

Abstract
Predicted secondary-structure elements encompassing the primer binding site in the 5′ untranslated region of Rous sarcoma virus (RSV) RNA play an integral role in multiple viral replications steps including reverse transcription, DNA integration, and RNA packaging (A. Aiyar, D. Cobrinik, Z. Ge, H. J. Kung, and J. Leis, J. Virol. 66:2464–2472, 1992; D. Cobrinik, A. Aiyar, Z. Ge, M. Katzman, H. Huang, and J. Leis, J. Virol. 65:3864–3872, 1991; J. T. Miller, Z. Ge, S. Morris, K. Das, and J. Leis, J. Virol. 71:7648–7656, 1997). These elements include the U5-Leader stem, U5-IR stem-loop, and U5-TΨC interaction region. Limited digestion of the 5′ untranslated region of wild-type and mutant RSV RNAs with structure- and/or sequence-specific RNases detects the presence of the U5-Leader stem and the U5-IR stem-loop. When a tRNATrp primer is annealed to wild-type RNAs in vitro, limited nuclease mapping indicates that the U5-IR stem becomes partially unwound. This is not observed when mutant RNAs with altered U5-IR stem-loop structures are substituted for wild-type RNAs. The U5-Leader stem also becomes destabilized when the tRNA primer is annealed to either wild-type or mutant RNA fragments. Nuclease mapping studies of tRNATrp, as well as the viral RNA, indicate that the U5-TΨC helix does form in vitro upon primer annealing. Collectively, these data suggest that the various structural elements near the RSV primer binding site undergo significant changes during the process of primer annealing.
Retroviral reverse transcription is the process by which a diploid viral RNA genome is converted into double-stranded DNA by the virus-encoded enzyme reverse transcriptase. Initiation of reverse transcription occurs in the 5′ untranslated region of the RNA genome at the primer binding site (PBS), which is annealed to the 3′ sequences of a cellular tRNA primer. The tRNA used to prime DNA synthesis is specific for each retrovirus. For Rous sarcoma virus (RSV), tRNATrp is used. RSV RNA sequences flanking the PBS are predicted to form specific secondary structures believed to be important for efficient initiation of reverse transcription. These predicted structures are the U5-Leader stem, composed of an inverted repeat sequence in U5 and Leader, and the U5-IR stem-loop, composed of an inverted repeat sequence in U5 (7, 8) (Fig. 1). An additional predicted interaction (U5-TΨC) between U5 sequences in RSV RNA and TΨC sequences in the tRNATrp primer also contributes to initiation of reverse transcription (1) (Fig. 1).
FIG. 1.

Schematic diagram of the predicted lowest-free-energy secondary structures formed when WT RSV RNA is complexed with tRNATrp (see Materials and Methods for a description of the structure prediction). Sequences are numbered from the 5′ end of the RNA genome. Only nucleotides 56 to 130 are shown, even though longer sequences were used for the predictions. tRNATrp is shown to interact with the viral RNA via two helices; one is formed between the 3′ 18 nucleotides of the tRNA and the PBS of the viral RNA, and the second is formed between the TΨC arm and loop of the tRNA and sequences in the U5 region of the viral RNA. tRNATrp sequences involved in these two interactions are shown in bold. All other tRNATrp sequences are labeled and represented by the circle-and-line drawing. The U5-IR and U5-Leader stems are also labeled.
A role for these predicted RNA structures in retroviral replication has been established by a genetic approach in which each individual structure was disrupted by multiple base substitution mutations, resulting in reverse transcription defects in vitro and in vivo. Restoration of these structures with alternative sequences partially rescues the defects (1, 7, 8). Sequences in the U5-IR stem-loop also contribute to DNA integration, since the first 15 nucleotides reverse transcribed become the U5 IN (integrase) recognition sequence (7). Recent studies also suggest that this region may be involved in RNA packaging (26).
Similar but not identical RNA secondary structures are predicted to exist in many retroelements, including retrotransposons like Tf1 (23), and retroviruses like Moloney murine leukemia virus (M-MuLV), feline immunodeficiency virus, and human immunodeficiency virus type 1 (HIV-1) (1, 8). For HIV-1, chemical-probing studies by Baudin et al. demonstrated that structures analogous to the U5-Leader stem and the U5-IR stem-loop exist in vitro in synthetic RNA fragments representing the 5′-terminal region of HIV-1 (5). With respect to the RSV U5-TΨC interaction, HIV-1 appears to have a different but analogous interaction between sequences in the U5-IR loop (also known as the A-rich loop) and sequences in the anticodon loop of tRNA3Lys (17, 19, 30). By using a variety of different assays, this loop-loop interaction has been shown to play a role in the initiation of HIV-1 reverse transcription (3, 18, 20, 22, 32).
Although the mutagenesis studies of RSV strongly suggest that the secondary structures surrounding the PBS in the genomic RNA exist and play a significant role in the initiation of reverse transcription, there is no direct physical evidence that these structures are present. In the following in vitro study, we have used sequence- and/or structure-specific RNases to map both the RSV RNA and tRNATrp secondary structures before and after formation of the viral RNA-tRNATrp complex. The mapping studies indicate that in uncomplexed viral RNA, both the U5-Leader stem and U5-IR stem-loop are formed; however, when tRNATrp is annealed to the viral RNA, both the U5-Leader and U5-IR stems are destabilized. Independent digestions of 32P-labeled viral RNA and 32P-labeled tRNATrp also indicate that both the PBS-tRNA and U5-TΨC interactions exist in vitro upon primer annealing. Additional mapping studies with mutant viral RNAs (defective in initiation of reverse transcription) complexed with tRNATrp support the above conclusions.
MATERIALS AND METHODS
Reagents.
T7 RNA polymerase (20 U/μl) and human placental RNase inhibitor (40 U/μl) were purchased from Ambion, and Vent DNA polymerase (2 U/μl) and T4 polynucleotide kinase (10 U/μl) were from New England Biolabs. NucTrap columns were obtained from Stratagene, and MicroSpin G-25 columns were obtained from Amersham Pharmacia Biotech. RNase T1 (100 U/μl), EcoRI (20 U/μl), T4 DNA ligase (1 U/μl), T4 RNA ligase (10 U/μl), glycogen (20 μg/μl), and deoxynucleoside and ribonucleoside triphosphates were purchased from Boehringer Mannheim. Shrimp alkaline phosphatase (1 U/μl) was purchased from Amersham Life Science, RNase V1 (720 U/ml) was purchased from Pharmacia Biotech, and RNase A was purchased from Sigma (no. R-5500). [γ-32P]ATP, [α-32P]GTP, and 5′-[32P]pCp (3,000 Ci/mmol; 10 μCi/μl) were obtained from New England Nuclear. Oligodeoxynucleotides were synthesized by either Midland Certified Reagent Co. (Midland, Tex.) or Genosys Biotechnologies, Inc. (The Woodlands, Tex.). Other chemicals were of the highest grade available and were purchased from Fisher Chemicals.
Plasmids.
pDC101S and the mutant vectors pDC101S-I12Lss and pDC101S-RDS have been described previously (26). All plasmids were transformed into Escherichia coli DH5α (obtained from Gibco BRL) and purified with the Qiagen Plasmid Maxi Kit as specified by the manufacturer.
Preparation of RSV DNA templates.
Plasmids pDC101S, pDC101S-I12Lss, and pDC101S-RDS were linearized with EcoRI and used as templates for PCR amplification with Vent polymerase. The following sets of primers were used: pDC101S amplified with T7-R and ASLV380–400, and pDC101S-I12Lss and pDC101S-RDS amplified with T7-R and ASLV270–240. T7-R is a plus-strand primer in which T7 promoter sequences are linked to sequences encompassing the first 18 nucleotides of R in the RSV RNA genome. ASLV380–400 is a minus-strand primer complementary to nucleotides 380 to 400 of the RSV RNA genome. ASLV270–240 is a minus-strand primer complementary to nucleotides 240 to 270 of the RSV RNA genome. The T7-R sequence is 5′CCCTAATACGACTCACTATAGCCATTTTACCATTCACC3′ (38-mer), the ASLV380–400 sequence is 5′CACCTTTATGACGGCTTCCAT3′ (21-mer), and the ASLV270–240 sequence is 5′CCTGCAGTAGAGCTCCCTCCGACGCCACTC3′ (30-mer). After amplification, bands of the expected sizes (420 bp for pDC101S, 302 bp for pDC101S-I12Lss and pDC101S-RDS) were purified from 1% agarose gels with the Qiaex II agarose gel extraction kit from Qiagen as specified by the manufacturer.
Preparation of tRNATrp DNA template.
tRNATrp DNA templates were prepared by ligating several overlapping synthetic oligodeoxynucleotides. An explanation of each oligomer, and its sequence, is as follows. (+)5′T7-tRNATrp is a plus-strand oligomer encompassing T7 promoter sequences linked to the first 15 nucleotides of the RSV primer tRNATrp (33). (+)Mid-tRNATrp is a plus-strand oligomer including nucleotides 16 to 45 of tRNATrp. (+)3′-tRNATrp is a plus-strand oligomer encompassing nucleotides 46 to 75 of tRNATrp. (−)5′-tRNATrp is a minus-strand oligomer complementary to nucleotides 22 to 75 of tRNATrp, while (−)3′-tRNATrp is a minus-strand oligomer complementary to nucleotides 1 to 21 of tRNATrp and the T7 promoter. The sequences of the oligomers are as follows: (+)5′T7-tRNATrp, 5′TAATACGACTCACTATAGACCTCGTGGCGCAA3′ (32-mer); (+)Mid-tRNATrp, 5′CGGTAGCGCGTCTGACTCCAGATCAGAAGG3′ (30-mer); (+)3′-tRNATrp, 5′CTGCGTGTTCGAATCACGTCGGGGTCACCA3′ (30-mer); (−)5′-tRNATrp, 5′TGGTGACCCCGACGTGATTCGAACACGCAGCCTTCTGATCTGGAGTCAGACGCG3′ (54-mer); and (−)3′-tRNATrp, 5′CTACCGTTGCGCCACGAGGTCTATAGTGAGTCGTATTA3′ (38-mer).
After synthesis, each oligomer was purified by denaturing polyacrylamide gel electrophoresis (PAGE) (10% polyacrylamide). Oligodeoxynucleotides needing 5′ phosphate groups for ligation [(+)Mid-tRNATrp, (+)3′-tRNATrp, and (−)3′-tRNATrp] were treated with T4 polynucleotide kinase as specified by the manufacturer. Equimolar amounts (100 pmol each) of the oligomers were mixed in the presence of 1× T4 DNA ligase buffer, heated to 95°C for 5 min, and allowed to cool slowly to room temperature. The annealed oligomers were then ligated by using T4 DNA ligase as specified by the manufacturer.
In vitro transcription of synthetic RNAs.
Viral RNA fragments were synthesized in a reaction mixture consisting of 20 nM DNA template, 8 mM MgCl2, 40 mM Tris-HCl (pH 7.9), 2 mM spermidine, 10 mM dithiothreitol, 20 mM NaCl, 1 mM each ribonucleoside triphosphate (ATP, CTP, GTP, and UTP), and 150 U of human placental RNase inhibitor. Each reaction was started by the addition of 140 U of T7 RNA polymerase, and the mixtures were incubated for 1 h at 37°C. Another 140 U of T7 RNA polymerase was added to each of the reaction mixtures, which were incubated for another 1 h at 37°C. At this point, the RNAs were extracted with phenol-chloroform and precipitated with 3 M sodium acetate (pH 5.2) and 95% ethanol. After centrifugation, the RNA pellets were air dried, suspended in STE buffer (100 mM NaCl, 20 mM Tris [pH 7.7], 10 mM EDTA [pH 8.0]), and passed through a NucTrap column as specified by the manufacturer. After removal of the unincorporated ribonucleotides by the column, the RNA fragments were again precipitated with 3 M sodium acetate (pH 5.2) and 95% ethanol. RNAs pelleted by centrifugation were suspended in 1 mM EDTA (pH 8.0) and quantified by measurement of UV absorbance at 260 nm.
tRNATrp primers were prepared in an identical fashion, except that the concentration of DNA template in the in vitro transcription reaction mixtures was 33 nM.
32P labeling and purification of viral RNA fragments used in viral RNA mapping experiments.
Wild-type (WT) and mutant viral RNA fragments were dephosphorylated for 1 h at 37°C in a reaction mixture consisting of 20 mM Tris-HCl (pH 8.0), 10 mM MgCl2, 5 U of shrimp alkaline phosphatase, and 2.5 μM RNA. The phosphatase was inactivated for 15 min at 65°C, and the dephosphorylated RNAs were 5′-end labeled for 1 h at 37°C in a mixture of 83 mM Tris-HCl (pH 7.6), 17 mM MgCl2, 5 mM dithiothreitol, 1.7 μM RNA, 40 U of T4 polynucleotide kinase, and 556 nM [γ-32P]ATP. After being labeled, the RNAs were precipitated with an equal volume of 5 M ammonium acetate, 20 μg of glycogen, and 2.5 volumes of 95% ethanol. The RNAs were pelleted by centrifugation, suspended in equal volumes of 1 mM EDTA (pH 8.0) and 90% formamide loading buffer (FLB) (90% formamide, 0.1% xylene cyanol, 0.1% bromophenol blue, 10 mM EDTA [pH 8.0]), and separated by denaturing PAGE (5% polyacrylamide). After electrophoresis, the full-length RNA bands were sliced from the gel and eluted with rotation for 6 to 8 h at 4°C in an elution buffer of 0.5 M sodium acetate (pH 5.2), 1 mM EDTA, 2.5% phenol, and 2.5% chloroform-isoamyl alcohol (24:1). Following elution, the RNAs were filtered through Millipore Ultrafree-MC filter units (0.45-μm-pore-size, low-protein-binding Durapore membrane). They were then extracted once with phenol-chloroform-isoamyl alcohol (25:24:1) and three times with chloroform-isoamyl alcohol (24:1), and precipitated with 1/10 volume of 3 M sodium acetate, 20 μg of glycogen, and 2.5 volumes of 95% ethanol. The purified RNAs were pelleted by centrifugation, suspended in 1 mM EDTA (pH 8.0), and quantified by measurement of UV absorbance at 260 nm. WT and mutant RNAs were divided into 0.7 pmol aliquots of 3 μl each and frozen at −20°C until used in viral RNA-mapping experiments.
32P labeling and purification of viral RNA fragments used in tRNATrp mapping experiments.
A small quantity of in vitro-transcribed viral RNA was dephosphorylated and was 5′-end labeled with [γ-32P]ATP in a manner similar to that described above. The small quantity of 32P-labeled viral RNA (approximately 50 pmol) was mixed with a large quantity of unlabeled viral RNA (approximately 2,000 pmol) of the same sequence and subjected to PAGE purification and elution as described above. The eluted RNAs were stored at −20°C in ethanol until they were needed for tRNATrp mapping experiments, at which point the purified RNAs were pelleted by centrifugation, suspended in 1 mM EDTA (pH 8.0), and quantified by measurement of UV absorbance at 260 nm.
32P labeling and purification of synthetic tRNATrp used in viral RNA-mapping experiments.
Synthetic tRNATrp was labeled internally with [α-32P]GTP during in vitro transcription. A reaction mixture containing 50 nM DNA template, 500 μM each ATP, UTP, and CTP, 21 μM [α-32P]GTP, 40 mM Tris-HCl (pH 8.0), 6 mM MgCl2, 2 mM spermidine, 20 mM dithiothreitol, 20 mM NaCl, 10 U of T7 RNA polymerase, and 10 U of RNase inhibitor was incubated for 1 h at 37°C. At this point, the RNA was precipitated with an equal volume of 5 M ammonium acetate, 20 μg of glycogen, and 2.5 volumes of 95% ethanol. The RNA was pelleted by centrifugation, suspended in 1 mM EDTA, and added to the nonradioactive tRNATrp synthesized above. The entire sample, consisting of tRNATrp synthesized with GTP or [α-32P]GTP, was subjected to electrophoresis through denaturing 10% polyacrylamide gels. RNA was recovered as described above. After quantification by measurement of UV absorbance at 260 nm, the synthetic tRNATrp was divided into aliquots with a concentration of 5 pmol/μl and used in the viral RNA-mapping experiments.
32P labeling and purification of synthetic tRNATrp used in tRNATrp-mapping experiments.
Synthetic tRNATrp was 3′-end labeled with 5′-[32P]pCp in a reaction mixture consisting of 50 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 10 mM dithiothreitol, 1 mM ATP, 30 μg of bovine serum albumin per ml, 10% dimethyl sulfoxide, 30 U of RNase inhibitor, 2.2 μM tRNATrp, 2.2 μM 5-[32P]pCp, and 20 U of T4 RNA ligase. The mixture was incubated overnight on ice and extracted once with an equal volume of phenol-chloroform-isoamyl alcohol (25:24:1), and the aqueous solution was centrifuged through a MicroSpin G-25 column to remove unincorporated nucleotides. The entire tRNATrp sample was mixed with an equal volume of 90% FLB and subjected to electrophoresis through denaturing 10% polyacrylamide gels as described above. RNA was recovered and quantified, and the sample was divided into 1-pmol aliquots of 2 μl each and frozen at −20°C until used in the tRNATrp-mapping experiments.
Nuclease mapping assays of 5′-32P-labeled viral RNA fragments with or without synthetic tRNATrp.
For each assay, 0.7 pmol of WT or mutant RNA was folded in a solution of 28 mM Tris-HCl (pH 8.0), 70 mM NaCl, 2.4 mM dithioerythritol, and 1 mM EDTA (pH 8.0) by incubating the reaction mixture for 2 min at 65°C and letting it cool slowly to room temperature. For assays with tRNATrp, 5 pmol of tRNA was added prior to the folding process. After cooling, MgCl2 was added to each reaction mixture to a final concentration of 7.4 mM, and the mixture was incubated at 40°C for 5 min. At this point, various concentrations of the RNase of choice (T1, V1, or A) were added, and the tubes were incubated for 5 min at 37°C. The digestion was stopped by placing the reaction mixtures on ice and adding 4 μg of rRNA and EDTA to a final concentration of 10 mM. The samples were immediately precipitated with 3 M sodium acetate, glycogen, and ethanol. For T1 digests, final enzyme concentrations of 0.003 or 0.0009 U/μl were used. For V1 digests, final enzyme concentrations of 1 × 10−4, 5 × 10−5, or 5 × 10−6 U/μl were used. For RNase A digests, final enzyme concentrations of 3, 1, and 0.3 pg/μl were used. For every set of digestion reaction mixtures, a negative control where no exogenous nuclease was added was also used. The enzyme concentrations mentioned above were selected after a dilution series of each enzyme was used in several digestion reactions and those concentrations resulting in less than 5% template digestion were chosen. This was done to ensure that each enzyme made only primary cleavages of the template.
Nuclease mapping assays of 3′-32P-labeled synthetic tRNATrp with or without wild-type viral RNA fragments.
For each assay, 1 pmol of tRNATrp was folded as described above. For assays with WT viral RNA, 3 pmol (400 ng) of viral RNA was added prior to the folding process. After cooling, 400 ng of unlabeled E. coli tRNA (Sigma R4251) was added to each reaction mixture where no viral RNA had been added. This was done to equalize the quantity of RNA in each tube. MgCl2 was then added to each reaction mixture to a final concentration of 7.4 mM, and the mixture was incubated at 40°C for 5 min. At this point, the RNAs were subjected to digestion with RNase T1 or V1 as described above. For T1 digests, final enzyme concentrations of 0.007, 0.002, or 0.02 U/μl were used. For V1 digests, final enzyme concentrations of 1 × 10−5, 3 × 10−5, or 1 × 10−4 U/μl were used. For every set of digestion reactions, a negative control where no exogenous nuclease was added was also used. Enzyme concentrations were chosen as described above.
Preparation of RNA sequence markers by RNase T1 digestion.
In a manner similar to that described by Donis-Keller et al. (11), 0.7 pmol of 5′-32P-labeled viral RNA or 1 pmol of 3′-32P-labeled tRNATrp was added to a reaction mixture composed of 6.7 M urea, 20 mM sodium citrate (pH 5.0), 50 ng of rRNA per μl, and 1 mM EDTA (pH 8.0). To facilitate the denaturing process, the reaction mixtures were incubated at 50 to 60°C for 5 min. RNase T1 at a final concentration of 0.5 U/μl was added directly to the reaction mixture, which was further incubated at 50 to 60°C for 5 min. The digestion was stopped and the samples were treated as described above.
Preparation of RNA markers by alkaline hydrolysis.
5′-32P-labeled viral RNA (0.7 pmol) or 3′-32P-labeled tRNATrp (1 pmol) was incubated at 95°C for 30 s in a solution of 133 ng of rRNA per μl, 33 mM NaOH, and 1 mM EDTA. The hydrolysis reaction was stopped by immediately precipitating the reaction products with 3 M sodium acetate, glycogen, and ethanol.
Visualization of digested RNAs.
After ethanol precipitation, the RNA samples were pelleted by centrifugation and suspended in equal volumes of 1 mM EDTA (pH 8.0) and 90% FLB. For the viral RNA-mapping experiments, one-third of each sample was analyzed by denaturing PAGE (5% polyacrylamide) and autoradiography, which visualized the 5′-32P-labeled viral RNA fragments. Since the majority of the tRNATrp was unlabeled, it was not seen on these films.
For the tRNATrp-mapping experiments, half of each sample was analyzed by denaturing PAGE (15% polyacrylamide) and autoradiography, which visualized the 3′-32P-labeled tRNATrp fragments. Since the majority of the viral RNA was unlabeled, it was not seen on these films.
Structure prediction.
The secondary structure of WT RSV RNA annealed to its tRNATrp primer (Fig. 1) has been predicted previously (1, 8). Wild-type and mutant RSV RNA secondary structures (without tRNATrp) were predicted by using Mfold version 3.0, which uses the RNA-folding energy parameters described by the Turner group (29). Prediction of the secondary structure of mutant RSV RNAs annealed to tRNATrp is based on the WT structure. All of the structures depicted throughout this paper are predicted to exist regardless of the length of the fragment used for the modeling exercise. Fragments ranging in length from 150 to 400 nucleotides were used to model these structures, and in every case the length of the fragment did not change the structures predicted to form from nucleotides 56 to 130 (WT sequence) or nucleotides 56 to 142 (mutant sequences).
RESULTS
Cleavage site assignment.
To identify specific sites of cleavage in the mapping experiments described below, either viral RNA (WT and mutant) or tRNATrp was subjected to limited alkaline hydrolysis such that an RNA ladder representing cleavage at every nucleotide was produced (Fig. 2A, lane 7; also see Fig. 7B, OH− lane). This ladder was then compared to the banding pattern produced from incomplete digestion of an aliquot of the same RNA with RNase T1 (specific for unpaired guanine residues) under denaturing conditions of 7 M urea and 50 to 60°C. Figures 2 and 4 show T1-digested marker lanes for the viral RNA molecules studied, while Fig. 7 depicts T1-digested marker lanes for tRNATrp. Note that the denaturing conditions used were not severe enough to cause every G residue in each type of viral RNA to be equally susceptible to T1 cleavage. This lack of cleavage implies that such G residues are in a stable secondary structure that is resistant to T1 cleavage.
FIG. 2.



PAGE analysis of 5′-32P-labeled WT RNAs subjected to limited nuclease digestion in the presence and absence of tRNATrp primer. Bands (arrows) in the marker lanes are WT RNA fragments digested with 5 U of T1 under denaturing conditions of 7 M urea and 60°C. Numbers to the left of each arrow describe the length of each digestion product from the 5′-32P label. To the left of each figure are brackets designating in which RNA secondary structure cleavage occurs. At the top of the figure are brackets designating in which lanes various components were added to the reaction mixture. (A) Synthetic WT RNA (0.7 pmol) with or without 5 pmol of synthetic tRNATrp was digested with increasing amounts of RNase T1. Lanes: 1 and 6, T1 concentration of 0.003 U/μl; 2 and 5, T1 concentration of 0.0009 U/μl; 3 and 4, negative controls where the RNAs were incubated in the absence of T1; 7, alkaline hydrolysis ladder of WT RSV RNA. (B) Synthetic WT RNA (0.7 pmol) with or without 5 pmol of synthetic tRNATrp was digested with increasing amounts of RNase A. Lanes: 1 and 8, RNase A concentration of 3 pg/μl; 2 and 7, RNase A concentration of 1 pg/μl; 3 and 6, RNase A concentration of 0.3 pg/μl; 4 and 5, negative controls where the RNAs were incubated in the absence of RNase A. (C) Synthetic WT RNA (0.7 pmol) with or without 5 pmol of synthetic tRNATrp was digested with increasing amounts of RNase V1. Lanes: 1 and 8, V1 concentration of 5 × 10−6 U/μl; 2 and 7, V1 concentration of 5 × 10−5 U/μl; 3 and 6, V1 concentration of 1 × 10−4 U/μl; 4 and 5, negative controls where no exogenous V1 was added.
FIG. 7.

PAGE analysis of 3′-32P-labeled tRNATrp subjected to limited nuclease digestion in the presence and absence of WT viral RNA. Bands (designated by arrows) in the marker lanes are fragments resulting from digestion of tRNATrp with 5 U of T1 under denaturing conditions of 7 M urea and 60°C. Numbers to the left of each arrow refer to which G residue in tRNATrp was cleaved by T1 (G residues numbered from the 5′ end, 32P label at the 3′ end). To the left of each figure are brackets designating in which tRNATrp secondary structure the cleavage occurs, and to the right of each figure are brackets designating in which viral RNA-tRNATrp structure the cleavage occurs. At the top of each figure are brackets designating in which lanes various components were added to the reaction mixture. (A) Synthetic tRNATrp (1 pmol) with or without 3 pmol of synthetic WT RNA was digested with increasing amounts of RNase T1. Lanes: 1 and 8, T1 concentration of 0.02 U/μl; 2 and 7, T1 concentration of 0.007 U/μl; 3 and 6, T1 concentration of 0.002 U/μl; 4 and 5, negative controls where the RNAs were incubated in the absence of T1. (B) Synthetic tRNATrp (1 pmol) with or without 3 pmol of synthetic WT RNA was digested with increasing amounts of RNase V1. Lanes: 1 and 8, V1 concentration of 1 × 10−4 U/μl; 2 and 7, V1 concentration of 3 × 10−5 U/μl; 3 and 6, V1 concentration of 1 × 10−5 U/μl; 4 and 5, negative controls where the RNAs were incubated in the absence of V1. The lane labeled OH− is an alkaline hydrolysis ladder of tRNATrp.
FIG. 4.

PAGE analysis of 5′-32P-labeled mutant RNAs subjected to limited RNase T1 digestion in the presence and absence of tRNATrp primer. Bands (arrows) in the marker lane resulted from digestion of either I12Lss or RDS mutant RNAs with 5 U of T1 under denaturing conditions of 7 M urea and 60°C. Notations are as in the legend to Fig. 2. In each panel, lanes 1 and 6 designate reactions where the T1 concentration was 0.003 U/μl, lanes 2 and 5 designate reactions where the T1 concentration was 0.0009 U/μl, and lanes 3 and 4 represent negative controls where the RNAs were incubated in the absence of T1. (A) Synthetic I12Lss mutant RNA (0.7 pmol) with or without 5 pmol of synthetic tRNATrp was digested with increasing amounts of T1. (B) Synthetic RDS mutant RNA (0.7 pmol) with or without 5 pmol of synthetic tRNATrp was digested with increasing amounts of T1.
Nuclease digestion of 5′ [32P]-labeled wild-type viral RNA fragments.
RNA fragments representing nucleotides 1 to 400 of WT RSV were subjected to limited digestion with three nucleases (RNase T1, RNase A, or RNase V1) as outlined in Materials and Methods. As mentioned above, T1 is specific for unpaired guanine residues, RNase A is specific for unpaired pyrimidine residues, and V1 has no sequence specificity but cleaves after bases involved in secondary or tertiary interactions. All digests described in this section, as well as throughout the rest of the paper, were performed such that only 1 to 5% of the full-length RNA product was cleaved by exogenous nuclease. Figure 2A shows the cleavage products visualized when WT RNAs were digested with increasing amounts of RNase T1 in the absence of tRNATrp. The data from this figure, as well as data from RNase A and V1 digests of WT fragments (Fig. 2B and C, respectively), are summarized in Fig. 3A. The predominant T1 cleavage site was at residue G90, which is predicted to lie within the U5-IR loop. As expected, G84, G87, and G94 were not cleaved since they lie within the predicted U5-IR stem, while G121 and G125 to G127 were also undigested because they form part of the U5-Leader stem. Cleavage of G80 and the lack of T1 cleavage at G103, G104, and G106 suggest that a short (4-bp) stem composed of nucleotides 75 to 78 and 103 to 106 (U5-PBS stem) also exists and is separated from the U5-IR stem by a bulge, as depicted in Fig. 3A. Moderate digestion of G112, G115, and G117 indicates that RNA sequences forming the 3′ portion of the PBS are fairly unstructured. It is unclear why G66, G74, and G93, all of which are predicted to lie within a bulge or loop, were relatively resistant to T1 cleavage.
FIG. 3.

Summary of nuclease digestion data for WT RSV RNA
fragments. Lowest-free-energy secondary-structure diagrams for WT
sequences are depicted (see Materials and Methods for a description of
structure prediction). G residues (target of T1 nuclease)
in the viral RNA are underlined. Nucleotide sequences are numbered
every 10 bases from the 5′ end of the RSV RNA genome. Brackets are used
to designate several RNA-RNA duplexes. (A) Digestion of uncomplexed WT
RNA fragments. A bold lowercase t indicates T1
cleavages, where t t t t was the strongest cleavage site
seen with 0.0009 U of T1 per μl and t t t
designates additional cleavage sites seen with 0.0009 U of
T1 per μl; t t describes moderate-cleavage
sites seen with 0.003 U of T1 per μl, and t
describes slight cleavages with 0.003 U of T1 per μl. A
bold lowercase v indicates V1 cleavages, where
v v v was the strongest cleavage seen with 0.0001 U of
V1 per μl, v v was a strong cleavage seen with
0.0001 U of V1 per μl, and v was a weak
cleavage seen with 0.0001 U of V1 per μl. A bold
lowercase a indicates RNase A cleavages, where a a a
a was the strongest cleavage seen with 0.3 pg of A per μl,
a a a describes additional cleavages seen with 0.3 pg of A
per μl, a a described additional cleavages seen with 1 pg
of A per μl, and a designates cleavages seen only with 3
pg of A per μl. (B) Digestion of WT RNA fragments complexed with
tRNATrp, where the tRNATrp sequences are shown
in bold capital letters. Description of cleavages is as for panel A.
(C) Changes in the digestion pattern when WT RNA fragments were
complexed with tRNATrp. Arrows indicate how the nuclease
digestion pattern changed as the WT fragments were complexed with the
tRNATrp primer. Upward arrows indicate increased cleavage,
while downward arrows indicate decreased cleavage. The number of arrows
indicates the magnitude of the change. ↑ and ↓ indicate changes in
the T1 pattern,
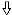 and
and
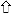 indicate changes in the
V1 pattern, and
and ↓ indicate changes in the A
pattern. Absence of arrows means there was no change in the digestion
pattern.
indicate changes in the
V1 pattern, and
and ↓ indicate changes in the A
pattern. Absence of arrows means there was no change in the digestion
pattern.
In general, RNase A and V1 digestion of WT RNAs (data shown in Fig. 2B and C and summarized in Fig. 3A) agreed with the T1 data described above. The most prominent RNase A cleavage was at C81, predicted to lie within the bulge between the U5-IR stem and the U5-PBS stem. Additional RNase A cleavages were seen scattered throughout those RNA sequences forming the unstructured region between the U5-PBS stem and the U5-Leader stem. RNase A did not digest any C’s or U’s in either the U5-IR or U5-PBS stem and displayed very limited cleavage of U62 and U122 in the U5-Leader stem. In contrast, all three stems (U5-Leader, U5-PBS, and U5-IR) showed moderate cleavage with V1, an enzyme with structural specificity opposite to that of RNase T1 and A. V1 also displayed very limited cleavage of U69 and U116, which are located in an unstructured region, as suggested by the RNase T1 and A data. This could indicate that the stem formed by nucleotides 69 to 72 and 114 to 117 exists transiently. This conclusion is supported by the fact that G72 was not susceptible to T1 cleavage and U69 was not susceptible to RNase A digestion.
Nuclease digestion of 5′-32P-labeled mutant viral RNA fragments.
I12Lss and RDS mutant viruses have been described previously (26). The I12Lss mutation is predicted to increase the size of the U5-IR loop by 12 bases, and the RDS mutation is predicted to increase the size of the U5-IR stem by 6 bp. Although both mutant viruses exhibited growth defects, the I12Lss mutation caused a mild reverse transcription initiation defect and resulted in normal levels of RNA packaging and DNA integration, while the RDS mutation caused a severe reverse transcription initiation defect and a severe RNA packaging defect but no defect in integration (26). Figure 4 shows the T1 cleavage products of I12Lss (Fig. 4A) and RDS (Fig. 4B) RNAs in the absence of tRNATrp. Figure 5A summarizes the T1 data from Fig. 4A as well as the RNase V1 and A cleavages (data not shown) for I12Lss. In an analogous fashion, Fig. 6A summarizes the T1 data from Fig. 4B as well as the RNase V1 and A cleavages (data not shown) for RDS. Overall, the nuclease digestion patterns of both I12Lss and RDS with all three RNases mimic those seen with WT RNAs.
FIG. 5.

Summary of nuclease digestion data for I12Lss mutant RSV
RNA fragments. Secondary-structure diagrams for I12Lss sequences are
depicted as described in Materials and Methods. Mutant nucleotides are
denoted by italicized lettering. Other notations are as described in
the legend to Fig. 3. (A) Digestion of uncomplexed I12Lss RNA
fragments. Description of cleavages is as in the legend to Fig. 3A.
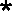 , the exact cleavage site was
unclear, although the data did indicate that cleavage was somewhere
within the U5-IR stem. (B) Digestion of I12Lss RNA fragments complexed
with tRNATrp, where the tRNATrp sequences are
shown in bold capital letters. Description of cleavages is as in the
legend to Fig. 3B, except for V1 cleavages, where there was
no dominant cleavage with 0.0001 U of V1 per μl;
therefore, there is no v v v designation. (C) Changes in the
digestion pattern when I12Lss RNA fragments were complexed with
tRNATrp. Description of changes is as in the legend to Fig.
3C.
, the exact cleavage site was
unclear, although the data did indicate that cleavage was somewhere
within the U5-IR stem. (B) Digestion of I12Lss RNA fragments complexed
with tRNATrp, where the tRNATrp sequences are
shown in bold capital letters. Description of cleavages is as in the
legend to Fig. 3B, except for V1 cleavages, where there was
no dominant cleavage with 0.0001 U of V1 per μl;
therefore, there is no v v v designation. (C) Changes in the
digestion pattern when I12Lss RNA fragments were complexed with
tRNATrp. Description of changes is as in the legend to Fig.
3C.
FIG. 6.

Summary of nuclease digestion data for RDS mutant RSV
RNA fragments. Secondary-structure diagrams for RDS sequences are
depicted as described in Materials and Methods. Mutant nucleotides are
denoted by italicized, shadowed lettering. Other notations are as
described in the legend to Fig. 3. (A) Digestion of uncomplexed RDS RNA
fragments. Description of cleavages is as in the legend to Fig. 3A. (B)
Digestion of RDS RNA fragments complexed with tRNATrp,
where tRNATrp sequences are shown in bold capital letters.
Description of cleavages is as outlined in the legend to Fig. 3B.
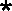 , the exact cleavage site was
unclear, but the data did indicate that the cleavage was somewhere
within the U5-IR stem. (C) Changes in digestion pattern when RDS RNA
fragments were complexed with tRNATrp. Changes are
described in the legend to Fig. 3C.
, the exact cleavage site was
unclear, but the data did indicate that the cleavage was somewhere
within the U5-IR stem. (C) Changes in digestion pattern when RDS RNA
fragments were complexed with tRNATrp. Changes are
described in the legend to Fig. 3C.
The major T1 cleavages of I12Lss (Fig. 4A and 5A) were all within the extended U5-IR loop at G93, G97, and G102. There were no T1 cleavages in the U5-IR, U5-PBS, and U5-Leader stems. The only T1 cleavages outside the U5-IR loop were at G124 and G129 (analogous to WT residues G112 and G117), indicating that the 3′ portion of the PBS in the mutant RNA is unstructured, analogous to the WT situation. Also like the WT, G66 and G105 (same as WT G93 with respect to the position of the residue relative to the U5-IR stem) were resistant to T1 digestion even though they are predicted to lie within a bulge and loop respectively. The major RNase A cleavage site of I12Lss was U131, which is predicted to lie in the bulge at the end of the U5-Leader stem. Additional RNase A cleavages were seen at U79 and C120 to C124, all of which are predicted to lie in single-stranded regions of the RNA structure. Surprisingly, there were several strong to moderate RNase A cleavages in all three stem structures (U62 and U135 in U5-Leader, C75 and C77 in U5-PBS, and U112 to U114 in U5-IR). The majority of the V1 cleavages occurred within the U5-Leader stem (C59, C61, U62, and A63), the U5-IR stem (G84, G85, U109, and U114), and the U5-PBS stem (C77 and G115). The bulged residue U79 was a moderate V1 target, and C123 was a weak V1 target. The observation that the single-strand-specific RNases T1 and A as well as the double-strand/tertiary-interaction-specific RNase V1 cleaved the predicted stem composed of nucleotides 69 to 72 and 126 to 129 support the conclusion drawn from the WT data that this stem is transiently formed.
As seen with both WT and I12Lss, the major T1 cleavages of RDS (Fig. 4B and 6A) were at G96 and G99, both of which are predicted to lie within the U5-IR loop. There were no T1 cleavages in the U5-Leader stem or the extended U5-IR stem, as expected. The only other T1 cleavages were seen at G124 and G129, again indicating that the 3′ portion of the PBS is unstructured. Similar to I12Lss, the major RNase A cleavage of RDS was at U131 within the bulge between the U5-Leader stem and the PBS. In fact, the majority of the RNase A cleavages were between the extended U5-IR stem and the U5-Leader stem, suggesting that this region is fairly unstructured. There was also RNase A digestion at either end of the U5-IR stem. There were strong V1 cleavages within both the U5-Leader stem and the U5-IR stem and mild cleavages in the RNA sequences connecting the two stems.
Digestion of 5′-32P-labeled WT viral RNA annealed to tRNATrp.
WT RNA fragments were annealed to synthetic tRNATrp and subjected to digestion with RNase T1, A, and V1 as described in Materials and Methods. Figure 2A shows the T1 cleavage data, while Fig. 3B summarizes this data, as well as that from the RNase A and V1 cleavages (Fig. 2B and C, respectively). Figure 3C summarizes the changes in digestion pattern when cleavages of the WT RNAs alone are compared to cleavages seen when the WT-tRNATrp complex is formed.
As with the uncomplexed WT RNA, the major T1 cleavage site in the WT-tRNATrp complex was at G90, within the U5-IR loop. Increased T1 susceptibility, indicating an increase in single-stranded character, was seen at several residues, i.e., G80, G84, and G103, when the WT RNA was annealed to its primer. Note that all three residues surround the junction of the U5-IR stem, the U5-TΨC interaction, and the 5′ portion of the PBS, a region where the 3′OH of the tRNATrp primer is presumably positioned. G121 and G125 to G127 within the U5-Leader stem also displayed slightly increased T1 sensitivity. Decreased T1 sensitivity, indicating a decrease in single-stranded character, was seen at residues G112, G115, and G117, where the 3′ portion of the PBS is predicted to base pair with tRNATrp. Surprisingly, there was an increase in T1 susceptibility for G72 and G74, which are predicted to form the U5-TΨC interaction.
With respect to RNase A, the strongest cleavage of the WT-tRNATrp complex was at C81, just as it was with the uncomplexed viral RNA. Immediately 5′ to C81 there was a significant increase in RNase A sensitivity (C77 and C79), which is indicative of increased single strandedness when the primer was annealed to the viral RNA. Residues within the U5-Leader stem (U62 and U122) also demonstrated a slight increase in RNase A sensitivity upon complex formation, comparable to the results seen with T1. There was a decrease in RNase A susceptibility for PBS nucleotides predicted to base pair with tRNATrp and an increase in RNase sensitivity for nucleotides predicted to form the U5-TΨC interaction (C75 and C77), data similar to that observed with T1.
In general, as expected, changes in V1 sensitivity seen with complex formation were opposite to those seen with RNase T1 and A. For example, when there was an increase in T1 or A sensitivity for nucleotides in the U5-Leader or U5-PBS stem, there was a concomitant decrease in V1 sensitivity (U62, A63, and C65 for U5-Leader, and C77, C78, and U79 for U5-PBS). The only site of significant disagreement between the T1 and A cleavage data and the V1 cleavage data was for residues involved in the U5-TΨC interaction region. For these RNA sequences (C71, G72, A73, G74, C75, and C77), there were increases in both T1 and A sensitivity and V1 sensitivity.
5′-32P-labeled mutant viral RNAs complexed with tRNATrp.
In experiments similar to those described above for WT RNA, the mutant RNAs, I12Lss and RDS, were annealed to synthetic tRNATrp and subjected to limited nuclease digestion. T1 cleavages of the two complexes, I12Lss-tRNATrp and RDS-tRNATrp, are shown in Fig. 4A and B, respectively. The digestion data for T1, A, and V1 (data not shown for RNase A and V1) is summarized in Fig. 5B for I12Lss and Fig. 6B for RDS. Changes observed in the digestion pattern upon complex formation are diagrammed in Fig. 5C (I12Lss) and Fig. 6C (RDS).
For the most part, formation of the mutant viral RNA-tRNATrp complex caused the same types of cleavage pattern changes as seen when WT RNAs were complexed with primer. When I12Lss was annealed to tRNATrp, the predominant T1 cleavages were in the extended U5-IR loop, similar to cleavages observed with I12Lss alone. However, there was an increase in T1 sensitivity for G80, which is predicted to lie near the 3′OH of the tRNA primer. A significant increase in T1 sensitivity was also observed for G70, at the base of the U5-TΨC interaction region, and a slight increase in T1 sensitivity was observed for G66, at the end of the U5-Leader stem. Similar to the WT complex, there was a slight increase in T1 susceptibility for G72 within the predicted U5-TΨC interaction region and a significant decrease in T1 sensitivity for nucleotides in the PBS. In general, differences in RNase A cleavages after complex formation paralleled those seen with T1; for example, there were decreases in sensitivity for nucleotides in the PBS and increases in sensitivity for C71 in the U5-TΨC interaction region and U69 5′ to the U5-TΨC interaction region. An increase in RNase A susceptibility was also observed for U135 in the U5-Leader stem. There were fewer V1 cleavages of the I12Lss-tRNATrp complex compared to I12Lss alone. Decreases in V1 cleavage were observed primarily in the U5-Leader stem. At the junction of the U5-IR stem, PBS, and U5-TΨC interaction region, not only were there increases in T1 and A sensitivity (noted above), but also there were decreases in V1 sensitivity (C77, U79, U114, and G115).
Formation of the RDS-tRNATrp complex caused several changes in the RNase cleavage patterns, the majority of which were similar to those seen with both WT and I12Lss complexes. Briefly, there were decreases in RNase T1 and A sensitivity for PBS sequences and decreases in V1 sensitivity for nucleotides in the U5-Leader stem. At the junction of the U5-IR stem, PBS, and U5-TΨC interaction region, G80 demonstrated significantly increased T1 sensitivity. As seen with both WT and I12Lss complexes, the data concerning the U5-TΨC interaction region was contradictory; i.e., increases in sensitivity to all three nucleases were seen. One of the most interesting changes in digestion pattern occurred in the U5-IR stem, where there was an increase in V1 sensitivity in the 3′ portion of the stem when RDS was complexed to tRNATrp. This change appears to be specific for the RDS mutant.
Nuclease digestion of synthetic 3′-32P-labeled tRNATrp.
tRNATrp was subjected to limited nuclease digestion with both T1 and V1 as described in Materials and Methods. Figure 7 shows the cleavage products visualized after T1 (Fig. 7A) and V1 (Fig. 7B) digestion. Note that only cleavages in the 3′ portion of tRNATrp are shown in these figures, since they are the most relevant to our discussion. All of the T1 and V1 cleavage data is summarized in Fig. 8A. The predominant T1 cleavage of tRNATrp was seen at G36, located in the anticodon loop. A moderate T1 cleavage was also seen at G56 in the TΨC loop. Weak T1 cleavages were scattered throughout the remainder of the tRNATrp sequence.
FIG. 8.

Summary of nuclease digestion data for tRNATrp. Secondary-structure diagrams for tRNATrp in the presence and absence of WT viral RNA fragments are depicted. The structure in panel A was adapted from reference 28. The structures in panels B and C are derived from the structure in panel A and the limited nuclease digestion data in Fig. 7. G residues (target of T1 nuclease) of the tRNA are underlined. Nucleotide sequences are numbered every 10 bases from the 5′ end of tRNATrp. Viral RNA sequences base paired to the tRNATrp are shown in bold capital letters, while viral sequences not involved in base pairing interactions are represented as lines. (A) Digestion of tRNATrp. A bold lowercase t indicates T1 cleavages, where t t t was the strongest cleavage site seen with 0.007 U of T1 per μl, t t was a moderate cleavage seen with 0.007 U/μl, and t was a moderate cleavage seen with 0.002 U/μl. A bold lowercase v indicates V1 cleavages, where v v v indicates moderate cleavages seen with 1 × 10−5 U of V1 per μl, v v indicates moderate cleavages seen with 3 × 10−4 U/μl, and v indicates moderate cleavages with 1 × 10−4 U/μl. (B) Digestion of tRNATrp annealed to WT RSV RNAs. Description of T1 cleavages is in panel A. Description of V1 cleavages is in panel A, except that v v v v indicates strong cleavages with 1 × 10−5 U/μl. (C) Changes in digestion pattern when tRNATrp was annealed to WT RSV RNAs. Description of changes is as in the legend to Fig. 3C.
The predominant V1 cleavage was at C27 in the anticodon stem (Fig. 8A); however, additional strong cleavages were seen in the 3′ portion of the acceptor stem at nucleotides 66 to 72. No significant V1 cleavages were seen in either the D stem or the TΨC stem, and no significant V1 cleavages were seen in any of the loop structures.
Digestion of 3′-32P-labeled tRNATrp annealed to WT RNA.
As described in Materials and Methods, tRNATrp was annealed to WT viral RNAs and subjected to limited nuclease digestion with RNase T1 and V1. Figure 7A shows the T1 cleavage data, Fig. 7B shows the V1 cleavage data, and Fig. 8B summarizes the data from these two experiments. In addition, Fig. 8C summarizes the changes in digestion pattern seen when cleavages of tRNATrp alone are compared to cleavages of tRNATrp annealed to WT viral RNA.
In general, the cleavage pattern of tRNATrp alone was similar to that of tRNATrp annealed to WT viral RNA. The two cleavage patterns differed primarily in the 3′ half of tRNATrp, where the tRNA is predicted to interact with the WT viral RNA. There were decreases in T1 sensitivity for G66 to G68 and G63 of tRNATrp, all of which are predicted to anneal to the PBS. G56 also showed a decrease in T1 sensitivity when tRNATrp was annealed to WT viral RNA, thereby supporting our prediction that this TΨC loop residue participates in the U5-TΨC interaction. Additional evidence supporting the U5-TΨC interaction comes from the observation that G48 demonstrated an increase in T1 sensitivity, presumably because the TΨC stem was disrupted during primer annealing. An increase in T1 sensitivity was also seen for G41 in the anticodon stem.
For the most part, the V1 cleavage data supports the tRNATrp–viral-RNA interactions modeled in Fig. 8B. There were increases in V1 sensitivity for nucleotides 70 to 72, which are predicted to form an internal portion of the tRNA-PBS duplex, and there was an increase in V1 sensitivity for U54, a TΨC loop residue predicted to base pair with viral U5 sequences. In addition, there was an increase in V1 sensitivity for U47, which is predicted to lie at the terminus of the U5-TΨC interaction. The only V1 data that appears to contradict our model was the decrease in V1 sensitivity seen for G66 to G68 and G56, all of which are predicted to form base pairs in our model. This apparent contradiction is explored in Discussion.
DISCUSSION
The present nuclease mapping studies not only provide the first direct physical evidence for the existence of a series of biologically relevant secondary structures in RSV RNA but also provide insight into how these structures change as tRNATrp is annealed. In uncomplexed WT and mutant viral RNAs, the predicted U5-IR stem-loop is formed as a discrete structure. This observation is supported by data showing the presence of strong T1 cleavages and the absence of V1 cleavages in U5-IR loop nucleotides and the absence of T1 cleavages and the presence of V1 cleavages in the U5-IR stem.
It is important to note that V1 does not cleave at every base pair in the U5-IR stem. This is particularly noteworthy for the extended RDS U5-IR stem and is consistent with other studies showing that RNase V1 cleaves selected base pairs (16, 24, 31) in the center of a helix consisting of a minimum of four to six contiguous stacked base pairs (4, 25). In addition, the structure of the helix may change once the initial V1 cut is made in the helical backbone, significantly reducing the affinity of the enzyme for substrate (13). V1 has also been shown to asymmetrically cleave certain helices so that one side is preferentially cleaved over the other (4, 16, 24). An alternative explanation for the RDS stem cleavage pattern is that the extended RDS U5-IR stem may participate in a tertiary interaction which sterically blocks the access of RNase V1 to the stem. This theoretical tertiary interaction may be relieved when RDS is annealed to the tRNATrp primer, since there is an increase in V1 cleavage of the U5-IR stem when the binary complex is formed.
Both the RDS and I12Lss U5-IR stems can adopt alternative conformations where the bulge (U79 in Fig. 5A and 6A) between the U5-IR stem and U5-PBS stem can occur at different positions. These alternative conformations explain why RNase A cleaves several residues predicted to lie within the RDS and I12Lss U5-IR and U5-PBS stems. For sequences outside the U5-IR stem-loop region, both the mutant RNA fragments and the WT RNA fragments exhibit similar nuclease digestion patterns. Since both RDS and I12Lss contain fairly large perturbations of the U5-IR stem-loop structure, it appears that mutagenesis of the stem does not affect the global structure of the viral RNA.
Nuclease-independent evidence for the existence of the U5-IR stem comes from the observation that there is band compression in the PAGE analysis for RNA fragments from this region when RDS RNA is partially alkaline hydrolyzed (data not shown). Band compression indicates that several RNA fragments are running abnormally in a denaturing polyacrylamide gel due to a highly stable incompletely denatured structure. The absence of significant T1 cleavages in the U5-IR stem of RDS under denaturing conditions (Fig. 4B, Marker lane) also confirms the existence of the highly stable, 14-bp RDS stem.
The absence of T1 cleavages and the presence of V1 cleavages in the U5-Leader stem of all three RNAs strongly suggest that the stem exists in vitro. It is unclear why RNase A, a single-strand-specific nuclease, also digests certain U residues found within this stem. Additional evidence for the U5-Leader structure comes from partial T1 digests of both WT and mutant RNAs under denaturing conditions (Fig. 2 and 4, Marker lanes). In these experiments, G residues involved in the stem are not T1 sensitive.
The region between the U5-Leader stem and U5-PBS stem is likely to be predominantly single stranded, since the majority of the cleavages in this region are by single-strand-specific nucleases. This is not completely unexpected, since the PBS nucleotides are found in this region. If the PBS nucleotides were locked into a rigid inflexible structure like a helix, it would be difficult for them to break those contacts and reestablish new contacts with the tRNATrp primer. However, since there are weak V1 cleavages at several of the nucleotides predicted to form a 4-bp stem between the U5-Leader stem and the U5-PBS stem, this stem may be formed transiently in the WT and mutant RNA fragments.
Confirmation of the accuracy of the nuclease digestion techniques used in the viral RNA-mapping experiments comes from application of these techniques to the uncomplexed tRNATrp molecule itself. Previous mapping studies of native tRNATrp indicate that V1 preferentially cleaves both the 5′ and 3′ portions of the anticodon stem and the 3′ terminus of the tRNA, results similar to our data summarized in Fig. 8A (15). However, neither our studies nor those of Garret et al. show significant V1 cleavages in any other region of the uncomplexed tRNATrp molecule (15). Overall, T1 digestion of tRNATrp confirms the cloverleaf structure portrayed in Fig. 8A, since significant T1 cleavages are seen primarily in the anticodon loop and the TΨC loop. Because the synthetic tRNATrp used in these studies lacks the base modifications seen in a native tRNA molecule, its structure may be less stable than the native structure, which could account for the minor T1 cleavages scattered throughout the stems. Similar structural mapping experiments with native tRNAVal and in vitro-transcribed tRNAVal demonstrated that the D stem of the unmodified tRNA is more susceptible to cleavage with single-strand-specific nucleases (10).
Binary-complex formation between WT or mutant RNAs and tRNATrp changes the nuclease digestion pattern of both the viral RNA and the tRNA. In general, both the viral RNA-mapping data and the tRNATrp-mapping data are consistent with the structural model depicted in Fig. 1. The most obvious changes in the viral RNA digestion patterns are seen in the PBS sequences, where there is a consistent decrease in RNase T1 and A sensitivity, as one would expect, when the unstructured PBS nucleotides are annealed to the 3′ 18 nucleotides of tRNATrp. Surprisingly, there is not a concomitant increase in V1 sensitivity for these nucleotides. In contrast, the tRNATrp-mapping data indicates that several of the tRNA nucleotides, predicted to anneal to the PBS, show either increased V1 sensitivity or decreased T1 sensitivity after complex formation. Although some tRNATrp nucleotides in this same region show decreases in V1 sensitivity, it is important to note that V1 still cleaves at these residues. This result suggests that the annealed tRNA nucleotides are still involved in a helical structure but that the structure is different from that seen in the uncomplexed tRNA.
As described in Materials and Methods, the viral RNA-tRNATrp complex mapped by the nuclease digestion assays is formed when the primer is annealed by heat to the PBS. Quantitative annealing of viral RNA to its tRNA primer has been confirmed by nondenaturing PAGE analysis, where the electrophoretic mobility of the viral RNA has been shown to decrease after tRNATrp annealing (data not shown). Other researchers have suggested that both HIV-1 and RSV NC (nucleocapsid) facilitate primer annealing of their respective primers to the corresponding viral RNA both in vitro and in vivo (6, 9, 21, 27). However, previous experiments in our laboratory with avian NC and an in vitro reverse transcription initiation assay suggest that NC only mildly stimulates reverse transcription under the described conditions (2). Since the limited nuclease digestion assays were performed under the same buffer conditions used in the in vitro reverse transcription assay, we did not use NC for primer annealing in these studies.
Other nuclease digestion pattern changes (outside the PBS) are seen in both the U5-Leader and U5-IR stems of the viral RNA after binary-complex formation. For all three RNA types, increased RNase T1 and A sensitivity and decreased V1 sensitivity indicate that the U5-Leader stem is destabilized after primer annealing. Destabilization of this duplex may be necessary to facilitate primer access and binding to PBS sequences. Formation of the WT-tRNATrp complex causes an increase in T1 sensitivity for G84, suggesting that the U5-IR stem is unwound when tRNATrp is bound. A similar increase in T1 sensitivity for G84 in I12Lss and RDS was not seen. This may be because the U5-IR stem is longer in both mutants and therefore more stable and resistant to unwinding. One striking observation seen in all three RNA-tRNATrp complexes is that the RNA sequences predicted to lie at the U5-IR stem and PBS-primer helix junction all show an increasingly single-stranded structure compared to the uncomplexed viral RNAs. Since this junction is the site of initiation of DNA synthesis at the 3′OH of tRNATrp, this region may be unstructured to facilitate RT access to the primer.
The remaining viral RNA and tRNATrp nuclease digestion pattern changes seen upon complex formation are found at the site of the predicted U5-TΨC interaction. Increases in V1 sensitivity for both viral RNA and tRNATrp nucleotides involved in the interaction suggest that it does exist in vitro. Decreases in T1 sensitivity for selected tRNATrp nucleotides support this observation. It is unclear why there are increases in RNase T1 and A sensitivity for several U5 nucleotides base paired to TΨC residues. One possible explanation is that the interaction is unstable and that, in vivo, the interaction is stabilized by modified bases in tRNATrp. This theory is supported by previous in vivo mutagenesis data, which indicates that the U5-TΨC interaction is required for initiation of reverse transcription (1). The modified tRNATrp nucleotides include 1-methyladenosine (m1A) in place of the A residue linking the tRNA sequences paired to the PBS and the tRNA sequences involved in U5-TΨC and pseudouridine residues in place of the U residues base paired to A73 and G74 (27). The importance of modified nucleotides in either allowing or preventing additional interactions (outside the PBS) between the tRNA primer and the retroviral RNA has recently been demonstrated for HIV-1 (17–19) and M-MuLV (14), respectively. The role of native tRNATrp in the formation of the RSV reverse transcription initiation complex is currently being investigated in vivo.
Mapping studies similar to those described above have provided structural data for two other retroviral RNA-tRNA complexes, HIV-1–tRNA3Lys and M-MuLV–tRNAPro. For HIV-1, Isel et al. demonstrated that binary-complex formation resulted in significant rearrangements in both the viral RNA and tRNA structures (17). In their model, tRNA3Lys formed multiple contacts (in addition to the standard PBS-tRNA duplex) with the viral RNA, including a 4-bp interaction between the anticodon loop of tRNA3Lys and the A-rich (U5-IR) loop of HIV-1. Although a U5-TΨC interaction does not appear to exist for HIV-1, subsequent analysis of this HIV-1 loop-loop contact by many different groups suggests that it is analogous to the U5-TΨC interaction of RSV, since both play a role in initiation of reverse transcription (3, 18, 20, 22, 32).
The additional (non-loop-loop) HIV-1–tRNA3Lys interactions proposed by Isel et al. have not been tested in an in vivo biological system, and so their relevance is unknown. In fact, select modeling studies of the three-dimensional structure proposed by Isel et al. suggest that these interactions result in a structure so “topologically knotted” that it may not be able to support the initiation of reverse transcription (12). However, recent enzymatic footprinting studies of HIV-1 RT complexed with HIV-1-tRNA3Lys suggest that the RNA model may be functional (19a). In contrast to the HIV-1 system, the structure of the M-MuLV–tRNAPro complex appears to be relatively simple, since no additional interactions outside the PBS-tRNA interaction appear to exist. Therefore, it seems that the RSV-tRNATrp structure depicted above is more complex than that proposed for M-MuLV and its tRNA primer but more simple than that described for HIV-1 and tRNA3Lys.
ACKNOWLEDGMENTS
This work was supported in part by National Cancer Institute grant CA-38046. S.M. is a Medical Scientist Trainee supported by grant GM-07250 from the National Institutes of Health.
We thank David McPheeters, Case Western Reserve University, for his invaluable advice on all things involving RNA.
REFERENCES
- 1.Aiyar A, Cobrinik D, Ge Z, Kung H J, Leis J. Interaction between retroviral U5 RNA and the T psi C loop of the tRNA(Trp) primer is required for efficient initiation of reverse transcription. J Virol. 1992;66:2464–2472. doi: 10.1128/jvi.66.4.2464-2472.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aiyar, A., and J. Leis. 1994. Unpublished data.
- 3.Arts E J, Stetor S R, Li X, Rausch J W, Howard K J, Ehresmann B, North T W, Wohrl B M, Goody R S, Wainberg M A, Grice S F. Initiation of (−) strand DNA synthesis from tRNA(3Lys) on lentiviral RNAs: implications of specific HIV-1 RNA-tRNA(3Lys) interactions inhibiting primer utilization by retroviral reverse transcriptases. Proc Natl Acad Sci USA. 1996;93:10063–10068. doi: 10.1073/pnas.93.19.10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Auron P E, Weber L D, Rich A. Comparison of transfer ribonucleic acid structures using cobra venom and S1 endonucleases. Biochemistry. 1982;21:4700–4706. doi: 10.1021/bi00262a028. [DOI] [PubMed] [Google Scholar]
- 5.Baudin F, Marquet R, Isel C, Darlix J L, Ehresmann B, Ehresmann C. Functional sites in the 5′ region of human immunodeficiency virus type 1 RNA form defined structural domains. J Mol Biol. 1993;229:382–397. doi: 10.1006/jmbi.1993.1041. [DOI] [PubMed] [Google Scholar]
- 6.Chan B, Musier-Forsyth K. The nucleocapsid protein specifically anneals tRNALys-3 onto a noncomplementary primer binding site within the HIV-1 RNA genome in vitro. Proc Natl Acad Sci USA. 1997;94:13530–13535. doi: 10.1073/pnas.94.25.13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cobrinik D, Aiyar A, Ge Z, Katzman M, Huang H, Leis J. Overlapping retrovirus U5 sequence elements are required for efficient integration and initiation of reverse transcription. J Virol. 1991;65:3864–3872. doi: 10.1128/jvi.65.7.3864-3872.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cobrinik D, Soskey L, Leis J. A retroviral RNA secondary structure required for efficient initiation of reverse transcription. J Virol. 1988;62:3622–3630. doi: 10.1128/jvi.62.10.3622-3630.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Rocquigny H, Gabus C, Vincent A, Fournie-Zaluski M C, Roques B, Darlix J L. Viral RNA annealing activities of human immunodeficiency virus type 1 nucleocapsid protein require only peptide domains outside the zinc fingers. Proc Natl Acad Sci USA. 1992;89:6472–6476. doi: 10.1073/pnas.89.14.6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Derrick W B, Horowitz J. Probing structural differences between native and in vitro transcribed Escherichia colivaline transfer RNA: evidence for stable base modification-dependent conformers. Nucleic Acids Res. 1993;21:4948–4953. doi: 10.1093/nar/21.21.4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Donis-Keller H, Maxam A M, Gilbert W. Mapping adenines, guanines, and pyrimidines in RNA. Nucleic Acids Res. 1977;4:2527–2538. doi: 10.1093/nar/4.8.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elgavish T, VanLoock M S, Harvey S C. Exploring three-dimensional structures of the HIV-1 RNA/tRNALys3 initiation complex. J Mol Biol. 1999;285:449–453. doi: 10.1006/jmbi.1998.2310. [DOI] [PubMed] [Google Scholar]
- 13.Favorova O O, Fasiolo F, Keith G, Vassilenko S K, Ebel J P. Partial digestion of tRNA–aminoacyl-tRNA synthetase complexes with cobra venom ribonuclease. Biochemistry. 1981;20:1006–1011. doi: 10.1021/bi00507a055. [DOI] [PubMed] [Google Scholar]
- 14.Fosse P, Mouge M, Gerard K, Westhof E, Ehresmann B, Ehresmann C. Modified nucleotides of tRNAPro restrict interactions in the binary primer/template complex of M-MuLV. J Mol Biol. 1998;275:731–746. doi: 10.1006/jmbi.1997.1487. [DOI] [PubMed] [Google Scholar]
- 15.Garret M, Romby P, Giege R, Litvak S. Interactions between avian myeloblastosis reverse transcriptase and tRNATrp. Mapping of complexed tRNA with chemicals and nucleases. Nucleic Acids Res. 1984;12:2259–2271. doi: 10.1093/nar/12.5.2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garrett R A, Olesen S O. Structure of eukaryotic 5S ribonucleic acid: a study of Saccharomyces cerevisiae5S ribonucleic acid with ribonucleases. Biochemistry. 1982;21:4823–4830. doi: 10.1021/bi00262a047. [DOI] [PubMed] [Google Scholar]
- 17.Isel C, Ehresmann C, Keith G, Ehresmann B, Marquet R. Initiation of reverse transcription of HIV-1: secondary structure of the HIV-1 RNA/tRNA(3Lys) (template/primer) J Mol Biol. 1995;247:236–250. doi: 10.1006/jmbi.1994.0136. [DOI] [PubMed] [Google Scholar]
- 18.Isel C, Lanchy J M, Le Grice S F, Ehresmann C, Ehresmann B, Marquet R. Specific initiation and switch to elongation of human immunodeficiency virus type 1 reverse transcription require the post-transcriptional modifications of primer tRNA3Lys. EMBO J. 1996;15:917–924. [PMC free article] [PubMed] [Google Scholar]
- 19.Isel C, Marquet R, Keith G, Ehresmann C, Ehresmann B. Modified nucleotides of tRNA(3Lys) modulate primer/template loop-loop interaction in the initiation complex of HIV-1 reverse transcription. J Biol Chem. 1993;268:25269–25272. [PubMed] [Google Scholar]
- 19a.Isel C, Westhof E, Massire C, Le Grice S F, Ehresmann B, Ehresmann C, Marquet R. Structural basis for the specificity of the initiation of HIV-1 reverse transcription. EMBO J. 1999;18:1038–1048. doi: 10.1093/emboj/18.4.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang S M, Zhang Z, Morrow C D. Identification of a sequence within U5 required for human immunodeficiency virus type 1 to stably maintain a primer binding site complementary to tRNA(Met) J Virol. 1997;71:207–217. doi: 10.1128/jvi.71.1.207-217.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li X, Quan Y, Arts E J, Li Z, Preston B D, de Rocquigny H, Roques B P, Darlix J L, Kleiman L, Parniak M A, Wainberg M A. Human immunodeficiency virus type 1 nucleocapsid protein (NCp7) directs specific initiation of minus-strand DNA synthesis primed by human tRNA(Lys3) in vitro: studies of viral RNA molecules mutated in regions that flank the primer binding site. J Virol. 1996;70:4996–5004. doi: 10.1128/jvi.70.8.4996-5004.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liang C, Li X, Rong L, Inouye P, Quan Y, Kleiman L, Wainberg M A. The importance of the A-rich loop in human immunodeficiency virus type 1 reverse transcription and infectivity. J Virol. 1997;71:5750–5757. doi: 10.1128/jvi.71.8.5750-5757.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin J H, Levin H L. Reverse transcription of a self-primed retrotransposon requires an RNA structure similar to the U5-IR stem-loop of retroviruses. Mol Cell Biol. 1998;18:6859–6869. doi: 10.1128/mcb.18.11.6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lockard R E, Kumar A. Mapping tRNA structure in solution using double-strand-specific ribonuclease V1 from cobra venom. Nucleic Acids Res. 1981;9:5125–5140. doi: 10.1093/nar/9.19.5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lowman H B, Draper D E. On the recognition of helical RNA by cobra venom V1 nuclease. J Biol Chem. 1986;261:5396–5403. [PubMed] [Google Scholar]
- 26.Miller J T, Ge Z, Morris S, Das K, Leis J. Multiple biological roles associated with the Rous sarcoma virus 5′ untranslated RNA U5-IR stem and loop. J Virol. 1997;71:7648–7656. doi: 10.1128/jvi.71.10.7648-7656.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prats A C, Sarih L, Gabus C, Litvak S, Keith G, Darlix J L. Small finger protein of avian and murine retroviruses has nucleic acid annealing activity and positions the replication primer tRNA onto genomic RNA. EMBO J. 1988;7:1777–1783. doi: 10.1002/j.1460-2075.1988.tb03008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schimmel P R, Söll D, Abelson N, editors. Transfer RNA: structure, properties, and recognition. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1979. [Google Scholar]
- 29.Serra M J, Turner D H. Predicting thermodynamic properties of RNA. Methods Enzymol. 1995;259:242–261. doi: 10.1016/0076-6879(95)59047-1. [DOI] [PubMed] [Google Scholar]
- 30.Skripkin E, Isel C, Marquet R, Ehresmann B, Ehresmann C. Psoralen crosslinking between human immunodeficiency virus type 1 RNA and primer tRNA3(Lys) Nucleic Acids Res. 1996;24:509–514. doi: 10.1093/nar/24.3.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vournakis J N, Celantano J, Finn M, Lockard R E, Mitra T, Pavlakis G, Troutt A, van den Berg M, Wurst R M. Sequence and structure analysis of end-labeled RNA with nucleases. Gene Amplif Anal. 1981;2:267–298. [PubMed] [Google Scholar]
- 32.Wakefield J K, Kang S M, Morrow C D. Construction of a type 1 human immunodeficiency virus that maintains a primer binding site complementary to tRNA(His) J Virol. 1996;70:966–975. doi: 10.1128/jvi.70.2.966-975.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Waters L C, Yang W K, Mullin B C, Nichols J L. Purification of tryptophan transfer RNA from chick cells and its identity with “spot 1” RNA of Rous sarcoma virus. J Biol Chem. 1975;250:6627–6629. [PubMed] [Google Scholar]