

ABSTRACT
Pathogenic species within the Rickettsia genus are transmitted to humans through arthropod vectors and cause a spectrum of diseases ranging from mild to life-threatening. Despite rickettsiae posing an emerging global health risk, the genetic requirements of their infectious life cycles remain poorly understood. A major hurdle toward building this understanding has been the lack of efficient tools for genetic manipulation, owing to the technical difficulties associated with their obligate intracellular nature. To this end, we implemented the Tet-On system to enable conditional gene expression in Rickettsia parkeri. Using Tet-On, we show inducible expression of antibiotic resistance and a fluorescent reporter. We further used this inducible promoter to screen the ability of R. parkeri to express four variants of the catalytically dead Cas9 (dCas9). We demonstrate that all four dCas9 variants can be expressed in R. parkeri and used for CRISPR interference (CRISPRi)-mediated targeted gene knockdown. We show targeted knockdown of an antibiotic resistance gene as well as the endogenous virulence factor sca2. Altogether, we have developed systems for inducible gene expression and CRISPRi-mediated gene knockdown for the first time in rickettsiae, laying the groundwork for more scalable, targeted mechanistic investigations into their infectious life cycles.
IMPORTANCE
The spotted fever group of Rickettsia contains vector-borne pathogenic bacteria that are neglected and emerging threats to public health. Due to the obligate intracellular nature of rickettsiae, the development of tools for genetic manipulation has been stunted, and the molecular and genetic underpinnings of their infectious lifecycle remain poorly understood. Here, we expand the genetic toolkit by introducing systems for conditional gene expression and CRISPR interference (CRISPRi)-mediated gene knockdown. These systems allow for relatively easy manipulation of rickettsial gene expression. We demonstrate the effectiveness of these tools by disrupting the intracellular life cycle using CRISPRi to deplete the sca2 virulence factor. These tools will be crucial for building a more comprehensive and detailed understanding of rickettsial biology and pathogenesis.
KEYWORDS: Rickettsia, Tet-On, CRISPRi, inducible expression
INTRODUCTION
Members of the Rickettsia genus are obligate intracellular Gram-negative bacteria with a broad host range (1–3). Several Rickettsia species are neglected human pathogens transmitted by arthropod vectors like ticks, mites, fleas, and lice, and some are among the oldest known vector-borne pathogens (4). Still, we know little about the genetic and molecular requirements of their infectious lifecycle. This knowledge gap is largely due to the challenges associated with studying obligate intracellular bacteria, including the lack of a modern toolkit to perform targeted genetic manipulation in these pathogens (3, 5, 6).
Over the course of their evolution as obligate intracellular bacteria, rickettsiae have undergone drastic genome reduction and rearrangement, giving rise to small, streamlined genomes (1, 7). Rickettsial genomes typically contain fewer than 1,500 predicted coding sequences in their 1.1–1.5 Mb genomes (7). Despite these small genomes, only a small fraction of rickettsial genes have been studied in detail, and even fewer have been directly studied in mutant strains of Rickettsia (1, 5, 6, 8, 9). As is the case in other obligate intracellular bacteria, the development of tools for genetic manipulation in rickettsiae has lagged behind many other model bacteria (5). Plasmid-driven transposon mutagenesis was only first reported in 2007 (10) and later adopted by others in the field (8, 9, 11, 12). It was not until 2011 that a shuttle vector was generated for use in rickettsiae (13), leading to the first genetic complementation of a mutant in 2016 (14). Reports of targeted genetic knockouts or silencing in rickettsiae have also emerged, using approaches based on allelic exchange (15, 16), group II intron mutagenesis (17), and peptide nucleic acids (18). However, these approaches are not always amenable to studying essential genes and are often low throughput. Additionally, no systems for conditional gene expression have been reported in rickettsiae. Thus, easy and scalable methods for targeted control of rickettsial gene expression would greatly advance the field’s ability to carry out detailed mechanistic analyses of the rickettsial infectious life cycle.
In the last decade, CRISPR-based tools have enabled genetic manipulation in many previously intractable organisms (19). One such tool is CRISPR interference (CRISPRi), which relies on a catalytically dead mutant of Cas9 (dCas9) to reversibly knock down genes of interest by physically blocking transcription initiation and/or elongation (20, 21). dCas9 is directed to the genomic loci of interest via sequence homology with a guide RNA (gRNA). This homology search between gRNA and the genome is licensed by direct interactions between the dCas9 protein and a protospacer adjacent motif (PAM) (22). Different variants of dCas9 recognize different PAMs (23), meaning that each dCas9 has a different repertoire of possible guide sequences that can be used to target a genome of interest. CRISPRi has been used for efficient and scalable gene knockdown in a wide variety of bacteria including Mycobacterium tuberculosis (24), Caulobacter crescentus (25), Chlamydia trachomatis (26), and Coxiella burnetii (27, 28).
Here, we expand the rickettsial genetic toolkit by introducing systems for conditional gene expression and targeted gene knockdown via CRISPRi in Rickettsia parkeri. We demonstrate the feasibility of conditional gene expression through inducible expression of an antibiotic-resistance gene and a fluorescent reporter gene using the Tet-ON system. We were subsequently able to use this conditional expression system to express four variants of dCas9 in R. parkeri, all of which enabled the knockdown of an antibiotic-resistance gene. We further show CRISPRi-mediated knockdown of the endogenous virulence gene sca2. Altogether, this work greatly expands the arsenal of genetic tools for rickettsiae, opening new avenues for mechanistic investigations into rickettsial biology and pathogenesis.
RESULTS
Development of an inducible promoter system for Rickettsia
We first set out to build a system for conditional gene expression in Rickettsia parkeri. We chose the Tet-On system developed from the Tn10 transposon of Escherichia coli (29, 30), based on the membrane permeability of tetracycline derivatives in mammalian host cells (31) and previous success of implementing a tetracycline-inducible promoter in Chlamydia trachomatis (32). To assess feasibility, we first needed to determine the viability of R. parkeri upon exposure to anhydrotetracycline (aTc), which is widely used as the inducer of Tet-On (33). To measure aTc toxicity, we infected Vero host cells with R. parkeri and added various concentrations of aTc at the time of infection. We then imaged and quantified plaque formation at 5 days post-infection (dpi). Plaques were observed at aTc concentrations from 0.1 to 250 ng/mL, indicating successful R. parkeri infection at these concentrations (Fig. 1A). At 100 and 250 ng/mL, plaque numbers began to trend downward but did not reach statistical significance, suggesting slight aTc toxicity may occur at these concentrations. At 500 ng/mL, no plaques were formed, indicating the sensitivity of R. parkeri to higher concentrations of aTc, similar to what was observed with C. trachomatis (32). These results demonstrate that aTc is well tolerated during infection, indicating that the Tet-On system may be suitable for inducible expression in R. parkeri.
Fig 1.

The Tet-On system enables conditional gene expression in R. parkeri. (A) Anhydrotetracycline toxicity curve in R. parkeri. Plaque assays were performed on Vero cell monolayers with varying concentrations of aTc indicated. The number of plaques formed at each aTc concentration was normalized to the no aTc control for each independent experiment (n = 3). ***P < 0.001 by ordinary one-way ANOVA with post hoc Tukey’s test. (B) Schematic of the Tet-On system cloned into pRAM18dSGA. The tet repressor, TetR, binds two tet operator sites (tetO) to block gene expression in the absence of aTc. The rparr-2 gene, which confers resistance to rifampicin, was placed under the control of Tet-On. Diagram not drawn to scale. (C) aTc induction of rifampicin resistance. Varying concentrations of aTc were added 30 mpi during plaque assays in Vero host cell monolayers. Each well shown had rifampicin added (200 ng/mL final concentration). All conditions shown were normalized to a no aTc and no rifampicin control well per independent experiment (n = 3). (D) Schematic of tagbfp cloned into the Tet-On system. The tagbfp gene was codon optimized for expression in Rickettsia conorii (14). Diagram not drawn to scale. (E and F) aTc induction of TagBFP during infection. A549 cell monolayers were infected with R. parkeri harboring a plasmid containing tagbfp under the control of Tet-On. aTc was added 24 hpi, and then samples were fixed at 48 hpi and subsequently imaged. (E) All images were set to the same minimum and maximum gray values per channel for comparison of BFP intensity. Scale bar, 2 µm. (F) Blue fluorescence from the expression of tagbfp was quantified for each bacterium across three independent experiments. **P < 0.01 using an ordinary one-way ANOVA.
We introduced the Tet-On system into R. parkeri by cloning the tetA/R bidirectional promoter into a pRAM18-based plasmid (13) (Fig. 1B). This bidirectional promoter drives the expression of the tet repressor (tetR) gene and a downstream gene of interest (tetA) in the reverse and forward directions, respectively. The promoter region contains two tet operator (tetO) sites, to which TetR binds to block expression in both directions. Upon binding of a tetracycline derivative like aTc, TetR undergoes a conformational change that causes it to release the tetO-binding site, thereby allowing gene expression from the tetA/R promoter (29). To test inducible expression from this pRAM-based Tet-On system, we cloned the rifampicin resistance gene rparr-2 (34) under the control of the forward tetA promoter (Fig. 1B). We then challenged an R. parkeri strain harboring this plasmid with rifampicin and varying concentrations of aTc and monitored plaque formation over 5 days. In the absence of aTc, the strain is sensitive to rifampicin treatment as expected, with no plaques observed in Vero cell monolayers (Fig. 1C). In contrast, plaques were formed upon induction with aTc at concentrations as low as 0.1 ng/mL. The number of plaques trended upward with increasing concentrations of aTc, peaking between 1 and 25 ng/mL aTc. At 50 ng/mL aTc and above, we noticed a downward trend in the number of plaques formed, likely due to the additive toxic effects of rifampicin and high concentrations of aTc. These results show that the Tet-On system enables the tunable and inducible expression of rparr-2 in R. parkeri with minimal leakiness.
Because plaque formation is an endpoint assay with a population-level readout, we sought to examine how inducible expression with the Tet-On system varied across individual bacteria. Therefore, we cloned the blue fluorescent protein TagBFP (tagbfp) under the control of the pRAM-based Tet-On system (Fig. 1D). We then carried out a 48-h infection of A549 cells, inducing with aTc at 24 hpi. With this 24-h induction, we detected dose-dependent increases in BFP signal at 1 and 25 ng/mL aTc (Fig. 1E and F). Furthermore, we observed consistent levels of BFP signal across individual bacterial cells (Fig. 1E), indicating that the Tet-On system can be used to conditionally express genes of interest uniformly throughout the population. Still, BFP expression was modest even at the highest induction condition, only increasing in pixel intensity by ~15% relative to WT R. parkeri lacking a BFP gene (Fig. 1F). In an attempt to increase BFP expression, we tested various concentrations of aTc above 25 ng/mL and reduced the induction time to 12 h to mitigate any toxic effects from high doses of aTc. However, we were unable to significantly increase BFP expression, even at aTc levels as high as 2,000 ng/mL (Fig. S1). This result suggested that the promoter, even if fully activated, does not give rise to high levels of gene expression. As an alternative approach, we attempted to increase BFP expression upon induction by engineering tetO sites into strong promoters from R. parkeri, like PompA and PompB (Fig. S2). Indeed, the intensity of BFP fluorescence with these engineered promoters was drastically higher relative to the original Tet-On system (Fig. S2). The inducible PompA and PompB systems exhibited ~100% and ~1,000% increases, respectively, in mean pixel intensity relative to WT R. parkeri lacking tagbfp. However, at the population level, BFP expression was starkly bimodal with only ~40% of the population strongly expressing BFP and the rest appearing BFP negative. As a point of comparison, we generated a strain with the same tetO-engineered PompA but lacking tetR, which, therefore, expresses tagbfp constitutively, and this strain displayed a ~ 490% increase in pixel intensity relative to WT (Fig. S2). The bimodality seen in the PompA and PompB inducible systems was not observed in the constitutive version of the tetO-engineered PompA, meaning that the bimodal expression is not inherent to the modified promoter. While further optimization will be required to develop inducible promoters with high levels of uniform gene expression, these data demonstrate the feasibility of conditional expression using the Tet-On system in R. parkeri.
Expression of dCas9 in Rickettsia parkeri via Tet-On
The lack of scalable and efficient methods to perform targeted genetic manipulation has been a major hurdle toward understanding the molecular details of the rickettsial infectious lifecycle. To address this issue and further expand the rickettsial genetic toolkit, we set out to develop a system for targeted gene knockdown in R. parkeri. Given its ease of use and successful application in numerous bacterial species, we chose CRISPRi (20, 21) as a candidate method for targeted genetic knockdown in R. parkeri. After numerous failed attempts to clone the CRISPRi components under the control of constitutive promoters on the pRAM18 backbone (data not shown), we decided to use our Tet-On inducible promoter system to express dCas9 and the constitutive promoter PrpsL to express the gRNA (Fig. 2A), similar to what was done in Caulobacter crescentus (25), another member of Alphaproteobacteria. Despite the low expression of tagbfp, we hypothesized that this system might be ideal for CRISPRi given that lower levels of dCas9 expression are better tolerated and sufficient for gene knockdown in other bacteria (19, 35, 36).
Fig 2.
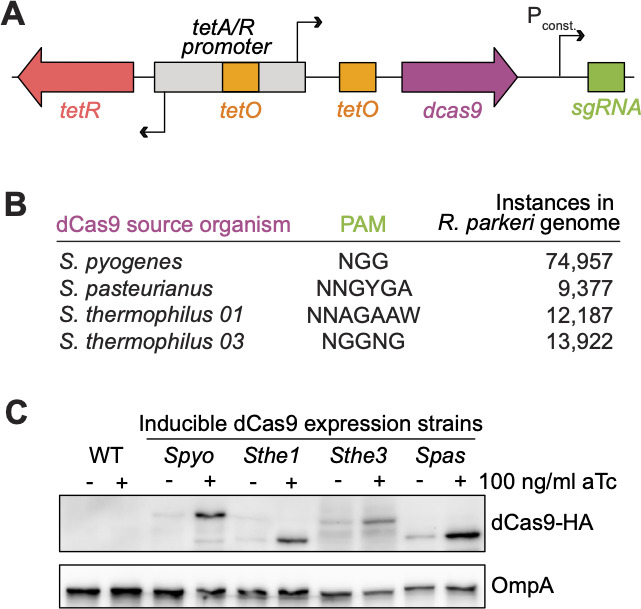
pRAM18-Tet-On can be used to express dCas9 in R. parkeri. (A) Schematic of pRAM18-based CRISPRi system. Expression of dCas9 is driven by the Tet-On promoter, and the sgRNA is driven by the constitutive promoter PrpsL. (B) Four dCas9 variants were cloned into pRAM18dSGA. Each dCas9 variant recognizes a distinct PAM, with each PAM found in varying instances in the R. parkeri genome. (C) Expression of dCas9 in R. parkeri. Each dCas9 variant was tagged with a C-terminal HA epitope and expression with or without aTc was visualized by Western blot, as well as OmpA as a loading control.
Since dCas9 proteins require binding to a protospacer adjacent motif to license DNA binding at the target sequence (22), the PAM limits the sequence space that is targetable via CRISPRi. Therefore, we reasoned that by selecting four candidate dCas9 variants that recognize different PAMs (Fig. 2B), we could maximize our chances of successful expression and expand the targetable range within the rickettsial genome. Two of these variants, Streptococcus pasteurianus (Spas) and Streptococcus thermophilus 03 (Sthe3), were successfully used in C. crescentus (25). In addition, we tested the dCas9s derived from Streptococcus pyogenes (Spyo), which has been commonly used in many other bacteria (19), and Streptococcus thermophilus 01 (Sthe1), which was successfully used for CRISPRi in M. tuberculosis (24). Based on the different PAMs of these dCas9s (Fig. 2B), there are 96,521 targetable sites in total in the R. parkeri genome, making much of the 1.3 Mb genome targetable if all four dCas9s were functional.
We next needed to determine if R. parkeri could express each of these dCas9 variants during infection. To this end, we generated strains harboring each variant of dCas9, to which we appended a C-terminal HA tag as previously described (37), and infected A549 cells for 72 h. We induced expression with 100 ng/mL aTc for the last 24 h of infection before harvesting cell lysates for Western blot analysis. We observed successful expression of all four dCas9 variants by Western blot (Fig. 2C). Each strain had elevated levels of dCas9 upon aTc induction but also displayed leaky expression in the uninduced condition. Altogether, these data demonstrate successful expression of dCas9 in R. parkeri during infection of human cells using the Tet-On system.
CRISPRi can be used to knockdown rifampicin resistance in R. parkeri
Given the successful expression of dCas9 during infection, we wanted to determine if CRISPRi could be used to knock down gene expression in R. parkeri. We chose to target rparr-2 as it allows easy measurement of knockdown efficiency by quantifying sensitivity to rifampicin (Fig. 3A). Because we needed to test four dCas9 variants with different PAMs and the optimal parameters for selecting gRNA target sites in R. parkeri were unknown, we modified the original rpsL promoter region of the rparr-2 locus in the Himar1 mariner-based transposon-containing plasmid pMW1650 (10) by enriching all four PAMs in the 100 bp immediately upstream of the predicted transcription start site. We introduced this test locus into the R. parkeri chromosome via random transposon insertion into the ompA locus, which has been documented to be dispensable during mammalian infection (17). We performed a plaque assay on Vero host cells using this transposon integrant and determined that it forms plaques like the wild-type parental strain, as expected (data not shown).
Fig 3.

CRISPRi knockdown of a rifampicin resistance gene. (A) Schematic of the engineered locus and screen to test knockdown of rifampicin resistance. The rpsL promoter driving the expression of rparr-2 in the pMW1650 plasmid was modified to include additional PAMs to allow for the testing of various dCas9 variants. Successful CRISPRi-mediated knockdown of rparr-2 would sensitize strains to treatment with rifampicin, while strains with nonfunctional CRISPRi would remain resistant to rifampicin. Spectinomycin selection ensures that the strains maintain the plasmid encoding the CRISPRi components. (B–E) Quantification of CRISPRi-mediated knockdown of rifampicin resistance via plaque assay. Vero cell monolayers were infected with R. parkeri strains encoding the S. pyogenes dCas9 (B), S. thermophilus 01 dCas9 (C), S. thermophilus 03 dCas9 (D), and S. pasteurianus dCas9 (E). For each dCas9 variant and sgRNA combination, the same volume of R. parkeri stock was added to each well, and then the number of plaques was normalized to the no aTc and no rifampicin condition for a total of n = 3 independent experiments. Statistical significance was determined by ordinary one-way ANOVA with post hoc Tukey’s test (*P < 0.05, **P < 0.005, and ****P < 0.0001). Schematics below each bar graph depict the relative locations of each sgRNA tested for each dCas9.
We next introduced the four dCas9s into this strain harboring the rparr-2 insertion. For each dCas9, we designed three different gRNAs targeting the promoter region of the rparr-2 test locus at varying distances from the promoter, covering both the template and nontemplate strands (Fig. 3). In this experimental setup, successful CRISPRi-mediated gene knockdown would sensitize the strain to rifampicin upon induction with aTc. In contrast, strains would remain resistant to rifampicin if CRISPRi knockdown failed (Fig. 3A). We tested each strain by monitoring plaque formation in Vero host cell monolayers at 5 dpi. Remarkably, all four dCas9s tested successfully knocked down rparr-2 expression, as observed by a decrease in the number of plaques formed relative to the non-target (NT) control gRNA plus aTc (Fig. 3B through E). For Spyo dCas9, two of the three guides tested yielded knockdown of rifampicin resistance (Fig. 3B). The remaining three dCas9s each had one successful gRNA out of the three guides tested (Fig. 3C through E). However, the majority of the gRNAs that exhibited successful knockdown of rparr-2 showed a reduction in plaque number both in the induced and uninduced conditions, indicating leakiness of the system. Two gRNAs, Spyo gRNA3 and Sthe1 gRNA1, showed only a partial reduction in plaques in the uninduced condition, compared to zero plaques observed in the induced condition, suggesting partial inducibility with these dCas9/gRNA combinations. Interestingly, all of the guides that yielded significant knockdown of rparr-2 targeted the nontemplate strand, similar to what was observed in other systems including C. crescentus (25). While further optimization will be required to decrease the leakiness of the inducible promoter system, our results demonstrate CRISPRi-mediated targeted gene knockdown for the first time in Rickettsia.
CRISPRi knockdown of the rickettsial virulence factor Sca2
Because the knockdown experiments described above targeted an exogenously introduced locus with an engineered promoter, we wanted to test the ability of our CRISPRi system to target endogenously encoded virulence factors in the R. parkeri genome. We chose to target the sca2 gene, which encodes a formin-like actin nucleator that mediates long actin tail formation (Fig. 4A) (11, 38, 39). Sca2 was an ideal target for several reasons: (i) the transposon mutant of sca2 has been well characterized (11, 39); (ii) loss of actin tail formation is easily observable via fluorescence microscopy; and (iii) sca2 does not appear to be encoded in an operon, making targeting with CRISPRi simpler.
Fig 4.

CRISPRi knockdown of the rickettsial virulence factor sca2. (A) Schematic of sca2 knockdown experiment. Sca2 is a formin-like actin nucleator responsible for forming long actin tails during R. parkeri infection. CRISPRi-mediated knockdown of sca2 should result in decreased actin tail formation. (B) CRISPRi targeting leads to decreased expression of Sca2 protein. A549 host cell monolayers were infected with R. parkeri. aTc was added to infections at 48 hpi, and lysates were harvested at 72 hpi. Sca2 and OmpA (loading control) protein levels were visualized via Western blotting (data are representative of three independent experiments). (C and D) Measurement of actin tail formation by immunofluorescence. A549 cell monolayers were infected with R. parkeri for 28 h, with aTc being added to appropriate wells at the time of infection. These samples were then fixed, stained, and imaged to visualize (C) and quantify (D) actin tail formation. Boxed region expanded in inset (arrowheads indicate bacteria with actin tails). Scale bar, 10 and 5 µm in inset. For each condition, at least 300 bacteria were quantified in each of n = 3 independent experiments. ***P < 0.001, determined by one-way ANOVA with post hoc Tukey’s test.
Based on our results from the knockdown of rifampicin resistance, we designed two gRNAs (gRNAs 1 and 2) for Spas dCas9, targeting the nontemplate strand upstream of the coding region in the endogenous sca2 locus. We first tested if we could detect obvious loss of sca2 expression using CRISPRi. We harvested R. parkeri infections of A549 host cells at 3 dpi, with 100 ng/mL aTc induction in the last 24 h before sample collection. Indeed, we were able to detect a decrease in Sca2 protein levels in both gRNA1 and gRNA2 relative to the NT control (Fig. 4B). Reduced Sca2 levels were also apparent in both the induced and uninduced conditions, in agreement with the data in Fig. 3 indicating leakiness of the system.
We then tested if CRISPRi-mediated silencing of sca2 also led to the expected reduction in actin tail frequency (11, 39, 40). We infected A549 host cells with WT and the strains harboring each of the gRNAs, with and without aTc, and fixed samples at 28 hpi for subsequent immunofluorescent staining and confocal microscopy (Fig. 4C). No differences were observed in tail frequencies between WT and NT control strains, suggesting that dCas9 expression and aTC dosage in this assay did not obviously alter infection under these conditions. As predicted from our Western blot data, both gRNA1 and gRNA2 resulted in a significant decrease in tail formation (Fig. 4D), corroborating the successful knockdown of sca2. Similar to the results from the experiments targeting rparr-2, no significant difference was observed between induced and uninduced, indicating leakiness of the inducible promoter system expressing dCas9. Taken together with the results above, our experiments demonstrate successful knockdown of the endogenously encoded sca2 virulence factor in R. parkeri using CRISPRi.
DISCUSSION
The Rickettsia genus is comprised of obligate intracellular bacteria, and several members are neglected and emerging human pathogens (41). Understanding their unique biology and mechanisms of pathogenesis has been hindered by the paucity of genetic tools available to carry out functional-genetic studies. Here, we have expanded the genetic toolkit by developing systems for conditional gene expression and targeted gene knockdown in rickettsiae. These tools will be invaluable for dissecting the genetic and molecular requirements of the rickettsial infectious life cycle and revealing novel biology at the host-pathogen interface.
The ability to control gene expression with a small molecule inducer will be a powerful tool for performing mechanistic investigations of key rickettsial virulence genes. For instance, by varying the timing and dosage of induction, this tool will allow us to study the kinetic requirements of a given virulence gene during the infectious life cycle. While conditional expression systems have been developed in C. burnetii and C. trachomatis, many obligate intracellular bacteria still lack them (42). Here, we have adapted the Tet-On system for use in R. parkeri, enabling conditional gene expression for the first time in a Rickettsia species. While this system uses aTc as the inducer molecule, our work suggests that other small molecule inducers could also potentially be implemented in rickettsiae, like isopropyl β-d-1-thiogalactopyranoside (IPTG) or arabinose. Further development of orthogonal conditional expression systems will open even more possibilities for genetic studies in rickettsiae.
Inducible promoter systems also offer other valuable applications including controlled expression of toxic proteins and conditional expression of other genetic tools like CRISPRi. In fact, heterologous expression of Cas9 and derivatives like dCas9 have been found to have toxic effects in various other bacteria, including model organisms like E. coli (19, 36). We were unable to clone dCas9 under a constitutive promoter into the pRAM18dSGA backbone in E. coli, which we speculate was due to dCas9 toxicity. Using the Tet-On system to control the expression of dCas9, we were able to successfully demonstrate CRISPRi-mediated targeted gene knockdown for the first time in the Rickettsiales order.
CRISPRi makes performing targeted gene knockdowns relatively simple as one only needs to design guide RNA sequences that target the locus of interest. However, the target space of CRISPRi is limited to sites within the genome with the appropriate PAM adjacent to the gRNA target sequence. Each dCas9 variant has a unique PAM that is recognized. Therefore, it is useful to have multiple variants of dCas9 to choose from to expand the range of targetable sites in the genome. Similar to previous reports (43), we also observed some flexibility in the S. pasteurianus dCas9 PAM requirements, with successful knockdown arising from gRNAs using both the NNGTGA and NNGCGA PAMs (e.g., sgRNA1 and sgRNA2, respectively, from the sca2 KD assay). In our study, we tested four variants of dCas9 that recognize different PAMs. Given previous work that found varying success with different dCas9 variants in different bacteria (24, 25), we were surprised that all four of the dCas9 variants we tested yielded successful gene knockdown. This variety of functional dCas9s affords us an expanded range of targetable sites within the rickettsial genome.
The design of optimal gRNA sequences also depends on other factors, including strand biases and proximity to the promoter. Similar to what was observed in several other bacteria, including the alphaproteobacterium C. crescentus (25), we found a stark preference for gRNAs targeting the nontemplate strand. Of the 14 gRNAs tested in this study, 9 of the gRNAs were complementary to the nontemplate strand. Incredibly, seven of these nine gRNAs targeting the nontemplate strand yielded significant gene knockdown. Most of the gRNAs were designed to target near the promoter, ideally within 100 bp of the predicted transcription start site of the target gene. Expanded testing of gRNAs targeting other rickettsial genes will be required to determine the precise parameters for optimal gRNA design in rickettsiae.
For both the inducible promoter and CRISPRi systems, there are limitations in their practical use in their current state. For example, the expression of bfp was difficult to detect in the presence of aTc, suggesting weak expression of certain genes in the “on” state. We attempted to overcome this by engineering tetO sites into known strong rickettsial promoters like PompA and PompB, but this yielded a starkly bimodal distribution of BFP expression (high BFP fluorescence vs no BFP signal) across the bacterial population. Given that these experiments were performed in the presence of antibiotic selection to maintain the plasmid, we do not believe this is due to plasmid loss. However, we cannot completely rule out the possibility of plasmid instability as a cause, and the observed distribution could be due to plasmid rearrangements or variability in plasmid copy number. Interestingly, similarly uneven expression from a Tet-On system was also observed in C. trachomatis, which was attributed to variable metabolic states at certain time points of infection during the chlamydial life cycle (32). We also attempted to implement the same tet-inducible promoter used in C. trachomatis (32), but this promoter was not functional in R. parkeri (data not shown). Additional work will be required to better understand what underlies the bimodal distribution we observed with our Tet-On system in R. parkeri.
Fortunately, despite the weak expression of bfp, the Tet-On system allows for sufficient expression of dCas9 for gene knockdown. It is possible that the low expression from our Tet-On system might be necessary to avoid toxicity in rickettsiae, as dCas9 has been shown to be toxic when expressed at high levels in other bacteria (19). Despite the Tet-On system displaying no detectable leakiness when controlling the expression of rparr-2 and bfp, the expression of dcas9 is less tightly controlled in the absence of aTc, with some variants showing more leaky expression than others. Notably, we observed some amount of inducibility for two of the gRNAs that yielded knockdown of rparr-2 (Spyo gRNA3 and Sthe1 gRNA1). This variability in leakiness between different gRNAs is similar to what was observed in Mycobacterium smegmatis, where they had to build and optimize a collection of new inducible promoters to enable the targeting of essential genes (24). We tested the optimized promoter that was implemented in M. tuberculosis (24), but it demonstrated weak and leaky expression in R. parkeri (data not shown). Alternatively, it might be possible to decrease the leakiness of the system by also placing the gRNA under the control of a separate inducible promoter (19). Beyond optimization of the inducible promoter system itself, it might also be possible to improve the inducibility of CRISPRi by introducing mismatches into non-seed regions of gRNAs to weaken their affinities to their respective target sites, similar to what has been done in other systems (44, 45). Another alternative approach could be to express a Cas9-specific anti-CRISPR protein to antagonize dCas9 in the absence of an inducer, but at a low enough level so that the anti-CRISPR could be overcome during the induction of dCas9 expression (46). Finally, care must be taken to avoid incorrectly interpreting potential off-target effects of the CRISPRi system, such as unintended downregulation of non-targeted genes. Selecting at least two different guides that result in the same phenotypic change can bolster support for a gene’s role in infection, similar to what we showed for silencing sca2. This approach is especially important given that our current CRISPRi system may be too large (currently ~15 kb) to easily support simultaneous knockdown and complementation, and dual plasmid transformation has not been done in Rickettsia spp. yet.
Nevertheless, the tools presented here open new roads for detailed investigations into the biology and pathogenesis of these important human pathogens. CRISPRi provides a platform for efficient and scalable targeted gene knockdown in rickettsiae, opening the possibility to directly probe the in vivo relevance of rickettsial genes that had previously only been studied through biochemical or exogenous expression assays. Our CRISPRi platform, combined with future improvements in transformation efficiency in rickettsiae, could allow for the first large-scale reverse genetic screens in Rickettsia. Moreover, sequence-specific binding of dCas9 can be used for other technological applications, such as CRISPR activation to increase the expression of endogenous loci (20). Overall, our work introduces two new methods for controlling gene expression in rickettsiae, which will ultimately be critical for gaining new insights into fundamental host-pathogen interactions and understanding how these neglected and emerging pathogens cause disease.
MATERIALS AND METHODS
Cell culture
Vero African green monkey kidney epithelial and A549 human lung epithelial cell lines were obtained from the University of California, Berkeley Cell Culture Facility (Berkeley, CA, USA). Vero cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Gibco catalog number 11965118) containing 5% fetal bovine serum (FBS). A549 cells were maintained in DMEM with 10% FBS. Assays measuring tagbfp expression, dcas9 expression, rparr-2 knockdown, and sca2 knockdown were conducted using DMEM containing tetracycline-negative FBS. Cell lines were confirmed to be mycoplasma-negative in a MycoAlert PLUS assay (Lonza catalog number LT07-710) performed by the Koch Institute High-Throughput Sciences Facility (Cambridge, MA, USA).
Plasmid construction
pRL0027 was generated from pRAM18dSGA[MCS] (13) (kindly provided by Ulrike Munderloh) by removing gfp and changing the promoter of the spectinomycin resistance cassette to PrpsL with an ompA terminator proceeding the gene. pRL0081 was generated from pRL0027 by cloning the Tet-On promoter from pdCas9-bacteria (Addgene catalog number 44249) along with rparr-2 and gfpuv under the control of Tet-ON with a single ompA terminator sequence. pRL0117, pRL0234, pRL0235, pRL0236, and pRL0237 were all generated similarly to pRL0081 but cloned downstream of the Tet-On promoter was a codon-optimized version of tagbfp for Rickettsia conorii (14), S. pyogenes dCas9 with constitutively expressed gRNA-SapI, S. thermophilus 01 dCas9 with constitutively expressed gRNA-SapI, S. thermophilus 03 dCas9 with constitutively expressed gRNA-SapI, and S. pasteurianus dCas9 with constitutively expressed gRNA-SapI, respectively. A C-terminal HA tag was appended to all dCas9 variants. pRL0200 was cloned via gene synthesis (Twist Biosciences) using the tagbfp sequence from pRL0117 and the ompA promoter from R. parkeri with a tetO1 operator sequence from pRL0081. pRL0203 was generated similarly to pRL0200 but with additional synthetic DNA fragments including the tetR gene with its promoter and gfpuv from pRL0081. pRL0202 was generated similarly to pRL0203 but with codon-optimized tagbfp under the control of a synthetic promoter constructed by adding two tetO2 sequences from pRL0081 to the ompB promoter from R. parkeri. pRL0057 was generated from pMW1650 (10) by replacing the rpsL promoter region with a 100-bp sequence that was modified to include additional PAM sequences.
Guide RNA plasmids were cloned via restriction cloning by digesting the pRL0234, pRL0235, pRL0236, and pRL0237 backbones with SapI and gel purifying the cut vector. Short oligonucleotides (Sigma) were designed to have compatible overhangs upon annealing and were ligated into the cut pRL0234-pRL0237 backbones. gRNA sequences were manually selected based on proximity and position relative to the predicted transcription start site and the likelihood of off-target effects. Potential off-target sites for each gRNA were screened for using Cas-OFFinder (47) (http://www.rgenome.net/cas-offinder/) and a modified Python script based on a previously published package (48). A full list of gRNA sequences is provided in Table S1.
Generation of R. parkeri strains
Wild-type R. parkeri strain Portsmouth (kindly provided by Chris Paddock) and all derivatives were propagated by infection and mechanical disruption of Vero cells grown in DMEM containing 2% FBS at 33°C as previously described (14). These bacterial stocks were further purified using 2 µm syringe filtering (Whatman) as previously described (40). Bacteria were clonally isolated from plaques formed from Vero host cell monolayer infection in the presence of agarose overlays as previously described (8). All bacterial stocks were stored as aliquots at −80°C in brain heart infusion media (Fisher Scientific, DF0037-17-8) to minimize freeze-thaw cycles. Titers were measured via plaque assay on Vero cells and quantified at 5 dpi.
Plasmids were introduced into R. parkeri via small-scale electroporation as previously described (8) with approximately 1 µg of dialyzed plasmid DNA. Selection was started 24 h after electroporation by overlaying a mixture of 0.5% agarose, DMEM with 2% FBS, and either rifampicin (200 ng/mL final concentration) or spectinomycin (50 µg/mL). The sites of transposon insertions for generating the modified rparr-2-containing strain for testing dCas9 knockdown were determined by semi-random nested PCR and Sanger sequencing as previously described (8).
Plaque assays
Plaque assays were conducted as previously described (8). Briefly, confluent Vero cell monolayers grown in 6-well plates were washed in phosphate-buffered saline (PBS) and subsequently infected with R. parkeri [multiplicity of infection (MOI) 0.001] in a humidified chamber and rocked for 30 min at 37°C. DMEM with 2% FBS and 0.5% agarose was overlaid on top of the infected cells, and this was incubated in a humidified chamber at 33°C with 5% CO2 for 5 days. Plaque assays were then imaged and analyzed using Fiji/ImageJ. For assays involving aTc induction, a small volume of concentrated aTc solution was added on top of the molten agarose overlays to give the appropriate final aTc concentration.
BFP expression assays
Confluent A549 host cell monolayers were grown on 12 mm coverslips in 24-well plates and infected at an MOI of approximately 0.05. R. parkeri was added to the media and centrifuged at 200 × g for 5 min at room temperature (RT). These infections were subsequently incubated at 33°C, and anhydrotetracycline was added to appropriate wells at 24 hpi. Samples were then fixed at 48 hpi by adding 4% paraformaldehyde in phosphate-buffered saline for 10 min at RT. Fixed samples were then washed in PBS, and residual paraformaldehyde was quenched by incubating samples with 0.1 M glycine for 10 min at RT. Next, samples were washed with PBS and incubated with blocking buffer (2% bovine serum albumin in PBS) for 30 min at RT. Samples were treated with primary and secondary antibodies suspended in blocking buffer for 1 h each, with three PBS washes after each incubation. Phalloidin conjugated to Alexa Fluor 647 (Invitrogen catalog # A22287) was used to detect actin and mouse anti-Rickettsia 14-13 (kindly provided by Ted Hackstadt) was used to detect R. parkeri. Coverslips were mounted with ProLong Gold Antifade mountant (Invitrogen catalog # P36934). For each condition, at least 300 bacteria were imaged using a 100× UPlanSApo (1.35 NA) objective. Images were processed with Fiji/ImageJ. CellProfiler (49) was used to measure blue fluorescence intensity within the bounds of individual bacteria as detected by anti-Rickettsia staining.
Actin tail assay
Confluent A549 host cell monolayers were infected and processed similarly to above with minor modifications. Infections were carried out at an MOI of approximately 0.1–0.5. Before infection, the media were replaced with fresh DMEM including appropriate antibiotics and anhydrotetracycline (100 ng/mL final concentration) in appropriate wells. Infected samples were fixed with paraformaldehyde at 28 hpi. Hoechst stain (Invitrogen catalog # H3570) was used to detect host cell nuclei. Image analysis was performed with Fiji/ImageJ. For every replicate of each strain and condition, at least three fields of view and at least 300 bacteria were analyzed to calculate the percentage of cytosolic bacteria with actin tails (>1 bacterial length). This was performed in triplicate.
Immunoblotting of Sca2 and dCas9-HA from infected host cell lysates
Fresh DMEM including appropriate antibiotics was added to confluent A549 cell monolayers, which were subsequently infected with strains of R. parkeri harboring plasmids with sca2 or dcas9-ha under the control of the Tet-On promoter. aTc was added 48 hpi. Then, at 72 hpi, the infected A549 host cell monolayers were resuspended in loading buffer [50 mM Tris-HCl (pH 6.8), 2% sodium dodecyl sulfate, 10% glycerol, 0.1% bromophenol blue, and 5% β-mercaptoethanol] and boiled for 20 min with vortexing. These samples were analyzed via Western blotting using rabbit anti-Sca2 (kindly provided by Matthew Welch), mouse anti-HA (BioLegend catalog number 901501), and mouse anti-OmpA 13-3 (kindly provided by Ted Hackstadt).
Statistical analyses
All statistical analyses were performed using GraphPad Prism 10. Graphical representations, statistical parameters, and significance are noted in figure legends. Statistical significance was defined as P < 0.05.
ACKNOWLEDGMENTS
We thank Ulrike Munderloh, Matthew Welch, Michael Laub, Chris Paddock, and Ted Hackstadt for sharing strains and reagents. We are grateful to members of the Lamason laboratory for helpful discussions.
Work in the Lamason laboratory is supported in part by the National Institutes of Health (R01 AI155489) and by the Office of the Assistant Secretary of Defense for Health Affairs through the Tick-Borne Disease Research Program (TB200032). Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense. J.M. is a Damon Runyon Fellow supported by the Damon Runyon Cancer Research Foundation (DRG-2396-20). D.L.E. was supported by the MIT Undergraduate Research Opportunities Program (UROP) Office, including through the Peter J. Eloranta Summer Undergraduate Research Fellowship, the John Reed UROP Fund, and the Joseph Woo (1988) UROP Fund for the Life Sciences.
Contributor Information
Rebecca L. Lamason, Email: rlamason@mit.edu.
Michael Y. Galperin, NCBI, NLM, National Institutes of Health, Bethesda, Maryland, USA
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/jb.00091-24.
Expression of TagBFP from Tet-On using higher concentrations of aTc.
Expression of TagBFP from engineered aTc-responsive rickettsial promoters.
gRNA sequences.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. 2012. Intracellular pathogens II: Rickettsiales. ASM Press, Washington, DC. [Google Scholar]
- 2. Biggs HM, Behravesh CB, Bradley KK, Dahlgren FS, Drexler NA, Dumler JS, Folk SM, Kato CY, Lash RR, Levin ML, Massung RF, Nadelman RB, Nicholson WL, Paddock CD, Pritt BS, Traeger MS. 2016. Diagnosis and management of tickborne rickettsial diseases: rocky mountain spotted fever and other spotted fever group rickettsioses, ehrlichioses, and anaplasmosis — United States: a practical guide for health care and public health professionals. MMWR Recomm Rep 65:1–44. doi: 10.15585/mmwr.rr6502a1 [DOI] [PubMed] [Google Scholar]
- 3. McGinn J, Lamason RL. 2021. The enigmatic biology of rickettsiae: recent advances, open questions and outlook. Pathog Dis 79:ftab019. doi: 10.1093/femspd/ftab019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Parola P, Paddock CD, Raoult D. 2005. Tick-borne rickettsioses around the world: emerging diseases challenging old concepts. Clin Microbiol Rev 18:719–756. doi: 10.1128/CMR.18.4.719-756.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McClure EE, Chávez ASO, Shaw DK, Carlyon JA, Ganta RR, Noh SM, Wood DO, Bavoil PM, Brayton KA, Martinez JJ, McBride JW, Valdivia RH, Munderloh UG, Pedra JHF. 2017. Engineering of obligate intracellular bacteria: progress, challenges and paradigms. Nat Rev Microbiol 15:544–558. doi: 10.1038/nrmicro.2017.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sit B, Lamason RL. 2024. Pathogenic Rickettsia spp. as emerging models for bacterial biology. J Bacteriol 206:e0040423. doi: 10.1128/jb.00404-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blanc G, Ogata H, Robert C, Audic S, Suhre K, Vestris G, Claverie J-M, Raoult D. 2007. Reductive genome evolution from the mother of Rickettsia. PLoS Genet 3:e14. doi: 10.1371/journal.pgen.0030014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lamason RL, Kafai NM, Welch MD. 2018. A streamlined method for transposon mutagenesis of Rickettsia parkeri yields numerous mutations that impact infection. PLoS One 13:e0197012. doi: 10.1371/journal.pone.0197012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim HK, Premaratna R, Missiakas DM, Schneewind O. 2019. Rickettsia conorii O antigen is the target of bactericidal Weil–Felix antibodies. Proc Natl Acad Sci U S A 116:19659–19664. doi: 10.1073/pnas.1911922116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu Z-M, Tucker AM, Driskell LO, Wood DO. 2007. Mariner-based transposon mutagenesis of Rickettsia prowazekii. Appl Environ Microbiol 73:6644–6649. doi: 10.1128/AEM.01727-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kleba B, Clark TR, Lutter EI, Ellison DW, Hackstadt T. 2010. Disruption of the Rickettsia rickettsii Sca2 autotransporter inhibits actin-based motility. Infect Immun 78:2240–2247. doi: 10.1128/IAI.00100-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Welch MD, Reed SCO, Lamason RL, Serio AW. 2012. Expression of an epitope-tagged virulence protein in Rickettsia parkeri using transposon insertion. PLoS One 7:e37310. doi: 10.1371/journal.pone.0037310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Burkhardt NY, Baldridge GD, Williamson PC, Billingsley PM, Heu CC, Felsheim RF, Kurtti TJ, Munderloh UG. 2011. Development of shuttle vectors for transformation of diverse Rickettsia species. PLoS One 6:e29511. doi: 10.1371/journal.pone.0029511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lamason RL, Bastounis E, Kafai NM, Serrano R, Del Álamo JC, Theriot JA, Welch MD. 2016. Rickettsia Sca4 reduces vinculin-mediated intercellular tension to promote spread. Cell 167:670–683. doi: 10.1016/j.cell.2016.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Driskell LO, Yu X, Zhang L, Liu Y, Popov VL, Walker DH, Tucker AM, Wood DO. 2009. Directed mutagenesis of the Rickettsia prowazekii pld gene encoding phospholipase D. Infect Immun 77:3244–3248. doi: 10.1128/IAI.00395-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nock AM, Clark TR, Hackstadt T. 2022. Regulator of actin-based motility (RoaM) downregulates actin tail formation by Rickettsia rickettsii and is negatively selected in mammalian cell culture. mBio 13:e0035322. doi: 10.1128/mbio.00353-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Noriea NF, Clark TR, Hackstadt T. 2015. Targeted knockout of the Rickettsia rickettsii OmpA surface antigen does not diminish virulence in a mammalian model system. mBio 6:e00323-15. doi: 10.1128/mBio.00323-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pelc RS, McClure JC, Kaur SJ, Sears KT, Rahman MS, Ceraul SM. 2015. Disrupting protein expression with peptide nucleic acids reduces infection by obligate intracellular Rickettsia. PLoS One 10:e0119283. doi: 10.1371/journal.pone.0119283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vigouroux A, Bikard D. 2020. CRISPR tools to control gene expression in bacteria. Microbiol Mol Biol Rev 84:e00077-19. doi: 10.1128/MMBR.00077-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bikard D, Jiang W, Samai P, Hochschild A, Zhang F, Marraffini LA. 2013. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res 41:7429–7437. doi: 10.1093/nar/gkt520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. 2013. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152:1173–1183. doi: 10.1016/j.cell.2013.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. doi: 10.1126/science.1225829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fonfara I, Le Rhun A, Chylinski K, Makarova KS, Lécrivain A-L, Bzdrenga J, Koonin EV, Charpentier E. 2014. Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR-Cas systems. Nucleic Acids Res 42:2577–2590. doi: 10.1093/nar/gkt1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rock JM, Hopkins FF, Chavez A, Diallo M, Chase MR, Gerrick ER, Pritchard JR, Church GM, Rubin EJ, Sassetti CM, Schnappinger D, Fortune SM. 2017. Programmable transcriptional repression in mycobacteria using an orthogonal CRISPR interference platform. Nat Microbiol 2:16274. doi: 10.1038/nmicrobiol.2016.274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guzzo M, Castro LK, Reisch CR, Guo MS, Laub MT. 2020. A CRISPR interference system for efficient and rapid gene knockdown in Caulobacter crescentus. mBio 11:e02415-19. doi: 10.1128/mBio.02415-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ouellette SP, Blay EA, Hatch ND, Fisher-Marvin LA. 2021. CRISPR interference to inducibly repress gene expression in Chlamydia trachomatis. Infect Immun 89:e0010821. doi: 10.1128/IAI.00108-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fu M, Liu Y, Wang G, Wang P, Zhang J, Chen C, Zhao M, Zhang S, Jiao J, Ouyang X, Yu Y, Wen B, He C, Wang J, Zhou D, Xiong X. 2022. A protein–protein interaction map reveals that the Coxiella burnetii effector CirB inhibits host proteasome activity. PLoS Pathog 18:e1010660. doi: 10.1371/journal.ppat.1010660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wachter S, Cockrell DC, Miller HE, Virtaneva K, Kanakabandi K, Darwitz B, Heinzen RA, Beare PA. 2022. The endogenous Coxiella burnetii plasmid encodes a functional toxin–antitoxin system. Mol Microbiol 118:744–764. doi: 10.1111/mmi.15001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bertrand KP, Postle K, Wray LV, Reznikoff WS. 1983. Overlapping divergent promoters control expression of Tn10 tetracycline resistance. Gene 23:149–156. doi: 10.1016/0378-1119(83)90046-x [DOI] [PubMed] [Google Scholar]
- 30. Bertrand KP, Postle K, Wray LV, Reznikoff WS. 1984. Construction of a single-copy promoter vector and its use in analysis of regulation of the transposon Tn10 tetracycline resistance determinant. J Bacteriol 158:910–919. doi: 10.1128/jb.158.3.910-919.1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gossen M, Bujard H. 1992. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci U S A 89:5547–5551. doi: 10.1073/pnas.89.12.5547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wickstrum J, Sammons LR, Restivo KN, Hefty PS. 2013. Conditional gene expression in Chlamydia trachomatis using the TET system. PLoS One 8:e76743. doi: 10.1371/journal.pone.0076743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bertram R, Neumann B, Schuster CF. 2022. Status quo of tet regulation in bacteria. Microb Biotechnol 15:1101–1119. doi: 10.1111/1751-7915.13926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Qin A, Tucker AM, Hines A, Wood DO. 2004. Transposon mutagenesis of the obligate intracellular pathogen Rickettsia prowazekii. Appl Environ Microbiol 70:2816–2822. doi: 10.1128/AEM.70.5.2816-2822.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cui L, Vigouroux A, Rousset F, Varet H, Khanna V, Bikard D. 2018. A CRISPRi screen in E. coli reveals sequence-specific toxicity of dCas9. Nat Commun 9:1912. doi: 10.1038/s41467-018-04209-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Riley LA, Guss AM. 2021. Approaches to genetic tool development for rapid domestication of non-model microorganisms. Biotechnol Biofuels 14:30. doi: 10.1186/s13068-020-01872-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hilton IB, D’Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, Gersbach CA. 2015. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol 33:510–517. doi: 10.1038/nbt.3199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Haglund CM, Choe JE, Skau CT, Kovar DR, Welch MD. 2010. Rickettsia Sca2 is a bacterial formin-like mediator of actin-based motility. Nat Cell Biol 12:1057–1063. doi: 10.1038/ncb2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Reed SCO, Lamason RL, Risca VI, Abernathy E, Welch MD. 2014. Rickettsia actin-based motility occurs in distinct phases mediated by different actin nucleators. Curr Biol 24:98–103. doi: 10.1016/j.cub.2013.11.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Harris EK, Jirakanwisal K, Verhoeve VI, Fongsaran C, Suwanbongkot C, Welch MD, Macaluso KR. 2018. Role of Sca2 and RickA in the dissemination of Rickettsia parkeri in Amblyomma maculatum. Infect Immun 86:e00123. doi: 10.1128/IAI.00123-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Parola P, Paddock CD, Socolovschi C, Labruna MB, Mediannikov O, Kernif T, Abdad MY, Stenos J, Bitam I, Fournier P-E, Raoult D. 2013. Update on tick-borne rickettsioses around the world: a geographic approach. Clin Microbiol Rev 26:657–702. doi: 10.1128/CMR.00032-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fisher DJ, Beare PA. 2023. Recent advances in genetic systems in obligate intracellular human-pathogenic bacteria. Front Cell Infect Microbiol 13:1202245. doi: 10.3389/fcimb.2023.1202245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tan SZ, Reisch CR, Prather KLJ. 2018. A robust CRISPR interference gene repression system in Pseudomonas. J Bacteriol 200:e00575-17. doi: 10.1128/JB.00575-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vigouroux A, Oldewurtel E, Cui L, Bikard D, van Teeffelen S. 2018. Tuning dCas9’s ability to block transcription enables robust, noiseless knockdown of bacterial genes. Mol Syst Biol 14:e7899. doi: 10.15252/msb.20177899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Collias D, Vialetto E, Yu J, Co K, Almási ÉDH, Rüttiger A-S, Achmedov T, Strowig T, Beisel CL. 2023. Systematically attenuating DNA targeting enables CRISPR-driven editing in bacteria. Nat Commun 14:680. doi: 10.1038/s41467-023-36283-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rauch BJ, Silvis MR, Hultquist JF, Waters CS, McGregor MJ, Krogan NJ, Bondy-Denomy J. 2017. Inhibition of CRISPR-Cas9 with bacteriophage proteins. Cell 168:150–158. doi: 10.1016/j.cell.2016.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bae S, Park J, Kim J-S. 2014. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30:1473–1475. doi: 10.1093/bioinformatics/btu048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Calvo-Villamañán A, Ng JW, Planel R, Ménager H, Chen A, Cui L, Bikard D. 2020. On-target activity predictions enable improved CRISPR–dCas9 screens in bacteria. Nucleic Acids Res 48:e64. doi: 10.1093/nar/gkaa294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McQuin C, Goodman A, Chernyshev V, Kamentsky L, Cimini BA, Karhohs KW, Doan M, Ding L, Rafelski SM, Thirstrup D, Wiegraebe W, Singh S, Becker T, Caicedo JC, Carpenter AE. 2018. CellProfiler 3.0: next-generation image processing for biology. PLoS Biol 16:e2005970. doi: 10.1371/journal.pbio.2005970 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expression of TagBFP from Tet-On using higher concentrations of aTc.
Expression of TagBFP from engineered aTc-responsive rickettsial promoters.
gRNA sequences.