

Abstract
Background and Aims:
Transforming growth factor-beta 1 (TGFβ1) induces HSC activation into metastasis-promoting cancer-associated fibroblasts (CAFs), but how the process is fueled remains incompletely understood. We studied metabolic reprogramming induced by TGFβ1 in HSCs.
Approaches and Results:
Activation of cultured primary human HSCs was assessed by the expression of myofibroblast markers. Glucose transporter 1 (Glut1) of murine HSC was disrupted by Cre recombinase/LoxP sequence derived from bacteriophage P1 recombination (Cre/LoxP). Plasma membrane (PM) Glut1 and glycolysis were studied by biotinylation assay and the Angilent Seahorse XFe96 Analyzer. S.c. HSC/tumor co-implantation and portal vein injection of MC38 colorectal cancer cells into HSC-specific Glut1 knockout mice were performed to determine in vivo relevance. Transcriptome was obtained by RNA sequencing of HSCs and spatialomics with MC38 liver metastases. TGFβ1-induced CAF activation of HSCs was accompanied by elevation of PM Glut1, glucose uptake, and glycolysis. Targeting Glut1 or Src by short hairpin RNA, pharmacologic inhibition, or a Src SH3 domain deletion mutant abrogated TGFβ1-stimulated PM accumulation of Glut1, glycolysis, and CAF activation. Mechanistically, binding of the Src SH3 domain to SH3 domain–binding protein 5 led to a Src/SH3 domain–binding protein 5/Rab11/Glut1 complex that activated Rab11-dependent Glut1 PM transport under TGFβ1 stimulation. Deleting the Src SH3 domain or targeting Glut1 of HSCs by short hairpin RNA or Cre recombinase/LoxP sequence derived from bacteriophage P1 recombination suppressed CAF activation in mice and MC38 colorectal liver metastasis. Multi-omics revealed that Glut1 deficiency in HSCs/CAFs suppressed HSC expression of tumor-promoting factors and altered MC38 transcriptome, contributing to reduced MC38 liver metastases.
Conclusion:
The Src SH3 domain–facilitated metabolic reprogramming induced by TGFβ1 represents a target to inhibit CAF activation and the prometastatic liver microenvironment.
INTRODUCTION
HSCs are the precursor of cancer-associated fibroblasts (CAFs) of liver cancer,[1–4] and this notion of HSC-to-CAF conversion was recently confirmed by genetic tracing, single-cell RNA sequencing, and ligand-receptor analyses.[3,5] Indeed, multiple lines of research revealed that in experimental liver metastasis mouse models, selective depleting HSCs or targeting CAF activation of HSCs by genetic approaches influenced desmoplastic liver metastatic growth and mortality of the mice.[2,5–7] Transforming growth factor-beta 1 (TGFβ1) is one of the most potent factors for CAF activation by activating intracellular signaling of HSCs, including ligation of TGFβ receptors at the plasma membrane (PM), endocytosis of TGFβ receptors, phosphorylation of mothers against decapentaplegic homolog 3 (SMAD) 2 and 3, nuclear translocation of SMAD complex, and gene transcription in the nucleus.[8,9] As a result, HSCs develop stress fibers positive for alpha-smooth muscle actin (αSMA) and express an increased amount of extracellular matrix proteins, characteristics of CAF activation.[7,10,11] CAF activation of HSCs therefore represents a target to inhibit the hepatic tumor microenvironment and colorectal liver metastasis.
The CAF activation process is an energy-dependent process. ATPs and other biomaterials required for CAF activation of HSCs are mainly produced by the breakdown of glucose, fatty acids, and amino acids by processes, such as glycolysis, the tricarboxylic acid cycle and oxidative phosphorylation, beta-oxidation, and amino acid catabolism.[12] Despite the fact that mitochondrial metabolism is the most efficient pathway for ATP generation, proliferating cells such as cancer cells and CAFs prefer the glycolytic pathways with lactate produced, termed “the Warburg effect” (Supplemental Figure 1A, http://links.lww.com/HEP/I231).[13] Although targeting glycolysis or glutaminolysis in mice suppressed liver fibrosis,[14–16] how CAF activation of HSCs is fueled in the hepatic tumor microenvironment remains poorly investigated.
Glucose transporters are multi-pass proteins at the PM that facilitate glucose transport into the cells. Glucose transporter 1 (Glut1), encoded by the SLC2A1 gene, is the predominant glucose transporter expressed by hepatic fibroblasts.[17] Similar to other PM proteins, Glut1 undergoes endocytosis and endosome-to-PM transport so that its PM level is tightly controlled by glucose and intracellular signaling. Upon endocytosis and in the early endosomes, PM proteins are either sorted to the lysosomes for degradation or Rab11-positive recycling endosomes for reuse. Although Glut1 gene expression is known to be transcriptionally upregulated by transforming growth factor-beta (TGFβ)1,[16,18] it is unknown whether TGFβ stimulation influences intracellular trafficking of Glut1.
Using various approaches, we found that TGFβ1 stimulation leads to PM accumulation of Glut1 for enhanced glucose uptake and glycolysis of HSCs and that Glut1 PM accumulation is facilitated by a protooncogene tyrosine-protein kinase Src (Src)/SH3 domain–binding protein 5 (SH3BP5)/Rab11/Glut1 protein complex induced by TGFβ1. Additionally, the Src homology 3 domain (SH3 domain) of Src is required for the complex formation, Rab11 activation, and PM Glut1 accumulation. Since HSCs are activated into CAFs under colorectal cancer (CRC) invasion of the liver,[2,5] functional studies were performed in mice, which revealed that deleting the Src SH3 domain or targeting HSC Glut1 by short hairpin RNA (shRNA) or Cre recombinase/LoxP sequence derived from bacteriophage P1 (Cre/LoxP) combination suppressed CAF activation of HSCs and CRC liver metastasis. Multi-omics revealed that targeting Glut1 altered HSC transcriptome and suppressed HSC expression of tumor-promoting factors. Moreover, spatial transcriptomics detected global transcriptomic changes in CRC cancer cells adjacent to Glut1-deficient CAFs. Together, our data uncover molecular and mechanistic insights into HSC metabolic reprogramming that impact the transcriptomes, CAF activation, and the pro-metastatic liver microenvironment.
METHODS
Cell culture
Primary human HSCs (#5300 ScienCell Research laboratories Carlsbad, CA) were used up to 9 passages. DMEM-low glucose medium (D6046 MilliporeSigma) with glucose adjusted to 1.5 g/L and L-glutamine to 1 mM was used for glycolysis-related studies. HT29 and HCT116 human colorectal cancer cells were purchased from ATCC (HTB-38™, Manassas, VA, USA) and authenticated by short tandem repeat DNA profiling by Genetica (Cincinnati, OH). MC38 mouse colorectal cancer cells were from Dr. Steven A. Rosenberg (National Cancer Institute).[2,6,7] Human liver cancer CAFs (HC-6019 Cell Biologics) and metastatic colorectal cancer CAFs (CAF115 Neuromics) were obtained commercially. Cells were frequently tested for mycoplasma infection and free of infection during the experiments.
Site-directed mutagenesis and viral transduction of HSCs
The standard PCR-based subcloning technique and Gibson Assembly (#E5510 New England Biolabs) were used to generate retroviral constructs expressing Rab11, Glut1, or wild-type Src. Q5 Site-Directed Mutagenesis (#E0554 New England Biolabs Ipswich, MA) was used to generate a Src SH3 domain deletion mutant. ShRNA lentiviral constructs were obtained from the MISSION shRNA library (MilliporeSigma). Packaging retroviral or lentiviral particles was done as described.[2,19,20] Details are in Supplemental Materials and Methods, http://links.lww.com/HEP/I230.
Plasma membrane (PM) Glut1, glucose uptake assay, and Agilent Seahorse XF96 Glycolysis Stress Test
Biotinylation followed by streptavidin agarose pulldown was performed to quantitate PM Glut1 of HSCs.[2,11,21] Glucose uptake was assessed by incubating HSCs with a fluorescent glucose analog (2-deoxy-2-[(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]-D-glucose [2-NBDG]) followed by flow cytometry with a BD LSRFortessa X-20 Cell Analyzer. ATP production and glycolysis were studied by the Agilent Seahorse XF96 analyzer. Details are in Supplemental Materials and Methods, http://links.lww.com/HEP/I230.
TGFβ transcriptome and spatial transcriptomics
TGFβ transcriptome was obtained by RNA sequencing and analyzed by the EdgeR package with the human genome (hg19) as the reference.[4,6,21] Reads were subjected to Gene Set Enrichment Analysis (GSEA) so differentially expressed genes (DEGs) and pathways were identified.[4,22] Heatmaps were generated by an online software from MD Anderson Cancer Center (https://build.ngchm.net/NGCHM-web-builder/View_HeatMap.html?adv=N).[4,6] The transcriptome of CAFs and MC38 cells on tumor sections was studied by NanoString GeoMx Digital spatial profiling (GeoMx DSP) with the GeoMx Mouse Whole Transcriptome Atlas Panel (20176 target probes). Experimental details and data analyses are in Supplemental Materials and Methods, http://links.lww.com/HEP/I230.
Study approval and portal vein tumor injection in mice
Human colorectal liver metastases from a Mayo Clinic Tissue collection[2,6] were subjected to immunofluorescence (IF) for αSMA, Glut1, Src, and Rab11, which was approved by the Institutional Review Board at the Mayo Clinic (IRB 10–005662). Tumor implantation protocols were approved by the Institutional Animal Care and Use Committee of The University of Minnesota. HT29/HSC co-implantation studies were done through subcutaneous injection of the cells into immunosuppressed nude mice. For portal vein tumor injection, Glut1 floxed mutant mouse line (Slc2a1tm1.1Stma/AbelJ, 031871) and Cre transgenic line (platelet-derived growth factor receptor beta (PDGFRβ)-P2A-CreERT2, 030201) from the Jackson Laboratory were bred for offspring. Ten days before portal vein tumor injection, mice received tamoxifen (T5648 MilliporeSigma) through i.p. injection once daily for consecutive 5 days (75 mg/kg of body weight, dissolved in corn oil) for Cre activation. Under general anesthesia by isoflurane (2%–5%), each mouse received 1 × 106 MC38 cells thrugh portal vein injection.[2,4,6] Mice received postoperative care for 3 days, and they were killed 11 days later. Details are in Supplemental Materials and Methods, http://links.lww.com/HEP/I230.
Statistical analysis
Data were expressed as mean ± SEM and subjected to two-tailed Student t-test or ANOVA using the GraphPad Prism 6 software (GraphPad Software, Inc., La Jolla, CA). p < 0.05 was considered statistically different. In WB for in vitro experiments, n is independent repeats; in IF data, n is either cell number or image number chosen for analysis; in animal studies, n is the number of animals for each group.
Additional reagents and methods are in Supplemental Materials and Methods, http://links.lww.com/HEP/I230.
RESULTS
TGFβ-induced myofibroblastic activation of HSCs is promoted by glucose
To determine the role of glucose in TGFβ1-stimulated HSC activation, HSCs in DMEM supplemented with different glucose concentrations (1 g/L and 4 g/L) were stimulated with TGFβ1 (5 ng/mL) for 24 hours. Western blot (WB) analysis for stellate cell activation markers, αSMA, fibronectin (FN), and connective tissue growth factor (CTGF) revealed that TGFβ1 elevated αSMA, FN, and CTGF expressions by HSCs cultured in 1 g/L glucose and that this effect of TGFβ1 was potentiated by 4 g/L of glucose (Figure 1A left, p < 0.05). Immunofluorescence (IF) for αSMA confirmed that the rate of activated HSC/myofibroblasts induced by TGFβ1 was 2 times higher for cells in 4 g/L glucose compared to cells in 1 g/L glucose (Figure 1A right, p < 0.01). Glucose uptake was assessed by fluorescent glucose analog 2-NBDG followed by flow cytometry for 2-NBDG, showing that TGFβ1 stimulation promoted glucose uptake by HSCs (Figure 1B left, p < 0.05). Agilent Seahorse-based assay revealed that ATP production by HSCs was increased by TGFβ1 as well (Figure 1B right, p < 0.05). Thus, TGFβ1-stimulated HSC activation is dependent on exogenous glucose.
FIGURE 1.
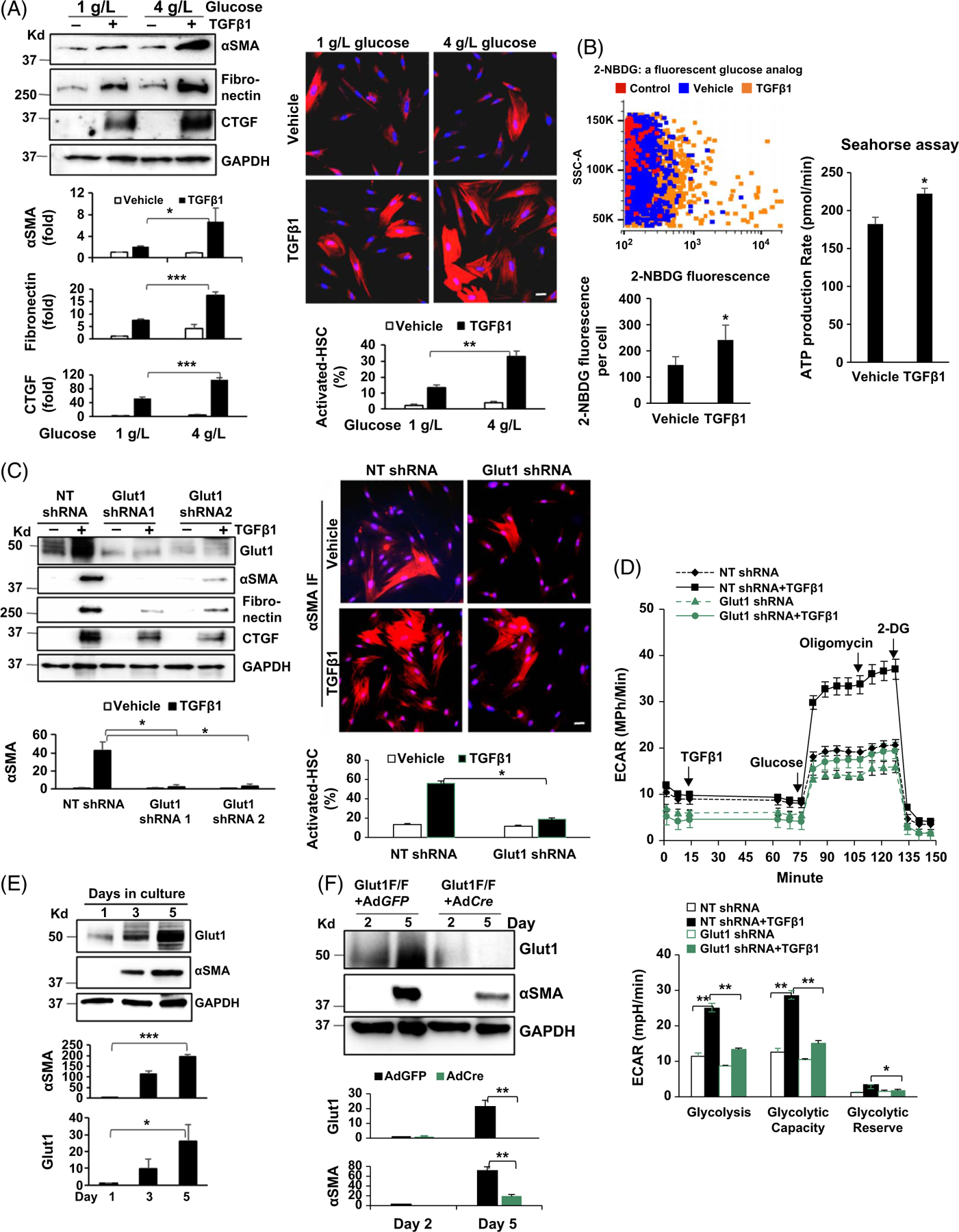
Myofibroblastic activation of HSCs is promoted by Glut1. (A) Left, primary human HSCs in different glucose concentrations were stimulated with TGFβ1 (5 ng/mL) for 24 hours and collected for WB analysis. TGFβ1 upregulation of the stellate cell activation markers was potentiated by 4 g/L glucose compared to 1 g/L glucose. *p < 0.05 ***p < 0.001 by ANOVA, n = 3 repeats. Right, HSCs as described above were collected for αSMA immunofluorescence (IF). The rate of activated HSCs/myofibroblasts was increased by 4 g/L glucose. **p < 0.01 by ANOVA, n = 12 microscopic fields per group, each containing more than 100 cells. Bar, 20 μm. (B) Left, HSCs were incubated with 2-NBDG and 2-NBDG fluorescence within the cells was quantified by flow cytometry. TGFβ1 (5 ng/mL) promoted glucose uptake by HSCs. *p < 0.05 by t-test, n > 3,000 cells per group. Right, Agilent Seahorse XF96 ATP Rate Assay was performed. ATP production was increased by TGFβ1 stimulation (5 ng/mL). *p < 0.05 by t-test, n = 5. (C) HSCs expressing NT shRNA, Glut1 shRNA1, or Glut1 shRNA2 by lentiviral transduction were stimulated with TGFβ1 and collected for WB and αSMA IF. Glut1 knockdown suppressed myofibroblastic activation of HSCs induced by TGFβ1. *p < 0.05 by ANOVA, n = 3 repeats for WB; n = 12 microscopic fields per group for IF, each containing more than 100 cells. Bar, 20 μm. (D) Real-time ECAR was obtained by the Agilent Seahorse Glycolysis Stress test. The times when TGFβ1, glucose, oligomycin, and 2-DG were added are shown. TGFβ1 (5 ng/mL) promoted glycolysis in control HSCs, but not in Glut1 knockdown HSCs. *p < 0.05 **p < 0.01 by ANOVA, n = 5. (E) Murine HSCs were collected for WB. Glut1 expression by HSCs increased time-dependently similar to αSMA expression. *p < 0.05; ***p < 0.001 by ANOVA, n = 3 repeats. (F) HSCs isolated from Slc2A1/Glut1 floxed mutant mice were transduced with GFP adenoviruses (control) or Cre adenoviruses. Cre/LoxP-mediated Glut1 gene deletion suppressed murine HSC activation. **p < 0.01 by ANOVA, n = 3 repeats. Abbreviations: αSMA, alpha-smooth muscle actin; Cre/LoxP, Cre recombinase/LoxP sequence derived from bacteriophage P1; 2-DG, 2-deoxy-D-glucose; ECAR, extracellular acidification rate; Glut1, glucose transporter 1; IF, immunofluorescence; NBDG, 2-deoxy-2-[(7-nitro-2,1,3-benzoxadiazol-4-yl) amino]-D-glucose; NT shRNA, nontargeting short hairpin RNA; TGFβ1, transforming growth factor-beta 1; WB, western blot.
Knockdown of glucose transporter 1 (Glut1) suppresses TGFβ1-stimulated glycolysis and HSC activation
Mammalian cells have 14 different glucose transporters; we therefore performed RNA sequencing to quantitate their transcripts in HSCs, which revealed that Glut1 transcript is the most abundant one (Supplemental Figure 1B, http://links.lww.com/HEP/I231). Glut1 was next knocked down by lentiviruses encoding Glut1 shRNA1 or Glut1 shRNA2, and cells transduced by nontargeting shRNA lentiviruses were used as the control. The knockdown effect was 72% ± 2% and 85% ± 5% for 2 shRNAs respectively (Supplemental Figure 1C left, http://links.lww.com/HEP/I231, p < 0.001). As confirmed by the 2-NBDG-based assay, glucose uptake by HSCs was significantly reduced by Glut1 knockdown (Supplemental Figure 1C right, http://links.lww.com/HEP/I231, p < 0.0001). WB showed that TGFβ1 (5 ng/mL) promoted expression of αSMA, FN, and CTGF in control cells, and this effect of TGFβ1 was suppressed by Glut1 knockdown (Figure 1C left, p < 0.05). αSMA IF confirmed that more than 50% of control HSCs were activated into myofibroblasts by TGFβ1 in contrast to less than 20% of Glut1 knockdown HSCs activated under a same condition (Figure 1C right, p < 0.05). Glut1 inhibitor BAY876 (20 nM) also inhibited HSC activation mediated by TGFβ1 (Supplemental Figure 1D, http://links.lww.com/HEP/I231, p < 0.01).
Lactate, a product of glycolysis, influences extracellular acidification rate (ECAR) once released from the cells (Supplemental Figure 1A, http://links.lww.com/HEP/I231). The changes of ECAR are therefore indicative of glycolytic changes inside of the cells, which is the basis of the Agilent Seahorse XF96 Glycolysis Stress Test. As revealed by real-time ECAR data, TGFβ1 (5 ng/mL) promoted ECAR of control HSCs (Figure 1D, black lines), and this effect of TGFβ1 was abrogated by Glut1 knockdown (Figure 1D, blue lines). The rates of glycolysis, glycolysis capacity, or glycolysis reserve, calculated on ECAR curves, supported that Glut1 knockdown inhibited glycolysis induced by TGFβ1 (Figure 1D lower, p < 0.05). Together, these data support that TGFβ1-stimulated glycolysis and CAF activation of HSCs in vitro is dependent on Glut1 of HSCs.
Targeting Glut1 by Cre/LoxP recombination suppresses myofibroblastic activation of murine HSC
Primary murine HSCs were isolated from the liver of mice, and their time-dependent spontaneous activation was revealed by WB for αSMA expression (Figure 1E, p < 0.001 and Supplemental Figure 1E, http://links.lww.com/HEP/I231). Similar to αSMA expression, Glut1 expression by murine HSCs increased time-dependently from day 1 to day 5 (Figure 1E, p < 0.05). To analyze the role of Glut1 in the activation of murine HSCs, HSCs isolated from Slc2a1/Glut1 floxed mutant mice were transduced by GFP adenoviruses (AdGFP; control) or Cre adenoviruses (AdCre) to knockout Slc2a1/Glut1. As revealed by WB, Glut1 was deleted by AdCre, and activation of murine HSCs was suppressed accordingly (Figure 1F, p < 0.01). Thus, targeting the Slc2a1/Glut1 gene by Cre/LoxP recombination suppresses murine HSC activation in vitro.
TGFβ1 induces endosome-to-PM translocation of Glut1 by Src
We next focused on Glut1 subcellular localization, as only PM Glut1 is able to transport glucose. IF and confocal microscopy demonstrated that in the absence of TGFβ1 stimulation, Glut1 (green) mainly localized at the perinuclear regions, and it translocated onto the peripheral PM under TGFβ1 stimulation (Figure 2A upper, arrows). Biotinylation of cell surface proteins confirmed that short-term TGFβ1 stimulation (1, 2, or 6 h) led to Glut1 accumulation at the PM of HSCs time-dependently (Figure 2A lower, p < 0.05). When we collected HSCs overexpressing Src-human influenza hemagglutinin (HA) fusion protein for IF, we detected Src-HA at the endosomes, which is consistent with the data reported.[23] Double IF revealed that Glut1 (green) and Src-HA (red) colocalized at the endosomes of HSCs (yellow) (Figure 2B left, arrowheads) and that 2 hours of TGFβ1 stimulation led to PM colocalization of Src and Glut1 in HSCs (Figure 2B right, arrows). TGFβ1-induced PM Glut1 was confirmed by biotinylation assay, which was blocked by Src shRNA (Figure 2C, p < 0.05). Dasatinib (200 nM), targeting tyrosine kinase activity of Src family proteins, also blocked PM targeting of Glut1 induced by TGFβ1 (Supplemental Figure 2A, http://links.lww.com/HEP/I232, p < 0.05).
FIGURE 2.
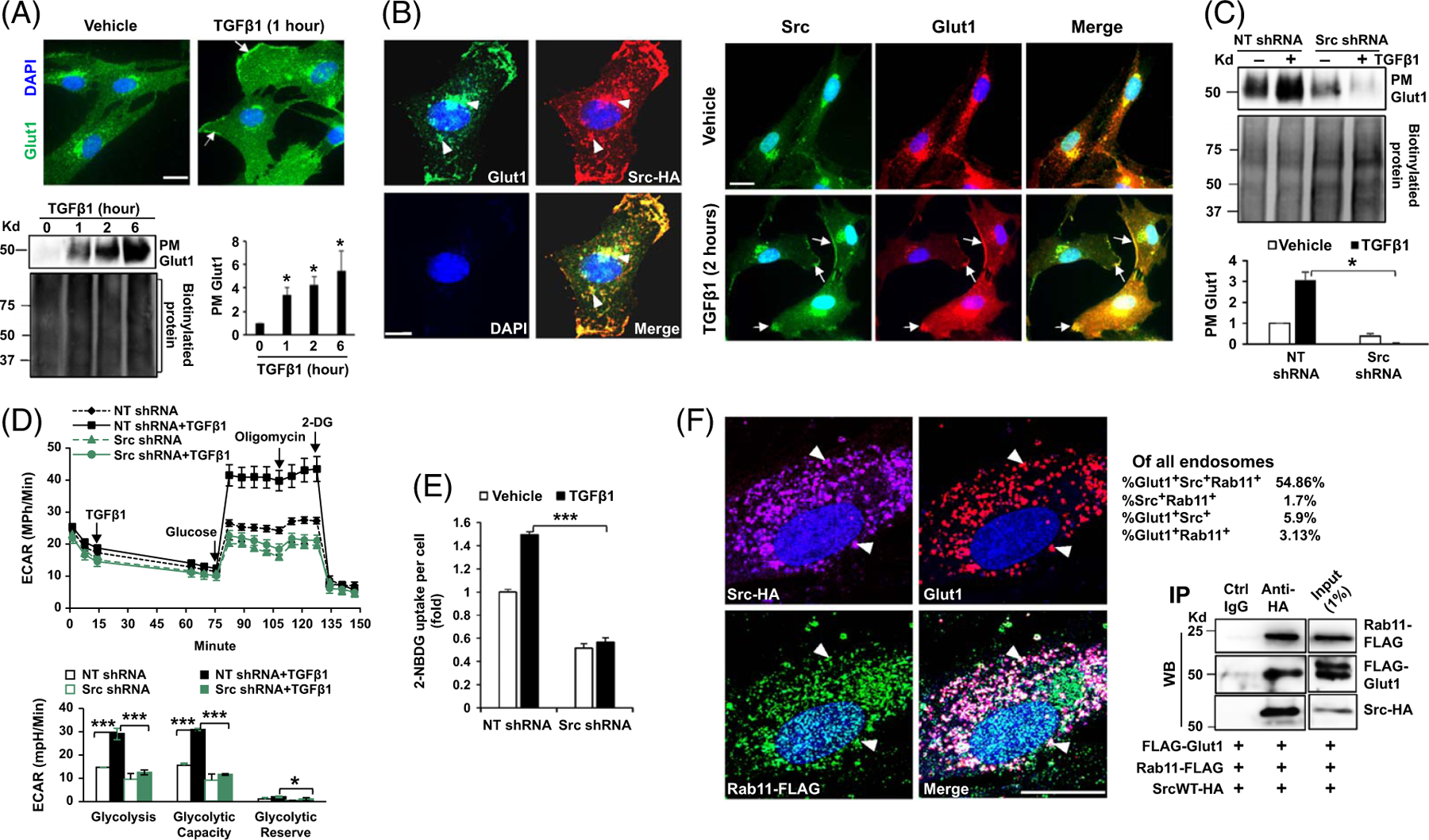
TGFβ1 promotes accumulation of Glut1 at the PM and glycolysis by Src. (A) Upper, HSCs stimulated with TGFβ1 for one hour were collected for IF for Glut1 (green) and cell nuclei were stained by DAPI (blue). TGFβ1 induced endosome-to-PM targeting of Glut1 in HSCs (arrows). Bar, 20 μm. Lower, biotinylation of cell surface proteins revealed that PM Glut1 increased by TGFβ1 time-dependently. *p < 0.05 by ANOVA, n = 3 repeats. (B) Left, double IF was performed with Src-HA–expressing HSCs. Glut1 (green) and Src-HA (red) colocalized at the endosomes of HSCs (yellow, arrowheads). Right, double IF revealed that TGFβ1 induced Src/Glut1 colocalization at the PM of HSC (arrows). Bar, 20 μm. (C) Biotinylation assay showed that TGFβ1 promoted PM Glut1 in control HSCs, but not in Src knockdown cells. *p < 0.05 by ANOVA, n = 3 repeats. (D) Real-time ECAR data revealed that TGFβ1 promoted glycolysis in control HSCs but not in Src knockdown HSCs. *p < 0.05 ***p < 0.001 by ANOVA, n = 5. (E) knockdown of Src reduced glucose uptake by HSCs. ***p < 0.001 by ANOVA, n = 20,000. (F) Left, triple IF and confocal microscopy demonstrated Src/Rab11/Glut1 colocalization at the endosomes of HSCs (arrowheads). The percentages of Src+Rab11+, Src+Glut1+, Glut1+Rab11+, and Src+Rab11+Glut1+ endosomes are shown. Bar, 20 μm. Right, coIP detected a Src/Rab11/Glut1 protein complex in HSCs. Data represent multiple repeats with similar results. Abbreviations: CoIP, co-immunoprecipitation; Glut1, glucose transporter 1; HA, human influenza hemagglutinin; IF, immunofluorescence; PM, plasma membrane; TGFβ1, transforming growth factor-beta 1.
Real-time ECAR and ATP production rate data confirmed that TGFβ1 enhanced glycolysis in control HSCs, but not in Src knockdown HSCs (Figure 2D and Supplemental Figure 2B, http://links.lww.com/HEP/I232, p < 0.05). 2-NBDG-based assay showed that TGFβ1-stimulated glucose uptake was inhibited by Src knockdown (Figure 2E, p < 0.001). As detected by RNA sequencing and WB, long-term TGFβ1 stimulation (24 h) elevated Src mRNA and protein levels of HSCs (Supplemental Figure 2C, http://links.lww.com/HEP/I232, p < 0.05). Consistent with that Glut1 phosphorylation at Serine 226 (pGlut1S226) is associated with Glut1 PM localization,[24] TGFβ1-promoted pGlut1S226 was abolished by Src shRNA or Dasatinib (Supplemental Figure 2D, http://links.lww.com/HEP/I232, p < 0.01). Moreover, WB revealed that TGFβ1-mediated upregulation of αSMA, FN, and CTGF in HSCs was abrogated by Src knockdown (Supplemental Figure 2E, http://links.lww.com/HEP/I232, p < 0.05).
To explore whether TGFβ1-induced Glut1 PM targeting requires SMAD2, we knocked down SMAD2 of HSCs by SMAD2 shRNA lentiviruses and found that TGFβ1 promoted PM Glut1 in control cells, but not in SMAD2 knockdown HSCs (Supplemental Figure 3A, http://links.lww.com/HEP/I233, p < 0.001). As revealed by the Agilent Seahorse glycolysis stress test, TGFβ1-stimulated glycolysis was suppressed by SMAD2 knockdown as well (Supplemental Figure 3B, http://links.lww.com/HEP/I233, p < 0.05). Moreover, TGFβ1-induced myofibroblastic activation of HSCs was suppressed by SMAD2 knockdown (Supplemental Figure 3C, http://links.lww.com/HEP/I233, p < 0.001). Thus, SMAD2 is required for Glut1 PM targeting and HSC activation induced by TGFβ1.
Glut1 forms a complex with Src and Rab11 at the recycling endosome
The small GTPase Rab11 is a master regulator for endosome-to-PM transport of proteins.[25] We therefore collected HSCs overexpressing Src-HA and Rab11-FLAG fusion proteins for triple IF for Src-HA (purple), Glut1 (red), and Rab11-FLAG (green), which revealed their colocalization in 54.86% of the endosomes of HSCs (white) (Figure 2F left, arrowheads). Next, we performed coimmunoprecipitation with HSCs coexpressing Src-HA, Rab11-FLAG, and FLAG-Glut1. Rab11, Glut1, and Src were coimmunoprecipitated, confirming a Glut1/Src/Rab11 protein complex in HSCs (Figure 2F right).
CRC invasion of the liver induces CAF activation of HSCs. To understand whether the Glut1/Src/Rab11 protein complex and metabolism of HSCs can be influenced by CRC cells, we collected conditioned medium (CM) from HT29 and HCT116 human CRC cancer cells; HSCs incubated with either CM overnight were subjected to WB. Both Glut1 and Rab11 protein levels of HSCs were elevated by CRC CM (Supplemental Figure 4A, http://links.lww.com/HEP/I234, p < 0.05), and glycolysis of HSCs was markedly promoted by either CM as well (Supplemental Figure 4B, http://links.lww.com/HEP/I234, p < 0.001), suggesting that CRC cells may release TGFβ1-like soluble factors to influence the Glut1/Src/Rab11 complex and glycolysis of HSCs. Moreover, we found that similar to HSCs, patient liver tumor CAFs were sensitive to Glut1 inhibition as BAY876 drastically suppressed glycolysis and their expression of αSMA and fibronectin (Supplemental Figures 4C and 4D, http://links.lww.com/HEP/I234, p < 0.05).
Deleting the SH3 domain of Src suppresses Rab11 activation, PM Glut1, glycolysis, and HSC activation induced by TGFβ1
SH3 domain–binding protein 5 (SH3BP5) is a guanine nucleotide exchange factor specific for the conversion of Rab11-GDP (inactive) to Rab11-GTP (active), which initiates vesicular trafficking.[26] Since Src contains an SH3 domain, we tested whether Src recruits SH3BP5 onto the endosomes for Rab11 activation through its SH3 domain. To this end, an HA-tagged Src SH3 domain deletion mutant was created ([Src with SH3 domain deleted]SrcSH3Del-HA), and wild-type Src was used as the control (SrcWT-HA). HSCs expressing SrcWT-HA or the SrcSH3Del-HA mutant were subjected to double IF for HA (red) and SH3BP5 (green), which revealed that deleting the Src SH3 domain indeed reduced Src/SH3BP5 colocalization in HSCs (yellow) (Figure 3A, p < 0.001).
FIGURE 3.
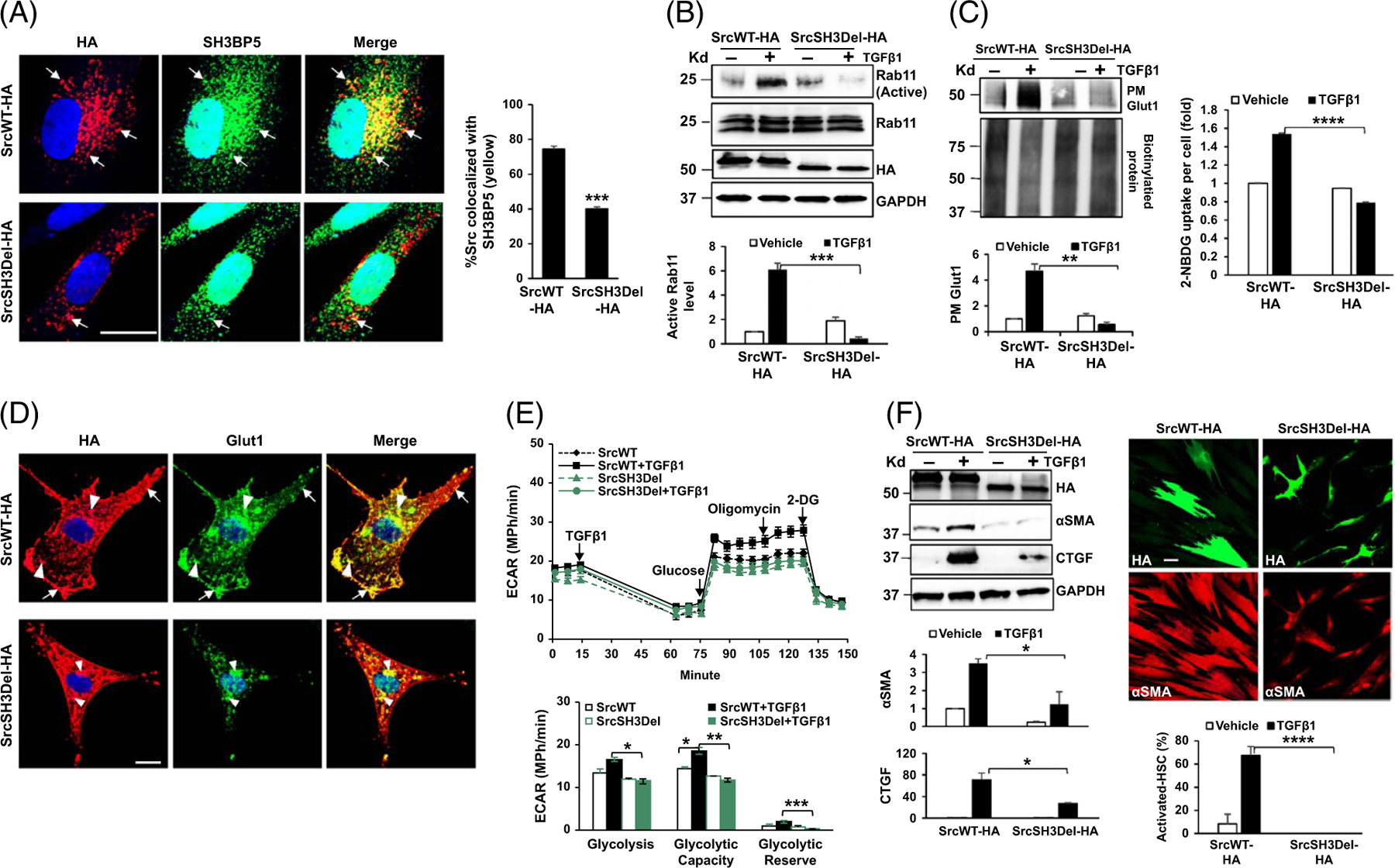
The SH3 domain of Src is required for Rab11 activation, PM Glut1, glycolysis, and HSC activation induced by TGFβ1. (A) HSCs expressing SrcWT-HA or SrcSH3Del-HA were collected for double IF for HA (red) and SH3BP5 (green). Deleting the Src SH3 domain reduced Src/SH3BP5 colocalization in HSCs. ***p < 0.001 by t-test, n = 15 cells. Bar, 20 μm. (B) Rab11 activity assay revealed that TGFβ1 promoted Rab11 activation in SrcWT-HA–expressing cells, but not in SrcSH3Del-HA–expressing cells. ***p < 0.001 by ANOVA, n = 3 repeats. (C) Left, biotinylation assay showed that PM Glut1 was increased by TGFβ1 in SrcWT-HA–expressing cells, but not in SrcSH3del-HA–expressing cells. **p < 0.01 by ANOVA, n = 3 repeats. Right, the Glucose uptake assay revealed that glucose uptake induced by TGFβ1 was abolished by the SrcSH3del-HA mutant. ****p < 0.0001 by ANOVA, n = 20,000. (D) Double IF and confocal microscopy demonstrated evident colocalization of Glut1 (green) and SrcWT-HA (red) at the endosomes (arrowheads) and PM of HSCs (arrows), and diminished colocalization of Glut1 (green) and SrcSH3Del-HA (red) at the PM. Bar, 20 μm. (E) Real-time ECAR data revealed that TGFβ1-promoted glycolysis was suppressed by the SrcSH3del-HA mutant. *p < 0.05; **p < 0.01 by ANOVA, n = 5. F. WB (left) and αSMA IF (red, right) for HSC activation markers showed that TGFβ1-induced HSC activation was suppressed by the SrcSH3del-HA mutant. *p < 0.05; ****p < 0.0001 by ANOVA, n = 3 for WB and n = 10 microscopic fields per group for IF. Bar, 50 μm. Abbreviations: αSMA, alpha-smooth muscle actin; ECAR, extracellular acidification rate; Glut1, glucose transporter 1; HA, human influenza hemagglutinin; IF, immunofluorescence; PM, plasma membrane; SH3BP5, SH3 domain–binding protein 5; SrcSH3Del-HA, HA-tagged Src SH3 domain deletion mutant; SrcWT-HA, HA-tagged wild-type Src; TGFβ1, transforming growth factor-beta 1; WB, western blot.
Rab11 activity assay demonstrated that TGFβ1 promoted Rab11-GTP level (active Rab11) in SrcWT-HA–expressing HSCs and that this effect of TGFβ1 was abolished in SrcSH3Del-HA–expressing HSCs (Figure 3B, p < 0.001). Biotinylation assay showed that TGFβ1 promoted PM Glut1 in SrcWT-HA–expressing HSCs, but not in SrcSH3Del-expressing HSCs (Figure 3C left, p < 0.01). TGFβ1 promoted uptake of 2-NBDG by SrcWT-HA–expressing HSCs, but not by SrcSH3Del-HA–expressing HSCs (Figure 3C right, p < 0.0001). Double IF confirmed diminished PM Glut1 in SrcSH3Del-HA–expressing HSCs than in SrcWT-HA–expressing HSCs (arrows, Figure 3D). Real-time ECAR data demonstrated that TGFβ1-stimulated glycolysis was abrogated by the SrcSH3Del-HA mutant compared to SrcWT-HA (Figure 3E, p < 0.05). Moreover, the mutant reduced SMAD2 phosphorylation and myofibroblastic activation of HSCs induced by TGFβ1 (Supplemental Figure 5A, http://links.lww.com/HEP/I235 and Figure 3F, p < 0.05), as detected by WB and/or IF for αSMA.
Morphologically, expression of the SrcSH3Del-HA mutant reduced the size of HSCs (Supplemental Figure 5B, http://links.lww.com/HEP/I235, p < 0.0001); SrcSH3del-HA–expressing HSCs were narrower and elongated with the most of them containing cytoplasmic vacuoles (arrows, Supplemental Figure 5B, http://links.lww.com/HEP/I235). Oil Red O staining revealed aberrant lipid droplets in SrcSH3del-HA–expressing HSCs with lipid droplets either accumulated or diminished at their cytoplasm compared to SrcWT-HA–expressing HSCs (arrows and arrowheads, Supplemental Figure 5C, http://links.lww.com/HEP/I235).
The Src SH3 domain mediates Src/SH3BP5/Rab11/Glut1 complex formation induced by TGFβ1
We tested whether the Src SH3 domain is required for binding of Rab11 to its activator SH3BP5. To this end, HSCs overexpressing Rab11-FLAG and SrcWT-HA and HSCs overexpressing Rab11-FLAG and the SrcSH3Del-HA mutant were subjected to co-immunoprecipitation. Rab11-FLAG was pulled down by anti-FLAG and co-precipitated SH3BP5 was determined by WB, which revealed that SH3BP5 bound to Rab11 was significantly lower in SrcSH3Del-HA–expressing HSCs than in SrcWT-HA–expressing HSCs (Figure 4A, p < 0.001). The level of SrcSH3Del-HA bound to Rab11 was also lower than that of SrcWT-HA (Figure 4A, p < 0.001). We next investigated whether TGFβ1 induced a Src/SH3BP5/Rab11/Glut1 complex in HSCs. HSCs coexpressing Rab11-FLAG, FLAG-Glut1, and SrcWT-HA or SrcSH3Del-HA were created for co-immunoprecipitation. Anti-HA was used to pull down SrcWT-HA or the SrcSH3Del-HA mutant and co-precipitated Rab11-FLAG, FLAG-Glut1, and SH3BP5 were quantitated by WB, which demonstrated that TGFβ1 indeed promoted a Src/SH3BP5/Rab11/Glut1 complex in SrcWT-HA–expressing HSCs, and this effect of TGFβ1 was abrogated in SrcSH3Del-HA–expressing HSCs (Figure 4B, p < 0.01). Thus, TGFβ1 induces a Src/SH3BP5/Rab11/Glut1 complex in HSCs, which is dependent on the Src SH3 domain.
FIGURE 4.
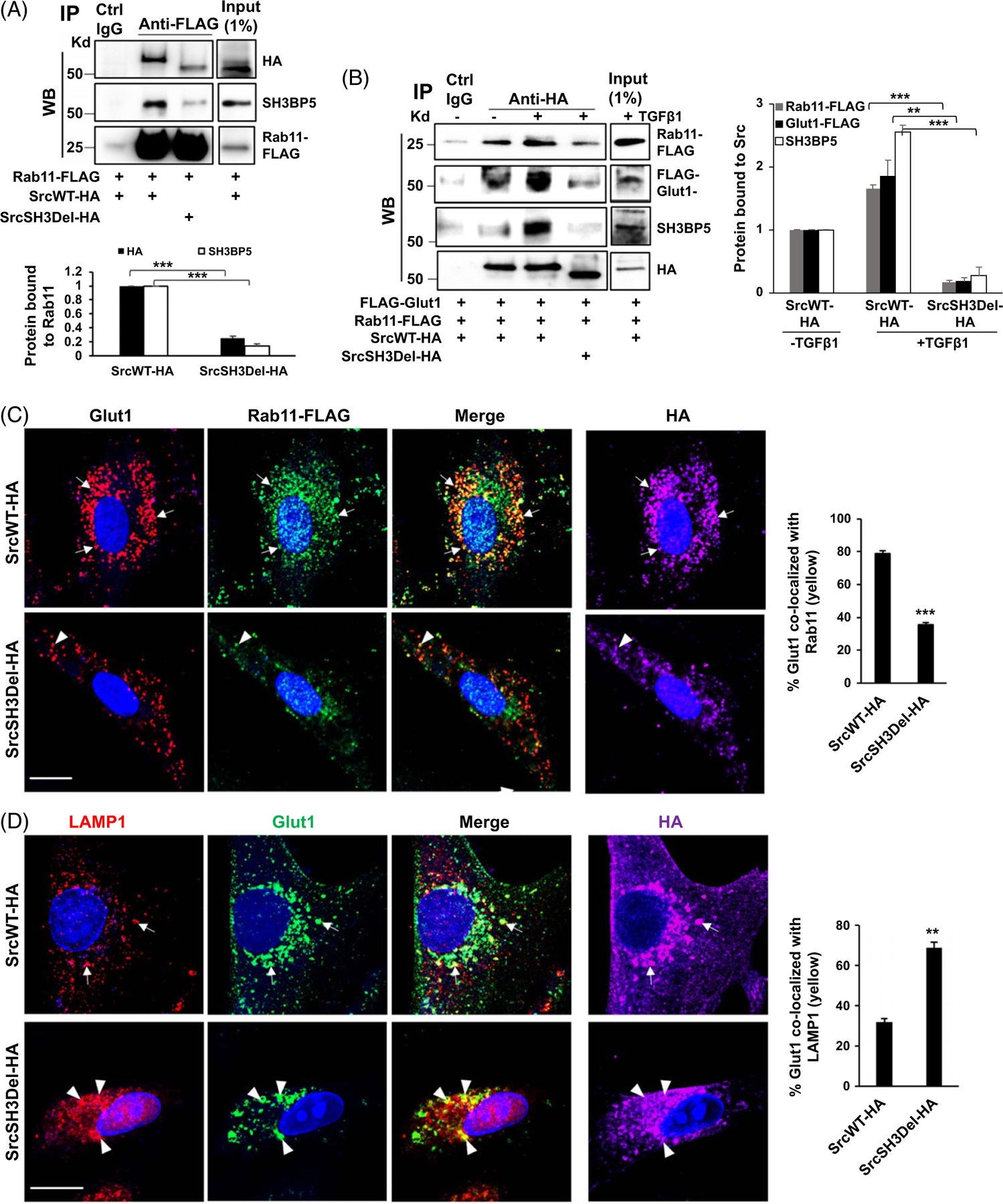
The SH3 domain of Src is required for an Src/SH3BP5/Rab11/Glut1 complex formation induced by TGFβ1. (A) HSCs expressing Rab11-FLAG in combination with SrcWT-HA or SrcSH3Del-HA mutant were collected for coIP. Rab11/SH3BP5 binding was reduced by the SrcSH3Del-HA mutant. ***p < 0.001 by ANOVA, n = 3 repeats. (B) HSCs expressing Rab11-FLAG, FLAG-Glut1, and SrcWT-HA or SrcSH3Del-HA were subjected to coIP. TGFβ1 promoted a Src/SH3BP5/Rab11/Glut1 complex in SrcWT-HA–expressing HSCs but not in SrcSH3Del-HA–expressing cells. **p < 0.01, ***p < 0.001 by ANOVA, n = 3. (C) Triple IF demonstrated that Glut1/Rab11 colocalization was reduced in SrcSH3del-HA–expressing cells (arrowheads) compared to SrcWT-HA–expressing cells (arrows). ***p < 0.001 by t-test, n = 20. Bar, 20 μm. (D) Triple IF revealed that Glut1/LAMP1 colocalization was increased by the SrcSH3del-HA mutant (arrowheads). **p < 0.01 by t-test, n = 20. Bar, 20 μm. Abbreviations: coIP, co-immunoprecipitation; HA, human influenza hemagglutinin; IF, immunofluorescence; LAMP1, lysosome-associated membrane glycoprotein 1; SH3BP5, SH3 domain–binding protein 5; SrcSH3Del-HA, HA-tagged Src SH3 domain deletion mutant; SrcWT-HA, HA-tagged wild-type Src; TGFβ1, transforming growth factor-beta 1.
We also investigated whether the Src SH3 domain is required for Glut1/Rab11 colocalization. As demonstrated by triple IF with TGFβ1-stimulated HSCs, the rate of Glut1/Rab11 colocalization was indeed reduced in SrcSH3del-HA–expressing HSCs than in SrcWT-HA–expressing cells (Figure 4C, p < 0.001). In contrast, the colocalization rate of Glut1 and lysosome-associated membrane glycoprotein 1, a marker of late endosomes/lysosomes, was elevated by the SrcSH3del-HA mutant (Figure 4D, p < 0.01). The results confirmed that the loss of the Src SH3 domain led to increased Glut1 localization at the late endosomes/lysosomes where proteins are degraded.
Targeting Glut1 influences TGFβ transcriptome and suppresses HSC expression of tumor-promoting factors
To explore how glycolysis influences TGFβ transcription, we performed RNA sequencing and identified 2,842 TGFβ1 targets as DEGs by Glut1 knockdown (Figure 5A false discovery rate <0.05). As revealed by GSEA with molecular signatures database (C2 gene sets), Glut1 knockdown altered transcripts related to cellular processes such as translation, metabolism, transcription and RNA processing, and others (Supplemental Figure 6A, http://links.lww.com/HEP/I236, Nominal p < 0.05, normalized enrichment score > 1). For example, Glut1 knockdown suppressed 295 transcripts in translation (Supplemental Figure 6B, http://links.lww.com/HEP/I236), 148 transcripts in epigenetic regulation of gene expression, 205 transcripts in rRNA processing, 111 transcripts in tRNA processing, and 188 transcripts in DNA replication (Supplemental Figure 7A–7D, http://links.lww.com/HEP/I237). As expected, 123 transcripts of TGFB1 targets up were reduced by Glut1 knockdown (Supplemental Figure 7E, http://links.lww.com/HEP/I237). Thus, Glut1 knockdown induced global transcriptomic changes in HSCs.
FIGURE 5.
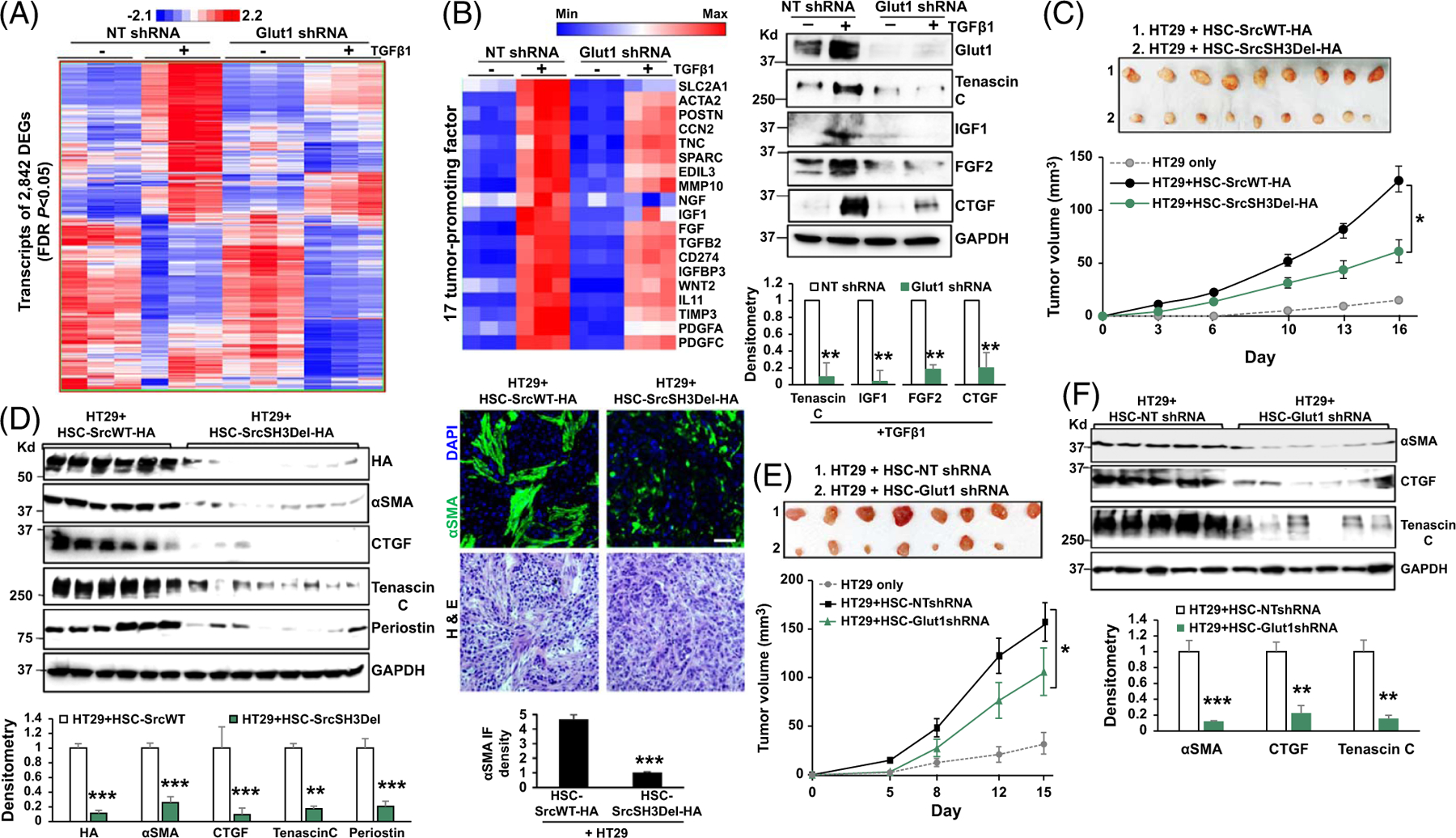
Glut1 knockdown suppresses HSC expression of paracrine factors that promote tumor growth. (A) RNA sequencing identified 2,842 TGFβ targets as DEGs with their expression shown in a heatmap. A color bar shows the minimum expression (blue) to the maximum expression value (red). (B) Left, 17 transcripts of tumor-promoting genes were reduced by Glut1 knockdown. Right, WB confirmed that TGFβ1 promoted HSC production of tenacin C, IGF1, FGF2, and CTGF, and this effect of TGFβ1 was inhibited by Glut1 knockdown. **p < 0.01 by t-test, n = 3. (C) HT29 cells mixed with SrcWT-HA–expressing or SrcSH3Del-HA–expressing HSCs were co-injected into immunosuppressed nude mice subcutaneously. HT29 tumors at the endpoint are shown on the top, and their growth curves are shown on the bottom. SrcSH3Del-HA–expressing HSCs were less effective at promoting HT29 tumor growth in mice compared to SrcWT-HA–expressing HSCs. *p < 0.05 by ANOVA, n = 9 per group. (D) Left, WB revealed that the levels of αSMA and HSC-derived tumor-promoting factors were reduced in tumors arising from HT29/HSC-SrcSH3Del-HA co-injections than in tumors arising from control co-injections. **p < 0.01, ***p < 0.001 by ANOVA, n = 6, 8. Right, αSMA IF showed that CAF density was reduced in tumors arising from HT29/HSC-SrcSH3Del-HA co-injections than in tumors arising from control co-injections. H&E staining is shown on the bottom. ***p < 0.001 by t-test, n = 4. Bar, 50 μm. (E) Co-injection of HT29 and HSCs into immunosuppressed nude mice revealed that Glut1 knockdown HSCs were less effective than control HSCs at promoting HT29 growth in mice. *p < 0.05 by ANOVA, n = 8 per group. (F) WB revealed that the levels of αSMA and HSC-derived tumor-promoting factors were reduced in tumors arising from HT29/Glut1 knockdown HSC co-injections compared to the tumors arising from control co-injections. **p < 0.01, ***p < 0.001 by ANOVA, n = 5, 6. Abbreviations: αSMA, alpha-smooth muscle actin; DEG, differentially expressed genes; FDR, false discovery rate; Glut1, glucose transporter 1; HA, human influenza hemagglutinin; H&E, Hematoxylin & Eosin; IF, immunofluorescence; SrcSH3del, Src with SH3 domain deleted; SrcSH3Del-HA, HA-tagged Src SH3 domain deletion mutant; SrcWT-HA, HA-tagged wild-type Src; WNT, wingless-related integration site.
Since HSC-derived CAFs are the dominant cell population interacting with cancer cells and modulating their growth,[3,5] we focused on DEGs encoding tumor-promoting paracrine factors, which led to the identification of a panel of 17 transcripts affected by Glut1 knockdown (Figure 5B left, p < 0.05). WB confirmed that TGFβ1 stimulation promoted HSCs to produce tenascin C, IGF1, FGF2, and CTGF, and this effect of TGFβ1 was abrogated by Glut1 knockdown (Figure 5B right, p < 0.01). Thus, targeting Glut1 altered the TGFβ1 transcriptome and HSC expression of tumor-promoting factors.
Targeting the Src SH3 domain or Glut1 reduces the tumor-promoting effect of HSCs in an HSC/tumor co-implantation mouse model
We next collected CM from SrcWT-expressing HSCs (control) and SrcSH3Del-expressing HSCs and assessed their role in the migration and proliferation of HT29 human CRC cells.[4,6] While control CM promoted HT29 migration and proliferation compared to the basal medium, this tumor-promoting effect of CM was reduced by the SrcSH3Del mutant (Supplemental Figures 8A and 8B, http://links.lww.com/HEP/I238, p < 0.05). To translate the finding into in vivo conditions, HT29 cells (0.5 × 106) were mixed with SrcWT-expressing or SrcSH3Del-expressing HSCs (0.5 × 106) followed by subcutaneous co-injection into immunosuppressed nude mice. Tumor size measurement revealed that SrcSH3Del-expressing HSCs were less effective at promoting HT29 growth in mice compared to SrcWT-expressing HSCs (Figure 5C, p < 0.05). αSMA is a marker of CAFs; reduced CAF density was detected by αSMA WB and IF in tumors arising from HT29/HSC-SrcSH3Del co-injections compared to those arising from control co-injections (Figure 5D, p < 0.001). The levels of HSC-derived factors, such as CTGF, tenascin C, and periostin, as well as collagen deposition, were all reduced in tumors arising from HT29/HSC-SrcSH3Del co-injections (Figure 5D and Supplemental Figure 8C, http://links.lww.com/HEP/I238 p < 0.01). Similarly, injection of HT29 cells, preconditioned with the CM of HSCs in vitro (45 min at 37°C), revealed that SrcSH3Del-HA–expressing HSCs were less effective than that of SrcWT-HA–expressing HSCs at promoting HT29 growth in immunosuppressed nude mice (Supplemental Figure 8D, http://links.lww.com/HEP/I238, p < 0.05). Thus, targeting the SH3 domain of Src suppresses the tumor-promoting effect of HSCs in vitro and in mice.
HT29 cells were also mixed with control or Glut1 knockdown HSCs for subcutaneous co-injection into immunosuppressed nude mice, which revealed that Glut1 knockdown HSCs were less effective than control HSCs at promoting HT29 growth, similar to the Src SH3 domain deletion mutant (Figure 5E, p < 0.05). Consistently, reduced CAF density was detected in tumors arising from HT29/HSC-Glut1 shRNA co-injections compared to those arising from control co-injections (Figure 5F and Supplemental Fig. 9A, http://links.lww.com/HEP/I240, p < 0.05), and HSC-derived CTGF and tenascin C as well as collagen deposition were also reduced in tumors arising from HT29/HSC-Glut1 shRNA co-injections (Figure 5F and Supplemental Fig. 9B, http://links.lww.com/HEP/I240, p < 0.05). Thus, targeting HSC Glut1 suppresses CAF activation and the tumor-promoting effect of HSCs in mice.
Targeting HSC Glut1 by Cre/LoxP suppresses CAF activation and MC38 CRC liver metastasis in mice
To validate Glut1/Src/Rab11 expression in the hepatic tumor microenvironment, we obtained liver biopsies of patients with CRC from a Mayo Clinic tissue collection for triple IF.[2,10,27] As shown in Supplemental Fig. 10 left, http://links.lww.com/HEP/I239, patient CRC cells were strongly positive for Glut1 (green), whereas the CAFs were strongly positive for αSMA (purple) and moderately positive for Glut1. Confocal microscopy with higher magnification and longer exposure demonstrated that Glut1 (green) indeed colocalized with Src (red, arrowheads) and Rab11 (red, arrows) in the cytoplasm (and at the PM) of the CAFs (yellow) (Supplemental Fig. 10 middle and right columns, http://links.lww.com/HEP/I239).
To target HSC Glut1, we set up a breeding of Slc2a1/Glut1 floxed mutant mice to tamoxifen-inducible Cre transgenic mice expressing Cre recombinase under the promoter of platelet-derived growth factor receptor β gene (PDGFRβ-P2A-CreERT2).[28] Age-matched Glut1F/F mice (control) and Glut1F/PDGFRβ-CreERT2 mice were selected for portal vein injection of MC38 CRC cells (1 × 106 cells per mouse) after tamoxifen injection for activation of Cre. At the endpoint, Glut1F/PDGFRβ-CreERT2 mice developed fewer MC38 liver metastases compared to Glut1F/F mice (Figure 6A, p < 0.01). Over 90% of the CAFs of CRC liver metastases were derived from HSCs,[5] so CAF densities of the liver metastases were compared. IF and WB detected reduced CAF density in MC38 liver metastases of Glut1F/PDGFRβ-CreERT2 mice compared to those of control mice (Figure 6B and C, p < 0.01). The levels of HSC-derived periostin, programmed death-ligand 1, and insulin-like growth factor–binding protein 3 were also reduced in those of Glut1F/PDGFRβ-CreERT2 mice (Figure 6B p < 0.01). IF demonstrated that the levels of Glut1 (red, Figure 6C), Src (red, Supplemental Fig. 11A, http://links.lww.com/HEP/I241), and Rab11 (red, Supplemental Fig. 11B, http://links.lww.com/HEP/I241) were reduced in Glut1F/PDGFRβ-CreERT2 CAFs (arrowheads) compared to Glut1F/F CAFs (arrows) (p < 0.01). Reduced collagen and lactate level were also detected in liver metastases of Glut1F/PDGFRβ-CreERT2 mice, whereas either TGFβ1 levels of the liver or TGFβ1 levels of MC38 tumors were comparable for 2 mouse groups (Supplemental Fig. 12A–12C, http://links.lww.com/HEP/I242). Consistently, co-injection of active TGFβ1 and MC38 into murine liver potentiated CAF activation and MC38 liver metastases, which was abrogated in Glut1F/PDGFRβ-CreERT2 mice as well (Supplemental Fig. 12D, http://links.lww.com/HEP/I242 p < 0.01). Thus, targeting HSC Slc2a1/Glut1 by Cre/LoxP suppresses HSC glycolysis, CAF activation, and CRC liver metastases in mice.
FIGURE 6.
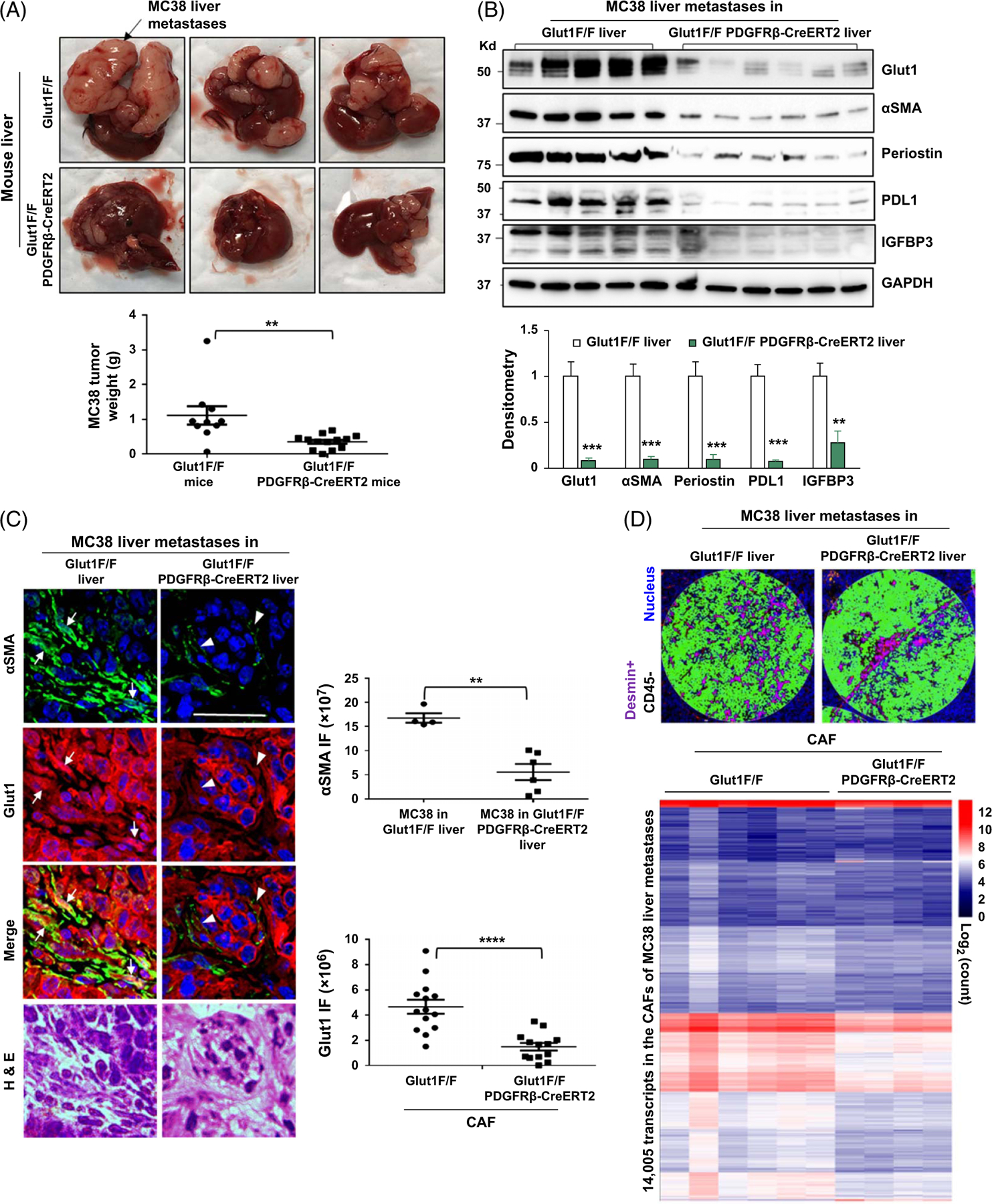
HSC-specific Glut1 knockout by Cre/LoxP suppresses CAF activation, CAF transcriptome, and colorectal liver metastasis in mice. (A) MC38 murine colorectal cancer cells were implanted into littermate-matched Glut1F/F (control) and Glut1F/PDGFRβ-CreERT2 mice by portal vein injection. Glut1F/PDGFRβ-CreERT2 mice developed fewer MC38 liver metastases compared to control mice. **p < 0.01 by t-test, n = 10, 14. (B) WB with tumor lysates revealed that protein levels of Glut1, αSMA, periostin, PD-L1, and IGFBP3 were reduced in liver metastases of Glut1F/PDGFRβ-CreERT2 mice compared to those of control mice. **p < 0.01; ***p < 0.001 by ANOVA, n = 5, 6. (C) Double IF revealed that αSMA IF was reduced in MC38 liver metastases of Glut1F/PDGFRβ-CreERT2 mice compared to that in MC38 liver metastases of control mice (green). **p < 0.01 by t-test, n = 4, 6 tumors. Glut1 IF was also reduced in Glut1 F/PDGFRβ-CreERT2 CAFs (arrowheads) compared to Glut1F/F CAFs (arrows) (red). ****p < 0.0001 by t-test, n = 14, 13. Bar, 50 μm. (D) Upper, 2 example ROIs within MC38 liver metastases selected for transcriptomic profiling are shown. Anti-desmin IF was used to label the CAFs (purple), and anti-CD45 IF was used to label immune cells (orange). Desmin+CD45- areas were selected for analysis. Lower, the transcripts detected from the CAFs of different tumor sections are shown by a heatmap with a scale bar on the right. Abbreviations: αSMA, alpha-smooth muscle actin; CAF, cancer-associated fibroblast; Cre/LoxP, Cre recombinase/LoxP sequence derived from bacteriophage P1; Glut1, glucose transporter 1; IF, immunofluorescence; IGFBP3, insulin-like growth factor–binding protein 3; PDGFRβ, platelet-derived growth factor receptor beta; PD-L1, programmed death-ligand 1; ROI, regions of interest; WB, western blot.
Spatialomics revealing global transcriptomic changes in Glut1F/PDGFRβ-CreERT2 CAFs and adjacent MC38 CRC cancer cells
To gain further insights into the role of HSC glycolysis on CRC liver metastasis, CAF transcriptome was studied on MC38 liver metastasis sections with the NanoString GeoMx digital spatialomics profiler (GeoMx DSP) and GeoMx Mouse Whole Transcriptome Atlas Panel targeting 20,177 murine transcripts. An anti-desmin antibody that labels CAFs (Supplemental Fig. 13A and 13B, http://links.lww.com/HEP/I243) and is compatible with the workflow of GeoMx DSP was used for IF. Anti-CD45 was used to label immune cells. Desmin+CD45- cells were enriched for transcriptomic profiling (purple, Figure 6D), which led to 14,005 transcripts and 1999 DEGs detected from the CAFs (Figure 6D and Supplemental Fig. 14A, http://links.lww.com/HEP/I244).
As analyzed by GSEA and mouse molecular signatures database (M2 gene sets), the transcriptomic changes in Glut1-deficient CAFs were related to processes such as translation, gene transcription and RNA processing, DNA replication, metabolism, and others (Figure 7A, Nominal p < 0.05, normalized enrichment score > 1). For example, 176 transcripts in rRNA processing and 117 transcripts in eukaryotic translation initiation were reduced in Glut1-deficient CAFs (Figure 7B). Thirteen transcripts in the mTOR pathway and 168 in the TCA cycle and respiratory electron transport were also reduced (Supplemental Fig. 14B and 14C upper, http://links.lww.com/HEP/I244). Consistent with the role of TGFβ in CAF activation, KARLSSON TGFB1 targets up (127 transcripts) exhibited a top score among all intracellular signaling pathways affected, followed by PDGF targets and signaling of wingless-related integration site, FGF, angiotensin II, interleukin, S1P, IGF, nerve growth factor, CTGF, and Hedgehog (Figure 7A and Supplemental Fig. 14C lower, http://links.lww.com/HEP/I244). The transcripts of genes related to HSC activation, Acta2, Fn1, Tgfbr1–3, Pdgfra-b, Fgfr1, Col1–4, or tumor-promoting factors, such as Tgfb1–3, Ccn2, Pdgfa-c, Postn, Tnc, Sparc, Timp3, Igfbp2–7, and Thbs2, were all reduced by Glut1 targeting (Figure 7C), consistent with RNA sequencing data of cultured HSCs.
FIGURE 7.
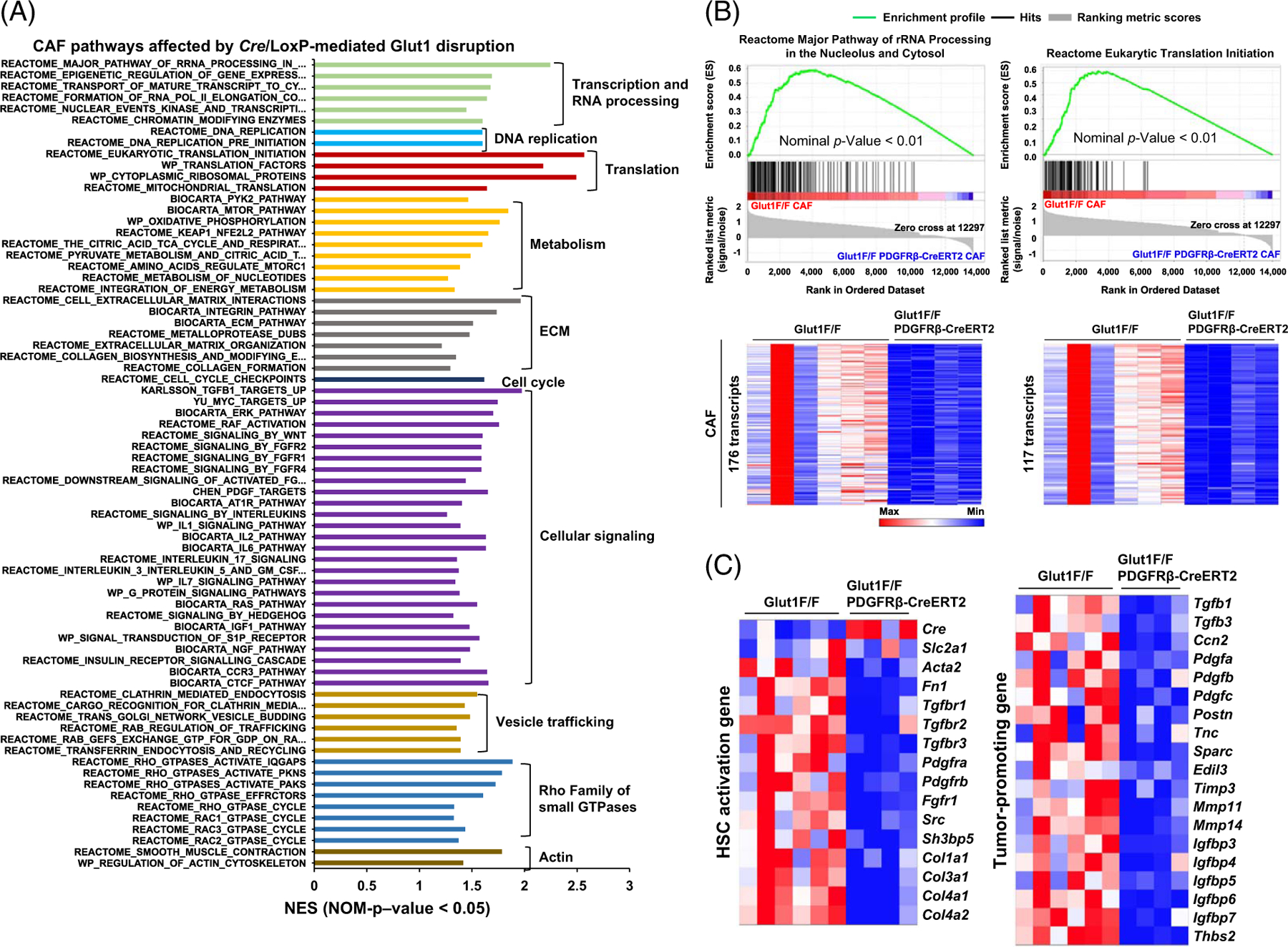
Spatialomics revealing transcriptomic changes in Glut1-deficient CAFs of MC38 liver metastases. (A) GSEA identified altered signaling pathways and cellular processes in Glut1-deficient CAFs compared to control CAFs based on NES > 1 and p < 0.05. (B) Two gene sets with transcription affected by Cre/LoxP-mediated Glut1 gene disruption are shown. Transcript enrichment is shown by the enrichment plots (upper), and transcript levels are shown by heatmaps (lower). The scale bar represents the minimum (blue) to the maximum expression level (red). (C) Two heatmaps revealed that Glut1 deficiency suppressed transcripts of 15 HSC activation genes (left) and 19 tumor-promoting genes (right) in the CAFs. Abbreviations: CAF, cancer-associated fibroblast; Cre/LoxP, Cre recombinase/LoxP sequence derived from bacteriophage P1; Glut1, glucose transporter 1; GSEA, Gene Set Enrichment Analysis; NES, normalized enrichment score.
Spatialomics also detected 16,658 transcripts and 2617 DEGs in MC38 cells caused by Glut1 targeting in adjacent CAFs (Figure 8A, false discovery rate p < 0.01). As analyzed by GSEA, the transcripts altered were related to processes such as cell cycle, metabolism, DNA replication, gene transcription, and others (Figure 8B). Strikingly, 390 transcripts in the M phase and 285 transcripts in cell cycle checkpoints were identified as the top pathways affected (Figure 8B, C upper, and Supplemental Fig. 15A upper, http://links.lww.com/HEP/I245). One hundred twenty-six transcripts in DNA replication (Supplemental Fig. 15A lower, http://links.lww.com/HEP/I245), 168 transcripts in the TCA cycle and respiratory electron transport (Figure 8C lower), and 22 transcripts in amino acids regulation of mammalian target of rapamycin complex 1 were reduced also (Supplemental Fig. 15B, http://links.lww.com/HEP/I245). Moreover, adenomatous polyposis coli MYC proto-oncogene pathway, wingless-related integration site signaling, E2F targets, as well as Rho GTPase effectors were identified as the top intracellular signaling affected (Supplemental Fig. 16A–C, http://links.lww.com/HEP/I246). Thus, targeting Glut1 of HSCs/CAFs led to transcriptomic changes in adjacent cancer cells that modified tumor behaviors and growth.
FIGURE 8.
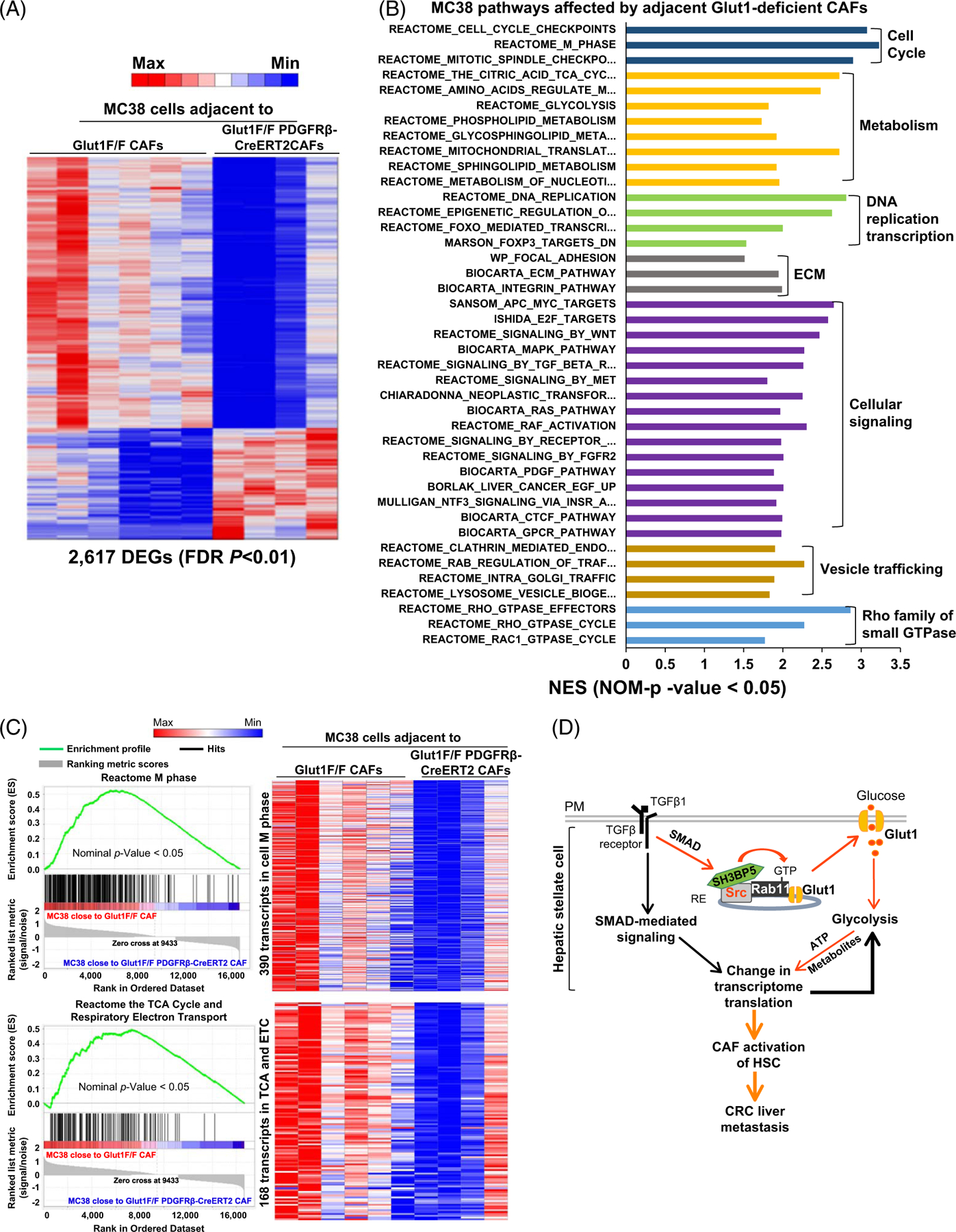
Targeting HSC/CAF Glut1 by Cre/LoxP leads to transcriptomic changes in adjacent MC38 cancer cells. (A) Spatial transcriptomic profiling identified 2617 DEGs in MC38 cancer cells adjacent to Glut1-deficient CAFs (FDR p < 0.01). A scale bar represents the lowest count (blue) to the highest count (red). (B) GSEA revealed pathways and cellular processes of MC38 cancer cells affected by adjacent Glut1-deficient CAFs (NES > 1 and p < 0.05). (C) 2 gene sets of MC38 cells with their expression affected by adjacent Glut1-deficient CAFs are shown. Transcript enrichment is shown by enrichment plots (left), and gene expression levels are shown by the heatmaps (right). (D) Schematic summary of this study. Abbreviations: APC, adenomatous polyposis coli; CAF, cancer-associated fibroblast; Cre/LoxP, Cre recombinase/LoxP sequence derived from bacteriophage P1; CRC, colorectal cancer; DEG, differentially expressed gene; FDR, false discovery rate; Glut1, glucose transporter 1; GSEA, Gene Set Enrichment Analysis; NES, normalized enrichment score; PM, plasma membrane; RE, recycling endosome; WNT, wingless-related integration site.
DISCUSSION
We found that TGFβ1 promotes PM Glut1 and glycolysis by inducing a Src/SH3BP5/Rab11/Glut1 protein complex in HSCs (Figure 8D). Functionally, targeting the Src SH3 domain or Glut1 of HSCs suppresses CAF activation of HSCs and CRC tumor growth in mice. Multi-omics reveals that targeting Glut1 alters HSC transcriptome and HSC expression of paracrine tumor-promoting factors, and additionally, the transcriptome of adjacent CRC cancer cells. Our data highlight an endosomal Src/SH3BP5/Rab11/Glut1 protein complex, induced by TGFβ1, as a target to inhibit CAF activation and CRC liver metastasis.
Although the transcripts of Glut3, Glut6, Glut10, and Glut13 were detected in HSCs/CAFs by multi-omics (Supplemental Figure 1B, http://links.lww.com/HEP/I231 and 14D), our study showed that targeting Glut1 markedly inhibited glucose uptake, glycolysis, and CAF activation of HSCs induced by TGFβ1, supporting Glut1 as the prominent glucose transporter controlling metabolism and CAF activation of HSCs. It is known that Glut1 is transcriptionally upregulated by TGFβ with its mRNA level increased at 3 hours and protein increased at 6 hours after TGFβ stimulation[16,18]; however, it is unknown whether Glut1 trafficking is influenced by TGFβ1. In this regard, our study is the only work determining whether and how TGFβ1 regulates Glut1 trafficking, glycolysis, and CAF activation in vitro and in the hepatic tumor microenvironment.
One study reported that upregulation of Glut1 expression by TGFβ requires SMAD, p38 MAPK, and P13K/AKT signaling pathways,[16] and another demonstrated that it occurs through the SMAD2/3 pathway and requires autocrine activation of the receptor tyrosine kinases (platelet-derived and EGF receptors), PI3K, MEK, and mammalian target of rapamycin complex.[18] Our biotinylation assay for SMAD2 knockdown HSCs also demonstrated a requirement of SMAD2 for the process of PM targeting of Glut1 induced by TGFβ1 (Supplemental Figure 3A, http://links.lww.com/HEP/I233). Additionally, we observed that SMAD2 phosphorylation induced by TGFβ1 was suppressed by the Src SH3 deletion mutant compared to wild-type Src (Supplemental Figure 5A, http://links.lww.com/HEP/I235). These data together suggest that Src may cross talk with the canonical TGFβ/SMAD signaling to modulate glycolysis and CAF activation of HSCs upon TGFβ1 stimulation, which may represent an interesting direction to pursue in the future studies.
“Translation” and “transcription and RNA processing” are the top cellular processes affected by Glut1 targeting as identified by multi-omics. It is possible that by altering these 2 important processes, further amplifications of the changes of the transcriptome and proteome occurred in the Glut1-deficient cells. Compared to RNA sequencing, Spatialomics detected more intracellular signaling pathways affected by Glut1 targeting, and additionally, it detected transcriptomic changes in adjacent MC38 cancer cells. Spatialomics therefore represents a unique and irreplaceable tool allowing us to study in vivo cross talks between cancer cells and the tumor microenvironment.
Our prior studies revealed that HSCs express TGFβ1 mRNA and its expression level is enhanced by TGFβ1.[4,21] We observed that targeting HSC Glut1 by Cre/LoxP led to reduced CAF densities and MC38 liver metastases in the portal vein tumor injection mouse model. The result of ELISA, however, showed that intratumoral latent TGFβ1 levels were comparable for both mouse groups (Supplemental Fig. 12C, http://links.lww.com/HEP/I242). These data suggest that in established liver metastases, other cells, in addition to CAFs, such as MC38, macrophages, and myeloid-derived cells, may contribute to intratumoral TGFβ1. Spatialomics indeed detected that in an ROI, the average count of TGFβ1 transcripts of MC38 origin was 6.8 folds as high as that of CAF origin (Supplemental Fig. 16D, http://links.lww.com/HEP/I246). These data led us to the hypothesis that TGFβ1 may be critical for CAF activation only at an early stage and before macro-metastases are established in the liver. This notion was indeed supported by our observation that co-injection of active TGFβ1 and MC38 into the portal vein of the mice potentiated CAF activation and liver metastasis compared to MC38 injection alone (Supplemental Fig. 12D, http://links.lww.com/HEP/I242).
In summary, we have uncovered a mechanism governing HSC activation whereby the Src SH3 domain facilitates the formation of a Src/SH3BP5/Rab11/Glut1 complex required for PM targeting of Glut1, glycolysis, and CAF activation induced by TGFβ1. The Src/SH3BP5/Rab11/Glut1 complex represents a target to inhibit CAF activation and the pro-metastatic liver microenvironment.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the H.I. Core Facility and Animal Facility staff for their assistance. They also thank Fernanda Rodriguez, Grant Barthel, and John Garbe at the UMN Genomic Center for Spatialomics and other staff for RNA sequencing.
FUNDING INFORMATION
This study was supported by the NIH grant R01CA160069 to Ningling Kang. The Mayo Clinic Hepatobiliary Cancer SPORE (P50 CA210964) Developmental Research Program, The Hormel Windfeldt Pilot, and Paint the Town Pink (PTTP) Award to Ningling Kang.
Abbreviations:
- αSMA
alpha-smooth muscle actin
- CAF
cancer-associated fibroblast
- CM
conditioned medium
- CRC
colorectal cancer
- Cre/LoxP
Cre recombinase/LoxP sequence derived from bacteriophage P1
- CTGF
connective tissue growth factor
- DEG
differentially expressed gene
- ECAR
extracellular acidification rate
- FN
fibronectin
- Glut1
glucose transporter 1
- GSEA
Gene Set Enrichment Analysis
- HA
human influenza hemagglutinin
- IF
immunofluorescence
- NBDG
2-deoxy-2-[(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]-D-glucose
- PDGFRbeta
Platelet-derived growth factor receptor beta
- PM
plasma membrane
- SH3 domain
Src homology 3 domain
- SH3BP5
SH3 domain–binding protein 5
- shRNA
short hairpin RNA
- SMAD
mothers against decapentaplegic homolog 3
- Src
proto-oncogene tyrosine-protein kinase Src
- SrcSH3del
Src with SH3 domain deleted
- SrcSH3Del-HA
HA-tagged Src SH3 domain deletion mutant
- SrcWT-HA
HA-tagged wild-type Src
- TGFβ1
transforming growth factor-beta 1
- WB
western blot
Footnotes
This study was presented at the Liver Meeting, AASLD, 2022.
Supplemental Digital Content is available for this article. Direct URL citations are provided in the HTML and PDF versions of this article on the journal’s website, www.hepjournal.com.
CONFLICT OF INTEREST
The authors have no conflicts to report.
DATA AVAILABILITY
Data from this study have been been deposited in the Gene Expression Omnibus (GEO) under accession numbers GSE223602 (RNA sequencing data) and GSE225458 (spatialomics data). Other data, analytic methods, and reagents will be made available to other researchers upon request.
REFERENCES
- 1.Kang N, Gores GJ, Shah VH. Hepatic stellate cells: Partners in crime for liver metastases? Hepatology. 2011;54:707–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu C, Billadeau DD, Abdelhakim H, Leof E, Kaibuchi K, Bernabeu C, et al. IQGAP1 suppresses TbetaRII-mediated myofibroblastic activation and metastatic growth in liver. J Clin Invest. 2013;123:1138–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Affo S, Nair A, Brundu F, Ravichandra A, Bhattacharjee S, Matsuda M, et al. Promotion of cholangiocarcinoma growth by diverse cancer-associated fibroblast subpopulations. Cancer Cell. 2021;39:866–882 e811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun L, Wang Y, Wang X, Navarro-Corcuera A, Ilyas S, Jalan-Sakrikar N, et al. PD-L1 promotes myofibroblastic activation of hepatic stellate cells by distinct mechanisms selective for TGF-beta receptor I versus II. Cell Rep. 2022;38:110349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhattacharjee S, Hamberger F, Ravichandra A, Miller M, Nair A, Affo S, et al. Tumor restriction by type I collagen opposes tumor-promoting effects of cancer-associated fibroblasts. J Clin Invest. 2021;131:e146987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dou C, Liu Z, Tu K, Zhang H, Chen C, Yaqoob U, et al. P300 acetyltransferase mediates stiffness-induced activation of hepatic stellate cells into tumor-promoting myofibroblasts. Gastroenterology. 2018;154:2209–221 e2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y, Tu K, Liu D, Guo L, Chen Y, Li Q, et al. p300 acetyltransferase is a cytoplasm-to-nucleus shuttle for SMAD2/3 and TAZ nuclear transport in transforming growth factor beta-stimulated hepatic stellate cells. Hepatology. 2019;70: 1409–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kang N, Shah VH, Urrutia R. Membrane-to-nucleus signals and epigenetic mechanisms for myofibroblastic activation and desmoplastic stroma: Potential therapeutic targets for liver metastasis? Mol Cancer Res. 2015;13:604–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Attisano L, Wrana JL. Signal transduction by the TGF-beta superfamily. Science. 2002;296:1646–7. [DOI] [PubMed] [Google Scholar]
- 10.Tu K, Li J, Verma VK, Liu C, Billadeau DD, Lamprecht G, et al. Vasodilator-stimulated phosphoprotein promotes activation of hepatic stellate cells by regulating Rab11-dependent plasma membrane targeting of transforming growth factor beta receptors. Hepatology. 2015;61:361–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu D, Fu X, Wang Y, Wang X, Wang H, Wen J, et al. Protein diaphanous homolog 1 (Diaph1) promotes myofibroblastic activation of hepatic stellate cells by regulating Rab5a activity and TGFbeta receptor endocytosis. FASEB J. 2020;34: 7345–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang F, Hilakivi-Clarke L, Shaha A, Wang Y, Wang X, Deng Y, et al. Metabolic reprogramming and its clinical implication for liver cancer. Hepatology. 2023;78:1602–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science. 2009;324:1029–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y, Choi SS, Michelotti GA, Chan IS, Swiderska-Syn M, Karaca GF, et al. Hedgehog controls hepatic stellate cell fate by regulating metabolism. Gastroenterology. 2012;143:1319–329 e1311–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Du K, Hyun J, Premont RT, Choi SS, Michelotti GA, Swiderska-Syn M, et al. Hedgehog-YAP signaling pathway regulates glutaminolysis to control activation of hepatic stellate cells. Gastroenterology. 2018;154:1465–479 e1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou MY, Cheng ML, Huang T, Hu RH, Zou GL, Li H, et al. Transforming growth factor beta-1 upregulates glucose transporter 1 and glycolysis through canonical and noncanonical pathways in hepatic stellate cells. World J Gastroenterol. 2021; 27:6908–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karim S, Liaskou E, Fear J, Garg A, Reynolds G, Claridge L, et al. Dysregulated hepatic expression of glucose transporters in chronic disease: Contribution of semicarbazide-sensitive amine oxidase to hepatic glucose uptake. Am J Physiol Gastrointest Liver Physiol. 2014;307:G1180–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andrianifahanana M, Hernandez DM, Yin X, Kang JH, Jung MY, Wang Y, et al. Profibrotic up-regulation of glucose transporter 1 by TGF-beta involves activation of MEK and mammalian target of rapamycin complex 2 pathways. FASEB J. 2016;30:3733–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang N, Yaqoob U, Geng Z, Bloch K, Liu C, Gomez T, et al. Focal adhesion assembly in myofibroblasts fosters a micro-environment that promotes tumor growth. Am J Pathol. 2010; 177:1888–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Decker NK, Abdelmoneim SS, Yaqoob U, Hendrickson H, Hormes J, Bentley M, et al. Nitric oxide regulates tumor cell cross-talk with stromal cells in the tumor microenvironment of the liver. Am J Pathol. 2008;173:1002–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Y, Li Q, Tu K, Wang Y, Wang X, Liu D, et al. Focal adhesion kinase promotes hepatic stellate cell activation by regulating plasma membrane localization of TGFbeta receptor 2. Hepatol Commun. 2020;4:268–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sandilands E, Brunton VG, Frame MC. The membrane targeting and spatial activation of Src, Yes and Fyn is influenced by palmitoylation and distinct RhoB/RhoD endosome requirements. J Cell Sci. 2007;120:2555–64. [DOI] [PubMed] [Google Scholar]
- 24.Lee EE, Ma J, Sacharidou A, Mi W, Salato VK, Nguyen N, et al. A protein kinase C phosphorylation motif in GLUT1 affects glucose transport and is mutated in GLUT1 deficiency syndrome. Mol Cell. 2015;58:845–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Welz T, Wellbourne-Wood J, Kerkhoff E. Orchestration of cell surface proteins by Rab11. Trends Cell Biol. 2014;24:407–15. [DOI] [PubMed] [Google Scholar]
- 26.Jenkins ML, Margaria JP, Stariha JTB, Hoffmann RM, McPhail JA, Hamelin DJ, et al. Structural determinants of Rab11 activation by the guanine nucleotide exchange factor SH3BP5. Nat Commun. 2018;9:3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu C, Li J, Xiang X, Guo L, Tu K, Liu Q, et al. PDGF receptor alpha promotes TGF-beta signaling in hepatic stellate cells via transcriptional and post transcriptional regulation of TGF-beta receptors. Am J Physiol Gastrointest Liver Physiol. 2014;307:G749–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cuervo H, Pereira B, Nadeem T, Lin M, Lee F, Kitajewski J, et al. PDGFRbeta-P2A-CreER(T2) mice: A genetic tool to target pericytes in angiogenesis. Angiogenesis. 2017;20:655–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data from this study have been been deposited in the Gene Expression Omnibus (GEO) under accession numbers GSE223602 (RNA sequencing data) and GSE225458 (spatialomics data). Other data, analytic methods, and reagents will be made available to other researchers upon request.