

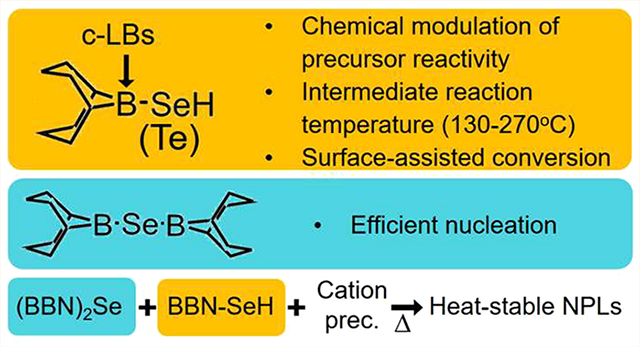
Abstract
To exploit the distinctive optoelectrical properties of nanomaterials, precise control over the size, morphology, and interface structure is essential. Achieving a controlled synthesis demands precursors with tailored reactivity and optimal reaction temperatures. Here, we introduce organoborane-based selenium and tellurium precursors borabicyclononane-selenol (BBN-SeH) and tellurol (BBN-TeH). The reactivity of these precursors can be modified by commercially available additives, covering a wide range of intermediate reactivity and filling significant reactivity gaps in existing options. By allowing systematic adjustment of growth conditions, they achieve the controlled growth of quantum dots of various sizes and materials. Operating via a surface-assisted conversion mechanism, these precursors rely on surface coordination for activation and undergo quantitative deposition on coordinating surfaces. These properties allow precise control over the radial distribution and density of different chalcogenide atoms within the nanoparticles. Diborabicyclononanyl selane ((BBN)2Se), an intermediate from the BBN-SeH synthesis, can also serve as a selenium precursor. While BBN-SeH suppresses nucleation, (BBN)2Se exhibits efficient nucleation under specific conditions. By leveraging these distinct activation behaviors, we achieved a controlled synthesis of thermally stable nanoplates with different thicknesses. This study not only bridges critical reactivity gaps but also provides a systematic methodology for precise nanomaterial synthesis.
Keywords: quantum dots, chalcogenide, precursors, reactivity, nanoplatelets
Graphical Abstract:
Nanomaterials have been extensively explored over the past few decades in a wide range of areas, including fundamental science, engineering, and commercial applications. Key characteristics of nanomaterials encompass the sensitive dependence of optoelectrical properties on their size and morphology, solution processability, and customizability of their properties. Prior research has highlighted the need for precise synthesis control. In 2D materials, atomic precision is required along the quantum-confined axis.1 In semiconductor nanocrystals, known as quantum dots (QDs), optoelectronic properties are sensitively affected not only by their size but also by detailed interfacial structure, such as alloy profile, and crystal defects.2–4 Critical properties to be controlled in nanomaterials include size, particularly along the quantum confined axis, size uniformity, shape, atomic arrangement at the interface, and surface properties. In general, solution synthesis is preferred due to its scalability and lower technical barriers, but it poses significant challenges in controlled synthesis. Solvothermal synthesis often involves entangled reactions with ill-defined intermediates, making it difficult to tune the reaction kinetics and methodically produce nanomaterials with desired structures.
Studies on QDs, one of the most extensively studied nanomaterials, highlight the importance of precursors with optimal reactivity and well-defined reaction pathways for controlled synthesis.5,6 Achieving suitable kinetics for solute production is critical for controlling size and morphology as well as achieving high crystallinity. Various strategies have been developed to tune precursor reactivity, such as leveraging the steric hindrance of precursors to modulate their reactivity toward counterion salts,7 selecting precursors with different bonding configurations such as thiol, thiourea, and elemental sulfur,8 and altering the bond polarity using distal groups.9,10 However, these approaches have fundamental limitations: each precursor covers a narrow range of solute production kinetics without a systematic method to identify the optimal precursor reactivity for high-quality QD production. The optimal conditions sensitively depend on the size and composition of the nanomaterials. Unfortunately, existing precursors often fail to achieve the desired reactivity, and optimal conditions for different systems are mostly identified by trial and error. Moreover, the need to synthesize different precursors to test various reactivities adds to the complexity. Here, we introduce a suite of chalcogenide precursors, characterized by well-defined reaction pathways and tunable reactivity across a wide spectrum, to enable systematic and controlled synthesis of high-quality nanomaterials. Building on our recently developed organoborane-based sulfur precursor,11,12 we have developed the organoborane-based selenium (Se) and tellurium (Te) precursor chemistries that would address the critical reactivity gap in the scarce Se and Te precursor selections and offer unparalleled control in the synthesis of chalcogenide-containing nanomaterials.
Se-incorporating QDs have broad applications, including HgSe QDs in near-infrared imaging13 and electroluminescence devices,14 PbSe QDs in photovoltaics,15 and ZnSe QDs in blue light-emitting diodes.16 However, the variety of Se precursors is limited, with a notable lack of options offering intermediate reactivity. The first-generation Se precursor, hydrogen selenide,17 has been phased out due to its high toxicity and reactivity and is now rarely used in the field. The most commonly used Se precursors are alkylphosphine-Se, such as trioctylphosphine-Se (TOP-Se) and tributylphosphine-Se (TBP-Se), but they have low reactivity and require high reaction temperatures, limiting their applications. For example, ZnSe growth on InP QD cores requires a reaction temperature of >340 °C.18 Due to the high reaction temperature requirement, ZnSe shell growth is not feasible on small InP QD cores with low melting temperatures (Tm). To tolerate high-temperature synthesis, additional cations like Zn,19 Ga,20 or Cu21 have been incorporated into InP cores. However, even with this strategy, uncontrolled alloying is often unavoidable, which alters their optophysical properties. Another limitation of trialkylphosphine-Se is a low conversion yield (~30%).22 The addition of dialkyl phosphine or diarylphosphine, which act as reducing agents, greatly improves the yield but they involve highly entangled reaction pathways making it difficult to fine-tune the conversion kinetics.23 Phosphine-free Se precursors, such as octadecene-Se (ODE-Se)24 and oleylamine (OLAm)-Se,25 have been developed to address the instability and toxicity of alkylphosphines. While these precursors become reactive at lower temperatures (200–250 °C), they rely on Se radical generation,26 which is inherently sensitive to impurities and involves ill-defined reaction pathways for QD synthesis. The selenourea precursors are activated around 240 °C and do not involve radicals serving as attractive Se precursors.27 However, there is still a wide gap in intermediate reactivity, failing to cover reaction temperatures below 240 °C. More importantly, altering the reactivity requires the synthesis of different precursor molecules. Furthermore, all existing precursors use common reaction pathways for nucleation and growth, inevitably coupling them and causing secondary nucleation under some reaction conditions.
While less common, Te-containing QDs are gaining attention for applications in optoelectronics,28 solar cells, and bioimaging. For example, PbTe QDs have been incorporated into solar cells,29 Ag2Te QDs30 and HgTe QDs31,32 have been used as active layers of photodetectors, and ZnSeTe alloyed QDs have been explored as blue and green light emitting materials in electroluminescence devices.33,34 The selection of Te precursors is more limited. Early precursors like polyanion tellurides prepared using superhydrides35 and hydrogen telluride (H2Te)36 fell out of favor due to their excessive reactivity and high toxicity. The superhydride-based synthesis suffers from reaction uncontrollability; the excess amount of strong reducing agents and polytellurides (Ten2−) induces the uncontrolled formation of Te nanoparticles along with the desired QDs due to their excessive reactivity. The widely used Te precursor is TOP-Te, as for other chalcogenides. However, its low reactivity necessitates high reaction temperatures and the use of excess amounts of phosphines, making precise control of QD shape and size difficult. Tris(dimethylamino)-phosphine telluride ((Me2N)3P:Te) has higher reactivity than TOP-Te due to the electron-donating character of the amino-N lone pairs.37 Activation of (Me2N)3P:Te via transamination has enabled the synthesis of CdTe nanoplatelets (NPLs) and nanowires at low reaction temperatures of ~100 °C. However, there remains a substantial reactivity gap for Te precursors that cannot be covered by TOP-Te, Ten2−, and (Me2N)3P:Te.
The Se and Te precursors reported here, (1s,5s)-9-borabicyclo[3.3.1]nonane-9-selenol (BBN-SeH) and (1s,5s)-9-borabicyclo[3.3.1]nonane-9-tellurol (BBN-TeH), parallel our recently developed 9-sulfanyl-9-borabicyclo[3.3.1]nonane (BBN-SH).11,12 As previously reported, the coordination of Lewis bases (LBs) to the empty orbital of the boron atom predictably modulates the strength of the boron-chalcogen bond. This modulation achieves controllable reactivity of BBN-EH (E:S, Se, and Te) precursors and precursor conversion across a wide range of synthesis temperatures (130–270 °C). Commercially available boron-coordinating LBs (c-LBs) with substituents of varying electron-donating strengths effectively regulate the reactivity of BBN-EH over a wide range of synthesis temperatures relevant to various QDs spanning a wide range of sizes and materials. Altogether, organoborane-based chalcogenide precursors represent a distinct precursor type where a single precursor covers a very wide reactivity range through reactivity modulation with chemical additives and enables controlled precursor deposition on QD cores of various sizes and compositions at mild temperatures. Additionally, unlike other existing precursors, they employ a surface-assisted quantitative conversion mechanism where the nucleation and growth reaction pathways are separated, eliminating secondary nucleation during growth. We leveraged these properties to grow high-quality ZnSe shells on small InP cores free of cation dopants and to incorporate Te atoms into the ZnSe QD matrix with precise control over their radial distribution and density. We have also utilized di((1S,5S)-9-borabicyclo[3.3.1]nonan-9-yl)selane ((BBN)2Se), an intermediate compound used for in situ generation of BBN-SeH, as a Se precursor for nanoplatelet (NPL) synthesis. By combining its nucleation capability with the nucleation inhibition property of BBN-SeH, we enabled the synthesis of highly thermostable NPLs, remaining stable up to 200 °C. We also showed that variation of the ligand composition leads to the production of different NPL species, likely due to modulation of the energy landscape of metastable NPL structures. Expanding the selection of precursors, particularly those with intermediate reactivity, marks a significant step forward in the controlled synthesis of diverse nanostructures, including both nanoparticles and 2D structures. Furthermore, the dual role of BBN-EH in feeding nanomaterial growth and surface stabilization provides distinct control over the growth kinetics and thermal stability of the resulting materials.
RESULTS AND DISCUSSION
Synthesis of BBN-SeH and Reactivity Modulation via c-LB Coordination.
Different from BBN-SH, which is synthesized by a one-step reaction from H2S under ambient conditions, BBN-SeH synthesis necessitates a two-step reaction under air-free conditions. Instead of using H2Se, which is highly toxic and volatile, we first synthesized (BBN)2Se using the 9,9′-bi-9-borabicyclo[3.3.1]nonane (BBN dimer) and Se powder. As (BBN)2Se forms, the reaction produces hydrogen gas, providing a visual indicator to monitor the progress of the reaction. The reaction was stopped after 1 h when H2 gas no longer evolved. (BBN)2Se was then converted to BBN-SeH by adding aniline (Figure 1a). Electrospray ionization-mass spectroscopy (ESI-MS) confirms the formation of (BBN)2Se (Figure S1) and BBN-SeH (M, m/z = 202.08, Figure S2a). The observed m/z distribution is in close agreement with the mixture of simulated data from [M]+ (C8H15BSe), [M + H]+ (C8H16BSe), and [M+3H]+ (C8H18BSe) (Figure S2b). 11B NMR spectra also show the conversion of (BBN)2Se (~59 ppm) to BBN-SeH (~52 ppm) and the aniline-BBN complex (~85 ppm) upon aniline addition (Figure S3). BBN-SeH exhibited extremely high susceptibility to oxidation, swiftly transitioning from the colorless solution (BBN-SeH) to white solids upon exposure to air. Even in air-free conditions, such as in a glovebox, BBN-SeH degraded rapidly. Thus, we developed an in situ production method in which BBN-SeH is produced during QD growth. To accommodate the air sensitivity of (BBN)2Se, we synthesized (BBN)2Se under air-free conditions and stored it in a glovebox until use. (BBN)2Se and c-LB of choice were transferred to the air-free reaction vessel containing reagents required for QD growth, including premade core seeds, cationic precursors, and QD ligands. The addition of aniline to the reaction vessel results in the in situ production of BBN-SeH, inducing QD growth upon heating.
Figure 1.
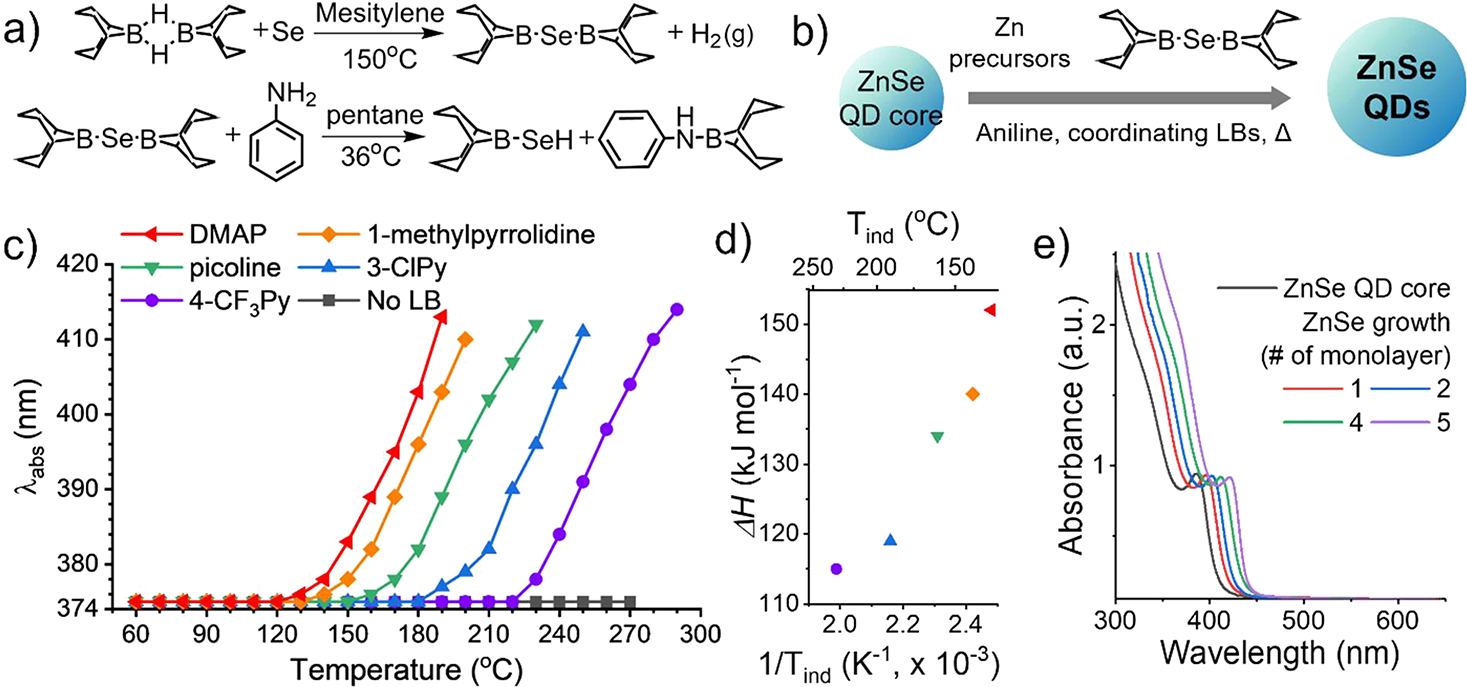
Synthesis of BBN-SeH and its application to ZnSe QD growth. (a) Two-step synthesis of BBN-SeH. (b) Scheme for BBN-SeH based ZnSe QD growth involving in situ production of BBN-SeH. (c) Identification of the induction temperature (Tind) of BBN-SeH upon the addition of c-LBs by monitoring the first absorption peak position of ZnSe QDs during growth. (d) Influence of c-LBs with varying BF3 affinity (ΔH) on Tind (red: dimethylaminopyridine (DMAP), orange: 1-methylpyrrolidine, green: picoline, blue: 3-chloropyridine (3-ClPy), purple: 4-(trimethylfluoro)-pyridine (4-CF3Py)). (e) Absorption spectra of ZnSe QDs during growth using BBN-SeH and picoline and (f) Evolution of the half width at half-maximum (HWHM) of the first absorption peak during growth.
We confirmed the controllable adjustment of the BBN-SeH reactivity by monitoring ZnSe QD growth after adding different c-LBs, mirroring our approach with BBN-SH (Figure 1b). For ZnSe QD growth, zinc oleate was used as a cationic precursor and oleic acid, OLAm, and TOP were used as ligands. Activation of BBN-SeH would cause QD growth, inducing the first absorption peak to red-shift; thus, the temperature that initiates the red-shift of the first absorption peak is defined as the induction temperature (Tind) (Figure 1c). For this experiment, we tested dimethylaminopyridine (DMAP), 1-methylpyrrolidine, picoline, 3-chloropyridine (3-ClPy), and 4-(trimethylfluoro)-pyridine (4-CF3Py) as c-LBs. With these c-LBs, BBN-SeH was activated at a wide range of temperatures (130–230 °C), covering the growth temperature (Tgrowth) of 130–250 °C for ZnSe QDs, the intermediate temperature range that is not accessible with existing Se precursors. Previous studies show that TOP-Se requires >340 °C for growing ZnSe shells on InP QD cores.18 Additionally, ODE-Se and OLAm-Se require ~320 °C for growing CdSe QDs,26 and selenourea achieves CdSe QD nucleation and growth at 240–290 °C.27 Notably, BBN-SeH remains unactivated up to 270 °C in the absence of c-LB, even in the presence of aniline, another type of LB. The inability to activate BBN-SeH using primary amine is consistent with our findings in the BBN-SH case.12 This observation also aligns with the fact that alkylamines (e.g., OLAm) and alkylphosphines (e.g., TOP), commonly employed as surface ligands in QD synthesis, do not activate BBN-SeH. While primary amine fails to activate BBN-SeH, aniline efficiently drives the proteolytic cleavage of (BBN)2Se, forming the BBN-aniline complex and BBN-SeH (Figure 1a).
Consistent with our previous work on BBN-SH, c-LBs exhibiting higher BF3 affinity (ΔH) resulted in lower Tind for ZnSe QD growth in a predictable manner (Figure 1d). Specifically, DMAP, 1-methylpyrrolidine, and picoline enable controlled ZnSe QD growth at 130–150, 140–160, and 160–180 °C, respectively, significantly lower than the conventional method using TOP-Se (340 °C) and ODE-Se (200–250 °C). Low Tgrowth enabled controlled shell growth on small QDs, which have a low Tm and are therefore difficult to grow controllably by using conventional precursors. Figure 1e presents the evolution of absorption spectra of ZnSe QDs during their growth from 3.0 nm in diameter core seeds with BBN-SeH and picoline. As additional ZnSe layers were deposited, the first absorption peak gradually red-shifted with slightly reduced the half-width at half-maximum (HWHM) (14 to 12 nm; 116 to 86 meV; Figure S4a, c). TEM images revealed a slight improvement in the monodispersity of the QDs after growth (3.0 ± 0.5 nm (17%) to 6.0 ± 0.7 nm (12%), Figure S6 and Table S1). We attribute this slight size focusing to the controlled addition of monomers38 via the surface-assisted growth mechanism of BBN-EH chemistry. The slight improvement in size monodispersity is consistent with BBN-SH based CdS QD growth.11 It is noteworthy that BBN-SeH based low-temperature growth maintained a narrow size distribution of small ZnSe QDs throughout, whereas TOP-Se based growth yielded a broad size distribution due to particle instability during high-temperature growth (Figure S5). The size growth probed by TEM images matches the expected values, suggesting quantitative conversion of BBN-SeH (Figure S6). The conversion efficiency of BBN-SeH for ZnSe QDs growth reached 98% (±20%) (see Supporting Information (SI) for details). In contrast, traditional Se precursors, including TOP-Se, ODE-Se, and OLAm-Se, exhibit low conversion efficiencies (~30%).22,24,25 The size measured from the TEM images before and after ZnSe QD growth well matched with the expected size from their first absorption peak (ZnSe cores: 3.0 ± 0.5 nm in diameter, ZnSe QDs after growth: 6.0 ± 0.7 nm in diameter, measured from 50 particles, Figure S7)39 and showed their isotropic growth (Table S1) with slight size focusing. This result confirms the quantitative incorporation of BBN-EH during growth.
Synthesis of BBN-TeH and Reactivity Modulation via c-LB Coordination.
Unlike the selenium case, bulk Te fails to produce the organoborane product when combined with BBN dimer40 due to the insufficient reduction potential of the BBN dimer for Te. Using a stronger reducing reagent is unfeasible, as it may potentially interfere with subsequent reactions and QD growth. Taking advantage of the relatively lower air sensitivity of BBN-TeH, we developed a two-step synthesis method employing common practices to minimize air exposure. We first added a strong reducing agent, LiAlH4, to Te powder, resulting in the rapid formation of LiAlTeH2, confirmed by hydrogen gas evolution. After purifying LiAlTeH2 by centrifugation, we introduced the BBN dimer to obtain the crude product of BBN-TeH. Further purification of BBN-TeH was carried out via sublimation (Figure 2a). Both ESI-MS (Figure S8) and 11B NMR (Figure S9) confirmed the formation of BBN-TeH. Since BBN-TeH was also susceptible to oxidation and water, the purified BBN-TeH was kept under a dry nitrogen environment.
Figure 2.
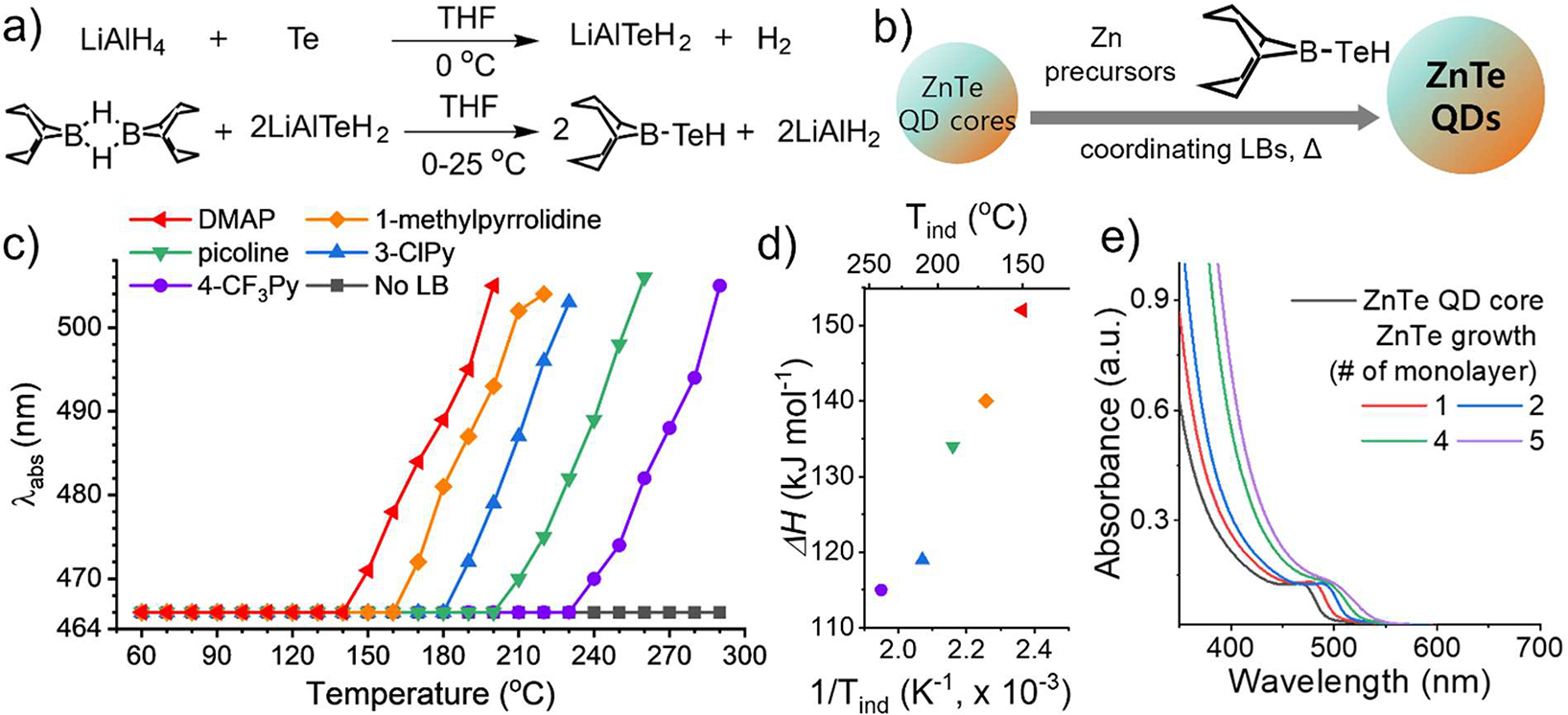
Synthesis of BBN-TeH and its application to ZnTe QD growth. (a) Two-step synthesis of BBN-TeH. (b) Scheme for ZnTe QD growth using BBN-TeH and coordinating Lewis bases. (c) Identification of Tind with different c-LBs by monitoring the position of the first absorption peak of ZnTe QDs, (d) Influence of c-LBs with varying BF3 affinity (ΔH) on Tind. (red: dimethylaminopyridine (DMAP), orange: 1-methylpyrrolidine, green: picoline, blue: 3-chloropyridine (3-ClPy), purple: 4-(trimethylfluoro)-pyridine (4-CF3Py)), (e) Absorption spectra of ZnSe QDs during growth using BBN-TeH and picoline.
We confirmed the controllable modulation of the BBN-TeH reactivity by monitoring ZnTe QD growth in the presence of various c-LBs. The zinc precursor (zinc oleate), solvents (ODE), and surface ligands (oleic acid, OLAm, and TOP) were kept the same as the ZnSe QD growth experiments (Figure 2b). Consistent with BBN-SH and BBN-SeH, c-LBs predictively modulated the precursor reactivity of BBN-TeH leading to a greater decrease in the Tind for c-LBs with a greater affinity for BF3 (Figure 2c–d). The BBN-TeH:c-LB chemistry covers the intermediate range of reactivity that existing Te precursors fail to achieve. For ZnTe QDs, the BBN-TeH:c-LB chemistry achieved controlled growth at 150 to 240 °C. In comparison, TOP-Te requires a reaction temperature of 250–300 °C33 and (Me2N)3P:Te grows ZnTe QDs at ~100 °C.37 Without c-LBs, the growth of ZnTe QDs was not observed up to 290 °C, reaffirming that primary amines do not activate the boron-chalcogenide bond in BBN-EH. We have reaffirmed the strong efficacy of intermediate reactivity precursors in achieving controlled growth of small QDs. Figure 2e presents the evolution of the absorption spectrum of ZnTe QDs during their growth from 2.8 nm core seeds with BBN-TeH and picoline at mild reaction temperature (190–210 °C). As the ZnTe layer is deposited, the first absorption peak progressively shifts toward longer wavelengths, showing controlled growth of ZnTe QDs. This result starkly differs from QD growth using TOP-Te, where the first absorption peak diminishes during the exposure of the ZnTe core seeds to high reaction temperatures, suggesting the disintegration of nanoparticles. Even after adding TOP-Te equivalent to several ZnTe monolayers (MLs), a distinct peak does not emerge and a high wavelength tail grows, indicating uncontrolled particle disintegration and secondary nucleation (Figure S10). This result highlights that the expansive range of intermediate reaction temperatures accessible with BBN-TeH (150–240 °C) allows for the controlled growth of nanoparticles having low to intermediate Tm. As shown, the BBN-TeH chemistry significantly broadens the scope for growing Te-containing QDs with high crystal quality and a precisely controlled morphology across a wider range of sizes and materials. The HWHM of the first absorption peak was marginally increased during the growth, confirming the size distribution of ZnTe QD was mostly preserved (Figure S4b, d). The size measured from the TEM images before and after ZnTe QD growth (ZnTe cores (in diameter): 2.8 ± 0.4 nm in diameter, ZnTe QDs after growth: 5.8 ± 0.8 nm in diameter, measured from 50 particles, Figure S11) and showed their isotropic growth (Table S1). Like BBN-SH and BBN-SeH, BBN-TeH also demonstrates quantitative conversion, confirmed by characterizing the size change during growth using TEM.
Comparison of BBN-EH Chemistries.
With all three organoborane chalcogenide precursors synthesized, we conducted a comprehensive comparison of their properties and the performance of each precursor. Our previous study has revealed that BBN-SH employs a surface-assisted conversion mechanism, necessitating coordination to the QD surface for activation.11 Similarly, both BBN-SeH and BBN-TeH remained inactive in the absence of nanoparticles, even in the presence of c-LBs (Figure 5b–c). The energetics involved in the surface-assisted conversion mechanism comprise the following four terms: the coordination energy of BBN-EH to the QD surface, the boron-chalcogenide bond strength, the formation energy of Zn-E nanoparticles, and the coordination energy of c-LBs to the boron atom of the precursor. All of these terms are significantly affected by the choice of chalcogenide. We compared the Tind of BBN-SeH and BBN-TeH during ZnE growth with different c-LBs (Figure 3a). In all cases, BBN-SeH exhibited a lower Tind compared to BBNTeH. Additionally, we examined the Tind of BBN-SH by monitoring the ZnS QD growth (Figure S12). When using picoline as a c-LB, the Tind of BBN-SH was 10 and 40 °C lower than that of BBN-SeH and BBN-TeH, respectively (Figure 3b). This trend aligns with predictions based on the formation energy of ZnS, ZnSe, and ZnTe QDs (ΔHf°: −205 kJ/mol, −160.0 kJ/mol, and −119.2 kJ/mol).41 However, decoupling how other energetics terms contribute to the activation energy remains challenging. Not surprisingly, the choice of cationic precursor also influences the precursors’ Tind. Consistent with the trend observed from previous studies, a zinc precursor induced higher Tind compared to that of a cadmium precursor. Specifically, when BBN-SH, picoline, and metal oleate were used, ZnS QD growth was induced at 10 °C higher than CdS QD growth.12
Figure 5.
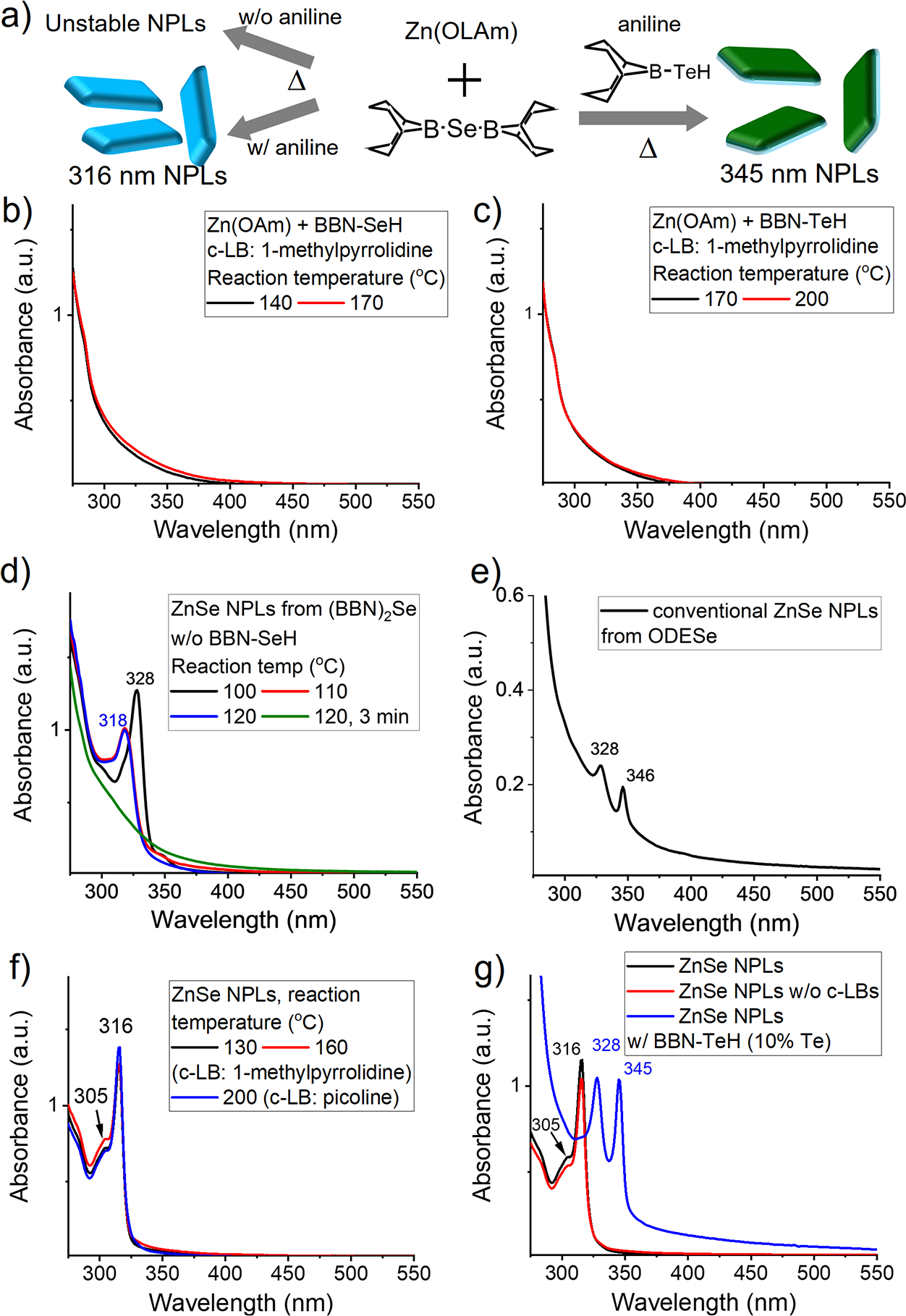
Synthesis of NPLs with exceptional thermal stability using BBN-EH. (a) Scheme for ZnSe NPLs synthesis using (BBN)2Se/BBN-EH chemistry. Addition of aniline generates BBN-SeH in situ and produces thermostable ZnSe NPLs. (b) Pure BBN-SeH and (c) BBN-TeH fail to induce NPL formation. (d) Without BBN-SeH, the formed NPLs are unstable, rapidly degrading and disappearing at 120 °C. (e) Absorption spectrum of ZnSe NPLs prepared by conventional protocol. (f) Presence of BBN-SeH renders ZnSe NPLs highly thermostable. (g) Addition of c-LBs does not induce surface-limited conversion due to vertical growth inhibition in NPLs (black and red). Addition of 10% BBN-TeH to (BBN)2Se produces thermostable 345 nm ZnSe NPLs.
Figure 3.
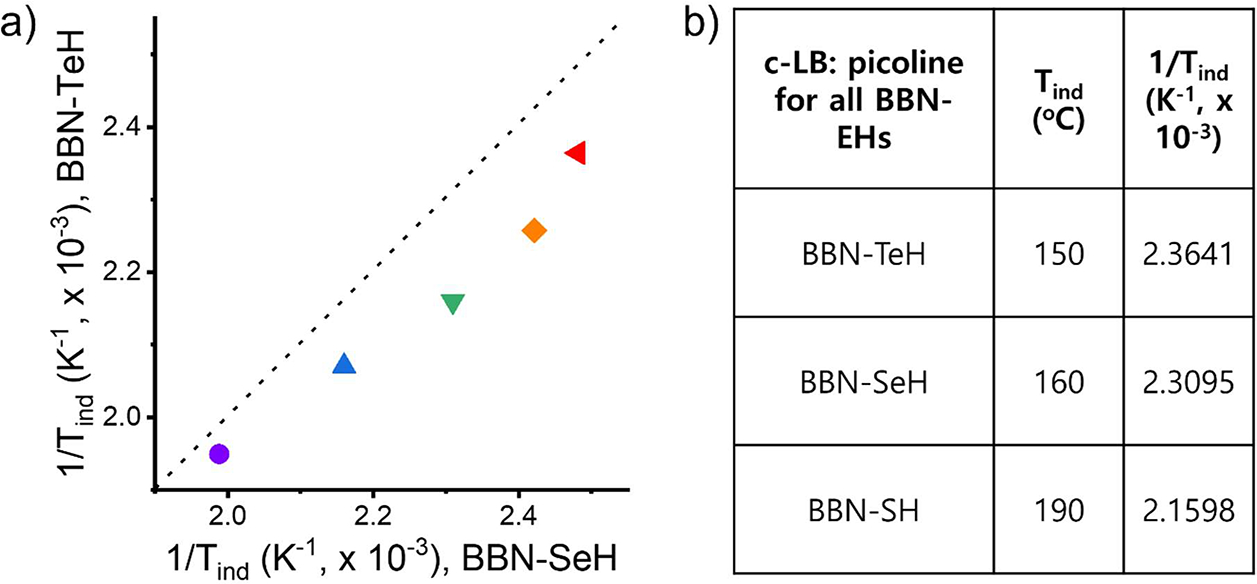
Comparative analysis of Tind for BBN-EH. (a) Tind of BBN-EH for ZnE (E: Se, Te) QD growth (red: DMAP, orange: 1-methylpyrrolidine, green: picoline, blue: 3-ClPy, purple: 4-CF3Py). The dotted line designated the same Tind for BBN-SeH and BBN-TeH. (b) Tind of BBN-EH for growing ZnE QDs (E: S, Se, and Te) using picoline as a c-LB.
The three organoborane precursors exhibited different levels of oxidation susceptibility. BBN-SH exhibited the highest stability in air among the three, allowing for common synthesis practices, including solvent removal using a rotary evaporator. In comparison, BBN-SeH was highly prone to oxidation, making storage impossible even in a glovebox, requiring in situ production during nanoparticle growth. BBN-TeH exhibited intermediate stability, allowing its purification and storage with routine air-free techniques.
Applying BBN-EH Chemistry: Controlled Shell Growth on Small QD Cores and Controlled Incorporation of Different Chalcogenide Ions into the QD Matrix.
The BBN-EH precursor chemistry offers unparalleled benefits for the controlled incorporation of precursor atoms into nanoparticles. First, each precursor covers a wide range of intermediate-range reactivity, providing access to low to intermediate reaction temperature ranges (100–240 °C) previously unattainable with existing Se and Te precursors. High reaction temperatures often overly destabilize the nanoparticle surface, inducing undesired alloying or even particle disintegration, especially for particles having low Tm, such as small particles or particles made of low Tm materials.42 BBN-EH greatly expands the synthetic capabilities by enabling controlled atomic deposition on particles of a wide range of sizes and materials. Second, their reactivity is controllably adjustable using commercially available chemical additives, allowing the systematic identification of optimal conditions for controlled deposition on cores of different sizes and materials. Third, the surface-assisted conversion mechanism and quantitative deposition enable precise control of the incorporation sites and density for different chalcogenide atoms during growth. Leveraging these properties, we successfully demonstrated controlled shelling on small InP cores (d: 1.7–2.4 nm) and controlled growth of anion-alloyed MLs, both of which have been challenging with existing precursors.
Indium phosphide (InP) QDs are widely used for light-emitting devices and have been incorporated into commercial products due to their minimal toxicity. Shelling is necessary to improve their photoelectric properties, and ZnSe has been a common choice. While high-quality InP/ZnSe QDs are routinely produced with InP cores larger than 2.7 nm in diameter, shelling on smaller cores has proven challenging. For cores smaller than 3 nm, it has been common practice to incorporate Zn or Ga atoms into the core to provide sufficient thermal stability during the high-temperature shelling process required for the existing Se precursors. Using BBN-SeH, we have successfully achieved high-quality ZnSe shell growth on pure InP cores of a small size (1.7–2.4 nm). In our prior research,12 we identified the optimal Tgrowth to be 10–20 °C lower than the Tm of cores. Thus, we first characterized the Tm of each core and selected an appropriate c-LB and reaction temperature accordingly (Figure S13). Specifically, for 1.7 nm cores with a Tm of 150 °C, we chose DMAP and 1-methylpyrrolidine as c-LBs, setting the Tgrowth at 130 °C (DMAP) or 140 °C (1-methylpyrrolidine). For 2.4 nm cores with a Tm of 200 °C, we selected 3-ClPy as the c-LB, setting the Tgrowth at 190 °C (Figure S14a and S14b).
Figure 4a–b presents the absorption and photoluminescence (PL) profiles of 1.7 nm InP cores before and after depositing 4 MLs of ZnSe shells using BBN-SeH and 1-methylpyrrolidine. Deposition of ZnSe shells is evident from the substantial increase in absorption below 450 nm, resulting in a significant enhancement of their PL, reaching a quantum yield (QY) of 30%. For the 2.4 nm InP QD cores, fold enhancement (~4) of absorbance at 350 nm after 4 ML ZnSe shell deposition matched with other’s work (Figure S14c)43 as well as confirming the size change after ZnSe shelling under the TEM images (Figure S14b). This successful shelling of extremely small InP cores achieved PL at 503 nm, a considerably bluer wavelength compared to previously reported InP/ZnSe QDs, highlighting the benefit of the intermediate reactivity of BBN-SeH.44 For InP/ZnSe QDs produced from 1.7 to 2.4 nm cores, QYs ranged 30–49% with the full width at half-maximum (FWHM) of the PL peak measured at 51–54 nm. These QY and FWHM values parallel the best results previously achieved without employing an etching process. The relatively low QY of InP/ZnSe QDs synthesized with 1.7 nm cores (30%) likely stems from reduced crystal quality at the core–shell interface due to the extremely small size of the cores.45 Prior studies have shown that acid-based etching of InP surfaces before shelling significantly improves QY by eliminating oxidized surfaces.18 Thus, we expect that combining the BBN-SeH-based ZnSe shelling with the etching process would extend the wavelength range covered by InP/ZnSe QDs exhibiting near-unity QY to wavelengths down to 500 nm. We also used TOP-Se, a conventional precursor, to grow ZnSe shells on small InP cores. Due to its low reactivity, shelling required a substantially higher reaction temperature (130 °C vs 340 °C). As expected, this high temperature induced substantial alloying, yielding greatly red-shifted PL at 584 nm (pink in Figure 4a). Moreover, the substantial particle instability during growth greatly increased the size distribution, resulting in a much larger peak width (80 nm). Even with the cation doping strategy developed to increase the Tm of InP cores, alloying was inevitable with extremely small cores.19–21 Notably, BBN-SeH, allows controlled growth of the ZnSe shell directly onto the small InP core (1.7 nm) even without cation doping, greatly simplifying the synthesis process as well as the photophysics of the resulting QDs. Without the InP QD cores, no significant change in the absorption spectrum was observed under the ZnSe shell deposition environment, confirming the surface-assisted growth of BBN-EH (Figure S14d).
Figure 4.
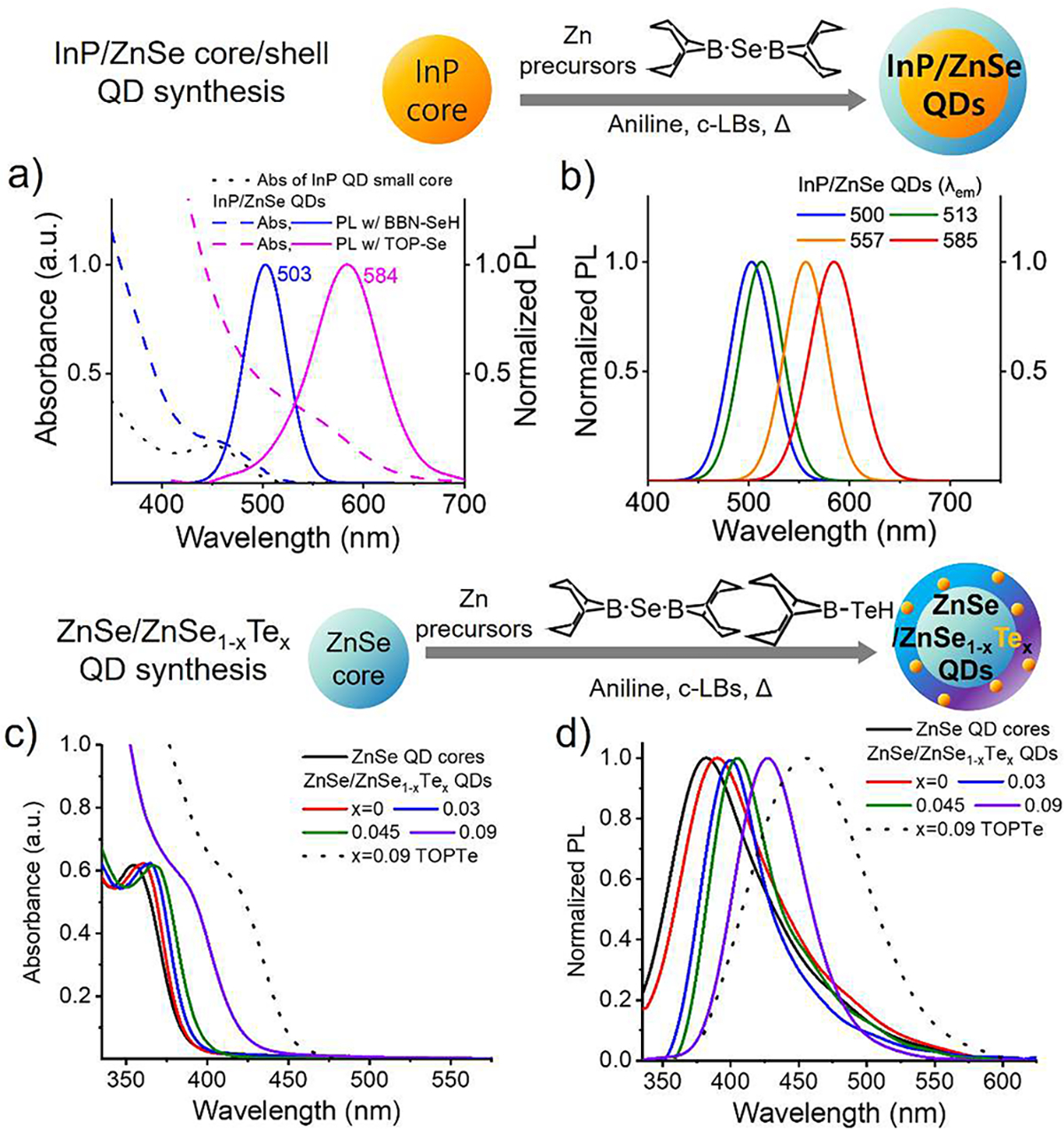
Utilizing BBN-EH for controlled shelling on small cores and controlled surface doping. (a) Absorption and photoluminescence(PL) spectra of InP/ZnSe core/shell QDs grown from small cores (1.7 nm) using DMAP (blue) and TOP-Se (pink). (b) PL profile of synthesized InP/ZnSe QDs covering the wide range of 500–585 nm. (c) Absorption and (d) PL spectrum of ZnSe core seed (black) and Te doped ZnSe (ZnSe/ZnSe1–xTex QDs where x = 0 (red), 0.03 (blue), 0.045 (green), and 0.09(purple)) QDs produced using BBN-SeH and BBN-TeH.
Further, we leveraged the surface-assisted, quantitative conversion properties of BBN-EH to precisely incorporate Te atoms into the ZnSe QD matrix, enabling controlled positioning and density of different chalcogenide atoms within QDs. Controlled incorporation of Te atoms into the ZnSe QD matrix was accomplished by adding a mixture of BBN-SeH and BBN-TeH during growth (Figure 4c, d). The amount of Te within the anion-alloyed shells was controlled by adjusting the ratio of BBN-SeH to BBN-TeH. Given that BBN-EH quantitatively deposits on the QD surface, we expect the Te % in the alloyed shell to align with the input ratio. In our experiments, we deposited two MLs of ZnSe1–xTex shells on 2.5 nm ZnSe QDs with different concentrations of BBN-TeH (x = 0%, 3%, 4.5%, and 9%). It is notable that the addition of a small amount of TOP-Se was necessary to improve the stability of the ZnSe QDs during growth. Volumetric calculations predicted Te-to-Se ratios of 0%, 2%, 3%, and 6% in the final QDs. Elemental analysis via ICP-AES (using Optima 8300) confirmed that these expected Te percentages were well-matched with the experimental values (Table S2). QDs were dissolved in aqua regia for ICP-AES measurements to ensure complete dissolution, while avoiding hydride formation. Due to the surface-assisted conversion mechanism of BBN-TeH and BBN-SeH and the mild reaction temperatures (i.e., 150–170 °C, c-LB: DMAP), we expect Te atoms to be incorporated at the growing surface rather than internally. Although imaging the radial distribution of Te atoms within the ZnSe QD matrix was infeasible due to their extremely small size (~3 nm), the optical properties of the produced QDs confirmed the outer layer distribution of Te atoms. Thus, we name these QDs as ZnSe/ZnSe1–xTex QDs where x = 0, 0.03, 0.045, and 0.09.
Figure 4c–d display the absorption and PL spectra of ZnSe QDs before and after the deposition of two MLs of ZnSe1–xTex alloy shells. The red shifts observed in Te-containing samples, compared to the 0% Te sample, verify the successful incorporation of Te atoms. As the Te content in the shell rose from 0% to 9%, the absorption and PL peaks shifted from 361 to 389 nm and 390 to 427 nm, respectively. A notable effect of increased Te level was the reduction of the broad red tail in the PL spectrum of bare ZnSe QDs, resulting in a symmetric Gaussian peak with a 60 nm FWHM for the ZnSe/ZnSe0.91Te0.09 QDs (Figure 4d). Additionally, Te incorporation increased the QY from minimal in the 0% Te sample to 18% for ZnSe/ZnSe0.91Te0.09 QDs. The optical characteristics of our ZnSe/ZnSe0.91Te0.09 QDs were markedly different from those of the previously reported internally alloyed ZnSe0.94Te0.06 QDs,16 despite having similar chemical compositions (Te-to-Se ratio: 6%) and sizes (3 nm). Our QDs exhibited a significantly bluer PL (420 vs 457 nm), aligning with the expected outer layer distribution of Te atoms. The absence of Te in the core results in a higher band gap than that for QDs with Te incorporated internally.46 The PL peak shapes also differed between the two QD types, confirming different internal structures. While internally alloyed ZnSe0.94Te0.06 QDs exhibited a significant red tail,16 similar to ZnSe QDs, our ZnSe/ZnSe0.91Te0.09 QDs exhibit a symmetric Gaussian PL profile. Several lines of evidence suggest that the extent of Te atom smearing into the core is low. First, previous studies showed that anion diffusion induces structural deformation, therefore, is inefficient.47,48 The low-temperature growth employed for BBN-EH is unlikely to cause efficient diffusion into the core. In contrast, high-temperature growth at 270–280 °C using TOP-Te induced a significantly higher red shift and extensive broadening of the fluorescence peak, indicating extensive smearing of Te atoms into the QD cores. Second, according to a prior DFT calculation, when Te atoms are uniformly dispersed near the surface of ZnSe/ZnSe1–xTex QDs, the density of states near the HOMO level closely resembles that of ZnSe QDs.46 The absorption spectra of our ZnSe/ZnSe1–xTex QDs produced with BBN-TeH retain the HWHM and the shape of the first absorption peak of bare ZnSe QDs, indicating a uniform surface distribution of Te atoms. Additionally, the QY was substantially higher for ZnSe/ZnSe0.91Te0.09 QDs (18% vs 4%), suggesting a protective effect of Te atoms on the QD surface. Finally, in our synthesis, adding more than 15% Te in the outermost layer caused a significant red shift, reduced PL intensity, and peak broadening. This points to a type-II core–shell energy alignment and further supports the outer layer incorporation of Te atoms in our QDs.
BBN-EH-based anion doping achieves a level of control inaccessible with conventional methods, including bottom-up synthesis with mixed precursors, postsynthesis ion diffusion, or layer-by-layer deposition with highly reactive precursors. A bottom-up synthesis produces QDs with uniformly distributed minor atoms.49 Moreover, differences in precursor reactivity and low reaction yields make it difficult to predict the incorporation efficiency. Ion diffusion has primarily been used for cation doping, as demonstrated in Ag+, Cu+, Mn2+ cation exchange in ZnSe QDs.50,51 Different from cation doping, anion doping is highly inefficient47,48 due to the substantial internal strains and lattice mismatch. Thus, anion doping requires excess precursors, high reaction temperatures,52 or specific surface ligands to promote the exchange process,53 making it difficult to control the position and density of the minor atoms.
Layer-by-layer deposition using a mixture of highly reactive precursors is comparable to the BBN-EH-based growth method in terms of quantitative incorporation and avoidance of undesired alloying.54 However, highly reactive precursors pose additional challenges, such as secondary nucleation, high toxicity, and poor morphology control. As shown, the BBN-EH chemistry offers several advantages for controlled QD growth. First, their intermediate reactivity and reactivity tunability enable systematic growth of high-quality shells on cores of various sizes and materials, even those with low Tm. Second, the surface-assisted conversion mechanism and quantitative deposition offer controlled growth without inducing secondary nucleation. For example, it allows for precise incorporation of minor atoms with controlled radial distribution and density within the QD matrix, thereby fine-tuning the optoelectronic properties. Third, the PL peak of same-size cores can be controllably tuned by varying the ratio of BBN-SeH and BBN-TeH. The wavelength range achieved with ZnSe/ZnSeTe QDs (λem: 390–505 nm) fills the gap in the literature, especially in the violet region.16,33 Lastly, Te incorporation on the surface improves the optical quality of QDs, including enhanced QY and a symmetric PL profile.
(BBN)2Se as Precursor: Controlled Synthesis of ZnSe Nanoplatelets with Excellent Thermal Stability.
We examined the properties of (BBN)2Se, an intermediate compound used for the in situ production of BBN-SeH, as a Se precursor. Unlike BBN-SeH and BBN-TeH, (BBN)2Se drives efficient nucleation under specific conditions. We envisioned combining the nucleation capability of (BBN)2Se and the strong surface coordination of BBN-SeH to synthesize thermostable ZnSe NPLs. NPLs have attracted attention as pure-color emitters and sensors due to their atomic precision in the quantum confinement axis. Previous studies have reported a quantized reaction pathway for ZnSe NPL synthesis in which different NPL species emerge sequentially via a surface-reaction-limited-nucleation and growth mechanism.55 In this mechanism, the emergence of a lower band gap NPL species is initiated by the dissolution of existing NPL species, nucleation of atomically precise species, and lateral growth where the feeding species came from the dissolved solutes. The nucleation barrier and energy slope for growth are different for different nanostructures, activating different reaction pathways at different temperatures. We hypothesized that strong coordination of BBN-SeH to NPLs would prevent the dissolution of NPLs and lock their structure, resulting in thermostable NPLs. We synthesized NPLs by injecting (BBN)2Se, c-LB, and aniline into a reaction vessel containing zinc nitrate, OLAm, and octylamine at 100 °C. Upon injection, (BBN)2Se rapidly nucleated and formed NPLs. The residual (BBN)2Se that did not participate in nucleation is converted to BBN-SeH via aniline, a form that does not nucleate, inhibiting secondary nucleation and serving as ligands for NPLs (Figure 5a).
Figure 5b–c confirms that BBN-SeH and BBN-TeH do not initiate nucleation even at temperatures well above Tind (BBN-SeH:1-methylpyrrolidine: 140 °C; BBN-TeH:1-methylpyrrolidine: 170 °C), as expected from their surface-assisted conversion mechanism. For this test, pure BBN-SeH was separately synthesized and added to the reaction vessel instead of in situ production to avoid the involvement of (BBN)2Se. In comparison, when elemental Se was used as the selenium precursor, two characteristic peaks at 328 and 346 nm emerged in the absorption profile at 150 °C, showing the formation of 345 nm ZnSe NPLs55 (Figure 5e). Yet, alongside these peaks, the absorption profile continuously increased below 400 nm, indicating the generation of heterogeneously sized ZnSe QDs via secondary nucleation. TEM images confirmed the presence of both NPLs and heterogeneously sized QDs (Figure S15). When (BBN)2Se was used alone, nucleation occurred rapidly, but the formed nanostructure was highly unstable. As shown in Figure 5d, a sharp peak at 328 nm emerged immediately upon injection at 100 °C. However, this peak rapidly shifted to 318 nm and completely disappeared within 3 min at 120 °C.
Injection of (BBN)2Se with aniline produced thermostable ZnSe NPLs. Upon injection and subsequent heating, a sharp peak at 316 nm appeared at 130 °C (Figure 5f). TEM analysis showed that this absorption profile corresponds to ZnSe NPLs (Figure S16). We determined that this population is 3 ML wurtzite ZnSe NPLs, as the observed transition energy of 3.92 eV (316 nm) and 4.06 eV (305 nm) match the calculated band energy for the heavy and light hole of 3 ML w-ZnSe, respectively.55,56 The thickness measured from TEM images aligns with the expected thickness (Figure S17). However, due to the low image contrast, our conclusion is mainly driven by their optical properties. The absorption peak of the produced NPLs remained stable up to 200 °C, indicating excellent thermal stability. These results are in stark contrast to conventionally synthesized ZnSe NPLs, which exhibit significant heat instability.55,58 As the temperature increases from 120 to 150 °C, thicker NPLs form and the NPLs completely dissolve eventually at higher temperatures (>150 °C). Previous studies have shown that the vertical growth of NPLs is inhibited. The formation of thicker NPLs involves the lateral dissolution of thinner NPLs followed by the nucleation of thicker NPLs and their lateral growth.57 The exceptional thermal stability of the (BBN)2Se/aniline-synthesized ZnSe NPLs indicates that the coordination of BBN-SeH strongly stabilizes the NPLs, preventing their lateral dissolution. Indeed, the TEM images of NPLs incubated at 200 °C reveal sizes similar to those of the as-synthesized NPLs, confirming our hypothesis (Figure S16a and S16c). In short, the introduction of aniline results in a partial conversion of (BBN)2Se to BBN-SeH, with the remaining (BBN)2Se driving NPL formation and BBN-SeH coordinating with the NPLs. The surface coordination of BBN-SeH effectively inhibits their lateral dissolution, thereby maintaining their thickness even at high temperatures. Inhibiting thermal dissolution and maintaining atomically defined thickness at high temperatures have been a critical challenge in the colloidal synthesis of 2D materials, including atomically thin 2D sheets, which are gaining attention for applications in flexible transistors59 and large-scale memristors.60 As such, BBN-EH chemistry would provide important insight into the design of solution-based synthetic methods for 2D materials.
Previous studies show that NPLs exclusively allow lateral growth, not vertical growth.55,57,58 Therefore, the production of NPLs of different thicknesses involves a process where existing NPLs dissolve and NPL seeds of different structures nucleate from these dissolved solutes and then undergo lateral growth. Given the strong inhibition of vertical growth in NPLs, we hypothesized that the surface-assisted precursor conversion pathway for BBN-SeH would be inhibited. To test this hypothesis, we added c-LBs (1-methylpyrrolidine and picoline) to the reaction solution. For all c-LB choices, the sharp absorption peak at 316 nm remained unchanged upon heating to temperatures substantially higher than the corresponding initiation temperatures (Figure 5f), confirming the inhibition of surface-assisted vertical growth.
The excellent thermal stability of 316 nm ZnSe NPLs produced with BBN-SeH presents challenges in accessing other populations using the conventional heat-up method. Rather than allowing for the formation of various NPL structures during heating, the use of BBN-SeH chemistry locks NPLs into the 316 nm variant. The previous study showed that the synthesis of different ZnSe NPL structures involves transitions between metastable wells in the potential energy landscape during heating.55 We hypothesized that changing the ligand composition would change the potential energy landscape and activate different reaction pathways, leading to the production of different NPL structures. Indeed, adding BBN-TeH to the (BBN)2Se solution at a ratio of 1:10 produced the previously reported 4 ML w-ZnSe NPLs, an ML thicker than the 316 nm NPLs (Figure 5g).55 Similar to the 316 nm ZnSe NPLs, 345 nm ZnSe NPLs also showed excellent thermal stability in the presence of BBN-TeH and BBN-SeH up to 200 °C (Figure S18). The 345 nm NPLs produced exhibit an absorption profile identical to that of previously reported 345 nm ZnSe NPLs, indicating the absence of Te in the produced NPLs. The observed inability of BBN-TeH to undergo nucleation (Figure. 5c) and the inhibition of vertical growth for NPLs57 explain the absence of Te incorporation in these NPLs. Moreover, the absorption profile and TEM images exhibited no secondary nucleation products, reaffirming the benefit of converting residual (BBN)2Se postnucleation to BBN-SeH, the nucleation-inhibiting form. Conversion of residual (BBN)2Se post nucleation to BBN-SeH, which inhibits nucleation, effectively eliminates the secondary nucleation pathway, greatly improving the purity of the desired products. This result contrasts with the conventional Se precursors, such as elemental Se, which produce a mixture of NPLs and QDs as shown in Figure S15.
CONCLUSION
We have developed organoborane-based Se and Te precursors, universal precursors whose reactivity can be controllably tuned by the addition of boron-coordinating LBs. These precursors not only diversify the limited selection of Se and Te sources but also encompass a medium reactivity range previously inaccessible with existing precursors. Commonly used Se precursors include alkylphosphine-Se, ODE-Se, and OLAm-Se, all of which have low reactivity and require high reaction temperatures (250–340 °C). The specific temperatures required for the reactions depend on multiple factors, including the size and material of the cores, the selection of the cation precursor, and the composition of the ligands. For example, ZnSe QD synthesis using TOP-Se requires 290–300 °C61 while ZnSe shell growth on InP QD cores using the same precursors requires 340 °C.18 Recently, a combination of diphenylphosphine, TOP-Se, and zinc oleate has been used to achieve mild reaction temperatures (140–185 °C).62 However, the catalytic property of diphenylphosphine makes it difficult to control the conversion kinetics, and its applications are limited. Commonly used Te precursors are TOP-Te, which has low reactivity and activates at high temperatures (280–300 °C), and ((Me2N)3P:Te), which is highly reactive and activates at 100 °C. The BBN-EH chemistry fills a large gap in the intermediate reactivity range, achieving precursor conversion at 130–270 °C. Moreover, the ability to predictively modulate the reactivity of universal precursors using commercially available chemical additives greatly lowers the technical barrier to achieving optimal reactivity for various nanomaterial syntheses. Similar to our prior work on BBN-SH,11,12 these precursors operate via a surface-assisted conversion mechanism, requiring a QD surface for activation and preventing secondary nucleation. Leveraging their intermediate reactivity and controllable nature, we enabled the systematic growth of high-quality shells on various cores, including those with low Tm incompatible with the high reaction temperatures typically required by conventional precursors. Specifically, we demonstrated high-quality ZnSe shelling on 1.7 nm pure InP cores at mild temperatures (130–240 °C). Moreover, we have shown that the surface-assisted quantitative conversion property of BBN-EH enables precise control of minor anion incorporation. By varying the ratio of different chalcogenide precursors and injection timing, we were able to precisely control the radial distribution and density of different chalcogenide anions, facilitating fine-tuning of the optoelectronic properties. Here, we incorporated Te atoms as minor atoms into the ZnSe QD matrix and showed that QDs with Te atoms in the outer layers exhibit distinct photophysical properties compared with volumetrically alloyed QDs. Finally, we investigated the properties of (BBN)2Se, an intermediate compound in BBN-SeH synthesis, as a precursor. Unlike BBN-SeH, (BBN)2Se undergoes nucleation. By combining the nucleation capability of (BBN)2Se with surface stabilization by BBN-SeH, we synthesized previously unreported 316 nm ZnSe NPLs. Surface coordination of BBN-SeH renders the NPLs highly thermostable up to 200 °C. Moreover, the nucleation inhibition property of BBN-SeH effectively prevented secondary nucleation, yielding pure NPLs with no secondary nucleation products. The introduction of BBN-TeH induces the formation of a different NPL species, 345 nm ZnSe NPLs, by modifying the energy landscape through altered ligand composition. Our study demonstrates that the organoborane-based Se and Te precursors effectively address the critical reactivity gap in the intermediate range by employing simple yet versatile chemistry, thereby providing an exceptional level of control in nanomaterial synthesis. Furthermore, their surface-assisted conversion mechanism and ability to stabilize surfaces offer additional handles to regulate nanomaterial growth and enhance their thermostability.
METHOD
Di((1S,5S)-9-borabicyclo[3.3.1]nonan-9-yl)selane ((BBN)2Se) Synthesis.
Di((1S,5S)-9-borabicyclo[3.3.1]-nonan-9-yl)selane ((BBN)2Se) was synthesized using a previously reported method with minor modification. (Köster, R.; Seidel, G.; Siebert, W.; Gangnus, B. in Inorg. Synth. Vol. 29 (Wiley, New Jersey, 1992).) Under the inert gas environment, 10 mmol (2.44 g) of 9-borabicyclo[3.3.1]nonane (9-BBN) dimer and 10.5 mmol (0.83 g) of selenium powder were placed in a flask. While the flask with the mixture of 9-BBN and selenium powder was connected under a nitrogen flow from the Schlenk line, 50 mL of freshly distilled mesitylene was slowly added and heated up to 150 °C. After the solution became clear, the solution was refluxed for 4 h. The residual solvent was removed by rotary evaporation. (1s,5s)-9-Borabicyclo[3.3.1]nonane-9-selenol (BBN-SeH) was freshly activated by adding aniline. Pure (BBN)2Se (2.72 g, 85% yield) was isolated by vacuum sublimation. 1H NMR (ppm, CDCl3, 400 Hz): δ = 2.20–1.13 (m, 28H, −BC8H14); 11B NMR (ppm, CDCl3, 400 MHz): δ = 59 (s).
(1s,5s)-9-Borabicyclo[3.3.1]nonane-9-tellurol (BBN-TeH) Synthesis.
Under the inert gas environment, 10.5 mmol (1.34 g) of tellurium powder was placed in a flask. While the flask with the tellurium powder was placed in an ice bath and connected under a nitrogen flow from the Schlenk line, 5 mL of a LiAlH4 solution (2 M in THF) was slowly added. Hydrogen gas was formed immediately. Pure LiAlTeH2 solution (supernatant) was purified by centrifugation. While the flask with 5 mmol (1.22 g) of 9-borabicyclo[3.3.1]nonane (9-BBN) dimer was placed in an ice bath and connected under a nitrogen flow from the Schlenk line, purified LiAlTeH2 solution was slowly added and heated up to room temperature. The residual solvent was removed by rotary evaporation. Pure BBN-TeH (2.24 g, 89%) was isolated by vacuum sublimation. 1H NMR (ppm, CDCl3, 400 Hz): δ = 2.20–1.13 (m, 14H, −BC8H14); 11B NMR (ppm, CDCl3, 400 MHz): δ = 62 (s).
ZnSe and ZnTe QD Synthesis.
ZnSe QDs were synthesized according to a literature method with minor modifications (Nano Lett. 2020, 20, 2387–2395). 5 mL of OLAm were loaded in the flask and degassed under vacuum at 100 °C for 30 min. Then the solution was heated to 300 °C under nitrogen, followed by a slow addition of 0.3 mL of TOP-Se (1.5 M). When the temperature reached 300 °C, 1 mL of diethylzinc solution (0.5 M, dissolved in TOP) was injected quickly into the flask under vigorous stirring. The growth temperature was kept at 290 °C. The core size was monitored by measuring the absorption spectra of aliquots. After the growth process was completed, an additional 5 mL of ODE was slowly introduced into the flask. When the targeted size of ZnSe QDs was obtained, the reaction was quenched by cooling the solution to room temperature as quickly as possible. ZnSe QDs were purified with hexane and ethanol and dispersed in hexane. For ZnTe QD synthesis, 0.45 mL of TOP-Te (1.0 M) was used instead of TOP-Se, while others were kept the same as the ZnSe QD synthesis conditions.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a start-up fund from the University of Illinois at Urbana–Champaign. We thank the SCS NMR Lab and Microanalysis Lab, University of Illinois, for its technical support. The Varian Inova 400-MHz NMR spectrometer was obtained with the financial support of the Roy J. Carver Charitable Trust. Transmission electron microscope measurements were carried out in part in the Materials Research Laboratory Central Research Facilities, University of Illinois.
Funding
This work was supported by the National Institutes for Health (R35GM147420; H.-S.H. and J.P) and National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (2021R1C1C2093095; J.P.). J.P. has been also supported by Basic Medical Science Facilitation Program through the Catholic Medical Center of The Catholic University of Korea funded by the Catholic Education Foundation.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.3c13159.
11B NMR spectra, mass spectra, TEM images, supplementary data, and a summary of the TEM and ICP-AES analysis (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acsnano.3c13159
Contributor Information
Joonhyuck Park, Department of Chemistry, University of Illinois at Urbana–Champaign, Urbana, Illinois 61801, United States; Department of Medical Life Sciences and Department of Biomedicine & Health Sciences, College of Medicine, The Catholic University of Korea, Seoul 06591, Republic of Korea.
Hee-Sun Han, Department of Chemistry, University of Illinois at Urbana–Champaign, Urbana, Illinois 61801, United States; Carl R. Woese Institute for Genomic Biology, University of Illinois at Urbana–Champaign, Urbana, Illinois 61801, United States.
REFERENCES
- (1).Giri A; Park G; Jeong U Layer-Structured Anisotropic Metal Chalcogenides: Recent Advances in Synthesis, Modulation, and Applications. Chem. Rev. 2023, 123, 3329–3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Tamang S; Lincheneau C; Hermans Y; Jeong S; Reiss P Chemistry of InP Nanocrystal Syntheses. Chem. Mater. 2016, 28, 2491–2506. [Google Scholar]
- (3).Lim J; Park Y-S; Wu K; Yun HJ; Klimov VI Droop-Free Colloidal Quantum Dot Light-Emitting Diodes. Nano Lett. 2018, 18, 6645–6653. [DOI] [PubMed] [Google Scholar]
- (4).Lee KJ; Kim G; Lim J; Nah S; Jeong KS; Cho M Tailoring Transition Dipole Moment in Colloidal Nanocrystal Thin Film on Nanocomposite Materials. Adv. Opt. Mater. 2022, 10, No. 2102050. [Google Scholar]
- (5).Campos MP; De Roo J; Greenberg MW; McMurtry BM; Hendricks MP; Bennett E; Saenz N; Sfeir MY; Abécassis B; Ghose SK; Owen JS Growth Kinetics Determine the Polydispersity and Size of PbS and PbSe Nanocrystals. Chem. Sci. 2022, 13, 4555–4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Harris DK; Bawendi MG Improved Precursor Chemistry for the Synthesis of III–V Quantum Dots. J. Am. Chem. Soc. 2012, 134, 20211–20213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Gary DC; Glassy BA; Cossairt BM Investigation of Indium Phosphide Quantum Dot Nucleation and Growth Utilizing Triarylsilylphosphine Precursors. Chem. Mater. 2014, 26, 1734–1744. [Google Scholar]
- (8).Chen L; Hu H; Chen Y; Li Y; Gao J; Li G Sulfur Precursor Reactivity Affecting the Crystal Phase and Morphology of Cu2–xS Nanoparticles. Chem. Eur. J. 2021, 27, 1057–1065. [DOI] [PubMed] [Google Scholar]
- (9).Hendricks MP; Campos MP; Cleveland GT; Jen-La Plante I; Owen JS A Tunable Library of Substituted Thiourea Precursors to Metal Sulfide Nanocrystals. Science 2015, 348, 1226–1230. [DOI] [PubMed] [Google Scholar]
- (10).Hamachi LS; Yang H; Jen-La Plante I; Saenz N; Qian K; Campos MP; Cleveland GT; Rreza I; Oza A; Walravens W; Chan EM; Hens Z; Crowther AC; Owen JS Precursor Reaction Kinetics Control Compositional Grading and Size of CdSe1–xSx Nanocrystal Heterostructures. Chem. Sci. 2019, 10, 6539–6552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Park J; Jayaraman A; Wang X; Zhao J; Han H-S Nanocrystal Precursor Incorporating Separated Reaction Mechanisms for Nucleation and Growth to Unleash the Potential of Heat-up Synthesis. ACS Nano 2020, 14, 11579–11593. [DOI] [PubMed] [Google Scholar]
- (12).Park J; Jayaraman A; Schrader AW; Hwang GW; Han H-S Controllable Modulation of Precursor Reactivity using Chemical Additives for Systematic Synthesis of High-Quality Quantum Dots. Nat. Commun. 2020, 11, 5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kamath A; Schaller RD; Guyot-Sionnest P Bright Fluorophores in the Second Near-Infrared Window: HgSe/CdSe Quantum Dots. J. Am. Chem. Soc. 2023, 145, 10809–10816. [DOI] [PubMed] [Google Scholar]
- (14).Shen X; Kamath A; Guyot-Sionnest P Mid-Infrared Cascade Intraband Electroluminescence with HgSe–CdSe Core–Shell Colloidal Quantum Dots. Nat. Photonics 2023, 17, 1042–1046. [Google Scholar]
- (15).Liu S; Xiong K; Wang K; Liang G; Li M-Y; Tang H; Yang X; Huang Z; Lian L; Tan M; Wang K; Gao L; Song H; Zhang D; Gao J; Lan X; Tang J; Zhang J Efficiently Passivated PbSe Quantum Dot Solids for Infrared Photovoltaics. ACS Nano 2021, 15, 3376–3386. [DOI] [PubMed] [Google Scholar]
- (16).Kim T; Kim K-H; Kim S; Choi S-M; Jang H; Seo H-K; Lee H; Chung D-Y; Jang E Efficient and Stable Blue Quantum Dot Light-Emitting Diode. Nature 2020, 586, 385–389. [DOI] [PubMed] [Google Scholar]
- (17).Mu J; Gao X Synthesis of CdSe Nanocrystals through a Reaction of H2Se Gas and Cd2+ Ions in Aqueous Medium and Their Optical and Structural Properties. J. Dispers. Sci. Technol. 2005, 26, 763–767. [Google Scholar]
- (18).Won Y-H; Cho O; Kim T; Chung D-Y; Kim T; Chung H; Jang H; Lee J; Kim D; Jang E Highly Efficient and Stable InP/ZnSe/ZnS Quantum Dot Light-Emitting Diodes. Nature 2019, 575, 634–638. [DOI] [PubMed] [Google Scholar]
- (19).Shen W; Tang H; Yang X; Cao Z; Cheng T; Wang X; Tan Z; You J; Deng Z Synthesis of Highly Fluorescent InP/ZnS Small-Core/Thick-Shell Tetrahedral-Shaped Quantum Dots for Blue Light-Emitting Diodes. J. Mater. Chem. C 2017, 5, 8243–8249. [Google Scholar]
- (20).Shen C; Zhu Y; Tao H; Li J; Zou J; Wang L; Liang J; Xiao X; Xu X; Xu G Blue-Emitting InP/GaP/ZnS Quantum Dots with Enhanced Stability by Siloxane Capping: Implication for Electroluminescent Devices. ACS Appl. Nano Mater. 2022, 5, 2801–2811. [Google Scholar]
- (21).Huang F; Bi C; Guo R; Zheng C; Ning J; Tian J Synthesis of Colloidal Blue-Emitting InP/ZnS Core/Shell Quantum Dots with the Assistance of Copper Cations. J. Phys. Chem. Lett. 2019, 10, 6720–6726. [DOI] [PubMed] [Google Scholar]
- (22).Frenette LC; Krauss TD Uncovering Active Precursors in Colloidal Quantum Dot Synthesis. Nat. Commun. 2017, 8, 2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Yu K; Liu X; Zeng Q; Leek DM; Ouyang J; Whitmore KM; Ripmeester JA; Tao Y; Yang M Effect of Tertiary and Secondary Phosphines on Low-Temperature Formation of Quantum Dots. Angew. Chem., Int. Ed. 2013, 52, 4823–4828. [DOI] [PubMed] [Google Scholar]
- (24).Bullen C; van Embden J; Jasieniak J; Cosgriff JE; Mulder RJ; Rizzardo E; Gu M; Raston CL High Activity Phosphine-Free Selenium Precursor Solution for Semiconductor Nanocrystal Growth. Chem. Mater. 2010, 22, 4135–4143. [Google Scholar]
- (25).Liu Y; Yao D; Shen L; Zhang H; Zhang X; Yang B Alkylthiol-Enabled Se Powder Dissolution in Oleylamine at Room Temperature for the Phosphine-Free Synthesis of Copper-Based Quaternary Selenide Nanocrystals. J. Am. Chem. Soc. 2012, 134, 7207–7210. [DOI] [PubMed] [Google Scholar]
- (26).Hou B; Benito-Alifonso D; Webster R; Cherns D; Galan MC; Fermín DJ Rapid Phosphine-Free Synthesis of CdSe Quantum Dots: Promoting the Generation of Se Precursors using a Radical Initiator. J. Mater. Chem. A 2014, 2, 6879–6886. [Google Scholar]
- (27).Campos MP; Hendricks MP; Beecher AN; Walravens W; Swain RA; Cleveland GT; Hens Z; Sfeir MY; Owen JS A Library of Selenourea Precursors to PbSe Nanocrystals with Size Distributions near the Homogeneous Limit. J. Am. Chem. Soc. 2017, 139, 2296–2305. [DOI] [PubMed] [Google Scholar]
- (28).VanOrman ZA; Conti CR III; Strouse GF; Nienhaus L Red-to-Blue Photon Upconversion Enabled by One-Dimensional CdTe Nanorods. Chem. Mater. 2021, 33, 452–458. [Google Scholar]
- (29).Böhm ML; Jellicoe TC; Tabachnyk M; Davis NJLK; Wisnivesky-Rocca-Rivarola F; Ducati C; Ehrler B; Bakulin AA; Greenham NC Lead Telluride Quantum Dot Solar Cells Displaying External Quantum Efficiencies Exceeding 120%. Nano Lett. 2015, 15, 7987–7993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ouyang J; Graddage N; Lu J; Zhong Y; Chu T-Y; Zhang Y; Wu X; Kodra O; Li Z; Tao Y; Ding J Ag2Te Colloidal Quantum Dots for Near-Infrared-II Photodetectors. ACS Appl. Nano Mater. 2021, 4, 13587–13601. [Google Scholar]
- (31).Chen M; Lan X; Tang X; Wang Y; Hudson MH; Talapin DV; Guyot-Sionnest P High Carrier Mobility in HgTe Quantum Dot Solids Improves Mid-IR Photodetectors. ACS Photonics 2019, 6, 2358–2365. [Google Scholar]
- (32).Zhang H; Peterson JC; Guyot-Sionnest P Intraband Transition of HgTe Nanocrystals for Long-Wave Infrared Detection at 12 μm. ACS Nano 2023, 17, 7530–7538. [DOI] [PubMed] [Google Scholar]
- (33).Lee S-H; Han C-Y; Song S-W; Jo D-Y; Jo J-H; Yoon S-Y; Kim H-M; Hong S; Hwang JY; Yang H ZnSeTe Quantum Dots As an Alternative to InP and Their High-Efficiency Electroluminescence. Chem. Mater. 2020, 32, 5768–5775. [Google Scholar]
- (34).Jang E-P; Han C-Y; Lim S-W; Jo J-H; Jo D-Y; Lee S-H; Yoon S-Y; Yang H Synthesis of Alloyed ZnSeTe Quantum Dots as Bright, Color-Pure Blue Emitters. ACS Appl. Mater. Interfaces. 2019, 11, 46062–46069. [DOI] [PubMed] [Google Scholar]
- (35).Detty MR; Seidler MD Bis(trialkylsilyl) Chalcogenides. 1. Preparation and Reduction of Group VIA Oxides. J. Org. Chem. 1982, 47, 1354–1356. [Google Scholar]
- (36).Zhang J; Jin S; Fry HC; Peng S; Shevchenko E; Wiederrecht GP; Rajh T Synthesis and Characterization of Wurtzite ZnTe Nanorods with Controllable Aspect Ratios. J. Am. Chem. Soc. 2011, 133, 15324–15327. [DOI] [PubMed] [Google Scholar]
- (37).Sun H; Wang F; Buhro WE Tellurium Precursor for Nanocrystal Synthesis: Tris(dimethylamino)phosphine Telluride. ACS Nano 2018, 12, 12393–12400. [DOI] [PubMed] [Google Scholar]
- (38).Vreeland EC; Watt J; Schober GB; Hance BG; Austin MJ; Price AD; Fellows BD; Monson TC; Hudak NS; Maldonado-Camargo L; Bohorquez AC; Rinaldi C; Huber DL Enhanced Nanoparticle Size Control by Extending LaMer’s Mechanism. Chem. Mater. 2015, 27, 6059–6066. [Google Scholar]
- (39).Toufanian R; Zhong X; Kays JC; Saeboe AM; Dennis AM Correlating ZnSe Quantum Dot Absorption with Particle Size and Concentration. Chem. Mater. 2021, 33, 7527–7536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Köuster R; Seidel G; Schüuβler W; Wrackmeyer B Die ersten Organobor-Tellur-Verbindungen[1]. Chem. Ber. 1995, 128, 87–89. [Google Scholar]
- (41).Sharma RC; Chang YA Thermodynamic Analysis and Phase Equilibria Calculations for the Zn-Te, Zn-Se and Zn-S Systems. J. Cryst. Growth 1988, 88, 193–204. [Google Scholar]
- (42).Zhang Z; Li JC; Jiang Q Modelling for Size-Dependent and Dimension-Dependent Melting of Nanocrystals. J. Phys. D: Appl. Phys. 2000, 33, 2653. [Google Scholar]
- (43).Toufanian R; Chern M; Kong VH; Dennis AM Engineering Brightness-Matched Indium Phosphide Quantum Dots. Chem. Mater. 2021, 33, 1964–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Cao F; Wang S; Wang F; Wu Q; Zhao D; Yang X A Layer-by-Layer Growth Strategy for Large-Size InP/ZnSe/ZnS Core–Shell Quantum Dots Enabling High-Efficiency Light-Emitting Diodes. Chem. Mater. 2018, 30, 8002–8007. [Google Scholar]
- (45).Fang X; Zhai W; Zhang K; Wang Y; Yao L; Tian C; Wan Y; Hou R; Li Y; Chen W; Ran G, Wide Range Tuning of the Size and Emission Color of CH3NH3PbBr3 Quantum Dots by Surface Ligands. AIP Adv. 2017, 7. DOI: 10.1063/1.4994995 [DOI] [Google Scholar]
- (46).Imran M; Paritmongkol W; Mills HA; Hassan Y; Zhu T; Wang Y-K; Liu Y; Wan H; Park SM; Jung E; Tam J; Lyu Q; Cotella GF; Ijaz P; Chun P; Hoogland S Molecular-Additive-Assisted Tellurium Homogenization in ZnSeTe Quantum Dots. Adv. Mater. 2023, 35, No. 2303528. [DOI] [PubMed] [Google Scholar]
- (47).Saruyama M; So Y-G; Kimoto K; Taguchi S; Kanemitsu Y; Teranishi T Spontaneous Formation of Wurzite-CdS/Zinc Blende-CdTe Heterodimers through a Partial Anion Exchange Reaction. J. Am. Chem. Soc. 2011, 133, 17598–17601. [DOI] [PubMed] [Google Scholar]
- (48).Choi D; Lee S; Lee J; Cho K-S; Kim S-W Disodium Diselenide in Colloidal Nanocrystals: Acting As an Anion Exchange Precursor, a Metal Selenide Precursor, and a Chalcogenide Ligand. Chem. Commun. 2015, 51, 899–902. [DOI] [PubMed] [Google Scholar]
- (49).Stavrinadis A; Konstantatos G Strategies for the Controlled Electronic Doping of Colloidal Quantum Dot Solids. ChemPhysChem 2016, 17, 632–644. [DOI] [PubMed] [Google Scholar]
- (50).Son DH; Hughes SM; Yin Y; Paul Alivisatos A Cation Exchange Reactions in Ionic Nanocrystals. Science 2004, 306, 1009–1012. [DOI] [PubMed] [Google Scholar]
- (51).Zhou Z-Q; Yang L-Y; Yan R; Zhao J; Liu Y-Q; Lai L; Jiang F-L; Maskow T; Liu Y Mn-Doped ZnSe Quantum Dots Initiated Mild and Rapid Cation Exchange for Tailoring the Composition and Optical Properties of Colloid Nanocrystals: Novel Template, New Applications. Nanoscale 2017, 9, 2824–2835. [DOI] [PubMed] [Google Scholar]
- (52).Park J; Zheng H; Jun Y -w.; Alivisatos, A. P. Hetero-Epitaxial Anion Exchange Yields Single-Crystalline Hollow Nanoparticles. J. Am. Chem. Soc. 2009, 131, 13943–13945. [DOI] [PubMed] [Google Scholar]
- (53).Li X; Ji M; Li H; Wang H; Xu M; Rong H; Wei J; Liu J; Liu J; Chen W; Zhu C; Wang J; Zhang J Cation/Anion Exchange Reactions toward the Syntheses of Upgraded Nanostructures: Principles and Applications. Matter 2020, 2, 554–586. [Google Scholar]
- (54).Tamang S; Lee S; Choi H; Jeong S Tuning Size and Size Distribution of Colloidal InAs Nanocrystals via Continuous Supply of Prenucleation Clusters on Nanocrystal Seeds. Chem. Mater. 2016, 28, 8119–8122. [Google Scholar]
- (55).Cunningham PD; Coropceanu I; Mulloy K; Cho W; Talapin DV Quantized Reaction Pathways for Solution Synthesis of Colloidal ZnSe Nanostructures: A Connection between Clusters, Nanowires, and Two-Dimensional Nanoplatelets. ACS Nano 2020, 14, 3847–3857. [DOI] [PubMed] [Google Scholar]
- (56).Basalaeva LS; Grafova VP; Duda TA; Kurus NN; Vasiliev RB; Milekhin AG Phonons of Atomically Thin ZnSe Nanoplatelets Grown by the Colloidal Method. J. Phys. Chem. C 2023, 127, 13112–13119. [Google Scholar]
- (57).Ott FD; Riedinger A; Ochsenbein DR; Knüsel PN; Erwin SC; Mazzotti M; Norris DJ Ripening of Semiconductor Nanoplatelets. Nano Lett. 2017, 17, 6870–6877. [DOI] [PubMed] [Google Scholar]
- (58).Park H; Chung H; Kim W Synthesis of Ultrathin Wurtzite ZnSe Nanosheets. Mater. Lett. 2013, 99, 172–175. [Google Scholar]
- (59).Carey T; Cassidy O; Synnatschke K; Caffrey E; Garcia J; Liu S; Kaur H; Kelly AG; Munuera J; Gabbett C; O’Suilleabhain D; Coleman JN High-Mobility Flexible Transistors with Low-Temperature Solution-Processed Tungsten Dichalcogenides. ACS Nano 2023, 17, 2912–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Bala A; So B; Pujar P; Moon C; Kim S In Situ Synthesis of Two-Dimensional Lateral Semiconducting-Mo:Se//Metallic-Mo Junctions Using Controlled Diffusion of Se for High-Performance Large-Scaled Memristor. ACS Nano 2023, 17, 4296–4305. [DOI] [PubMed] [Google Scholar]
- (61).Ji B; Koley S; Slobodkin I; Remennik S; Banin U ZnSe/ZnS Core/Shell Quantum Dots with Superior Optical Properties through Thermodynamic Shell Growth. Nano Lett. 2020, 20, 2387–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Long Z; Liu M; Wu X.-g.; Gu K; Yang G; Chen Z; Liu Y; Liu R; Zhong H A Reactivity-Controlled Epitaxial Growth Strategy for Synthesizing Large Nanocrystals. Nat. Synthesis 2023, 2, 296–304. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.