

Abstract
Staphylococcus aureus is a facultative intracellular pathogen of human macrophages, which facilitates chronic infection. The genotypes, pathways, and mutations influencing that phenotype remain incompletely explored. Here, we used two distinct strategies to ascertain S. aureus gene mutations affecting pathogenesis in macrophages. First, we analyzed isolates collected serially from chronic cystic fibrosis (CF) respiratory infections. We found that S. aureus strains evolved greater macrophage invasion capacity during chronic human infection. Bacterial genome-wide association studies (GWAS) identified 127 candidate genes for which mutation was significantly associated with macrophage pathogenesis in vivo. In parallel, we passaged laboratory S. aureus strains in vitro to select for increased infection of human THP-1 derived macrophages, which identified 15 candidate genes by whole-genome sequencing. Functional validation of candidate genes using isogenic transposon mutant knockouts and CRISPR interference (CRISPRi) knockdowns confirmed virulence contributions from 37 of 39 tested genes (95%) implicated by in vivo studies and 7 of 10 genes (70%) ascertained from in vitro selection, with one gene in common to the two strategies. Validated genes included 17 known virulence factors (39%) and 27 newly identified by our study (61%), some encoding functions not previously associated with macrophage pathogenesis. Most genes (80%) positively impacted macrophage invasion when disrupted, consistent with the phenotype readily arising from loss-of-function mutations in vivo. This work reveals genes and mechanisms that contribute to S. aureus infection of macrophages, highlights differences in mutations underlying convergent phenotypes arising from in vivo and in vitro systems, and supports the relevance of S. aureus macrophage pathogenesis during chronic respiratory infection in CF. Additional studies will be needed to illuminate the exact mechanisms by which implicated mutations affect their phenotypes.
Author summary
Staphylococcus aureus is a significant cause of disease and can cause chronic infections that are extremely difficult to treat. One of the reasons that S. aureus persists so effectively in the human body is its ability to enter and survive within macrophages, which protects the bacteria from immune clearance and antibiotic therapies. A better understanding of the genes and mechanisms that S. aureus uses to infect macrophages could lead to new, more effective treatments for chronic infection. Here, we identified gene mutations associated with improved macrophage pathogenesis in S. aureus by studying isolates from chronic respiratory infection in vivo and using a laboratory macrophage model in vitro. We identified multiple, previously unknown genes and functions that can influence S. aureus macrophage pathogenesis.
Introduction
Staphylococcus aureus is an important cause of chronic disease affecting a variety of organ systems, and incurs significant morbidity and mortality worldwide [1]. Chronic S. aureus infections can persist or relapse for years to decades, even despite therapeutic intervention with antibiotics [2–7]. The tenacity of S. aureus during chronic infection is attributable in part to the emergence of adaptations that increase the organism’s fitness over time [8–11]. Among other features, studies have shown that changes promoting the invasion and persistence of S. aureus within host cells, particularly macrophages [1,12–18], occur frequently in chronic diseases [16,19–23]. As a facultative intracellular pathogen, S. aureus is able to evade the immune system and establish a microbial reservoir able to perpetuate persistent infection in a variety of clinical conditions [1,12–15,17,18,21,24,25].
Factors and pathways important for intracellular invasion and survival of S. aureus have been identified in prior studies [12–14,26]. These comprise a diverse range of mechanisms affecting intracellular survival and subsequent dispersal, including manipulation of host cell autophagy, metabolism, programmed cell death, and self-modulation of virulence gene expression [27]. Nevertheless, the genes contributing to such phenotypes remain incompletely characterized, owing to the inherent complexity of these processes and their dependence on both strain- and host-encoded features [26]. Moreover, prior studies have focused on identifying genes that are essential to S. aureus intracellular pathogenesis, rather than spontaneous chromosomal mutations that are selected during infection and that drive such phenotypes in vivo.
Here, we employed a multiphasic approach to investigate mutations affecting intracellular pathogenesis of S. aureus within human macrophages, using cystic fibrosis (CF) as a paradigm for chronic infection [1,18,21]. In order to explore pathogenesis-enhancing mutations arising in vivo during human infection, we conducted genome-wide association studies (GWAS) of clonally related S. aureus isolates longitudinally obtained from CF patient airways at the time of initial isolation and late in the course of disease [4]. To identify mutations relevant to those same phenotypes in defined genetic backgrounds and under well-controlled experimental conditions, we separately performed artificial selection of two phylogenomically distinct S. aureus laboratory strains for intracellular pathogenesis using a scalable human macrophage cell line model (THP-1) [28,29]. Using a combination of isogenic transposon mutant knockouts [30] and a newly developed CRISPR interference (CRISPRi) [31] knockdown vector, we subsequently performed functional validation of genes affecting S. aureus macrophage pathogenesis identified by both strategies.
Materials and methods
Laboratory S. aureus strains, cell lines, and growth conditions
S. aureus strain ATCC29213 was obtained from the American Type Culture Collection. Strain JE2 and JE2 transposon mutants were from the Biodefense and Emerging Infections Research Resources Repository (BEI Resources). All bacteria in this study were maintained in LB medium supplemented with hemin, menadione, and thymidine (sup-LB) in order to support potentially relevant auxotrophic mutants [32].
THP-1 cells were obtained from ATCC and cultured according to the supplier’s recommendations at 37°C in a humidified 5% (v/v) CO2 air atmosphere in RPMI 1640 medium (Gibco) supplemented with 0.1 mg/ml l-glutamine, 0.1 mg/ml streptomycin, 100 U/ml penicillin (PAA) and 20% (v/v) Nu-Serum Serum Replacement (Corning).
S. aureus clinical isolates
Control clinical isolates of methicillin-sensitive S. aureus (MSSA) and methicillin-resistant S. aureus (MRSA), derived from individuals without CF and from sites not involved in active disease, were obtained from fully de-identified surveillance swabs [33] at the University of Washington Clinical Microbiology Laboratory following the completion of clinical testing. MSSA isolates were obtained by streaking swabs onto mannitol-salt agar, and MRSA isolates from streaking onto chromogenic MRSA selective plates (bioMerieux, Marcy-l’Étoile, France). Control clinical isolates (n = 180) were subjected to whole-genome sequencing as described below.
Clinical isolates from children with CF were previously described and characterized by whole-genome sequencing [4], constituting 1,382 longitudinally-collected S. aureus isolates collected from 246 participants. For clonally related isolates derived from the same patient (n = 237 lineages), we selected the first S. aureus isolate that was initially cultured from the patient and the last isolate temporally collected during the study period [4]. Secondary data, but no human subjects, were analyzed in this study. From this collection of paired clinical isolates, we further reduced the set analyzed in this study to a cohort of 57 lineages identified from the phylogeny of that collection [4] which spanned the genomic diversity present within the collection.
For each such lineage, we identified a matching, phylogenomically similar control strain to minimize lineage-specific effects [34] in comparisons between clinical and control strains. To accomplish this, whole-genome sequencing data from CF and control isolates were subjected to de novo assembly using ABySS 2.0 [35]. We generated a core-genome alignment containing the entire control population and the first isolate from each CF lineage using ROARY [36]. SNP-sites [37] was then used to calculate pairwise core-genome distances between CF-derived and control isolates. For each CF-derived isolate, a phylogenomically similar match from the control population was then identified using an optimal matching strategy to minimize the global distance between all matched cases and controls in the set [38] with a maximum core-genome SNP caliper of 6% and a maximum control to clinical isolate matching ratio of two. This process resulted in matching of the 57 CF-derived isolate pairs to 33 distinct control isolates. All isolates (57 CF first-collected isolates, 57 CF last-collected isolates, and 33 matched control isolates) were subjected to characterization by intracellular pathogenesis assays as described above (S1 Table).
Intracellular pathogenesis assays and selection procedures
48 hours prior to bacterial infection, THP-1 cells were differentiated into monocytes with PMA (Sigma) solubilized in DMSO at a final concentration of 10 ng/ml [39–41]. PMA-containing medium was removed from cells 24 hours after treatment. Cells were washed with incubation medium (IM, RPMI 1640 containing no amendments) and incubated for 24 additional hours.
Bacteria were expanded overnight in sup-LB broth, washed twice in Dulbecco’s phosphate-buffered saline (DPBS) (Difco) to remove soluble toxins, and cell density assessed by optical density at 600 nm (OD600). Six replicates were independently passaged in parallel for each strain.
Intracellular pathogenesis assays were based on previously described lysostaphin protection assays [42]. Briefly, bacteria were combined with differentiated THP-1 cells at a multiplicity of infection (MOI) of 10 in IM for 1 h, with aliquots of inoculum plated to empirically determine the number of bacteria applied. The medium was replaced with RPMI 1640 containing 50 μg/ml lysostaphin (Sigma), and selections were performed at 37°C with 5% atmospheric CO2. 3 h or 48 h post-inoculation, host cells were washed with DPBS and lysed with water. Serial dilutions of lysate were plated onto sup-LB agar to evaluate the number of viable bacteria. At least 4 independent replicates were used to assess the number of viable cells at both 3 and 48 hours. Viable bacteria counted at 3 h were normalized to empiric counts of the bacteria initially applied to determine the proportion of inoculated cells remaining viable after early macrophage entry (measurement of “macrophage invasion”). Viable organisms counted at 48 h were normalized to the number of viable organisms counted at 3 hours for the same isolate (“macrophage survival index”). Measurements of invasion phenotypes were normalized to in-run controls of the parental JE2 or ATCC29213 strains, as appropriate, to control for possible batch effects, whereas the macrophage survival index was inherently normalized for invasion efficiency. Invasion testing of clinical isolates and lineage-matched controls were normalized to an in-run JE2 control. Parental JE2 and ATCC29213 strain invasion was measured as absolute (non-normalized) values collected in a single experiment, due to lack of an external comparator.
For passaging experiments, procedures were completed essentially as described for intracellular pathogenesis assays, except that an initial MOI of 100 was used, lysate was collected only at 48 h, and bacteria were transferred to sup-LB broth for overnight expansion prior to the next passage. As the bacteria became more efficient at infection with increased passaging, it was necessary to empirically reduce the MOI over time to prevent host cells from undergoing lysis during infection while maintaining selective pressure for mutants with increased cytotoxic capability. Consequently, for the first 8 passages, an MOI of 100 was used, after which the MOI was reduced to 10.
Whole-genome sequencing and variant analysis
Whole-genome sequencing was performed as previously described [43] using MiSeq and NextSeq500 platforms with 300 cycle chemistries (Illumina, San Diego, CA). Briefly, variant calling was conducted as before [43] utilizing reference genomes identified as those most closely matching the de novo assemblies of sequenced isolates. Mutations were manually verified using the Integrative Genomics Viewer [44] prior to annotation of sequence features. Common gene names were established using BLAST [45] searches of target gene sequences against S. aureus reference genomes and by cross-correlating gene identifiers against pan-genome identifiers in the AureoWiki database [46]. MLST was determined using mlst-2.0.3 (https://github.com/tseemann/mlst) in conjunction with the PubMLST database [47].
Bacterial genome-wide association studies
Three complementary classes of bacterial GWAS were conducted using clinical isolates and matched control strains to identify associations between bacterial genotypes and macrophage pathogenesis phenotype measurements.
To assess the association of specific genotypes with phenotypes in this phylogenetically diverse collection, we performed reference-free unitig analysis with pyseer (v1.3.10) [48]. We adjusted for population structure using a mixed effects model with an approximate maximum likelihood phylogeny constructed from the ROARY core-genome alignment using Fastree2.1.11 [49]. Phenotype measurements for macrophage invasion and macrophage survival index were provided as continuous variables. Both CF isolates and matched controls were included in this analysis.
Different mutations occurring in a gene may confer equivalent phenotypic effects encoded by distinct genotypes. Unitig-based analysis has less power to detect this class of genotype-phenotype association, which is better assessed using kernel-based models of association between the collective “burden” of mutations within a gene and a phenotype. We therefore performed burden testing using the pyseer rare variants method, considering all non-synonymous mutations as equally weighted and using a mixed effects model incorporating the same phylogenetic tree as above. Both CF isolates and matched controls were included in this analysis.
To identify de novo mutations occurring within clonally related S. aureus lineages from an individual host, whole-genome sequences of the first- and last-collected clinical isolate from each patient were compared. For each gene in the pangenome, a multivariable linear regression analysis was performed in R version 4.2.2 to assess the independent association between the presence of a non-synonymous de novo mutation in that gene (primary predictor variable) with the absolute value of change in measured phenotype between first and last isolates (outcome variable), with adjustment for changes in two other phenotypes (covariates): biofilm formation (measured as previously [50], S1 Table) and the alternate macrophage invasion phenotype measured in this study (i.e., intracellular survival for analysis for invasion activity, and vice versa). Only CF isolates were included in this analysis.
Candidate genes implicated in invasion and survival phenotypes in vivo by GWAS were selected for functional testing by prioritizing those with high levels of association in a single GWAS model and those that were independently identified in more than one model, including associations with lower individual levels of significance. For unitig analyses, we selected genes having three or more component unitigs achieving Bonferroni-adjusted levels of significance with alpha values of 0.05. For burden testing analyses of both phenotypes, all genes achieving Bonferroni-adjusted significance using an alpha value of 0.05 were selected. For analysis of de novo mutation within clinical isolate lineages, we selected all genes with p-values<0.05 and association scores in the top 5%. Lastly, candidate genes showing overlap between GWAS analyses for a phenotype (those with a one or two unitigs meeting Bonferroni-adjusted significance and further identified as undergoing de novo mutation between a first-last pair) were selected for validation.
S. aureus CRISPRi knockdown
We generated a CRISPRi [31] vector for use in S. aureus by modifying our earlier system for CAS9-mediated counterselection [51]. Briefly, dCAS9 from pdCAS9 [52] was cloned under expression of the constitutive p23 promoter into plasmid pCN50wt [53] as described previously [51], followed by addition of a synthetic sgRNA under control of the PRAB17 promoter as previously [51]. To facilitate screening for successful guide RNA cloning, a GFP expression cassette was ultimately cloned into the BSAI site of the sgRNA to generate plasmid pCRISPRi (S2 Table). Guide RNAs were designed against coding sequences using CRISPOR [54] with screening for off-target effects performed against the S. aureus USA300_FPR3757 genome, and selecting guide RNAs designed in the “reverse” orientation of the chosen coding sequence. Guide RNA templates were synthesized as ssDNA oligonucleotides (IDT, S2 Table) with appropriate tail sequences, then phosphorylated, annealed, and cloned into the BSAI site of pCRISRPi as described elsewhere [51]. After transformation into E. coli, plasmid was purified from a transformant lacking fluorescence and silencing vectors were subsequently transformed into S. aureus JE2 as described elsewhere [51]. Gene knockdown was assessed using gene-targeted real-time PCR (S2 Table) of cDNA prepared from mid-log phase growth cells, normalizing gene-specific expression to that of gyrA. Gene expression levels in individual CRISPRi mutants were compared to relative gene expression obtained for a CRISRPi knockdown targeted to a biologically irrelevant sequence (GFP) [55] using the ΔΔCt method (S3 Table).
Quantitative macrophage pathogenesis phenotypes of CRISPRi mutants were assessed as above, but including 10 μg/mL chloramphenicol in all culture media to select for retention of plasmids, and comparing metrics to those of a GFP-silenced CRISPRi strain [56]. The GFP-silenced CRISPRi strain was found to be equivalent to those of the wild-type strain carrying a GFP-expressing plasmid built on the same vector backbone.
Complementation studies
Genes for complementation studies were PCR amplified from JE2 genomic DNA (S2 Table) and cloned into vector pCN50wt. Where possible, the native promoters of cloned genes were used to drive gene expression by including the intergenic DNA falling between the gene of interest and the upstream gene. Three genes (saeS, B7H15_07805, and B7H15_05310) were assumed to be expressed within an operon on the basis of a gene in the same orientation terminating <30 bp upstream, and were therefore expressed under control of the constitutive Pcap promoter using PCR to add that element [57]. Transposon mutants were transformed with matched complementation vectors and with pCN50wt expressing superfolder GFP (ordered as a synthetic gene cassette, S2 Table) [58], which served as a comparator for phenotypic studies. Measurements of macrophage invasion and macrophage survival index were directly compared between complemented and GFP vector-transformed transposon mutant strains assayed in parallel, at least in quadruplicate.
Macrophage viability assessments
The number of viable and dead macrophages was assessed 48 hours after S. aureus infection using tryptan blue exclusion assays as described elsewhere [59], with measurements taken at least in triplicate.
Growth rate analyses
Growth rates were monitored by absorbance at OD600 during incubation at 37°C in sup-LB broth, using a BioTek Synergy H1 Plate reader (Agilent), with measurements taken in 10 minute intervals and experiments performed at least in triplicate.
Results
Intracellular pathogenesis of macrophages is elevated in S. aureus isolates from people with CF
We ascertained whether S. aureus from chronic airway infections in people with CF were subject to selection for enhanced intracellular macrophage pathogenesis in vivo. We utilized a previously described collection of clonally related S. aureus isolate pairs collected longitudinally over time from pediatric participants with CF [4], selecting a subset of 57 distinct clinical first- and last-collected isolate pairs that represented the genomic diversity present in the collection (S1 Table). To provide phylogenetically matched control strains for comparison, we compared each CF clinical isolate against a population of S. aureus control strains that were not derived from active disease and which originated from individuals without CF in order to identify those with the most closely matched genomes (n = 33).
CF isolates and lineage matched controls were assayed for intracellular pathogenesis phenotypes in differentiated THP-1 cells, which have previously been used as a model for S. aureus macrophage interactions [28,56,60]. Macrophage intracellular pathogenesis can be regarded either with respect to the proportion of bacteria that are able to enter and evade initial killing within host cells after phagocytosis (which in this study we term “invasion”), or to the rate of ongoing bacterial survival within host cells over longer periods of time (which here we term “survival”). We therefore assessed these properties separately: invasion was measured as the fraction of viable intracellular bacteria recovered from the initial inoculum 3 hours after sterilization of the extracellular compartment, whereas survival was assessed 48 hours after initial THP-1 host cell infection as the fraction of viable intracellular bacteria recovered at that time relative to the number ascertained at 3 hours.
We found that early S. aureus isolates obtained from individuals with CF by serial surveillance (i.e., those from the time of initial detection) exhibited capacities for macrophage invasion similar to those measured in phylogeny-matched control isolates from the general population. However, the final isolates collected from those same patient lineages after persistent respiratory infection (median 22.9 months, interquartile range 5.1 months), had significantly higher metrics of invasion than the early isolates (Fig 1A). 32 of the isolate pairs showed significantly (p<0.05, two-tailed T test) increased invasion phenotypes over time, while 14 demonstrated decreases, and 11 showed no significant change (S1 Table and S2A Fig). The aggregate increase in intracellular invasion phenotypes result was significant both when comparing groups of later isolates against the first clonally-related isolate collected from the same patient (p = 0.041, paired two-tailed T test) and against phylogeny matched controls from unaffected individuals (p = 0.005, paired two-tailed T test). In contrast, 20 of the isolate pairs showed significantly (p<0.05, two-tailed T test) increased levels of macrophage intracellular survival over time, while 20 evidenced decreases and 11 had no significant change in that phenotype. (S1 Table and S2B Fig). Differences between groups of first, last, and control isolates did not differ significantly among comparator populations in macrophage survival when considered in aggregate (Fig 1B).
Fig 1. Macrophage pathogenesis phenotypes of S. aureus lineages from individuals with CF over time and phylogenetically matched control strains.
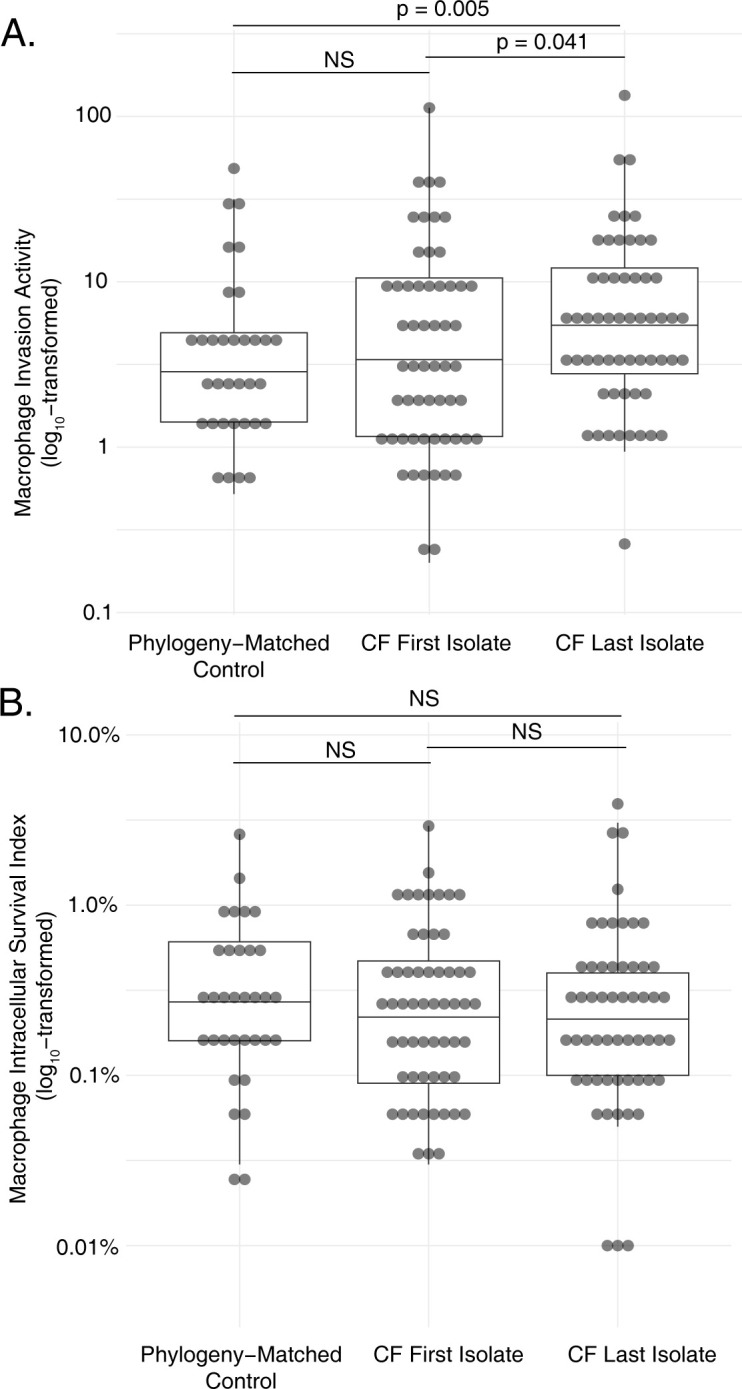
Phenotypes of S. aureus strains isolated from respiratory cultures of individuals with CF at the time of initial isolation during serial surveillance are compared to the last-collected strains from same patient, and to phylogeny-matched strains from unaffected controls. Measures of macrophage invasion, relative to in-batch testing of the JE2 control strain (A), and macrophage intracellular survival (B) are separately reported. Statistical significance was assessed using paired two-tailed T tests.
These findings indicate that, at the time of initial detection, S. aureus strains infecting individuals with CF begin with capacities for macrophage invasion similar to strains from general population that are not involved in chronic disease, but that selective pressure for that phenotype occurs in the CF airway in vivo. Persistent survival within macrophages does not appear to be as strongly selected in CF respiratory infections, although it may be important in a subset of individual strains or clinical cases.
Genomic analysis identifies candidate genes associated with intracellular macrophage pathogenesis in vivo
In order to identify genetic variation associated with enhanced macrophage pathogenesis phenotypes in vivo, we performed bacterial GWAS [48]. For both invasion and survival phenotypes, three complementary models of genetic association were applied (S1 Fig).
First we performed unitig-based testing, a reference-free method which identifies unique, variable-length sequences that are associated with a phenotype and incorporates statistical adjustment for bacterial population structure [61]. This analysis (S1A Fig) identified 179 sequences in 110 genes significantly associated with macrophage invasion (S4 Table). However, the unitigs most highly associated with macrophage invasion comprised an apparent haplotype, indicating insufficient sample diversity to resolve a specific candidate risk allele by this method and requiring prioritization of genes comprising multiple significantly associated unitigs for functional validation. No unitig sequences were significantly associated with survival.
Second, we conducted burden testing [62], which considers the collective impact of independent, non-synonymous variants occurring within a single annotated gene. This analysis (S1B Fig, S5 Table and S6 Table) identified five genes significantly associated with macrophage invasion (purR, folB, sirA, B7H15_03165, B7H15_06690) and three with macrophage survival (recG, B7H15_09405, B7H15_06245).
Last, we examined non-synonymous de novo mutations occurring between temporally first- and last-collected CF clinical isolates. This method of comparison provides the most robust control for bias related to residual population structure and reference annotation. This strategy (S1C Fig) identified 16 (S7 Table) and four genes (clfA, fnbA, sdrE, and a hypothetical protein, S8 Table) whose spontaneous mutation in vivo was associated with changes in macrophage invasion and survival, respectively. To assess the functional impact of such mutations, we examined the proportion of stop-gain mutations and indels (which are more likely to confer loss-of-function effects) to missense changes (which could confer carry either loss-of-function or gain-of-function effects) to assess for statistical imbalance between these classes for each gene. We found that three of the 16 invasion-associated genes (19%) and three of four of the survival-associated genes (75%) exhibited a statistically significant (p<0.05, Fisher Exact Test) excess of loss-of-function associated mutation types, although the missense mutations occurring in other genes may also exert similar effects. No genes demonstrated a proportional excess of missense mutation or patterns of mutation at common residues that would be expected from gain-of-function effects.
Together, these analyses implicate variation in 127 unique candidate genes as being correlated with macrophage pathogenesis in vivo, and suggest that mutations occurring in vivo frequently confer loss-of-function effects.
Serial passaging selects mutants with elevated intracellular pathogenesis in macrophages in vitro
To select for mutants having greater capacity to invade and survive within host macrophages under well-controlled laboratory conditions, and in well-defined bacterial genetic backgrounds, we separately conducted serial passaging experiments of S. aureus strains in the presence of THP-1 cell line-derived macrophages. We characterized the initial capacity for macrophage pathogenesis of two well-defined and phylogenomically distinct S. aureus reference strains, JE2 (Sequence Type 8, Clonal Complex 8) and ATCC29213 (Sequence Type 5, Clonal Complex 5). Viable bacteria from both strains were recovered three hours after macrophage invasion; however, a significantly greater proportion of JE2 were internalized at three hours relative to ATCC29213 (Fig 2A, p<0.0001, unpaired two-tailed T test). After normalizing for these differences in initial invasion, the natural capacity of these strains for ongoing intracellular survival at 48 hours also differed, with a significantly greater fraction of initially internalized JE2 surviving relative to ATCC29213 (Fig 2B, p<0.0001, unpaired two-tailed T test). To assess the fate of infected macrophages, we quantified their absolute number and the proportion of dead cells 48 hours after S. aureus was applied. Compared to an uninfected macrophage control, neither significant reduction in the number of macrophages (p>0.12, two-tailed T test) nor significant increases in the proportion of dead macrophages (p>0.45, two-tailed T test) were observed after infection with either S. aureus strain, indicating that S. aureus did not kill infected macrophages by lytic or non-lytic mechanisms.
Fig 2. Macrophage pathogenesis phenotypes of S. aureus laboratory strains ATCC29213 and JE2.
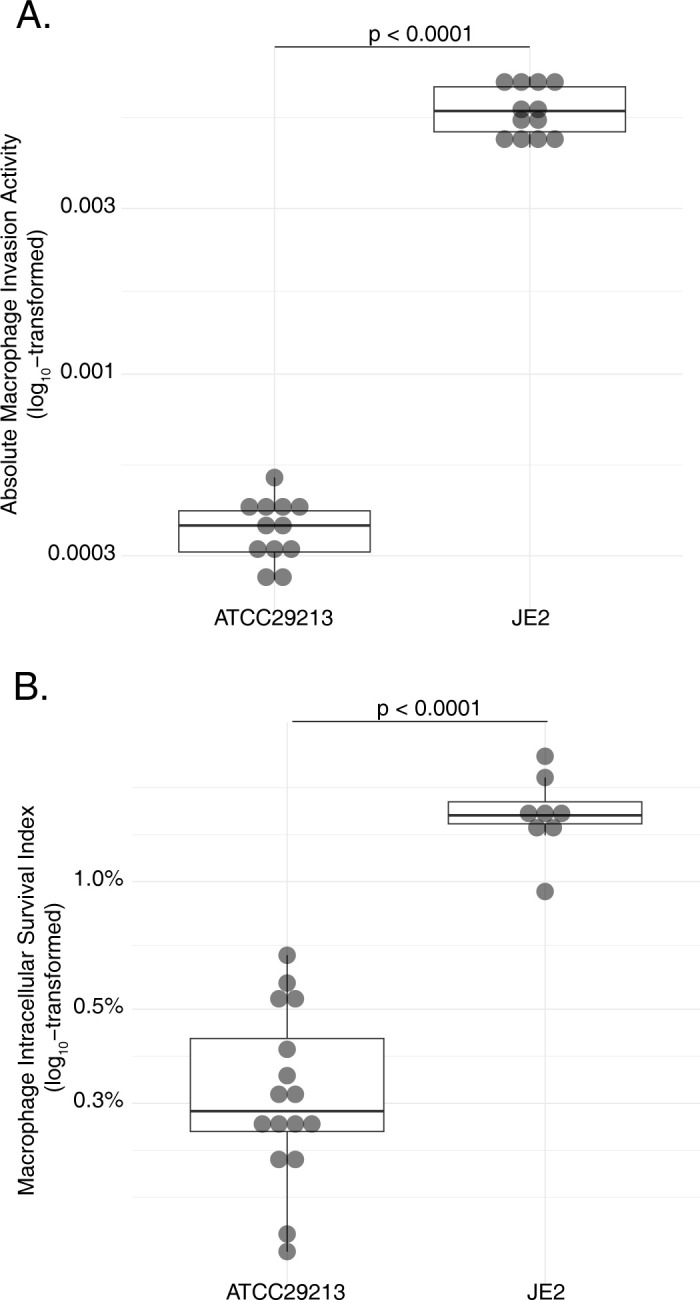
Phenotypes of strains were assessed for macrophage invasion (A) and macrophage intracellular survival (B). Statistical significance of measurements was assessed using unpaired two-tailed T tests. Macrophage invasion in this analysis was performed in a single experiment and determined as an absolute value, due to a lack of an external comparator strain for normalization.
From these differing natural baselines, six independent replicates from both strains were then passaged to select for intracellular macrophage pathogenesis phenotypes using lysostaphin protection assays [42,56], which selectively kill bacteria that have not been internalized by eukaryotic cells. After 12 passages, isolates were obtained from each expanded population and subjected to further characterization (S9 Table).
While strain JE2 showed greater baseline capacity for macrophage invasion than ATCC29213, that phenotype increased with passaging of both strains, derivatives of which ultimately achieved similar levels of activity (Fig 3A). In contrast, we observed significant strain-level differences in intracellular survival after selection (Fig 3B): ATCC29213, but not JE2, showed markedly increased survival in macrophages after passaging. For passaged ATCC29213 isolates, the fraction of bacteria recovered from macrophages after 48 hours exceeded that at the time of invasion, indicating some degree of bacterial replication following macrophage infection. The survival capacity of two JE2 derivatives decreased slightly with passaging, possibly indicating greater selective pressure for invasion phenotypes and mutual exclusivity of invasion and survival traits in that strain. Growth rate analysis of passaged mutants was conducted (S10 Table). Half of the replicates (two ATCC29213 derivatives and four JE2 mutants) had growth rates not significantly different from their parental strains. Four replicates (three ATCC29213 and one JE2) had doubling times significantly greater than their parents, whereas only one replicate from each lineage replicated significantly faster.
Fig 3. Macrophage pathogenesis phenotypes of passaged S. aureus laboratory strains.

Derivatives of JE2 and ATCC29213 after passaging with selective pressure for macrophage pathogenesis assessed for macrophage invasion, relative to in-batch testing of the matched parental strain (A), and macrophage intracellular survival (B). Statistical significance was assessed using unpaired two-tailed T tests.
Whole-genome sequencing detected a total of 15 genes that were mutated in passaged isolates (S11 Table). Most genes (n = 10) were mutated in only a single isolate, but five (femA, putative Abi-domain protease B7H15_05310, putative autolysin B7H15_09680, vraS, and ccpA) were each independently mutated in two to five evolved ATCC29213 isolates. Six of the mutated genes were affected by at least one unequivocally inactivating mutation (i.e., stop-gain or frameshift), consistent with loss-of-function effects, while the remainder were affected by missense mutations whose effects were less interpretable. No genes were mutated in common between ATCC29213 and JE2 over the course of passaging. Two JE2 replicates did not have detectable coding sequence or promoter mutations, suggesting epigenetic changes or mutations in remote regulatory elements.
Comparison of candidate genes identified by in vivo and in vitro approaches
Among 127 candidate genes identified by GWAS and 15 identified by passaging, only a single gene (clpX) was implicated by both in vivo and in vitro approaches. Given that 5,854 unique genes were considered in the GWAS analysis, this degree of overlap did not exceed that expected by random chance (p = 0.228; significance threshold for number of overlapping genes for by SuperExactTest [63], method 2). We conclude that distinct gene subsets were identified by in vivo and in vitro analyses.
Validation of candidate genes affecting macrophage intracellular pathogenesis
We next functionally evaluated candidate genes identified by in vitro and in vivo strategies for their ability to influence intracellular pathogenesis phenotypes in the THP-1 macrophage model. All genes identified by in vitro studies were targeted; however, given the large number of candidates implicated by analysis of in vivo isolates, we prioritized genes identified by GWAS for validation as those with the highest levels of association in a single analysis and those that were identified by more than one GWAS model. In total, the most significant 35 of 121 (28.9%) candidate invasion genes and all seven (100%) candidate survival genes met prioritization criteria for functional testing.
When available, we first tested the phenotypic impact of candidate genes using defined, isogenic transposon knockout mutants of strain JE2 [30]. Transposon mutants existed for 30 of 41 candidate genes prioritized from in vivo selection and 10 of the 15 of candidate in vitro genes. To further control for assay variability, we included transposon mutants for three genes that GWAS identified as having no association with intracellular pathogenesis, and which displayed similar intracellular pathogenesis phenotypes to the wild-type parental strain. We subsequently considered candidate genes to have a measurable impact on macrophage pathogenesis phenotypes if their corresponding transposon mutant effected a change that was statistically different from replicate measurements of the parental JE2 strain (defined as having a median measurement outside the 95% CI of controls).
Transposon disruption of 28 (93%) of in vivo candidates, six (60%) of in vitro candidates, and none of the uninvolved control genes significantly increased S. aureus invasion of THP-1 macrophages, while the remainder did not measurably alter that phenotype (Fig 4A). In contrast, S. aureus survival within macrophages was significantly decreased by disruption of only four (13%) of the in vivo candidates, none of the in vitro candidates, and none of the uninvolved control genes, with no enhancement of that phenotype observed (Fig 4B). Cumulatively, transposon disruptions of all but five candidate genes impacted macrophage pathogenesis. All such mutations improved macrophage invasion; however, four also negatively affected S. aureus survival within macrophages.
Fig 4. Transposon disruption of candidate macrophage pathogenesis genes.

Fold-difference in measurements of macrophage invasion, relative to in-batch testing of the parental JE2 strain (A), and macrophage intracellular survival (B) for specified transposon mutants. Candidate genes are grouped according to the strategy by which they were identified, and sorted within groups by median value. Values from in-run JE2 strain controls are shown, and JE2 transposon mutants in genes not implicated in macrophage pathogenesis were used as an additional control group. Boxes indicate median, 25th, and 75th percentiles with whiskers extending to the most distant value < 1.5 times the interquartile range. Red horizontal lines indicate the 95% confidence interval of all JE2 control measurements, with significant mutants defined as having a median measurement outside this range. Values higher than the 95% confidence intervals indicate increased macrophage pathogenesis phenotypes, and those below the interval correspond to decreased macrophage pathogenesis phenotypes. Genome-wide association study (GWAS).
Given the large number of targets identified, we performed genetic complementation studies on a subset of genes showing the largest effect sizes for the invasion phenotype, and the complete, but more limited number of genes exhibiting significant survival effects (S12 Table). We transformed transposon mutant strains with appropriate complementation vectors and compared relevant phenotypes against matched controls transformed with a GFP-expressing plasmid. Complementation reverted the expected macrophage invasion phenotype in 12 of the 16 transposon mutants tested and the macrophage survival phenotype of three of the four mutants tested, providing further evidence for their biological impact. Failure of some mutants to yield the expected phenotype after complementation may reflect mismatches in the dosage or regulation of plasmid-borne genes, and thus does not necessarily indicate that the tested gene lacks contributions to macrophage pathogenesis.
To expand our scope of testing to factors not available as transposon knockouts, including potentially essential genes, we developed a constitutively expressed CRISPRi [31] gene silencing vector for use in S. aureus. Using high specificity guide RNA designs [54], nine additional isogenic knockdowns were successfully generated in JE2 (S3 Table) and their effects on intracellular pathogenesis assessed relative to CRISPRi knockdown of an irrelevant control target.
As anticipated, knockdowns did not quantitatively influence phenotypes as prominently as gene knockouts. Nevertheless, all tested candidate genes significantly impacted one or both macrophage pathogenesis phenotypes. One in vivo candidate gene significantly increased invasion and five others significantly decreased it (Fig 5A). Similarly, two of the in vivo candidates positively impacted macrophage survival, while five reduced it (Fig 5B). Knockdown of the single gene identified in common between in vivo analyses and passaging, clpX, also notably impaired survival.
Fig 5. CRISPRi knockdown of candidate macrophage pathogenesis genes.
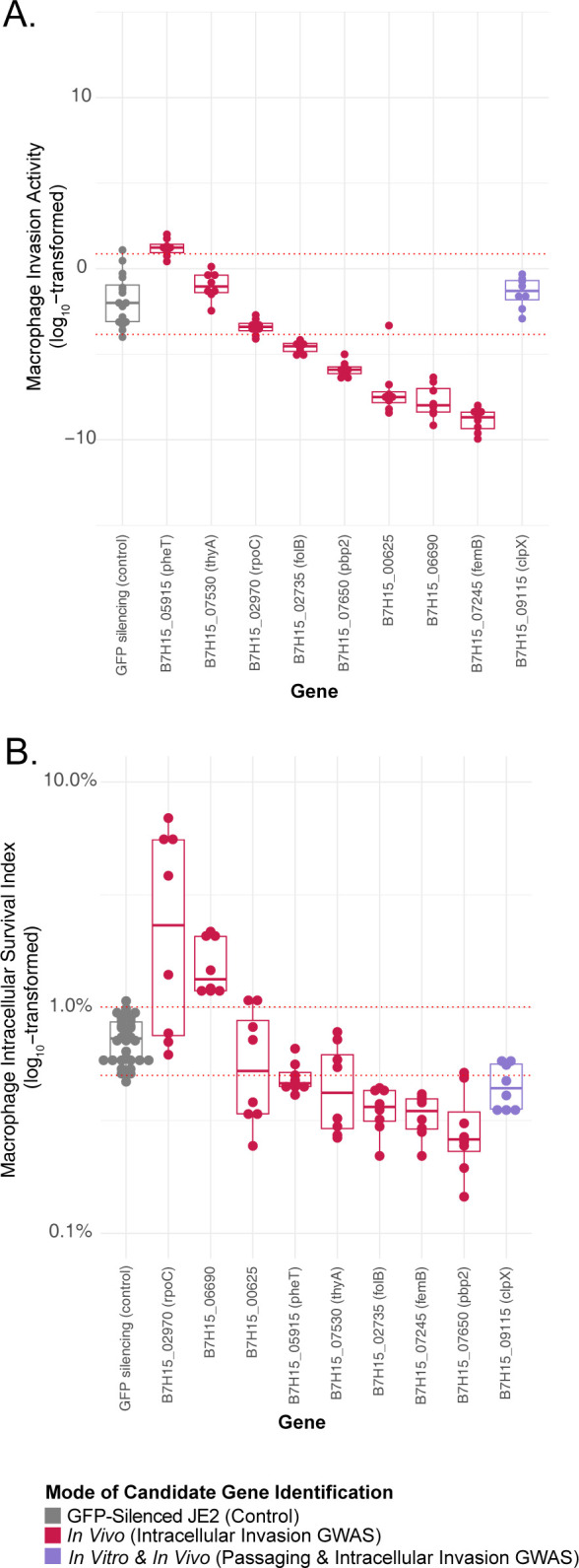
Fold-difference in measurements of macrophage invasion, normalized to in-batch testing of the parental JE2 strain carrying a GFP-targeted CRISPRi construct(A), and macrophage intracellular survival (B) for specified CRISPRi gene knockdowns. Candidate genes are grouped according to the strategy by which they were identified, and sorted within groups by median value. JE2 carrying a GFP-expressing plasmid built on the same vector backbone as silencing constructs was used as an additional control group. Boxes indicate median, 25th, and 75th percentiles with whiskers extending to the most distant value < 1.5 times the interquartile range. Red horizontal lines indicate the 95% confidence interval of all control measurements from JE2 with GFP-targeted CRISPRi, with significant mutants defined as having a median measurement outside this range. Values higher than the 95% confidence intervals indicate increased macrophage pathogenesis phenotypes, and those below the interval correspond to decreased macrophage pathogenesis phenotypes. Genome-wide association study (GWAS).
Between the two validation approaches, 37 of the 39 tested candidate genes from in vivo analysis and seven of 11 genes from in vitro passaging impacted one or both macrophage pathogenesis phenotypes, with a single gene shared in common between those two sets.
Features of S. aureus isogenic mutants with altered survival phenotypes
Counts of intracellular bacteria could be impacted by changes in bacterial survival, host-cell killing, or bacterial replicative capacity. We therefore assessed these features in isogenic mutants exhibiting differences in the macrophage survival phenotype. Relative to wild-type JE2, no mutant strain evidenced significant differences in the total number of macrophages (p>0.29, two-tailed T test) or the proportion of dead host cells (p>0.20, two-tailed T test) observed 48 hours after infection, indicating no measurable alterations to host cell death by lytic or non-lytic mechanisms. With only a single exception (saeS), isogenic mutants did not have significantly slower growth rates than matched comparator strains (S13 Table). We conclude that reduced intracellular survival phenotypes observed in isogenic mutants largely reflect differences in bacterial viability within macrophages, rather than reduced bacterial replication or impaired killing of the host cell population.
Discussion
S. aureus must overcome different stressful conditions to establish and subsequently maintain an infection. During acute phases, bacteria must withstand innate immunity and destroy superficial host cells, whereas during chronic phases, persistent survival depends on resistance to lysosomal degradation and adaptation to low-nutrient conditions in intracellular environments [9]. Although some elements of phenotype switching [64] can be coordinated through changes in gene expression [27], prior work has shown that S. aureus undergoes genomic evolution (mutational adaptation) during infection to stably modify its initial phenotypes and better suit survival in the human host [4,65,66]. Here, we used parallel in vivo and in vitro experimental designs to identify genes contributing to two S. aureus phenotypes important to persistence: invasion, which is the ability to able to enter and survive within host cells after phagocytosis, and survival, the ability to persist intercellularly within macrophages over time.
Using chronic respiratory infection in people with CF as an exemplar of S. aureus persistence in vivo, we analyzed clonally related S. aureus isolate pairs taken from individuals early and late in the course of infection [4]. We found that the phenotypes of S. aureus isolated from patients with CF at the time of initial detection were equivalent to controls sharing a similar genetic background, consistent with the understanding that diverse S. aureus strains may be acquired from the community by individuals with CF without a requirement for specific virulence traits [4]. However, we observed that strains’ capacity for macrophage invasion increased significantly over time (Fig 1A), consistent with selective pressure for that feature being important during chronic S. aureus disease in individuals with CF [20,21]. In contrast, intracellular macrophage survival did not consistently increase during chronic infection, suggesting that trait may not be as universally selected. This could indicate that the inherent ability of S. aureus to survive within macrophages [1,67] is already adequate to sustain chronic persistence in CF [16,21,56], and that further improvements to that phenotype might incur disadvantageous fitness costs. Subsequent genome-wide association studies identified mutations in a total of 127 unique genes tracking with these phenotypic changes in vivo (Fig 2).
In parallel, we employed selection of laboratory S. aureus strains to model evolutionary pressures for macrophage pathogenesis in vitro (Fig 4) and used whole-genome analysis to identify associated mutations. Although that approach did not allow us to disambiguate mutations driving macrophage invasion from those influencing survival, it provided for well-controlled and well-defined experimental conditions. Whole-genome sequencing revealed 15 unique genes mutated in passaged isolates (S11 Table). Interestingly, we observed no overlap in the genes mutated during passaging of the two strains examined in this work, potentially indicating strain-specific mutation profiles or gene effects, consistent with our prior studies [56].
Using a combination of isogenic transposon mutant knockouts and CRISPRi knockdowns, we subsequently assessed the contributions of prioritized candidate genes identified by in vitro and in vivo analytic strategies (Figs 4 and 5). Forty-four of the 49 tested genes (90%) significantly affected one or both macrophage intracellular pathogenesis phenotypes. The majority of validated genes (n = 35, 80%) enhanced macrophage invasion when disrupted (Table 1), consistent with our finding that mutation of these genes is associated with increased macrophage pathogenesis in vivo (Fig 1) largely from the accumulation of loss-of-function variants. Only seven of the tested genes (14%) had an entirely negative phenotypic impact on S. aureus macrophage pathogenesis, although it is possible those factors may confer advantages in vivo that are not measured by our in vitro assay, or could prove beneficial when occurring in concert with secondary mutations or when affected by gain of function mutations not modeled in our validation.
Table 1. Validated genes in S. aureus impacting macrophage pathogenesis.
| Category | Gene name or JE2 Locus ID | Gene Function | Evidence for macrophage pathogenesis | Identification strategy | Validation Strategy | Invasion Effect | Survival effect |
|---|---|---|---|---|---|---|---|
| metabolism | arcR | arginine deiminase operon repressor | [68] | in vitro | Knockout | Increase | NS† |
| purR * | purine biosynthesis operon repressor | [69] | in vivo invasion | Knockout | Increase | NS | |
| purH | Bifunctional purine biosynthesis protein | [70] | in vivo invasion | Knockout | Increase | NS | |
| B7H15_07365 | hydrolase | This study | in vivo invasion | Knockout | Increase | NS | |
| kbl | glycine C-acetyltransferase | This study | in vivo invasion | Knockout | Increase | NS | |
| thiD | hydroxymethylpyrimidine/phosphomethylpyrimidine kinase | This study | in vivo invasion | Knockout | Increase | NS | |
| thyA | thymidylate synthase | [71] | in vivo invasion | CRISPRi | NS | Decrease | |
| cdr | CoA-disulfide reductase | This study | in vivo invasion | Knockout | Increase | Decrease | |
| pheT | Phenylalanine—tRNA ligase beta subunit | This study | in vivo invasion | CRISPRi | Increase | Decrease | |
| folB | Dihydroneopterin aldolase | This study | in vivo invasion | CRISPRi | Decrease | Decrease | |
| ansA | L-asparaginase, type II | This study | in vivo invasion | Knockout | Increase | NS | |
| ptaA | PTS glucose transporter subunit IIBC | This study | in vivo survival | Knockout | Increase | NS | |
| gntK | Gluconokinase | This study | in vivo invasion | Knockout | Increase | NS | |
| mnhD * | Na+/H+ antiporter subunit D | This study | in vivo invasion | Knockout | Increase | NS | |
| biofilm | nreC | Oxygen regulatory protein | [72] | in vitro | Knockout | Increase | NS |
| yycI * | regulatory protein | [73] | in vitro | Knockout | Increase | NS | |
| adherence and autolysis | atl * | bifunctional autolysin | [74] | in vivo invasion | Knockout | Increase | NS |
| fnbA * | fibronectin-binding protein A | [75,76] | in vivo survival | Knockout | Increase | NS | |
| clfA | clumping factor A | [77] | in vivo survival | Knockout | Increase | NS | |
| yycI * | regulatory protein | [78] | in vitro | Knockout | Increase | NS | |
| invasion | atl * | bifunctional autolysin | [79] | in vivo invasion | Knockout | Increase | NS |
| fnbA * | fibronectin-binding protein A | [75,76] | in vivo | Knockout | Increase | NS | |
| B7H15_05310 | putative Abi-domain protein | This study | in vitro | Knockout | Increase | NS | |
| iron metabolism | sirA | iron ABC transporter substrate-binding protein | [80] | in vivo invasion | Knockout | Increase | NS |
| virulence gene regulation | vraS | two-component sensor histidine kinase | [81] | in vitro | Knockout | Increase | NS |
| kdpD | sensor histidine kinase | [82] | in vivo invasion | Knockout | Increase | NS | |
| saeS | two-component sensor histidine kinase | [83–85] | in vivo invasion | Knockout | Increase | Decrease | |
| purR * | purine biosynthesis operon repressor | [69,86] | in vivo invasion | Knockout | Increase | NS | |
| yjbH * | transcriptional regulator | [87] | in vivo invasion and survival | Knockout | Increase | NS | |
| B7H15_06690 | transcriptional regulator | This study | in vivo invasion | CRISPRi | Decrease | Increase | |
| immune cell survival | clpX | protease | [89] | in vitro | CRISPRi | NS | Decrease |
| immune evasion | sdrE | MSCRAMM family adhesin | [88] | in vivo survival | Knockout | Increase | NS |
| lytM | glycyl-glycine endopeptidase | This study | in vivo invasion | Knockout | Increase | NS | |
| stress response | yjbH * | transcriptional regulator | [87] | in vivo invasion and survival | Knockout | Increase | NS |
| nfrA | NADPH-dependent oxidoreductase | This study | in vivo invasion | Knockout | Increase | NS | |
| B7H15_12300 | ClpA-like protein | This study | in vitro | Knockout | Increase | NS | |
| mnhD * | Na+/H+ antiporter subunit D | This study | in vivo invasion | Knockout | Increase | NS | |
| betT | choline transporter | This study | in vivo invasion | Knockout | Increase | Decrease | |
| peptidoglycan metabolism | fmhA | methicillin resistance protein | This study | in vivo invasion | Knockout | Increase | NS |
| femA | aminoacyltransferas | This study | in vitro | CRISPRi | Decrease | NS | |
| femB | aminoacyltransferase | This study | in vivo invasion | CRISPRi | Decrease | Decrease | |
| pbp2 | penicillin-binding protein 2 | This study | in vivo invasion | CRISPRi | Decrease | Decrease | |
| mutation repair | mutS | DNA mismatch repair protein | This study | in vivo invasion | Knockout | Increase | NS |
| unknown | murQ | N-acetylmuramic acid 6-phosphate etherase | [90] | in vivo invasion | Knockout | Increase | NS |
| rumA | 23S rRNA (uracil(1939)-C(5))-methyltransferase | This study | in vivo invasion | Knockout | Increase | NS | |
| rpoC | DNA-directed RNA polymerase subunit beta | This study | in vivo invasion | CRISPRi | NS | Increase | |
| B7H15_00625 | Hypothetical protein | This study | in vivo invasion | CRISPRi | Decrease | NS | |
| B7H15_08855 | RecD/TraA family helicase | This study | in vivo invasion | Knockout | Increase | NS | |
| B7H15_03805 | Hypothetical protein | This study | in vivo invasion | Knockout | Increase | Decrease | |
| B7H15_11520 | Hypothetical protein | This study | in vivo invasion | Knockout | Increase | NS |
* = gene with multiple functional categories
† NS = Not significant
Macrophage viability was not affected by the presence of infecting wild-type or mutant S. aureus strains, indicating that changes in macrophage pathogenesis reflect alterations in bacterial intracellular survival, rather than killing of the host-cell population. This is consistent with observations that S. aureus may not incur substantial macrophage death during infection [1,67].
Previously described virulence factors (n = 17) comprised 39% of all genes in our study having experimentally validated macrophage pathogenesis phenotypes, supporting the effectiveness of the overall experimental approach. These known factors contributed to diverse, well-described pathogenesis functions in CF and other diseases, comprising cellular metabolism (arcR [68], purR [69], purH [70], thyA [71]), biofilm formation (nreC [72], yycI [73]) and other adhesion mechanisms (atl [74], fnbA [75,76], clfA [77]), autolysis (yycI [78], atl [74]), invasins (atl [79], fnbA [75,76]), iron metabolism (sirA [80]), virulence gene regulation (vraS [81], kdpD [82], saeS [83–85], purR [69,86], yjbH [87]), and immune evasion (sdrE [88]). Other functions were more specialized to intracellular pathogenesis within immune cells, including protease clpX [89] and factors promoting protection against oxidative stress (yjbH(87)) which is a prominent feature of the macrophage intracellular environment. One virulence gene (murQ) has previously been reported to influence S. aureus hemolysis, by unknown mechanisms [90]. Notably, some virulence gene mutations that were found to positively influence macrophage invasion are known to attenuate S. aureus virulence in biological models (purH, atl, fnbA, vraS, clpX), illustrating that some mutations which are deleterious in acute infection states can be beneficial for chronic infection phenotypes.
A larger number of S. aureus macrophage pathogenesis genes (n = 27) are newly implicated by this effort, comprising 61% of genes passing experimental validation. The putative or documented roles of many mirror the functions of known virulence factors, including Abi-domain protein (B7H15_05310 [91]), immune evasion (lytM [92,93]), transcriptional regulation (B7H15_06690), cellular metabolism (thiD, ptaA, pheT, folB, gntK, mnhD [94], kbl, cdr, ansA, B7H15_07365), and response to oxidative (nfrA [95], clpA [96]-like gene B7H15_12300), pH (mnhD [97]), or osmotic (betT [98]) stress. However, other newly validated genes offer insight into previously unappreciated mechanisms affecting intracellular macrophage pathogenesis. The most substantial constellation of functionally related genes include fmhA [99], femA [100], femB [100], and pbp2 [100], all of which have functions in peptidoglycan assembly. This finding potentially indicates an important role for cell wall metabolism in S. aureus persistence, either due to direct changes in host cell interaction [101–103], or specific structural changes that modulate fitness within macrophages [104]. The well-characterized mismatch repair gene mutS conferred increased invasion phenotypes when inactivated by transposon mutagenesis, potentially reflecting greater population diversity carrying phenotypically advantageous mutations [105] and not acting as a virulence factor directly. Although the remaining validated genes had hypothetical functions or non-obvious roles in pathogenesis, some, such as rumA, had pronounced phenotypic effects and merit dedicated investigations to understand their contributions to S. aureus macrophage pathogenesis.
In addition to illuminating previously overlooked factors relevant to intracellular macrophage pathogenesis and chronic infection, these studies highlight pronounced differences between the results of in vivo and in vitro investigatory strategies. Macrophage pathogenesis phenotypes quantitatively increased during both chronic human infection and serial passaging, and most genes implicated by in vivo and in vitro strategies were confirmed to impact macrophage pathogenesis in the same functional laboratory model. Yet, despite these phenotypic similarities, only a single candidate gene was identified in common by parallel in vivo and in vitro studies. Although some disparities could be attributed to strain-specific differences [56], the consistency of particular mutations occurring in vivo among independent strains from different patients argues against this phenomenon being widespread. We conclude that mutations identified even in highly controlled in vitro studies may fail to identify changes enhancing the same phenotypes that arise naturally in human disease. This schism, which we previously observed in the evolution of P. aeruginosa antibiotic resistance [106], may reflect differences in fitness constraints, selective pressures, and population sizes occurring in vivo and in vitro. Although both strategies identify genes measurably contributing to pathogenesis in our cultured macrophage model, this finding illustrates the value of analyzing clinical isolates to understand factors driving infectious disease in vivo.
Our work is subject to several limitations. Cell lines cultured in vitro are not physically or biochemically identical to analogous cells or tissues found in vivo [34], and macrophages from individuals with CF have some impaired functionality [21,107], making both our in vitro selection schema and macrophage pathogenesis assays necessarily contrived. However, our finding that metrics of THP-1 invasion employed here and by others [28,60] increased during chronic infection offers support that the model reasonably approximates in vivo conditions and is clinically relevant. Separately, our approach may not identify legitimate, but less frequently occurring mutational adaptations due to the sample sizes available. Effector genes whose contributions to intracellular pathogenesis are not quantitatively large may have similarly been overlooked by our strict functional validation criteria. It is also possible that critical adaptations in our in vivo study population occurred prior to collection of the earliest CF isolates, limiting identification of key genes that are subject to rapid selection: however, functional measures of intracellular pathogenesis in these first isolates were similar to phylogenetically matched control strains not associated with infection, indicating that the impact of early adaptation not captured in this study was small. Additionally, CF isolates were derived from sputum and oropharyngeal swabs, as bronchoalveolar lavage is not commonly performed in children, and thereby may not fully reflect the microbiology of the lower airways [4]. Lastly, we are unable to isolate specific selective pressures occurring in vivo or to correlate them with isolate phenotypes: S. aureus is able to invade and infect a wide variety of cell types [12–15,21], and it is possible that selective pressures for invasion of non-phagocytic host cells, or less likely, for phenotypes unrelated to host cell invasion, occur in vivo and the enhancement of macrophage invasion is merely coincidental.
Although this effort focused on a model of respiratory infection in CF, S. aureus is capable of causing infection in a variety of organ systems [7,14,19,108,109] and host-cell invasion is a hallmark of many S. aureus chronic disease states [12–15,21]. The effector genes identified here may therefore have generalized importance in other infective processes, and indeed, show activity in a macrophage cell line that is not affected by CFTR mutation. Future work will seek to more comprehensively catalog the intracellular pathogenesis mechanisms present across S. aureus genetic lineages and to understand similarities and differences in the gene content promoting pathogenesis of additional host cell types. This knowledge may enable development of novel therapeutic strategies to prevent the establishment of bacterial intracellular reservoirs or to disrupt persistent populations that sustain chronic infection and that are resistant to conventional forms of treatment.
Supporting information
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Quantile-quantile plots for in vivo mutations associated with macrophage invasion (blue) and survival (red) phenotypes by various implementations of genome-wide association studies. Association between phenotypes by analysis of (A) reference-free sequence differences (“unitigs”), (B) non-synonymous mutations with gene kernels (“burden testing”), and (C) de novo mutations arising within clonally related patient lineages over time (“first-last comparison”). Significant and non-significant results are shaded differentially.
(PDF)
Phenotypes of S. aureus strains isolated from respiratory cultures of individuals with CF at the time of initial isolation during serial surveillance are compared to the last-collected strains from same patient. Colored lines in this connected dot plot indicate the direction and significance of changes in individual first-last isolate pairs (red = significant increase over time, blue = significant decrease over time, grey = no-significant change) for measures of macrophage invasion relative to in-batch testing of the JE2 control strain (A), and macrophage intracellular survival (B). Statistical significance was assessed using paired two-tailed T tests.
(PDF)
Data Availability
Sequence data generated in support of this study are available from the NCBI Sequence Read Archive under accessions PRJNA860616 and PRJNA1029652.
Funding Statement
This work was supported by grants from Vertex Pharmaceuticals (Vertex Cystic Fibrosis Independent Research Innovation Award to SJS, https://www.vrtx.com), the Cystic Fibrosis Foundation (SINGH19R0 to SJS, https://www.cff.org/), and NIH (NIDDK P30 DK089507 to SJS, NIAMS K23 AR080209 to DRL, https://www.nih.gov/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Pidwill GR, Gibson JF, Cole J, Renshaw SA, Foster SJ. The Role of Macrophages in Staphylococcus aureus Infection. Front Immunol. 2021. Jan 19;11:620339. doi: 10.3389/fimmu.2020.620339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hirschhausen N, Block D, Bianconi I, Bragonzi A, Birtel J, Lee JC, et al. Extended Staphylococcus aureus persistence in cystic fibrosis is associated with bacterial adaptation. Int J Med Microbiol. 2013. Dec;303(8):685–92. doi: 10.1016/j.ijmm.2013.09.012 [DOI] [PubMed] [Google Scholar]
- 3.Branger C, Gardye C, Lambert-Zechovsky N. Persistence of Staphylococcus aureus strains among cystic fibrosis patients over extended periods of time. J Med Microbiol. 1996. Oct;45(4):294–301. doi: 10.1099/00222615-45-4-294 [DOI] [PubMed] [Google Scholar]
- 4.Long DR, Wolter DJ, Lee M, Precit M, McLean K, Holmes E, et al. Polyclonality, Shared Strains, and Convergent Evolution in Chronic Cystic Fibrosis Staphylococcus aureus Airway Infection. Am J Respir Crit Care Med. 2021. May 1;203(9):1127–37. doi: 10.1164/rccm.202003-0735OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Al-Zubeidi D, Hogan PG, Boyle M, Burnham CAD, Fritz SA. Molecular epidemiology of methicillin-resistant Staphylococcus aureus isolated in serial cultures from the respiratory tract of children with cystic fibrosis. Pediatr Infect Dis J. 2014. Jun;33(6):549–53. doi: 10.1097/INF.0000000000000204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andersen C, Kahl BC, Olesen HV, Jensen-Fangel S, Nørskov-Lauritsen N. Intravenous antibiotics given for 2 weeks do not eradicate persistent Staphylococcus aureus clones in cystic fibrosis patients. Clin Microbiol Infect. 2014. May;20(5):O285–291. doi: 10.1111/1469-0691.12406 [DOI] [PubMed] [Google Scholar]
- 7.Conlon BP. Staphylococcus aureus chronic and relapsing infections: Evidence of a role for persister cells: An investigation of persister cells, their formation and their role in S. aureus disease. Bioessays. 2014. Oct;36(10):991–6. doi: 10.1002/bies.201400080 [DOI] [PubMed] [Google Scholar]
- 8.Goerke C, Wolz C. Adaptation of Staphylococcus aureus to the cystic fibrosis lung. Int J Med Microbiol. 2010. Dec;300(8):520–5. doi: 10.1016/j.ijmm.2010.08.003 [DOI] [PubMed] [Google Scholar]
- 9.Tuchscherr L, Bischoff M, Lattar SM, Noto Llana M, Pförtner H, Niemann S, et al. Sigma Factor SigB Is Crucial to Mediate Staphylococcus aureus Adaptation during Chronic Infections. PLoS Pathog. 2015. Apr;11(4):e1004870. doi: 10.1371/journal.ppat.1004870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suligoy CM, Lattar SM, Noto Llana M, González CD, Alvarez LP, Robinson DA, et al. Mutation of Agr Is Associated with the Adaptation ofStaphylococcus aureusto the Host during Chronic Osteomyelitis. Front Cell Infect Microbiol. 2018;8:18. doi: 10.3389/fcimb.2018.00018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Howden BP, Davies JK, Johnson PDR, Stinear TP, Grayson ML. Reduced Vancomycin Susceptibility in Staphylococcus aureus, Including Vancomycin-Intermediate and Heterogeneous Vancomycin-Intermediate Strains: Resistance Mechanisms, Laboratory Detection, and Clinical Implications. Clinical Microbiology Reviews. 2010. Jan 1;23(1):99–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fraunholz M, Sinha B. Intracellular Staphylococcus aureus: live-in and let die. Front Cell Infect Microbiol. 2012;2:43. doi: 10.3389/fcimb.2012.00043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lowy FD. Is Staphylococcus aureus an intracellular pathogen? Trends Microbiol. 2000. Aug;8(8):341–3. doi: 10.1016/s0966-842x(00)01803-5 [DOI] [PubMed] [Google Scholar]
- 14.Garzoni C, Kelley WL. Staphylococcus aureus: new evidence for intracellular persistence. Trends Microbiol. 2009. Feb;17(2):59–65. doi: 10.1016/j.tim.2008.11.005 [DOI] [PubMed] [Google Scholar]
- 15.Moldovan A, Fraunholz MJ. In or out: Phagosomal escape of Staphylococcus aureus. Cellular Microbiology. 2019. Mar;21(3):e12997. [DOI] [PubMed] [Google Scholar]
- 16.Lacoma A, Cano V, Moranta D, Regueiro V, Domínguez-Villanueva D, Laabei M, et al. Investigating intracellular persistence of Staphylococcus aureus within a murine alveolar macrophage cell line. Virulence. 2017. Nov 17;8(8):1761–75. doi: 10.1080/21505594.2017.1361089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thwaites GE, Gant V. Are bloodstream leukocytes Trojan Horses for the metastasis of Staphylococcus aureus? Nat Rev Microbiol. 2011. Mar;9(3):215–22. doi: 10.1038/nrmicro2508 [DOI] [PubMed] [Google Scholar]
- 18.Kubica M, Guzik K, Koziel J, Zarebski M, Richter W, Gajkowska B, et al. A potential new pathway for Staphylococcus aureus dissemination: the silent survival of S. aureus phagocytosed by human monocyte-derived macrophages. PLoS ONE. 2008. Jan 9;3(1):e1409. doi: 10.1371/journal.pone.0001409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kalinka J, Hachmeister M, Geraci J, Sordelli D, Hansen U, Niemann S, et al. Staphylococcus aureus isolates from chronic osteomyelitis are characterized by high host cell invasion and intracellular adaptation, but still induce inflammation. Int J Med Microbiol. 2014. Nov;304(8):1038–49. doi: 10.1016/j.ijmm.2014.07.013 [DOI] [PubMed] [Google Scholar]
- 20.Tan X, Coureuil M, Ramond E, Euphrasie D, Dupuis M, Tros F, et al. Chronic Staphylococcus aureus Lung Infection Correlates With Proteogenomic and Metabolic Adaptations Leading to an Increased Intracellular Persistence. Clinical Infectious Diseases. 2019. Nov 13;69(11):1937–45. doi: 10.1093/cid/ciz106 [DOI] [PubMed] [Google Scholar]
- 21.Li C, Wu Y, Riehle A, Ma J, Kamler M, Gulbins E, et al. Staphylococcus aureus Survives in Cystic Fibrosis Macrophages, Forming a Reservoir for Chronic Pneumonia. Infect Immun. 2017. May;85(5). doi: 10.1128/IAI.00883-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trouillet-Assant S, Lelièvre L, Martins-Simões P, Gonzaga L, Tasse J, Valour F, et al. Adaptive processes of Staphylococcus aureus isolates during the progression from acute to chronic bone and joint infections in patients: S. aureus adaptation during BJI. Cellular Microbiology. 2016. Oct;18(10):1405–14. [DOI] [PubMed] [Google Scholar]
- 23.Siwczak F, Cseresnyes Z, Hassan MIA, Aina KO, Carlstedt S, Sigmund A, et al. Human macrophage polarization determines bacterial persistence of Staphylococcus aureus in a liver-on-chip-based infection model. Biomaterials. 2022. Aug;287:121632. doi: 10.1016/j.biomaterials.2022.121632 [DOI] [PubMed] [Google Scholar]
- 24.Hamza T, Dietz M, Pham D, Clovis N, Danley S, Li B. Intra-cellular Staphylococcus aureus alone causes infection in vivo. Eur Cell Mater. 2013. Jul 8;25:341–50; discussion 350. doi: 10.22203/ecm.v025a24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Plouin-Gaudon I, Clement S, Huggler E, Chaponnier C, François P, Lew D, et al. Intracellular residency is frequently associated with recurrent Staphylococcus aureus rhinosinusitis. Rhinology. 2006. Dec;44(4):249–54. [PubMed] [Google Scholar]
- 26.Vozza EG, Mulcahy ME, McLoughlin RM. Making the Most of the Host; Targeting the Autophagy Pathway Facilitates Staphylococcus aureus Intracellular Survival in Neutrophils. Front Immunol. 2021. Jun 16;12:667387. doi: 10.3389/fimmu.2021.667387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soe YM, Bedoui S, Stinear TP, Hachani A. Intracellular Staphylococcus aureus and host cell death pathways. Cellular Microbiology [Internet]. 2021. May [cited 2023 Aug 21];23(5). Available from: https://onlinelibrary.wiley.com/doi/10.1111/cmi.13317. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen HA, Denis O, Vergison A, Theunis A, Tulkens PM, Struelens MJ, et al. Intracellular Activity of Antibiotics in a Model of Human THP-1 Macrophages Infected by a Staphylococcus aureus Small-Colony Variant Strain Isolated from a Cystic Fibrosis Patient: Pharmacodynamic Evaluation and Comparison with Isogenic Normal-Phenotype and Revertant Strains. Antimicrob Agents Chemother. 2009. Apr;53(4):1434–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chanput W, Mes JJ, Wichers HJ. THP-1 cell line: an in vitro cell model for immune modulation approach. Int Immunopharmacol. 2014. Nov;23(1):37–45. doi: 10.1016/j.intimp.2014.08.002 [DOI] [PubMed] [Google Scholar]
- 30.Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, Bose JL, et al. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. MBio. 2013. Feb 12;4(1):e00537–00512. doi: 10.1128/mBio.00537-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larson MH, Gilbert LA, Wang X, Lim WA, Weissman JS, Qi LS. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat Protoc. 2013. Nov;8(11):2180–96. doi: 10.1038/nprot.2013.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Precit MR, Wolter DJ, Griffith A, Emerson J, Burns JL, Hoffman LR. Optimized In Vitro Antibiotic Susceptibility Testing Method for Small-Colony Variant Staphylococcus aureus. Antimicrob Agents Chemother. 2016. Jan 4;60(3):1725–35. doi: 10.1128/AAC.02330-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dantes R, Mu Y, Belflower R, Aragon D, Dumyati G, Harrison LH, et al. National burden of invasive methicillin-resistant Staphylococcus aureus infections, United States, 2011. JAMA Intern Med. 2013 Nov 25;173(21):1970–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strobel M, Pförtner H, Tuchscherr L, Völker U, Schmidt F, Kramko N, et al. Post-invasion events after infection with Staphylococcus aureus are strongly dependent on both the host cell type and the infecting S. aureus strain. Clin Microbiol Infect. 2016. Sep;22(9):799–809. [DOI] [PubMed] [Google Scholar]
- 35.Jackman SD, Vandervalk BP, Mohamadi H, Chu J, Yeo S, Hammond SA, et al. ABySS 2.0: resource-efficient assembly of large genomes using a Bloom filter. Genome Res. 2017. May;27(5):768–77. doi: 10.1101/gr.214346.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MTG, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015. Nov 15;31(22):3691–3. doi: 10.1093/bioinformatics/btv421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Page AJ, Taylor B, Delaney AJ, Soares J, Seemann T, Keane JA, et al. SNP-sites: rapid efficient extraction of SNPs from multi-FASTA alignments. Microb Genom. 2016. Apr;2(4):e000056. doi: 10.1099/mgen.0.000056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stuart EA. Matching methods for causal inference: A review and a look forward. Stat Sci. 2010. Feb 1;25(1):1–21. doi: 10.1214/09-STS313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsuchiya S, Kobayashi Y, Goto Y, Okumura H, Nakae S, Konno T, et al. Induction of maturation in cultured human monocytic leukemia cells by a phorbol diester. Cancer Res. 1982. Apr;42(4):1530–6. [PubMed] [Google Scholar]
- 40.Aldo PB, Craveiro V, Guller S, Mor G. Effect of culture conditions on the phenotype of THP-1 monocyte cell line. Am J Reprod Immunol. 2013. Jul;70(1):80–6. doi: 10.1111/aji.12129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maeß MB, Wittig B, Cignarella A, Lorkowski S. Reduced PMA enhances the responsiveness of transfected THP-1 macrophages to polarizing stimuli. J Immunol Methods. 2014. Jan 15;402(1–2):76–81. doi: 10.1016/j.jim.2013.11.006 [DOI] [PubMed] [Google Scholar]
- 42.Kim JH, Chaurasia AK, Batool N, Ko KS, Kim KK. Alternative Enzyme Protection Assay To Overcome the Drawbacks of the Gentamicin Protection Assay for Measuring Entry and Intracellular Survival of Staphylococci. Infect Immun. 2019. Mar;87(5):e00119–19. doi: 10.1128/IAI.00119-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Werth BJ, Ashford NK, Penewit K, Waalkes A, Holmes EA, Bryan A, et al. Evolution of cefiderocol resistance in Stenotrophomonas maltophilia using in vitro serial passage techniques. JAC Antimicrob Resist. 2022. Mar;4(1):dlac011. doi: 10.1093/jacamr/dlac011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011. Jan;29(1):24–6. doi: 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990. Oct 5;215(3):403–10. doi: 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- 46.Fuchs S, Mehlan H, Bernhardt J, Hennig A, Michalik S, Surmann K, et al. Aureo Wiki – The repository of the Staphylococcus aureus research and annotation community. International Journal of Medical Microbiology. 2018. Aug;308(6):558–68. [DOI] [PubMed] [Google Scholar]
- 47.Jolley KA, Maiden MC. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics. 2010. Dec;11(1):595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lees JA, Galardini M, Bentley SD, Weiser JN, Corander J. pyseer: a comprehensive tool for microbial pangenome-wide association studies. Bioinformatics. 2018. Dec 15;34(24):4310–2. doi: 10.1093/bioinformatics/bty539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Price MN, Dehal PS, Arkin AP. FastTree 2 –Approximately Maximum-Likelihood Trees for Large Alignments. Poon AFY, editor. PLoS ONE. 2010. Mar 10;5(3):e9490. doi: 10.1371/journal.pone.0009490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Long DR, Penewit K, Lo HY, Almazan J, Holmes EA, Bryan AB, et al. In Vitro Selection Identifies Staphylococcus aureus Genes Influencing Biofilm Formation. Infect Immun. 2023. Mar 15;91(3):e0053822. doi: 10.1128/iai.00538-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Penewit K, Holmes EA, McLean K, Ren M, Waalkes A, Salipante SJ. Efficient and Scalable Precision Genome Editing inStaphylococcus aureusthrough Conditional Recombineering and CRISPR/Cas9-Mediated Counterselection. MBio. 2018. Feb 20;9(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bikard D, Jiang W, Samai P, Hochschild A, Zhang F, Marraffini LA. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 2013. Aug;41(15):7429–37. doi: 10.1093/nar/gkt520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lewis JD, Salipante SJ. Development of advanced control material for reverse transcription-mediated bacterial nucleic acid amplification tests. J Clin Microbiol. 2024. May 8;62(5):e0024324. doi: 10.1128/jcm.00243-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Concordet JP, Haeussler M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Research. 2018. Jul 2;46(W1):W242–5. doi: 10.1093/nar/gky354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.de Bakker V, Liu X, Bravo AM, Veening JW. CRISPRi-seq for genome-wide fitness quantification in bacteria. Nat Protoc. 2022. Feb;17(2):252–81. doi: 10.1038/s41596-021-00639-6 [DOI] [PubMed] [Google Scholar]
- 56.Lo HY, Long DR, Holmes EA, Penewit K, Hodgson T, Lewis JD, et al. Transposon sequencing identifies genes impacting Staphylococcus aureus invasion in a human macrophage model. Infect Immun. 2023. Oct 17;91(10):e0022823. doi: 10.1128/iai.00228-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schwendener S, Perreten V. New shuttle vector-based expression system to generate polyhistidine-tagged fusion proteins in Staphylococcus aureus and Escherichia coli. Appl Environ Microbiol. 2015. May 1;81(9):3243–54. doi: 10.1128/AEM.03803-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pédelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol. 2006. Jan;24(1):79–88. doi: 10.1038/nbt1172 [DOI] [PubMed] [Google Scholar]
- 59.Strober W. Trypan Blue Exclusion Test of Cell Viability. Curr Protoc Immunol. 2015. Nov 2;111:A3.B.1-A3.B.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gabryszewski SJ, Wong Fok Lung T, Annavajhala MK, Tomlinson KL, Riquelme SA, Khan IN, et al. Metabolic Adaptation in Methicillin-Resistant Staphylococcus aureus Pneumonia. Am J Respir Cell Mol Biol. 2019. Aug;61(2):185–97. doi: 10.1165/rcmb.2018-0389OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.San JE, Baichoo S, Kanzi A, Moosa Y, Lessells R, Fonseca V, et al. Current Affairs of Microbial Genome-Wide Association Studies: Approaches, Bottlenecks and Analytical Pitfalls. Front Microbiol. 2020. Jan 30;10:3119. doi: 10.3389/fmicb.2019.03119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee S, Abecasis GR, Boehnke M, Lin X. Rare-variant association analysis: study designs and statistical tests. Am J Hum Genet. 2014. Jul 3;95(1):5–23. doi: 10.1016/j.ajhg.2014.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang M, Zhao Y, Zhang B. Efficient Test and Visualization of Multi-Set Intersections. Sci Rep. 2015. Nov 25;5(1):16923. doi: 10.1038/srep16923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Edwards AM. Phenotype switching is a natural consequence of Staphylococcus aureus replication. J Bacteriol. 2012. Oct;194(19):5404–12. doi: 10.1128/JB.00948-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Proctor RA, Balwit JM, Vesga O. Variant subpopulations of Staphylococcus aureus as cause of persistent and recurrent infections. Infect Agents Dis. 1994. Dec;3(6):302–12. [PubMed] [Google Scholar]
- 66.Fitzgerald JR. Evolution of Staphylococcus aureus during human colonization and infection. Infect Genet Evol. 2014. Jan;21:542–7. doi: 10.1016/j.meegid.2013.04.020 [DOI] [PubMed] [Google Scholar]
- 67.Koziel J, Maciag-Gudowska A, Mikolajczyk T, Bzowska M, Sturdevant DE, Whitney AR, et al. Phagocytosis of Staphylococcus aureus by macrophages exerts cytoprotective effects manifested by the upregulation of antiapoptotic factors. PLoS ONE. 2009;4(4):e5210. doi: 10.1371/journal.pone.0005210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Makhlin J, Kofman T, Borovok I, Kohler C, Engelmann S, Cohen G, et al. Staphylococcus aureus ArcR controls expression of the arginine deiminase operon. J Bacteriol. 2007. Aug;189(16):5976–86. doi: 10.1128/JB.00592-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sause WE, Balasubramanian D, Irnov I, Copin R, Sullivan MJ, Sommerfield A, et al. The purine biosynthesis regulator PurR moonlights as a virulence regulator in Staphylococcus aureus. Proc Natl Acad Sci U S A. 2019. Jul 2;116(27):13563–72. doi: 10.1073/pnas.1904280116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lan L, Cheng A, Dunman PM, Missiakas D, He C. Golden pigment production and virulence gene expression are affected by metabolisms in Staphylococcus aureus. J Bacteriol. 2010. Jun;192(12):3068–77. doi: 10.1128/JB.00928-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kriegeskorte A, Block D, Drescher M, Windmüller N, Mellmann A, Baum C, et al. Inactivation of thyA in Staphylococcus aureus attenuates virulence and has a strong impact on metabolism and virulence gene expression. mBio. 2014. Jul 29;5(4):e01447–01414. doi: 10.1128/mBio.01447-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Somerville GA, Proctor RA. At the Crossroads of Bacterial Metabolism and Virulence Factor Synthesis in Staphylococci. Microbiol Mol Biol Rev. 2009. Jun;73(2):233–48. doi: 10.1128/MMBR.00005-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wu S, Zhang J, Peng Q, Liu Y, Lei L, Zhang H. The Role of Staphylococcus aureus YycFG in Gene Regulation, Biofilm Organization and Drug Resistance. Antibiotics (Basel). 2021. Dec 19;10(12):1555. doi: 10.3390/antibiotics10121555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Porayath C, Suresh MK, Biswas R, Nair BG, Mishra N, Pal S. Autolysin mediated adherence of Staphylococcus aureus with Fibronectin, Gelatin and Heparin. Int J Biol Macromol. 2018. Apr 15;110:179–84. doi: 10.1016/j.ijbiomac.2018.01.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shinji H, Yosizawa Y, Tajima A, Iwase T, Sugimoto S, Seki K, et al. Role of fibronectin-binding proteins A and B in in vitro cellular infections and in vivo septic infections by Staphylococcus aureus. Infect Immun. 2011. Jun;79(6):2215–23. doi: 10.1128/IAI.00133-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lei MG, Gudeta DD, Luong TT, Lee CY. MgrA Negatively Impacts Staphylococcus aureus Invasion by Regulating Capsule and FnbA. Freitag NE, editor. Infect Immun. 2019. Dec;87(12):e00590–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Higgins J, Loughman A, Van Kessel KPM, Van Strijp JAG, Foster TJ. Clumping factor A of Staphylococcus aureus inhibits phagocytosis by human polymorphonuclear leucocytes. FEMS Microbiology Letters. 2006. May;258(2):290–6. [DOI] [PubMed] [Google Scholar]
- 78.Gajdiss M, Monk IR, Bertsche U, Kienemund J, Funk T, Dietrich A, et al. YycH and YycI Regulate Expression of Staphylococcus aureus Autolysins by Activation of WalRK Phosphorylation. Microorganisms. 2020. Jun 9;8(6):E870. doi: 10.3390/microorganisms8060870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hirschhausen N, Schlesier T, Schmidt MA, Götz F, Peters G, Heilmann C. A novel staphylococcal internalization mechanism involves the major autolysin Atl and heat shock cognate protein Hsc70 as host cell receptor. Cell Microbiol. 2010. Dec;12(12):1746–64. doi: 10.1111/j.1462-5822.2010.01506.x [DOI] [PubMed] [Google Scholar]
- 80.Oogai Y, Matsuo M, Hashimoto M, Kato F, Sugai M, Komatsuzawa H. Expression of virulence factors by Staphylococcus aureus grown in serum. Appl Environ Microbiol. 2011. Nov;77(22):8097–105. doi: 10.1128/AEM.05316-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Barua N, Yang Y, Huang L, Ip M. VraSR Regulatory System Contributes to the Virulence of Community-Associated Methicillin-Resistant Staphylococcus aureus (CA-MRSA) in a 3D-Skin Model and Skin Infection of Humanized Mouse Model. Biomedicines. 2021. Dec 24;10(1):35. doi: 10.3390/biomedicines10010035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Freeman ZN, Dorus S, Waterfield NR. The KdpD/KdpE two-component system: integrating K+ homeostasis and virulence. PLoS Pathog. 2013. Mar;9(3):e1003201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Goerke C, Fluckiger U, Steinhuber A, Bisanzio V, Ulrich M, Bischoff M, et al. Role of Staphylococcus aureus global regulators sae and sigmaB in virulence gene expression during device-related infection. Infect Immun. 2005. Jun;73(6):3415–21. doi: 10.1128/IAI.73.6.3415-3421.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.DelMain EA, Moormeier DE, Endres JL, Hodges RE, Sadykov MR, Horswill AR, et al. Stochastic Expression of Sae-Dependent Virulence Genes during Staphylococcus aureus Biofilm Development Is Dependent on SaeS. mBio. 2020. Jan 14;11(1):e03081–19. doi: 10.1128/mBio.03081-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yang D, Ho YX, Cowell LM, Jilani I, Foster SJ, Prince LR. A Genome-Wide Screen Identifies Factors Involved in S. aureus-Induced Human Neutrophil Cell Death and Pathogenesis. Front Immunol. 2019. Jan 31;10:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xiong YQ, Li Y, Goncheva MI, Elsayed AM, Zhu F, Li L, et al. The Purine Biosynthesis Repressor, PurR, Contributes to Vancomycin Susceptibility of Methicillin-resistant Staphylococcus aureus in Experimental Endocarditis. J Infect Dis. 2024. Jan 31;jiad577. doi: 10.1093/infdis/jiad577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Paudel A, Panthee S, Hamamoto H, Grunert T, Sekimizu K. YjbH regulates virulence genes expression and oxidative stress resistance in Staphylococcus aureus. Virulence. 2021. Dec;12(1):470–80. doi: 10.1080/21505594.2021.1875683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Herr AB, Thorman AW. Hiding in plain sight: immune evasion by the staphylococcal protein SdrE. Biochemical Journal. 2017. Jun 1;474(11):1803–6. doi: 10.1042/BCJ20170132 [DOI] [PubMed] [Google Scholar]
- 89.Kim GL, Akoolo L, Parker D. The ClpXP Protease Contributes to Staphylococcus aureus Pneumonia. J Infect Dis. 2020. Sep 14;222(8):1400–4. doi: 10.1093/infdis/jiaa251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Connolly J, Boldock E, Prince LR, Renshaw SA, Whyte MK, Foster SJ. Identification of Staphylococcus aureus Factors Required for Pathogenicity and Growth in Human Blood. Infect Immun. 2017;85(11). doi: 10.1128/IAI.00337-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Marroquin S, Gimza B, Tomlinson B, Stein M, Frey A, Keogh RA, et al. MroQ Is a Novel Abi-Domain Protein That Influences Virulence Gene Expression in Staphylococcus aureus via Modulation of Agr Activity. Infect Immun. 2019. Mar;87(5):e00002–19. doi: 10.1128/IAI.00002-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.O’Halloran DP, Wynne K, Geoghegan JA. Protein A is released into the Staphylococcus aureus culture supernatant with an unprocessed sorting signal. Infect Immun. 2015. Apr;83(4):1598–609. doi: 10.1128/IAI.03122-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Becker S, Frankel MB, Schneewind O, Missiakas D. Release of protein A from the cell wall of Staphylococcus aureus. Proc Natl Acad Sci U S A. 2014. Jan 28;111(4):1574–9. doi: 10.1073/pnas.1317181111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bayer AS, McNamara P, Yeaman MR, Lucindo N, Jones T, Cheung AL, et al. Transposon Disruption of the Complex I NADH Oxidoreductase Gene (snoD) in Staphylococcus aureus Is Associated with Reduced Susceptibility to the Microbicidal Activity of Thrombin-Induced Platelet Microbicidal Protein 1. J Bacteriol. 2006. Jan;188(1):211–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Streker K, Freiberg C, Labischinski H, Hacker J, Ohlsen K. Staphylococcus aureus NfrA (SA0367) Is a Flavin Mononucleotide-Dependent NADPH Oxidase Involved in Oxidative Stress Response. J Bacteriol. 2005. Apr;187(7):2249–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Loughlin MF, Arandhara V, Okolie C, Aldsworth TG, Jenks PJ. Helicobacter pylori mutants defective in the clpP ATP-dependant protease and the chaperone clpA display reduced macrophage and murine survival. Microb Pathog. 2009. Jan;46(1):53–7. doi: 10.1016/j.micpath.2008.10.004 [DOI] [PubMed] [Google Scholar]
- 97.Vaish M, Price-Whelan A, Reyes-Robles T, Liu J, Jereen A, Christie S, et al. Roles of Staphylococcus aureus Mnh1 and Mnh2 Antiporters in Salt Tolerance, Alkali Tolerance, and Pathogenesis. J Bacteriol. 2018. Mar 1;200(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Casey D, Sleator RD. A genomic analysis of osmotolerance in Staphylococcus aureus. Gene. 2021. Jan;767:145268. doi: 10.1016/j.gene.2020.145268 [DOI] [PubMed] [Google Scholar]
- 99.Willing S, Dyer E, Schneewind O, Missiakas D. FmhA and FmhC of Staphylococcus aureus incorporate serine residues into peptidoglycan cross-bridges. J Biol Chem. 2020. Sep 25;295(39):13664–76. doi: 10.1074/jbc.RA120.014371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Berger-Bächi B, Tschierske M. Role of fem factors in methicillin resistance. Drug Resist Updat. 1998;1(5):325–35. doi: 10.1016/s1368-7646(98)80048-4 [DOI] [PubMed] [Google Scholar]
- 101.Weidenmaier C, Peschel A. Teichoic acids and related cell-wall glycopolymers in Gram-positive physiology and host interactions. Nat Rev Microbiol. 2008. Apr;6(4):276–87. doi: 10.1038/nrmicro1861 [DOI] [PubMed] [Google Scholar]
- 102.van Dalen R, Peschel A, van Sorge NM. Wall Teichoic Acid in Staphylococcus aureus Host Interaction. Trends in Microbiology. 2020. Dec;28(12):985–98. doi: 10.1016/j.tim.2020.05.017 [DOI] [PubMed] [Google Scholar]
- 103.Wang M, Buist G, van Dijl JM. Staphylococcus aureus cell wall maintenance—the multifaceted roles of peptidoglycan hydrolases in bacterial growth, fitness, and virulence. FEMS Microbiol Rev. 2022. Oct 28;46(5):fuac025. doi: 10.1093/femsre/fuac025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sutton JAF, Carnell OT, Lafage L, Gray J, Biboy J, Gibson JF, et al. Staphylococcus aureus cell wall structure and dynamics during host-pathogen interaction. PLoS Pathog. 2021. Mar;17(3):e1009468. doi: 10.1371/journal.ppat.1009468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Oliver A, Mena A. Bacterial hypermutation in cystic fibrosis, not only for antibiotic resistance. Clinical Microbiology and Infection. 2010. Jul;16(7):798–808. doi: 10.1111/j.1469-0691.2010.03250.x [DOI] [PubMed] [Google Scholar]
- 106.Mclean K, Lee D, Holmes E, Penewit K, Waalkes A, Ren M, et al. Genomic analysis identifies novel Pseudomonas aeruginosa resistance genes under selection during inhaled aztreonam therapy in vivo. Antimicrobial Agents and Chemotherapy. doi: 10.1128/AAC.00866-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Porto PD, Cifani N, Guarnieri S, Di Domenico EG, Mariggiò MA, Spadaro F, et al. Dysfunctional CFTR Alters the Bactericidal Activity of Human Macrophages against Pseudomonas aeruginosa. Nigou J, editor. PLoS ONE. 2011. May 18;6(5):e19970. doi: 10.1371/journal.pone.0019970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shah PL, Mawdsley S, Nash K, Cullinan P, Cole PJ, Wilson R. Determinants of chronic infection with Staphylococcus aureus in patients with bronchiectasis. Eur Respir J. 1999. Dec;14(6):1340–4. doi: 10.1183/09031936.99.14613409 [DOI] [PubMed] [Google Scholar]
- 109.Clement S, Vaudaux P, Francois P, Schrenzel J, Huggler E, Kampf S, et al. Evidence of an intracellular reservoir in the nasal mucosa of patients with recurrent Staphylococcus aureus rhinosinusitis. J Infect Dis. 2005. Sep 15;192(6):1023–8. doi: 10.1086/432735 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Quantile-quantile plots for in vivo mutations associated with macrophage invasion (blue) and survival (red) phenotypes by various implementations of genome-wide association studies. Association between phenotypes by analysis of (A) reference-free sequence differences (“unitigs”), (B) non-synonymous mutations with gene kernels (“burden testing”), and (C) de novo mutations arising within clonally related patient lineages over time (“first-last comparison”). Significant and non-significant results are shaded differentially.
(PDF)
Phenotypes of S. aureus strains isolated from respiratory cultures of individuals with CF at the time of initial isolation during serial surveillance are compared to the last-collected strains from same patient. Colored lines in this connected dot plot indicate the direction and significance of changes in individual first-last isolate pairs (red = significant increase over time, blue = significant decrease over time, grey = no-significant change) for measures of macrophage invasion relative to in-batch testing of the JE2 control strain (A), and macrophage intracellular survival (B). Statistical significance was assessed using paired two-tailed T tests.
(PDF)
Data Availability Statement
Sequence data generated in support of this study are available from the NCBI Sequence Read Archive under accessions PRJNA860616 and PRJNA1029652.