

Abstract
Background: Deep learning models for patch classification in whole-slide images (WSIs) have shown promise in assisting follicular lymphoma grading. However, these models often require pathologists to identify centroblasts and manually provide refined labels for model optimization. Objective: To address this limitation, we propose PseudoCell, an object detection framework for automated centroblast detection in WSI, eliminating the need for extensive pathologist's refined labels. Methods: PseudoCell leverages a combination of pathologist-provided centroblast labels and pseudo-negative labels generated from undersampled false-positive predictions based on cell morphology features. This approach reduces the reliance on time-consuming manual annotations. Results: Our framework significantly reduces the workload for pathologists by accurately identifying and narrowing down areas of interest containing centroblasts. Depending on the confidence threshold, PseudoCell can eliminate 58.18-99.35% of irrelevant tissue areas on WSI, streamlining the diagnostic process. Conclusion: This study presents PseudoCell as a practical and efficient prescreening method for centroblast detection, eliminating the need for refined labels from pathologists. The discussion section provides detailed guidance for implementing PseudoCell in clinical practice.
Keywords: Centroblast cell detection, deep convolutional neural network, follicular lymphoma, hard negative mining, morphological features
I. Introduction
Follicular lymphoma (FL) is the second most prevalent lymphoid malignancy in Western and Asian countries. It is responsible for 5--35% of non-Hodgkin lymphoma (NHL) [1], [2], [3]. Most FL carries the translocation t(14;18), which causes the overexpression of the BCL-2 protein. FL patients usually present with lymphadenopathy, infrequent B-symptoms, systemic fever symptoms, night sweats, and weight loss. The progression of a disease can be predicted using a combination of clinical and laboratory findings and the histopathological grade of the disease [4].
The World Health Organization (WHO) classification system is currently the gold standard for grading follicular lymphoma (FL). This system relies on the number of centroblast cells (CBs), large neoplastic cells, identified within a tissue sample [5]. Traditionally, pathologists manually count CBs under a microscope using hematoxylin and eosin (H&E) stained tissue sections. However, this process is time-consuming and laborious due to the vast size difference between whole slide images (WSIs) and individual cells (as illustrated in Fig. 1). Additionally, it suffers from subjectivity and variability among experts, leading to inter- and intra-observer variability ranging from 61--73% [6]. This high variability introduces sampling bias, hinders reproducibility, and ultimately impacts patient care due to a lack of consensus among pathologists [6]. Consequently, there is a crucial need to enhance the precision, reliability, and reproducibility of histological grading in FL.
Fig. 1.

Automated detection of centroblast cells in whole-slide images. The tiny red square within WSI represents the patch image. Experts examined WSI under a patch-by-patch microscope to identify CB. In contrast, our proposed model can immediately identify CB with a confidence score.
Numerous studies have proposed automated methods to localize and classify FL by using whole-slide images (WSI), scanned images from the tissue samples, aiming to facilitate the work of pathologists [7], [8], [9], [10], [11], [12], [13], [14], [15], [16], [17]. The techniques can be categorized into two groups: 1) machine learning (ML)-based approaches with human-engineered features [7], [8], [9], [10], [11], [12], [13] and 2) deep learning (DL)-based approaches [14], [15], [16], [17]. ML-based approaches have been explored for classifying and detecting CBs. However, their performance was often limited by the reliance on hand-engineered features, particularly those based on color distributions and morphological characteristics. This approach can lead to overfitting, high false-positive (FP) prediction rates, and difficulties in generalizing to new datasets [8], [9], [10], [11], [12]. The heavy dependence on the specific combination of chosen features further restricts model performance. Consequently, recent research has shifted towards a DL-based approach, which eliminates the need for hand-engineered features and can automatically extract essential features from the training data.
DL-based models, especially Convolutional Neural Networks (CNN), have been recently applied to detect and classify lymph nodes on H&E-stained WSIs. To detect lymphocytes in breast cancer (BC), Liu et al. [14] addressed the tumor class imbalance problem by applying random sampling and data augmentation on patches (i.e., cropped images from WSI) before training the InceptionV3 [18]. Their method achieved the best sensitivity on the Camelyon16 dataset. Then, Lu et al. [15] proposed an automated pipeline to achieve a robust model for a new cohort. Their approach employed cascade training on a U-Net architecture [19], involving an iterative process of model fine-tuning. Specifically, the model was initially trained on a source dataset and then fine-tuned on the new cohort using its predicted lymphocyte masks. These masks were subsequently evaluated and refined by pathologists before being used for further model training. This cascade training process was repeated for two iterations, ultimately yielding a model with an F1-score of 0.927. However, while demonstrating promising results, this method introduces additional workload for pathologists during the mask refinement stage, which contradicts the goal of reducing their workload.
In contrast to BC research, where DL has been extensively explored for WSI analysis, most DL studies in FL have primarily focused on patch-level classification, specifically identifying whether patches contain CBs. This patch-level approach offers limited interpretability for directly grading FL. Somaratne et al. [16] addressed this by proposing a one-class training approach to minimize the generalization gap between two FL datasets. Their method involved incorporating images from the target dataset into the training set and then applying transfer learning with AlexNet [20]. This resulted in a 12% improvement in patch classification accuracy compared to training from scratch. Syrykh et al. [17] employed a CNN-based model to differentiate between FL and follicular hyperplasia (FH) at four different resolutions. While their model achieved accurate patch-level classification at the highest resolution, the study also highlighted the sensitivity of DL approaches to pre-processing steps, particularly stain normalization (SN). This was evidenced by a significant drop in the area under the curve (AUC) from 0.92--0.99 on the internal dataset to 0.63-0.69 on an external dataset. This accentuates the importance of stain normalization in the pre-processing phase for robust and generalizable DL models in FL.
According to the limitations mentioned above: (1) Deep Learning (DL) is sensitive to the variation of stain color in WSIs; (2) the need for expert-refined labels during training, and (3) class imbalance between CB and non-CB cells. These limitations restricted DL's improvement on FL WSIs to cell-level prediction.
To overcome these limitations, we proposed a framework called PseudoCell to explore the feasibility of DL-based object detection models on CB detection tasks. We aim to use the state-of-the-art object detection model, YOLOv8 [21], as our backbone model. Firstly, we compare the consistency of two Stain Normalization (SN) methods on our dataset to prevent the effect of color variation from WSI. Secondly, the need for expertly refined labels during training will be imitated through the hard negative mining technique (HNM) [22], i.e., retrieving false-positive (FP) predictions from the trained model, afterward incorporating them into the training set as pseudo-negative labels (non-CB class), then use the new training set to train a new model. Since the number of pseudo-negative labels is higher than the number of CB labels from pathologists, the imbalance class issue must be addressed before incorporating pseudo-negative labels. Thirdly, three distinct undersampling approaches were explored to mitigate the class imbalance issue before incorporating pseudo-negative labels into the training set.
To our knowledge, HNM was initially introduced in the field of computer vision and has yet to be utilized in the context of digital histopathological image recognition. While previous work on cancer cells sought refined labels from experts to enhance the model, we instead attempted to imitate it through the HNM. This framework allows us to improve the model autonomously without relying on additional work from pathologists. Therefore, the comparison between different HNM approaches was mainly investigated.
Lastly, we have provided a practical guideline based on high-power field selection and CB identification in WSI for applying our PseudoCell as a pre-screening tool for FL patients. Integrating this framework with histopathological workflow can reduce experts' workload by narrowing down the region experts focus on while examining the tissue. Potential real-world applications (such as quality control, training, and education tools) are also discussed to benefit human-machine collaboration.
II. Materials and Methods
A. Data Collection
This study included 75,245 patches (512x512 pixels) of follicular lymphoma (FL) admitted for treatment at the Faculty of Medicine Siriraj Hospital between 2016 and 2020. No significant correlation between clinicopathological parameters was observed (data not shown). The Siriraj Institutional Review Board (SIRB) (MU-MOU CoA No. 973/2020) has approved the procedures for obtaining and using tissue. Formalin-fixed paraffin-embedded (FFPE) tissue samples with a thickness of 3-5 microns were prepared for automated hematoxylin and eosin (H&E) staining and scanned at a resolution of 0.12 microns per pixel using a 3Dhistech Panoramic 1000 microscope with a 40x objective lens. The resulting images were saved in NRXS format.
From a total of 75,245 patches, 1203 patches contain Centroblast (CB) cells, and 3045 patches without CB were selected and annotated by a consensus of two doctors (one of them is a pathologist). The annotation is manually drawn around CB as a bounding box (bbox), Fig. 2(a).
Fig. 2.
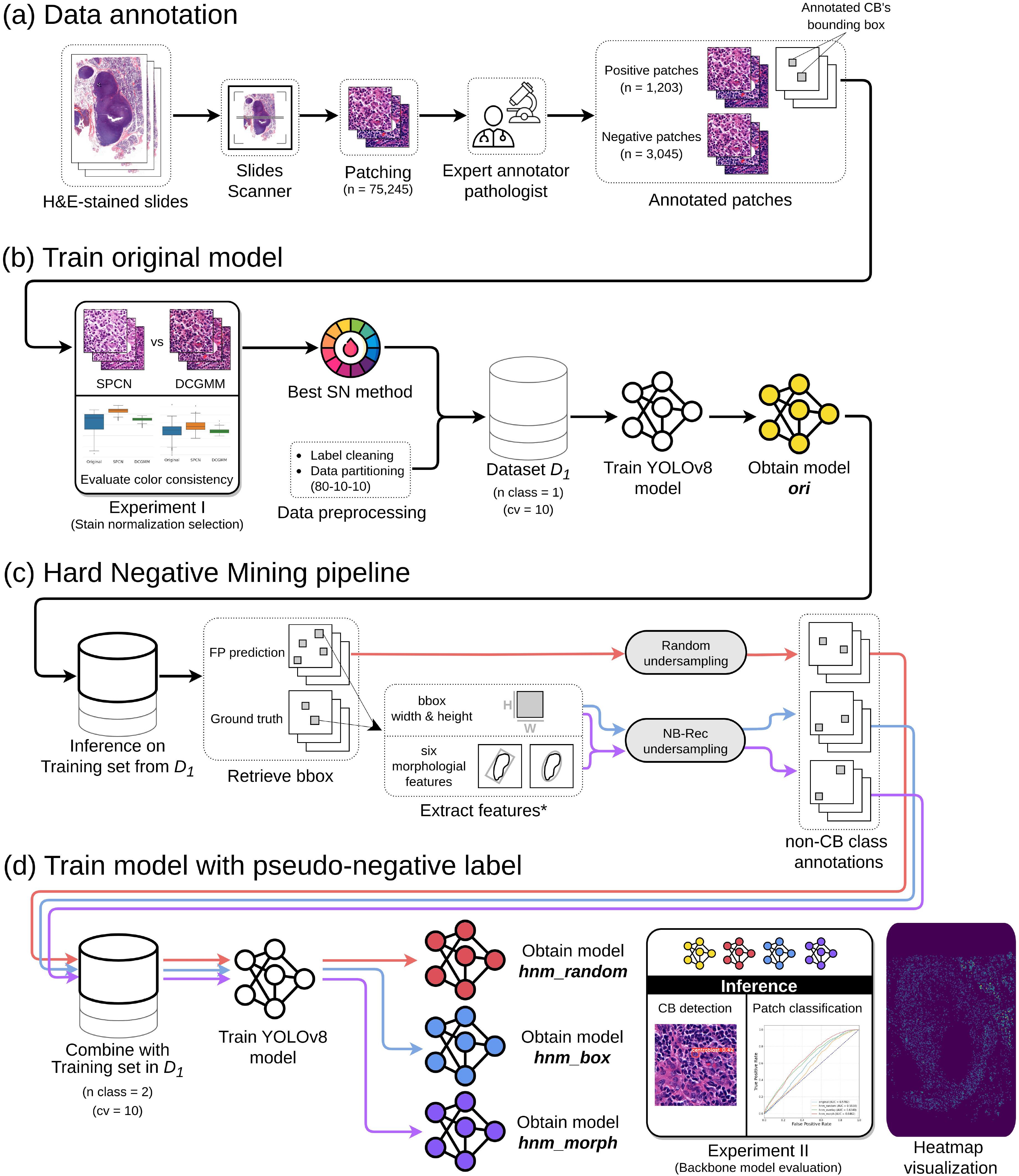
Overview of the PseudoCell framework. (a) Data annotation: H&E-stained slides were scanned and patched into 512x512 pixels. An expert pathologist annotated centroblast (CB) cells on selected patches by locating a rectangle bound to each CB while another expert pathologist reviewed the annotations. (b) Train original model: The CB-annotated patches were cleaned and then normalized the color using the best stain normalization method from Experiment I. Then, 80%, 10%, and 10% of the data were separated into train, validation, and test sets to create dataset 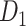 . Five methods augmented the training set before being fed into YOLOv8. YOLOv8 was trained and validated in a 10-fold cross-validation manner to generate ori model. (c) Hard Negative Mining pipeline: The ori model was then applied to infer the training set to retrieve false-positive (FP) samples. We then employed three undersampling strategies (red, blue, and purple paths) to avoid the imbalance class issue. *: the six morphological features [28] were calculated using a binary image segmented by a trained HoverNet model [29]. (d) Train model with pseudo-negative label: The undersampled FP samples from each path were combined with the training set of dataset
. Five methods augmented the training set before being fed into YOLOv8. YOLOv8 was trained and validated in a 10-fold cross-validation manner to generate ori model. (c) Hard Negative Mining pipeline: The ori model was then applied to infer the training set to retrieve false-positive (FP) samples. We then employed three undersampling strategies (red, blue, and purple paths) to avoid the imbalance class issue. *: the six morphological features [28] were calculated using a binary image segmented by a trained HoverNet model [29]. (d) Train model with pseudo-negative label: The undersampled FP samples from each path were combined with the training set of dataset 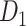 as a new class. Consequently, each path had its own training set of two classes with identical validation and test sets from
as a new class. Consequently, each path had its own training set of two classes with identical validation and test sets from 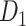 . YOLOv8 was trained and validated with similar manner as ori model to obtain hnm_random, hnm_box, and hnm_morph models. Finally, all models were compared in Experiment II, and the best approach was applied to visualize the heatmap of WSI.
. YOLOv8 was trained and validated with similar manner as ori model to obtain hnm_random, hnm_box, and hnm_morph models. Finally, all models were compared in Experiment II, and the best approach was applied to visualize the heatmap of WSI.
B. The Proposed Framework
Based on the challenge of CB cell detection, we proposed a framework in Fig. 2(b)–2(d) that gives reproducible cell-level predictions. Our proposed framework comprises three parts: 1) Train original model, 2) Hard Negative Mining pipeline, and 3) Train model with pseudo-negative label.
1). Train Original Model
As shown in Fig. 2(b), three steps comprise this part to obtain a one-class dataset and a CB detection model: 1.1) Stain normalization selection; 1.2) Data preprocessing; 1.3) Model training.
a). Stain normalization selection
Even though our WSIs came from the same lab and scanner, the WSIs still have different stain colors. So, Stain normalization was applied to our preprocessing step.
Stain normalization (SN) is the color distribution transformation from a source image 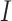 into a target image
into a target image 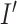 . The transformation can be described through the operation
. The transformation can be described through the operation 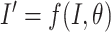 where
where 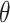 is a collection of parameters derived from the template image, and
is a collection of parameters derived from the template image, and 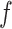 is the function that maps the visual appearance of a given image
is the function that maps the visual appearance of a given image 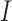 to the template image. Generally,
to the template image. Generally, 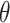 is designed to capture the color information of the primary stain components (e.g., hematoxylin and eosin). Consequently, stain-normalized images will have a color distribution similar to the template image [23].
is designed to capture the color information of the primary stain components (e.g., hematoxylin and eosin). Consequently, stain-normalized images will have a color distribution similar to the template image [23].
In this work, we consider two state-of-the-art SN methods:
-
•
Structure Preserving Color Normalization (SPCN): Vahadane et al. proposed in [24], which tackled the stain separation problem with the assumption that stain density is non-negative, and the color basis is sparse. The sparseness constraint reduces the solution space of the color decomposition problem. Then, the color basis of a source image is replaced with those from a template image while maintaining its original stain concentrations.
-
•
Deep convolutional Gaussian mixture models (DCGMM): Zanjani et al. proposed in [25]. This method first converts the source image into the HSD color system. Then, a GMM is fitted to the color distribution individually per tissue class. To train the DCGMM, E-step and M-step of the EM-algorithm are replaced by gradient descent and the back-propagation algorithm. The advantage of this approach is that it does not need any assumptions about the H&E image content.
We conduct an experiment, detailed in Section II-C1, to compare and select the most appropriate SN method for our dataset (i.e., one that produces processed images with low color variation and minimal background error).
b). Data preprocessing
Due to the considerable human errors during annotation, label cleaning was necessary before feeding data into the model. Our dataset's two most prevalent errors were 1) bbox annotations with zero areas and 2) repeated bbox annotations on a single CB cell. Since the annotator may have accidentally generated a bbox with zero areas by clicking the mouse, we removed all box annotations with zero areas from our dataset. Regarding the second error, we first calculated the center of each bbox and then retrieved the groups of bounding boxes whose center-to-center distance is within a constant. If bbox annotations share the same CB cell, we select the bbox that best fits the cell based on manual inspection of each bbox group.
Then, we will apply the stain normalization method from the previous experiment to the annotated positive patches to standardize our dataset's color variation. Lastly, 80%, 10%, and 10% of the normalized positive patches were separated into train, validation, and test sets to create dataset 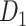 .
.
c). Model training
Before feeding the training set into the model, five augmentation methods (flip up-down, flip left-right, rotate 90 degrees, rotate 180 degrees, and rotate 270 degrees) were applied to the training set.
We trained and validated a YOLOv8 model with the X6 architecture using 10-fold cross-validation on the augmented dataset 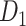 . The model, consisting of 350 layers and approximately 97 million parameters, was trained under the default hyperparameter configuration. We employed stochastic gradient descent (SGD) to minimize cross-entropy loss during training, which ran for a maximum of 500 epochs with early stopping implemented to prevent overfitting. This resulted in the original model, referred to as “ori” throughout this work.
. The model, consisting of 350 layers and approximately 97 million parameters, was trained under the default hyperparameter configuration. We employed stochastic gradient descent (SGD) to minimize cross-entropy loss during training, which ran for a maximum of 500 epochs with early stopping implemented to prevent overfitting. This resulted in the original model, referred to as “ori” throughout this work.
2). Hard Negative Mining Pipeline
In histopathological image recognition, pathologists typically annotate only target cells (i.e., CB cells) and leave other cells unannotated to minimize the annotation cost. It causes DL-based models to typically perform poorly due to many false-positive (FP) predictions.
We hypothesize that distinguishing CB cells from other cells that look like CB cells (non-CB cells) is the key to improving the model. One approach is incrementing the non-CB labels as a new class in the dataset. In practice, we retrieve the FP bbox (i.e., non-CB annotation) from the ori model inference on the training set and add them to the training set as a new class. As shown in Fig. 2(c), the following three steps were employed to generate a dataset with pseudo-negative labels: 2.1) Retrieve FP predictions; 2.2) Undersample; and 2.3) Combine the non-CB class with the training set.
a). Retrieve FP predictions
To obtain FP annotations, we applied the ori model to the training set within each cross-validation fold using a low confidence threshold of 0.001. This ensured the model predicted all possible negative cases. Since CB bounding boxes (bbox) in the training set had a long side smaller than 100 pixels, we further filtered out any predicted FP bbox with a side length exceeding this threshold. This step helped to eliminate spurious detections of large objects that were unlikely to be true CBs.
b). Undersample
As the number of identified FP predictions remained significantly higher than the actual number of CB cells, directly incorporating these negatives into the training set would lead to a class imbalance problem. To address this issue and ensure balanced representation during model training, we considered two undersampling strategies: Random undersampling and Neighborhood-based Recursive search undersampling (NB-Rec) [26].
-
•
Random undersampling is a popular non-heuristic technique due to its simplicity of application. Despite its simplicity, there is a significant disadvantage that must be considered. Given that balanced class, distribution is a stopping criterion, random undersampling may eliminate potentially useful samples to achieve this balance [27].
-
•
NB-Rec eliminates the majority class sample, which may overlap with the minority class. As described in Algorithm 1, the majority sample is considered overlapping when it is in the neighborhood of more than one minority sample. Since the NB-Rec uses K-Nearest Neighbor (KNN), we must search for
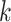 before execution to produce several negatives approximately equal to the actual CB.
before execution to produce several negatives approximately equal to the actual CB.
As the NB-Rec undersampling method requires coordinate information, we extracted features from both the ground truth and FP bbox. The width and height of each bbox were directly obtained. Additionally, six morphological features, as described in [28], were calculated for each cell within the bbox. To achieve this, we first segmented the cells using a trained HoverNet model based on the PanNuke architecture [29], resulting in binary images of individual cells within each bbox. These binary images were then used to compute the morphological features.
As depicted in Fig. 2(c) by the red, blue, and purple paths, we obtain three sets of undersampled FP predictions: (1) the set from random undersampling, (2) the set from applied NB-Rec undersampling to bbox width and height, and (3) the set from NB-Rec undersampling applied to the first- and second-principal components of bbox width, bbox height, and six morphological features using the Principal Component Analysis (PCA) method.
c). Combine the non-CB class with the training set
Following the undersampling procedures described in the previous step, we created three new datasets by incorporating the respective sets of undersampled FP predictions into the original training set of 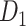 . These new datasets have similar images to
. These new datasets have similar images to 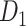 but contain two classes (CB and non-CB) in the training set. However, to maintain consistency and evaluate the models' ability to generalize to unseen data, we retained only the CB class in the validation and testing sets for all three datasets.
but contain two classes (CB and non-CB) in the training set. However, to maintain consistency and evaluate the models' ability to generalize to unseen data, we retained only the CB class in the validation and testing sets for all three datasets.
Algorithm 1: NB-Rec.

3). Train Model With Pseudo-Negative Label
As shown in Fig. 2(d), similar to the part 1) Training original model, we use the same setup model training step on the dataset of red, blue, and purple paths to get model hnm_random, hnm_box, and hnm_morph, respectively.
C. Experiment Setup:
All experiments were performed with an NVIDIA Tesla V100-SXM2 graphic card.
1). Experiment I: Stain Normalization Selection
This experiment aimed to ensure color consistency within our dataset by comparing two stain normalization methods: SPCN and DCGMM. We selected a template image, preferred by an expert, from the available patches. Both stain normalization methods were then applied to the remaining images in the dataset. The resulting normalized images were evaluated using metrics described in Supplementary Materials, Section I-A1. The stain normalization method that demonstrated superior performance will be employed in the pre-processing phase of this work.
2). Experiment II: Backbone Model Evaluation
This experiment aims to compare the performance of models from different training approaches (i.e., conventional and HNM approaches) on both object detection and image classification tasks. We use the training pipeline described in Section II-B to obtain four models:
-
•
Original (ori) model: Conventional object detection approach with one class annotation.
-
•
Model trained with random HNM (hnm_random): Randomly add FP samples from ori prediction on the training set into the training set as a new class. Then, it trains the model with the same setup as ori.
-
•
Model trained with HNM of bbox features (hnm_box): Instead of randomly sampling, this approach undersamples the FP samples using NB-Rec on the width and height of the FP bounding box, then adds them into the training set.
-
•
Model trained with HNM of morphological features (hnm_morph): Similar to hnm_box but using NB-Rec on first- and second-principal components from six morphological features and width and height of FP bounding box.
We use metrics from Supplementary Materials, Section I-A2 to evaluate the performance models on object-level prediction.
To evaluate the model's performance on the image classification task, we mapped the cell-level predictions to patch-level classifications using the following criteria: a patch was classified as positive if it contained at least one predicted CB cell; otherwise, it was classified as negative. Given that the test set of each cross-validation folds comprised 120-121 images containing CBs (positive images), we augmented each test set with additional negative images randomly selected from our database. This ensured a balanced class distribution for robust model classification performance evaluation.
III. Results
A. Experimental Results
1). Experiment I. Stain Normalization Selection
Deep convolutional Gaussian mixture models (DCGMM) yielded the lowest standard deviation (SD) and coefficient of variation (CV) for both Normalized Median Intensity (NMI) and Normalized Median Hue (NMH) metrics, as indicated in Table I. Since NMI qualifies the color consistency of the nuclei [30] and NMH quantifies the global color variation of an image population [31]. Thus, the results indicate that DCGMM provides qualitatively similar color distributions for nuclei with less color variation within the image population (see Fig. 3). Comparing the original and the Structure Preserving Color Normalization (SPCN), the box plots in Fig. 4 demonstrate that DCGMM has the smallest spread of NMI and NMH values around the median (inter-quartile range) with variance statistical significance (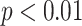 ).
).
TABLE I. Standard Deviation and Coefficient of Variation for NMI and NMH, Along With Mean Values and Standard Deviation for AMCE, for Three Different Image Populations.
| Method | NMI | NMH | AMCE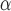
|
AMCE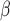
|
||
|---|---|---|---|---|---|---|
| SD | CV | SD | CV | Mean 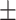 SD SD |
||
| Original | 0.082 | 0.137 | 0.039 | 0.042 | 5.30 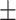 5.06 5.06 |
17.28 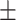 9.34 9.34 |
| SPCN | 0.057 | 0.086 | 0.012 | 0.012 |
4.67
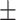 3.34 3.34 |
1.64
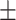 1.33 1.33 |
| DCGMM | 0.026 | 0.044 | 0.008 | 0.008 | 6.43 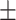 2.69 2.69 |
18.47 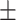 1.33 1.33 |
The bold values indicate the best score among the three stain normalization methods (Original, SPCN, DCGMM) in each metric.
Fig. 3.

Illustration of the performance of different stain normalization methods. The top-left image is the template image. The next column is the images sampled from the original images, followed by the results of normalization using SPCN and DCGMM, respectively.
Fig. 4.

Box plots of NMI, NMH, AMCE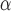 and AMCE
and AMCE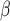 values for all stain normalization methods in Experiment I. * denotes the statistical significance of
values for all stain normalization methods in Experiment I. * denotes the statistical significance of 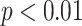 , and ns denotes not statistical significance.
, and ns denotes not statistical significance.
Next, we evaluate the background error of each image population. DCGMM has a significantly higher mean Absolute Mean Color Error in 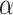 space (AMCE
space (AMCE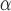 ) than the original image population. For
) than the original image population. For 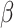 space (AMCE
space (AMCE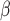 ), DCGMM does not have statistical significance compared to the original. It suggests that the DCGMM-processed images contain more or equal background errors than the original images, contradicting the goal of reducing color variations.
), DCGMM does not have statistical significance compared to the original. It suggests that the DCGMM-processed images contain more or equal background errors than the original images, contradicting the goal of reducing color variations.
Even though SPCN does not have statistical significance with the original at AMCE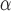 , at AMCE
, at AMCE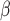 , SPCN provides significantly less error than the original. Moreover, both values of SPCN' are statistically significantly less than DCGMM.
, SPCN provides significantly less error than the original. Moreover, both values of SPCN' are statistically significantly less than DCGMM.
Therefore, we decided to implement SPCN in our framework pipeline, as it offers lower SD and CV in NMI and NMH values than the original and better AMCE values for both 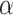 and
and  spaces than DCGMM.
spaces than DCGMM.
2). Experiment II. Backbone Model Evaluation
This experiment aimed to benchmark the performance of models trained using both conventional and HNM approaches for object detection and image classification tasks. Prior to comparing their performance, we first analyzed the optimization process of each model during training to gain insights into their convergence behavior and stability. As indicated in Supplementary Fig. S1, it was observed that the validation loss of the original (ori) model reached the lowest value when converged. Other models (i.e., all HNM approaches) exhibited a similar converging pattern more rapidly than the ori model, albeit with a higher loss. Despite the model trained with HNM of morphological features (hnm_morph) going with the same trend as other HNM approaches, it achieved the highest performance in terms of mean average precision (mAP@50) and accuracy, as shown in Figs. 5 and 6, respectively.
Fig. 5.
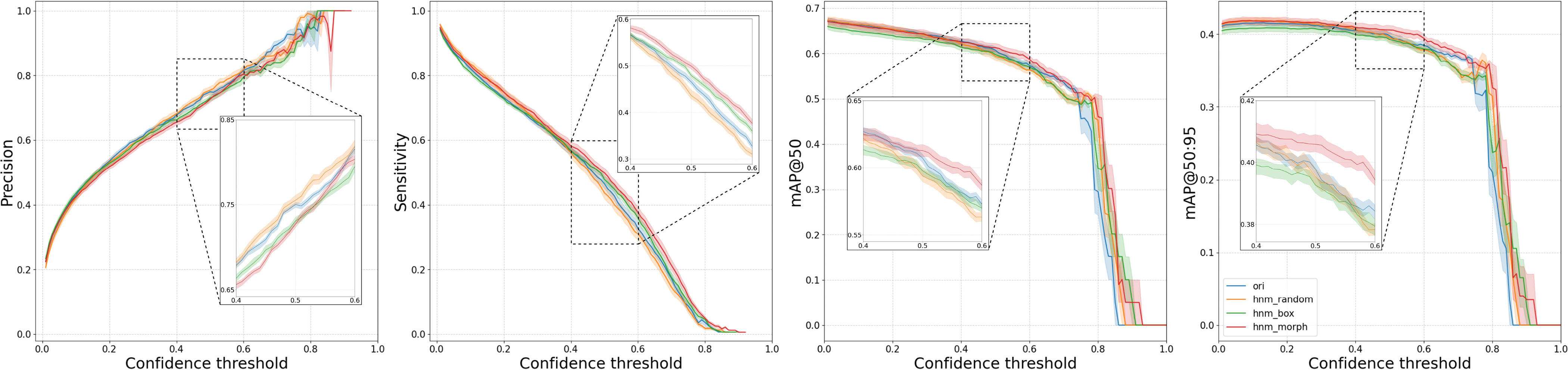
Centroblast detection results: Precision, Sensitivity, mAP at 0.5 IOU threshold (mAP@50), and mAP at 0.5 to 0.95 IOU threshold (mAP@50:95) of each model on test dataset with standard deviation error bar.
Fig. 6.
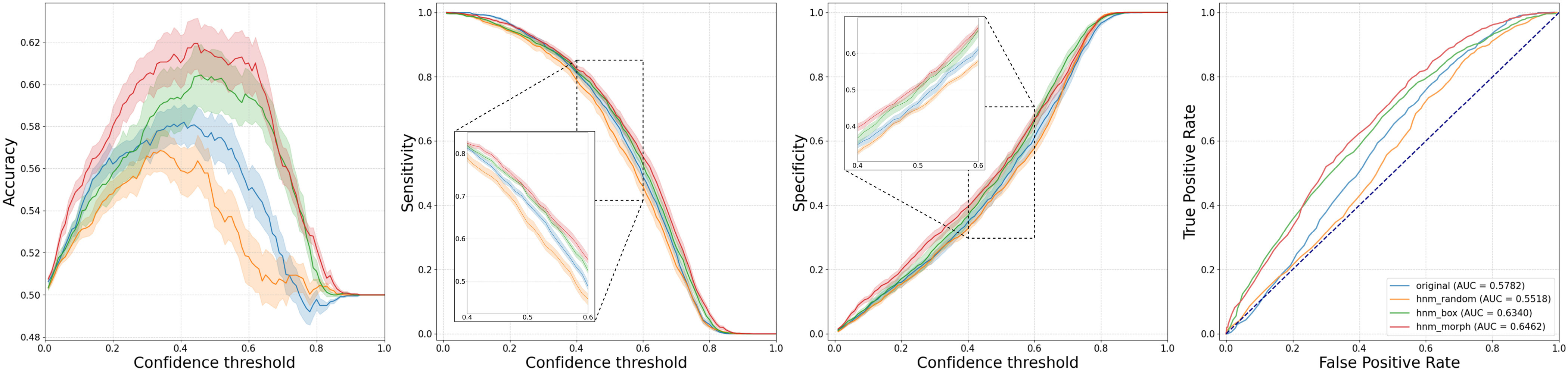
Patch classification results: Accuracy, Sensitivity, Specificity, and Receiver Operating Characteristics (ROC) curves of each model on test dataset with standard deviation error bar.
For the object detection task, Fig. 5 provided an overview of model performance on each metric with a confidence interval. All models follow the same trend. The hnm_morph achieved slightly better sensitivity, but in contrast, the trade-off appears to be on precision. Nevertheless, its performance is superior to other models in mAP@50 and mAP@50:95.
For the image classification task, the hnm_morph outperformed all other models, especially the model trained with random HNM (hnm_random). Notice that the performance of hnm_random dropped from ori on all metrics. In contrast, hnm_morph, which was trained with the same approach but with a more reasonable undersampling method, improved its performance over the ori.
IV. Discussion
A. Effect of Training With Pseudo-Negative Labels on the Model Performance
Concerning the impact of training with pseudo-negative labels from the hard negative mining (HNM) technique over the conventional training approach, we demonstrated improvements in centroblast (CB) detection and patch classification. However, the validation loss of these models is higher than that of the conventional training method (Supplementary Materials, Fig. S1). This is because the softmax function divides the total probability mass across multiple classes (i.e., CB and non-CB) rather than just one class of CB. When computing each class's confidence score in the bounding box using the softmax function, the number of divisors is increased to two, resulting in the confidence scores for each class becoming smaller on average as the number of classes increases. Finally, the class loss of the YOLO model, which is the cross-entropy loss, in models trained with HNM is higher than in the ori model.
B. Effect of Undersampling Approaches on the Model Performance
Since our framework was designed to imitate the training loop with the pathologist's refined labels, identifying CB based on cell color and morphology, we retrieved false-positive samples and fed them to the model. We obtain the following models based on three undersampling approaches: hnm_random, hnm_box, and hnm_morph. As the result of object detection and image classification tasks, the model with the morphological features (hnm_morph) performs best. Suggests that the design of the undersampling approach is essential to take advantage of the HNM technique and that pathologists' intuition still provides some information for deep learning to distinguish between non-CB cells and actual CB cells.
C. Guidance for Clinical Implementation and Future Works
In conventional histopathological workflow, pathologists count the number of centroblasts (CB) in ten randomly selected high-power fields (HPF), leading to high inter- and intra-observer variability and being vulnerable to sampling bias. The inter- and intra-observer variability among pathologists is crucial since it directly impacts patient grading and management [6]. To reduce the variability between pathologists, a solid guideline for finding potential HPF in WSI is one solution.
With PseudoCell framework, pathologists will obtain two guidelines: (i) heatmap visualization for potential CB regions in WSI and (ii) CB annotations at the HPF level for identifying CB. Pathologists' remaining job is to select the HPF and then accept the annotation or self-identify CB cells. Pathologists must only set the confidence threshold (conf_thres), ranging from zero to one when using PseudoCell. The conf_thres parameter determines the initial confidence level of CB annotations reported to pathologists. A low conf_thres (conf_thres = 0.2) produces a dense heatmap, as in Fig. 7, whereas a high conf_thres (conf_thres = 0.8) produces a sparse heatmap that more closely resembles the expert-annotation.
Fig. 7.

Heatmap visualization of detected centroblast in each patch on an unseen WSI. As conf_thres increases, the bright patch becomes sparser and more prone to expert annotation.
We will divide the histopathological process into the HPF selection and CB identification phases. In each phase, the real-world adjustment of conf_thres to facilitate pathologist preference could take the form of the following suggestion:
1). HPF Selection Phase
With a high conf_thres, PseudoCell offers a sparse HPF that is still sufficient to grade FL, which is suitable for pathologists who wish to complete the grading task rapidly. In contrast, when conf_thres is low, PseudoCell’ generates a dense heatmap that identifies the region containing intensive CB and regions with less CB. This approach is suitable for pathologists who wish to determine the HPF independently.
2). CB Identification Phase
A high conf_thres is advantageous for pathologists who prefer to self-identify on CB with some framework-suggested CB annotation. In contrast, a low conf_thres will enable the framework to recommend more CB annotation, which is ideal for pathologists who wish to check off the annotation.
The pathologists' workload can be reduced by PseudoCell accurately narrowing down the areas requiring their attention during examining tissue as in Fig. 7. From all 24,757 patches with tissue in WSI, the framework highlights 10,353 and 161 patches that contain potential CB candidates based on conf_thres with an inference time of approximately 0.03 seconds per patch. In other words, the framework can eliminate 58.18 to 99.35% of all WSI patches that do not appear to be CB candidates at the conf_thres. Pathologists can, therefore, focus on identifying CB on the slide. We anticipated that the inter- and intra-variability of pathologists would decrease after implementing our framework in the real world. Meanwhile, the machine can benefit from pathologists's actual CB as refined labels. These labels can be used to improve the model's performance in the future. This cycle leads to human-machine collaboration in the real world, which is one of the objectives of this work.
Pseudocell can also provide a second opinion that offers additional patient safety and instills greater confidence in doctors, enhancing their efficiency and reducing the likelihood of errors. For instance, when there is a need to distinguish between an infection and follicular cell lymphoma, particularly in its early stages, Thai pathologists, already handling a heavy workload, could issue a false negative, especially when the pathological area is small.
Integrating PseudoCell into the histopathological workflow offers several benefits. Firstly, the model assists pathologists by highlighting regions or suggesting potential CB cell candidates within the tissue, narrowing the examination focus. It serves as an additional quality control mechanism, flagging areas that may contain CB cells and assisting pathologists in not overlooking significant findings, thereby reducing diagnostic errors. In addition, pathologists can use the model's predictions as a benchmark to compare and contrast their observations. This iterative process improves their recognition of centroblast cells, enhancing diagnostic precision. Incorporating PseudoCell contributes to improved efficiency, quality control, and training and education for identifying centroblast cells in histopathology.
While implementing the PseudoCell framework offers a cost-effective alternative to the ongoing expenses of maintaining a pathology team, its integration into clinical practice faces workflow optimization and trust-building challenges. The complexity of healthcare systems necessitates careful integration to ensure seamless operation, potentially requiring adjustments to existing infrastructure and the deep learning pipeline. Additionally, addressing the “black box” nature of deep learning models through enhanced interpretability and transparency is crucial for gaining the confidence of clinicians and patients in PseudoCell’s predictions and recommendations.
In future work, if additional object detection models or updated versions exist, the PseudoCell framework permits their implementation by modifying the backbone model. Furthermore, dealing with data limitations and model transparency is crucial for pathologists to understand and have confidence in the model's decision-making. Combining weakly supervised paradigms (e.g., MIL or Attention Map) with explainability techniques (e.g., LIME, SHAP, and CAM) is a promising next step to investigate.
V. Conclusion
In conclusion, our study introduces the PseudoCell framework for centroblast (CB) cell detection, which enhances the performance of the backbone model by using false-positive samples from the Hard Negative Mining (HNM) method as pseudo-negative labels. PseudoCell effectively distinguishes between actual CB and non-CB cells in patches from whole-slide images (WSI). Our experiments and evaluations demonstrate that model training from HNM on Neighborhood-based Recursive search undersampling using morphological features achieves the best results in CB detection and patch classification tasks. PseudoCell can reduce pathologists' workload by accurately identifying tissue areas requiring attention during examination. Depending on the confidence threshold, PseudoCell can eliminate 58.18--99.35% of non-CB tissue areas on WSI. Furthermore, PseudoCell can serve as a second opinion to differentiate between infection and follicular cell lymphoma, particularly in the early stages, making it cost-efficient for quality control and educational purposes in CB recognition. This study presents a practical centroblast prescreening method that does not rely on pathologists' refined labels for improvement. It suggests the potential for human-machine collaboration in CB identification, alleviating the burden on clinicians by focusing their labeling efforts on regions suggested by PseudoCell rather than manual labeling as conventionally done.
Appendix.
Although our findings are promising, the limited sample size used for model training and testing may affect its accuracy and generalizability. Additionally, the PseudoCell model demonstrates sensitivity to WSI stain color variations, leading to false positives and false negatives in cell detection, as illustrated in Fig. 8. For example, Fig. 8(c) shows a false positive identification by the hnm_morph model compared to the ori model. In contrast, Fig. 8(d) demonstrates some false negatives, potentially due to stain variations that cause missing cell structures. Further investigation is needed to fully understand the cause of these false negatives and explore potential mitigating strategies. Despite these limitations, the model accurately identified CBs in blurred images and those with mixed cell types, Fig. 8(a) and 8(b). This suggests that data augmentation techniques and a larger, more diverse dataset could improve the model's robustness. Our study demonstrates the potential of object detection models trained with pseudo-negative labels to enhance cell- and slide-level prediction performance, ultimately aiming to reduce pathologist workload.
Fig. 8.

Comparision between ori, hnm_morph, and pathologist's annotation in identifying centroblast cells. The illustration shows that ori and hnm_morph correctly identify CB in (a) and (b), which are partially blurred and mixed types of cells, respectively. For (c), hnm_morph had a false positive. For (d), hnm_morph performed better than ori but still had some false negative predictions.
Supplementary Materials
Supplementary materials explain all evaluation metrics, visualize validation losses for each model during training, and illustrate the models performance in challenging cases.
Acknowledgment
The authors wish to thank Surat Phumphuang for coordinating the research.
Funding Statement
This work was supported in part by the New Discovery and Frontier Research under Grant (R016420005, Fund 3), Mahidol University and in part by the NSRF via the Program Management Unit for Human Resources & Institutional Development, Research and Innovation under Grant B38G670007.
Contributor Information
Narongrid Seesawad, Email: theerawit.w@vistec.ac.th.
Piyalitt Ittichaiwong, Email: piyalitt.itt@mahidol.ac.th.
Chanitra Thuwajit, Email: chanitra.thu@mahidol.ac.th.
Theerawit Wilaiprasitporn, Email: theerawit.w@vistec.ac.th.
References
- [1].Suzumiya J., “Current status and progress of lymphoma research in east asian countries: Introduction and planning,” Int. J. Hematol., vol. 107, no. 4, pp. 392–394, 2018. [DOI] [PubMed] [Google Scholar]
- [2].Swerdlow S. H. et al. , “The 2016 revision of the world health organization classification of lymphoid neoplasms,” Blood, J. Amer. Soc. Hematol., vol. 127, no. 20, pp. 2375–2390, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Intragumtornchai T. et al. , “Non-hodgkin lymphoma in south east asia: An analysis of the histopathology, clinical features, and survival from Thailand,” Hematol. Oncol., vol. 36, no. 1, pp. 28–36, 2018. [DOI] [PubMed] [Google Scholar]
- [4].Mozas P., Rivero A., and López-Guillermo A., “Past, present and future of prognostic scores in follicular lymphoma,” Blood Rev., vol. 50, 2021, Art. no. 100865. [DOI] [PubMed] [Google Scholar]
- [5].Swerdlow S. H. et al. , WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, vol. 2, Lyon, France: Int. Agency Res. Cancer, 2008, pp. 266–277. [Google Scholar]
- [6].Lozanski G. et al. , “Inter-reader variability in follicular lymphoma grading: Conventional and digital reading,” J. Pathol. Inform., vol. 4, no. 1, 2013, Art. no. 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Belkacem-Boussaid K., Sertel O., Lozanski G., Shana'aah A., and Gurcan M., “Extraction of color features in the spectral domain to recognize centroblasts in histopathology,” in Proc. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc., 2009, pp. 3685–3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cheng J., Veronika M., and Rajapakse J. C., “Identifying cells in histopathological images,” in Proc. Int. Conf. Pattern Recognit., 2010, pp. 244–252. [Google Scholar]
- [9].Michail E., Kornaropoulos E. N., Dimitropoulos K., Grammalidis N., Koletsa T., and Kostopoulos I., “Detection of centroblasts in H&E stained images of follicular lymphoma,” in Proc. IEEE 22nd Signal Process. Commun. Appl. Conf., 2014, pp. 2319–2322. [DOI] [PubMed] [Google Scholar]
- [10].Dimitropoulos K., Michail E., Koletsa T., Kostopoulos I., and Grammalidis N., “Using adaptive neuro-fuzzy inference systems for the detection of centroblasts in microscopic images of follicular lymphoma,” Signal, Image Video Process., vol. 8, no. 1, pp. 33–40, 2014. [Google Scholar]
- [11].Sertel O., Lozanski G., Shana'ah A., and Gurcan M. N., “Computer-aided detection of centroblasts for follicular lymphoma grading using adaptive likelihood-based cell segmentation,” IEEE Trans. Biomed. Eng., vol. 57, no. 10, pp. 2613–2616, Oct. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Belkacem-Boussaid K., Pennell M., Lozanski G., Shana'ah A., and Gurcan M., “Computer-aided classification of centroblast cells in follicular lymphoma,” Anal. Quantitative Cytol. Histol Int. Acad. Cytol. Amer. Soc. Cytol., vol. 32, no. 5, Art. no. 254, 2010. [PMC free article] [PubMed] [Google Scholar]
- [13].Dimitropoulos K., Barmpoutis P., Koletsa T., Kostopoulos I., and Grammalidis N., “Automated detection and classification of nuclei in pax5 and H&E-stained tissue sections of follicular lymphoma,” Signal, Image Video Process., vol. 11, no. 1, pp. 145–153, 2017. [Google Scholar]
- [14].Liu Y. et al. , “Detecting cancer metastases on gigapixel pathology images,” 2017, arXiv:1703.02442.
- [15].Lu Z. et al. , “Deep-learning–based characterization of tumor-infiltrating lymphocytes in breast cancers from histopathology images and multiomics data,” JCO Clin. cancer Inform., vol. 4, pp. 480–490, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Somaratne U. V., Wong K. W., Parry J., Sohel F., Wang X., and Laga H., “Improving follicular Lymphoma identification using the class of interest for transfer learning,” in Proc. IEEE Digit. Image Comput., Techn. Appl., 2019, pp. 1–7. [Google Scholar]
- [17].Syrykh C. et al. , “Accurate diagnosis of lymphoma on whole-slide histopathology images using deep learning,” NPJ Digit. Med., vol. 3, no. 1, pp. 1–8, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Szegedy C. et al. , “Going deeper with convolutions,” in Proc. IEEE Conf. Comput. Vis. Pattern Recognit., 2015, pp. 1–9. [Google Scholar]
- [19].Ronneberger O., Fischer P., and Brox T., “U-Net: Convolutional networks for biomedical image segmentation,” in Proc. 18th Int. Conf. Med. Image Comput. Computer-Assist. Interv., 2015, pp. 234–241. [Google Scholar]
- [20].Krizhevsky A., Sutskever I., and Hinton G. E., “ImageNet classification with deep convolutional neural networks,” Commun. ACM, vol. 60, no. 6, pp. 84–90, 2017. [Google Scholar]
- [21].Jocher G., Chaurasia A., and Qiu J., “Ultralytics YOLO,” Jan. 2023. [Online]. Available: https://github.com/ultralytics/ultralytics
- [22].Shrivastava A., Gupta A., and Girshick R., “Training region-based object detectors with online hard example mining,” in Proc. IEEE Conf. Comput. Vis. Pattern Recognit., 2016, pp. 761–769. [Google Scholar]
- [23].Ciompi F. et al. , “The importance of stain normalization in colorectal tissue classification with convolutional networks,” in Proc. IEEE 14th Int. Symp. Biomed. Imag., 2017, pp. 160–163. [Google Scholar]
- [24].Vahadane A. et al. , “Structure-preserving color normalization and sparse stain separation for histological images,” IEEE Trans. Med. Imag., vol. 35, no. 8, pp. 1962–1971, Aug. 2016. [DOI] [PubMed] [Google Scholar]
- [25].Zanjani F. G. et al. , “Histopathology stain-color normalization using deep generative models,” Med. Imag. Deep Learn., 2018. [Online]. Available: https://openreview.net/forum?id=SkjdxkhoG
- [26].Vuttipittayamongkol P. and Elyan E., “Neighbourhood-based undersampling approach for handling imbalanced and overlapped data,” Inf. Sci., vol. 509, pp. 47–70, 2020. [Google Scholar]
- [27].López V., Fernández A., García S., Palade V., and Herrera F., “An insight into classification with imbalanced data: Empirical results and current trends on using data intrinsic characteristics,” Inf. Sci., vol. 250, pp. 113–141, 2013. [Google Scholar]
- [28].Vrabac D. et al. , “DLBCL-Morph: Morphological features computed using deep learning for an annotated digital DLBCL image set,” Sci. Data, vol. 8, no. 1, 2021, Art. no. 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Graham S. et al. , “Hover-Net: Simultaneous segmentation and classification of nuclei in multi-tissue histology images,” Med. Image Anal., vol. 58, 2019, Art. no. 101563. [DOI] [PubMed] [Google Scholar]
- [30].Bejnordi B. E. et al. , “Stain specific standardization of whole-slide histopathological images,” IEEE Trans. Med. Imag., vol. 35, no. 2, pp. 404–415, Feb. 2016. [DOI] [PubMed] [Google Scholar]
- [31].Pontalba J. T., Gwynne-Timothy T., David E., Jakate K., Androutsos D., and Khademi A., “Assessing the impact of color normalization in convolutional neural network-based nuclei segmentation frameworks,” Front. Bioeng. Biotechnol., vol. 7, 2019, Art. no. 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.