. 2018, 20, 16138–16150. 10.1039/C8CP01750F. [DOI] [PubMed] [Google Scholar]
- Enami S. Fates of Organic Hydroperoxides in Atmospheric Condensed Phases. J. Phys. Chem. A 2021, 125, 4513–4523. 10.1021/acs.jpca.1c01513. [DOI] [PubMed] [Google Scholar]
- Qiu J.; Ishizuka S.; Tonokura K.; Colussi A. J.; Enami S. Reactivity of Monoterpene Criegee Intermediates at Gas-Liquid Interfaces. J. Phys. Chem. A 2018, 122, 7910–7917. 10.1021/acs.jpca.8b06914. [DOI] [PubMed] [Google Scholar]
- Enami S.Interfacial Criegee Chemistry. In Multiphase Environmental Chemistry in the Atmosphere; American Chemical Society: 2018; Vol. 1299, pp 35–47. [Google Scholar]
- Enami S.; Colussi A. J. Criegee Chemistry on Aqueous Organic Surfaces. J. Phys. Chem. Lett. 2017, 8, 1615–1623. 10.1021/acs.jpclett.7b00434. [DOI] [PubMed] [Google Scholar]
- Enami S.; Colussi A. J. Reactions of Criegee Intermediates with Alcohols at Air-Aqueous Interfaces. J. Phys. Chem. A 2017, 121, 5175–5182. 10.1021/acs.jpca.7b04272. [DOI] [PubMed] [Google Scholar]
- Jr-Min Lin J.; Chao W. Structure-Dependent Reactivity of Criegee Intermediates Studied with Spectroscopic Methods. Chem. Soc. Rev. 2017, 46, 7483–7497. 10.1039/C7CS00336F. [DOI] [PubMed] [Google Scholar]
- Enami S.; Colussi A. J. Efficient Scavenging of Criegee Intermediates on Water by Surface-Active cis-Pinonic Acid. Phys. Chem. Chem. Phys. 2017, 19, 17044–17051. 10.1039/C7CP03869K. [DOI] [PubMed] [Google Scholar]
- Li X.; Hede T.; Tu T.; Leck C.; Agren H. Surface-Active cis-Pinonic Acid in Atmospheric Droplets: A Molecular Dynamics Study. J. Phys. Chem. Lett. 2010, 1, 769–773. 10.1021/jz9004784. [DOI] [Google Scholar]
- Qiu J.; Ishizuka S.; Tonokura K.; Sato K.; Inomata S.; Enami S. Effects of pH on Interfacial Ozonolysis of α-Terpineol. J. Phys. Chem. A 2019, 123, 7148–7155. 10.1021/acs.jpca.9b05434. [DOI] [PubMed] [Google Scholar]
- Qiu J.; Liang Z.; Tonokura K.; Colussi A. J.; Enami S. Stability of Monoterpene-Derived α-Hydroxyalkyl-Hydroperoxides in Aqueous Organic Media: Relevance to the Fate of Hydroperoxides in Aerosol Particle Phases. Environ. Sci. Technol. 2020, 54, 3890–3899. 10.1021/acs.est.9b07497. [DOI] [PubMed] [Google Scholar]
- Qiu J.; Tonokura K.; Enami S. Proton-Catalyzed Decomposition of α-Hydroxyalkyl-Hydroperoxides in Water. Environ. Sci. Technol. 2020, 54, 10561–10569. 10.1021/acs.est.0c03438. [DOI] [PubMed] [Google Scholar]
- Enami S.; Hoffmann M. R.; Colussi A. J. Criegee Intermediates React with Levoglucosan on Water. J. Phys. Chem. Lett. 2017, 8, 3888–3894. 10.1021/acs.jpclett.7b01665. [DOI] [PubMed] [Google Scholar]
- Tobias H. J.; Ziemann P. J. Kinetics of the Gas-Phase Reactions of Alcohols, Aldehydes, Carboxylic Acids, and Water with the C13 Stabilized Criegee Intermediate Formed from Ozonolysis of 1-Tetradecene. J. Phys. Chem. A 2001, 105, 6129–6135. 10.1021/jp004631r. [DOI] [Google Scholar]
- Rocha I. M.; Galvão T. L. P.; Sapei E.; Ribeiro da Silva M. D. M. C.; Ribeiro da Silva M. A. V. Levoglucosan: A Calorimetric, Thermodynamic, Spectroscopic, and Computational Investigation. J. Chem. Engineer. Data 2013, 58, 1813–1821. 10.1021/je400207t. [DOI] [Google Scholar]
- Hu M.; Qiu J.; Tonokura K.; Enami S. Aqueous-Phase Fates of α-Alkoxyalkyl-Hydroperoxides Derived from the Reactions of Criegee Intermediates with Alcohols. Phys. Chem. Chem. Phys. 2021, 23, 4605–4614. 10.1039/D0CP06308H. [DOI] [PubMed] [Google Scholar]
- Endo Y.; Sakamoto Y.; Kajii Y.; Enami S. Decomposition of Multifunctionalized α-Alkoxyalkyl-Hydroperoxides Derived from the Reactions of Criegee Intermediates with Diols in Liquid Phases. Phys. Chem. Chem. Phys. 2022, 24, 11562–11572. 10.1039/D2CP00915C. [DOI] [PubMed] [Google Scholar]
- Enami S.; Morino Y.; Sato K. Mechanism of Fenton Oxidation of Levoglucosan in Water. J. Phys. Chem. A 2023, 127, 2975–2985. 10.1021/acs.jpca.3c00512. [DOI] [PubMed] [Google Scholar]
- Gupta V.; Carroll K. S. Sulfenic Acid Chemistry, Detection and Cellular Lifetime. Biochim. Biophys. Acta 2014, 1840, 847–875. 10.1016/j.bbagen.2013.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddie K. G.; Carroll K. S. Expanding the Functional Diversity of Proteins through Cysteine Oxidation. Curr. Opin. Chem. Biol. 2008, 12, 746–754. 10.1016/j.cbpa.2008.07.028. [DOI] [PubMed] [Google Scholar]
- Helin A.; Sietiö O. M.; Heinonsalo J.; Bäck J.; Riekkola M. L.; Parshintsev J. Characterization of Free Amino Acids, Bacteria and Fungi in Size-Segregated Atmospheric Aerosols in Boreal Forest: Seasonal Patterns, Abundances and Size Distributions. Atmos. Chem. Phys. 2017, 17, 13089–13101. 10.5194/acp-17-13089-2017. [DOI] [Google Scholar]
- Jaenicke R. Abundance of Cellular Material and Proteins in the Atmosphere. Science (New York, N.Y.) 2005, 308, 73–73. 10.1126/science.1106335. [DOI] [PubMed] [Google Scholar]
- Cantin A. M.; North S. L.; Hubbard R. C.; Crystal R. G. Normal Alveolar Epithelial Lining Fluid Contains High Levels of Glutathione. J. Appl. Physiol. 1987, 63, 152–7. 10.1152/jappl.1987.63.1.152. [DOI] [PubMed] [Google Scholar]
- Enami S.; Hoffmann M. R.; Colussi A. J. Simultaneous Detection of Cysteine Sulfenate, Sulfinate, and Sulfonate During Cysteine Interfacial Ozonolysis. J. Phys. Chem. B 2009, 113, 9356–9358. 10.1021/jp904316n. [DOI] [PubMed] [Google Scholar]
- Enami S.; Hoffmann M. R.; Colussi A. J. Ozone Oxidizes Glutathione to a Sulfonic Acid. Chem. Res. Toxicol. 2009, 22, 35–40. 10.1021/tx800298j. [DOI] [PubMed] [Google Scholar]
- Kim H. I.; Kim H. J.; Shin Y. S.; Beegle L. W.; Jang S. S.; Neidholdt E. L.; Goddard W. A.; Heath J. R.; Kanik I.; Beauchamp J. L. Interfacial Reactions of Ozone with Surfactant Protein B in a Model Lung Surfactant System. J. Am. Chem. Soc. 2010, 132, 2254–2263. 10.1021/ja908477w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giamalva D.; Church D. F.; Pryor W. A. A Comparison of the Rates of Ozonation of Biological Antioxidants and Oleate and Linoleate Esters. Biochem. Biophys. Res. Commun. 1985, 133, 773–779. 10.1016/0006-291X(85)90971-4. [DOI] [PubMed] [Google Scholar]
- Zannoni N.; Lakey P. S. J.; Won Y.; Shiraiwa M.; Rim D.; Weschler C. J.; Wang N.; Ernle L.; Li M.; Bekö G.; et al. The Human Oxidation Field. Science (New York, N.Y.) 2022, 377, 1071–1077. 10.1126/science.abn0340. [DOI] [PubMed] [Google Scholar]
- Wang S.; Zhao Y.; Chan A. W. H.; Yao M.; Chen Z.; Abbatt J. P. D. Organic Peroxides in Aerosol: Key Reactive Intermediates for Multiphase Processes in the Atmosphere. Chem. Rev. 2023, 123, 1635–1679. 10.1021/acs.chemrev.2c00430. [DOI] [PubMed] [Google Scholar]
- Rovelli G.; Jacobs M. I.; Willis M. D.; Rapf R. J.; Prophet A. M.; Wilson K. R. A Critical Analysis of Electrospray Techniques for the Determination of Accelerated Rates and Mechanisms of Chemical Reactions in Droplets. Chem. Sci. 2020, 11, 13026–13043. 10.1039/D0SC04611F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito S.; Numadate N.; Teraoka H.; Enami S.; Kobayashi H.; Hama T. Impurity Contribution to Ultraviolet Absorption of Saturated Fatty Acids. Sci. Adv. 2023, 9, eadj6438 10.1126/sciadv.adj6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto Y.; Yabushita A.; Kawasaki M.; Enami S. Direct Emission of I2 Molecule and IO Radical from the Heterogeneous Reactions of Gaseous Ozone with Aqueous Potassium Iodide Solution. J. Phys. Chem. A 2009, 113, 7707–7713. 10.1021/jp903486u. [DOI] [PubMed] [Google Scholar]
- Vieceli J.; Roeselova M.; Potter N.; Dang L. X.; Garrett B. C.; Tobias D. J. Molecular Dynamics Simulations of Atmospheric Oxidants at the Air-Water Interface: Solvation and Accommodation of OH and O3. J. Phys. Chem. B 2005, 109, 15876. 10.1021/jp051361+. [DOI] [PubMed] [Google Scholar]
- Chang Y.-P.; Wu S.-J.; Lin M.-S.; Chiang C.-Y.; Huang G. G. Ionic-Strength and pH Dependent Reactivities of Ascorbic Acid toward Ozone in Aqueous Micro-Droplets Studied Using Aerosol Optical Tweezers. Phys. Chem. Chem. Phys. 2021, 23, 10108–10117. 10.1039/D0CP06493A. [DOI] [PubMed] [Google Scholar]
- Anglada J. M.; Martins-Costa M.; Ruiz-López M. F.; Francisco J. S. Spectroscopic Signatures of Ozone at the Air-Water Interface and Photochemistry Implications. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 11618–11623. 10.1073/pnas.1411727111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artiglia L.; Edebeli J.; Orlando F.; Chen S. Z.; Lee M. T.; Arroyo P. C.; Gilgen A.; Bartels-Rausch T.; Kleibert A.; Vazdar M. A Surface-Stabilized Ozonide Triggers Bromide Oxidation at the Aqueous Solution-Vapour Interface. Nat. Commun. 2017, 10.1038/s41467-017-00823-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taatjes C. A.; Meloni G.; Selby T. M.; Trevitt A. J.; Osborn D. L.; Percival C. J.; Shallcross D. E. Direct Observation of the Gas-Phase Criegee Intermediate (CH2OO). J. Am. Chem. Soc. 2008, 130, 11883–11885. 10.1021/ja804165q. [DOI] [PubMed] [Google Scholar]
- Weschler C. J.; Nazaroff W. W. Human Skin Oil: A Major Ozone Reactant Indoors. Environ. Sci. Atmos. 2023, 3, 640–661. 10.1039/D3EA00008G. [DOI] [Google Scholar]
- Zhou Z.; Abbatt J. P. D. Formation of Gas-Phase Hydrogen Peroxide via Multiphase Ozonolysis of Unsaturated Lipids. Environ. Sci. Technol. Lett. 2021, 8, 114–120. 10.1021/acs.estlett.0c00757. [DOI] [Google Scholar]
- Heine N.; Houle F. A.; Wilson K. R. Connecting the Elementary Reaction Pathways of Criegee Intermediates to the Chemical Erosion of Squalene Interfaces During Ozonolysis. Environ. Sci. Technol. 2017, 51, 13740–13748. 10.1021/acs.est.7b04197. [DOI] [PubMed] [Google Scholar]
- Zhang R.; Wu W. Studies on the Structures and Interactions of Glutathione in Aqueous Solution by Molecular Dynamics Simulations and Nmr Spectroscopy. J. Mol. Liq. 2011, 162, 20–25. 10.1016/j.molliq.2011.05.010. [DOI] [Google Scholar]
- Lampela O.; Juffer A. H.; Rauk A. Conformational Analysis of Glutathione in Aqueous Solution with Molecular Dynamics. J. Phys. Chem. A 2003, 107, 9208–9220. 10.1021/jp030556j. [DOI] [Google Scholar]
- George C.; Brüggemann M.; Hayeck N.; Tinel L.; Donaldson J.. Chapter 14 - Interfacial Photochemistry. In Physical Chemistry of Gas-Liquid Interfaces; Faust J. A., House J. E., Eds.; Elsevier: 2018; pp 435–457. [Google Scholar]
- Bertram T. H.; Cochran R. E.; Grassian V. H.; Stone E. A. Sea Spray Aerosol Chemical Composition: Elemental and Molecular Mimics for Laboratory Studies of Heterogeneous and Multiphase Reactions. Chem. Soc. Rev. 2018, 47, 2374–2400. 10.1039/C7CS00008A. [DOI] [PubMed] [Google Scholar]
- Burkholder J. B.; Abbatt J. P. D.; Cappa C.; Dibble T. S.; Kolb C. E.; Orkin V. L.; Wilmouth D. M.; Sander S. P.; Baker J. R.; Crounse J. D.; et al. Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies; JPL Publication, 2020; Vol. 19–5. [Google Scholar]
- Keller-Rudek H.; Moortgat G. K.; Sander R.; Sörensen R. The MPI-Mainz UV/Vis Spectral Atlas of Gaseous Molecules of Atmospheric Interest. Earth Syst. Sci. Data 2013, 5, 365–373. 10.5194/essd-5-365-2013. [DOI] [Google Scholar]
- Atkinson R.; Baulch D. L.; Cox R. A.; Crowley J. N.; Hampson R. F.; Hynes R. G.; Jenkin M. E.; Rossi M. J.; Troe J. Evaluated Kinetic and Photochemical Data for Atmospheric Chemistry: Volume I - Gas Phase Reactions of Ox, Hox, NOx and SOx Species. Atmos. Chem. Phys. 2004, 4, 1461–1738. 10.5194/acp-4-1461-2004. [DOI] [Google Scholar]
- Rapf R. J.; Perkins R. J.; Dooley M. R.; Kroll J. A.; Carpenter B. K.; Vaida V. Environmental Processing of Lipids Driven by Aqueous Photochemistry of α-Keto Acids. ACS Cent. Sci. 2018, 4, 624–630. 10.1021/acscentsci.8b00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura E.; Hasegawa T.; Umemura J. Quantitative Analysis of Molecular Orientation in Chlorophyll a Langmuir Monolayer: A Polarized Visible Reflection Spectroscopic Study. Biophys. J. 1995, 69, 1142–1147. 10.1016/S0006-3495(95)79988-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya Y.; Hasegawa T.; Okada T.; Ogawa N.; Kawai E.; Abe K.; Ogasawara M.; Kato S.; Nakata S. Analysis of Gibbs Monolayer Adsorbed at the Toluene/Water Interface by UV- Visible Partial Internal Reflection Spectrometry. Anal. Chem. 2006, 78, 7850–7856. 10.1021/ac061501r. [DOI] [PubMed] [Google Scholar]
- Yamaguchi S.; Tahara T. Development of Electronic Sum Frequency Generation Spectroscopies and Their Application to Liquid Interfaces. J. Phys. Chem. C 2015, 119, 14815–14828. 10.1021/acs.jpcc.5b02375. [DOI] [Google Scholar]