

Abstract
Background:
Telomeres prevent damage to coding DNA as end-nucleotides are lost during mitosis. Mutations in telomere maintenance genes cause excessive telomere shortening, a condition known as short telomere syndrome (STS). One hepatic manifestation documented in STS is porto-sinusoidal vascular disorder (PSVD).
Methods:
As the etiology of many cases of PSVD remains unknown, this study explored the extent to which short telomeres are present in patients with idiopathic PSVD.
Results:
This monocentric cross-sectional study included patients with histologically defined idiopathic PSVD. Telomere length in 6 peripheral blood leukocyte subpopulations was assessed using fluorescent in situ hybridization and flow cytometry. Variants of telomere-related genes were identified using high-throughput exome sequencing. In total, 22 patients were included, of whom 16 (73%) had short (9/22) or very short (7/22) telomeres according to age-adjusted reference ranges. Fourteen patients (64%) had clinically significant portal hypertension. Shorter telomeres were more frequent in males (p = 0.005) and patients with concomitant interstitial lung disease (p < 0.001), chronic kidney disease (p < 0.001), and erythrocyte macrocytosis (p = 0.007). Portal hypertension (p = 0.021), low serum albumin level (p < 0.001), low platelet count (p = 0.007), and hyperbilirubinemia (p = 0.053) were also associated with shorter telomeres. Variants in known STS-related genes were identified in 4 patients with VSTel and 1 with STel.
Conclusions:
Short and very short telomeres were highly prevalent in patients with idiopathic PSVD, with 31% presenting with variants in telomere-related genes. Telomere biology may play an important role in vascular liver disease development. Clinicians should consider measuring telomeres in any patient presenting with PSVD.
INTRODUCTION
Telomeres are repetitive DNA sequences at the ends of human chromosomes composed of 6 nucleotides (TTAGGG).1,2 They act as shields to preserve coding regions because DNA replication inherently leads to the erosion of chromosome ends. This mechanism is counteracted by telomerase, an enzyme that adds new telomeric DNA to the ends of chromosomes.2,3 Telomerase is a multienzyme complex with 2 core units, the reverse transcriptase (telomerase reverse transcriptase) and the RNA template (telomerase RNA component), as well as additional proteins.4 Telomerase is highly expressed in germinal and early embryonic cells but suppressed with cellular differentiation.5 Therefore, the average telomere length (TL) shortens during one’s lifetime. However, physiological TL attrition is not linear but more pronounced at early and late ages.2,6,7 If a cell’s telomeres become too short, a DNA damage response is activated, mobilizing a molecular pathway that results in cellular senescence or apoptosis.1,8
Mutations in genes responsible for telomere maintenance and repair lead to critically short telomeres and stem cell deficiency, a condition known as short telomere syndrome (STS).1,9,10 It is important to note that even in patients diagnosed with STS based on TL measurement, a predisposing mutation in the telomere-related gene is often not identified. Indeed, mutations in telomere-related genes have been identified in only ~half of these patients.11,12 Shortened telomeres result in a wide range of clinical manifestations affecting various organ systems, including the lungs, liver, bone marrow, immune system, and skin. The age of onset and severity of symptoms varies widely, with younger individuals typically experiencing more severe manifestations.2,13,14
The diagnosis of STS is based on clinical findings and the pattern of short telomeres measured in multiple subpopulations of peripheral blood leukocytes using fluorescent in situ hybridization combined with flow cytometry.4,15,16,17 The average TL per cell subpopulation is compared to a cell type-specific reference range derived from a population of over 400 healthy individuals aged 0–102 years.7,18 Importantly, telomere measurements in leukocytes are directly proportional to the TL measured in other tissues of the body.15
Patients’ telomeres are considered “normal” if their average length is above the 10th percentile (normal telomere length [NTel]), “short” if the average length is between the first and 10th percentile (short telomere length [STel]), and “very short” if it is below the first percentile (very short telomere length [VSTel]).5,6 Patients with VSTel are commonly referred for genetic counseling to investigate the presence of potentially predisposing mutations, whereas those with STel are considered to be in the gray zone, requiring additional clinical information to define follow-up.10
Common clinical features of STS include pulmonary fibrosis, liver disease, bone marrow failure, and increased risk of cancer. Interstitial lung disease (ILD), particularly idiopathic pulmonary fibrosis, appears most often after the age of 50 years and is recognized as one of the common clinical manifestations of STS in adults.19,20 Hematological manifestations are also frequent.11 They generally precede lung disease by several decades and vary in severity from subtle cytopenias to bone marrow failure to clonal progression toward myelodysplastic syndrome and acute myeloid leukemia.20,21,22,23 Patients with STS have a lifetime cancer risk of 15%, with hematological malignancies making up two-thirds of cases.2,24
Liver disease occurs in up to 40% of patients with STS, often before the age of 40 years. It may manifest as hepatopulmonary syndrome, cryptogenic cirrhosis, or nodular regenerative hyperplasia (NRH).6,25,26 Many cases lead to the development of portal hypertension (PH).
Porto-sinusoidal vascular disorder (PSVD) is a group of hepatic vascular diseases characterized by lesions involving the portal venules and sinusoids in the absence of cirrhosis, with or without associated PH.27,28,29,30 According to the current consensus, diagnosis of PSVD requires a liver biopsy to rule out cirrhosis. Once cirrhosis is excluded, the diagnosis is based on the presence of a specific clinical sign of PH (gastric, esophageal, or ectopic varices; portal hypertensive bleeding; or portosystemic collaterals on imaging), a specific histologic lesion (formerly obliterative portal venopathy, now designated as portal vein stenosis; NRH; or incomplete septal fibrosis), or a nonspecific sign of PH (ascites; platelet count < 150,000/mm3; spleen size ≥ 13 cm in the largest axis) coupled with a nonspecific histologic lesion of PSVD (herniated portal vein branches, hypervascularized portal tracts, periportal abnormal vessels, nonzonal sinusoidal dilation, and mild perisinusoidal fibrosis).27
The pathogenesis of PSVD remains unclear. Commonly associated comorbidities are identified in roughly 50% of cases, including DILI, prothrombotic conditions, autoimmune diseases, common variable immune deficiency, HIV infection, and hereditary diseases, such as familial obliterative portal venopathy.28,30 More recently, PSVD was also found to occur in the extremely rare Mahvash disease.31 PSVD may be diagnosed in the absence of PH in up to 70% of cases, with a 20% risk of developing gastroesophageal varices within 10 years of diagnosis.28 Hence, the pathological documentation of PSVD in the absence of PH may indicate a preclinical disease state.28
If none of the above-mentioned associated conditions can be identified, PSVD is considered idiopathic. Based on previous reports demonstrating the presence of NRH in patients with proven STS, we hypothesized that some of these patients may have developed PSVD secondary to STS.25
Here, we examined the TL in patients with idiopathic PSVD and explored the telomere-related gene profile of those with short telomeres. Second, we aimed to characterize the distinguishing features of patients who developed otherwise unexplained PSVD in the presence of short telomeres.
METHODS
Patients
This single-center cross-sectional study was approved by the local ethics committee (CER-VD protocol number 2022-00587 and amendment #1 for genetic testing). Participants were included from September 1, 2022 to April 1, 2023. Those with well-documented PSVD were recruited from the outpatient liver clinic of the Division of Gastroenterology and Hepatology of Lausanne University Hospital and identified using a multiparametric clinical data warehouse search engine. Research is reported in line with the STROBE guidelines.32 Keywords used for the identification of study participants included “porto-sinusoidal vascular disorder,” “nodular regenerative hyperplasia,” “incomplete septal fibrosis,” “obliterative portal venopathy,” “noncirrhotic portal hypertension,” and “portal hypertension in the absence of cirrhosis.” The identified patients were checked for inclusion and exclusion criteria.
Inclusion criteria were (1) age between 18 and 80 years and (2) idiopathic PSVD with histopathological documentation, regardless of the presence of PH. The idiopathic nature was retained in the absence of linked DILI, prothrombotic conditions, autoimmune diseases, common variable immune deficiency, and HIV infection. Exclusion criteria were as follows: (1) pregnancy; (2) refusal to consent to participate in the study; and (3) the following chronic liver diseases: chronic viral hepatitis, autoimmune hepatitis, primary biliary cholangitis, primary sclerosing cholangitis, alcohol-associated liver disease, NAFLD, and hereditary liver diseases (hemochromatosis, alpha-1 antitrypsin deficiency, Wilson disease, and other rare inborn errors of metabolism). Patients who received drugs known to be associated with PSVD development were excluded.33 Patients with active prothrombotic conditions or past thromboembolic events were excluded.
Demographic, clinical, laboratory, radiological, and histological data were collected from digital medical records and anonymized before analysis. Demographic data included age, gender, and ethnicity. Clinical data included tobacco use, alcohol abuse, drug history, personal/family history of liver disease, lung disease, hematopoietic disease, immunodeficiency, early onset osteoporosis, cancer, organ transplant, and early hair greying. Laboratory data included, at the time of liver biopsy, liver function tests (aspartate transaminase, alanine transaminase, alkaline phosphatase, γ-glutamyl transferase, and total and direct bilirubin), international normalized ratio, serum albumin, factor V, C-reactive protein, leukocyte counts with differential, platelet count, hemoglobin, mean corpuscular volume, ferritin, and transferrin saturation.
Radiological data included any techniques assessing liver shape and size as well as signs of PH, including Doppler ultrasound, MRI, and 3-phase CT. Liver stiffness was measured using transient elastometry (FibroScan; Echosens, Paris, France).
Hepatic outcomes, including the presence of PH and its complications (variceal bleeding, ascites, etc.), jaundice, and need for liver transplantation, were documented from time of histological PSVD confirmation until last clinical follow-up at time of TL assessment.
Liver histology
The histological specimens included archived samples obtained from transparietal, transjugular, or surgical liver biopsies. Stains available for all cases included hematoxylin and eosin, Masson’s trichrome to exclude cirrhosis, reticulin stain to investigate NRH, and immunohistochemical staining for cytokeratin 7. Histological slides were independently reevaluated for specific and nonspecific histologic lesions of PSVD by 2 pathologists, 1 junior (Chiara Saglietti) and 1 senior (Christine Sempoux), who were blinded to the clinical history.
Diagnosis of porto-sinusoidal vascular disorder
According to the recent consensus on the PSVD definition, the diagnosis was confirmed if the patient presented with specific signs of PH in the absence of cirrhosis.28 Additionally, PSVD was retained if the biopsy revealed a histological lesion specific for PSVD or if it revealed a nonspecific histological lesion in a patient with a nonspecific sign of PH.28
Fluorescent in situ hybridization combined with flow cytometry
TL was measured using multicolor fluorescent in situ hybridization combined with flow cytometry technology on peripheral blood leukocytes as described previously.34,35 In brief, red blood cells were lysed using ammonium chloride, and the DNA of the leukocytes was denatured and hybridized with a fluorescently labelled telomere-specific peptic nucleic acid probe, as well as with leukocyte subset-specific antibodies. After several washing steps, cell type-specific telomere fluorescence in 6 different subsets of leukocytes (granulocytes, lymphocytes, naive T cells [CD45RA+/CD20−], memory T cells [CD45RA−/CD20−], B cells [CD20+], and natural killer cells [CD57+]) was analyzed using a flow cytometer. Telomere length was assessed as an absolute value (kilo base pairs, kb) and calculated as delta telomere length (dTL=difference of the absolute value to the age-related reference value of the 50th percentile in kb) to compare values independent of age.17,34
Patients were considered to fall into the NTel category if at least 3/6 leukocyte subsets had NTel and the average dTL was below 1.5 kb. Patients were assigned to the VSTel category if at least out of 3/6 leukocyte subsets had VSTel. The remaining patients were considered to have STel.
Genetic testing
With appropriate informed consent from the participants, exome sequencing was conducted on NextSeq 500 sequencing from Illumina using the comprehensible library from Twist Biosciences® on genomic DNA extracted from leukocytes of patients with STel or VSTel. The raw NGS reads were aligned to the human reference genome GRCh37/hg19 using Novoalign (Novocraft Technologies; v4.02.02), and bioinformatics analysis was carried out using an in-house pipeline (v.8.3). Variant calling followed genome analysis toolkit Best Practices recommendations (https://gatk.broadinstitute.org/). copy number variation analysis was based on the ExomeDepth tool.36 The analysis targeted a virtual panel of genes related to STS, including ACD, CTC1, DKC1, NAF1, NHP2, NOP10, PARN, POT1, RPA1, RTEL1, STN1, TERT, TINF2, WRAP53, ZCCHC8. Sequencing of TERC was performed using Sanger sequencing. The results were transmitted to patients within the framework of a dedicated genetic consultation in the case of significant genetic variant detection. The patients whose genetic analyses did not reveal any abnormalities continued their planned follow-up with an attending hepatologist.
Statistical analyses
Descriptive statistics included the median and range for continuous variables, as well as frequencies and percentages for binary and categorical variables. Categorical data were compared using chi-squared or Fisher exact test and continuous variables by t-test and ANOVA, or nonparametric testing (Mann-Whitney U test or Kruskal-Wallis test), as appropriate. Statistical analyses were performed using STATA/SE version 18.
RESULTS
Cohort description
A total of 22 patients met the inclusion criteria (Figure 1). Their primary characteristics are presented in Table 1 and Supplemental Table S1, http://links.lww.com/HC9/A978. The median patient age was 54 years (range, 27–71 y). Eight patients (36%) were female. The median age at the first documentation of PSVD was 36 years (range, 16–63 y). Patients were diagnosed with PSVD at a median of 8 years before inclusion into the study (range, 1–36 y). Eighteen patients were of Caucasian, 2 of Asian and 2 of African ethnicity. Three patients (14%) had a history of cancer (renal cell carcinoma, bladder carcinoma, and prostate adenocarcinoma); none of them had received systemic chemotherapy. Six patients (27%) presented with coexisting ILD. Thirteen patients (59%) were known for hematological disease, including 9 cases of uni-lineage or multi-lineage cytopenias of variable severity, 1 case of myelodysplastic syndrome, 2 cases of idiopathic thrombocytosis, and 1 monoclonal gammopathy. Four patients had chronic kidney disease (18%). Before the study, STS was suspected in 6 patients who presented with a typical clinical picture and, notably, an associated ILD.
FIGURE 1.
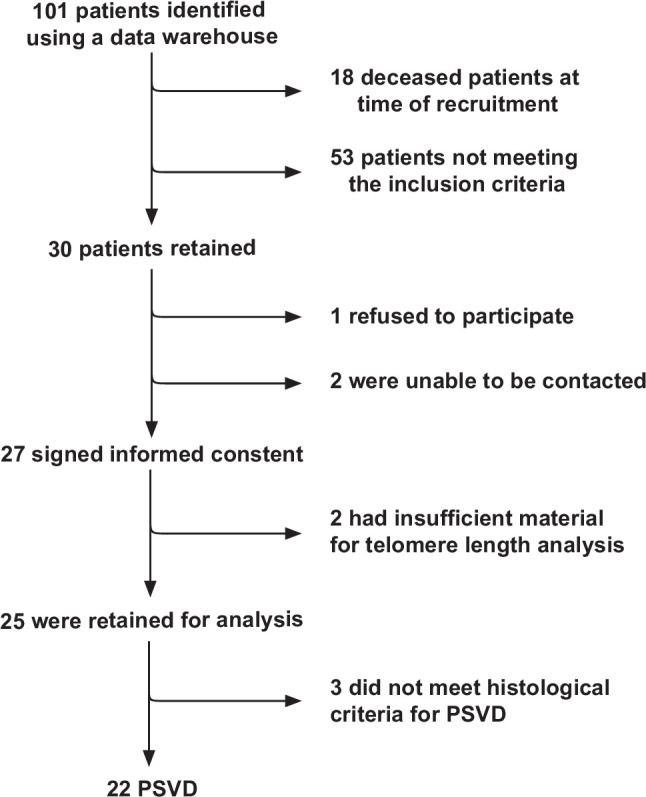
Flowchart portraying the patient selection process. Of the 101 patients identified, 18 were deceased at the time of recruitment, 2 were unable to be contacted, 1 refused participation, and 53 did not meet inclusion criteria (18 patients presented evident causes of PSVD listed in the exclusion criteria; 18 had other underlying chronic liver diseases listed in the exclusion criteria; 17 patients had no available liver biopsy or were below 18 y of age). Of the 27 retained patients, 2 had insufficient biological material for analysis, and 3 did not meet the diagnostic histological criteria for the idiopathic PSVD as defined in ref. 28. Abbreviation: PSVD, porto-sinusoidal vascular disorder.
TABLE 1.
Overview of results by telomere length (TL) group
| Total n = 22 | NTel n = 6 | STel n = 9 | VSTel n = 7 | |
|---|---|---|---|---|
| Women (n, %) | 8 (36) | 4 (67) | 4 (44) | 0 (0%) |
| Age at time of analysis (y, median, range) | 54 (27–71) | 42 (27–59) | 47 (35–64) | 57 (28–71) |
| Age at documentation of liver disease (y, median, range) | 36 (16–63) | 38 (16–56) | 34 (25–58) | 51 (21–63) |
| PSVD classification | ||||
| Nodular regenerative hyperplasia (n, %) | 5 (23) | 1 (17) | 2 (22) | 2 (29) |
| Portal vein stenosis (n, %) | 16 (73) | 5 (83) | 7 (78) | 4 (57) |
| Noncirrhotic portal hypertension (n, %) | 1 (5) | 0 (0) | 0 (0) | 1 (14) |
| Signs of portal hypertension (n, %) | 14 (64) | 2 (33) | 5 (56) | 7 (100) |
| Ascites (n, %) | 7 (32) | 0 (0) | 3 (33) | 4 (57) |
| Esophageal varices (n, %) | 11 (50) | 1 (17) | 5 (56) | 5 (71) |
| Splenomegaly (n, %) | 8 (36) | 1 (17) | 5 (56) | 2 (29) |
| Platelet count (G/l, median, range) | 182 (25–526) | 219 (165–526) | 284 (25–500) | 106 (40–189) |
| Available HVPG measurement (n) | 11/22 | 2/6 | 3/9 | 6/7 |
| Gradient (mm Hg, median, range) | 11 (2–16) | 4 (2–6) | 12 (3–15) | 12 (2–16) |
| Other conditions | ||||
| Interstitial lung disease (n, %) | 6 (27) | 0 (0) | 0 (0) | 6 (86) |
| Hematological manifestations (n, %)a | 13 (59) | 4 (67) | 4 (44) | 5 (71) |
| Chronic kidney disease (n, %)b | 4 (18) | 0 (0) | 0 (0) | 4 (57) |
| Laboratory analyses | ||||
| Liver function tests | ||||
| ALT (U/l, median, range) | 40 (12–495) | 44 (12–495) | 55 (16–289) | 38 (22–56) |
| ALP (U/l, median, range) | 125 (40–239) | 109 (40–239) | 125 (63–185) | 117 (72–160) |
| R-value (median, range)c | 1.1 (0.3–6.9) | 1.0 (0.4–6.9) | 1.6 (0.3–6.5) | 1.1 (0.4–2.3) |
| Liver function | ||||
| Total bilirubin (µmol/l, median, range) | 10 (3–43) | 9 (5–16) | 10 (3–35) | 15 (5–43) |
| Albumin (g/l, median, range) | 45 (27–48) | 47 (46–48) | 45 (39–47) | 33 (27–41) |
| INR (median, range) | 1.0 (1.0–3.4) | 1.0 (1.0–1.2) | 1.0 (1.0–3.4)d | 1.1 (1.0–1.4) |
| Imaging analyses | ||||
| Dysmorphic liver (n, %) | 10/21 (48) | 0/6 (0) | 4/8 (50) | 6/7 (86) |
| Portal venous flow < 15 cm/s (n, %) | 9/20 (45) | 1/6 (17) | 3/8 (38) | 5/6 (83) |
Note: Results of the cohort stratified by TL classification (cf. Methods section).
Hematological manifestations included unexplained cytopenia, myelodysplastic syndrome, monoclonal gammopathy, iron-deficiency anemia, and unexplained thrombocytosis.
Chronic kidney disease patients had an estimated glomerular filtration rate inferior to 60 mL/min/1.73 m2.
([ALT/ULN] / [ALP/ULN]); ULN for ALT, 40 U/l; ULN for ALP, 120 U/l.
Two patients taking vitamin K inhibitors, both presented normal factor V levels.
Abbreviations: ALP, alkaline phosphatase; ALT, alanine aminotransferase; INR, international normalized ratio; NRH, nodular regenerative hyperplasia; NTel, normal telomere length; PVS, portal vein stenosis; PSVD, porto-sinusoidal vascular disorder; STel, short telomere length; ULN, upper limit of the norm; VSTel, very short telomere length.
At the time of liver biopsy, liver function tests were elevated in ~half of the patients, with a median alanine transaminase of 40 U/l (range, 12–495 U/l, aspartate transaminase of 38 U/l (range, 13–201 U/l), alkaline phosphatase of 125 U/l (range, 40–239 U/l, γ-glutamyl transferase of 112 U/l (range, 12–867 U/l, and total bilirubin level of 10 µmol/l (range, 3–43 µmol/l). The median R-value, a ratio of alanine transaminase to alkaline phosphatase, which aids in differentiating cytolytic from cholestatic hepatic injury, was 1.11 (range 0.36–6.91).37 Fifteen patients (68%) had an R-value below 2 (cholestatic pattern), 5 (22%) had an R-value between 2 and 5 (mixed pattern), and 2 (9%) had a ratio above 5 (hepatocellular pattern). Median international normalized ratio was 1.0 (range, 1.0–3.4) and albumin 45 g/l (range, 27–48 g/l). Thrombocytopenia was present in 9 patients (41%), with a median platelet count of 182 G/l (range, 25–526 G/l). On imaging, 10/22 (45%) patients were considered to have a dysmorphic liver, as assessed by expert liver radiologists.
In total, 14 patients (64%) presented with PH, including 11 (50%) patients with gastroesophageal varices, 8 (36%) with splenomegaly, 7 (32%) with ascites, 7 (32%) with portal hypertensive gastropathy, and 3 with other complications (portal hypertensive colopathy or portosystemic shunts on imaging). HVPG was measured in 11 patients, the median being 10 mm Hg (range, 2–16 mm Hg). PH was more common in males than females (12/14 males vs. 2/8 females, p = 0.008), as were higher total bilirubin (p = 0.099) and lower platelet counts (p = 0.018). Additionally, clinically significant PH was present in all patients with ILD (p = 0.040), and ILD was associated with lower serum albumin levels (p = 0.001) and platelet counts (p = 0.010).
One patient was biopsied in 1986 for clinically significant PH with ascites and esophageal varices; the corresponding histological samples were no longer available for review, but the histology reports documented the absence of cirrhosis.
Histological samples from 21 patients were available for review. The absence of cirrhosis was confirmed in all cases, and specific histologic lesions of PSVD were identified (NRH = 5; portal vein stenosis = 16). None of the patients presented with incomplete septal fibrosis. Figure 2 exemplifies histological lesions observed in the patient cohort.
FIGURE 2.

Histological features of PSVD observed in our patient cohort. (A) and (B) Nodular regenerative hyperplasia, with parenchymal nodularity highlighted by reticulin stain in panel B (× 10). (C) Portal vein stenosis, characterized by a portal space with no clearly visible portal venous branch (× 40). (D) Herniated portal vein in direct contact with periportal parenchyma (× 40). (E) Hypervascularized portal space with multiple small-caliber venous branches (× 20). Abbreviation: PSVD, porto-sinusoidal vascular disorder.
Three patients (14%) underwent liver transplantation for complications of PH, one of which received a combined liver and lung transplantation.38 Another patient with prior lung transplantation died of PH complications. A fifth patient with ILD and PSVD died of acute respiratory distress syndrome before being placed on the transplant list for combined liver and lung transplantation.
Telomere lengths
Six patients (27%) fell into the NTel, 9 (41%) into STel, and 7 (32%) into the VSTel category. TL was significantly shorter (p = 0.039), and the average dTL was significantly larger (2.45 vs. 1.25, p = 0.005) in male patients than in female patients.
Table 1 summarizes the results stratified by TL classification. Figure 3 shows the TL of all patients. Table 2 shows the TL in all 6 subsets of leukocytes for each patient, classified as VSTel, STel, or NTel, together with their dTL. Granulocytes showed the highest dTL. The dTL is summarized as a heat map in Supplemental Table S2, http://links.lww.com/HC9/A978.
FIGURE 3.

Telomere length in leukocyte subsets from the 22 patients with porto-sinusoidal vascular disorder considered idiopathic. The 6 leukocyte subsets tested are lymphocytes, granulocytes, naive T cells (CD45RA pos), memory T cells (CD45RA neg), B cells (CD20 pos), and NK/NKT cells (CD57 pos). The colored lines represent percentiles of telomere length measurements in kilobases (kb) from over 400 healthy individuals [ref.7,17,18,35]: red, 99th percentile; yellow, 90th percentile, dark green, 50th percentile (median); light green, 10th percentile; turquoise, 1st percentile. Symbols represent individual patients: blue diamonds, normal telomere length; red squares, short telomere length; and green triangles, very short telomere length. Abbreviations: NK, natural killer; NKT, natural killer T.
TABLE 2.
Telomere length (in kb) in each patient for 6 subsets of peripheral blood leukocytes
| Patient | Lymphocytes | deltaTL (kb) | Granulocytes | deltaTL (kb) | Naive T cells | deltaTL (kb) | Memory T cells | deltaTL (kb) | B cells | deltaTL (kb) | NK/NKT cells | deltaTL (kb) | Average deltaTL (kb) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Normal telomere length group (above 10th percentile of reference range) | |||||||||||||
| 1 | NTel | 1.14 | NTel | 1.49 | STel | 1.46 | NTel | 0.70 | NTel | 0.98 | NTel | 0.96 | 1.12 |
| 6 | NTel | 1.13 | STel | 1.82 | STel | 1.57 | NTel | 0.43 | STel | 1.28 | NTel | 0.94 | 1.19 |
| 7 | NTel | 0.40 | NTel | 1.18 | NTel | 0.82 | NTel | -0.12 | STel | 1.23 | NTel | -0.21 | 0.55 |
| 9 | NTel | -0.28 | NTel | 0.81 | NTel | 0.13 | NTel | -0.67 | NTel | 0.64 | NTel | -0.69 | -0.01 |
| 13 | STel | 1.23 | STel | 1.86 | STel | 1.37 | NTel | 0.92 | STel | 1.42 | NTel | 0.76 | 1.26 |
| 15 | NTel | 0.86 | STel | 1.54 | NTel | 1.03 | NTel | 0.37 | STel | 1.39 | NTel | 0.31 | 0.92 |
| Short telomere length group (between 1st and 10th percentile of reference range) | |||||||||||||
| 2 | STel | 1.61 | STel | 2.23 | STel | 2.07 | NTel | 1.07 | STel | 1.82 | NTel | 1.28 | 1.68 |
| 3 | STel | 1.30 | STel | 2.20 | STel | 1.59 | NTel | 0.95 | STel | 1.71 | NTel | 1.25 | 1.50 |
| 4 | STel | 1.50 | STel | 1.99 | STel | 1.87 | STel | 1.46 | STel | 1.91 | STel | 1.72 | 1.74 |
| 8 | STel | 1.46 | STel | 2.53 | STel | 1.96 | NTel | 1.14 | VSTel | 2.37 | STel | 1.73 | 1.86 |
| 10 | STel | 1.80 | STel | 2.44 | STel | 2.27 | STel | 1.40 | VSTel | 2.49 | NTel | 1.59 | 2.00 |
| 11 | STel | 1.74 | VSTel | 2.69 | STel | 2.25 | STel | 1.25 | VSTel | 2.54 | NTel | 1.15 | 1.94 |
| 12 | STel | 1.99 | VSTel | 2.87 | STel | 2.32 | STel | 1.68 | VSTel | 3.01 | NTel | 1.59 | 2.24 |
| 14 | STel | 1.56 | STel | 2.24 | STel | 2.31 | NTel | 0.97 | STel | 2.15 | NTel | 1.39 | 1.77 |
| 16 | STel | 1.44 | STel | 2.14 | STel | 1.83 | STel | 1.27 | VSTel | 2.30 | NTel | 1.46 | 1.74 |
| Very short telomere length group (below 1st percentile of reference range) | |||||||||||||
| 5 | STel | 2.11 | VSTel | 2.90 | VSTel | 2.62 | STel | 1.90 | VSTel | 2.79 | STel | 1.99 | 2.39 |
| 17 | VSTel | 3.38 | VSTel | 4.22 | VSTel | 3.73 | VSTel | 3.03 | STel | 1.91 | VSTel | 3.28 | 3.26 |
| 18 | VSTel | 3.91 | VSTel | 4.09 | VSTel | 4.35 | VSTel | 3.37 | Not available | VSTel | 3.61 | 3.87 | |
| 19 | VSTel | 2.94 | VSTel | 4.21 | VSTel | 3.33 | VSTel | 2.81 | VSTel | 3.31 | STel | 2.91 | 3.25 |
| 20 | VSTel | 3.13 | VSTel | 3.65 | VSTel | 3.66 | VSTel | 2.54 | VSTel | 5.27 | STel | 3.05 | 3.55 |
| 21 | VSTel | 2.73 | VSTel | 4.15 | VSTel | 3.28 | VSTel | 2.35 | VSTel | 3.27 | STel | 3.06 | 3.14 |
| 22 | VSTel | 3.37 | Not available | VSTel | 3.91 | VSTel | 3.02 | VSTel | 3.62 | STel | 2.87 | 3.36 | |
Note: TL and delta TL for each cell type in each patient, as well as the average dTL.
Abbreviations: NK, natural killer, NKT, natural killer T-cell; NTel, normal telomere length; STel, short telomere length; TL, telomere length; VSTel, very short telomere length.
Patients with PH more often had short or very short TL (7/7 VSTel, 5/9 STel, 2/6 NTel, p = 0.039) and a significantly greater average dTL (2.39 vs. 1.37 kb in those without PH, p = 0.021) in all subsets of leukocytes.
The presence of PH complications was associated with an increased average dTL, including ascites (p = 0.023), decreased platelet counts (p = 0.007), and slowed portal venous flow, defined as < 15 cm/s, on Doppler ultrasound (p = 0.027). Patients with a shorter TL were more likely to have abnormal liver function. A higher average dTL was associated with a higher bilirubin value (p = 0.053) and a lower serum albumin level (p < 0.001).
Several comorbidities classically described in STS were significantly more prevalent in patients with STel and VSTel. The coexistence of ILD was strongly associated with VSTel (p < 0.001), and the average dTL in patients with lung disease was more than twice that of patients without (3.40 vs. 1.49 kb, p < 0.001). All 4 patients with chronic kidney disease had VSTel in most subsets of leukocytes, and their dTL was significantly larger than that of those without (3.48 vs. 1.69 kb, p < 0.001). Erythrocyte macrocytosis was associated with shorter telomeres in all leukocyte subsets (p = 0.007). In imaging studies, dysmorphic livers and slowed portal vein flow were more commonly described in patients with shorter TL (p = 0.004 and p = 0.027, respectively).
Genetic analysis
Genetic testing was performed in the 16 patients presenting with STel or VSTel. Of the 16 patients, 5 (31%) showed variants in telomere-related genes. One patient with STel presented with a variant classified as a variant of uncertain significance (VUS) in DKC1. This patient showed NRH histologically and clinically significant PH. Of the 7 patients with VSTel, 4 (71%) harbored variants within a known STS-related gene. One patient presented with a VUS in the RTEL1 gene, 2 presented with a likely pathogenic variant in the RTEL1 gene (of which 1 presented an additional VUS in the same gene), and 1 patient presented with a likely pathogenic TERT variant. The genetic results are summarized in Table 3.39
TABLE 3.
Genetic analyses of patients with STel and VSTel
| Patient ID | Telomere length | TRG mutation | Mutated gene | Zygosity | Variant | ACMG classificationa |
|---|---|---|---|---|---|---|
| 5 | VSTel | Absent | — | — | — | — |
| 17 | VSTel | Present | TERT | Heterozygous | NM_198253.2, c.2213C>T, (p.Thr738Met) | Likely pathogenic |
| 18 | VSTel | Present | RTEL1 | Heterozygous Heterozygous |
1) NM_032957.4, c.1553 + 5G>A, (p?) 2) NM_032957.4, c.3037C>T, (p.Arg1013Trp) |
1) Likely pathogenic 2) VUS |
| 19 | VSTel | Present | RTEL1 | Heterozygous | NM_032957.4, c.2320C>T, p.(Arg774Cys) | Likely pathogenic |
| 20 | VSTel | Present | RTEL1 | Heterozygous | NM_032957.4, c.1720C>T, (p.Arg574Cys) | VUS |
| 21 | VSTel | Absent | — | — | — | — |
| 22 | VSTel | Absent | — | — | — | |
| 2 | STel | Absent | — | — | — | — |
| 3 | STel | Absent | — | — | — | — |
| 4 | STel | Present | DKC1 | Heterozygous | NM_001363.5, c.1462G>A, (p.Glu488Lys) | VUS |
| 8 | STel | Absent | — | — | — | — |
| 10 | STel | Absent | — | — | — | — |
| 11 | STel | Absent | — | — | — | — |
| 12 | STel | Absent | — | — | — | — |
| 14 | STel | Absent | — | — | — | — |
| 16 | STel | Absent | — | — | — | — |
See reference39.
Abbreviations: STel, short telomere length; TRG, telomere-related gene; VSTel, very short telomere length; VUS, variant of unknown significance.
DISCUSSION
Abnormally shortened telomeres are a hallmark of STS, which is characterized by a wide spectrum of clinical manifestations. Previous reports have documented a liver disease prevalence of up to 40%.1,6,8 In a recent retrospective study, Sidali et al assessed the prevalence of liver disease in patients with telomere-related gene mutations. In their experience, up to 72% of these patients showed evidence of liver disease. When liver biopsy was available, PSVD was observed in 48% of the cases.26 Moreover, in a population study in the United Kingdom involving 472,732 participants with a PCR-based lymphocyte TL analysis, PH was the fifth most overrepresented condition in individuals with short lymphocyte telomeres.20
Identifying STS in a patient with idiopathic PSVD is crucial not only for enhancing our understanding of disease pathogenesis but also for its significant clinical implications. First, it paves the way for novel targeted therapeutic approaches, exemplified by danazol, a recently emphasized agent that functions by stimulating telomerase activity and promoting telomere elongation.6 Hence, managing these patients should extend beyond solely addressing portal hypertension complications and proactive screening for frequently associated comorbidities like pulmonary, hematological, and dermatological conditions.
NRH and portal vein stenosis are the 2 specific histological lesions for PSVD diagnosis in the absence of cirrhosis. NRH has been described in patients with proven STS.25 In daily clinical practice, many PSVD cases are considered idiopathic after investigations ruled out drug toxicity, a hypercoagulable state, autoimmunity, common variable immune deficiency, and HIV infection. To date, it is unknown to which extent short telomeres may underlie these so-called idiopathic forms of PSVD.
We assessed the average TL in 6 subsets of peripheral blood leukocytes in 22 well-characterized patients with idiopathic PSVD. To limit the role of confounding factors on TL measurement, we excluded any patient presenting chronic liver diseases that could directly impact TL. We documented a high rate of shortened TL in these individuals, 41% with STel and 32% with VSTel, as compared to reference ranges from a large population consisting of over 400 healthy individuals.7,18 Importantly, only a minor proportion of the patients (6/22) included in the present study were clinically suspected to have STS before inclusion in this study.40
Some elements of interest emerge when examining the characteristics of patients with confirmed STel or VSTel. First, when combining the STel and VSTel groups, the median age of onset of liver disease in patients was 35 years, which is in line with previous descriptions of STS.25 Second, males were significantly more likely to present shorter TL and increased dTL than females. This finding is consistent with large population studies on TL.20 In addition, all patients carrying variants were male. One potential explanation is that males are more prone to carry or accumulate risk factors for TL shortening, either mutations in inherited STS-related or yet unknown genes or acquired risk factors such as tobacco use and dyslipidemia. The high guanine content of telomeric DNA makes it highly susceptible to oxidative damage.1,41,42 In addition, sex hormones, especially estrogen, seem to play an important role in maintaining telomere length. Indeed, numerous studies have shown that estrogen exposure is associated with greater TL, and estrogen deficiency leads to increased telomere attrition.43,44,45
In our experience, the presence of concomitant ILD is strongly associated with reduced TL. ILD is one of the most common manifestations of STS, and telomere-associated gene mutations are present in up to one-third of familial interstitial pulmonary fibrosis cases.6,19 Hence, there is a significant prevalence of short telomeres in patients with both ILD and PSVD. Therefore, this clinical association should prompt early TL assessment. Furthermore, it is noteworthy that all patients with concomitant ILD had a more severe liver phenotype, notably clinically significant PH.
All 4 patients with chronic kidney disease also had VSTel and genetic variants. Various studies suggest that kidney disease may be another prominent feature of STS, possibly following an endothelial pathogenesis similar to that of PSVD.1,46,47 Nevertheless, renal involvement requires more precise characterization. In our study, the presence of clinically significant PH was a potential confounding factor, as it may contribute to kidney dysfunction.
Another clinical feature of interest is the presence of erythrocyte macrocytosis. Indeed, STS patients frequently develop bone marrow dysfunction and failure, often initially presenting as isolated macrocytosis.48 In our study, a higher mean corpuscular volume was strongly linked with short telomeres. This additional marker may be useful for assessing patients with idiopathic PSVD.
In terms of liver function test profiles, we predominantly observed a cholestatic or mixed pattern, corroborated by an R-value < 2 in 68% and < 5 in an additional 22% of patients, as expected in PSVD.28 Furthermore, when stratified for TL, patients with shorter telomeres more often presented predominant cholestasis.
Patients with VSTel and STel had significantly higher rates of clinically significant PH and its associated complications. Laboratory signs of liver dysfunction and dysmorphia on imaging were also significantly more prevalent among patients with VSTel and STel. Altogether, these observations suggest a relationship between shorter telomeres and a more severe PSVD phenotype. Hence, analysis of TL could potentially aid in elucidating the pathogenesis of PSVD, recognizing an underlying STS, and categorizing the risk level of patients with PSVD. Furthermore, we cautiously suggest that shortened telomeres might have prognostic significance in the context of PSVD, potentially foreseeing advancement to PH and its associated complications.
Despite the high rate of STel and VSTel in our patients with PSVD, identifiable variants in telomere-related genes were mostly observed in patients with very short telomeres. In total, 4 VSTel patients harbored one or more variants in a known STS-related gene. Two of them had a likely pathogenic variant in the RTEL1 gene, which was considered causative of their clinical phenotype, whereas variants classified as VUS with an uncertain association with their hepatic disease were identified in the RTEL1 and TERC genes in the remaining two patients. In the STel group, 1 patient had a VUS in DKC1.
The RTEL1 (regulator of telomere elongation 1) gene encodes a DNA helicase that plays a crucial role in telomere maintenance and DNA repair.49 The enrichment of variants in this gene in the present cohort aligns with a previously documented association between RTEL1 impairment and liver disease.50
It is noteworthy to emphasize that this is in line with previous reports showing that a significant proportion of patients with established STS remain without evidence of genetic mutations.11 Importantly, the present genetic study was restricted to the analysis of the coding regions of genes with a known association with STS. Nonetheless, it is highly plausible that some patients in our cohort may harbor variants in as-of-yet-unknown genes implicated in telomere maintenance. A subsequent step for further investigating the genetic background of STS and PSVD would be to broaden the search to a larger panel of genes and include intronic regions. This could involve employing a systematic whole-genome sequencing approach, as recently reported by Shan et al.51 The possibility of polygenic inheritance should also be considered.
In contrast, PSVD itself may cause short telomeres in peripheral leukocytes as a result of an inflammatory state, which is known to play a role in PSVD pathogenesis.28,30 Thus, the presence of shorter TL may be the result of an increased leukocyte turnover.
Our study has some limitations. Firstly, due to the rarity of idiopathic PSVD, the patient cohort is relatively small. Second, this study may underestimate the prevalence of STS in PSVD due to the strict inclusion criteria for patients with idiopathic PSVD. Indeed, cases linked to identifiable causes, such as drugs, procoagulant conditions, or autoimmune diseases, may also have an undetected link with shortened TL. The inclusion of such patients, or even patients with cirrhosis, as a control group may have strengthened the relevance of our results. Indeed, few studies to date have focused on TL in patients with different chronic liver diseases. The lack of patients with other liver diseases as controls has been partially mitigated by measuring TL using a highly accurate and reproducible technique that inherently compares patients with age-related data from a large reference population of healthy individuals.
In conclusion, we report a remarkably high rate of shortened telomeres in patients with idiopathic PSVD, with around one-third harboring telomere-related gene variants. We hypothesize that telomere shortening plays an important role in the pathogenesis and prognosis of PSVD. Hence, clinicians should consider measuring TL in patients presenting with idiopathic PSVD. In instances of STel or VSTel, this could prompt further investigations into other prevalent manifestations of STS, thereby potentially improving patient management and outcomes. Further studies with larger prospective cohorts, including patients whose PSVD diagnosis is linked to an established clinical risk factor, as well as patients with other liver diseases, are needed.
Supplementary Material
Acknowledgments
ACKNOWLEDGMENTS
The authors thank Professor Andrea De Gottardi for his support in the development of this project.
FUNDING INFORMATION
This study was supported by a bursary from the Department of Medicine of Lausanne University Hospital to Alexander Coukos and a “Bourse Pro-Femmes” of the Faculty of Biology and Medicine of the University of Lausanne to Montserrat Fraga.
CONFLICTS OF INTEREST
The authors have no conflicts to report.
Footnotes
Abbreviations: DTL, delta telomere length; ILD, Interstitial lung disease; NRH, nodular regenerative hyperplasia; NTel, normal telomere length; PH, portal hypertension; PSVD, porto-sinusoidal vascular disorder; STS, short telomere syndrome; STel, short telomere length; TL, telomere length; VSTel, very short telomere length; VUS, variant of uncertain significance.
Jean-Marc Good and Gabriela M. Baerlocher share second-to-last authorship.
Supplemental Digital Content is available for this article. Direct URL citations are provided in the HTML and PDF versions of this article on the journal’s website, www.hepcommjournal.com.
Contributor Information
Alexander Coukos, Email: alexcoukos@gmail.com.
Chiara Saglietti, Email: chiara.saglietti@chuv.ch.
Christine Sempoux, Email: christine.sempoux@chuv.ch.
Monika Haubitz, Email: monika.haubitz@unibe.ch.
Thomas Greuter, Email: thomas.greuter@chuv.ch.
Laureane Mittaz-Crettol, Email: laureane.mittaz-crettol@chuv.ch.
Fabienne Maurer, Email: fabienne.maurer@chuv.ch.
Elise Mdawar-Bailly, Email: elise.mdawar-bailly@chuv.ch.
Darius Moradpour, Email: Darius.Moradpour@chuv.ch.
Lorenzo Alberio, Email: lorenzo.alberio@chuv.ch.
Jean-Marc Good, Email: jean-marc.good@chuv.ch.
Gabriela M. Baerlocher, Email: gabriela.baerlocher@hematology.ch.
Montserrat Fraga, Email: montserrat.fraga@chuv.ch.
REFERENCES
- 1.Rossiello F, Jurk D, Passos JF, d’Adda di Fagagna F. Telomere dysfunction in ageing and age-related diseases. Nat Cell Biol. 2022;24:135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armanios M. The role of telomeres in human disease. Annu Rev Genomics Hum Genet. 2022;23:363–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet. 2012;13:693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Armando RG, Mengual Gomez DL, Maggio J, Sanmartin MC, Gomez DE. Telomeropathies: Etiology, diagnosis, treatment and follow-up. Ethical and legal considerations. Clin Genet. 2019;96:3–16. [DOI] [PubMed] [Google Scholar]
- 5.Barbaro PM, Ziegler DS, Reddel RR. The wide-ranging clinical implications of the short telomere syndromes. Intern Med J. 2016;46:393–403. [DOI] [PubMed] [Google Scholar]
- 6.Kam MLW, Nguyen TTT, Ngeow JYY. Telomere biology disorders. NPJ Genom Med. 2021;6:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alter BP, Baerlocher GM, Savage SA, Chanock SJ, Weksler BB, Willner JP, et al. Very short telomere length by flow fluorescence in situ hybridization identifies patients with dyskeratosis congenita. Blood. 2007;110:1439–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Di Micco R, Krizhanovsky V, Baker D, d’Adda di Fagagna F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. 2021;22:75–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calado RT, Young NS. Telomere diseases. N Engl J Med. 2009;361:2353–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Penrice DD, Simonetto DA. Short telomeres: Cause and consequence in liver disease. Semin Liver Dis. 2020;40:385–391. [DOI] [PubMed] [Google Scholar]
- 11.Grill S, Nandakumar J. Molecular mechanisms of telomere biology disorders. J Biol Chem. 2021;296:100064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Savage SA. Dyskeratosis congenita and telomere biology disorders. Hematology Am Soc Hematol Educ Program. 2022;2022:637–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schratz KE. Extrahematopoietic manifestations of the short telomere syndromes. Hematology Am Soc Hematol Educ Program. 2020;2020:115–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alter BP, Rosenberg PS, Giri N, Baerlocher GM, Lansdorp PM, Savage SA. Telomere length is associated with disease severity and declines with age in dyskeratosis congenita. Haematologica. 2012;97:353–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Demanelis K, Jasmine F, Chen LS, Chernoff M, Tong L, Delgado D, et al. Determinants of telomere length across human tissues. Science. 2020;369:eaaz6876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aubert G, Hills M, Lansdorp PM. Telomere length measurement-caveats and a critical assessment of the available technologies and tools. Mutat Res. 2012;730:59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baerlocher GM, Vulto I, de Jong G, Lansdorp PM. Flow cytometry and FISH to measure the average length of telomeres (flow FISH). Nat Protoc. 2006;1:2365–2376. [DOI] [PubMed] [Google Scholar]
- 18.Aubert G, Baerlocher GM, Vulto I, Poon SS, Lansdorp PM. Collapse of telomere homeostasis in hematopoietic cells caused by heterozygous mutations in telomerase genes. PLoS Genet. 2012;8:e1002696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alder JK, Armanios M. Telomere-mediated lung disease. Physiol Rev. 2022;102:1703–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schneider CV, Schneider KM, Teumer A, Rudolph KL, Hartmann D, Rader DJ, et al. Association of telomere length with risk of disease and mortality. JAMA Intern Med. 2022;182:291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feurstein S, Adegunsoye A, Mojsilovic D, Vij R, West DePersia AH, Rajagopal PS, et al. Telomere biology disorder prevalence and phenotypes in adults with familial hematologic and/or pulmonary presentations. Blood Adv. 2020;4:4873–4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamaguchi H, Baerlocher GM, Lansdorp PM, Chanock SJ, Nunez O, Sloand E, et al. Mutations of the human telomerase RNA gene (TERC) in aplastic anemia and myelodysplastic syndrome. Blood. 2003;102:916–918. [DOI] [PubMed] [Google Scholar]
- 23.Ly H, Calado RT, Allard P, Baerlocher GM, Lansdorp PM, Young NS, et al. Functional characterization of telomerase RNA variants found in patients with hematologic disorders. Blood. 2005;105:2332–2339. [DOI] [PubMed] [Google Scholar]
- 24.Mangaonkar AA, Patnaik MM. Short telomere syndromes in clinical practice: bridging bench and bedside. Mayo Clin Proc. 2018;93:904–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kapuria D, Ben-Yakov G, Ortolano R, Cho MH, Klachiem-Dekel O, Takyar V, et al. The spectrum of hepatic involvement in patients with telomere disease. Hepatology. 2019;69:2579–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sidali S, Borie R, Sicre de Fontbrune F, El Husseini K, Rautou PE, Lainey E, et al. Liver disease in germline mutations of telomere-related genes: Prevalence, clinical, radiological, pathological features, outcome, and risk factors. Hepatology. 2024;79:1365–1380. [DOI] [PubMed] [Google Scholar]
- 27.Gioia S, Nardelli S, Ridola L, Riggio O. Causes and management of non-cirrhotic portal hypertension. Curr Gastroenterol Rep. 2020;22:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Gottardi A, Sempoux C, Berzigotti A. Porto-sinusoidal vascular disorder. J Hepatol. 2022;77:1124–35. [DOI] [PubMed] [Google Scholar]
- 29.de Franchis R, Bosch J, Garcia-Tsao G, Reiberger T, Ripoll C, Baveno VII Faculty . Baveno VII - Renewing consensus in portal hypertension. J Hepatol. 2022;76:959–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Gottardi A, Rautou PE, Schouten J, Rubbia-Brandt L, Leebeek F, Trebicka J, et al. Porto-sinusoidal vascular disease: Proposal and description of a novel entity. Lancet Gastroenterol Hepatol. 2019;4:399–411. [DOI] [PubMed] [Google Scholar]
- 31.Robbins J, Halegoua-DeMarzio D, Basu Mallick A, Vijayvergia N, Ganetzky R, Lavu H, et al. Liver Transplantation in a woman with Mahvash disease. N Engl J Med. 2023;389:1972–1978. [DOI] [PubMed] [Google Scholar]
- 32.von Elm E, Altman DG, Egger M, Pocock SJ, Gøtzsche PC, Vandenbroucke JP, STROBE Initiative . The strengthening the reporting of observational studies in epidemiology (STROBE) statement: Guidelines for reporting observational studies. Lancet. 2007;370:1453–1457. [DOI] [PubMed] [Google Scholar]
- 33.Ghabril M, Vuppalanchi R. Drug-induced nodular regenerative hyperplasia. Semin Liver Dis. 2014;34:240–245. [DOI] [PubMed] [Google Scholar]
- 34.Baerlocher GM, Mak J, Tien T, Lansdorp PM. Telomere length measurement by fluorescence in situ hybridization and flow cytometry: Tips and pitfalls. Cytometry. 2002;47:89–99. [DOI] [PubMed] [Google Scholar]
- 35.Baerlocher GM, Lansdorp PM. Telomere length measurements in leukocyte subsets by automated multicolor flow-FISH. Cytometry A. 2003;55:1–6. [DOI] [PubMed] [Google Scholar]
- 36.Royer-Bertrand B, Cisarova K, Niel-Butschi F, Mittaz-Crettol L, Fodstad H, Superti-Furga A. CNV detection from exome sequencing data in routine diagnostics of rare genetic disorders: Opportunities and limitations. Genes (Basel). 2021;12:1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chalasani NP, Maddur H, Russo MW, Wong RJ, Reddy KR. ACG Clinical Guideline: Diagnosis and management of idiosyncratic drug-Induced liver injury. Am J Gastroenterol. 2021;116:878–98. [DOI] [PubMed] [Google Scholar]
- 38.Moschouri E, Vionnet J, Giostra E, Daccord C, Lazor R, Sciarra A, et al. Combined lung and liver transplantation for short telomere syndrome. Liver Transpl. 2020;26:840–844. [DOI] [PubMed] [Google Scholar]
- 39.Richards S Aziz N Bale S Bick D Das S Gastier-Foster J et al.ACMG Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coukos A, Daccord C, Lazor R, Blum S, Naveiras O, Unger S, et al. [Short telomere syndrome in adults: A rare entity that should be evoked] (article in French). Rev Med Suisse. 2022;8:1606–1613. [DOI] [PubMed] [Google Scholar]
- 41.Hoare M, Das T, Alexander G. Ageing, telomeres, senescence, and liver injury. J Hepatol. 2010;53:950–961. [DOI] [PubMed] [Google Scholar]
- 42.Butt HZ, Atturu G, London NJ, Sayers RD, Bown MJ. Telomere length dynamics in vascular disease: A review. Eur J Vasc Endovasc Surg. 2010;40:17–26. [DOI] [PubMed] [Google Scholar]
- 43.Lin J, Kroenke CH, Epel E, Kenna HA, Wolkowitz OM, Blackburn E, et al. Greater endogenous estrogen exposure is associated with longer telomeres in postmenopausal women at risk for cognitive decline. Brain Res. 2011;1379:224–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bayne S, Li H, Jones ME, Pinto AR, van Sinderen M, Drummond A, et al. Estrogen deficiency reversibly induces telomere shortening in mouse granulosa cells and ovarian aging in vivo. Protein Cell. 2011;2:333–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yeap BB, Knuiman MW, Divitini ML, Hui J, Arscott GM, Handelsman DJ, et al. Epidemiological and Mendelian randomization studies of dihydrotestosterone and estradiol and leukocyte telomere length in men. J Clin Endocrinol Metab. 2016;101:1299–1306. [DOI] [PubMed] [Google Scholar]
- 46.Park S, Lee S, Kim Y, Cho S, Kim K, Kim YC, et al. A Mendelian randomization study found causal linkage between telomere attrition and chronic kidney disease. Kidney Int. 2021;100:1063–70. [DOI] [PubMed] [Google Scholar]
- 47.Lata S, Marasa M, Li Y, Fasel DA, Groopman E, Jobanputra V, et al. Whole-exome sequencing in adults with chronic kidney disease: A pilot study. Ann Intern Med. 2018;168:100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fogarty PF, Yamaguchi H, Wiestner A. Late presentation of dyskeratosis congenita as apparently acquired aplastic anaemia due to mutations in telomerase RNA. Lancet. 2003;362:1628–1630. [DOI] [PubMed] [Google Scholar]
- 49.Vannier JB, Sandhu S, Petalcorin MI, Petalcorin MIR, Wu X, Nabi Z, et al. RTEL1 is a replisome-associated helicase that promotes telomere and genome-wide replication. Science. 2013;342:239–242. [DOI] [PubMed] [Google Scholar]
- 50.Chiu V, Hogen R, Sher L, Wadé N, Conti D, Martynova A, et al. Telomerase variants in patients with cirrhosis awaiting liver transplantation. Hepatology. 2019;69:2652–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shan J, Megarbane A, Chouchane A, Karthik D, Temanni R, Romero AR, et al. Genetic predisposition to porto-sinusoidal vascular disorder: A functional genomic-based, multigenerational family study. Hepatology. 2023;77:501–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.