

Abstract
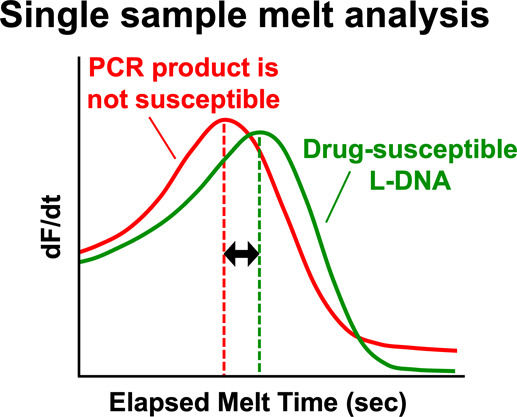
The melt analysis feature in most real-time polymerase chain reaction (PCR) instruments is a simple method for determining if expected or unexpected products are present. High-resolution melt (HRM) analysis seeks to improve the precision of melt temperature measurements for better PCR product sequence characterization. In the area of tuberculosis (TB) drug susceptibility screening, sequencing has shown that a single base change can be sufficient to make a first-line TB drug ineffective. In this study, a reagent-based calibration strategy based on synthetic left-handed (L)-DNA, designated LHRM, was developed to confirm validation of a PCR product with single base resolution. To test this approach, a constant amount of a double-stranded L-DNA melt comparator was added to each sample and used as a within-sample melt standard. The performance of LHRM and standard HRM was used to classify PCR products as drug-susceptible or not drug-susceptible with a test bed of nine synthetic katG variants, each containing single or multiple base mutations that are known to confer resistance to the first-line TB drug isoniazid (INH). LHRM achieved comparable classification to standard HRM relying only on within-sample melt differences between L-DNA and the unknown PCR product. Using a state-of-the-art calibrated instrument and multiple sample classification analysis, standard HRM was performed at 66.7% sensitivity and 98.8% specificity. Single sample analysis incorporating L-DNA for reagent-based calibration into every sample maintained high performance at 77.8% sensitivity and 98.7% specificity. LHRM shows promise as a high-resolution single sample method for validating PCR products in applications where the expected sequence is known.
Introduction
Sequencing of tuberculosis (TB) drug-resistant strains has shown that many drug-resistant variants have one or more single nucleotide polymorphisms (SNPs) often clustered in contiguous regions of the drug-resistant genome.1−3 These resistance-related characteristic changes provide potential biomarkers for drug treatment decisions. However, requiring sequencing for every positive TB sample as part of a clinical treatment algorithm remains cost-prohibitive in many resource-constrained settings.
When the presence of drug-susceptible cases is high, a follow-on test to confirm drug susceptibility provides a pragmatic first step in a clinical treatment algorithm. The goal is to confirm drug susceptibility for the majority of samples and focus the limited resources on more complex testing for the small number of cases that are not drug-susceptible.
Based on known TB sequencing data, amplification-based susceptibility testing has been used to characterize samples and inform the drug treatment algorithm. These approaches are divided into two categories, direct or indirect testing. Direct testing confirms the presence of one or more specific SNPs that make the strain untreatable by a particular drug. Alternatively, indirect testing, based on the SNP clustering observation, broadly seeks to confirm the presence of the nonmutated, susceptible sequence that makes the strain treatable by a particular drug.
In the presence of multiple SNPs that independently confer resistance, the former SNP-targeted approach requires either implementation of a separate test for each SNP or a multiplexed design including all SNPs. Although this direct testing approach shows promise for particular TB strains of concern,4,5 it remains difficult to scale as the number of drug-resistant SNPs increases.
The latter cluster-based strategy for susceptibility testing is based on polymerase chain reaction (PCR) detection. This approach seeks to validate that the PCR product has the known drug-susceptible sequence by melt analysis. Melt analysis is based on the observation that any given double-stranded DNA sequence dissociates at a characteristic melt temperature (Tm). This property is used to compare the melt temperature of an unknown PCR product to the characteristic melt temperature of the known drug-susceptible wild-type sequence. Any shift from this wild-type melt temperature implies that the unknown test sample contains one or more SNPs. Melt analysis capitalizes on the hardware capabilities of PCR instrumentation and is often available in real-time PCR instruments. Some real-time instruments also offer high-resolution melt (HRM) capabilities by including a temperature calibration feature. Because of many variables that affect the melt properties, standard HRM classifies an unknown sample by comparing the melt temperature of the unknown PCR product to the melt temperature of a known PCR product,4−10 usually included in as additional samples in the assay. The requirement for instrument calibration to enable the comparison of two or more samples is a major source of complexity in these approaches. In the case of current PCR and melt-based TB drug susceptibility tests Xpert MTB/RIF Ultra11 and Xpert MTB/XDR,12 unknown samples are compared to an algorithm-based reference library of Tm signatures from a set of known mutations.13 Proprietary designs around this relatively expensive technology, however, continue to limit its utility, particularly where it is most needed. Since a limited number of point-of-care diagnostics currently offer low-cost TB drug resistance testing by HRM,14−17 there is a demonstrated need for simpler validation of drug susceptibility in the TB treatment algorithm using more widely available real-time PCR instruments.
In this report, a potential single sample approach based on reagent-based calibration is proposed to simplify HRM and avoid the requirement for multiple sample comparisons in every assay. In this design, left helical L-DNA is added to every sample as a standard melt comparator. The approach is based on the assumption that both double-stranded L-DNA additive and double-stranded D-DNA PCR product in the same well are affected by hybridization melt characteristics in the same way. If the melt characteristics of the L-DNA additive and the D-DNA from the PCR amplicon of a drug-susceptible sample are set identical, any difference in melt characteristics between L-DNA and an unknown PCR product is attributed to a change in PCR product sequence. In other words, the drug-susceptible reference sequence is included for comparison to the sample PCR amplicon, not in a separate well as D-DNA, but rather within each sample as L-DNA.
Two key features of L-DNA support the feasibility of this approach. First, published reports suggest that L-DNA does not interfere or participate in PCR reactions18−23 and has been employed in applications where it does not interact with normal biological processes, such as intracellular biosensing24−27 and PCR control.22,23 Second, several reports suggest that L-DNA and naturally occurring D-DNA with identical sequences have identical melt characteristics,24,28,29 suggesting that matching melt characteristics should be possible.
Performance of L-DNA-based HRM (LHRM) was compared to standard HRM using a state-of-the-art HRM instrument for both methods. The assays were applied to drug susceptibility screening for isoniazid (INH), a first-line prodrug therapeutic for TB. The internal comparator L-DNA was synthesized as a 56-nucelotide sequence from the drug-susceptible TB katG gene where over 250 INH-resistance-related mutations are clustered.30 The nine synthetic variants were selected to provide product validation challenges that ranged from relatively easy, due to multibase mutations, to very difficult, due to only a single base mutation. PCR products of these synthetic targets were classified as drug-susceptible or not drug-susceptible and used to compare the LHRM and standard HRM methods.
Experimental Section
DNA Oligonucleotide Design
The melt analysis test bed was developed using the drug-susceptible TB katG gene.30 A single primer set was designed to cover the most prevalent variant S315T found in 94% of INH-resistant clinical isolates31,32 and a subset of the many single or multibase variants in the neighboring region that also confer INH resistance.1,32−35 Single-stranded PCR targets were synthesized with D-DNA sequences of drug-susceptible wild-type katG (H37Rv: 2153889–2156111) and nine clinically relevant drug-resistant katG mutants (Table 1). The selected variants included a range of melt differences from wild type, offering both easy and challenging drug susceptibility classification cases. The theoretical Tm spread of variants was 2.8 °C based on a nearest neighbor oligonucleotide calculator36 with automated settings. Detailed information on the DNA oligonucleotide sequences used in these studies is shown in Table S1. All DNA oligonucleotides employed for development and testing of the assay were synthesized by Integrated DNA Technologies (Coralville, Iowa, USA) or biomers.net (Ulm, Baden-Württemberg, Germany).
Table 1. Sequences (Written 5′-to-3′) of the Drug-Susceptible Wild-Type katG and Nine katG Variantsa.

Each variant has INH-resistance-related mutations (red) that induce theoretical melt differences (Tm diff.) from wild-type (right column).
Standard HRM Approach
The standard HRM approach for drug susceptibility screening is based on a two-sample comparison of Tm’s between an unknown PCR product and a known drug-susceptible PCR product (Figure S1). Reactions were performed in the QuantStudio 5 real-time PCR thermal cycler (Thermo Fisher Scientific #A28137). This highly capable instrument was selected to facilitate standard HRM performance as a state-of-the-art comparison method for LHRM.37Reactions had a 20 μL final volume containing 1X of SensiFAST Probe No-ROX Kit (Bioline #BIO-86005), 1X LCGreen Plus (BioFire Defense, LLC #BCHM-ASY-005), and 250 nM of each katG-specific primer (MEP176 and MEP177). Each target sample contained a final concentration of wild-type (MEP183) or mutant (MEP184–189,197–199) single-stranded DNA target at 2 × 106 copies per reaction. An example of a standard HRM reaction setup is outlined in Table S2. PCR reactions were initiated with a 95 °C hold for 2 min followed by 40 cycles of 95 °C for 5 s and 59 °C for 20 s. A HRM was performed immediately following PCR by annealing 95–50 °C at 0.1 °C/s followed by melting 65–95 °C at 0.025 °C/s (continuous acquisition mode).38−41 Double-stranded DNA PCR product fluorescence was monitored during PCR and during the melt reaction using LCGreen Plus on the green optical channel (excitation 470 ± 15/emission 520 ± 15). Complete details are included in Supporting Information (see page S3).42
Standard HRM Analysis and Statistics
PCR quantification cycle (Cq) was determined with the QuantStudio 5 Design and Analysis Software. Nonamplifying samples did not report Cq and were excluded from the data analysis. Amplifying samples with Cq over 35 were excluded from the data analysis because they did not achieve the PCR plateau phase. Representative PCR amplification curves of samples are included in Figure S11. Tm was calculated with the proprietary QuantStudio 5 Design and Analysis Software based on the first derivative of fluorescence with respect to temperature. Based on Tm analysis of all samples, Tm cutoff points were established to maximize test specificity when classifying standard HRM analyzed samples as drug-susceptible or not. Specificity was maximized to decrease the false-positive rate, i.e., decrease the misdiagnosis of variant samples as drug-susceptible. This maximized specificity strategy is often used for HRM classification of TB samples with drug resistance.10,43,44 Each test sample was individually classified. A sample was classified as drug-susceptible when PCR product Tm was within the drug-susceptible Tm cutoff range of 82.4 and 82.5 °C. Since true positives are known, standard HRM was assessed for its sensitivity and specificity using this Tm cutoff range to classify drug susceptibility among 9 true drug-susceptible samples (n = 3 trials of wild-type in triplicate) and 81 true not drug-susceptible samples (n = 3 trials of 9 variant types in triplicate). In the experiment testing heating variability, significance was evaluated using Tm comparison (unpaired t test, significance level of α = 0.95) of 96-well plate quadrants of S315T as compared to wild-type (n = 1 trial with 24 replicates per sample type). All statistics were performed in Microsoft Excel 2022 except for the sensitivity and specificity analysis that was performed in Python. Complete details are included in Supporting Information (see page S3).
LHRM Approach
LHRM for drug susceptibility screening is based on elapsed melt time (tm) comparison between an unknown PCR product and a drug-susceptible L-DNA comparator within a single sample (Figure S2). To ensure a fair comparison between LHRM and standard HRM, both methods were tested using the same QuantStudio 5 instrument. LHRM used identical PCR cycling, PCR fluorescence monitoring, PCR quantification, melt reaction cycling, reaction loading placement, and heating variability test setup as standard HRM. LHRM statistics were identical to that of standard HRM, except for a data subset testing heating variability. Key changes from standard HRM are the inclusion of an additional reagent (L-DNA), monitoring melt reaction fluorescence on a second optical channel, and analysis of fluorescence changes as a function of time from the start of the QuantStudio 5 continuous mode melt instead of melt temperature provided by the instrument’s calibration.
A double-stranded L-DNA drug-susceptible comparator was synthesized using left helical enantiomeric DNA bases (i.e., L-DNA)45 with an identical sequence to the known drug-susceptible katG sequence. The 56-base L-DNA was synthesized with the same length and sequence as the drug-susceptible PCR amplicon. The double-stranded L-DNA was end-labeled with Texas Red (TXR) fluorophore and Black Hole Quencher 2 (BHQ2) quencher to monitor its behavior during melting on the orange fluorescence channel (excitation 580 ± 10/emission 623 ± 14). Detailed information on the L-DNA oligonucleotide sequences used in these studies is shown in Table S1. LHRM reactions included 2 μL of L-DNA mix with final copy counts of 1 × 1011 copies TXR-labeled forward strand L-DNA (23FEB_katGf56_TXR) and 3 × 1011 copies BHQ2-labeled reverse strand L-DNA (23FEB_katG_56_Rcmp+5_BHQ2) per reaction. An example reaction setup containing the L-DNA additive is outlined in Table S3.
To ensure identical melt characteristics of D-DNA and end-labeled L-DNA, additional experiments were performed varying L-DNA strand concentration and strand ratio. In experiments varying L-DNA strand concentrations, reaction component deviations included 100 nM final concentration of each katG-specific primer and 1 × 1011, 2 × 1011, and 4 × 1011 copies of L-DNA strands (forward and reverse) per reaction. In experiments varying L-DNA forward to reverse strand ratio, reaction component deviations included 100 nM final concentration of each katG-specific primer and 2 μL of L-DNA mix at 1:1, 1:2, and 1:3 ratios of forward to reverse strands for final L-DNA copy numbers of 1 × 1011 copies of forward strand plus 1 × 1011, 2 × 1011, and 3 × 1011 copies of reverse strand, respectively. Linear interpolation of three different L-DNA strand ratios was used to determine the relationship between copies of L-DNA reverse strands per reaction and L-DNA melt measurement. The L-DNA reverse strand copy number with a melt measurement matching that of wild-type PCR product was selected. Complete details are included in Supporting Information (see page S4).
LHRM Analysis and Statistics
Representative PCR amplification curves of samples containing L-DNA are included in Figure S12. Elapsed melt time (tm) was calculated from the second degree Savitsky–Golay polynomials46 at each point (performed in MATLAB 2023A) based on the first derivative of fluorescence with respect to elapsed melt time. Elapsed melt time is a means of Tm reporting derived from the uncalibrated QuantStudio 5 raw data. Here, tm is defined as the elapsed melt time (in seconds) to reach the maximum derivative of fluorescence with respect to elapsed melt time. Significant differences between wild-type PCR product and L-DNA within each sample were assessed using paired t tests (of tm) with a significance level of α = 0.95 (n = 3 trials in triplicate). Test samples were classified as drug-susceptible when sample tm difference was zero. LHRM classification criteria are based on our assumption that L-DNA and PCR product melt characteristics are identical if and only if their sequences match. Specificity was maximized to decrease the false-positive rate. Since true positives are known, LHRM was assessed for its sensitivity and specificity using a tm difference of zero to classify drug susceptibility among 9 true drug-susceptible samples (n = 3 trials of wild type in triplicate) and 79 true not drug-susceptible samples (n = 3 trials of 9 variant types in triplicate, except variant S315T+G316D+A312V which had one trial with a single replicate due to Cq exclusion).
To directly compare time-based LHRM analysis within a single sample and temperature-based standard HRM analysis between samples, L-DNA-containing samples were also analyzed using standard HRM analysis. Sample Tm was calculated with the proprietary QuantStudio 5 Design and Analysis Software. Based on Tm analysis of all samples, Tm cutoff points were established to maximize test specificity when classifying each test sample as drug-susceptible or not. A sample was classified as drug-susceptible when PCR product Tm was within the drug-susceptible Tm cutoff range of 82.4 and 82.5 °C. Since true positives are known, classification sensitivity and specificity were assessed using this Tm cutoff range to classify drug susceptibility among 9 true drug-susceptible samples (n = 3 trials of wild type in triplicate) and 79 true not drug-susceptible samples (n = 3 trials of 9 variant types in triplicate, except variant S315T+G316D+A312V of one trial with a single replicate due to Cq exclusion).
Alternative strategies exist to establish drug-susceptible classification cutoff points for HRM analysis, and this was explored in Supporting Information (see pages S10–S11). This supplemental work used a maximized Youden J Statistic47 to establish drug-susceptible classification cutoff points for the same data sets across standard HRM and LHRM analysis strategies (see pages S10–S11). This alternative cutoff strategy generally improved sensitivity and decreased specificity.
In the experiment testing heating variability, significance was evaluated using melt measurement comparison (Mann–Whitney U test, significance level of α = 0.95) of 96-well plate quadrants of S315T as compared to wild type (n = 1 trial with 24 replicates). The heating variability Mann–Whitney U test was performed twice, once using Tm as the melt measurement and once using tm difference as the melt measurement. All statistics were performed in Microsoft Excel 2022 except for the sensitivity and specificity analysis that was performed in Python. Complete details are included in Supporting Information (see pages S4–S5).
Results and Discussion
An initial classification of the test variants by standard HRM confirmed that classification was successful for most variants but that the small melt difference in the single base mutation S315T was near the limits of this calibrated instrument approach. As predicted by the theoretical melt differences (Table 1), all variant samples analyzed by standard HRM had lower melt temperatures compared to the known wild-type samples, except S315G which had a higher melt temperature (Figure 1). The nine selected variants had a Tm spread of 2.43 °C, offering both easy and challenging classification cases against wild type (Figure 1). Average sample Tm’s and Tm differences are reported in Table S4. Standard HRM correctly classified 6/9 wild-type katG samples as drug-susceptible and 80/81 variant samples as not drug-susceptible; the most clinically prevalent variant S315T was misclassified once. Although S315G has the smallest theoretical melt difference (0.33 °C) from wild type and was initially thought to be the most difficult variant to correctly classify, S315T was experimentally the most difficult case because under the sample salt conditions, it induced the smallest melt difference (−0.18 °C) from wild type among all nine variants. Standard HRM relied on Tm analysis of data from multiple samples to form the drug-susceptible classification Tm cutoff range of 82.4 to 82.5 °C. Using a state-of-the-art calibrated instrument, standard HRM performed at 66.7% sensitivity and 98.8% specificity when classifying drug susceptibility. Sample classification accuracy and relationships between sample type Tm’s are shown in Figure 2. In particular, standard HRM misclassification is illustrated by three wild-type samples above the upper drug-susceptible cutoff range and one S315T sample within the drug-susceptible cutoff range.
Figure 1.
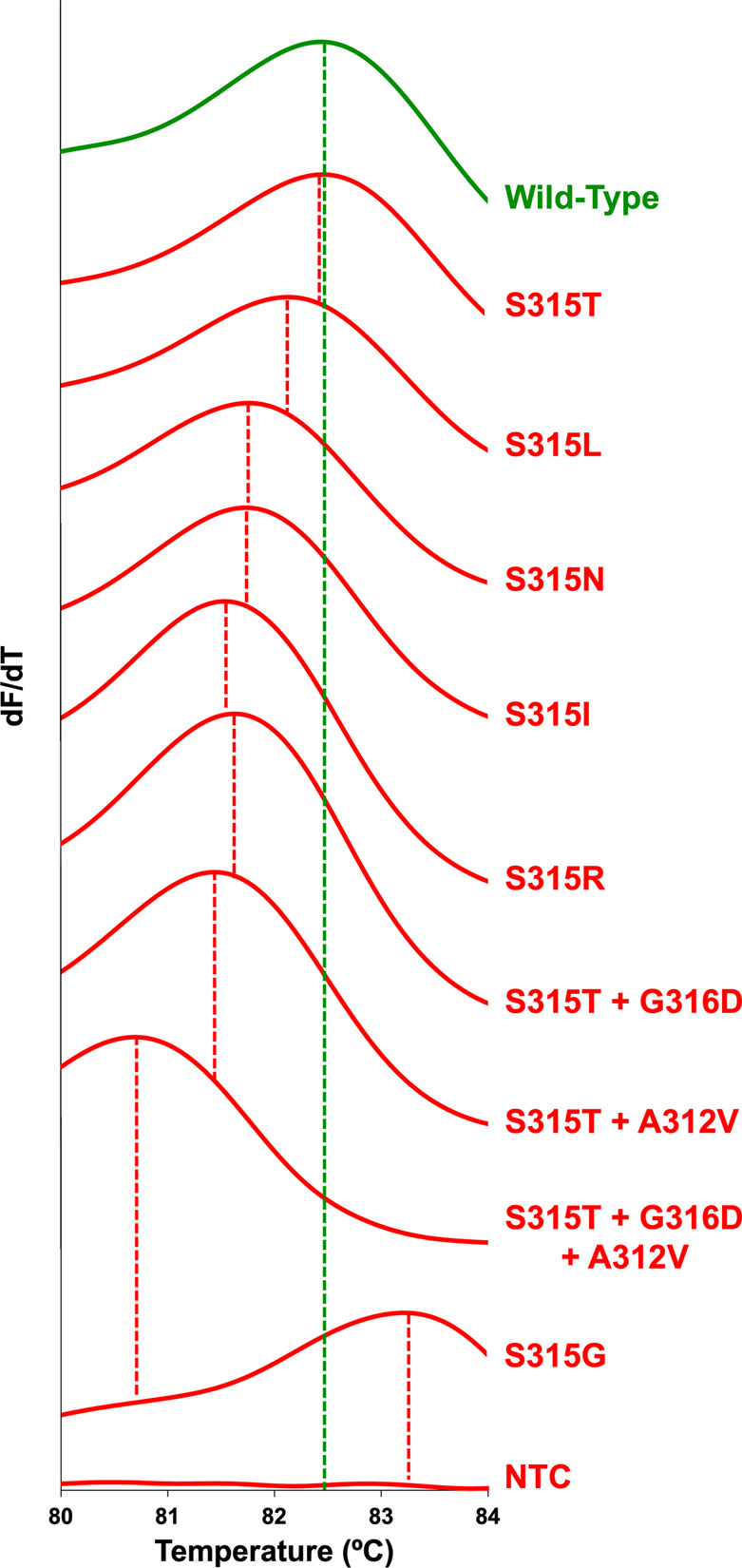
Multisample comparison of Tm obtained from standard high-resolution melt of 11 representative samples containing wild-type (green), one of nine variants (red), or no template control polymerase chain reaction products. Dashed vertical lines indicate the Tm for each sample on the horizontal axis.
Figure 2.
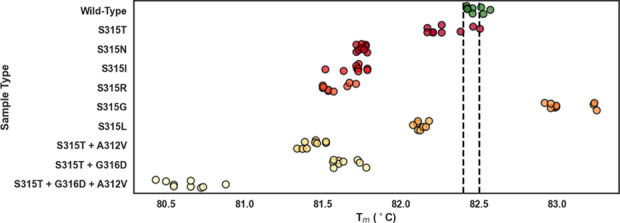
Polymerase chain reaction product melt temperatures of samples analyzed by standard high-resolution melt across wild-type (green) and nine variants. Samples were classified as drug-susceptible or not by comparing sample Tm to the drug-susceptible Tm cutoff range of 82.4 and 82.5 °C (indicated by dashed black lines). Each point represents an individual test sample.
This S315T classification error was thought to be due to instrument heating variability that has been observed in many plate-based real-time PCR instruments.48 Nonuniform heating limits Tm comparison accuracy48 and is only partially compensated by instrument calibration.37 In a direct test of this, the QuantStudio 5 was found to exhibit heating variability across the 96-well plate (Figure S3) and this impacted standard HRM classification of the most difficult case, S315T. Standard HRM failed to distinguish between wild type and S315T when using PCR product melt characteristics since there was no significant difference between wild-type and S315T sample Tm’s (p > 0.05, unpaired t test, 24 replicates per sample type). The 24 identical samples exhibited thermal edge effects of generally higher Tm’s and zone-based Tm’s for two samples in shared heating elements, with trends holding for both wild-type and S315T sample types (Figure S3). Specifically, wild-type average PCR product Tm (mean ± SD) was 82.46 ± 0.08 °C across all wells and 82.53 ± 0.07 °C across edge wells. S315T average PCR product Tm (mean ± SD) was 82.42 ± 0.10 °C across all wells and 82.50 ± 0.09 °C across edge wells. The heating variability of the QuantStudio 5 (Figure S3) is presumably due to variations in the instrument’s six independent temperature heating zones (pairwise vertical columns)37 that are not completely corrected by the instrument calibration procedures.
We used this simple experimental design to test the initial feasibility of the L-DNA approach. Repeating this heating experiment but incorporating L-DNA into each sample did not change the experimental outcome. The approach still failed to distinguish between wild type and S315T because there was no statistical difference between PCR product Tm’s (p > 0.05, Mann–Whitney U test, 24 replicates per sample type, column 2 in Table S7). These results confirmed that the addition of L-DNA itself does not affect the between-sample analysis based on temperature and indirectly supports the initial assumption that L-DNA does not interfere with the PCR reaction. However, reanalysis using PCR product to L-DNA melt time differences did successfully overcome effects from nonuniform heating and distinguished between variant S315T and wild type. The additional L-DNA elapsed time data in this experimental design were used to show that a reanalysis incorporating a within-sample melt difference between L-DNA and PCR product distinguishes between these two sequences with a single base difference. Using tm differences, there was a significant difference between quadrants of wild-type and S315T samples (p < 0.05, Mann–Whitney U test, 24 replicates per sample type, column 4 in Table S7).
Due to the success of the limited data set employing L-DNA, the full variant test bed (Table 1) previously performed (Figure 2) was run incorporating an L-DNA comparator into every sample. These data were analyzed by two different methods: standard HRM analysis using temperature-based melt characteristics between PCR products of multiple samples and LHRM analysis using time-based melt differences between the PCR product and L-DNA comparator within each sample.
As shown in Figure 3, standard HRM analysis classification accuracy of L-DNA-containing samples was similar to the original standard HRM experiment without L-DNA (Figure 2). Average sample Tm’s and Tm differences are reported in Table S5. 3/9 wild-type katG samples were correctly classified as drug-susceptible and 77/79 variant samples were correctly classified as not drug-susceptible. Standard HRM misclassification is illustrated by six wild-type samples above the upper drug-susceptible cutoff range and two S315T samples within the drug-susceptible cutoff range (Figure 3). Standard HRM analysis of L-DNA-containing samples performed at 33.3% sensitivity and 97.5% specificity when classifying drug susceptibility. Samples containing L-DNA and analyzed by standard HRM had decreased sensitivity and comparable specificity metrics as compared to samples without L-DNA and analyzed by standard HRM.
Figure 3.
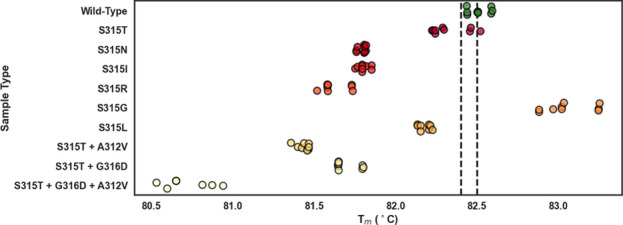
Polymerase chain reaction product melt temperatures of samples containing an L-DNA comparator in every sample but analyzed by standard HRM across wild-type (green) and nine variants. Samples were classified as drug-susceptible or not by comparing sample Tm to the drug-susceptible Tm cutoff range of 82.4 and 82.5 °C (indicated by dashed black lines).
Samples containing L-DNA were reanalyzed via LHRM analysis. The LHRM and standard HRM analysis strategies resulted in similar melt behavior across the wild-type and full variant test bed (Figure S4). Using LHRM, all variant samples had lower elapsed melt times compared to the drug-susceptible comparator, except S315G which had a higher elapsed melt time (Figure 4). L-DNA and DNA PCR product had nearly identical melt characteristics when the sequences matched (wild-type katG) but differed if there was a sequence mismatch (katG variants) (Figure 4 and Table S6). Using melt time differences between PCR product and L-DNA within each sample, LHRM correctly classified 7/9 wild-type katG samples as drug-susceptible and 78/79 variant samples as not drug-susceptible.
Figure 4.

Within-sample comparison of tm obtained from LHRM for 11 representative samples containing both wild-type L-DNA (green) and wild-type, variant, or NTC PCR products (red). Dashed vertical lines indicate the two tm’s within each sample on the horizontal axis.
Unlike standard HRM, LHRM drug-susceptible classification criteria were established without relying on analysis of data from multiple samples. Instead, LHRM classified samples as drug-susceptible when a sample’s tm difference was zero. This simple classification strategy enabled single sample classification without requiring data from other samples. LHRM performed at 77.8% sensitivity and 98.7% specificity when classifying drug susceptibility. Sample classification accuracy and relationships between sample type tm differences are illustrated in Figure 5, which can be directly compared to Figure 3. In particular, LHRM misclassification is illustrated by two wild-type samples below the drug-susceptible cutoff and one S315T sample within the drug-susceptible cutoff (Figure 5). S315T was the only variant misclassified by LHRM analysis, as similarly observed in standard HRM analysis classification (Figure 3). LHRM correctly classified drug susceptibility with specificity very similar to and with improved sensitivity over standard HRM analyzed samples. This trend held for LHRM as compared to standard analyzed samples with and without L-DNA. Notably, LHRM accomplished high success metrics without requiring multiple sample classification data and without relying on temperature-based melt reporting determined by instrument calibration.
Figure 5.
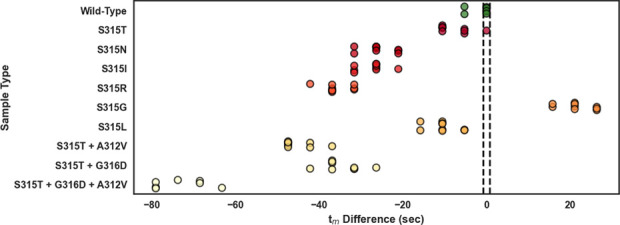
Within-sample tm differences of samples containing an L-DNA comparator in every sample and analyzed by LHRM across wild-type (green) and nine variants. Samples were classified as drug-susceptible or not by comparing sample tm difference to the drug-susceptible classification criteria of tm difference = 0 (indicated by dashed black lines).
Heating variation was identified as a major methodological artifact that reduced classification performance for S315T by HRM (Figure S3). While no other methodological artifacts were identified in HRM testing, it was speculated that some types of sample preparation errors could introduce systematic hybridization changes that could also be corrected using L-DNA. Possible errors may include culture media carryover, extraction errors, kit-to-kit master mix differences, or sample-to-sample salt concentration variability resulting from reagent pipetting errors.8 A contrived study of salt differences is included in Supporting Information (see page S12) to demonstrate an example of how LHRM can correct for these types of errors. In particular, this supplemental study verified that a sample’s salt content has a detrimental potential impact on classification accuracy and that within-assay differences can be overcome by LHRM to ultimately restore classification capabilities.
Three key features are critical for the success of the LHRM approach. First, LHRM reactions must include the same amount of double-stranded L-DNA in every sample to provide a signature comparator hybridization event. Constant L-DNA concentration in every sample was maintained by adding L-DNA from a stock into the master mix. Since L-DNA and the PCR product hybridization events were changed by heating (Figure S3) and salt variation errors (see Supporting Information, page S12) in the same way, the L-DNA to PCR product melt difference corrected for any artifact-induced melt shifts. Small mutation-induced melt shifts in the PCR product were then detectable due to the reduction in between-sample errors and ultimately facilitated LHRM classification performance with specificity very similar to and improved sensitivity over the standard method.
The second key design challenge for LHRM was determining how to discriminate between double-stranded L-DNA and D-DNA melting behavior in a single sample. Apparently, all readily available intercalating dyes do not discriminate between enantiomeric DNA (see Supporting Information, Table S9). Therefore, if double-stranded L-DNA and double-stranded D-DNA are combined in a single reaction, the intercalation melt signal reports a composite melt curve. To overcome this L-DNA and D-DNA intercalating crosstalk, L-DNA was end-labeled with Texas Red. The goal was to measure only double-stranded DNA PCR product fluorescence signal on the green optical channel using LCGreen intercalating dye and measure only double-stranded L-DNA fluorescence signal on the orange optical channel using Texas Red fluorophore and quencher end-labeling. Figure 6A demonstrates intercalating crosstalk (i.e., detectable melt signals on both fluorophore-quencher and intercalator channels) when samples contained 4 × 1011 copies of double-stranded L-DNA. Samples with 1 × 1011 copies of double-stranded L-DNA (Figure 6C) produced sufficient fluorophore-quencher signal for accurate L-DNA melt measurements with little crosstalk contribution detectable in the intercalator channel, and therefore, this L-DNA copy count was selected for further LHRM development.
Figure 6.
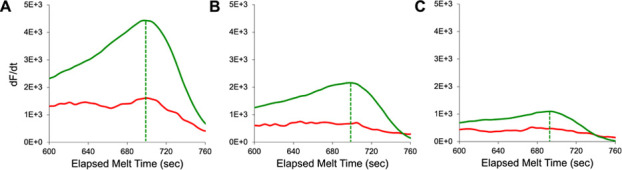
Melt curve derivatives for LHRM samples with 1:1 double-stranded L-DNA at (A) 4 × 1011, (B) 2 × 1011, and (C) 1 × 1011 copies per strand (forward and reverse) per reaction. As total L-DNA copy number decreased, both fluorophore-quencher signal (green) and intercalator signal (red) decreased in magnitude. End-labeled strands retained tm identification even when strand copy count decreased. Dashed lines indicate L-DNA tm measured by a fluorophore-quencher signal.
The third design challenge was matching the melt characteristics of the L-DNA melt comparator to the melt characteristics of the wild-type PCR product to support the initial melt matching assumption. Several factors made matching the melt characteristics of the two difficult. It is well-known that the L-DNA sequence end-labeling used to overcome single tube detection also changes the DNA’s melt temperature, even with a 5-base spacer on the quencher strand.49 Previous reports have also established that total DNA concentration and strand ratio affect the melt temperature.50−52 In addition to these well-known factors, even when they have the same sequence, for unknown reasons, unlabeled double-stranded L-DNA and D-DNA have a small difference in melt temperatures measured by intercalation (Table S9). Since the strand ratio was the easiest to adjust, the strand ratio of the added L-DNA was empirically modified to compensate for these other factors and ultimately match the L-DNA and D-DNA wild-type melt characteristics.33 The theory behind this report’s experimental tuning strategy is detailed in a complementary work by Spurlock et al., in 2024.23 Spurlock et al., in 2024, detailed the theory behind the effects of DNA concentration and strand ratio on annealing,23 while this report experimentally demonstrates its use in melt analysis. As Figure 7A demonstrates, different L-DNA forward to reverse strand ratios shift L-DNA tm (Table S8). This phenomenon was used to compensate for all factors discussed above and achieve an empirical melt match between drug-susceptible L-DNA and D-DNA (right most panel in Figure 7A). As reverse strand L-DNA copy count increased, L-DNA tm increased (Figure 7A and Table S8). A positive linear relationship between the number of L-DNA reverse strand copies per reaction and L-DNA tm indicated that 2.79 × 1011 L-DNA reverse strand copies (and 1 × 1011 L-DNA forward strand copies) per reaction would produce the 695 s tm matching average wild-type PCR product tm (Figure 7B). The optimal L-DNA strand ratio was rounded up from 1:2.79 to 1:3 (1 × 1011 L-DNA forward strand copies and 3 × 1011 L-DNA reverse strand copies per reaction) for ease of sample preparation in LHRM. This method produced an average melt difference of approximately 1 s between drug-susceptible L-DNA and wild-type PCR product (top row in Table S6). In further support of melt matching, there was no statistical difference between tm’s of drug-susceptible L-DNA and wild-type PCR product (p > 0.05, paired t test, n = 3 trials in triplicate). It is important to note that although this study sought to match the L-DNA melt and PCR product melt, it is not critical to do so. Even without tuning L-DNA to make the melt difference zero, for a fixed concentration of L-DNA in every sample, the tm difference will still be a constant in the system and samples can be classified as drug-susceptible when sample tm difference equals that constant. Alternatively, intentionally tuning for an excessively large melt mismatch between L-DNA and the wild-type PCR product may help to minimize intercalating crosstalk.
Figure 7.

(A) Representative wild-type D-DNA polymerase chain reaction (PCR) product (red) and internal comparator L-DNA (green) derivative melt plots analyzed by LHRM analysis containing double-stranded L-DNA at 1:1, 1:2, and 1:3 ratios of forward to reverse L-DNA strands with 1 × 1011 forward strand copies and 1 × 1011, 2 × 1011, and 3 × 1011 reverse strand copies per reaction, respectively. Dashed lines indicate D-DNA PCR product tm (red) and L-DNA tm (green). (B) There is a positive linear relationship (dashed blue line) between number of L-DNA reverse strand copies and L-DNA tm measured via fluorophore-quencher signal. This relationship suggested that an L-DNA forward to reverse strand ratio of 1:2.79 (1 × 1011 forward strand copies and 2.79 × 1011 reverse strand copies per reaction) would match drug-susceptible L-DNA tm to the drug-susceptible D-DNA PCR product average tm of 695 s (dashed red lines).
L-DNA tuning and time-scale melt analysis contribute to the simplicity of LHRM single sample classification, i.e., a sample is susceptible if its tm difference equals zero. A given LHRM sample’s tm difference can only consist of discrete values in multiples of 5.27 s. Discrete melt data every 5.27 s enable easier LHRM classification as melt data are inherently grouped into distinct time points and will clearly have tm differences of zero or not (Figure 5). The discrete nature of LHRM melt data is an artifact of the instrument melting ramp rate of 0.025 °C/s and the fluorescence sampling rate of one acquisition per 5.27 s. In contrast to discrete LHRM melt data, sample Tm’s resulting from standard HRM analysis are continuous in nature. As a result, standard HRM analysis produces a spread of data points with less separation between sample types (Figures 2 and 3). Unlike LHRM’s discrete melt data, this continuous spread of data requires grouping into subsets using multiple samples to form the drug-susceptible classification cutoffs.
In this initial LHRM report, a QuantStudio 5 was used to show that LHRM can provide similar performance to standard HRM with use of a single sample. It is important to note, however, that the instrument itself is not critical for the conclusions of this work. Although not confirmed in this report, some features of LHRM suggest this strategy would work with less capable instrument designs. For example, LHRM does not use temperature-based melt reporting determined by instrument calibration. Instead, LHRM utilizes time-defined melt analysis. Measurements were quantified using time instead of temperature for two reasons. First, time-quantified melt measurements are based on raw data unaffected by instrument calibration errors present in the Tm.37 Second, this strategy facilitates future LHRM measurements in other types of real-time PCR instruments that are not as well-calibrated as the QuantStudio 5 instrument. To use an available real-time PCR instrument, it must at a minimum have melt analysis capabilities built into its software. Although existing real-time PCR instruments commonly have melt analysis capabilities for product assessment purposes, the instruments generally lack the necessary resolution for high sensitivity SNP scanning by melting.53 Performing LHRM on a real-time PCR instrument requires access to sample times and fluorescence values recorded during heating of the melt procedure. The resolution of LHRM classification within a single sample is still limited by correct differentiation between an L-DNA comparator sequence and a PCR product sequence that can differ by only a single base. Among the many factors that make this differentiation challenging is the ratio of the fluorescence sampling rate compared to the instrument’s heating rate. A higher ratio makes differentiation easier. In this report, the ratio is 7.59 fluorescent samples collected per °C. Since it is relatively easy to set the instrument’s desired continuous ramp rate, the maximum sampling rate of the instrument is likely the determining factor that limits the resolution of this within-sample method. Future work is required to determine if the L-DNA reagent-based strategy can be implemented on other instruments with real-time PCR and built-in melt analysis data generation capabilities.
Conclusions
By including L-DNA for reagent-based calibration in every sample, LHRM successfully classifies PCR melt products as INH-susceptible or not based on within-sample differences between an L-DNA comparator and an unknown PCR product. LHRM achieves comparable classification specificity and sensitivity to standard HRM with single sample analysis. With further development, LHRM shows promise as an initial drug susceptibility screen that may be incorporated into the TB clinical treatment algorithm where real-time PCR instruments are available.
Acknowledgments
We thank Logan Tsukiyama for preliminary investigational support. This work was supported in part by the National Institutes of Health (R21AI152497, R01AI157827, R01AI135937), the National Science Foundation Graduate Research Fellowship Program under Grant No. 1937963 awarded to N.A.M., and the National Science Foundation Graduate Research Fellowship Program under Grant No. 1937963 awarded to D.J.N. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.4c01611.
Additional experimental details, methods, and materials, including oligonucleotide sequences for this work; complete details regarding approach, analysis, and statistics of standard HRM and LHRM; standard HRM and LHRM tabulated melt data; experiments exploring heating variability of the QuantStudio 5; comparison of LHRM and standard HRM analysis strategies; alternative classification strategies that establish drug-susceptible cutoff points for standard HRM and LHRM using a maximized Youden J Statistic; experiments exploring sample-to-sample salt variability on standard HRM and LHRM classification; experimental characterization of L-DNA; comparison of equal concentration 1:1 D-DNA and unlabeled L-DNA; and representative PCR amplification curves of samples with and without L-DNA (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- WHO . Catalogue of Mutations in Mycobacterium Tuberculosis Complex and Their Association with Drug Resistance, 2021.
- Papaventsis D.; Casali N.; Kontsevaya I.; Drobniewski F.; Cirillo D. M.; Nikolayevskyy V.. Systematic Review Whole Genome Sequencing of Mycobacterium Tuberculosis for Detection of Drug Resistance: A Systematic Review, 2016. [DOI] [PubMed] [Google Scholar]
- Jabbar A.; Phelan J. E.; Florez De Sessions P.; Khan T. A.; Rahman H.; Khan S. N.; Cantillon D. M.; Wildner L. M.; Ali S.; Campino S.; Waddell S. J.; Clark T. G.. Whole Genome Sequencing of Drug Resistant Mycobacterium Tuberculosis Isolates from a High Burden Tuberculosis Region of North West Pakistan. [DOI] [PMC free article] [PubMed]
- Choi G. E.; Lee S. M.; Yi J.; Hwang S. H.; Kim H. H.; Lee E. Y.; Cho E. H.; Kim J. H.; Kim H. J.; Chang C. L. High-Resolution Melting Curve Analysis for Rapid Detection of Rifampin and Isoniazid Resistance in Mycobacterium Tuberculosis Clinical Isolates. J. Clin Microbiol 2010, 48 (11), 3893–3898. 10.1128/JCM.00396-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galarza M.; Fasabi M.; Levano K. S.; Castillo E.; Barreda N.; Rodriguez M.; Guio H. High-Resolution Melting Analysis for Molecular Detection of Multidrug Resistance Tuberculosis in Peruvian Isolates. BMC Infect. Dis. 2016, 16 (1), 260. 10.1186/s12879-016-1615-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar J. S.; Wittwer C. T. High-Resolution Melting Curve Analysis for Molecular Diagnostics. Molecular Diagnostics: Third Edition 2017, 79–102. 10.1016/B978-0-12-802971-8.00006-7. [DOI] [Google Scholar]
- Hu S.; Li G.; Li H.; Liu X.; Niu J.; Quan S.; Wang F.; Wen H.; Xu Y.; Li Q. Rapid Detection of Isoniazid Resistance in Mycobacterium Tuberculosis Isolates by Use of Real-Time-PCR-Based Melting Curve Analysis. J. Clin. Microbiol. 2014, 52 (5), 1644–1652. 10.1128/JCM.03395-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew M.; Pryor R.; Palais R.; Meadows C.; Erali M.; Lyon E.; Wittwer C. Genotyping of Single-Nucleotide Polymorphisms by High-Resolution Melting of Small Amplicons. Clin Chem. 2004, 50 (7), 1156–1164. 10.1373/clinchem.2004.032136. [DOI] [PubMed] [Google Scholar]
- Erali M.; Voelkerding K. V.; Wittwer C. T. High Resolution Melting Applications for Clinical Laboratory Medicine. Experimental and Molecular Pathology. 2008, 85, 50–58. 10.1016/j.yexmp.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]; August
- Thant Y. M.; Saikaew S.; Tharinjaroen C. S.; Phunpae P.; Pongsararuk R.; Preechasuth K.; Butr-Indr B.; Intorasoot S.; Tragoolpua K.; Chaiprasert A.; Wattananandkul U. Combined Locked Nucleic Acid Probes and High-Resolution Melting Curve Analysis for Detection of Rifampicin-Resistant Tuberculosis in Northern Thailand. Diagnostics 2022, 12 (10), 2307. 10.3390/diagnostics12102307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravorty S.; Marie Simmons A.; Rowneki M.; Parmar H.; Cao Y.; Ryan J.; Banada P. P.; Deshpande S.; Shenai S.; Gall A.; Glass J.; Krieswirth B.; Schumacher S. G.; Nabeta P.; Tukvadze N.; Rodrigues C.; Skrahina A.; Tagliani E.; Cirillo D. M.; Davidow A.; Denkinger C. M.; Persing D.; Kwiatkowski R.; Jones M.; Alland D.. The New Xpert MTB/RIF Ultra: Improving Detection of Mycobacterium Tuberculosis and Resistance to Rifampin in an Assay Suitable for Point-of-Care Testing, 2017; p 10. [DOI] [PMC free article] [PubMed]
- Cao Y.; Parmar H.; Gaur R. L.; Lieu D.; Raghunath S.; Via N.; Battaglia S.; Cirillo D. M.; Denkinger C.; Georghiou S.; Kwiatkowski R.; Persing D.; Alland D.; Chakravorty S. Xpert MTB/XDR: A 10-Color Reflex Assay Suitable for Point-of-Care Settings to Detect Isoniazid, Fluoroquinolone, and Second-Line-Injectable-Drug Resistance Directly from Mycobacterium Tuberculosis-Positive Sputum. J. Clin Microbiol 2021, 59 (3), e02314 10.1128/JCM.02314-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinricka B. Genotyping of Mycobacterium Tuberculosis Rifampin Resistance-Associated Mutations by Use of Data from Xpert MTB/RIF Ultra Enables Large-Scale Tuberculosis Molecular Epidemiology Studies. J. Clin. Microbiol. 2020, 58 (1), e01504 10.1128/JCM.01504-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castan P.; De Pablo A.; Fernández-Romero N.; Rubio J. M.; Cobb B. D.; Mingorance J.; Toro C. Point-of-Care System for Detection of Mycobacterium Tuberculosis and Rifampin Resistance in Sputum Samples. J. Clin Microbiol 2014, 52 (2), 502–507. 10.1128/JCM.02209-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clutter D. S.; Mazarei G.; Sinha R.; Manasa J.; Nouhin J.; LaPrade E.; Bolouki S.; Tzou P. L.; Hannita-Hui J.; Sahoo M. K.; Kuimelis P.; Kuimelis R. G.; Pinsky B. A.; Schoolnik G. K.; Hassibi A.; Shafer R. W. Multiplex Solid-Phase Melt Curve Analysis for the Point-of-Care Detection of HIV-1 Drug Resistance. Journal of Molecular Diagnostics 2019, 21 (4), 580–592. 10.1016/j.jmoldx.2019.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yenice C. P.; Chahin N.; Jauset-Rubio M.; Hall M.; Biggs P.; Dimai H. P.; Obermayer-Pietsch B.; Ortiz M.; O’Sullivan C. K. Semiautomated Electrochemical Melting Curve Analysis Device for the Detection of an Osteoporosis Associated Single Nucleotide Polymorphism in Blood. Anal. Chem. 2023, 95 (38), 14192–14202. 10.1021/acs.analchem.3c01668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahin N.; Escobar-Nassar S.; Osma J.; Bashammakh A. S.; Alyoubi A. O.; Ortiz M.; O’sullivan C. K. Low-Cost Platform for Multiplexed Electrochemical Melting Curve Analysis. ACS Meas. Sci. Au 2021, 2, 147–156. 10.1021/acsmeasuresciau.1c00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson D. J.; Reischer R. J.; Taylor A. J.; Wechter W. J. Preparation and Characterization of Oligonucleotides of D- and L-2’ Deoxyuridine. Nucleosides Nucleotides 1984, 3 (5), 499–512. 10.1080/07328318408081285. [DOI] [Google Scholar]
- Damha M. J.; Giannari P. A.; Marfeyb P.; Reid’ L. S. Oligodeoxynucleotides Containing Unnatural L-2’-Deoxyribose. Tetrahedron Lett. 1991, 32 (23), 25732576 10.1016/S0040-4039(00)78788-0. [DOI] [Google Scholar]
- Urata H.; Ogura E.; Shinohara K.; Ueda Y.; Akagi M. Synthesis and Properties of Mirror-Image DNA. Nucleic Acids Res. 1992, 20 (13), 3325–3332. 10.1093/nar/20.13.3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbesi A.; LCapobianco M.; PColonna F.; Tondelli L.; Arcamone F.; Manzini G.; WHilbers C.; MEAelen J.; JJBIommers M.; Ricerche Sud M. Sette Santi, via. L-DNAs as Potential Antimessenger Oligonucleotides: A Reassessment. Nucleic Acids Res. 1993, 21 (18), 4159. 10.1093/nar/21.18.4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams N. M.; Gabella W. E.; Hardcastle A. N.; Haselton F. R. Adaptive PCR Based on Hybridization Sensing of Mirror-Image l -DNA. Anal. Chem. 2017, 89 (1), 728–735. 10.1021/acs.analchem.6b03291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurlock N.; Gabella W. E.; Nelson D. J.; Evans D. T.; Pask M. E.; Schmitz J. E.; Haselton F. R. Implementing L-DNA Analogs as Mirrors of PCR Reactant Hybridization State: Theoretical and Practical Guidelines for PCR Cycle Control. Anal. Methods 2024, 16 (18), 2769–2974. 10.1039/D4AY00083H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young B. E.; Kundu N.; Sczepanski J. T. Mirror-Image Oligonucleotides: History and Emerging Applications. Chem.—Eur. J. 2019, 25 (34), 7981–7990. 10.1002/chem.201900149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L.; Peng R.; Fu T.; Zhang X.; Wu C.; Chen H.; Liang H.; Yang C. J.; Tan W. Biostable L-DNAzyme for Sensing of Metal Ions in Biological Systems. Anal. Chem. 2016, 88 (3), 1850–1855. 10.1021/acs.analchem.5b04170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabza A. M.; Young B. E.; Sczepanski J. T. Heterochiral DNA Strand-Displacement Circuits. J. Am. Chem. Soc. 2017, 139 (49), 17715–17718. 10.1021/jacs.7b10038. [DOI] [PubMed] [Google Scholar]
- Kim K.-R.; Hwang D.; Kim J.; Lee C.-Y.; Lee W.; Sung Yoon D.; Shin D.; Min S.-J.; Chan Kwon I.; Suk Chung H.; Ahn D.-R.. Streptavidin-Mirror DNA Tetrahedron Hybrid as a Platform for Intracellular and Tumor Delivery of Enzymes, 2018. [DOI] [PubMed] [Google Scholar]
- Urata H.; Shinohara K.; Ogura E.; Yoshiaki U.; Akagi M. Mirror-Image DNA. J. Am. Chem. Soc. 1991, 113, 8174–8175. 10.1021/ja00021a057. [DOI] [Google Scholar]
- Hauser N. C.; Martinez R.; Jacob A.; Rupp S.; Hoheisel D.; Matysiak S.. Utilising the Left-Helical Conformation of L-DNA for Analysing Different Marker Types on a Single Universal Microarray Platform. [DOI] [PMC free article] [PubMed]
- Unissa A. N.; Selvakumar N.; Narayanan S.; Suganthi C.; Hanna L. E.. Investigation of Ser315 Substitutions within KatG Gene in Isoniazid-Resistant Clinical Isolates of Mycobacterium Tuberculosis from South India, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Heym B.; Allen B.; Young D.; Cole S. The Catalase-Peroxidase Gene and Isoniazid Resistance of Mycobacterium Tuberculosis. Nature 1992, 358 (6387), 591–593. 10.1038/358591a0. [DOI] [PubMed] [Google Scholar]
- Vilchèze C.; Jacobs W. R. JR. Resistance to Isoniazid and Ethionamide in Mycobacterium Tuberculosis: Genes, Mutations, and Causalities. Microbiol. Spectrum 2014, 2 (4), MGM2-0014-2013 10.1128/microbiolspec.mgm2-0014-2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barozi V.; Musyoka T. M.; Amamuddy O. S.; Zlem O.; Bishop T. Deciphering Isoniazid Drug Resistance Mechanisms on Dimeric Mycobacterium Tuberculosis KatG via Post-Molecular Dynamics Analyses Including Combined Dynamic Residue Network Metrics. ACS Omega 2022, 7, 13313–13332. 10.1021/acsomega.2c01036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J. H.; Nam J. S.; Kim K. J.; Choi Y.; Lee H.; Cho S. N.; Ro Y. T. Molecular Characterization of Drug-Resistant and -Susceptible Mycobacterium Tuberculosis Isolated from Patients with Tuberculosis in Korea. Diagn Microbiol Infect Dis 2012, 72 (1), 52–61. 10.1016/j.diagmicrobio.2011.09.010. [DOI] [PubMed] [Google Scholar]
- Hazbón M. H.; Brimacombe M.; Del Valle M. B.; Cavatore M.; Guerrero M. I.; Varma-Basil M.; Billman-Jacobe H.; Lavender C.; Fyfe J.; García-García L.; León C. I.; Bose M.; Chaves F.; Murray M.; Eisenach K. D.; Sifuentes-Osornio J.; Cave M. D.; De León A. P.; Alland D. Population Genetics Study of Isoniazid Resistance Mutations and Evolution of Multidrug-Resistant Mycobacterium Tuberculosis. Antimicrob. Agents Chemother. 2006, 50 (8), 2640. 10.1128/AAC.00112-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibbe W. OligoCalc: An Online Oligonucleotide Properties Calculator. Nucleic Acids Res. 2007, 35, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thermo Fisher Scientific Inc. QuantStudio 3 and QuantStudio 5 Real-Time PCR Systems, 2019. https://tools.thermofisher.com/content/sfs/brochures/quantstudio-3-and-5-real-time-pcr-systems.pdf (accessed September 10, 2023).
- Sun L.; Wang L.; Zhang C.; Xiao Y.; Zhang L.; Zhao Z.; Ren L.; Peng J. Rapid Detection of Predominant SARS-CoV-2 Variants Using Multiplex High-Resolution Melting Analysis. Microbiol Spectr 2023, 11 (3), e0005523 10.1128/spectrum.00055-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentaleb E. M.; El Messaoudi M. D.; Abid M.; Messaoudi M.; Yetisen A. K.; Sefrioui H.; Amzazi S.; Ait Benhassou H. Plasmid-Based High-Resolution Melting Analysis for Accurate Detection of RpoB Mutations in Mycobacterium Tuberculosis Isolates from Moroccan Patients. BMC Infect Dis 2017, 17 (1), 548. 10.1186/s12879-017-2666-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshikawa T.; Miyoshi H.. High-Resolution Melting Analysis to Discriminate between the SARS-CoV-2 Omicron Variants BA.1 and BA.2, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi H.; Ichinohe R.; Koshikawa T.. High-Resolution Melting Analysis after Nested PCR for the Detection of SARS-CoV-2 Spike Protein G339D and D796Y Variations, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thermo Fisher Scientific Inc. QuantStudio 3 and 5 Real-Time PCR Systems Installation, Use, and Maintenance Guide, 2021.
- Hristea A.; Otelea D.; Paraschiv S.; MacRi A.; Baicus C.; Moldovan O.; Tinischi M.; Arama V.; Streinu-Cercel A.. Detection of Mycobacterium Tuberculosis Resistance Mutations to Rifampin and Isoniazid by Real-Time PCR. Indian Journal of Medical Microbiology; Medknow Publications and Media Pvt. Ltd., July 1, 2010; pp 211–216. [DOI] [PubMed] [Google Scholar]
- Liu Z.-B.; Cheng L.-P.; Pan H.-Q.; Wu X.-C.; Lu F.-H.; Cao J.; Wang L.; Wei W.; Chen H.-Y.; Sha W.; Sun Q. Performance of the MeltPro TB Assay as Initial Test for Diagnosis of Pulmonary Tuberculosis with Drug-Resistance Detection. Mol. Med. 2023, 29 (1), 153. 10.1186/s10020-023-00743-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson D. J.; Reischer R. J.; Taylor A. J.; Wechter W. J. Preparation and Characterization of Oligonucleotides of D- and L-2’ Deoxyuridine. Nucleosides Nucleotides 2007, 3 (5), 499–512. 10.1080/07328318408081285. [DOI] [Google Scholar]
- Wittwer C. T.; Reed G. H.; Gundry C. N.; Vandersteen J. G.; Pryor R. J. High-Resolution Genotyping by Amplicon Melting Analysis Using LCGreen. Clin Chem. 2003, 49 (6), 853–860. 10.1373/49.6.853. [DOI] [PubMed] [Google Scholar]
- Youden W. J. Index for Rating Diagnostic Tests. Cancer 1950, 3 (1), 32–35. . [DOI] [PubMed] [Google Scholar]
- Herrmann M. G.; Durtschi J. D.; Wittwer C. T.; Voelkerding K. V. Expanded Instrument Comparison of Amplicon DNA Analysis for Mutation Scanning and Genotyping. Clin. Chem. 2007, 53 (8), 1541–1544. 10.1373/clinchem.2007.088120. [DOI] [PubMed] [Google Scholar]
- Zimmers Z. A.; Adams N. M.; Gabella W. E.; Haselton F. R. Fluorophore-Quencher Interactions Effect on Hybridization Characteristics of Complementary Oligonucleotides. 2019, 11, 2862. 10.1039/C9AY00584F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber-Gosche S.; Edwards R. A. Thermodynamics of Oligonucleotide Duplex Melting. J. Chem. Educ. 2009, 86 (5), 644–650. 10.1021/ed086p644. [DOI] [Google Scholar]
- Wu P.; Nakano S. I.; Sugimoto N. Temperature Dependence of Thermodynamic Properties for DNA/DNA and RNA/DNA Duplex Formation. Eur. J. Biochem. 2002, 269 (12), 2821–2830. 10.1046/j.1432-1033.2002.02970.x. [DOI] [PubMed] [Google Scholar]
- You Y.; Tataurov A. V.; Owczarzy R. Measuring Thermodynamic Details of DNA Hybridization Using Fluorescence. Biopolymers 2011, 95 (7), 472. 10.1002/bip.21615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed G. H.; Wittwer C. T. Sensitivity and Specificity of Single-Nucleotide Polymorphism Scanning by High-Resolution Melting Analysis. Clin Chem. 2004, 50 (10), 1748–1754. 10.1373/clinchem.2003.029751. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.