

Abstract
Recessive dystrophic epidermolysis bullosa (RDEB) is a severely debilitating disorder caused by pathogenic variants in COL7A1 and is characterized by extreme skin fragility, chronic inflammation, and fibrosis. A majority of patients with RDEB develop squamous cell carcinoma, a highly aggressive skin cancer with limited treatment options currently available. In this study, we utilized an approach leveraging whole-genome sequencing and RNA sequencing across 3 different tissues in a single patient with RDEB to gain insight into possible mechanisms of RDEB-associated squamous cell carcinoma progression and to identify potential therapeutic options. As a result, we identified PLK-1 as a possible candidate for targeted therapy and discovered microsatellite instability and accelerated aging as factors potentially contributing to the aggressive nature and early onset of RDEB squamous cell carcinoma. By integrating multitissue genomic and transcriptomic analyses in a single patient, we demonstrate the promise of bridging the gap between genomic research and clinical applications for developing tailored therapies for patients with rare genetic disorders such as RDEB.
Keywords: Accelerated aging, Microsatellite instability, Multiomic analyses, Recessive dystrophic epidermolysis bullosa, Squamous cell carcinoma
INTRODUCTION
Recessive dystrophic epidermolysis bullosa (RDEB) is a severely debilitating disorder characterized by extreme skin fragility, wide-spread blistering of the skin and mucous membranes, chronic inflammation, and fibrosis leading to severe scarring, joint contractures, and in some cases, vision loss. Up to 90% of RDEB patients with the severe-generalized form develop squamous cell carcinoma (SCC) (Fine et al, 2009), which is frequently highly aggressive and life threatening. RDEB is caused by loss-of-function pathogenic variants in COL7A1, the gene that encodes COLVII (Hovnanian et al, 1992). COLVII itself does not appear to be a tumor suppressor because COL7A1 heterozygotes do not exhibit increased cancer risk, and variations in COL7A1 are not currently associated with any non-RDEB cancers (Tartaglia et al, 2021). Instead, extreme epidermal fragility, trauma-induced blistering, and impaired wound healing feed into a progressive cycle of scarring and fibrosis that culminates at an early age in SCC that is highly metastatic and lethal with a 5-year survival rate approaching 0% (Fine et al, 2009). Exceptions exist, as several patients with RDEB have survived metastasis free after developing multiple cutaneous SCCs over many years (Tartaglia et al, 2021).
Current treatment for RDEB is mainly palliative, focusing on symptom-relief therapies that include bandaging, pain and itch control, and management of bacterial and fungal infections, although several curative treatments are currently being explored. Studies have shown that increased TGF-ß expression and activity contribute to RDEB fibrosis (Fritsch et al, 2008; Küttner et al, 2013; Ng et al, 2012) and that reduction of TGF-ß signaling using the angiotensin II receptor blocker losartan can ameliorate long-term symptoms in a murine model of RDEB (Nyström et al, 2015). A phase II clinical trial called REFLECT (symptom-RElieF with Losartan – EB Clinical Trial) was recently completed, with losartan receiving orphan drug designation by the European Medicines Agency and Food and Drug Administration for treatment of RDEB. Other curative therapies that aim to replace COLVII are in progress. Gene therapies include topical COL7A1 delivery through the nonintegrating viral vector Beremagene Gepervavae (NCT03536143) (Gurevich et al, 2022) and transplantation of autologous epidermal sheets transduced with full-length COL7A1 (NCT01263379) (Siprashvili et al, 2016). Cellular therapies include allogeneic bone marrow transplantation (NCT02582775 and NCT01033552) (Wagner et al, 2010) and transfusion of ABCB5-positive mesenchymal stem cells to reduce RDEB-induced inflammation and tissue scarring (NCT03529877) (Kiritsi et al, 2021). Because RDEB-associated SCC is positively correlated with the severity of RDEB, a reduction in RDEB symptoms should reduce the risk of developing RDEB-associated SCC.
Currently, no specific therapies have been approved for RDEB-associated SCC. Studies of the development of SCC in patients with RDEB are complicated by the relative patient scarcity because RDEB is a rare disease, and RDEB-associated SCC is even rarer. In recent years, the use of microarray and next-generation DNA and RNA sequencing (RNA-seq) has provided insight into the molecular basis of RDEB-associated SCC (Breitenbach et al, 2015; Chacón-Solano et al, 2019; Chang and Shain, 2021; Cho et al, 2018; Ng et al, 2012; Zhang et al, 2018). On the basis of these studies, PLK1 was found overexpressed in RDEB-associated SCC, and targeting PLK1 by the specific inhibitor rigosertib is currently being investigated in 2 clinical trials (NCT03786237 and NCT04177498). Meanwhile, off-label use of ruxolitinib, a Jak1/2 inhibitor, was limited by significant side effects (Mittapalli et al, 2020). The largest sequencing study to date looked at the genomes of 27 RDEB-associated SCC tumors and compared them with those of 38 UV-induced cutaneous SCC and 279 head and neck SCC tumors from the general population (Cho et al, 2018). They found head and neck SCC to be the most similar to RDEB-associated SCC and identified APOBEC mutational processes in RDEB-associated SCC as causative of accelerated accumulation of mutation burden in these patients. However, no specific genetic differences were found that accounted for the highly aggressive nature of RDEB-associated SCC because all driver genes were shared with other SCC subtypes. In addition, no genetic elements besides COL7A1 distinguished RDEB-associated SCC from other SCC subtypes. The variants found in RDEB-associated SCC were found to be highly similar to the ones found in aged sun-exposed Caucasian skin (Cho et al, 2018).
In this study, we examined genomic mutational signatures and gene expression in a female patient with generalized-severe RDEB who presented with her seventh recurrence of SCC. In addition to the excised SCC tumor, we also evaluated nonblistered skin and blood. To investigate the mutational evolution and changes in gene expression culminating in RDEB-associated SCC in this individual, we performed whole-genome sequencing (WGS) of all 3 tissues and RNA-seq of the skin and tumor samples. Similar to previous reports, we identified enhanced SCC PLK-1 expression compared with that in the skin, raising the possibility that the PLK-1 inhibitor rigosertib might offer a therapeutic benefit. In addition, we discovered microsatellite instability (MSI) and accelerated aging as factors potentially contributing to the aggressive nature and early onset of RDEB-associated SCC.
RESULTS
Patient presentation and clinical course
The patient is a Caucasian female aged 32 years who presented with blisters at birth, prompting a diagnostic skin biopsy and genetic testing. COL7A1 gene sequencing revealed 2 compound heterozygous variants: a pathogenic variant c.1564C>T (p.Q522*) in exon 12 and a likely pathogenic variant c.3551-3T>G in intron 26 (Kern et al, 2006). Her clinical course was consistent with generalized-severe RDEB. Between the ages of 22 and 31 years, the patient developed a total of seven unique bilateral lower extremity SCCs. All were in chronic wound or blister sites: 5 hyperkeratotic in appearance and 2 erosive. All 7 were well-differentiated on histopathology, and none were metastatic. All but 1 SCC were amenable to resection with Mohs procedures and grafting with skin grafts. The 1 SCC not amenable to surgical resection was treated consecutively with acitretin, capecitabine, and pembrolizumab. The patient developed osteomyelitis in the area adjacent to the SCC and required below-the-knee amputation. The seventh SCC examined in greater depth in this study arose on the left dorsal foot and was treated with Mohs resection and xenograft placement (Figure 1a). Histopathological analysis of the tumor at the time of initial biopsy revealed irregular epidermal hyperplasia with pale-staining epithelium and mild keratinocyte atypia, an endophytic and undermining architectural growth pattern, and dermal islands of mildly atypical keratinizing squamous epithelium (Figure 1b). Underlying dermal fibrosis consistent with scarring from RDEB was also present. The final diagnosis was of well-differentiated SCC with features typical to those seen in patients with RDEB extending to the deep and lateral margins. At the time of the initial biopsy, 3 tissue samples of blood, blister-adjacent skin, and the SCC were taken for WGS and RNA-seq analyses (Figure 1c).
Figure 1. Clinical images and histology for the patient RDEB-associated SCC and overview of analysis approach.
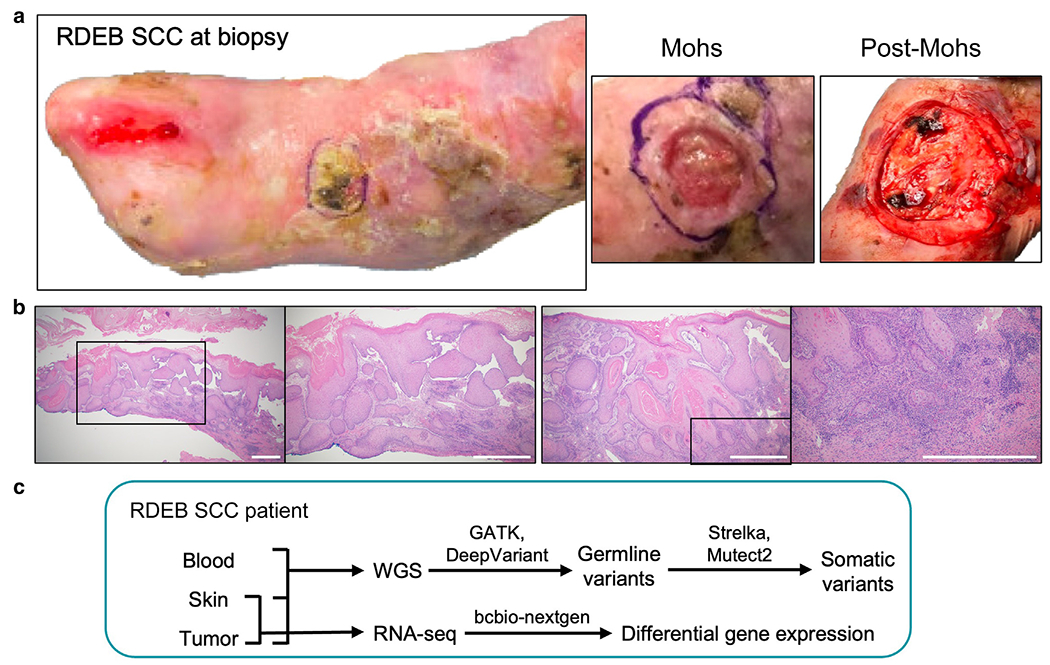
(a) Photographs of the patient RDEB-associated SCC at the time of biopsy, at Mohs and post-Mohs procedure. (b) Representative H&E images of the biopsied SCC shown at ×20 and ×40 magnification (left panel) and ×40 and ×100 magnification (right panel). Black squares indicate the areas shown at the higher magnification in the adjacent images. Bar = 100 μm. (c) Schematic illustration of experimental design: blood, skin, and SCC tumor tissues were collected at the time of surgery and analyzed using WGS and RNA-seq. RDEB, recessive dystrophic epidermolysis bullosa; RNA-seq, RNA sequencing; SCC, squamous cell carcinoma; WGS, whole-genome sequencing.
Detection of COL7A1 variants by WGS
WGS was performed for the blood, skin, and tumor samples, and the WGS reads were visualized using Integrative Genomics Viewer. An overview of WGS mapping and coverage is provided in Table 1. As expected, WGS analyses detected the 2 COL7A1 variants found by the germline genetic testing at birth (Figure 2a), and these variants were present in blood, skin, and tumor (Figure 2b). The c.1564C>T variant is a nonsense pathogenic variant that changes glutamine to a premature termination codon (p.Q522*) in exon 12. The second, the c.3551-3T>G likely pathogenic variant, is predicted to affect an upstream splice site (Kern et al, 2006).
Table 1.
Summary Statistics from WGS of Blood, Skin, and Tumor Tissues Obtained from the Patient with RDEB
| Sample | Coverage | Total Reads | Genome Territory | Number of SNV | Number of MNV | Number of Insertions | Number of Deletions | Number of Mixed |
|---|---|---|---|---|---|---|---|---|
| Blood | 35.80× | 716,133,302 | 2,864,957,046 bp | 3,708,264 | 147,308 | 362,587 | 396,283 | 69,903 |
| Skin | 42.65× | 852,532,666 | 2,864,957,046 bp | 3,740,217 | 148,901 | 389,199 | 420,041 | 74,853 |
| SCC | 81.95× | 1,639,133,214 | 2,864,957,046 bp | 3,685,080 | 152,000 | 407,568 | 436,358 | 78,920 |
Abbreviations: MNV, multiple nucleotide variant; RDEB, recessive dystrophic epidermolysis bullosa; SCC, squamous cell carcinoma; WGS, whole-genome sequencing.
Figure 2. Confirmation of COL7A1 pathogenic genetic variants by WGS.
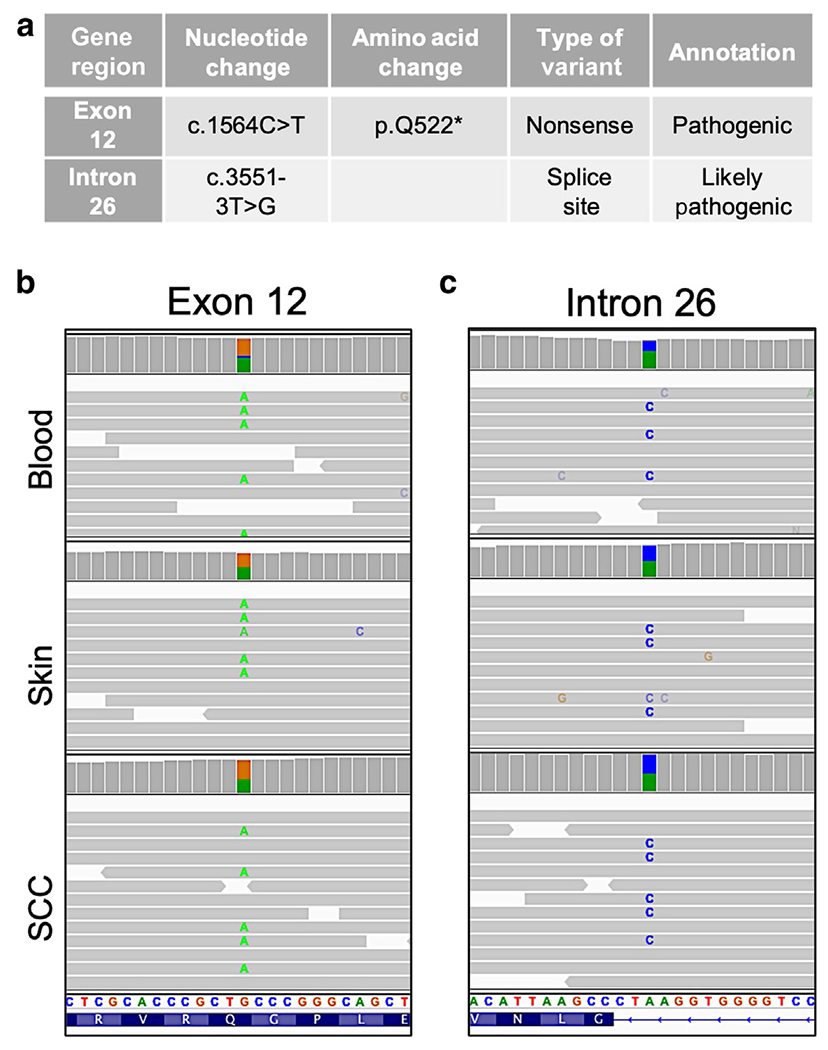
(a) The table depicts the COL7A1 variants diagnosed by clinical genetic testing. IGV plot illustrates (b) c.1564 C>T variant in exon 12 and (c) c.3551-3T>G variant in intron 26. IGV, Integrative Genomics Viewer; SCC, squamous cell carcinoma; WGS, whole-genome sequencing.
RNA-seq confirms abnormal splicing caused by the c.3551-3T>G COL7A1 variant
At the time of initial diagnosis, the pathogenicity and role of the c.3551-3T>G COL7A1 variant in aberrant RNA splicing could only be suggested by 2 splicing prediction algorithms (Kern et al, 2006). In our study, using the RNA-seq data from the skin and tumor samples visualized in Integrative Genomics Viewer with a sashimi plot, we documented that the c.3551-3T>G COL7A1 variant in intron 26 results in the inclusion of intron 26 and intron 27 in the final transcript (Figure 3a). To determine whether normally spliced transcripts are derived from the allele with the c.3551-3T>G variant, we used SpliceAI (Jaganathan et al, 2019). This algorithm returned an acceptor loss of 0.82, strongly predicting the occurrence of some regular splicing from this allele. However, because the acceptor loss score was not 1, long-read RNA-seq would be needed to confirm that the normally spliced transcripts observed in the sashimi plot are not derived from the opposite allele.
Figure 3. RNA-seq analyses of the patient skin and SCC samples.
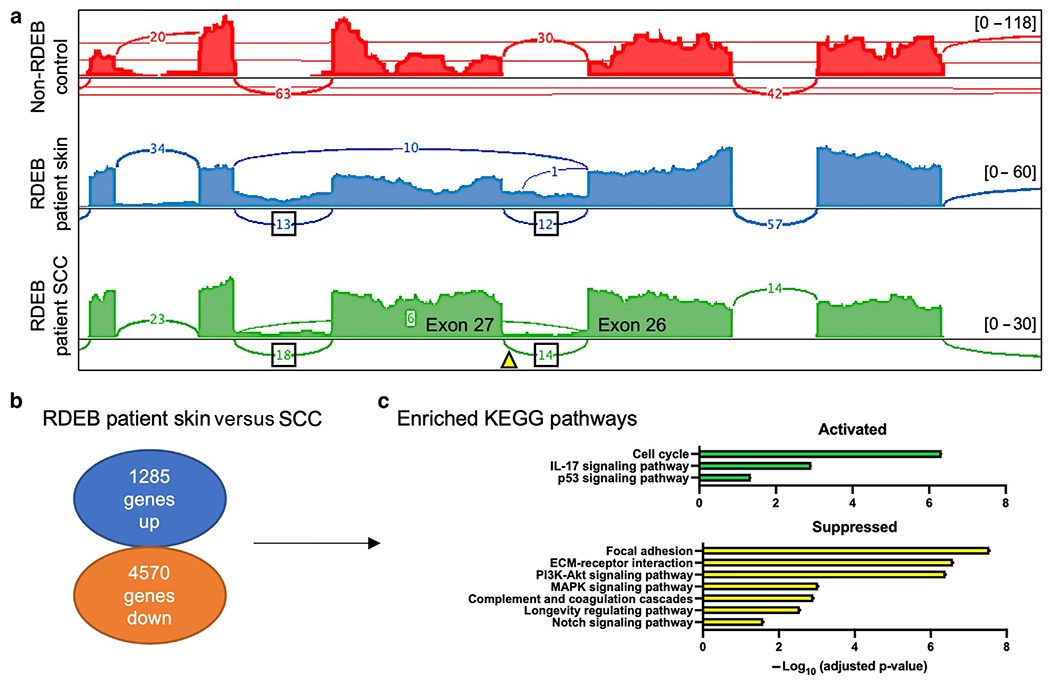
(a) Read coverages are represented as histograms for normal skin (red), RDEB skin (blue), and RDEB-associated SCC tumor (green). The arch depicts the splicing sites, and the number in the arch indicates RNA-seq reads in this region. The triangle indicates the position of the c.3551-3T>G variant. Track heights are indicated in brackets. (b) Fold change analysis of gene expression in the RDEB-associated SCC tumor compared with that in the skin. (c) KEGG pathways enriched among the differentially expressed genes. KEGG, Kyoto Encyclopedia of Genes and Genomes; RDEB, recessive dystrophic epidermolysis bullosa; RNA-seq, RNA sequencing; SCC, squamous cell carcinoma.
RNA-seq reveals therapeutically targetable genes and pathways
Comparative RNA-seq analysis identified 5855 differentially expressed genes in the patient SCC compared with those in the skin, of which 1285 demonstrated increased mRNA levels, and 4570 demonstrated decreased mRNA levels (Figure 3b). Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis revealed enrichment of cell cycle (adjusted P = 4.71 × 10−7), IL-17 signaling (adjusted P = 0.0012), and p53 signaling (adjusted P = 0.045) pathways and suppression of the focal adhesion (adjusted P = 2.86 × 10−8), extracellular matrix–receptor interaction (adjusted P = 2.62 × 10−7), phosphoinositide 3-kinase–Akt signaling (adjusted P = 4.06 × 10−7), MAPK signaling (adjusted P = 0.0009), complement and coagulation cascades (adjusted P = 0.0012), longevity regulating (adjusted P = 0.0028), and NOTCH signaling (adjusted P = 0.026) pathways (Figure 3c) (complete list from Kyoto Encyclopedia of Genes and Genomes analysis is shown in Supplementary Figure S1). Among the upregulated genes, we observed a 6.56-fold increase in PLK1 expression in the tumor sample compared with that in the skin. This finding might be of clinical relevance because PLK1 inhibitors are now being investigated in clinical trials for the treatment of solid tumors (Hagege et al, 2021).
RDEB-associated SCC somatic variants identified by WGS
The WGS of blood, skin, and SCC generated 716,133,302; 852,532,666; and 1,639,133,214 reads, resulting in 35.80-fold, 42.65-fold, and 81.95-fold coverage, respectively (Table 1). A pairwise comparison revealed a similar non-synonymous mutational burden in the SCC tumor compared with that in blood or skin (0.260 per Mbp or 0.263 per Mbp, respectively), whereas a significantly lower mutation burden was observed in skin compared with that in blood (0.027 per Mbp) (Supplementary Table S1). Examination of the proband SCC tumor mutational profile, constructed by subtracting the somatic mutations in the skin from the somatic mutations in the tumor, revealed variants in the NOTCH, p53, SWI/SNF, Hippo, cell stress, and Ras MAPK/phosphoinositide 3-kinase pathways, whereas no variants were seen in the Rb pathway (Figure 4a). Variants in these pathways have been reported by Chang and Shain (2021) as the key driver mutations in RDEB-associated SCC. Specifically, we detected 2 missense variants in NOTCH1—c.5976G>T (p.M1992I) and c.928G>T (p.G310W)—that were also reported in the study by Cho et al (2018) as well as an intragenic variant in NOTCH2. In addition, we found a missense variant in USP28, c.2682G>T (p.L894F), and an intragenic variant in MDM2, both components of the p53 pathway involved in DNA repair (Eischen, 2017; Lambrus et al, 2016). We also discovered variants in the members of the Hippo signaling pathway, including 2 in FAT1—c.9695_9696insGTA (p.3231_3232ins*) and c.283T>G (p.F95V)—also seen in the cohort of patients studied by Cho et al (2018) as well as an intragenic variant in YAP1. In the cell stress pathway, we detected a missense variant, variant c.339C>A (p.S113R), in CASP8, a mediator of apoptosis during inflammation, previously noted as enriched in RDEB-associated SCC compared with that in other SCC types (Chang and Shain, 2021).
Figure 4. WGS analyses of the patient’s skin and SCC.
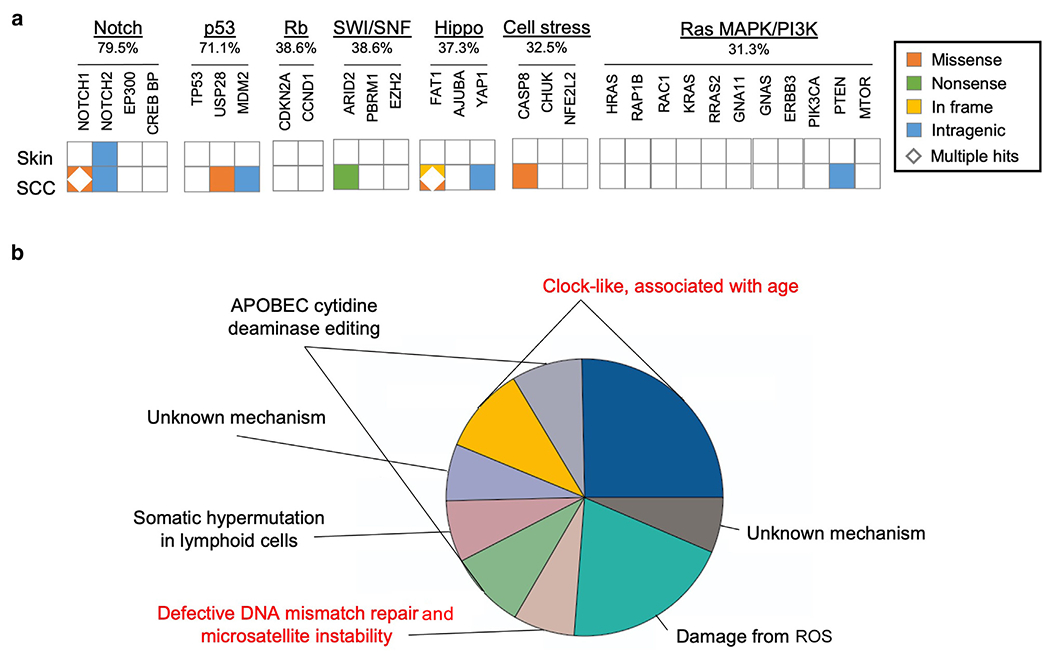
(a) Tiling plot of somatic variants detected in RDEB skin and SCC grouped by oncogenic pathways. The percentages of SCC samples with a variant in the given pathway were reported by Chang and Shain (2021). (b) Pie chart showing the proportions of single-nucleotide variants corresponding to specific COSMIC mutational signatures. The previously unreported defective DNA mismatch repair and microsatellite instability and clock-like associated with age signatures are highlighted in red. COSMIC, Catalogue of Somatic Mutations in Cancer; PI3K, phosphoinositide 3-kinase; RDEB, recessive dystrophic epidermolysis bullosa; SCC, squamous cell carcinoma; WGS, whole-genome sequencing.
Mutational profiling of RDEB-associated SCC
Mutational signatures represent unique sequence motifs found in cancer genomes that are associated with different mutational processes. Using the Catalogue of Somatic Mutations in Cancer SigProfiler search tool, we detected 9 signatures in the patient’s SCC compared with that in the skin. Specifically, we identified the clock-like associated with age (signatures 1 and 5), APOBEC cytidine deaminase editing (signatures 2 and 13), a defective DNA mismatch repair and MSI (signature 15), a damage from ROS (signature 18), and a somatic hypermutation in lymphoid cells (signature 9) signatures (Figure 4b). The clock-like signatures, the APOBEC signatures, and the damage from ROS signature were also observed in the Cho et al (2018) patient cohort. We did not detect signature 7 associated with UV damage in our tumor sample, whereas this signature was found by Cho et al (2018) to contribute to more than a third of their RDEB-associated SCC mutational signature profiles in some patients. However, our study uncovered the defective DNA mismatch repair and MSI signature (signature 15), which was not previously reported in RDEB-associated SCC.
Detection of high MSI in RDEB-associated SCC
DNA mismatch repair plays an important role in excising DNA mismatch errors that occur during DNA replication. Defects in mismatch repair lead to MSI resulting in increased mutational burden and enhanced tumor immunogenicity (Kauffmann et al, 2008; Wagner et al, 2016; Wilczak et al, 2017). High MSI has been previously reported in non-RDEB cutaneous SCC (Chang and Shain, 2021) and head and neck SCC (Cilona et al, 2020) but not in RDEB-associated SCC. Using the RNA-seq–based PreMSIm algorithm, we found that in our patient, the SCC tumor sample was classified as MSI high, whereas the skin sample was MSI low. RNA-seq analyses revealed increased expression of DNA mismatch repair–related transcripts such as MSH6, MLH3, RFC, PCNA, EXO1, and POLQ in RDEB-associated SCC compared with those in the skin. Interestingly, high genomic instability was also observed in the Cho et al (2018) cohort. However, the authors concluded that accelerated mutagenesis was driven by APOBECs and not defective DNA repair. High MSI status of the patient tumor might provide additional treatment options such as immunotherapy, which was approved by the United States Food and Drug Administration for patients with MSI-high tumors regardless of tumor type and tumor PD-L1 expression levels (Karpel et al, 2023).
Accelerated aging seen in both RDEB skin and SCC
To explore the functional impact of the variants identified in the patient’s SCC, we correlated 10,085 somatic variants with 5885 differentially expressed transcripts in the tumor compared with those in the skin. We identified 748 differentially expressed genes with somatic variants. To further explore the relevance of these findings to RDEB pathological processes, we compared these data with 3444 previously described differentially expressed transcripts from patients with RDEB and their age- and sex-matched (sex referring to the biological categories of female or male) healthy controls (Breitenbach et al, 2015). We identified 80 transcripts that were shared between these datasets (Figure 5a). Gene Ontology analyses of these 80 transcripts revealed significant enrichment of categories such as positive regulation of cellular senescence (Gene Ontology: 2000774, adjusted P = 0.022) and positive regulation of cell aging (Gene Ontology: 0090343, adjusted P = 0.024), which included the genes ARG2 and HMGA2 (Figure 5b). ARG2 regulates nitric oxide synthesis and plays a role in promoting cell senescence and inflammation (Yepuri et al, 2012). HMGA2 promotes cancer cell proliferation, inhibits apoptosis, and influences DNA damage repair mechanisms (Mansoori et al, 2021).
Figure 5. Detection of age-related transcriptional changes in patient skin and SCC.
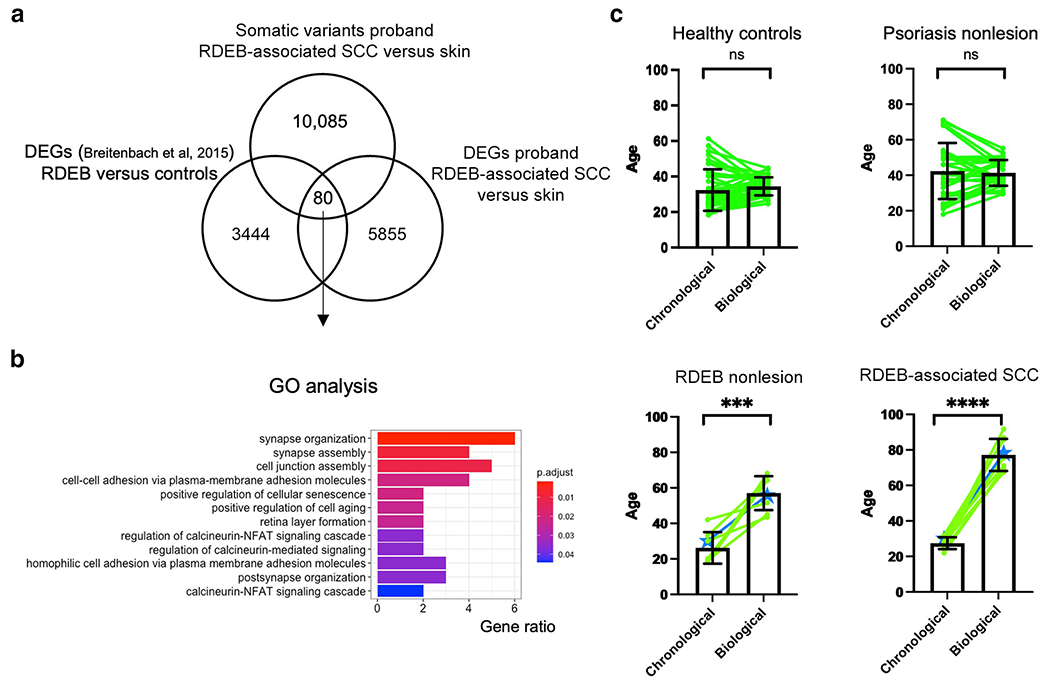
(a) Overlap between somatic variants from comparing WGS tumor with skin (our patient, n = 1), differentially expressed genes upregulated when comparing RNA-seq of tumor with skin (our patient, n = 1), and differentially expressed genes upregulated when comparing microarray of RDEB with normal controls (n = 3 for each). (b) GO analysis on 80 overlapping genes from a. (c) Comparison of chronological with biological age for healthy control skin (n = 38), nonlesional skin from patients with psoriasis (n = 27), nonlesional skin from patients with RDEB (n = 8), and RDEB-associated SCC tumors (n = 9). Mean ± SD is shown. Pairing of samples is depicted in green; discovery patient samples are in blue. GO, Gene Ontology; ns, not significant; RDEB, recessive dystrophic epidermolysis bullosa; RNA-seq, RNA sequencing; SCC, squamous cell carcinoma; WGS, whole-genome sequencing.
Diverse environmental and genetic factors may result in differences in an individual’s chronological and biological age. On the basis of the RNA-seq results described earlier and our detection of the clock-like associated with age mutational signatures (signatures 1 and 5) in the patient tumor sample, we hypothesized that RDEB might lead to accelerated aging in skin and SCC. Recent studies have shown that biological age can be determined using a curated set of DNA methylation marks (Bernabeu et al, 2023; Horvath, 2013) or RNa expression data using RNAAgeCalc (Ren and Kuan, 2020). RNAAgeCalc is a tissue-specific transcriptional age calculator trained on RNA-seq data from healthy individuals. We employed RNAAgeCalc to examine the biological age of our patient’s skin and tumor samples. Because the training dataset for RNAAgeCalc only included individuals between the ages of 18 and 65 years, we limited the RDEB and RDEB-associated SCC patient samples we curated (Chacón-Solano et al, 2019; Cho et al, 2018) to the same age group. As a comparison, we used RNA-seq data from healthy control skin and from nonlesional psoriasis skin (another inflammatory skin disease) from a previously published study (Tsoi et al, 2019). A highly statistically significant difference was seen when chronological age was compared with biological age for the RDEB skin (P = 0.007) (chronological age = 26.25 ± 8.86 years, biological age = 57.11 ± 9.55 years) and RDEB-associated SCC (P < 0.001) (chronological age = 27.44 ± 3.32 years, biological age = 77.19 ± 9.04 years) samples but not for the healthy control skin (P = .25) (chronological age = 32.63 ± 11.64 years, biological age = 34.70 ± 5.12 years) or nonlesional psoriasis skin (P = 0.67) (chronological age = 42.41 ± 15.80 years, biological age = 41.36 ± 7.23 years) samples (Figure 5c). These results potentially implicate accelerated biological aging in RDEB-associated skin disease and SCC development.
DISCUSSION
In this study, we employed integrated somatic and germline WGS and transcriptomic analyses across 3 different tissues in a single patient with RDEB to gain insight into possible patient-specific mechanisms of RDEB-associated SCC progression and potential therapeutic options for our discovery patient. We confirmed that the c.3551-3T>G COL7A1 variant results in abnormal splicing, as was previously predicted (Kern et al, 2006). The observation of high PLK1 RNA expression in the patient’s SCC points to the potential role of the clinically approved PLK1 inhibitor, rigosertib.
WGS of the skin and SCC from this patient revealed non-synonymous variants in the NOTCH, p53, SWI/SNF, Hippo, cell stress, and Ras MAPK/phosphoinositide 3-kinase pathways. This was echoed by the RNA-seq analysis, where the p53 signaling pathway was activated, and the NOTCH, MAPK, and phosphoinositide 3-kinase/Akt signaling pathways were suppressed, according to Kyoto Encyclopedia of Genes and Genomes enrichment analysis. Cho et al (2018) noted that RDEB-associated SCC develops at much lower mutation burden rates than UV-induced SCC but acquires similar driver mutations. In our study, we also observed a relatively low tumor mutation burden rate on the basis of the somatic variants detected by WGS. It is conceivable that the inflammatory microenvironment and increased cell turnover resulting from excessive wound healing accelerates tumor progression regardless of the relatively low number of non-synonymous variants.
WGS identified several mutational signatures that have also been reported in other RDEB-associated SCC cases (Cho et al, 2018). We detected an additional signature, defective DNA mismatch repair and MSI, which was in line with the MSI-high status of SCC by transcriptional analysis. This observation suggests potential therapeutic options such as the use of the PD-1 inhibitor Keytruda, which was recently approved by the United States Food and Drug Administration for the treatment of solid tumors with high MSI status regardless of the tumor PDL-1 expression levels (Karpel et al, 2023; Migden et al, 2018; Sharon and Bell, 2022). Notably, another PD-1 inhibitor, cemiplimab, has already been successfully used in a male patient with RDEB aged 32 years with metastatic SCC (Khaddour et al, 2020).
From the original RNAAgeCalc publication (Ren and Kuan, 2020), the authors noted that transcriptional age is associated with mutation burden in some The Cancer Genome Atlas cancers. However, the relatively low mutation burden rate in our discovery patient and other patients with RDEB implies that accelerated aging as measured by the transcriptional age calculator RNAAgeCalc is not simply attributed to tumor mutation burden for RDEB-associated SCC. Our previously unreported finding that biological age is accelerated in RDEB skin and RDEB-associated SCC has several implications. Notably, the accelerated biological age of skin from individuals with RDEB (57.11 years) and those with RDEB-associated SCC tumors (77.16 years) is comparable with the age of onset for UV-induced SCC (70 years [Garcovich et al, 2017]) and head and neck SCC (66 years [Johnson et al, 2020]) in the general population, which might explain the extremely early onset of SCC seen in patients with RDEB.
In summary, our study highlights the importance of an integrated, multiomics approach to caring for patients with rare disorders such as RDEB. Owing to limited patient availability, clinical trials investigating potential therapies are relatively scarce in these populations. Multitissue multiomics analyses in a single patient may help point to new therapeutic avenues to explore.
MATERIALS AND METHODS
Study approval
Biospecimen collection was completed with written informed patient consent on a University of Minnesota Institutional Review Board–approved protocol (MT2013-01R). Evaluation of the specimens was determined not to constitute human subject research by the same Institutional Review Board, although the patient provided written informed consent for use of medical photography and histopathology images in this manuscript.
WGS and variant calling
WGS was performed on the Broad Institute Genomics Platform. Reads were aligned to the hg19 genome build. Integrative Genomics Viewer (Robinson et al, 2011) (version 2.11.2) was used for sequencing visualization. Germline variants were called using the GATK common practices workflow (DePristo et al, 2011) and Freebayes (Garrison and Marth, 20121) (version 1.3.2). Genetic variants were filtered at a minimum quality score of 20 using vcftools (Danecek et al, 2011) (version 0.1.16). Variant annotations and functional effect predictions were obtained using snpEff (Cingolani et al, 2012) (version 3.6). Somatic single-nucleotide variations and insertions/deletions in tumor against skin, tumor against blood, and skin against blood were detected using MuTect1 (Cibulskis et al, 2013) and Strelka (Saunders et al, 2012).
RNA-seq
RNA-seq was performed on the Broad Institute Genomics Platform. Paired-end reads were aligned to hg19/build 37 using Burrows-Wheeler Aligner (Li and Durbin, 2009). To generate FASTQ files, the BAM files were sorted using bio-samtools (version 1.43) sort -n -o followed by bedtools (version 2.27.1) bamtofastq (version 1.3.2) conversion using -fq2 to output paired-end FASTQ files. Samtools (Li et al, 2009) (version 1.9) was used for bam file sorting and indexing. Integrative Genomics Viewer (Robinson et al, 2011) (version 2.11.2) was used for sequencing visualization and sashimi plot generation. FASTQ files from a normal skin sample were downloaded from the National Center for Biotechnology Information Sequence Read Archive (GSM4127073) as a control for the splice junction sashimi plot. The FASTQ files were processed using the bcbio-nextgen pipeline (Ewels et al, 2016) from the Harvard Bioinformatics Core. Within this pipeline, quality control was performed using fastqc (version 0.11.5), and sequence reads were aligned to the human reference genome (hg19) using STAR (Spliced Transcripts Alignment to a Reference) (Dobin et al, 2013) (version 2.5.2b). Log2 fold change > 1.5 in gene expression between tumor and skin was used as a cutoff for differentially expressed genes.
Mutational signature analysis
Mutational signature analysis was conducted on nonsynonymous SNVs in tumor against skin in the R environment using deconstructSigs (Rosenthal et al, 2016) (version 1.8.0). The proportion of mutations in each of the possible 96 trinucleotide contexts was calculated to create a sample signature profile (Supplementary Figure S2). A multiple linear regression model was fitted to determine the linear combination of Catalogue of Somatic Mutations in Cancer consensus mutational signatures.
MSI prediction
MSI status was predicted on the basis of mRNA gene expression using PreMSIm (Li et al, 2020) (version 1.0). Transcripts Per Kilobase Million–normalized values of SCC and normal skin RNA-seq datasets, along with a training dataset of 1383 pancancer RNA-seq samples from The Cancer Genome Atlas, were added 1, log2 transformed, and rescaled to the range (0, 1). Each sample was classified as MSI high or MSI low on the basis of their expression of a 15-gene panel, which included the genes DDX27, EPM2AIP1, HENMT1, LYG1, MLH1, MSH4, NHLRC1, NOL4L, RNL2, RPL22L1, SHROOM4, SMAP1, TTC30A, and ZSWIM3.
Biological age calculation
RNAAgeCalc (Ren and Kuan, 2020) (version 1.2.0) was used to calculate the biological age from the non-SCC and SCC RNA-seq data from this study as well as normal skin, nonlesional psoriatic skin, RDEB non-SCC skin, and RDEB-associated SCC samples from the following National Center for Biotechnology Information Gene Expression Omnibus datasets: GSE121212, GSE111582, and GSE119501. The RNA age predictor is trained by all tissues automatically using the GTExAge signature.
Statistical analysis
To compare biological to chronological age, a paired t-test was used, with α < 0.05 used as the cutoff for significance.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Maegen Harden at the Broad Institute for assistance with data management. We would also like to thank Po-Ruh Loh and Arezou Ghazani of Brigham and Women’s Hospital for their helpful comments and discussion. This work was supported by the National Institutes of Health (NIH)/National Institute of Biomedical Imaging and Bioengineering T32EB016652-06 to CAAL, NIH/National Center for Advancing Translational Sciences KL2TR002492 and UL1TR002494 to CLE, a DoD Physician Research Award (W81XWH-21-1-0084, PC200150) and a DoD Idea Development Award - Early-Career Investigator (KC210042/W81XWH-22-1-0455) to SHA, NIH/National Institute of Arthritis and Musculoskeletal and Skin Diseases 1R01AR063070 to JT, NIH/National Eye Institute grants 1R01EY025794 and R24EY028767, NIH/National Institute on Aging grant 1P01AG071463-01A1, NIH/National Heart, Lung, and Blood Institute grant 1R01HL161087 to NYF and MHF, a VA R&D Merit Review Award (1I01RX000989), BLR&D Review Award (1I01BX006004), and a Harvard Stem Cell Institute seed grant award to NYF.
Abbreviations:
- MSI
microsatellite instability
- RDEB
recessive dystrophic epidermolysis bullosa
- RNA-seq
RNA sequencing
- SCC
squamous cell carcinoma
- WGS
whole-genome sequencing
Footnotes
Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. arXiv 2012.
CONFLICT OF INTEREST
MHF and NYF are inventors or coinventors of the United States and international ABCB5-related patents assigned to Brigham and Women’s Hospital, Boston Children’s Hospital, and/or VA Boston Healthcare System (Boston, MA) and licensed to Rheacell GmbH & Co. KG (Heidelberg, Germany). MHF serves as a scientific advisor and holds stock in Rheacell GmbH & Co. KG. The remaining authors state no conflict of interest.
SUPPLEMENTARY MATERIAL
Supplementary material is linked to the online version of the paper at http://www.jidonline.org, and at https://doi.org/10.1016/j.jid.2023.11.025.
DATA AVAILABILITY STATEMENT
The RNA-sequencing data have been deposited to the National Center for Biotechnology Information Gene Expression Omnibus under accession number GSE236849.
REFERENCES
- Bernabeu E, McCartney DL, Gadd DA, Hillary RF, Lu AT, Murphy L, et al. Refining epigenetic prediction of chronological and biological age. Genome Med 2023;15:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitenbach JS, Rinnerthaler M, Trost A, Weber M, Klausegger A, Gruber C, et al. Transcriptome and ultrastructural changes in dystrophic epidermolysis bullosa resemble skin aging. Aging (Albany NY) 2015;7: 389–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacón-Solano E, León C, Díaz F, García-García F, García M, Escámez MJ, et al. Fibroblast activation and abnormal extracellular matrix remodelling as common hallmarks in three cancer-prone genodermatoses. Br J Dermatol 2019;181:512–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang D, Shain AH. The landscape of driver mutations in cutaneous squamous cell carcinoma. NPJ Genom Med 2021;6:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho RJ, Alexandrov LB, den Breems NY, Atanasova VS, Farshchian M, Purdom E, et al. APOBEC mutation drives early-onset squamous cell carcinomas in recessive dystrophic epidermolysis bullosa. Sci Transl Med 2018;10:eaas9668. [DOI] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31:213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cilona M, Locatello LG, Novelli L, Gallo O. The mismatch repair system (MMR) in head and neck carcinogenesis and its role in modulating the response to immunotherapy: a critical review. Cancers (Basel) 2020;12: 3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics 2011;27:2156–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eischen CM. Role of Mdm2 and Mdmx in DNA repair. J Mol Cell Biol 2017;9: 69–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016;32:3047–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine JD, Johnson LB, Weiner M, Li KP, Suchindran C. Epidermolysis bullosa and the risk of life-threatening cancers: the National EB Registry experience, 1986–2006. J Am Acad Dermatol 2009;60:203–11. [DOI] [PubMed] [Google Scholar]
- Fritsch A, Loeckermann S, Kern JS, Braun A, Bösl MR, Bley TA, et al. A hypomorphic mouse model of dystrophic epidermolysis bullosa reveals mechanisms of disease and response to fibroblast therapy. J Clin Invest 2008;118:1669–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcovich S, Colloca G, Sollena P, Andrea B, Balducci L, Cho WC, et al. Skin cancer epidemics in the elderly as an emerging issue in geriatric oncology. Aging Dis 2017;8:643–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich I, Agarwal P, Zhang P, Dolorito JA, Oliver S, Liu H, et al. In vivo topical gene therapy for recessive dystrophic epidermolysis bullosa: a phase 1 and 2 trial. Nat Med 2022;28:780–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagege A, Ambrosetti D, Boyer J, Bozec A, Doyen J, Chamorey E, et al. The Polo-like kinase 1 inhibitor onvansertib represents a relevant treatment for head and neck squamous cell carcinoma resistant to cisplatin and radiotherapy. Theranostics 2021;11:9571–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S DNA methylation age of human tissues and cell types [published correction appears in Genome Biol 2015;16:96. Genome Biol 2013;14: R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovnanian A, Duquesnoy P, Blanchet-Bardon C, Knowlton RG, Amselem S, Lathrop M, et al. Genetic linkage of recessive dystrophic epidermolysis bullosa to the type VII collagen gene. J Clin Invest 1992;90:1032–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, et al. Predicting splicing from primary sequence with deep learning. Cell 2019;176:535–48.e24. [DOI] [PubMed] [Google Scholar]
- Johnson DE, Burtness B, Leemans CR, Lui VWY, Bauman JE, Grandis JR. Head and neck squamous cell carcinoma [published correction appears in Nat Rev Dis Primers 2023;9:4]. Nat Rev Dis Primers 2020;6:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpel HC, Slomovitz B, Coleman RL, Pothuri B. Treatment options for molecular subtypes of endometrial cancer in 2023. Curr Opin Obstet Gynecol 2023;35:270–8. [DOI] [PubMed] [Google Scholar]
- Kauffmann A, Rosselli F, Lazar V, Winnepenninckx V, Mansuet-Lupo A, Dessen P, et al. High expression of DNA repair pathways is associated with metastasis in melanoma patients. Oncogene 2008;27:565–73. [DOI] [PubMed] [Google Scholar]
- Kern JS, Kohlhase J, Bruckner-Tuderman L, Has C. Expanding the COL7A1 mutation database: novel and recurrent mutations and unusual genotype-phenotype constellations in 41 patients with dystrophic epidermolysis bullosa. J Invest Dermatol 2006;126:1006–12. [DOI] [PubMed] [Google Scholar]
- Khaddour K, Gorell ES, Dehdashti F, Tang JY, Ansstas G. Induced remission of metastatic squamous cell carcinoma with an immune checkpoint inhibitor in a patient with recessive dystrophic epidermolysis bullosa. Case Rep Oncol 2020;13:911–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiritsi D, Dieter K, Niebergall-Roth E, Fluhr S, Daniele C, Esterlechner J, et al. Clinical trial of ABCB5+ mesenchymal stem cells for recessive dystrophic epidermolysis bullosa. JCI Insight 2021;6:e151922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Küttner V, Mack C, Rigbolt KT, Kern JS, Schilling O, Busch H, et al. Global remodelling of cellular microenvironment due to loss of collagen VII. Mol Syst Biol 2013;9:657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrus BG, Daggubati V, Uetake Y, Scott PM, Clutario KM, Sluder G, et al. A USP28–53BP1-p53-p21 signaling axis arrests growth after centrosome loss or prolonged mitosis. J Cell Biol 2016;214:143–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/Map format and SAMtools. Bioinformatics 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Feng Q, Wang X. PreMSIm: an R package for predicting microsatellite instability from the expression profiling of a gene panel in cancer. Comput Struct Biotechnol J 2020;18:668–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansoori B, Mohammadi A, Ditzel HJ, Duijf PHG, Khaze V, Gjerstorff MF, et al. HMGA2 as a critical regulator in cancer development. Genes (Basel) 2021;12:269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migden MR, Rischin D, Schmults CD, Guminski A, Hauschild A, Lewis KD, et al. PD-1 blockade with cemiplimab in advanced cutaneous squamouscell carcinoma. N Engl J Med 2018;379:341–51. [DOI] [PubMed] [Google Scholar]
- Mittapalli VR, Kühl T, Kuzet SE, Gretzmeier C, Kiritsi D, Gaggioli C, et al. STAT3 targeting in dystrophic epidermolysis bullosa. Br J Dermatol 2020;182:1279–81. [DOI] [PubMed] [Google Scholar]
- Ng YZ, Pourreyron C, Salas-Alanis JC, Dayal JH, Cepeda-Valdes R, Yan W, et al. Fibroblast-derived dermal matrix drives development of aggressive cutaneous squamous cell carcinoma in patients with recessive dystrophic epidermolysis bullosa. Cancer Res 2012;72:3522–34. [DOI] [PubMed] [Google Scholar]
- Nyström A, Thriene K, Mittapalli V, Kern JS, Kiritsi D, Dengjel J, et al. Losartan ameliorates dystrophic epidermolysis bullosa and uncovers new disease mechanisms. EMBO Mol Med 2015;7:1211–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X, Kuan PF. RNAAgeCalc: a multi-tissue transcriptional age calculator. PLoS One 2020;15:e0237006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol 2011;29:24–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. Decon-structSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol 2016;17:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka:accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 2012;28:1811–7. [DOI] [PubMed] [Google Scholar]
- Sharon S, Bell RB. Immunotherapy in head and neck squamous cell carcinoma: a narrative review. Front oral maxillofac Med 2022;4:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siprashvili Z, Nguyen NT, Gorell ES, Loutit K, Khuu P, Furukawa LK, et al. Safety and wound outcomes following genetically corrected autologous epidermal grafts in patients with recessive dystrophic epidermolysis bullosa. JAMA 2016;316:1808–17. [DOI] [PubMed] [Google Scholar]
- Tartaglia G, Cao Q, Padron ZM, South AP. Impaired wound healing, fibrosis, and cancer: the paradigm of recessive dystrophic epidermolysis bullosa. Int J Mol Sci 2021;22:5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoi LC, Rodriguez E, Degenhardt F, Baurecht H, Wehkamp U, Volks N, et al. Atopic dermatitis is an IL-13-dominant disease with greater molecular heterogeneity compared to psoriasis. J Invest Dermatol 2019;139:1480–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner JE, Ishida-Yamamoto A, McGrath JA, Hordinsky M, Keene DR, Woodley DT, et al. Bone marrow transplantation for recessive dystrophic epidermolysis bullosa [published correction appears in N Engl J Med 2010;363:1383]. N Engl J Med 2010;363:629–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner VP, Webber LP, Salvadori G, Meurer L, Fonseca FP, Castilho RM, et al. Overexpression of MutSalpha complex proteins predicts poor prognosis in oral squamous cell carcinoma. Medicine (Baltimore) 2016;95:e3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilczak W, Rashed S, Hube-Magg C, Kluth M, Simon R, Büscheck F,et al. Up-regulation of mismatch repair genes MSH6, PMS2 and MLH1 parallels development of genetic instability and is linked to tumor aggressiveness and early PSA recurrence in prostate cancer. Carcinogenesis 2017;38:19–27. [DOI] [PubMed] [Google Scholar]
- Yepuri G, Velagapudi S, Xiong Y, Rajapakse AG, Montani JP, Ming XF, et al. Positive crosstalk between arginase-II and S6K1 in vascular endothelial inflammation and aging. Aging Cell 2012;11:1005–16. [DOI] [PubMed] [Google Scholar]
- Zhang H, Lee CAA, Li Z, Garbe JR, Eide CR, Petegrosso R, et al. A multitask clustering approach for single-cell RNA-seq analysis in recessive dystrophic epidermolysis bullosa. PLoS Comput Biol 2018;14: e1006053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-sequencing data have been deposited to the National Center for Biotechnology Information Gene Expression Omnibus under accession number GSE236849.