

SUMMARY
Activating mutations in PIK3CA are frequently found in estrogen-receptor-positive (ER+) breast cancer, and the combination of the phosphatidylinositol 3-kinase (PI3K) inhibitor alpelisib with anti-ER inhibitors is approved for therapy. We have previously demonstrated that the PI3K pathway regulates ER activity through phosphorylation of the chromatin modifier KMT2D. Here, we discovered a methylation site on KMT2D, at K1330 directly adjacent to S1331, catalyzed by the lysine methyltransferase SMYD2. SMYD2 loss attenuates alpelisib-induced KMT2D chromatin binding and alpelisib-mediated changes in gene expression, including ER-dependent transcription. Knockdown or pharmacological inhibition of SMYD2 sensitizes breast cancer cells, patient-derived organoids, and tumors to PI3K/AKT inhibition and endocrine therapy in part through KMT2D K1330 methylation. Together, our findings uncover a regulatory crosstalk between post-translational modifications that fine-tunes KMT2D function at the chromatin. This provides a rationale for the use of SMYD2 inhibitors in combination with PI3Kα/AKT inhibitors in the treatment of ER+/PIK3CA mutant breast cancer.
In brief
Blawski et al. show that KMT2D is methylated by the lysine methyltransferase SMYD2 at K1330. SMYD2 loss attenuates PI3K inhibitor-induced KMT2D chromatin binding at estrogen-receptor (ER) loci and ER-dependent transcription. SMYD2 inhibition sensitizes cells, patient-derived organoids, and tumors to PI3K/AKT inhibition and endocrine therapy through KMT2D K1330 methylation.
Graphical Abstract
INTRODUCTION
Breast cancer is the most commonly diagnosed cancer in the world. Over 70% of breast cancers express the estrogen receptor (ER), which they rely on for their growth and proliferation. These cancers are treated with inhibitors targeting the ER signaling pathway, such as selective ER modulators (e.g., tamoxifen), aromatase inhibitors (AIs; e.g., anastrozole, letrozole) and selective ER degraders such as fulvestrant and, more recently, elacestrant. The addition of CDK4/6 inhibitors has markedly prolonged progression-free survival compared to endocrine therapy alone.1
Forty percent of ER+, HER2— cancers harbor alterations in the phosphoinositide 3-kinase (PI3K) pathway, such as activating mutations in PIK3CA, the gene encoding the p110ɑ catalytic subunit of PI3K. We have previously observed that PI3Kɑ inhibition results in enhanced ER function, which confers resistance to PI3Kα inhibitors and can be reversed with the addition of endocrine therapies.2 The combination of the PI3K inhibitor alpelisib with the ER degrader fulvestrant is approved by the US Food and Drug Administration (FDA) for the treatment of patients with ER+ PIK3CA-mutated breast cancer.3
Mechanistically, we demonstrated that PI3K inhibition elevates ER activity to drive tumor growth in ER+/PIK3CA mutant breast cancer, through the lysine methyltransferase KMT2D. KMT2D is directly phosphorylated at S1331 and inactivated by PI3K pathway effectors AKT and/or SGK, resulting in loss of binding of KMT2D and ER to the chromatin and consequent transcriptional repression.4,5 Pharmacological blockade of PI3Kɑ releases this inhibitory mechanism, resulting in the recruitment of KMT2D to the chromatin and subsequent activation of ER-dependent gene programs that fuel breast cancer growth, suggesting that inhibition of KMT2D could be a therapeutic strategy in combination with PI3K inhibitors for treatment of ER+/PIK3CA mutant breast cancer.
KMT2D (also known as MLL2 or MLL4), a member of the complex of proteins associated with Set1 (COMPASS)-like family, is known to catalyze histone 3 lysine 4 mono- and di-methylation, an epigenetic modification that facilitates gene expression.6 Recent studies have shown that deficiency of KMT2D induces abnormal epigenomic reprogramming and lung tumorigenesis, conferring therapeutic vulnerability to glycolytic inhibitors or SHP2 and pan-ERBB inhibition in lung cancer.7,8 Disruption of KMT2D also perturbs germinal center B cell development, promoting lymphomagenesis.9,10 In addition, CRISPR screens have identified KMT2D as a key determinant of immune checkpoint blockade and indicate that KMT2D loss may sensitize multiple cancer types to immunotherapy.11
Despite the increasing evidence pointing to a critical role of KMT2D in cancer, the precise post-translational regulation mediating these effects remains largely unclear, likely due to the large size of KMT2D (~593 kDa), which renders biochemical characterization of post-translational modifications challenging. Recent evidence suggests that methylation may affect gene expression and cellular functions, not only by modifying histone tails but also by altering the activity of transcription factors such as YY2,12 ER,13 E2F-1,14 p65,15 STAT3,16 RUNX1,17 and p5318 or epigenetic modifiers such as EZH2.19 However, the extent of KMT2D methylation in cancer cells and its consequences on KMT2D function are unknown. Given that, to our knowledge, potent and selective KMT2D inhibitors are not yet available, the study of such mechanisms may lead to the identification of novel therapeutic opportunities in ER+ breast cancer. Additionally, there has been long-term interest in the intersection between signaling and epigenetics, but the mechanistic links between key components of these pathways have often been lacking. Here, we provide a comprehensive connection between oncogenic PI3K pathway signaling and SMYD2-mediated regulation of the KMT2D-ER axis, underscoring the relevance of the chromatin state in connecting oncogenic signaling responses with gene activity.
Specifically, we uncovered a methylation site on KMT2D at K1330, directly adjacent to the phosphorylated S1331, which we identified to be catalyzed by the lysine methyltransferase SMYD2. Knockdown of SMYD2 sensitizes ER+/PIK3CA mutant breast cancer cell lines and tumors to PI3K/AKT inhibition. Mechanistically, SMYD2 knockdown attenuates alpelisibinduced KMT2D chromatin binding globally and at ER loci. Accordingly, loss of SMYD2 abrogates alpelisib-induced changes in gene expression, including ER-dependent transcription. Our data suggest a role for epigenetic therapy using SMYD2 inhibitors in combination with alpelisib for the treatment of ER+/PIK3CA mutant breast cancer, supporting the pursuit of SMYD2 as a potential target for cancer therapy in this setting.
RESULTS
KMT2D is methylated by SMYD2 in breast cancer in vitro and in cells
While performing mass spectrometry (MS) studies to identify the phosphorylation of KMT2D at S1331 by AKT/SGK,4 we unexpectedly observed yet another post-translational modification of KMT2D, a mono- and di-methylation event at the adjacent lysine, K1330, of KMT2D immunoprecipitated from HEK293T cell lysates (Figures 1A and S1A).
Figure 1. KMT2D is methylated by SMYD2 at K1330.

(A) Schematic showing the domain structure of lysine methyltransferase KMT2D and the conserved AGC consensus site containing both the S1331 phosphorylation site modified by AKT/SGK1 (previously published) and the identified methylation site at K1330. Sequence alignments for the indicated species were performed using Clustal W2.
(B) In vitro screen of recombinant protein lysine methyltransferases (PKMTs) to identify the methyltransferase able to catalyze K1330me. Radiometric assays were conducted using 3H-S-adenosyl-L-methionine (SAM) and a KMT2D peptide (amino acids [aa] 1,321–1,343) and activity was measured as 3H counts per minute (3H-CPM) using liquid scintillation counting. Peptide only and enzyme only controls were included to indicate background signal from the KMT2D peptide and PKMTs respectively (n = 3 biological replicates, representative shown).
(C) Radiometric methylation assay as in (B), using the wild-type KMT2D peptide and KMT2D peptides with the K1330 site mutated to alanine (K1330A) or arginine (K1330R).
(D) Fragment constructs of KMT2D (aa 1,222–1,819) with a c-terminal V5 tag were overexpressed in HEK293T cells and immunoprecipitated using V5-conjugated agarose beads. Wild-type KMT2D was compared to K1330A/R mutant constructs generated with site-directed mutagenesis. Site-specific antibodies (Eurogentec) were used to detect K1330me1 and K1330me2.
(E) Full-length (FL) wild-type or mutant KMT2D constructs with an N-terminal HA tag were expressed in HEK293T cells and immunoprecipitated using HA-conjugated agarose. Site-specific antibodies were used to detect K1330me1/2 modifications on full-length KMT2D.
(F) Lysates from stable T47D cell lines with two different doxycycline-inducible shRNAs were used to detect changes in K1330me1/2 upon loss of SMYD2.
(G) Wild-type V5-tagged KMT2D constructs were co-expressed with wild-type SMYD2 in HEK293T cells and treated with the indicated doses of LLY507, a catalytic inhibitor of SMYD2, for 48 h before collection. Following immunoprecipitation, samples were subjected to immunoblot with K1330me1/2 antibodies.
(H) Same as in (G) but with a different catalytic inhibitor of SMYD2, AZ506.
(I) Kinetic analysis of recombinant AKT1 activity on KMT2D wild-type, mutant, or modified peptides. Reactions were conducted using 33P-ATP and detected using a radiometric filter-binding assay (Reaction Biology).
(J) V5-tagged fragment wild-type KMT2D and point mutants were overexpressed and immunoprecipitated from HEK293T cells, followed by immunoblot with the pS1331 antibody. One representative western blot is shown and the differences in pS1331 are quantified on the right (n = 3 biological replicates, mean, SD). Indicated p values were calculated using an unpaired, two-tailed Student’s t test.
(K) In vitro methylation assay using recombinant SMYD2 and wild-type, mutant, or modified KMT2D peptides (aa 1,321–1,343), conducted as in Figures 1B and 1C.
(L) V5-tagged fragment wild-type KMT2D and point mutants were overexpressed and immunoprecipitated from HEK293T cells, followed by immunoblot with K1330me1/me2 antibodies.
To identify which methyltransferase is responsible for methylating K1330 of KMT2D, we performed methylation assays using a panel of recombinant lysine methyltransferases that could be reconstituted in vitro, including EZH2, EZH1, SMYD2, SMYD3, SETD8, and NSD2, and found SMYD2 to methylate the KMT2D peptide (Figures 1B, S1B, and S1C). Moreover, additional methylation assays using recombinant SMYD2 and wild-type or K1330A, K1330R mutant peptides of KMT2D demonstrated that SMYD2 can successfully methylate wild-type KMT2D, but not the KMT2D K1330A or K1330R point mutants of the target lysine (Figure 1C).
We next established antibodies that recognize the mono- or di-methylation of KMT2D at K1330 to determine whether SMYD2 methylates KMT2D at K1330 in cells. We transfected HEK293T cells with wild-type V5-KMT2D (amino acids 1,222–1,819) or the K1330A/R mutants and probed the immunoprecipitated V5-KMT2D proteins with the anti-KMT2D-K1330me1 or me2 antibodies. While these antibodies recognized immunoprecipitated wild-type KMT2D, they failed to recognize the SMYD2-methyl-dead mutants (K1330A and K1330R) of KMT2D (Figure 1D). Similar results were also obtained when full-length wild-type HA-KMT2D or the K1330A and K1330R mutants were immunoprecipitated from HEK293T cells and probed with mono- and di-methylated antibodies (Figure 1E).
To further verify that this methylation event is SMYD2 dependent in cells, SMYD2 expression in ER+/PIK3CA mutant T47D breast cancer cells was stably knocked down in a doxycycline-induced manner. Upon SMYD2 knockdown, KMT2D K1330 mono- and di-methylation was shown to be impaired (Figure 1F). Mono- and di-methylated KMT2D was also detected in HEK293T cells upon SMYD2 wild-type overexpression but not upon expression of a catalytic dead mutant of SMYD2 (Figure S1D). We next targeted the catalytic activity of SMYD2 pharmacologically using distinct SMYD2 inhibitors such as LLY507, AZ506, or AZ39, and we then assessed the methylation status of KMT2D in cells.20,21 In all cases, inhibition of SMYD2 was accompanied by a dose-dependent decrease of KMT2D methylation at K1330 (Figures 1G, 1H, and S1E). Collectively, these data support that KMT2D is methylated at K1330 by SMYD2.
Given the close proximity of SMYD2-mediated methylation at K1330 of KMT2D with the AKT/SGK-dependent phosphorylation of KMT2D at S1331, we next examined any possible interplay between these two post-translational modifications. We first performed in vitro enzyme kinetics assays using recombinant full-length AKT1 in the presence of wild-type KMT2D, K1330A/R mutant, or methylated K1330 peptides to characterize the kinetic properties of S1331 phosphorylation in each case. We discovered that AKT1 phosphorylated S1331 in all peptides in vitro, following a Michaelis-Menten model, albeit at a lower catalytic efficiency for the K1330A/R peptide mutants or the methylated KMT2D peptide as reflected by kcat/Km values in comparison to wild type (Figure 1I).
To further dissect whether the methylation of KMT2D affects KMT2D phosphorylation, we also performed in vitro kinase assays in the presence of AKT1 recombinant kinase using immunoprecipitated wild-type full-length KMT2D, the phospho-dead mutant S1331A, or methyl-dead mutants (K1330A and K1330R) as substrates. Western blot analyses followed using an antibody for KMT2D pS1331. As shown in Figure S1F, AKT was able to phosphorylate wild-type and the methyl-dead mutants of KMT2D (K1330A, K1330R) but not the phospho-dead mutant (S1331A). We also performed in vitro kinase assays using a shorter fragment of KMT2D (amino acids 1,222–1,819) immunoprecipitated from HEK293T as a substrate with recombinant active AKT isoforms (AKT1, AKT2, or AKT3) or SGK1. In all cases, these PI3K effectors could phosphorylate wild-type KMT2D at S1331 as detected by phosphorylated S1331 antibody but not the phospho-dead mutant (S1331A) (Figures S1F and S1G).
To test the interplay between SMYD2-mediated methylation at K1330 and KMT2D-S1331 phosphorylation in cells, we overexpressed fragment constructs (amino acids 1,222–1,819) of wild-type KMT2D, the K1330A/R mutants, and S1331A mutant in 293T cells and probed the immunoprecipitated KMT2D proteins with the phosphorylated S1331 antibody. While this antibody failed to recognize the S1331A mutant as expected, it showed a significant increase of phosphorylation of SMYD2 methyl-dead mutants (K1330A/R) of KMT2D (Figure 1J). When we overexpressed full-length KMT2D or the methyl-dead mutants in 293T cells, we also observed an increase in phosphorylation of KMT2D in the methyl-dead mutants in comparison to wild type (Figure S1H).
To identify whether phosphorylation of KMT2D affects SMYD2-mediated methylation of KMT2D, we next performed in vitro methylation assays using recombinant SMYD2 and phosphorylated KMT2D peptides (pS1331) or the phospho-dead mutant of KMT2D (S1331A). SMYD2 could methylate the phospho-dead mutant KMT2D (S1331A) but was unable to methylate the peptide that was phosphorylated at S1331 (Figure 1K). Consistent with this, immunoblotting of immunoprecipitated wild-type KMT2D or K1330A, K1330R, and S1331A mutants led to ablation of methylation of KMT2D for the K1330A/R mutants but not wild type or the phospho-dead mutant (S1331A) of KMT2D (Figure 1L). Taken together, our in vitro and in vivo data suggest a potential two-way crosstalk between S1331 phosphorylation and K1330 methylation of KMT2D.
SMYD2 methylation positively regulates KMT2D function in breast cancer
Given the important regulatory role we had previously identified for S1331 phosphorylation in suppressing the coactivator function of KMT2D at ER-target genes, we sought to understand the functional impact of SMYD2-catalyzed methylation at K1330. KMT2D encodes a conserved protein belonging to the SET1 family of histone lysine methyltransferases, which catalyze the methylation of lysine 4 on histone 3 (H3K4), a marker associated with transcriptionally active chromatin.6,22 Although the K1330 modification is not in the catalytic domain of KMT2D, we wanted to understand if K1330 methylation could regulate the activity of KMT2D on its H3 substrate. To this end, we immunoprecipitated full-length wild-type KMT2D, the K1330A and K1330R mutants of KMT2D that cannot be methylated by SMYD2, and a catalytic-dead point mutation of KMT2D Y5426A or R5432W as a positive control and assessed their activity on an H3 peptide using scintillation assays. While the Y5426A and R5432W mutant had significantly impaired enzymatic activity compared to the wild-type protein, the K1330A and K1330R mutant catalyzed H3K4 methylation at similar levels to wild-type KMT2D (Figures 2A and S2A). These data show that KMT2D K1330A/R mutants are capable of methylating H3K4, suggesting that SMYD2-mediated methylation of KMT2D is not affecting KMT2D enzymatic activity on H3.
Figure 2. SMYD2 methylation of KMT2D affects KMT2D binding genome-wide and cofactor associations in breast cancer.

(A) In vitro methylation assay to assess activity of immunoprecipitated full-length KMT2D wild type and mutants on an H3 peptide substrate (aa 1–21). A catalytic-dead mutant of KMT2D (Y5426A) was used as a control. Substrate only and enzyme only samples were used to control for background signal from the H3 peptide and KMT2D mutants respectively (n = 3 biological replicates, representative shown).
(B) Western blot of SMYD2 and vinculin in MCF7 cells transduced with shSMYD2–1 or shSMYD2–2.
(C) Tornado plot of KMT2D ChIP-seq showing global binding sites of KMT2D in MCF7 cells with doxycycline-inducible SMYD2 knockdown. Cells were treated with DMSO or 1 μM PI3Kα inhibitor alpelisib for 8 h before crosslinking.
(D) KMT2D binding at estrogen receptor (ER) target loci (GEO: GSE59530) in all aforementioned conditions.
(E–G) Cluster analysis of KMT2D ChIP-seq peaks comparing SMYD2 knockdown alone (E), alpelisib treatment alone (F), and the combination of SMYD2 knockdown and alpelisib treatment (G). HOMER motif analysis for the clusters with differential KMT2D binding was performed with top significant motifs shown to the right of their respective heatmaps.
(H) KMT2D binding at ER-FOXA1 target genes IGFBP4 and GREB1 with SMYD2 knockdown and/or alpelisib treatment.
We next sought to determine if SMYD2 knockdown influences KMT2D genome-wide binding to the chromatin in breast cancer. To this end, we performed chromatin immunoprecipitation followed by sequencing (ChIP-seq) of KMT2D in ER+/PIK3CA mutant MCF7 breast cancer cells with doxycycline-inducible SMYD2 knockdown (Figure 2B), treated with vehicle or the PI3K inhibitor alpelisib. KMT2D binding showed a marked increase upon PI3K inhibition, confirming that the phosphorylation of KMT2D by AKT/SGK affects KMT2D chromatin recruitment in breast cancer. Of note, we have also previously shown that alpelisib treatment leads to an increase in H3K4me1, the histone mark catalyzed by KMT2D, at ER loci. Conversely, overexpression of the kinase SGK1, which phosphorylates KMT2D, results in global downregulation of H3K4me1.4,5
Ablation of SMYD2 attenuated the PI3K inhibitor-induced occupancy of KMT2D at the chromatin. This phenotype was observed for global KMT2D binding and also at the chromatin peaks where KMT2D co-localizes with ER or the ER cofactor, FOXA1 (Figures 2C–2D and S2B). We further dissected the chromatin occupancy of KMT2D upon SMYD2 knockdown, alpelisib treatment, or the combination using cluster analyses (cluster 1 unchanged peaks; cluster 2 lost peaks; cluster 3 gained peaks). SMYD2 knockdown in basal conditions altered KMT2D binding genome-wide, with peaks lost in cluster 2 showing enrichment of binding motifs for ER and AP1 transcription factors (TFs) and gained peaks in cluster 3 displaying enrichment for GRHL2, TEAD, and AP1 TFs (Figure 2E). Indeed, KMT2D peaks in cluster 3 overlapped with GRHL2 ChIP-seq23 (14%) (Figure S2C) with the majority of these peaks found at putative enhancers (70%) (Figures S2D and S2E).24
PI3K inhibition also led to widespread changes in KMT2D occupancy, with the majority of sites being gained. In these sites, we observed enrichment of binding motifs for FOXA1, ER, AP1, or GRHL2 TFs, confirming that PI3K inhibition increases KMT2D binding at ER-FOXA1 sites (Figure 2F). Notably, knockdown of SMYD2 under alpelisib treatment reversed alpelisib-induced KMT2D binding, with the majority of KMT2D peaks attenuated by SMYD2 knockdown. In these lost peaks, we identified enrichment of binding motifs for AP1, TEAD, AP2 alpha, and ER, among others (Figure 2G). Consistent with this, alpelisib-induced KMT2D binding was attenuated at ER-FOXA1 loci, including CCND1, IGFBP4, and GREB1 as representative examples (Figures 2H and S2F).
To understand whether KMT2D and SMYD2 are found at the chromatin, we next performed cell fractionation assays in MCF7 cells where we overexpressed V5-KMT2D or FLAG-SMYD2. This assay demonstrated that KMT2D and SMYD2 are indeed present at the chromatin (Figure S2G). Moreover, a CUT&RUN assay for FLAG-SMYD2 showed that SMYD2 and KMT2D are co-recruited at the chromatin and that these overlapped sites are enriched with the ERE or forkhead motifs (Figures S2H and S2I).
To further elucidate the functional impact of SMYD2-catalyzed methylation at K1330 of KMT2D, we also performed tandem mass tag (TMT) MS in cells where we immunoprecipitated empty vector, wild-type KMT2D, and K1330-A/R mutants. We identified a significant number of proteins enriched or lost in both K1330 mutants compared to the wild type (Figures S3A–S3C; Table S1). Grouping the interactors based on their known functions, we found components of the INO80 chromatin remodeling complex, ESCRT complex, and histone acetyltransferase complexes, among others, to differentially interact with wild-type KMT2D in comparison to the methyl-dead mutants (Figure S3D). All the identified proteins involved in transcription were mapped in Figure S3E and they included proteins such as INO80, INO80D, CBX1, and CBX8, known components of chromatin-modifying complexes, along with other TFs and histone variants. This suggests that K1330 methylation may play an important role in the recruitment of cofactors involved in regulating chromatin accessibility and transcriptional machinery at KMT2D target genes, which remain to be further delineated.
Loss of SMYD2 abrogates alpelisib-induced changes in gene expression, including ER-dependent transcription
Given that SMYD2 knockdown affected alpelisib-induced chromatin recruitment of KMT2D to ER loci, we hypothesized that SMYD2 knockdown also affects the alpelisib-mediated transcriptome. We next performed RNA sequencing (RNA-seq) in MCF7 cells treated with DMSO or alpelisib after doxycycline-induced SMYD2 knockdown. RNA-seq showed a clear separation of gene expression between control and SMYD2 knockdown in basal conditions (2,308 upregulated genes; 2,138 downregulated genes) and upon alpelisib treatment (1,539 upregulated genes; 1,626 downregulated genes) with an overlap of 29% of differentially expressed genes upon SMYD2 knockdown in basal and alpelisib treatment (Figures S4A and S4B). Indeed, knockdown of SMYD2 led to altered transcription of thousands of genes upon PI3K inhibition. Out of 3,995 genes that were either downregulated or upregulated by PI3K inhibition, 1,248 were affected by SMYD2 knockdown in MCF7 cells (Figure 3A), indicating that more than 30% of all alpelisib-regulated gene expression requires SMYD2. Gene set enrichment analysis (GSEA) in the basal or PI3K inhibition setting identified estrogen response as one of the top gene signatures to be negatively regulated by loss of SMYD2 (Figure 3C). GSEA also identified cellular proliferation (G2M, cell cycle, E2F targets) as significantly downregulated pathways in SMYD2 knockdown cells compared to the control (Figure 3B). Moreover, the expression of ER canonical target genes such as MYC, GREB1, PGR, IGFBP4, whose expression is increased upon PI3K inhibition, was attenuated upon SMYD2 knockdown (Figure S4C).
Figure 3. SMYD2 knockdown abrogates alpelisib-induced changes in gene expression, including ER-dependent transcription.

(A) Heatmap of RNA-seq data showing the subset of genes in MCF7 cells regulated by alpelisib treatment (3,995), with and without SMYD2 knockdown (1,248 differential, log2 fold change >0.5 and adjusted p value <0.1). Cells were treated with 1 μM alpelisib for 24 h before collection (n = 3 technical replicates).
(B) Plot of C2 pathways from gene set enrichment analysis (GSEA) in SMYD2 knockdown compared to control in DMSO and alpelisib treatment conditions. All gene sets shown were significant with a p value of <0.05. Nominal p values and false discovery rate (FDR) adjusted p values were calculated using the GSEA package. Normalized enrichment score (NES) for each gene set is indicated on the x axis.
(C) GSEA plot of the Dutertre estrogen response gene set in SMYD2 knockdown compared to control in both DMSO and alpelisib treatment conditions. Nominal p values and FDR adjusted p values were calculated using the GSEA package.
(D) Gene set variation analysis (GSVA) results of estrogen-responsive gene sets in ER+ patient samples from the TCGA and METABRIC datasets corresponding with median SMYD2 mRNA level. Data are shown as median with interquartile range and analyzed using a two-tailed Student’s t test.
We next investigated whether SMYD2 expression might be linked with ER activity using ER gene signatures in clinical samples from The Cancer Genome Atlas (TCGA) and the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC). We stratified ER+ tumors into two groups, SMYD2-low and SMYD2-high, according to their mRNA expression levels. We found that SMYD2-low ER+ patients had lower ER transcriptional activity compared to SMYD2-high patients (Figure 3D). Consistent with our preclinical results, inverse ER transcriptional activities for a subset of ER target genes were observed in SMYD2 mRNA expression low and high samples.
SMYD2 loss improves the therapeutic response of PI3K/AKT inhibitors in breast cancer in vitro and in vivo
We next examined the effect of SMYD2 knockdown on the therapeutic response of breast cancer cells to PI3K inhibition. In vitro proliferation assays using the doxycycline-inducible SMYD2 shRNAs in MCF7 and T47D cells showed that SMYD2 knockdown sensitizes breast cancer cells to PI3K inhibition by alpelisib (Figures 4A and 4B). Crystal violet assays upon SMYD2 knockdown and alpelisib treatment in both MCF7 and T47D cells also confirmed these results (Figures S5A and S5B).
Figure 4. SMYD2 inhibition improves the therapeutic response of breast cancer to PI3K/AKT inhibitors in vitro and in vivo.

(A) MTT proliferation assays with doxycycline-inducible SMYD2-knockdown MCF7 cell lines using a dose response of alpelisib for a treatment period of 5 days. Significance at individual doses was assessed using an unpaired, two-tailed Student’s t test. p values are indicated as follows: *p < 0.05,** p < 0.01,***p < 0.001 and ****p < 0.0001 (n = 3 biological replicates, representative shown, mean, SD).
(B) MTT proliferation assays with doxycycline-inducible SMYD2-knockdown T47D cell lines using a dose response of alpelisib for a treatment period of 5 days. p values are indicated as in (A) (n = 3 biological replicates, representative shown, mean, SD).
(C) MTT assays with a shSMYD2 MCF7 cell line treated with fulvestrant, alpelisib, and the combination for a treatment period of 10 days. Drug concentrations were as follows: fulvestrant 100 nM, alpelisib 1 μM. Indicated p values were generated with an unpaired, two-tailed Student’s t test (n = 3 biological replicates, representative shown).
(D) MTT proliferation assays with shSMYD2 MCF7 cell lines using a dose response of the AKT inhibitor capivasertib for a treatment period of 5 days. p values are indicated as in (A) (n = 3 biological replicates, representative shown, mean, SD).
(E) MTT assays in MCF7 cells to assess the combination of a single dose of SMYD2 catalytic inhibitors AZ506 (10 μM) and AZ39 (10 μM) with a dose response of alpelisib for a treatment period of 5 days. p values are indicated as in (A) (n = 3 biological replicates, representative shown, mean, SD).
(F) MTT assays in T47D cells to assess the combination of a single dose of SMYD2 catalytic inhibitors AZ506 (5 μM) and AZ39 (10 μM) with a dose response of alpelisib for a treatment period of 5 days. p values are indicated as in (A) (n = 3 biological replicates, representative shown, mean, SD).
(G) CTG assays of PDX-derived organoids treated for 14 days with 5 μM AZ506, alpelisib 1 μM, fulvestrant 1 μM, or the combination of AZ506 and alpelisib. Statistical analysis was conducted using an unpaired, two-tailed Student’s t test (n = 3 biological replicates, mean, SEM).
(H) Images using 4× magnification microscopy of organoids treated as described in (G). Scale bar: 200 μm.
(I) Xenograft study using the doxycycline-inducible shSMYD2 cell line. Mice were treated daily with vehicle or alpelisib (25 mg/kg) for the indicated time course. p values were calculated using a two-sided Mann-Whitney U test (n = 5 per group, mean, SEM).
(J) Western blot of xenograft tumor samples validating doxycycline-induced knockdown of SMYD2 and catalytic inhibition of PI3Kα as measured by AKT pS473.
(K) Kaplan-Meier curves showing survival of TCGA ER+ breast cancer patients stratified by the presence or absence of SMYD2 amplification. p value calculated using log rank test.
Combinatory treatment of fulvestrant with alpelisib is a standard-of-care treatment for metastatic PIK3CA mutant/ER+ breast cancer. In this regard, we also tested the effect of SMYD2 knockdown on the sensitivity of MCF7 cells to these clinically approved drugs. SMYD2 knockdown significantly increased therapeutic sensitivity to fulvestrant and alpelisib monotherapy as well as the combination treatment (Figures 4C and S5D). SMYD2 knockdown alone had a modest but significant effect on the growth of MCF7 or T47D breast cancer cells, with a stronger effect in T47D cells.
Phase III studies led to the recent approval of the AKT inhibitor capivasertib in combination with fulvestrant in HR+/HER2 advanced breast cancer patients with PIK3CA/AKT1/PTEN alterations.25 To this end, we also performed in vitro proliferation assays using a dose response of capivasertib. Similarly, to PI3K inhibition, knockdown of SMYD2 also further sensitized MCF7 cells to treatment with capivasertib (Figures 4D and S5E).
In addition to genetic knockdown of SMYD2, we also performed proliferation assays using pharmacological inhibition of SMYD2 with AZ506, AZ39, LLY507, or EPZ033294 inhibitors in MCF7 cells. Of note, treatment of cells with these inhibitors led to a reduction in methylated KMT2D at K1330 (Figures 1G, 1H, and S1E). Similar to knockdown of SMYD2, pharmacological inhibition of SMYD2 had a modest effect on the growth of cells and a stronger phenotype when combined with PI3K inhibition (Figures 4E and S5F). Similar findings were also found when T47D cells were treated with the SMYD2 inhibitors, AZ506 or AZ39 (Figure 4F), suggesting that SMYD2 inhibitors sensitize breast cancer cells further to PI3K inhibitors. The triple combination of fulvestrant, alpelisib, and AZ506 also had a more significant effect in growth than the combinatory treatment (Figure S5G).
We next took advantage of multiple ER+/PIK3CA mutant PDX-derived organoids, namely HCI-011, HCI-017, and HCI-018, developed from Welm and colleagues, which model their original tumors with high fidelity.26 We demonstrated that these organoid models were sensitive to fulvestrant and alpelisib treatment, confirming their dependency on the ER and PI3K signaling pathways, respectively. Notably, significant growth attenuation was observed upon treatment with the SMYD2 inhibitor AZ506, and the combination of AZ506 with alpelisib was significantly more efficacious in attenuating the growth of organoids than either treatment alone (Figures 4G and 4H). A dose-response curve of AZ506 in HCI-017 organoids demonstrated a strong antitumor activity of AZ506 even at low doses, which was more significant in combination with alpelisib (Figure S5H).
We next investigated the in vivo activity of alpelisib in combination with SMYD2 knockdown by injecting MCF7 human breast cancer cells with doxycycline-inducible SMYD2 shRNAs into nude mice. SMYD2 silencing by doxycycline diet and alpelisib treatment alone had a modest effect on the growth of tumor xenografts (Figure 4I). However, SMYD2 knockdown markedly increased the antitumor activity of alpelisib in vivo, with the combination of SMYD2 loss and alpelisib providing robust and durable tumor regression in the ER+/PIK3CA mutant xenograft tumor model. In these tumors, we also confirmed the knockdown of SMYD2 upon doxycycline treatment and the reduction in phosphorylated AKT at S473 upon alpelisib treatment (Figure 4J).
We also studied the prevalence of SMYD2 alterations in ER+ breast cancer using the TCGA cohort. These analyses indicated that ~70% of breast cancer have alterations in SMYD2, with the vast majority displaying gained copy number or amplification of SMYD2 (Figures S5I and S5J). Kaplan-Meier survival analysis of SMYD2 showed that amplification of SMYD2 was associated with reduced overall survival in patients with ER+ breast cancer (Figure 4K).
Altogether, our data point to SMYD2 as a promising candidate for cancer therapy in combination with PI3K/AKT inhibitors for the treatment of ER+/PIK3CA mutated breast cancer.
SMYD2-mediated methylation of KMT2D alters the therapeutic response of PI3K/AKT inhibitors in breast cancer
SMYD2 is known to catalyze the lysine methylation of multiple substrates such as H3K36 and H3K4,27 as well as non-histone proteins such as p53,18 ERα,13 EZH2,19 p65,15 STAT3,16 RUNX1,17 YY2,12 and E2F14 and may also exert its function in breast cancer through mechanisms independent of KMT2D methylation that remain to be identified. To determine whether the effect of SMYD2 inhibition or knockdown on breast cancer growth or gene expression programs are due in part to the methylation of KMT2D at K1330, we generated KMT2D methyl-dead mutant (K1330A) knockin (KI) MCF7 cells (Figures S6A and S6B). We first performed proliferation assays using two distinct K1330A clones, which showed that the KI cells grow slower than the wild-type KMT2D cells. Notably, the KI cells were more sensitive to alpelisib or capivasertib treatment (Figures 5A and 5B). In addition, both KI clones were also more sensitive to fulvestrant or the combination of fulvestrant with alpelisib (Figure 5C), mimicking the effect of SMYD2 knockdown or pharmacological inhibition of SMYD2. We next investigated the in vivo growth of the KMT2D KI by injecting MCF7 KI breast cancer cells into nude mice. These tumors grew significantly slower than the wild-type KMT2D tumors, suggesting that SMYD2 is exerting its effects on breast cancer growth in cells and tumors also by KMT2D-mediated methylation (Figure 5D). Of note, the KI or wild-type KMT2D tumors did not grow at equal tumor volume at any time point throughout the experiment, which precluded the investigation of the in vivo activity of alpelisib.
Figure 5. SMYD2-mediated methylation of KMT2D affects the therapeutic response of breast cancer to PI3K/AKT inhibitors.

(A) PrestoBlue assays with K1330A CRISPR KI clones compared to unmodified MCF7 cells using a dose response of the PI3K inhibitor alpelisib for a treatment period of 5 days. Significance at individual doses was assessed using an unpaired, two-tailed Student’s t test. p values are indicated as follows: *p < 0.05,**p < 0.01,***p < 0.001, and ****p < 0.0001 (n = 3 biological replicates, mean, SEM).
(B) PrestoBlue assays with K1330A CRISPR KI clones compared to unmodified MCF7 cells using a dose response of the AKT inhibitor capivasertib for a treatment period of 5 days (n = 3 biological replicates, mean, SEM).
(C) PrestoBlue assays using K1330A CRISPR KI clones compared to unmodified MCF7 cells to assess the effect of fulvestrant, alpelisib, and the combination. All drugs were used at 1 μM concentration. Treatments were conducted for 7 days and statistical analysis was conducted using a two-tailed unpaired Student’s t test (n = 3 biological replicates, mean, SEM).
(D) Xenograft study using unmodified MCF7 and the K1330A CRISPR KI Clone-2. Tumor volume was measured for the indicated time course. p values were calculated using a two-sided Mann-Whitney U test (n = 15 per group, mean, SEM).
(E) Heatmap of RNA-seq conducted in the K1330A CRISPR KI clones with and without alpelisib treatment. Genes were filtered using an adjusted p value <0.1 and the heatmap was generated using k-means clustering. The overlapped differential genes for the two K1330A clones were used for the analysis. The number of genes in each cluster (1–5) is indicated on the left (n = 3 technical replicates).
(F) Heatmap showing the differential expression of genes regulated by alpelisib treatment in the K1330A CRISPR KI clones compared to the unmodified MCF7 control. (total of 436 genes, 137 differentially expressed with log2fold change >0.5 and adjusted p value <0.1) Cells were treated with 1 μM alpelisib for 24 h before collection.
(G) RT-qPCR analysis of ER target genes in MCF7 K1330A CRISPR KI clones compared to wild type, treated with ethanol or 100 nM estradiol (E2) for 24 h. p values were generated using a two-tailed Student’s t test (n = 3 biological replicates, representative shown, mean, SEM).
(H) Venn diagrams indicating the overlapped genes from the MCF7 shSMYD2 RNA-seq and the MCF7 K1330A CRISPR KI RNA-seq in each treatment condition (DMSO or alpelisib). Differential genes from each RNA-seq dataset were used for the knockdown (shSMYD2 versus control) and for the KI (overlap between K1330A clones versus wild type), filtered by adjusted p value <0.1.
To determine the role of SMYD2-mediated methylation of KMT2D on gene expression, we next performed RNA-seq in wild-type and KMT2D KI cells treated with DMSO or alpelisib. Focusing on the overlapped differentially expressed genes between the two KI clones, we observed widespread downregulation in gene expression between KI and wild-type cells (cluster 4 and 5) and upregulation in gene expression (cluster 1, 2, and 3), suggesting that SMYD2-mediated methylation of KMT2D is markedly affecting transcriptional output in breast cancer (Figure 5E). Similar to what we observed with knockdown of SMYD2, over 30% of the genes that were downregulated or upregulated by PI3K inhibition were affected by the K1330A KI clones of KMT2D, indicating that KMT2D methylation at K1330 is required for regulation of PI3K inhibitor-induced transcriptional programs (Figure 5F). Indeed, the transcription of canonical ER-target genes such as AREG, BCL2, SGK3, and TFF2, whose expression is altered by alpelisib, was attenuated in KMT2D K1330A KI clones (Figure S6C). Similarly, the estrogen-dependent transcription of ER-target genes such as GREB1, TFF1, and WISP2 was also downregulated in the KI cells (Figure 5G).
To differentiate between SMYD2-mediated KMT2D methylation and other functions of SMYD2, we next compared the gene expression changes mediated by the KI of KMT2D with those induced by the knockdown of SMYD2 in basal conditions or with PI3K inhibitor treatment. These analyses determined that a substantial number of differentially expressed genes are in common between both (2,158 genes in DMSO settings and 1,564 genes in alpelisib condition) (Figure 5H), suggesting that the effect of SMYD2 knockdown on gene expression is in part phenocopied by the KI of KMT2D. Gene Ontology analyses of these overlapped genes identified cell cycle and hormone response pathways, among others, in both the DMSO and alpelisib settings (Figure S6D).
Altogether, our findings demonstrate that SMYD2-mediated methylation of KMT2D phenocopies in part the effect that SMYD2 inhibition has on cell proliferation, tumor growth, drug response, and transcriptional output, suggesting an important role for SMYD2-mediated methylation of KMT2D in the therapeutic response of breast cancer.
DISCUSSION
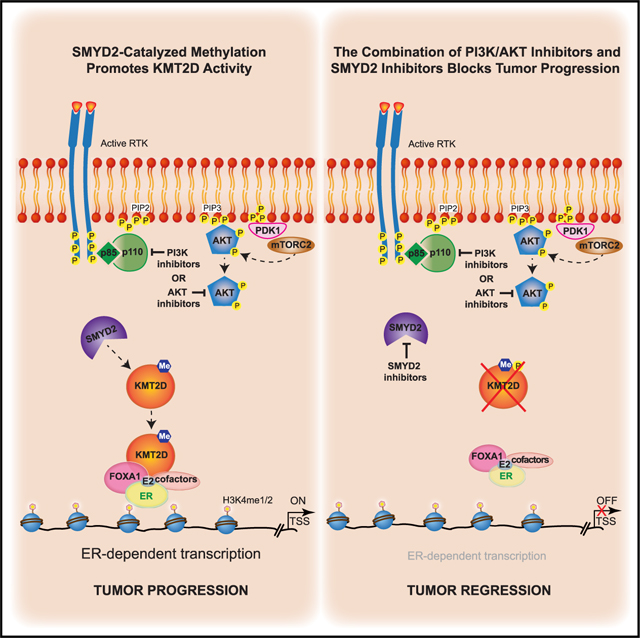
We had previously uncovered a critical role for the lysine methyltransferase KMT2D in regulating the interplay between ER and the PI3K/AKT pathway in breast cancer through the direct phosphorylation of KMT2D by the PI3K effectors AKT/SGK.4,5 AKT/SGK negatively regulate KMT2D through direct S1331 phosphorylation. When PI3K is inhibited, this downstream regulatory event is lost, leading to increased chromatin recruitment of KMT2D and an upregulation of ER-dependent transcription.4,5 Here, we discovered a previously unknown regulatory mechanism for KMT2D function mediated by the direct methylation of KMT2D by the lysine methyltransferase SMYD2. While the catalytic activity of KMT2D as a H3K4 histone methyltransferase is not affected by SMYD2-mediated methylation, ablation of SMYD2 abrogates alpelisib-induced chromatin binding of KMT2D genome-wide and at ER cis-regulatory elements. Accordingly, loss of SMYD2 attenuates alpelisib-induced gene expression changes, including ER transcriptional output (Figure 6).
Figure 6. Proposed model.

We had previously uncovered a critical role for the lysine methyltransferase KMT2D in regulating the interplay between ER and the PI3K/AKT pathway in breast cancer through the direct phosphorylation of KMT2D by the PI3K effectors AKT/SGK4,5 (first panel). AKT/SGK negatively regulate KMT2D through direct S1331 phosphorylation. When PI3K is inhibited, this downstream regulatory event is lost, leading to increased chromatin recruitment of KMT2D and an upregulation of ER-dependent transcription.4,5 Here we discovered a previously unknown regulatory mechanism for KMT2D function mediated by the direct methylation of KMT2D by the lysine methyltransferase SMYD2 (middle panel). Ablation of SMYD2 abrogates alpelisib-induced chromatin binding of KMT2D genome-wide and at ER cis-regulatory elements. Accordingly, loss of SMYD2 attenuates alpelisib-induced gene expression changes, including ER transcriptional output. Knockdown or pharmacological inhibition of SMYD2 sensitizes breast cancer cells, patient-derived organoids, and tumors to PI3K/AKT inhibition and endocrine therapy through KMT2D K1330 methylation (last panel).
SMYD2 is a member of the SET and MYND domain-containing family of lysine methyltransferases and is known to catalyze the lysine methylation of H3K36 and H3K4,27 as well as non-histone proteins such as p53,18 ERα,13 EZH2,19 p65,15 STAT3,16 RUNX1,17 YY2,12 and E2F.14 Overexpression and deregulation of SMYD2 has been correlated with progression of different tumors, making it a promising therapeutic target in cancer even if its role in tumors is not yet fully understood. To this end, research efforts have been aimed at developing therapeutic agents targeting SMYD2, and multiple SMYD2 inhibitors have been reported.20,21,28 However, most of these inhibitors have been tested in only in vitro systems with modest activity as single agents, and whether they have an effect on tumors and the underlying mechanisms are unknown.20,21 Our data uncovered for the first time that the knockdown or pharmacological inhibition of SMYD2 by multiple inhibitors affects the growth of breast cancer cells and patient-derived organoids and sensitizes them to PI3K/AKT inhibition or endocrine therapy. Importantly, knockdown of SMYD2 in a xenograft model also further sensitized tumors to alpelisib treatment.
Loss of SMYD2 or pharmacological inhibition of SMYD2 also sensitized breast cancer cells to current standard-of-care treatments such as the selective ER downregulator fulvestrant. This was observed with fulvestrant alone as well as the clinically approved combination of fulvestrant with alpelisib. In addition, the AKT inhibitor capivasertib was recently approved in combination with fulvestrant after phase III studies demonstrated significant improvement in progression-free survival among ER+ breast cancer patients whose disease had progressed on aromatase inhibitor therapy with or without a CDK4/6 inhibitor.25 Notably, SMYD2 inhibitors or knockdown also sensitized breast cancer cells to capivasertib.
Importantly, our results also show that the role of SMYD2 in ER+ breast cancer is partly dependent on the methylation of KMT2D, as the K1330A KI cells display common gene expression changes, similar growth profiles, and treatment responses to that of inhibition of SMYD2. However, we speculate that the role of SMYD2 in ER+ breast cancer is also attributed to additional substrates and unknown mechanisms that remain to be delineated.
Regulation of the KMT2D protein by post-translational modification has not been previously uncovered, likely due to the large size of KMT2D (~593 kDa), which renders biochemical assays challenging. Our recent findings have uncovered a multi-faceted regulatory mechanism for KMT2D controlled by AKT/SGK1-mediated phosphorylation at S1330 and SMYD2-catalyzed methylation of KMT2D at the adjacent lysine. While the phosphorylation event negatively regulates the function of KMT2D, the methylation modification positively affects KMT2D function in the context of PI3K inhibition. However, future experiments will need to address the functional interplay of these two modifications and how they alter the function of KMT2D in breast cancer and other settings. This will likely involve association with unknown cofactors that may be disease specific. CRISPR-CAS9 technology in combination with cryoelectron microscopy studies of full-length KMT2D will also provide important structural and functional insights on the regulatory mechanisms of KMT2D post-translational modifications.
Altogether, our findings uncover a mechanism that controls and fine-tunes KMT2D function at tissue-specific enhancers through post-translational modification by SMYD2, providing a rationale for epigenetic therapy using SMYD2 inhibitors in combination with PI3K/AKT inhibitors in ER+/PIK3CA mutant breast cancer (Figure 6). We speculate that methylated KMT2D may function as a biomarker of sensitivity to therapeutic agents targeting SMYD2 in breast cancer. It is likely that SMYD2 potentially regulates other unknown substrates that could mediate breast cancer progression and serve as novel drug targets. Our study also highlights the importance of identifying the tumor subtypes that are highly addicted to chromatin-based mechanisms in order to target the patient population where combination therapy with epigenetic drugs will be the most efficacious. To this end, our study, which connects oncogenic PI3K signaling with epigenetic-based control of gene expression, supports the clinical evaluation of SMYD2 inhibitors in combination with PI3Kα or AKT inhibitors in breast cancer, which may bring transformational benefits to breast cancer patients.
Limitations of the study
In this study, we demonstrate that the histone methyltransferase KMT2D is methylated by SMYD2 at K1330, adjacent to the S1331 site phosphorylated by AKT/SGK. Loss of SMYD2 attenuates PI3K inhibitor-induced chromatin binding of KMT2D at ER loci, downregulating ER-dependent transcription. Inhibition or genetic perturbation of SMYD2 sensitizes cells, patient-derived organoids, and tumors to PI3K/AKT inhibitors. While methyldead KI clones of KMT2D phenocopy many of the effects of SMYD2 inhibition on cell and tumor growth, drug response, and transcriptional output, we hypothesize that SMYD2 effects in ER+ breast cancer may also be attributed to additional substrates of SMYD2. Mechanistically, we have also identified epigenetic interactors that differentially associate with wild-type or methyl-dead KMT2D, which remain to be further characterized. Further investigation is also needed using SMYD2 inhibitors in combination with PI3K/AKT or anti-ER inhibitors in in vivo xenograft models. This could provide additional insights into the clinical potential of SMYD2 inhibitors in combination with PI3K/AKT and endocrine therapy in PI3K-driven, ER+ breast cancer.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Eneda Toska, etoska1@jhmi.edu.
Materials availability
Inhibitors of SMYD2, plasmids, antibodies, engineered cell lines, and other reagents generated in this study are itemized in the key resources table. When applicable, these resources will be made available by contacting the lead contact.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| V5 tag (D3H8Q) | Cell Signaling Technology | Cat# 13202; Lot 4 RRID: AB_2687461 |
| KMT2D K1330me1/me2 | Eurogentec | Custom |
| KMT2D pS1331 | Eurogentec | Custom |
| Vinculin | Cell Signaling Technology | Cat# 4650 RRID: AB_10559207 |
| KMT2D | Fortis LifeSciences | Cat# A300-BL1185, RB X-ALR |
| phospho-AKT (Ser473) (D9E) | Cell Signaling Technology | Cat# 4060 RRID: AB_2315049 |
| phospho-RXRXX(S/T) (23C8D2) | Cell Signaling Technology | Cat# 10001 RRID: AB_10950819 |
| HA tag (C29F4) | Cell Signaling Technology | Cat# 3724 RRID: AB_1549585 |
| SMYD2 (D14H7) | Cell Signaling Technology | Cat #9734 RRID: AB_10889559 |
| DYKDDDDK Tag (D6W5B) Rabbit mAb | Cell Signaling Technology | Cat #14793; Lot 7 RRID: AB_2572291 |
| ECL™ Anti-Rabbit IgG Horseradish Peroxidase linked whole antibody (from donkey) | GE Healthcare | Cat# NA934V; Lot 17010251 RRID: AB_772206 |
| KMT2D ChIP antibody | Sigma Aldrich | Cat# HPA035877 RRID: AB_10670673 |
| V5 antibody | Abcam | Cat#ab9116 RRID:AB_307024 |
| H3 Antibody | Abcam | Cat #ab1791 RRID: AB_302613 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| NEB®5-alpha Competent E. coli (High Efficiency) | NEB | Cat# C2987H |
|
| ||
| Chemicals, peptides, and recombinant proteins 3H-SAM |
Perkin Elmer | NET155V250UC |
|
| ||
| Ultima Gold Scintillation Cocktail | Revvity | Cat# 6013321 |
| α-Cyano-4-hydroxycinnamic acid | Sigma Aldrich | Cat#C8982 |
| KMT2D peptides (unmodified, K1330me1, K1330me2, pS1331) Sequence: AHGGRGRGRARL KSTASSIETLVVAD |
GenScript | Custom |
| V5-agarose beads | Millipore Sigma | Cat# A7345 |
| EZviewTM RED Anti-HA agarose beads | Millipore Sigma | Cat# E6779 |
| H3 peptide (aa1–21) | Genscript | Custom |
| MK2206 | Selleckchem | Catalog No.S1078 |
| Alpelisib | Medchem Express | Cat# HY-15244 |
| Capivasertib | AstraZeneca | N/A |
| Fulvestrant | Selleckchem | Cat# S1191 |
| LLY-507 | Sigma Aldrich | Cat# SML1279 |
| AZ506 | Medchem Express | Cat# HY-134828 |
| EPZ033294 | Viva Biotech | N/A |
| AZ Compound 39 | Astra Zeneca | N/A |
| Doxycycline hydrochloride Dimethyl Sulfoxide |
Millipore Sigma Corning |
Cat #D3447 Cat# 25–950-CQC; Lot 01019003 |
| Crystal Violet | Sigma Aldrich | Cat #V5265 |
| PrestoBlue | ThermoFisher Scientific | Cat #A13262 |
| Y-27632 | Tocris | Cat# 1254 |
| TrypLE | ThermoFisher Scientific | Cat# 12605036 |
| Protein G Dynabeads | ThermoFisher Scientific | Cat #10004D |
| AMPure beads XP | Beckman Coulter | A63880 |
| SYBR™ Select Master Mix for CFX | Applied Biosystems | Cat# 4472942; Lot 00815825 |
| Matrigel | Corning | Cat# 354262 |
| Lipofectamine 3000 | ThermoFisher Scientific | Cat# L3000015 |
| Recombinant AKT1 | SignalChem | Cat#A16–10G |
| Recombinant AKT2 | SignalChem | Cat #A17–10H |
| Recombinant AKT3 | SignalChem | Cat #A18–10G |
| Recombinant SGK1 | SignalChem | Cat #S06–10G |
|
| ||
| Critical commercial assays | ||
|
| ||
| QuikChange Lightning Site-Directed Mutagenesis Kit |
Agilent | Cat#210518 |
| Q5 mutagenesis kit | NEB | Cat #E0552S |
| Roche MTT kit | Millipore Sigma | Cat# 11465007001 |
| Cell-Titer Glo 3D | Promega | Cat# G9681 |
| Qiagen RNeasy® Mini Kit (250) | Qiagen | Cat# 74106; Lot 163029673 |
| iScript™ cDNA Synthesis Kit | Bio-Rad | Cat# 1708891 |
| CUTANA CUT & RUN Kit | EpiCypher | Cat# 14–1048 |
| CUTANA CUT & RUN Library Prep Kit | EpiCypher | Cat# 14–1001 |
|
| ||
| Deposited data | ||
|
| ||
| ChIP-seq/RNA-seq/Cut&Run fastq files | This paper | GSE255295, GSE253957, GSE253958 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| h-MCF7 | ATCC | ATCC® HTB-22™ |
| h-T47D | ATCC | ATCC® HTB-133™ |
| h-293T | ATCC | ATCC® CRL-3216™ |
| HCI-011 | Welm Laboratory | N/A |
| HCI-017 | Welm Laboratory | N/A |
| HCI-018 | Welm Laboratory | N/A |
| Homozygous KMT2D K1330A (AAG>gcG) clones | Synthego | Custom |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Female NSG mice | Antitumor assessment core (MSKCC) | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
|
h-GREB1 F: GACAGGACTGTGGACTGGTG R: ATTTGTTTCCAGCCCTCCTT |
IDT | N/A |
|
h-TFF1 F: TTGTGGTTTTCCTGGTGTCA R: CCGAGCTCTGGGACTAATCA |
IDT | N/A |
|
h-WISP2 F: CAGGGGTCGCAGTCCACAAAA R: AGGCAGTGAGTTAGAGGAAAGG |
IDT | N/A |
|
h-GAPDH
F: AACAGCGACACCCATCCTG R: CATACCAGGAAATGAGCTTGACAA SMYD2 Y240A Mutagenesis Primers F: TTTTACCAGCGCCATTGATCTC CTGTACCCAACG R: ACCTCCTCTCCCGGCTTG |
IDT | N/A |
| SMYD2 Y240A Mutagenesis Primers F: TTTTACCAGCGCCATTGATCTC CTGTACCCAACG R: ACCTCCTCTCCCGGCTTG |
IDT | N/A |
| KMT2D Y5426A Mutagenesis Primers F:GCACACAATGGTTATCGAGGCCAT TGGCACCATCATTCGG R:CCGAATGATGGTGCCAATGGCCT CGATAACCATTGTGTGC |
IDT | N/A |
| KMT2D K1330A Mutagenesis Primers F:AGGACGGGCCCGGCTAGCGTC AACTGCTTCTTC R:GAAGAAGCAGTTGACGCTAGC CGGGCCCGTCCT |
IDT | N/A |
| KMT2D K1330R Mutagenesis Primers F:GACGGGCCCGGCTAAGGTC AACTGCTTCTT R:AAGAAGCAGTTGACCTTAGCC GGGCCCGTC |
IDT | N/A |
| shSMYD2–1 Antisense Guide Sequence TTTGATCTCAGAAATATATGGA |
This paper | N/A |
| shSMYD2–2 Antisense Guide Sequence TAAATGAAAACTGAAATGCTGC |
This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| plx302 V5-tagged KMT2D | Toska et al., 20174 | N/A |
| pCMV HA-tagged KMT2D | Zhang et al., 20159 | N/A |
| pcDNA3.1 FLAG SMYD2 | Generated in the Luo Laboratory | N/A |
| LT3 REPIR | MSKCC RNAi core | N/A |
| pCMV-VSVG | Stewart et al., 200329 | Addgene 8454 |
| pCMV-dR8.2 | Stewart et al., 200329 | Addgene 8455 |
|
| ||
| Software and algorithms | ||
|
| ||
| Gen5 software v1.02 | Agilent | https://www.agilent.com/en/product/cell-analysis/cell-imaging-microscopy/cell-imaging-microscopy-software/biotek-gen5-software-for-imaging-microscopy-1623226 |
| Adobe Illustrator 2024 | Adobe | https://www.adobe.com/products/illustrator/free-trial-download.html |
| Graphpad Prism v9.4.1 | Graphpad Software, Inc. | https://www.graphpad.com/scientific-software/prism/ |
| cBioportal | Cerami et al., 201230 Gao et al., 201331 de Bruijn et al., 202332 |
https://www.cbioportal.org/ |
| FastQC v0.11.9 | Andrews, S. (2010) | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Salmon v1.10.0 | Patro et al., 201733 | https://github.com/COMBINE-lab/Salmon |
| DESeq2 (v1.28.1) | Love et al., 201434 | http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html |
| GSEA software v4.3.2 | Subramanian, Tamayo et al., 200535 and Mootha, Lindgren et al. 200336 |
https://www.gsea-msigdb.org/gsea/index.jsp |
| MSigDB (v7.0) | Subramanian et al., 200535; Liberzon et al., 201537 |
http://software.broadinstitute.org/gsea/msigdb |
| bowtie2 v2.4.2 bedtools v2.30.0 |
Ben Langmead & Steven Salzberg, 201238 Quinian and Hall, 201039 |
https://www.nature.com/articles/nmeth.1923) https://github.com/arq5x/bedtools2 |
| MACS2 v2.1.1 | Zhang et al., 200840 | https://liulab-dfci.github.io/software/ |
| GenomicAlignments v1.36.0 | Lawrence M et al., 201341 | https://bioconductor.org/packages/release/bioc/html/GenomicAlignments.html |
| deepTools | Ramirez et al., 201642 | http://deeptools.ie-freiburg.mpg.de/ |
| HOMER v4.11 | Heinz et al., 201043 | |
|
| ||
| Other | ||
|
| ||
| Tri-Carb 2910TR liquid scintillation counter | Perkin Elmer | N/A |
| BioTek Cytation 5 microplate reader | Agilent | N/A |
| Omega FLUOstar luminometer | BMG Labtech | BMG-415–103 |
| QuantStudio™ 5 | Applied Biosystems | A28570 |
Data and code availability
The next generation sequencing data generated in this manuscript has been deposited at GEO, under GSE255295, GSE253957, and GSE253958. These datasets are publicly available as of the date of publication.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
In vivo animal studies
Animals were maintained based on the institutional guidelines of Memorial Sloan Kettering Cancer Center by the Antitumor Assessment Core Facility. For the shSMYD2 xenograft study 10 × 106 MCF7 cells in 1:1 DF12 media/Matrigel (Corning) were injected subcutaneously into four to six week old female NSG mice maintained in the presence of exogenous estradiol added into the drinking water and doxycycline feed. Mice were randomized when tumors reached ~130 (mm3) of volume. 5 mice (10 tumors) per group were then treated with alpelisib, 25 (mg x kg-1) in 0.5% in carboxymethylcellulose (Sigma-Aldrich) daily or vehicle for the indicated times. Tumors were collected at the end of the experiments, two to 4 h after the last treatment.
For the MCF7 KI cell line xenograft study 10 × 106 MCF7 cells in 1:1 supplemented MEM media/Matrigel (Corning) were injected subcutaneously into four to six week old female NSG mice (n = 15) maintained in the presence of exogenous estradiol added into the drinking water. Tumors were measured twice weekly.
Cell lines
All cell lines were obtained from ATCC and used at low passages. T47D breast cancer cells were maintained in RPMI 1640 with 10% FBS, 1% L-glutamine, and 1% penicillin-streptomycin. MCF7 breast cancer cells were maintained in DF12 DMEM Dulbecco medium with 10% FBS, 1% L-glutamine, and 1% penicillin-streptomycin. HEK 293T cells were maintained in DMEM Dulbecco medium supplemented with 10% FBS, 1% L-glutamine and 1% penicillin-streptomycin. Synthego KI cell lines were maintained in MEM with 1% sodium pyruvate, 1% Glutamax, 10% FBS, 1% penicillin-streptomycin and .01 mg/ml recombinant human insulin. All cell lines were maintained in 5% CO2 at 37°C.
Patient-derived organoids
Organoid models (HCI-011, HCI-017, and HCI-018) were obtained via MTA from Alana Welm and were maintained as previously described.24
METHOD DETAILS
Plasmids & lentiviral transfection
The pCMV-HA-KMT2D plasmid was a gift from Dr. Laura Pasqualucci from Columbia University. The plx302 V5-KMT2D fragment (aa1222–1819) plasmid was generated previously.4 The pcDNA3.1 FLAG-SMYD2 plasmid was generated by restriction enzyme-based cloning in the Luo laboratory. The KMT2D mutants and SMYD2 mutants were generated using a PCR-based site-directed mutagenesis approach. All plasmids were verified by Sanger sequencing. Doxycycline-inducible SMYD2 knockdown cell lines were generated using the LT3REPIR (pRRL) vector (T3G-dsRED-mirE/shRNA-PGK-PURO-IRES-rtTA3), pCMV-VSVG, pCMV-dR8.2 vectors as previously described.4 All shRNAs were designed using the SplashRNA algorithm (PMID:28263295) and expressed from miR-E backbone vectors (PMID: 24332856). Guide sequences are shared in the key resources table.
In vitro methylation assays: Liquid scintillation counting (LSC) and matrix-assisted laser desorption/ionization mass spectrometry (MALDI-TOF)
Scintillation assay reactions were set up using .75μM 3H-SAM (PerkinElmer, 5–15 Ci/mmol in 9:1 v/v of sulfuric acid and ethanol). For the lysine methyltransferase screen enzyme concentrations were optimized in previous assays: 500nM for EZH1, EZH2, SMYD2, SMYD3, Set7/9, SETD2, SUV39H2 and NSD2, GLP (100nM), G9a (200nM) and SETD8 (1mM). All recombinant methyltransferases were purified from E.coli with the exception of the EZH1 (BPS BioScience 51006) and EZH2 (BPS BioScience 51003) complexes. KMT2D peptides were synthesized by GenScript and were used at a final concentration of 5mM. Reactions were run overnight at room temperature. Samples were then spotted on Whatman P81 phosphocellulose filter paper (GE Life Sciences) in triplicate. After drying, filter paper was washed with 50mM sodium bicarbonate buffer to remove any unreacted cofactor. Individual spotted samples were cut into 1.2 × 1.2 cm squares and prepared for liquid scintillation counting by the addition of 500 mL distilled water and 5mL Ultima Gold scintillation cocktail. Samples were allowed to equilibrate for at least 30 min and then CPM (counts per minute) were detected using a Tri-Carb 2910TR liquid scintillation counter (PerkinElmer).
Kinetic measurements (Figure 1I) were performed by Reaction Biology using recombinant Akt1 (SignalChem) in assay buffer (20 mM HEPES pH 7.5, 10 mM MgCl2,1mM EGTA, 0.02% Brij35, 0.02 mg/mL BSA, 0.1mMNa3VO4, 2mMDTT,1%DMSO) Enzyme was added to substrate in reaction buffer, followed by 33P-ATP to initiate the reactions. 6 ATP concentrations were used: 1, 3, 10, 30, 100, 300 μM. Activity was measured using a filter-binding assay.
For the Matrix Assisted Laser Desorption/Ionization (MALDI) mass spectrometry experiment reactions were set up with 500nM SMYD2 enzyme, 20μM peptide and 50μM SAM cofactor. Reactions were run overnight at room temperature. After reactions peptides were separated out of the reaction mixture using ZipTip pipette tips (Millipore Sigma) containing a C18 resin. Samples were pipetted 5–10 times and then the tips were washed with .1% TFA water and subsequently eluted with 50% acetonitrile/.1% TFA water, according to the manufacturer’s protocol. The samples were spotted on a MALDI plate using the dried droplet method; the elution sample was mixed with a solution of α-cyano-hydroxy-cinnamic acid (CHCA) matrix (Protea Biosciences) dissolved in 50% acetonitrile/.1% TFA water. After drying the samples were analyzed on a Voyager-DE STR Biospectrometry instrument (Applied Biosystems) at the Rockefeller University Proteomics Core Facility. Desorption/ionization was obtained using a delayed-extraction, positive ion mode with a 337-nm nitrogen laser.
Immunoprecipitation (IP), immunoblot, in vitro kinase assay, and IP methylation assay
For immunoprecipitation assays, HEK293T cells were transfected with V5-KMT2D fragment (aa1222–1819) or full-length HA-KMT2D using polyethylenimine for 48 h. For experiments with SMYD2 inhibitors the compounds were added 24 h after transfection and cells were collected 72 h after transfection. Cells were washed with PBS and lysed in RIPA buffer with protease inhibitors and phosphatase inhibitors. V5-fragment KMT2D was immunoprecipitated with V5-agarose beads (Sigma A7345) for 2 h at 4°C. After washing, samples were eluted from the beads by heating in SDS loading buffer. Samples were run on 4–12% SDS PAGE gels, transferred to a nitrocellulose membrane at 70V for 2 h and blotted with an anti-V5 antibody (CST D3H8Q), KMT2D K1330me1/me2, and KMT2D pS1331 antibodies generated for this project by Eurogentec. HA-KMT2D was immunoprecipitated with EZviewTM RED Anti-HA agarose beads (Sigma-Aldrich). Samples for western with endogenous KMT2D or HA-KMT2D were resolved on 3–8% gels and transferred to PVDF membranes.
The membranes were probed using specific antibodies-V5 (D3H3Q #13202, CST), Vinculin (#4650, CST), KMT2D (A300-BL1185, RB X-ALR, Fortis LifeSciences), phospho-AKT (Ser473) (D9E #4060, CST), phospho-RXRXX(S/T) (23C8D2 #10001, CST), HA (C29F4 #3724, CST), SMYD2 (D14H7 #9734, CST). pKMT2D (S1331) antibodies were generated by Eurogentec (third bleed) and affinity purified against GRARLKSTASSIC and GRARLKS(PO3H2)TASSIC peptides. K1330me1/2 antibodies were generated by Eurogentec.
For in vitro kinase assays, HEK293T cells were transfected with HA-KMT2D or V5-KMT2D fragment (aa1222–1819) plasmids using polyethylenimine. Cells were collected 48 h after transfection and treated with 2μM Akt1 inhibitor MK2206 for 1 h before collection. Immunoprecipitation was conducted as described above. After washes in lysis buffer, beads were resuspended in kinase reaction buffer (25 mM MOPS, pH 7.2, 12.5 mM β-glycerolphosphate, 25 mM MgCl2, 5 mM EGTA, 2 mM EDTA, and 0.25 mM DTT) and the kinase (Akt1/2/3 or SGK1, SignalChem) and ATP were added at final concentrations of 50nM and 200 μM respectively. Reactions were run for 30 min at 30°C, followed by western blotting with the indicated antibodies.
For IP methylation assays, HA-KMT2D was immunoprecipitated as described above. On-bead enzyme activity was assessed using .75μM 3H-SAM and an H3 peptide (aa1–21) substrate using the scintillation assay. Methylation reactions were conducted in 50mM Tris pH 8, .005% Tween 20 (v/v), .0005% BSA, and 1mM TCEP.
Fractionation assay
Cell pellets were lysed in hypotonic buffer (10mM Hepes-NaOH pH 7.9, 10mM KCl, 1.5 mM KCl, 1.5mM MgCl2, .5mM β-mercaptoethanol with protease inhibitors). After lysis a final volume of 2% NP-40 was added to each sample. After incubation for several minutes on ice, samples were centrifuged 4 °C at 2,400g/rcf for 15 min. The supernatant was collected as the cytoplasmic fraction. The remaining pellet was washed twice with 500ul cold hypotonic buffer and resuspended in 2X cell pellet volume of nuclear lysis buffer (10 mM Tris-HCl pH 7.6,150 mM NaCl, 0.5% NP-40, 1 mM DTT, 2 mM MgCl2, 0.5 mM EDTA with protease inhibitors) for 1 h at 4 °C rotating. Lysed nuclei were pelleted at 16,000g/rcf for 15 min at 4 °C and supernatant was collected as the soluble chromatin fraction (nuclear). The insoluble pellet was resuspended in 500ul shearing buffer (10 mM Tris-HCl, pH 7.6, 1mM EDTA, 0.1% SDS with protease inhibitors) and sonicated with a Covaris E220 ultrasonicator for 720 s. The lysate was centrifuged at 16,000g/rcf for 15 min and the supernatant was collected as the chromatin fraction.
MTT viability assays
For the alpelisib and capivasertib dose curve experiments, 2,500 MCF7 cells expressing doxycycline-inducible shRNAs against SMYD2 were seeded per well in 96-well plates. +Dox conditions were pre-treated for 3 days with 1 μg/mL doxycycline prior to seeding. 24 h after seeding, cells were treated with DMSO or a 2-fold serial dilution of alpelisib or capivasertib, starting at 8 μM and 4μM respectively. Dox was refreshed at this time. Cells were treated for 5 days before MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenylte-trazolium bromide) solution was added and viability was measured.
For the drug combination experiment, 1,000 MCF7 cells expressing dox-inducible shRNAs against SMYD2 were seeded per well in 96-well plates. +Dox conditions were pre-treated for 3 days prior to seeding. 24 h after seeding, cells were treated with DMSO, 100nM Fulvestrant, 1μM Alpelisib, 1μM Palbociclib, Alpelisib + Fulvestrant, or Palbociclib + Fulvestrant. Dox was refreshed at this time. Cells were treated for 10 total days, refreshing dox and drug after 5 days, before MTT solution was added and viability was measured.
For MTT assays with SMYD2 inhibitors, cells were seeded in 96 well plates at 2,500 cells per well and treated with drug after 24 h. DMSO or SMYD2 inhibitors were added at a single dose (5, 10 μM) in combination with a 2-fold serial dilution of alpelisib starting at 8 μM. Five days after inhibitor treatment MTT reagent was added and viability was measured.
Crystal violet assays
For crystal violet assays MCF7 and T47D cells were plated at 20,000 or 40,000 cells per well in 12-well plates. Cells were treated with either BYL719 (125nM), LLY-507 (5μM), or the combination with DMSO as a negative control 24 h after plating. Fresh media with inhibitor was added every three days. Cells were stained with crystal violet after 8 days of inhibitor treatment. For SMYD2 inducible knockdown cell lines cells were treated with 1 μg/mL of doxycycline for 3 days prior to seeding.
K1330A knockin clone generation
Homozygous KMT2D K1330A (AAG>gcG) clones were generated by Synthego. Specific guide RNAs, spCas9 and donor templates were delivered to MCF7 cells via electroporation. Successfully edited pools were subjected to single cell dilution and clonal expansion, with final verification by Sanger sequencing. Cells from the WT MCF7 pool underwent mock transfection.
Proliferation assays with K1330A knockin clones
At day one, 2,500 viable cells were seeded in 180ul of full medium as technical triplicates on a 96-well plate. The day after, cells were treated with either DMSO or the indicated drugs. For drug combination experiments all drugs were used at a dose of 1μM. For the alpelisib and capivasertib dose curves cells were treated with a 2-fold serial dilution starting at 8 μM. At day 7, 20μL of PrestoBlue (#A13262, ThermoFisher) was added to the wells and cells were stained for 30 min in the incubator (37°C, 5% CO2). Then the emitted fluorescence was read on a BioTek Cytation 5 microplate reader (Agilent) at 540/590nm and analyzed using the Gen5 software v1.02. Raw values were corrected for media and normalized to DMSO controls. Normalized values plotted using Prism5 v9.4.1 are means ± SEM of three biological replicates, and p-values were calculated with two-sided unpaired Student’s t tests.
Growth assays with human PDX-derived organoids
Organoids were cultured in matrigel (Corning, 354262) in adequate full media as detailed previously.26 Organoids were then harvested and subjected to two TrypLE treatments (ThermoFisher, 12605036) of 5 min at 37°C with 250 rpm rotation. 5,000 single cells were then embedded in 40μL-domes of matrigel in 12-well plates, flipped and incubated for 15 min at 37°C to allow polymerization. Full medium with 10μM Y-27632 (Tocris, 1254) containing 1μM Fulvestrant, 1μM Alpelisib, 5μM AZ506 or DMSO vehicle control and treated media were refreshed every 3 days for 2 weeks. At day 14, domes were resuspended in 100μL of PBS and 100μL of Cell-Titer Glo 3D (Promega, G9681). Lysates were distributed in black 96-well plates and incubated for 15 min at 37°C before the luminescence was read using the autogain setting on the Omega FLUOstar luminometer (BMG Labtech, BMG-415–103). Raw values were normalized to DMSO controls and normalized values plotted using Prism 5 v9.4.1 are means ± SEM of three biological replicates and pp-values were calculated with two-sided unpaired Student’s t tests.
Survival analysis
TCGA Breast Invasive Carcinoma patients copy number variation data in Nature cohort44 was downloaded from cbioportal. Patients without overall survival information recorded were filtered out and ER positive patients were extracted for survival analysis. Survival of 27 patients with a SMYD2 copy number amplification (CNV = 2) was compared with survival of 167 patients without any copy number alteration (CNV = 0) of SMYD2. Kaplan-Meier curves were used to describe the survival times of patients of both groups. Log rank test was used to compare survival times between two groups. R package “survival” and “survminer” were used to perform and visualize the analysis. The oncoprint was plotted for ER positive patients from the same dataset using Oncoprinter from cbioportal.
Signature activity score
The transcriptomic data of breast cancer patients from TCGA and the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) breast cancer cohorts was obtained from the cBioPortal (http://cbioportal.org).32,30,31 For the analysis, only breast cancer patients with ER-positive status were included. These patients were divided into groups based on the median expression of SMYD2. Estradiol response-related signatures were obtained from MSigDB.35,37 R package GSVA 1.40.1 was used to calculate the activity scores of these signatures. The significance of the activity scores was assessed using a T test.
RNA extraction, cDNA synthesis, and quantitative real-time PCR
Total RNA was extracted from cells using RNeasy Mini kit (Qiagen). cDNA synthesis was performed using the Bio-Rad iScript cDNA synthesis kit according to the manufacturer’s instructions. The qPCR SYBR green master mix (Applied Biosystems) was used to amplify specific cDNA fragments with the oligonucleotides listed below using QuantStudio 5 (Applied Biosystems). The data was analyzed by the change-in-threshold (2—ΔΔCT) method using GAPDH as a housekeeping genes to obtain relative RNA expression.
RNA-seq analysis and gene set enrichment analysis (GSEA)
FastQC v0.11.9 (Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) was used to perform quality control for all sequencing fastq files. These fastq files were then quantified using Salmon v1.10.0.33 Salmon provides fast and bias-aware quantification of transcript expression with the human reference genome hg38. The differentially expressed genes were identified by DESeq2 v1.40.1,34 filtered by pp-values adjusted for multiple tests inflation. The downstream PCA plot, heatmaps, and volcano plots were created based on DESeq2 results with a log-transformed expression matrix.
To perform gene set enrichment analysis, DESeq2 was used to obtain the normalized input gene expression matrix for the GSEA software v4.3.35,36
The reference gene sets for GSEA, specifically the hallmark gene sets (H), positional gene sets (C1), curated gene sets (C2), regulatory target gene sets (C3), computational gene sets (C4), ontology gene sets (C5), oncogenic signature gene sets (C6), immunologic signature gene sets (C7), and cell type signature gene sets (C8), are from The Molecular Signatures Database (MSigDB).35,37
ChIP-seq
Cells were crosslinked in adherent conditions with paraformaldehyde added at a final concentration of 1%, and incubated at room temperature for 15 min. The crosslinked cells were quenched with ice-cold glycine for 5 min, washed with PBS and collected. The cells were then lysed with SDS lysis buffer [10 (mL) of 1% SDS, 10 (mM) EDTA, 50 (mM) Tris-HCl, pH 8.1) containing protease and phosphatase inhibitors (Roche) for 15 min prior to sonication. Cells were sonicated in 10 s pulses for a total of 10 min. The sheared chromatin was diluted with ChIP Dilution buffer [0.01% SDS, 1.1% Triton X-100, 1.2 (mM) EDTA, 16.7 (mM) Tris-HCl, pH 8.1, 167 (mM) NaCl] and incubated overnight with Protein G Dynabeads (Thermo Fisher Scientific), which were pre-incubated with specific antibodies. KMT2D (HPA035877) ChIP antibody was obtained from Sigma-Aldrich. The immune complexes were washed twice with low salt wash buffer [0.1% SDS, 1% Triton X-100, 2 (mM) EDTA, 20 (mM) Tris-HCl, pH 8.1, 150 (mM) NaCl], high salt wash buffer [0.1% SDS, 1% Triton X-100, 2 (mM) EDTA, 20 (mM) Tris-HCl, pH 8.1, 500(mM) NaCl], LiCl wash buffer [0.25 (M) LiCl, 1% NP40, 1% deoxycholate, 1 (mM) EDTA, 10(mM) Tris-HCl, pH 8.1] and 1x TE buffer [10 (mM) Tris-HCl, 1 (mM) EDTA pH 8.0]. The complexes were eluted with elution buffer [1% SDS, 0.1 (M) NaHCO3]. The eluates were reverse crosslinked at 65 (°C) for 4 h by adding 20 (μM) of 5 (M) NaCl followed by proteinase K treatment for 1 h at 4 (°C). DNA fragments were eluted using AMPure beads and generated libraries were high-throughput sequenced using HiSeq 2500 (Illumina).
ChIP-seq analysis
The adaptors in ChIP-seq FASTQ files were trimmed using Trimmomatic. The remaining reads were aligned to hg38 using bowtie2 v2.4.2.38 PCR duplicates were removed using the MarkDuplicates function in Picard. Then, the bw files were generated by the genomeCoverageBed function in bedtools v2.30.0 with a scaling factor of Reads Per Kilobase per Million mapped reads (RPKM). Peaks were called using MACS2 v2.1.1.40 The peak count for each sample was determined using the countOverlaps function from the R package GenomicAlignments v1.36.0.41
Regarding the cluster analyses, for each comparison group, the peaks from two samples were merged together. The scores per peak (SPP) matrix was then calculated on those peaks using corresponding normalized bigwig files with deepTools multiBigwigSummary. The peaks were clustered into three groups by the difference of SPP of two samples (cluster1/unchanged: treated - control <3, cluster2/up-regulated: treated - control ≤ −3, cluster3/down-regulated: treated - control ≥ 3). Heatmaps were plotted on three clusters of reference peaks using computeMatrix and plotHeatmap in deepTools.42
For the overlap of KMT2D and GRHL2 ChIP-seq peaks were called using macs2 and peaks from two replicates (GSM2171834, GSM2171835) were retained and merged together. Merged GRHL2 peaks were intersected with KMT2D peaks using findOverlapsOfPeaks function in R. The overlapped peaks were annotated using HOMER and the pie plots were made to visualize the genomic distribution of the annotations.43 Overlap with H3K27ac ChIP-seq (GSM3526008, GSM3526009) was performed in the same manner.
CUT&RUN
MCF7 cells were transfected with either empty vector or FLAG-SMYD2 for 48 h. FLAG-SMYD2 CUT&RUN were performed with Epicypher’s CUTANA CUT&RUN kit v2.1 and library preparation kit v1.0 following the manufacturer’s instructions using an FLAG antibody (CST 14793). No E. coli spike-in was used. Library quality and concentrations were assessed using the D1000 TapeStation system (Agilent). Libraries were sequenced for 25 cycles in 75-bp paired-end mode on Illumina Novaseq 6000 and data were analyzed as described.45 Briefly, adapters were clipped and paired-end Homo sapiens reads were mapped to UCSC hg38 using Bowtie2 (with parameters: –end-to-end –very-sensitive –no-mixed –no-discordant -q –phred33 -I 10 -X 700). Continuous-valued data tracks (bed-Graph and bigWig) were generated using genomecov in bedtools v.2.30.0 (-bg option) and normalized as fraction of total counts. Genomic tracks were displayed using Integrated Genome Browser. SMYD2 CUT&RUN peaks were called by SEACR (v.1.3) using the stringent settings. Tornado plots and metaprofiles were generated using deepTools v.3.5.1. De novo motif discovery was performed using HOMER v3.4 findMotifsGenome.pl script with default settings.43
Immunoprecipitation-mass spectrometry (IP-MS)
Preparation of lysate, immunoprecipitation, and tryptic peptides: Cell pellets were resuspended in 500 μL of Lysis Buffer with 50mM EPPS((4-(2-Hydroxyethyl)-1-piperazinepropanesulfonic acid), Sigma, cat#H3375), pH 7.5, 150mM NaCl, 1% v/v triton-100 with protease and phosphatase inhibitors and 1 μL of benzonase (334U/μl EMD Millipore, cat# 71206–3) Lysates were clarified by centrifuging at 20,000 g at 4C for 5 min. The supernatants were filtered through 1μm glass fiber plate (Pall Corporation Acropep Advance, cat# 8231) at 1000 X g for 1 min. V5-tagged KMT2D was immunoprecipitated using V5 antibody (ab9116) and protein A DynaBeads. The supernatants were removed and the beads were washed 5 times with ice-cold wash buffer with 50mM EPPS pH 7.5, 150mM NaCl. The beads were digested with 30μL of 10mM EPPS buffer pH 8.5, with 20 ng/μl of trypsin and 10 ng/μl of LysC for 1400 rpm at 37C for 2 h. The digests were transferred into new tubes and digested at 37 C, 1400rpm overnight.
TMT labeling: TMTpro 16 plex label reagent (Thermo, cat#A44520#) were added directly to the digests with anhydrous Acetonitrile (sigma, cat271004) adjusting to 30 percent. The labeling reactions were performed at room temperature for 1 h. The left over TMT tags were quenched with 10μL of 0.5% hydroxylamide (Aldrich, cat#467804) and final concentration to be 0.2% for 15 min. The samples were pulled together and were dried by speed-vacuum.
High pH peptide fractionation: The dried TMT labeled peptides were desalted by a Sep-Pak Vac 1cc (50mg) tC18 Cartridges (Waters, cat WAT054960). The desalted TMT labeled peptides were fractionated by high pH spin column (Thermo Scientific, cat 84868, following vendor’s instruction) into 8 fractions. Four final fractions were combined from the previous 8 fractions (i.e., fractions 1, 5 were pooled, followed by fractions 2, 6, etc.).
LC-MS/MS data collection: Pooled fractions were vacuum-centrifuged then reconstituted in 1% ACN/0.1% FA and 1μg of sample was injected for LC-MS/MS. Four final fractions were analyzed by LC-MS/MS using an Easy-nLC 1200 HPLC (Thermo Fisher Scientific) with a 50 cm (inner diameter 75mm) EASY-Spray Column (PepMap RSLC, C18, 2μm, 100Å ) heated to 60°C coupled to a Orbitrap Fusion Lumos Tribrid Mass Spectrometer (Thermo Fisher Scientific). Peptides were separated at a flow rate of 300 nL/min using a linear gradient of 1–35% acetonitrile (0.1% FA) in water (0.1% FA) over 4 h and analyzed by SPS-MS3. MS1 scans were acquired over a range of m/z 375–1500, 120K resolution, AGC target of 4×105, and maximum IT of 50 ms. MS2 scans were acquired on MS1 scans of charge 2–7 using an isolation of 0.7 m/z, collision induced dissociation with activation of 35%, turbo scan and max IT of 50 ms. MS3 scans were acquired using specific precursor selection (SPS) of 10 isolation notches, m/z range 100–1000, 50K resolution AGC target of 1×105, HCD activation of 65%, and max IT of 150 ms. The dynamic exclusion was set at 38 s.
Raw data files were processed using Proteome Discoverer (PD) version 2.4.1.15 (Thermo Scientific). For each of the TMT experiments, raw files from all fractions were merged and searched with the SEQUEST HT search engine with a Homo sapiens UniProt protein database downloaded on 2019/01/09 (176,945 entries). cysteine carbamidomethylation was specified as fixed modifications, while Methionine oxidation, acetylation of the protein N terminus, TMTpro (K) and TMTpro (N-term) and Deamidation (NQ) were set as variable modification. The precursor and fragment mass tolerances were 10 ppm and 0.6 Da respectively. A maximum of two trypsin missed cleavages were permitted. Searches used a reversed sequence decoy strategy to control peptide false discovery rate (FDR) and 1% FDR was set as threshold for identification.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses and number of replicates are demonstrated in the appropriate figure legends and the method details section. Briefly, p values were calculated using Student’s t test and error bars are ± STD or SEM as indicated. p values for the in vivo experiments were calculated using a two-sided Mann-Whitney U-test.
Supplementary Material
Highlights.
KMT2D is methylated at K1330 by SMYD2
There is crosstalk between phosphorylation of KMT2D at S1331 and methylation at K1330
Loss of SMYD2 abrogates PI3Ki-induced KMT2D chromatin binding and ER-dependent transcription
Inhibition of KMT2D methylation sensitizes cells and tumors to PI3K/AKT inhibition
ACKNOWLEDGMENTS
We would like to acknowledge the MSKCC Proteomics Core for their work on the immunoprecipitation/MS experiments. We would also like to acknowledge Jason Lin for his work in purifying recombinant proteins for the PMT screen. This work has been supported by R01HG010889 to H.J. and R35GM131858 to M.L. It has also been supported by Innovation in Cancer Informatics, an AstraZeneca grant, a Jayne Koskinas Ted Giovanis grant, NCI K22CA245487, R21CA252530, R01CA276187, and Breast Cancer Alliance to E.T.
DECLARATION OF INTERESTS
M. Sallaku is an employee of Loxo Oncology. M. Scaltriti has ownership interest (including patents) in Medendi.org, reports other support from AstraZeneca, and is an employee of AstraZeneca. H.M.C. and T.C. are employees of AstraZeneca. P.C. reports personal fees from Venthera outside the submitted work. P.R. has received institutional grants/funding from Grail, Novartis, AstraZeneca, EpicSciences, Invitae/ArcherDx, Biothernostics, Tempus, Neogenomics, Biovica, Guardant, Personalis, and Myriad and has shares/ownership interest in Odyssey Biosciences as well as consultation/ad-board/honoraria from Novartis, AstraZeneca, Pfizer, Lilly/Loxo, Prelude Therapeutics, Epic Sciences, Daiichi-Sankyo, Foundation Medicine, Inivata, Tempus, SAGA Diagnostics, Paige.ai, Guardant, and Myriad. E.T. reports grants and personal fees from AstraZeneca and consulting from Menarini.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114174.
REFERENCES
- 1.Hanker AB, Sudhan DR, and Arteaga CL (2020). Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell 37, 496–513. 10.1016/j.ccell.2020.03.009 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bosch A, Li Z, Bergamaschi A, Ellis H, Toska E, Prat A, Tao JJ, Spratt DE, Viola-Villegas NT, Castel P, et al. (2015). PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor-positive breast cancer. Sci. Transl. Med 7, 283ra51. 10.1126/scitranslmed.aaa4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.André F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, Iwata H, Conte P, Mayer IA, Kaufman B, et al. (2019). Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med 380, 1929–1940. 10.1056/NEJMoa1813904. [DOI] [PubMed] [Google Scholar]
- 4.Toska E, Osmanbeyoglu HU, Castel P, Chan C, Hendrickson RC, Elkabets M, Dickler MN, Scaltriti M, Leslie CS, Armstrong SA, and Baselga J. (2017). PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science 355, 1324–1330. 10.1126/science.aah6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toska E, Castel P, Chhangawala S, Arruabarrena-Aristorena A, Chan C, Hristidis VC, Cocco E, Sallaku M, Xu G, Park J, et al. (2019). PI3K Inhibition Activates SGK1 via a Feedback Loop to Promote Chromatin-Based Regulation of ER-Dependent Gene Expression. Cell Rep. 27, 294–306.e5. 10.1016/j.celrep.2019.02.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu D, Gao X, Morgan MA, Herz H-M, Smith ER, and Shilatifard A. (2013). The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol. Cell Biol 33, 4745–4754. 10.1128/MCB.01181-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pan Y, Han H, Hu H, Wang H, Song Y, Hao Y, Tong X, Patel AS, Misirlioglu S, Tang S, et al. (2023). KMT2D deficiency drives lung squamous cell carcinoma and hypersensitivity to RTK-RAS inhibition. Cancer Cell 41, 88–105.e8. 10.1016/j.ccell.2022.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alam H, Tang M, Maitituoheti M, Dhar SS, Kumar M, Han CY, Ambati CR, Amin SB, Gu B, Chen T-Y, et al. (2020). KMT2D Deficiency Impairs Super-Enhancers to Confer a Glycolytic Vulnerability in Lung Cancer. Cancer Cell 37, 599–617.e7. 10.1016/j.ccell.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang J, Dominguez-Sola D, Hussein S, Lee J-E, Holmes AB, Bansal M, Vlasevska S, Mo T, Tang H, Basso K, et al. (2015). Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat. Med 21, 1190–1198. 10.1038/nm.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, Jiang M, Hu D, Agirre X, Niesvizky I, et al. (2015). The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat. Med 21, 1199–1208. 10.1038/nm.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang G, Chow RD, Zhu L, Bai Z, Ye L, Zhang F, Renauer PA, Dong MB, Dai X, Zhang X, et al. (2020). CRISPR-GEMM Pooled Mutagenic Screening Identifies KMT2D as a Major Modulator of Immune Checkpoint Blockade. Cancer Discov. 10, 1912–1933. 10.1158/2159-8290.CD-19-1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu X-N, Shi T-T, He Y-H, Wang F-F, Sang R, Ding J-C, Zhang W-J, Shu X-Y, Shen H-F, Yi J, et al. (2017). Methylation of transcription factor YY2 regulates its transcriptional activity and cell proliferation. Cell Discov. 3, 17035. 10.1038/celldisc.2017.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang X, Tanaka K, Yan J, Li J, Peng D, Jiang Y, Yang Z, Barton MC, Wen H, and Shi X. (2013). Regulation of estrogen receptor α by histone methyltransferase SMYD2-mediated protein methylation. Proc. Natl. Acad. Sci. USA 110, 17284–17289. 10.1073/pnas.1307959110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kontaki H, and Talianidis I. (2010). Lysine methylation regulates E2F1-induced cell death. Mol. Cell 39, 152–160. 10.1016/j.molcel.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 15.Ea C-K, and Baltimore D. (2009). Regulation of NF-kappaB activity through lysine monomethylation of p65. Proc. Natl. Acad. Sci. USA 106, 18972–18977. 10.1073/pnas.0910439106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dasgupta M, Dermawan JKT, Willard B, and Stark GR (2015). STAT3-driven transcription depends upon the dimethylation of K49 by EZH2. Proc. Natl. Acad. Sci. USA 112, 3985–3990. 10.1073/pnas.1503152112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao X, Jankovic V, Gural A, Huang G, Pardanani A, Menendez S, Zhang J, Dunne R, Xiao A, Erdjument-Bromage H, et al. (2008). Methylation of RUNX1 by PRMT1 abrogates SIN3A binding and potentiates its transcriptional activity. Genes Dev. 22, 640–653. 10.1101/gad.1632608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang J, Perez-Burgos L, Placek BJ, Sengupta R, Richter M, Dorsey JA, Kubicek S, Opravil S, Jenuwein T, and Berger SL (2006). Repression of p53 activity by Smyd2-mediated methylation. Nature 444, 629–632. 10.1038/nature05287. [DOI] [PubMed] [Google Scholar]
- 19.Zeng Y, Qiu R, Yang Y, Gao T, Zheng Y, Huang W, Gao J, Zhang K, Liu R, Wang S, et al. (2019). Regulation of EZH2 by SMYD2-Mediated Lysine Methylation Is Implicated in Tumorigenesis. Cell Rep. 29, 1482–1498.e4. 10.1016/j.celrep.2019.10.004. [DOI] [PubMed] [Google Scholar]
- 20.Nguyen H, Allali-Hassani A, Antonysamy S, Chang S, Chen LH, Curtis C, Emtage S, Fan L, Gheyi T, Li F, et al. (2015). LLY-507, a Cell-active, Potent, and Selective Inhibitor of Protein-lysine Methyltransferase SMYD2. J. Biol. Chem 290, 13641–13653. 10.1074/jbc.M114.626861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cowen SD, Russell D, Dakin LA, Chen H, Larsen NA, Godin R, Throner S, Zheng X, Molina A, Wu J, et al. (2016). Design, Synthesis, and Biological Activity of Substrate Competitive SMYD2 Inhibitors. J. Med. Chem 59, 11079–11097. 10.1021/acs.jmedchem.6b01303. [DOI] [PubMed] [Google Scholar]
- 22.Herz H-M, Mohan M, Garruss AS, Liang K, Takahashi Y-H, Mickey K, Voets O, Verrijzer CP, and Shilatifard A. (2012). Enhancer-associated H3K4 monomethylation by Trithorax-related, the Drosophila homolog of mammalian Mll3/Mll4. Genes Dev. 26, 2604–2620. 10.1101/gad.201327.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jozwik KM, Chernukhin I, Serandour AA, Nagarajan S, and Carroll JS (2016). FOXA1 Directs H3K4 Monomethylation at Enhancers via Recruitment of the Methyltransferase MLL3. Cell Rep. 17, 2715–2723. 10.1016/j.celrep.2016.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu G, Chhangawala S, Cocco E, Razavi P, Cai Y, Otto JE, Ferrando L, Selenica P, Ladewig E, Chan C, et al. (2020). ARID1A determines luminal identity and therapeutic response in estrogen-receptor-positive breast cancer. Nat. Genet 52, 198–207. 10.1038/s41588-019-0554-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turner NC, Oliveira M, Howell SJ, Dalenc F, Cortes J, Gomez Moreno HL, Hu X, Jhaveri K, Krivorotko P, Loibl S, et al. (2023). Capivasertib in Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med 388, 2058–2070. 10.1056/NEJMoa2214131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guillen KP, Fujita M, Butterfield AJ, Scherer SD, Bailey MH, Chu Z, DeRose YS, Zhao L, Cortes-Sanchez E, Yang C-H, et al. (2022). A human breast cancer-derived xenograft and organoid platform for drug discovery and precision oncology. Nat. Cancer 3, 232–250. 10.1038/s43018-022-00337-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown MA, Sims RJ, Gottlieb PD, and Tucker PW (2006). Identification and characterization of Smyd2: a split SET/MYND domain-containing histone H3 lysine 36-specific methyltransferase that interacts with the Sin3 histone deacetylase complex. Mol. Cancer 5, 26. 10.1186/1476-4598-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomenius MJ, Totman J, Harvey D, Mitchell LH, Riera TV, Cosmopoulos K, Grassian AR, Klaus C, Foley M, Admirand EA, et al. (2018). Small molecule inhibitors and CRISPR/Cas9 mutagenesis demonstrate that SMYD2 and SMYD3 activity are dispensable for autonomous cancer cell proliferation. PLoS One 13, e0197372. 10.1371/journal.pone.0197372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen ISY, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. 10.1261/rna.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404. 10.1158/2159-8290.CD-12-0095 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1. 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Bruijn I, Kundra R, Mastrogiacomo B, Tran TN, Sikina L, Mazor T, Li X, Ochoa A, Zhao G, Lai B, et al. (2023). Analysis and Visualization of Longitudinal Genomic and Clinical Data from the AACR Project GENIE Biopharma Collaborative in cBioPortal. Cancer Res. 83, 3861–3867. 10.1158/0008-5472.CAN-23-0816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrå le M, Laurila E, et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 34, 267–273. 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 37.Liberzon A, Birger C, Thorvaldsdó ttir H, Ghandi M, Mesirov JP, and Tamayo P. (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425. 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, and Carey VJ (2013). Software for computing and annotating genomic ranges. PLoS Comput. Biol 9, e1003118. 10.1371/journal.pcbi.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T. (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165. 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cancer Genome Atlas Network (2012). Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meers MP, Tenenbaum D, and Henikoff S. (2019). Peak calling by Sparse Enrichment Analysis for CUT&RUN chromatin profiling. Epigenet. Chromatin 12, 42. 10.1186/s13072-019-0287-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The next generation sequencing data generated in this manuscript has been deposited at GEO, under GSE255295, GSE253957, and GSE253958. These datasets are publicly available as of the date of publication.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.