

Abstract
Computational screening for potentially bioactive molecules using advanced molecular modeling approaches including molecular docking and molecular dynamic simulation is mainstream in certain fields like drug discovery. Significant advances in computationally predicting protein structures from sequence information have also expanded the availability of structures for non-model species. Therefore, the objective of this work was to develop an analysis pipeline to harness the power of these bioinformatics approaches for cross-species extrapolation for evaluating chemical safety. The Sequence Alignment to Predict Across Species Susceptibility (SeqAPASS) tool compares protein-sequence similarity across species for conservation of known chemical targets, providing an initial line of evidence for extrapolation of toxicity knowledge. However, with the development of structural models from tools like the Iterative Threading ASSEmbly Refinement (ITASSER), analyses of protein structural conservation can be included to add additional lines of evidence and generate protein models across species. Models generated through such a pipeline could then be used for advanced molecular modeling approaches in the context of species extrapolation. Two case examples illustrating this pipeline from SeqAPASS sequences to ITASSER generated protein structures were created for human liver fatty acid binding protein (LFABP) and androgen receptor (AR). Ninety-nine LFABP and 268 AR protein models representing diverse species, were generated and analyzed for conservation using TM-align. The results from the structural comparisons were in line with the sequence-based SeqAPASS workflow adding further evidence of LFABL and AR conservation across vertebrate species. This analysis lays the foundation for expanding the capabilities of the web-based SeqAPASS tool to include structural comparisons for species extrapolation, facilitating more rapid and efficient toxicological assessments among species with limited or no existing toxicity data.
INTRODUCTION
The global regulatory landscape regarding chemical safety is rapidly realizing the need to shift away from animal testing and instead capitalize on existing empirical data and advances in computational and high-throughput (e.g., cell-based; transcriptomic) approaches to fulfill data needs for decision-makers (Daston et al., 2022). As efforts are underway to reduce, refine, and replace animal tests and new chemicals continue to be created, it is anticipated that over time there will be less whole organism toxicity data generated to use for chemical hazard and risk assessments. Therefore, it is critical that scientifically grounded emerging technologies in the areas of predictive, comparative, and systems toxicology are poised to fill the knowledge gaps. Further, case examples must convincingly demonstrate the utility of these approaches, defining both strengths and weaknesses, with well-characterized domains of applicability for regulatory bodies to transition to their use in decision-making with confidence.
The challenge to fill knowledge gaps in chemical safety evaluation when animal testing is impractical is exacerbated by the need to consider not only protection of human health, but also the environment. As in the past, surrogate organisms are selected or in use for cell- or transcriptomic-based assays and in the development of computational models, such as physiologically-based toxicokinetic models, quantitative structure activity relationship models, and high-throughput toxicokinetic methods (Armitage et al., 2021; Dawson et al., 2021; Harrill et al., 2020; Kavlock et al., 2012; Thiel et al., 2015). Just as with historic whole animal studies, as new approach methods (i.e., approaches that are designed to reduce or eliminate animal use (van derZalm et al., 2022)) are developed and employed, there are needs to understand their effectiveness in protecting the health of all species in the environment. It has been demonstrated that understanding of biological pathway conservation, and even more specifically, gene or protein conservation, can aid in the extrapolation of toxicity data and knowledge across species at the molecular and cellular level (e.g., Jensen et al., 2022; Gunnarsson et al., 2008; LaLone et al., 2016; Verbruggen et al., 2018). Taking knowledge from one (or a handful of) species and using the information to predict or infer the effect in another species for which there is no empirical data is termed cross-species extrapolation. Methods developed to inform species extrapolation have more recently relied on advances in bioinformatics and concepts in evolutionary biology to make significant developments in this important area of toxicology (LaLone et al., 2021; Rivetti et al., 2020; van den Berg et al., 2021; Colbourne et al. 2022). These advances contribute to address the complex challenge of predicting chemical susceptibility across species, which ultimately will need to consider factors beyond pathway conservation including stressor exposure, organism life stage, life history, etc. With the recognition that conservation is not the only consideration in species sensitivity, such assessments of similarities at the molecular level can be applied with other existing pathway knowledge to enhance species extrapolation.
The field of bioinformatics continues to advance rapidly, where sequence comparisons, homology modeling, molecular docking, molecular dynamics simulations, ligand- and structure-based virtual screening have become common-place in computational studies evaluating and predicting protein-ligand interactions (Salo-Ahen et al., 2020). These strategies are employed, in a large part, by researchers and practitioners in the biomedical, pharmacological, and medicinal fields to identify drug candidates and other treatments for human disease (Adelusi et al., 2022). Environmental toxicologists have come to recognize that such advances could prove advantageous for chemical safety evaluations relative to human and environmental health for identification of candidate chemicals that have the potential to lead to adverse outcomes for certain species, taxa, or populations (Thomas et al., 2017).
These advances in computational methods allow for the strategic development of a semi-automated pipeline for the comparison of protein structural conservation across species that can expand the predictive capabilities of existing sequence-based extrapolation tools, specifically, the U.S. Environmental Protection Agency Sequence Alignment to Predict Across Species Susceptibility tool (SeqAPASS; seqapass.epa.gov/seqapass/; (LaLone et al., 2016) ). The SeqAPASS tool was developed for both expert and non-expert users to rapidly compare protein sequences across species to predict chemical susceptibility and understand pathway conservation. The approach takes advantage of the large and continuously growing databases of protein sequences (e.g., NCBI, UniProt), to capitalize on what is known about a protein-ligand, chemical-protein, or protein-protein interaction for one species to predict the potential interaction in hundreds to thousands of species, most of which would never (or could never) be used as toxicity test organisms.
The SeqAPASS methodology continues to evolve as the field of bioinformatics advances and the capabilities to automate complex comparative protein evaluations are realized. Protein sequence comparisons are commonly used to understand differences in the protein target due to point mutations, substitutions, insertions, or gaps in sequences across species and to identify orthologs, or sequences that have diverged from a speciation event but maintained similar function. Such information can indicate divergence or similarity in protein function and has been useful for understanding species differences at the molecular level. However, molecular modeling allows for investigation of the geometry of the protein in 3-dimensions. Such understanding can allow for evaluation of likelihood for physical and chemical interactions with the protein structure, which could lead to more informed understanding of chemical interactions across species. Therefore, with the adage in protein biology that structure determines function there has been a desire to take sequence-based predictions of chemical susceptibility from SeqAPASS and move into structural comparisons to add lines of evidence toward protein conservation to ultimately improve cross species predictive capabilities for decision-makers.
Since 1994 the Protein Structure Prediction Center sponsored by the US National Institute of General Medical Sciences (NIH/NIGMS) has organized the Critical Assessment of Protein Structure Prediction (CASP) experiments which has been aimed at defining the state of the science in protein structural prediction and identifying knowledge gaps for focused research efforts (predictioncenter.org/). From 2006 to 2020, the Iterative Threading ASSEmbly Refinement (I-TASSER) algorithm for automated structural prediction has been consistently ranked as one of the top methods. The I-TASSER pipeline includes threading-based fold recognition, fragment-based structure assembly and refinement to generate accurate 3D protein structure predictions and functional annotation (Roy et al., 2010). This computationally powerful method for developing protein structural models based on protein sequence was selected as the open source, publicly accessible platform to integrate with the SeqAPASS pipeline to develop structural models across species for the purpose of predicting chemical susceptibly to inform research and regulatory decision-making. Notably, implementation of artificial intelligence in protein structural predictions was realized recently in 2022 with the announcement by AlphaFold, providing profound expansion in available protein structural models to represent the diversity of the species. Therefore, available quality structures generated from multiple resources will continue to enhance the ability to compare across species and will be explored as the current work described herein progresses.
The objectives of this work are to expand the SeqAPASS evaluation to generate protein structures across species that can be used for making predictions of chemical susceptibility based on structural conservation and to develop a semi-automated pipeline for this approach that takes advantage of existing and open-source tools and databases, including I-TASSER and TM-align (Figure 1) (Roy et al., 2010; Zhang & Skolnick, 2005). Case examples were expanded from previously published work to demonstrate the utility of these approaches in the context of understanding conservation of chemical molecular protein targets. Specifically, previously published work focused on cross species protein structural comparisons to human liver fatty acid binding protein and androgen receptor, demonstrating how the SeqAPASS sequence-based predictions could be used to consider structural conservation through generating structures for use in more advanced molecular modeling techniques. These published case examples addressed challenges in cross species extrapolation relative to per- and poly-fluoro alkylated substances (PFAS) bioaccumulation relative to liver fatty acid binding protein (LFABP) and extrapolation of high-throughput screening assay results using human androgen receptor (AR). These protein targets were also selected to represent differing functions and amino acid sequence lengths, with LFABP being a transport protein with a relatively short sequence of 127 amino acids and AR being a well-studied nuclear receptor with a longer sequence of 914 amino acids and a well resolved ligand binding domain (LBD) important for chemical interactions. It is anticipated that this work in concert with previously published results will lead to further exploration of chemical-protein interactions with a broader representation of species.
Figure 1.

Diagram showing the pipeline of data flowing from Sequence Alignment to Predict Across Species Susceptibility (SeqAPASS) protein sequences to the generation of protein structure models across species for use in advanced molecular in silico approaches and structural comparisons in the context of species extrapolation. The process described here is envisioned to produce Level 4 of the SeqAPASS tool evaluation and will be developed as components of the web-based tool in upcoming public version releases. The SeqAPASS query initiates the pipeline using a query species protein and generates hit sequences, representing the diversity of species. Hit sequences from SeqAPASS Level 1 output are then prioritized using PERL script identifying sequences more likely to generate quality structures. The list of priority sequences is then passed to Iterative Threading ASSEmbly Refinement (I-TASSER) to generate structural models and output describing the quality of the models. Protein structural models are then compared between the query species and each hit species individually generating metrics for the alignment of structures. The structural conservation can then be used as another line of evidence toward conservation in the SeqAPASS analysis. The 3D protein structure models generated for several species representing diverse taxa can then be refined and used in a variety of computational approaches including molecular docking, as an example. Acronym: PDB – Protein Data Bank (i.e., files with x, y, z coordinates for 3D visualization of the proteins).
MATERIALS AND METHODS
Sequence-Based Predictions of Protein Conservation
The US EPA Sequence Alignment to Predict Across Species Susceptibility (SeqAPASS v5.0) tool was used to evaluate sequence similarity to predict conservation and chemical susceptibility for two case examples. There are three levels available to construct a SeqAPASS query dependent on how much information exists regarding a chemical-protein interaction. For creating the structural models in the developed pipeline, only results from Level 1, primary amino acid comparisons, are necessary. However, since SeqAPASS data for the LFABP and AR case studies have been evaluated previously (Cheng et al., 2021 and Vliet et al., 2022), other SeqAPASS data from Level 2, functional domain comparisons, and Level 3, critical individual amino acid comparisons, were generated as previously described.
Two query protein accessions were selected as query sequences to submit to the SeqAPASS tool as appropriate species for each case example. Human LFABP (NP_001434.1) and AR (AAI32976.1) were submitted to SeqAPASS Level 1. Level 2 comparisons were completed for the AR LBD (cd_07073). A Level 2 comparison was not submitted for LFABP as the only specific hit domain identified was nearly the full length of the primary amino acid sequence. In these instances, the Level 2 results would not yield any additional taxonomic resolution in the SeqAPASS analyses. Level 3 individual amino acid residues were evaluated for both proteins according to those deemed critical from literature review and those published previously (Table 1). Output from SeqAPASS was collected. (Supplemental Data, S1).
Table 1.
SeqAPASS query information for the evaluation of human liver fatty acid binding protein and androgen receptor
| Level 1 | Level 2 | Level 3 | ||||
|---|---|---|---|---|---|---|
| Query Protein | Accession | Query Domain | Accession | Template Sequence | Amino acids | References |
| Human fatty acid-binding protein, liver | NP_001434.1 | Not Applicable | Not Applicable | Human NP_00143.1 | Phenylalanine (F)50 | Cheng et al., 2021 |
| Human androgen receptor | AAI32976.1 | Ligand binding domain of the nuclear androgen receptor, ligand activated transcription regulator | cd07073 | Human AAI32976 | Asparagine (N)700 Glutamine (Q)706 Arginine (R)747, Threonine (T)872 |
Vliet et al., 2022 |
Accessions are National Center for Biotechnology Information (NCBI) protein identifiers. The SeqAPASS Level 1 evaluation compares primary amino acid sequences, Level 2 compares the functional domain identified as a specific hit in NCBI Common Domains Database, and Level 3 compares critical (close contact) amino acids across species.
The FASTA formatted sequences (i.e., text-based sequence format that begins with a single-line description, followed by lines of sequence data) for each protein accession from NCBI were collected based on the SeqAPASS Level 1 Primary Report output generating a list for the query and each hit protein. The protein FASTA sequences were then prioritized for creating structures in I-TASSER. For each taxonomic group, high priority sequences were identified by removing predicted, hypothetical, partial, low quality, unnamed accessions, or dissimilar annotation to the query protein (based on the protein name). Such sequence alignments are typical to occur as many species have limited annotation, due to a lack of homology to other proteins with known functional annotation. Therefore, in developing structural models for comparative purposes it is important to understand if the protein sequence is indeed more or less likely to be similar to the query protein. Similarity of annotations are a consideration in this determination. However, if the taxonomic group only had predicted hypothetical, partial, low quality, or unnamed proteins, these would be labeled as low priority but kept and tagged to include those sequences with greatest sequence similarity to represent each taxonomic group.
Modeling Protein Structures Across Species
The I-TASSER standalone program v5.1 (https://zhanggroup.org/I-TASSER/) was used to generate protein structural models by submitting the aligned FASTA sequences from the SeqAPASS output from the respective proteins, LFABP and AR, and setting a restraint (i.e., a specific structure suggested by the user as a template to be considered in the threading process) using the identified Protein Data Bank structure (RSCB PDB; https://www.rcsb.org/).
Specifically, the PDB, which archives available protein structural data, was queried to identify any available crystal structures that would be suitable for representing the query proteins from the SeqAPASS analysis, as well as assist in identifying ligand binding coordinates on the proteins of interest. Specifically, PDB IDs 3STM for LFABP and 2AMA for AR were the structures identified and used as the restraints for the respective I-TASSER queries. Priority protein sequences from SeqAPASS output were then submitted as FASTA to I-TASSER with PDB restraints to generate protein structural models and collect metrics relative to the quality of the protein structure generated.
Tables were automatically generated using a custom PERL script which imports a list of proteins and results from SeqAPASS Level 1 report, assigns priorities and submits the selected proteins to I-TASSER (See Figure 1; Supplemental Data, S2). The script then captures metrics associated with each protein structure model from I-TASSER output, as described below. Outputs included confidence score (C-score) for estimating the quality of predicted models based on the significance of threading template alignments and the convergence parameters of the structure assembly simulations (Roy et al., 2010). The C-score is commonly between −5 and 2, where the greater the value the higher the confidence in the model. TM-score is a metric for assessing the topological similarity of protein structures and has a value between 0 and 1, where 1 indicates a perfect match between two structures. A TM-score > 0.5 indicates a model of correct topology whereas a TM-score < 0.17 indicates a random similarity (Roy et al., 2010). The Root Mean Square Deviation (RMSD) of atomic positions is the measure of the average distance between the atoms of superimposed proteins. Therefore, the lower RMSD, the closer the model is to the target structure. The number of protein structure decoys, which are the artificial structural conformations of proteins used to guide the design, testing, and training of the protein folding force fields, are reported for each model. The cluster density is also reported and used to define the number of structure decoys at a unit of space in the cluster. A higher cluster density means the structure occurs more often in the simulation trajectory and is therefore likely a higher-quality model (Roy et al., 2010). Models were generated and saved as PDB files for further analysis.
Aligning protein structural models across species
The human protein structural models for both LFABP and AR, were then compared using TM-align (https://zhanggroup.org/TM-align/) with the respective structural models generated from I-TASSER representing diverse species. TM-align compares two protein structures generating optimized amino acid residue alignment based on structural similarity using heuristic dynamic programming iterations. Output from TM-align includes optimal superpositions of the two compared structures as PDB files and the TM-score for each structural alignment. TM-scores below 0.2 correspond to randomly chosen unrelated proteins while those higher than 0.5 assume generally the same fold. Because the AR models generated by I-TASSER represented the full protein sequences and provide relatively low average TM-align Scores, a few species were selected to modify and focus solely on the LBD (as is represented by the existing human crystal structure, 2AMA). TM-align was executed for the human model and the LBD representing the different species to demonstrate that as models are refined to specific functional regions they align more closely and are therefore likely to be more useful for more advanced modeling approaches.
Protein structure conservation
To determine whether the models generated for LFABP and AR could be used as an additional line of evidence toward protein conservation, results from ITASSER were evaluated to ensure they met the quality criteria for C-score, TM-score, and RMSD as described above. In addition, to account for differences in sequence lengths used to generate the structures, the absolute value of amino acid length difference, (i.e., human - comparison species), was determined and expressed as percent. A density plot was generated displaying the absolute value of length difference versus the human (query species) length as a percent. On the density plot, the first local minimum greater than the global maximum was identified and used as the length cutoff to determine those more likely or less likely to be similar to the query species. For LFABP and AR the cut-offs were within 10% and 42% of the human sequence length, respectively. This evaluation provided a means to isolate species with lengths that differ from human and flag those species to check prior to moving forward with advance molecular modeling approaches. All high-quality protein models were those that maintained similar structure to the human and therefore could be used as another line of evidence for conservation of the protein in that species. Low quality structures did not meet I-TASSER scoring requirements. These results do not indicate that the protein is not conserved in that species, but that the sequence was not able to yield a quality structure. At the protein structure level, the only evidence gained is for conservation, not lack thereof, because the prioritization process eliminated poor quality/differently annotated sequences for generating structures.
RESULTS
SeqAPASS Results
Using the human LFABP as the query sequence in SeqAPASS, predictions for 1112 species (139 ortholog candidate sequences) representing 42 taxonomic groups were generated from Level 1 primary amino acid sequence comparisons. Results from Level 1 predict that chemicals that interact with the LFABP in humans are likely to interact with LFABP in all other vertebrates, due to evidence of structural conservation. A thorough evaluation of human LFABP conservation across species, including an exploratory Level 3 evaluation with hypothesized critical amino acids was previously reported by Cheng et al., 2021 (Supplemental Data, S1).
Querying the human AR in SeqAPASS Levels 1 and 2 yielded predictions for 1336 species (157 ortholog candidates) representing 54 taxonomic groups. Sequence comparisons at the level of the primary amino acid and LBD predict that most vertebrate species have similar susceptibility to chemicals that act on the human AR. Of those vertebrate species that aligned in Level 1, sequences that were annotated as hypothetical, unnamed, partial, low-quality protein, or with a different annotation than androgen receptor (e.g., progesterone receptor) were not evaluated in Level 3. Comparing the four amino acids involved in hydrogen bonding with testosterone, dihydrotestosterone, tetrahydrogestrinone, and two investigational synthetic androgens, 571 of the 598 species evaluated in Level 3 individual amino acid comparison were predicted to have similar chemical susceptibility as humans based on evidence of conservation of the AR (See Supplemental Data, S1 for species specific predictions of susceptibility). Lines of evidence across all three levels of SeqAPASS evaluation suggest that AR is well conserved across vertebrate species, as previously reported (Vliet et al., 2022).
Proteins prioritized for structural modeling
Prioritization of liver fatty acid binding protein output from SeqAPASS
Of the 1112 protein accessions from Level 1 primary report for LFABP, 100 were identified as sequences for structural modeling. There were 91 high priority sequences and 9 identified as low priority sequences (but included as the only representatives for a taxonomic group) with similar annotation to the query sequence that were selected for modeling. Of the 42 taxonomic groups represented in the SeqAPASS Level 1 results, 26 taxonomic groups were included. The remaining 1,012 protein accessions were categorized as low priority sequences that did not meet criteria for protein structural modeling. From the prioritization effort, 100 LFABP sequences were submitted to I-TASSER (Supplemental Data, S3).
Prioritization of androgen receptor protein output from SeqAPASS
From the AR SeqAPASS Level 1 output, protein accessions of 1336 species were found to align with the human sequence. Of those, 273 sequences were identified as high priority for creating structural models and 3 were determined to be low priority sequences but were selected for modeling because they were the only sequences for a particular taxonomic group and had similar annotation. Eleven vertebrate taxonomic groups were represented by the sequences selected to move on to develop structural models (Supplemental Data, S3).
I-TASSER Output for Liver Fatty Acid Binding Protein
Of the 100 LFABP proteins prioritized and submitted to I-TASSER, 99 protein structures were generated (Supplemental materials: PDB files of LFABP models). Evaluating Model#1 from the I-TASSER output yielded C-Scores between −4.02 and 1.78, indicating confidence in the models. The TM-scores ranged from 0.28 ± 0.09 to 0.97 ± 0.05, with 91 structures > 0.80 ± 0.09. The RMSD was between 1.2 ± 1.2 and 15.9 ± 3.2, where the lower the value, the better the model. Finally, the cluster density was in the range of 0.006 to 1.25, where the higher the value, the higher the quality of the model (Supplemental Data, S4). From these parameters only three Roundworm structures (Models generated from sequences with NCBI accessions KRX58332.1, KRX21621.1, KRY37637.1 were identified as low-quality structures as their TM-scores were <0.5.
I-TASSER Output for Androgen Receptor
From the 276 AR sequences selected for submission to I-TASSER, 268 protein structures were created (Supplemental materials: PDB files of AR models). The I-TASSER Model#1 output produced C-scores between −4.11 and −0.56, within the typical range of models. The range for TM-score was from 0.28 ± 0.09 to 0.64 ± 0.13, with 84 structures > 0.50 indicative of correct topology. The RMSD scores were in the range of 8.3 ± 4.5 to 19.3 ± 2.0 and density was between 0.003 to 0.132 (Supplemental Data, S4). Therefore, of the structural models generated, 184 were identified as low-quality structures, therefore, these models would not move forward for further evaluation.
TM-align Results Aligning Structures to Human LFABP and Human AR
The TM-align protein structural alignment of I-TASSER generated models resulted in structural similarity between 63.71% and 96.70% comparing human LFABP to the 98 species (Supplemental Data, S4). Of note, the human structure was made up of 127 amino acids, whereas the LFABP of other species ranged between 97 and 277 amino acids. Although when taking into account the length of the proteins, only two sequences from the Band-tailed pigeon (Patagioenas fasciata monilis; NCBI accession OPJ70771.1), and a fluke (Fasciola gigantica; NCBI accession TPP64087.1) were >10% different in length of the full sequence compared to human. The full-atom structure of the entire chain for selected species were superposed with the human LFABP (PDB:3STM) to demonstrate conservation of structure for those identified to meet defined quality criteria (Figure 2). For LFABP the model generated for Firefly (Abscondita cerata; NCBI accession AEM45872.1) had the lowest average TM-align score at 0.72 and through visual examination does not align as well with the experimentally derived human LFABP (Figure 2F).
Figure 2.
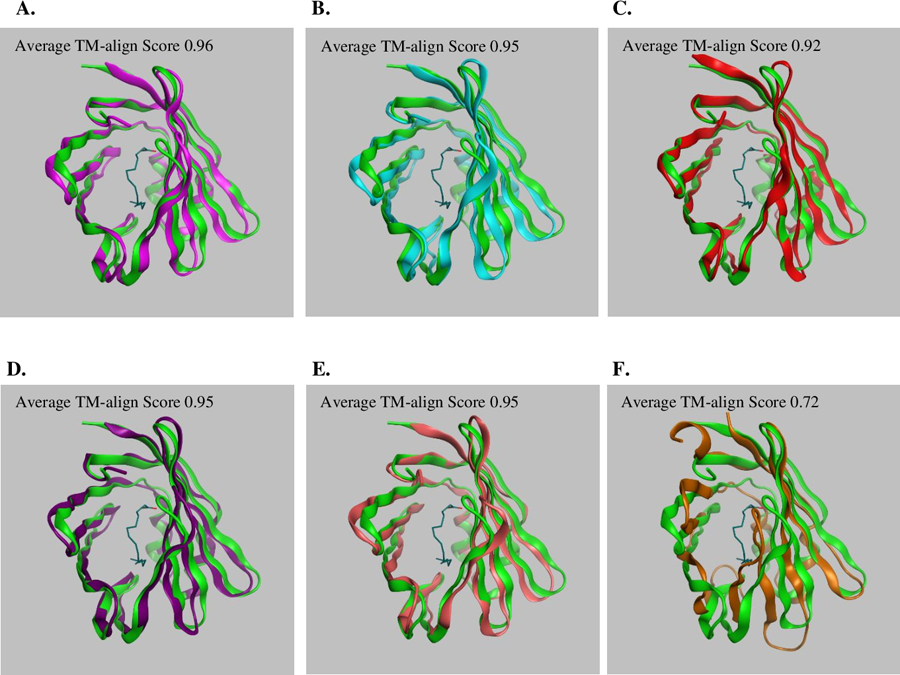
Superposed structures of the crystal structure of human (Homo sapiens) Liver Fatty Acid Binding Protein (LFABP) in complex with one molecule of palmitic acid (PDB ID 3STM; green) with A. House mouse (Mus musculus) fatty acid-binding protein, liver (NCBI Accession NP_059095.1; magenta), B. Chicken (Gallus gallus) fatty acid-binding protein, liver (NCBI Accession NP_989523.1; light blue), C. Zebrafish (Danio rerio) fatty acid binding protein 1-A, liver (NCBI Accession NP_001038177.1; red), D. Grey whale (Eschrichtius robustus) Fatty acid-binding protein, liver (NCBI Accession MBW01623.1; purple), E. Turkey vulture (Cathartes aura) Fatty acid-binding protein, liver (NCBI Accession KFP50005.1; pink), and F. Firefly (Abscondita cerata) fatty acid-binding protein (NCBI Accession AEM45872.1; brown). The Average TM-align Score calculated by taking the average of the TM-align Score normalized to the House mouse, Chicken, Zebrafish, Grey Whale, Turkey vulture, or Round worm LFABP model and the TM-align Score normalized to the Human 3STM, between the pairwise comparisons (Supplemental Data, S4). TM-align score is a metric for assessing the topological similarity of protein structures and has a value between 0 and 1, where 1 indicates a perfect match between two structures. Illustrations made using Molecular Operating Environment (MOE) software (Chemical Computing Group, Montreal, QC, Canada).
For AR TM-align evaluation, the percent similarity comparing human AR to the 267 structures from other species resulted in structural similarity between 21.61% and 52.89% (Supplemental Data, S4). The human AR structure was comprised of 914 amino acids whereas other species ranged between 381 to 918 amino acids. Of the 84 high quality structures, eight avian species were >42% different in length of the full sequence compared to human. Those that include shorter sequence lengths encompassed the LBD of the AR. Comparing the full-length human androgen receptor to dog (Canis lupus familiaris; NCBI Accession BCD56309.1), chicken (Gallus gallus, NCBI Accession NP_001035179.1), and African clawed frog (Xenopus laevis, NCBI Accession AAC97386.1) AR demonstrated that the conserved region was the LBD with the majority of the models not aligning well (Figure 3). Therefore, the full-atom structures for selected species were superposed with the crystal structure of human AR LBD in complex with dihydrotestosterone (PDB:2AMA) and then modified to focus on conservation of the LBD specifically (Figure 4). For AR, the model generated for Amur sturgeon (Acipenser schrenckii, NCBI Accession AGN52747.1) had the lowest average TM-align score at 0.30 when the full structure was aligned to the human structure. However, upon focusing on the LBD, the average TM-align score improves to 0.94 and aligns well with the experimentally derived human AR LBD when examined visually (Figure 4F).
Figure 3.

Superposed structures of the full human androgen receptor (AR) model generated using I-TASSER (green) with the full protein model for A. dog (Canis lupus familiaris) canine androgen receptor (NCBI Accession BCD56309.1; orange), B. chicken (Gallus gallus) androgen receptor (NCBI Accession NP_001035179.1; purple), C. African clawed frog (Xenopus laevis) androgen receptor alpha isoform (NCBI Accession AAC97386.1; magenta). The Average TM-align Score calculated by taking the average of the TM-align Score normalized to the dog, chicken, African clawed frog AR model and the TM-align Score normalized to the Human 2AMA, between the pairwise comparisons (Supplemental Data, S4). TM-align score is a metric for assessing the topological similarity of protein structures and has a value between 0 and 1, where 1 indicates a perfect match between two structures. Illustrations made using Molecular Operating Environment (MOE) software (Chemical Computing Group, Montreal, QC, Canada).
Figure 4.

Superposed structures of the crystal structure of human Androgen Receptor (AR) ligand binding domain (LBD) in complex with dihydrotestosterone (PDB ID 2AMA; green) with A. Dog (Canis lupus familiaris) canine androgen receptor (NCBI Accession BCD56309.1; orange; modified to only include LBD), B. Chicken (Gallus gallus) androgen receptor (NCBI Accession NP_001035179.1; purple; modified to only include LBD), C. African clawed frog (Xenopus laevis) androgen receptor alpha isoform (NCBI Accession AAC97386.1; magenta; modified to only include LBD), D. Chinese alligator (Alligator sinensis) androgen receptor (NCBI Accession AXF36050.1; red; modified to only include LBD), E. Terrapins Florida red-bellied turtle (Pseudemys nelsoni) androgen receptor (NCBI Accession BAF91192.1; light blue), and F. Amur sturgeon (Acipenser schrenckii) androgen receptor (NCBI Accession AGN52747.1; pink; modified to only include LBD). The Average TM-align Score calculated by taking the average of the TM-align Score normalized to the Dog, Chicken, African clawed frog, Chinese alligator, red-bellied turtle, or Amur sturgeon AR model and the TM-align Score normalized to the Human 2AMA, between the pairwise comparisons (Supplemental Data, S4). TM-align score is a metric for assessing the topological similarity of protein structures and has a value between 0 and 1, where 1 indicates a perfect match between two structures. Illustrations made using Molecular Operating Environment (MOE) software (Chemical Computing Group, Montreal, QC, Canada).
Line of Evidence for Structural Conservation Across Species
Of the LFABP models that were generated, 97 were determined to be high quality models in I-TASSER. After evaluating TM-align scores and comparing lengths of the proteins and determining which structures were within 10% of the absolute value of amino acid length difference, those 97 high quality structures would be considered conserved, and 53 targets have data to increase evidence of conservation captured in Level 1 and 3. (Supplemental Data, S5). These structural evaluations (which will correspond to the development of Level 4 evaluations in SeqAPASS) can be used as another line of evidence for structural conservation in combination with the previous sequence-based results from SeqAPASS. Such conservation indicates that these species are likely to share bioaccumulation potential similar to the human in consideration of PFAS.
There were 84 high quality AR structures evaluated in TM-align (Supplemental Data, S5). From these data another line of evidence was created indicating that AR is conserved across vertebrates. The results from Levels 1, 2, and 3 from SeqAPASS along with the new Level 4 data for structural conservation across these species indicate that those chemicals screened using human cell-based high-throughput methods are likely similarly able to modulate other vertebrates. These 84 structures can be used for advanced molecular modeling approaches, though results indicate that focusing on the LBD would provide better models for comparisons across species.
DISCUSSION
Advances in bioinformatics approaches continue to improve computer-aided drug design and during the recent COVID-19 pandemic were consequently in the limelight as a means to computationally aid in the identification of potential therapeutics for SARS-CoV-2 virus (Barghash et al., 2021). Through the efforts of scientists rapidly pushing the field forward in response to current needs, the strengths and limitations of molecular modeling approaches such as protein modeling, molecular docking, molecular dynamic simulations, and virtual screening have been illuminated (Haddad et al., 2020). As these bioinformatics approaches are being vetted within the fields of biotechnology, pharmacology, and medicine, it is not surprising that they are being recognized in other fields for potentially unique applications. In the field of environmental toxicology, it is important to understand interactions of biomolecules with chemicals that may enter the environment. Bioinformatics approaches allow for the utilization of knowledge from one or more species to predict or infer the likelihood for effect in another species. In chemical safety, cross species extrapolation is typically referring to predicting toxicity in other species.
The SeqAPASS tool is recognized as an approach for cross species extrapolation that provides lines of evidence toward protein conservation across species to inform predictions of chemical susceptibility. As described, the current version of the web-based tool (v6.1) aligns protein sequences and provides output that answers a very specific question of whether a known chemical target in one species is likely to be present in another species and interact with that chemical in a similar manner. The actual output from SeqAPASS provides predictions of “yes” a species is likely susceptible or “no” a species is not likely susceptible to a chemical. Sequence alignment can advance our understanding of species similarities and provide valuable insights into likely interactions with chemicals across large numbers of species very rapidly, particularly when looking at comparisons of critical individual amino acids involved in chemical-protein interactions. With the progress in computational methods, new insights can be gained from protein structural alignments and the use of more advanced molecular modeling approaches for species extrapolation. Therefore, to advance the SeqAPASS approach, a command line analysis pipeline was created to automate the process for prioritization of sequences from SeqAPASS output which could then be fed into I-TASSER to generate protein structural models with metrics to understand the quality of the structure. The models generated for several species representing diverse taxa could then be used (with additional refinement) for further and more complex molecular docking, molecular dynamic simulations, and virtual screening (e.g., Galli et al., 2014). Using TM-align, models can then be compared to generate another line of evidence toward structural conservation between the query species and hit species evaluated in SeqAPASS (Figure 1). This initial pipeline from sequence prioritization to model generation and structural alignment is the foundation for the creation of SeqAPASS Level 4.
During the completion of the development of the pipeline and evaluation of protein structures described above, advances in another exciting protein modeling program were announced. Specifically, DeepMind’s program AlphaFold (alphafold.ebi.ac.uk/), which takes advantage of artificial intelligence to predict protein structures was described. AlphaFold was identified as the most accurate protein prediction tool during the Critical Assessment of Protein Structure Prediction14 (CASP14; predictioncenter.org/), was used recently to predict more than 200,000 million protein structures representing the diversity of species (Jumper, J., et al., 2021). The accuracy of these predicted proteins has been shown to rival physical experiments and have therefore been placed in the European Molecular Biology Laboratory’s (EMBL’s) European Bioinformatics Institute (EBI) database for public accessibility. Therefore, as development of the SeqAPASS pipeline continues for Level 4 (i.e., structural comparisons across species) the utility of the AlphaFold structures will be further explored along with those generated from I-TASSER.
Evaluating protein structures using computational approaches to understand chemical interactions in diverse species relative to environmental health is not a new concept. There are a number of studies using these bioinformatics approaches including, as examples, exploring differences in aryl hydrocarbon receptor activation among sturgeon species via homology modeling (Doering et al., 2015), evaluation of drug targets such as cyclooxygenase 2 and progesterone with molecular docking to explore the likelihood for interactions of diverse species with diclofenac and ibuprofen and levonorgestrel (Walker and McEldowney et al., 2013), examining the interactions of microplastics and their additives through molecular docking with zebrafish (Chen et al., 2021), assessing the interactions of tebufenozide with the LBD of the ecdysone receptor of insect species (Zotti et al., 2012), and assessing binding affinities generated from molecular docking of per- and polyfluoroalkyl substances to the LBD of Baikal seal and human peroxisome proliferator-activated receptor alpha to understand interspecies differences in binding (Ishibashi et al., 2019). Studies such as those described above continue to expand in the environmental toxicology space exploring the utility of bioinformatics. Therefore, any advancement to the process of generating quality protein models across species is likely to expedite the use of computational approaches for understanding chemical interactions. To demonstrate the utility of the developed pipeline, two case examples focused on LFABP and AR were considered.
The first case example was an extension from a published study computationally examining LFABP in multiple vertebrate species (i.e., human, rat, chicken, zebrafish, rainbow trout, fathead minnow, and Japanese medaka) using a combination of bioinformatics approaches including SeqAPASS, homology modeling, molecular docking, and molecular dynamic simulations in the context of understanding bioaccumulation potential of per- and polyfluoroalkyl substances (Cheng et al., 2021). From this evaluation it was demonstrated that each of these computational approaches have strengths and weaknesses. However, when used in combination they provide the greatest insights into chemical-protein interactions and can be valuable for understanding cross species differences in chemical susceptibility, and more specifically chemical binding affinity. The work by Cheng et al. provided the foundation to begin exploring the potential for advancing the SeqAPASS workflow by automatically generating quality protein structures for several species, across a diversity of taxa. Through the described pipeline, the structure of LFABP for selected species were compared with the human protein, demonstrating conservation of structure for those species, and adding an additional line of evidence toward structural conservation between the query species and hit species evaluated in SeqAPASS (Figure 2). Upon preparation of protein structural models across species/taxa through this pipeline, the process setting up for the evaluation of chemical interactions using more advanced computational techniques such as docking and molecular dynamic simulations across many species becomes more expedited. These data can then be considered in comparative approaches as another line of evidence toward predictions of protein conservation that can be useful for predicting chemical susceptibility (Cheng et al., 2021).
The androgen receptor was used for the second case example because it is a large ligand-dependent nuclear receptor with a range of functions including crucial roles in developmental and reproductive systems. The AR, as a key target for drug development and an important molecular target relative to endocrine disruption (Kleinstreuer et al., 2022; LaLone et al., 2018), has been primarily used in molecular docking experiments relative to agonist interactions (Galli et al., 2014), as the structure of antagonist-bound receptor has yet to be elucidated experimentally. This highlights the importance of having X-ray crystallography, nuclear magnetic resonance, and cryo-electron microscopy resolved structures with high resolution (<2.0 angstroms) to select as a template when generating quality structural models. With I-TASSER, a composite approach to protein structure prediction, a combination of techniques, such as threading, ab initio modeling, and atomic-level structure refinement are used to generate models. However, as mentioned, the AlphaFold program is likely to advance protein modeling capabilities even when high resolution structures are not available. The present evaluation demonstrated, through comparison of the human AR LBD with that of the dog, chicken, African clawed frog, Chinese alligator, Terrapins Florida red-bellied turtle, and Amur sturgeon, that structural conservation is observed when compared to the structure bound to the strong agonist dihydrotestosterone (Figure 3). Work by Vliet et al. (2022) compiled a weight of evidence for conservation of AR modulated pathways across non-mammalian vertebrate species using functional inhibitor and in vivo data in combination with SeqAPASS analyses. This current structural comparison provides an additional line of evidence that the AR is conserved in vertebrate taxa. For example, such results can aid in understanding the utility of human high-throughput results for informing potential chemical interactions in other species as well as potential to inform the biologically plausible taxonomic domain of applicability for adverse outcome pathways (LaLone et al., 2018; Jensen et al., 2022).
Certainly, there are structure refinement steps when preparing protein models for more advanced molecular approaches. However, regardless of the intent for protein modeling, a first step is to generate quality protein structural models for more thorough evaluation. Here, it has been demonstrated that models for two very different proteins, the LFABP and AR, can be generated for 99 and 268 species, respectively, in a systematic, semi-automated, transparent, and consistent manner using SeqAPASS output, I-TASSER, and TM-align. This initial work described here sets the stage for interface development and next steps for expanding the features and capabilities of the web-based SeqAPASS interface. With the public release of SeqAPASS v7.0 scheduled for 2023, it is projected to include automated prioritization of sequences for structural alignment in preparation for protein structure creation using tools like I-TASSER. The intent will be to take the first steps in creating this semi-automated pipeline within the SeqAPASS tool interface itself.
As the SeqAPASS workflow continues to advance, it is essential to also describe challenges associated with these computational bioinformatics-based approaches in species extrapolation. First, it is noteworthy to understand that the SeqAPASS workflow is focused on chemical-protein or protein-protein interactions, with the focus on collecting lines of evidence for structural conservation. However, the challenge of extrapolating biological pathway knowledge or chemical susceptibility across species is much broader and encompasses considerations of chemical exposure, absorption, distribution, metabolism, and elimination, the life history of the organism and other modifying factors outside conservation analyses. Another primary consideration is the level of protein expression that can drive differences in sensitivity. Therefore, SeqAPASS and the continued development of the workflow is intended to be used in line with other weight of evidence approaches and is well-suited to aid in defining the biologically plausible taxonomic domain of applicability in the context of the adverse outcome pathway framework (Jensen et al., 2022). Further, the reliance on protein sequence information for predictive approaches necessitates improvements of both available protein sequence for a wide range of species, beyond mammalian species and common model organisms, as well as quality annotation of those sequences. In addition, the resolution of crystal structures bound with diverse ligands enhances the capabilities for generating structural models. As the availability of both protein sequences and structures continues to expand, improvements in the quality of information for a diversity of species, as well as what can be learned about chemical-protein and protein-protein interactions remain priorities for the continued advancement of approaches like SeqAPASS for cross species extrapolation. In addition, the greater the understanding of how chemicals interact with molecular targets, the better such comparative approaches can be applied.
Therefore, as new chemicals are being developed, it is extremely useful to employ methods to understand molecular interactions with human and/or environmental receptors by harnessing the power of computers. This greater understanding not only allows for extrapolation of knowledge, but also contributes to avoiding potentially harmful interactions with environmental receptors. Any advances that improve computational processes to reduce the need for super computers to run computationally expensive bioinformatics methods will enhance the utility of these approaches for environmental toxicology, where a better understanding of chemical-protein interactions and binding affinities, across many species and many chemicals is needed. As structural modeling approaches continue to advance through the use of artificial intelligence, and docking approaches are combined with molecular dynamic simulations to both identify likely binding poses and more accurately predict binding affinities, there is promise that these approaches will not only advance drug discovery, but also the field of environmental toxicology (Saho-Ahen et al., 2020; Jumper et al., 2021; Barghash et al., 2021; Cheng et al., 2021). The intent is to continue to incorporate methods within the SeqAPASS workflow that allow for users to take advantage of state-of-the-science techniques for examining how chemicals may interact with proteins across multiple species. The advancement of the SeqAPASS workflow will continue to inform chemical susceptibility predictions more rapidly and among species where no existing toxicity data exists.
Supplementary Material
Supplemental Materials, PDB files of LFABP models. Protein Databank formatted files (i.e., files with x, y, z coordinates for 3D visualization of the proteins) for all Liver Fatty Acid Binding Protein (LFABP) structural models generated from Iterative Threading ASSEmbly Refinement (I-TASSER).
Supplemental Materials, PDB files of AR models: Protein Databank formatted files (i.e., files with x, y, z coordinates for 3D visualization of the proteins) for all Androgen Receptor (AR) structural models generated from Iterative Threading ASSEmbly Refinement (I-TASSER).
Supplemental Data, S2. Text to describe PERL Script for prioritizing Level 1 Sequence Alignment to Predict Across Species Susceptibility (SeqAPASS) Protein accessions to be submitted to Iterative Threading ASSEmbly Refinement (I-TASSER) and further processed to submit to TM-align.
Supplemental Data, S4. Iterative Threading ASSEmbly Refinement (I-TASSER) and TM-align outputs from running and evaluating prioritized sequences and structures for Liver Fatty Acid Binding Protein (LFABP) and Androgen Receptor (AR).
Supplemental Data, S3. Proteins from the Sequence Alignment to Predict Across Species Susceptibility (SeqAPASS) output from Level 1 human Liver Fatty Acid Binding Protein (LFABP, NCBI protein accession: NP_001434.1) and Androgen Receptor (AR, NCBI protein accession: AAI32976.1) that have been prioritized for submission to Iterative Threading ASSEmbly Refinement (I-TASSER).
Supplemental Data, S5. Evaluation of Iterative Threading ASSEmbly Refinement (I-TASSER) and TM-align outputs for determining protein structural conservation. Level 1 (L1), Level 2 (L2), and Level 3 (L3) for human Liver Fatty Acid Binding Protein (LFABP, NCBI protein accession: NP_001434.1) and Androgen Receptor (AR, NCBI protein accession: AAI32976.1) are combined with the newly generated Level 4 (L4) structural comparisons to add lines of evidence toward conservation.
Supplemental Data, S1. Sequence Alignment to Predict Across Species Susceptibility (SeqAPASS) output from Level 1 (L1), Level 2 (L2), and Level 3 (L3) for human Liver Fatty Acid Binding Protein (LFABP, NCBI protein accession: NP_001434.1) and Androgen Receptor (AR, NCBI protein accession: AAI32976.1) including Summary reports.
Acknowledgement:
We thank J. Haselman and K. Coady for providing comments on this paper. This manuscript has been reviewed in accordance with the requirements of the US Environmental Protection Agency (EPA) Office of Research and Development. The authors declare no conflicts of interest.
Footnotes
Publisher's Disclaimer: Disclaimer: The views expressed in this work are those of the authors and do not necessarily reflect the views or policies of the US EPA, nor does the mention of trade names or commercial products constitute endorsement or recommendation for use.
Data Availability statement:
All data are provided in the Supplemental Data. For all other enquiries, please contact the corresponding author (LaLone.Carlie@epa.gov).
REFERENCES
- Adelusi TI, Oyedele AQK, Boyenle ID, Ogunlana AT, Adeyemi RO, Ukachi CD, Idris MO, Olaoba OT, Adedotun IO, Kolawole OE, Xiaoxing Y, & Abdul-Hammed M (2022). Molecular modeling in drug discovery. In Informatics in Medicine Unlocked (Vol. 29). 10.1016/j.imu.2022.100880 [DOI] [Google Scholar]
- Armitage JM, Hughes L, Sangion A, & Arnot JA (2021). Development and intercomparison of single and multicompartment physiologically-based toxicokinetic models: Implications for model selection and tiered modeling frameworks. Environment International, 154, 106557. 10.1016/J.ENVINT.2021.106557 [DOI] [PubMed] [Google Scholar]
- Barghash RF, Fawzy IM, Chandrasekar V, Singh AV, Katha U, Mandour AA, & Raffaini G (2021). coatings In Silico Modeling as a Perspective in Developing Potential Vaccine Candidates and Therapeutics for COVID-19. 10.3390/coatings11111273 [DOI] [Google Scholar]
- Cheng W, Doering JA, LaLone C, & Ng C (2021). Integrative computational approaches to inform relative bioaccumulation potential of per- And polyfluoroalkyl substances across species. Toxicological Sciences, 180(2). 10.1093/toxsci/kfab004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbourne JK, Shaw JR, Sostare E, Rivetti C, Derelle R, Barnett R, Campos B, LaLone C, Viant M, Hodges G (2022). Toxicity by descent: a comparative approach for chemical hazard assessment. Environmental Advances. In Review. [Google Scholar]
- Daston GP, Mahony C, Thomas RS, & Vinken M (2022). Assessing Safety Without Animal Testing: The Road Ahead. Toxicological Sciences, 187(2), 214–218. 10.1093/toxsci/kfac039 [DOI] [PubMed] [Google Scholar]
- Dawson DE, Ingle BL, Phillips KA, Nichols JW, Wambaugh JF, & Tornero-Velez R (2021). Designing QSARs for Parameters of High-Throughput Toxicokinetic Models Using Open-Source Descriptors. Environ. Sci. Technol, 55, 6505–6517. 10.1021/acs.est.0c06117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doering JA, Farmahin R, Wiseman S, Beitel SC, Kennedy SW, Giesy JP, & Hecker M (2015). Differences in activation of aryl hydrocarbon receptors of white sturgeon relative to lake sturgeon are predicted by identities of key amino acids in the ligand binding domain. Environmental Science & Technology, 49(7), 4681–4689. 10.1021/acs.est.5b00085 [DOI] [PubMed] [Google Scholar]
- Galli CL, Sensi C, Fumagalli A, Parravicini C, & Marinovich M (2014). A Computational Approach to Evaluate the Androgenic Affinity of Iprodione, Procymidone, Vinclozolin and Their Metabolites. PLoS ONE, 9(8), 104822. 10.1371/journal.pone.0104822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad Y, Adam V, & Heger Z (2020). Ten quick tips for homology modeling of high-resolution protein 3D structures. PLoS Computational Biology, 16 (4). 10.1371/JOURNAL.PCBI.1007449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrill JA, Everett LJ, Haggard DE, Sheffield T, Bundy JL, Willis CM, Thomas RS, Shah I and Judson RS, 2021. High-throughput transcriptomics platform for screening environmental chemicals. Toxicological Sciences, 181(1), 68–89. 10.1093/toxsci/kfab009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunnarsson L, Jauhiainen A, Kristiansson E, Nerman O and Larsson DJ, 2008. Evolutionary conservation of human drug targets in organisms used for environmental risk assessments. Environmental science & technology, 42(15), 5807–5813. 10.1021/es8005173 [DOI] [PubMed] [Google Scholar]
- Jensen MA, Blatz DJ, LaLone CA, Defining the Biologically Plausible Taxonomic Domain of Applicability of an Adverse Outcome Pathway: A Case Study Linking Nicotinic Acetylcholine Receptor Activation to Colony Death (2022). Environmental Toxicology and Chemistry. In review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, Bridgland A, Meyer C, Kohl S, Ballard A, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior A, Kavukcuoglu K, Kohli P, Hassabis D, Hassabis D 2021. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavlock R, Chandler K, Houck K, Hunter S, Judson R, Kleinstreuer N, Knudsen T, Martin M, Padilla S, Reif D and Richard A, 2012. Update on EPA’s ToxCast program: providing high throughput decision support tools for chemical risk management. Chemical research in toxicology, 25(7), 1287–1302. 10.1021/tx3000939 [DOI] [PubMed] [Google Scholar]
- Kleinstreuer NC, Ceger P, Watt ED, Martin M, Houck K, Browne P, Thomas RS, Casey WM, Dix DJ, Allen D, Sakamuru S, Xia M, Huang R, & Judson R (2022). 2017, 30, 946–964 Chem. Res. Toxicol, 14, 3. 10.1021/acs.chemrestox.6b00347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaLone CA, Basu N, Browne P, Edwards SW, Embry M, Sewell F, & Hodges G (2021). International Consortium to Advance Cross-Species Extrapolation of the Effects of Chemicals in Regulatory Toxicology. Environmental Toxicology and Chemistry, 40(12), 3226–3233. 10.1002/etc.5214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaLone CA, Villeneuve DL, Doering JA, Blackwell BR, Transue TR, Simmons CW, Swintek J, Degitz SJ, Williams AJ, & Ankley GT (2018). Evidence for Cross Species Extrapolation of Mammalian-Based High-Throughput Screening Assay Results. Environ Sci Technol 2018, 52 (23), 13960–13971. 10.1021/acs.est.8b04587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaLone CA, Villeneuve DL, Lyons D, Helgen HW, Robinson SL, Swintek JA, Saari TW, & Ankley GT (2016). Sequence alignment to predict across species susceptibility (SeqAPASS): A web-based tool for addressing the challenges of cross-species extrapolation of chemical toxicity. Toxicological Sciences, 153(2). 10.1093/toxsci/kfw119 [DOI] [PubMed] [Google Scholar]
- Rivetti C, Allen TEH, Brown JB, Butler E, Carmichael PL, Colbourne JK, Dent M, Falciani F, Gunnarsson L, Gutsell S, Harrill JA, Hodges G, Jennings P, Judson R, Kienzler A, Margiotta-Casaluci L, Muller I, Owen SF, Rendal C, … Campos B (2020). Vision of a near future: Bridging the human health–environment divide. Toward an integrated strategy to understand mechanisms across species for chemical safety assessment. Toxicology in Vitro, 62, 104692. 10.1016/J.TIV.2019.104692 [DOI] [PubMed] [Google Scholar]
- Roy A, Kucukural A, & Zhang Y (2010). I-TASSER: A unified platform for automated protein structure and function prediction. Nature Protocols, 5(4). 10.1038/nprot.2010.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salo-Ahen OMH, Alanko I, Bhadane R, Bonvin AMJJ, Vargas Honorato R, Hossain S, Juffer AH, Kabedev A, Lahtela-Kakkonen M, Larsen AS, Lescrinier E, Marimuthu P, Mirza MU, Mustafa G, Nunes-Alves A, Pantsar T, Saadabadi A, Singaravelu K, & Vanmeert M (2020). processes Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development. 10.3390/pr9010071 [DOI] [Google Scholar]
- Thiel C, Schneckener S, Krauss M, Ghallab A, Hofmann U, Kanacher T, Zellmer S, Gebhardt R, Hengstler JG, & Kuepfer L (2015). A Systematic Evaluation of the Use of Physiologically Based Pharmacokinetic Modeling for Cross-Species Extrapolation. Journal of Pharmaceutical Sciences, 104(1), 191–206. 10.1002/JPS.24214 [DOI] [PubMed] [Google Scholar]
- Thomas RS, Cheung R, Westphal M, Krewski D, & Andersen ME (2017). Risk science in the 21st century: A data-driven framework for incorporating new technologies into chemical safety assessment. International Journal of Risk Assessment and Management, 20(1). 10.1504/IJRAM.2017.082560 [DOI] [Google Scholar]
- van den Berg SJP, Maltby L, Sinclair T, Liang R, & van den Brink PJ (2021). Cross-species extrapolation of chemical sensitivity. Science of The Total Environment, 753, 141800. 10.1016/J.SCITOTENV.2020.141800 [DOI] [PubMed] [Google Scholar]
- van der Zalm AJ, Barroso J, Browne P, Casey W, Gordon J, Henry TR, Kleinstreuer NC, Lowit AB, Perron M and Clippinger AJ (2022). A framework for establishing scientific confidence in new approach methodologies. Archives of Toxicology, 1–15. 10.1007/s00204-022-03365-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbruggen B, Gunnarsson L, Kristiansson E, Tobias Tobias Österlund T, Owen SF, Snape JR, & Tyler CR (2018). ECOdrug: a database connecting drugs and conservation of their targets across species. Nucleic Acids Research, 46. 10.1093/nar/gkx1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vliet SM, Markey K, Lynn S, Adetona A, Ceger P, Choski N, Hamm J, Karmaus A, Watson A, Ewans A, Daniels A, Rooney J, Fallacara D, Vitense K, Wolf K, Thomas A, LaLone CA (2022). Weight-of-Evidence for Cross-Species Conservation of Androgen Receptor-Based Chemical Toxicity. In journal review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, & Skolnick J (2005). TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Research, 33(7). 10.1093/nar/gki524 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Materials, PDB files of LFABP models. Protein Databank formatted files (i.e., files with x, y, z coordinates for 3D visualization of the proteins) for all Liver Fatty Acid Binding Protein (LFABP) structural models generated from Iterative Threading ASSEmbly Refinement (I-TASSER).
Supplemental Materials, PDB files of AR models: Protein Databank formatted files (i.e., files with x, y, z coordinates for 3D visualization of the proteins) for all Androgen Receptor (AR) structural models generated from Iterative Threading ASSEmbly Refinement (I-TASSER).
Supplemental Data, S2. Text to describe PERL Script for prioritizing Level 1 Sequence Alignment to Predict Across Species Susceptibility (SeqAPASS) Protein accessions to be submitted to Iterative Threading ASSEmbly Refinement (I-TASSER) and further processed to submit to TM-align.
Supplemental Data, S4. Iterative Threading ASSEmbly Refinement (I-TASSER) and TM-align outputs from running and evaluating prioritized sequences and structures for Liver Fatty Acid Binding Protein (LFABP) and Androgen Receptor (AR).
Supplemental Data, S3. Proteins from the Sequence Alignment to Predict Across Species Susceptibility (SeqAPASS) output from Level 1 human Liver Fatty Acid Binding Protein (LFABP, NCBI protein accession: NP_001434.1) and Androgen Receptor (AR, NCBI protein accession: AAI32976.1) that have been prioritized for submission to Iterative Threading ASSEmbly Refinement (I-TASSER).
Supplemental Data, S5. Evaluation of Iterative Threading ASSEmbly Refinement (I-TASSER) and TM-align outputs for determining protein structural conservation. Level 1 (L1), Level 2 (L2), and Level 3 (L3) for human Liver Fatty Acid Binding Protein (LFABP, NCBI protein accession: NP_001434.1) and Androgen Receptor (AR, NCBI protein accession: AAI32976.1) are combined with the newly generated Level 4 (L4) structural comparisons to add lines of evidence toward conservation.
Supplemental Data, S1. Sequence Alignment to Predict Across Species Susceptibility (SeqAPASS) output from Level 1 (L1), Level 2 (L2), and Level 3 (L3) for human Liver Fatty Acid Binding Protein (LFABP, NCBI protein accession: NP_001434.1) and Androgen Receptor (AR, NCBI protein accession: AAI32976.1) including Summary reports.
Data Availability Statement
All data are provided in the Supplemental Data. For all other enquiries, please contact the corresponding author (LaLone.Carlie@epa.gov).