

Abstract
Neuropsychiatrie symptoms (NPS) are common manifestations of neurodegenerative disorders and are often early signs of those diseases. Among those neurodegenerative diseases, TDP-43 proteinopathies are an increasingly recognized cause of early neuropsychiatrie manifestations. TDP-43-related diseases include frontotemporal dementia (FTD), amyotrophic lateral sclerosis (ALS), and Limbic-Predominant Age-Related TDP-43 Encephalopathy (LATE). The majority of TDP-43-related diseases are sporadic, but a significant proportion is bereditary, with progranulin (GRN) mutations and C9orf72 repeat expansions as the most common genetic etiologies. Studies reveal that NPS can be the initial manifestation of those diseases or can complicate disease course, but there is a lack of awareness among clinicians about TDP-43-related diseases, which leads to common diagnostic mistakes or delays. There is also emerging evidence that TDP-43 accumulations could play a role in late-onset primary psychiatric disorders. In the absence of robust biomarkers for TDP-43, the diagnosis remains primarily based on clinical assessment and neuroimaging. Given the association with psychiatric symptoms, clinical psychiatrists have a key role in the early identification of patients with TDP-43-related diseases. This narrative review provides a comprehensive overview of the pathobiology of TDP-43, resulting clinical presentations, and associated neuropsychiatric manifestations to help guide clinical practice.
Keywords: Frontotemporal dementia, neuropsychiatric symptoms, differential diagnosis, amyotrophic lateral sclerosis, limbic predominant age related, TDP-43 encephalopathy (LATE)
INTRODUCTION
Neuropsychiatric symptoms (NPS) are common manifestations of neurodegenerative disorders, and are often early signs of those diseases.1,2 NPS of dementia include apathy, agitation, depression, delusions, and hallucinations, and frequently present to psychiatrists as late-onset (>40 years of age) behavioral change. The prevalence and nature of NPS vary according to dementia subtypes and the underlying neuropathology.3 In particular, NPS are highly prevalent in frontotemporal dementia (FTD), often preceding cognitive symptoms,4,5 and can lead to high rates of misdiagnosis, significant diagnostic delays, and may prevent patients from accessing appropriate resources.6–9 In this context, distinguishing FTD from primary psychiatric disorders (PPD) is particularly challenging, even among specialists, due to overlapping clinical presentations, heterogeneous clinical presentation, and the lack of molecular biomarkers for FTD.6,10 A deeper understanding of the relationship between NPS and the specific underlying neuropathology of FTD may improve diagnostic accuracy, disease management, clinical trial recruitment, and ultimately, access to emerging disease-specific therapies.
FTD is an umbrella term that includes the clinical syndromes of behavioral variant frontotemporal dementia (bvFTD), semantic variant primary progressive aphasia (svPPA), and nonfluent variant PPA (nfvPPA).11,12 The P ID-related disorders of amyotrophic lateral sclerosis (ALS) and the Parkinsonian-like syndromes of progressive supranuclear palsy (PSP) and corticobasal syndrome (CBS) can appear with different combinations of behavioral, cognitive, and language deficits.13–15 At the pathological levels, these syndromes are secondary to frontotemporal lobar degeneration (FTLD), which is a comprehensive term for progressive neurodegenerative changes largely in the frontal and temporal lobes. Among FTLD entities, pathological inclusions of transactive response DNA-binding protein 43 (TDP-43) can be seen in at least half of bvFTD cases, with significant neurodegeneration and atrophy of the frontal and temporal lobes as well as in subcortical structures.16,17 Up to 98% of all ALS cases have TDP-43 pathology,18 and TDP-43 pathological inclusions are reported postmortem in other neurodegenerative diseases that can present with NPS.19,20 As the common denominator across a wide range of neurodegenerative diseases, interest has increased in TDP-43 proteinopathies and how they manifest as NPS, with emerging evidence suggesting that TDP-43 may contribute to severe mental illnesses.21 Increasing awareness of FTD disorders and other TDP-43 proteinopathies among psychiatrists is critical to improving clinical recognition of TDP-43-related diseases among patients presenting with NPS.22 In this narrative review, we will 1) overview mechanisms of TDP-43 proteinopathy and clinical syndromes resulting from TDP-43 proteinopathy, 2) examine the prevalence of NPS among TDP-43 proteinopathies and genetic and molecular links to psychiatric disorders, 3) provide recommendations to improve the differential diagnosis of FTD vs PPD, and 4) discuss treatments in development for TDP-43 proteinopathies.
Normal Structure and Function of TDP-43
TDP-43 is a heterogeneous nuclear ribonucleoprotein containing 414 amino adds encoded by the TARDBP gene. TDP-43 is comprised of an N-terminal domain with a nuclear localization signal, 2 highly conserved RNA recognition motifs (RRM1 and RRM2) that permit sequence-specific binding to RNA, and a glycine-rich, intrinsically disordered C-terminal domain whose cleaved fragments contribute to prion-like aggregation in vulnerable cells in the disease state.23 These TDP-43 structural domains mediate its binding to DNA, RNA, and proteins, including other TDP-43 proteins.24–26 While TDP-43 predominantly resides in the nucleus, it continuously shuttles between the nucleus and the cytoplasm to perform diverse functions in both compartments.27 TDP-43 regulates several aspects of RNA metabolism, including transcription, mRNA splicing, and translation.25,28 Cellular levels of TDP-43 are tightly controlled through a negative feedback mechanism in which TDP-43 regulates production of its own mRNA.29 TDP-43 is also involved in the formation of stress granules, which are cytoplasmic assemblies of translationally stalled mRNAs, translation initiation factors, and RNA-binding proteins formed in response to various cellular stressors.30 TDP-43 contributes to normal, noncell-autonomous physiological functions of glia, while deletion of glial TDP-43 argues for cell-autonomous roles for TDP-43.31
PATHOLOGY
The histopathological hallmark of TDP-43 proteinopathy is the mislocalization and accumulation of hyperphosphorylated, ubiquitinated, and N-terminally truncated TDP-43 in neurons and glial cells.18,25 C-terminal domain fragments of TDP-43 exit the nucleus to form TDP-43 cytoplasmic inclusions.32 TDP-43 histopathology is classified into subtypes A-E based on inclusion morphology and subcellular distribution,33,34 and each subtype is associated with particular causal gene mutations or clinical syndromes.35,36
Genes Associated With TDP-43 Proteinopathies
Genetic mutations in TARDBP, which encodes TDP-43, were first found to be a cause of sporadic and familial ALS and FTD disorders, providing a direct link between TDP-43 and disease pathogenesis.37,38 However, TARDBP mutations are a rare cause of ALS and FTD, accounting for only 1% of ALS cases and <1% of FTD cases.39 TDP-43 is found in both genetic and sporadic FTD and ALS. Apolipoprotein E ε4 (APOE4), the most common genetic risk factor for AD, has an increased frequency of TDP-43 pathology,40 and reactive astrocytes carrying the APOE4 risk allele have been reported in ALS.41 Glial cell inclusions of TDP-43 appear characteristic of most ALS cases and can be sporadic or associated with C9orf72, TARDBP, or optineurin (OPTN) mutations.42
TDP-43 Pathogenesis
Multiple disease pathways have been proposed, and evidence supports both loss-of-function (LOF) and gain-of-toxic function TDP-43 disease mechanisms.43,44 Under disease conditions, TDP-43 mislocalizes from the nucleus to the cytoplasm, which is recognized as a critical event initiating the disease cascade.18,45,46 TDP-43 binding to RNA enables it to play a critical role in transcription regulation and stress granule formation, and to have both positive and negative effects on the transcription of key mRNAs, such as neurofilament light and progranulin, in motor neurons.47 As a component of RNA granules in neuronal dendrites, TDP-43 can regulate local translation critical for synaptic plasticity and cognitive function.48 Under stress conditions, TDP-43 can partition into stress granules along with RNA and other proteins, and a defective stress response in neurons may promote the conversion of stress granules into pathological TDP-43 inclusions.25,49
Clinical Syndromes
FTD
BvFTD is the most common clinical presentation of FTD, representing approximately 50% to 60% of all FTD disorders.50,51 It features pronounced changes in personality, including social disinhibition, apathy, loss of empathy, and perseverative or compulsive behaviors. SvPPA is typified by difficulties in single word comprehension and impaired word-finding, while deficits in nfvPPA language production present as effortful speech and agrammatism.11,12,13 TDP-43 causes approximately half of all sporadic bvFTD cases, and therefore one cannot identify with a high degree of certainty which patients have TDP-43-vs tau-related diseases. At present, it is unclear if there is dominance of one proteinopathy vs another in terms of behavioral features such as apathy, loss of empathy, and socially inappropriate behaviors.54,55 The proportion of svPPA and right temporal variant FTD cases that are due to TDP-43 is as high as 90%,20 and therefore clinicians can be more certain of a TDP-43 disease in this context.54,55
Approximately 15% of patients with FTD show comorbid motor neuron disease (FTD-MND), while an additional 27% of patients show some evidence of motor dysfunction during the disease course56. Autosomal dominant gene mutations in progranulin (GRN), microtubule associated protein tau (MAPI), and chromosome 9 open reading frame 72 (C9orf72) with hexanucleotide repeat expansions are the major heritable forms of FTD. FTD-GRN and FTD-C9orf72 are associated with TDP-43 pathology, while FTD-MAPT is associated with tau pathology (Fig. 1).57
FIGURE 1.
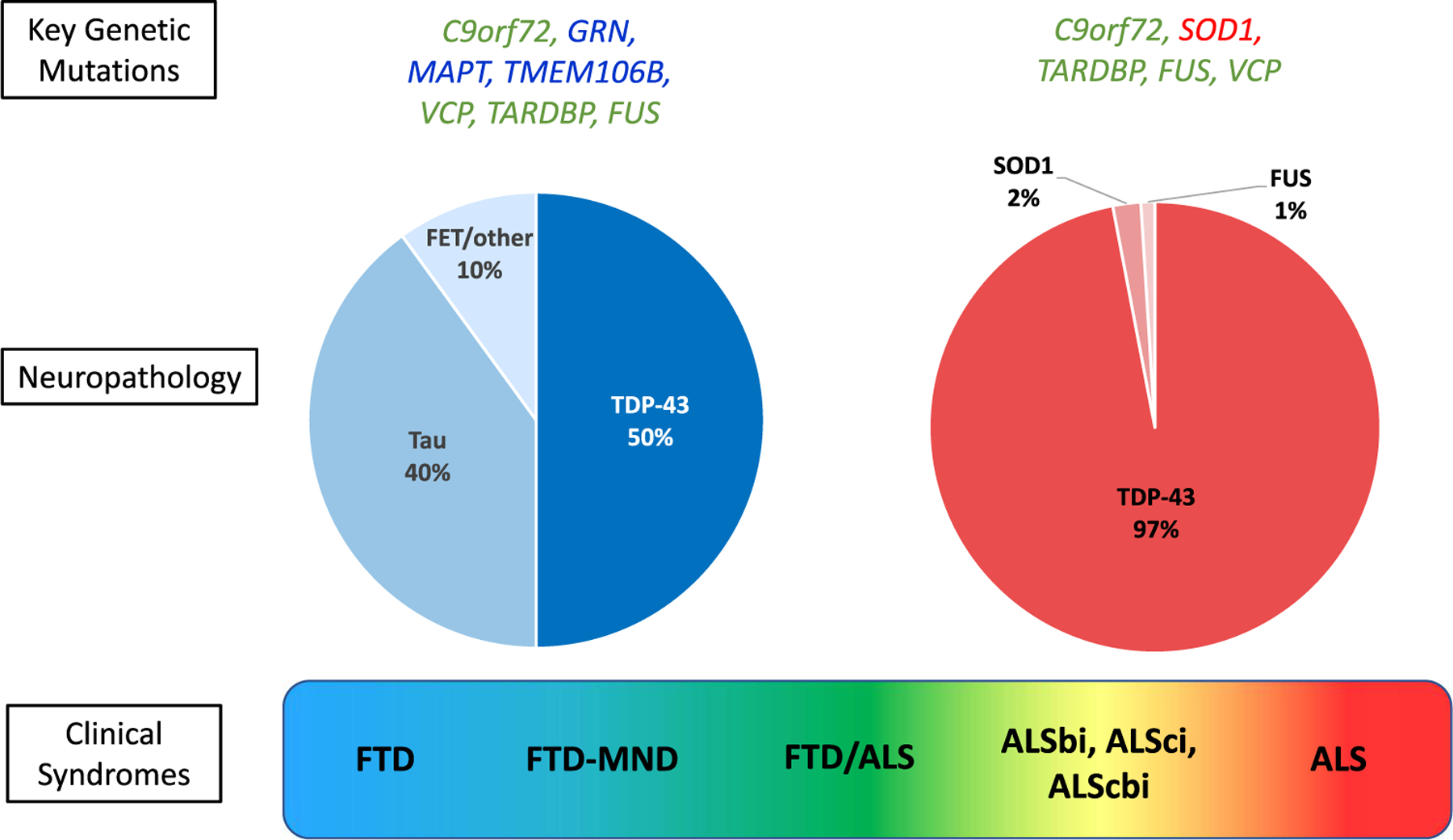
Pathological and genetic distribution in FTD and ALS. Visual representation of the continuum of clinical syndromes from FTD to ALS (bottom). The prevalence of pathological subtypes is depicted in pie charts with FTD on the left and ALS on the right. The most common genetic mutations are listed above the pie charts. ALS, amyotrophic lateral sclerosis; ALS-bi, ALS with behavioral impairment; ALS-cbi, ALS with combined cognitive and behavioral impairment; ALS-ci, ALS with cognitive impairment; FTD, frontotemporal dementia; MND, motor neuron disease.
ALS
ALS is a progressive neurodegenerative disease affecting cortical motor neurons as well as lower motor neurons at the bulbar and spinal level; appearing first as muscle weakness, it eventually leads to muscle atrophy and respiratory failure.58 TDP-43-positive inclusions are present in most ALS cases (98%), with the exception of familial ALS resulting from gene mutations in superoxide dismutase 1 (SOD1) and fused in sarcoma (FUS), which contain SOD1- or FUS-positive inclusion bodies, respectively.59,60 Most cases (90%−95%) of ALS are considered sporadic,61,62 and median age of onset is between 51 and 66 years.63 Approximately half of ALS patients develop deficits in executive function and behavior and can be classified by the revised ALS with FTD spectrum criteria.64 Typical ALS motor deficits can present with various combinations of mixed FTD behaviors and cognitive changes, or in some cases with nfvPPA language deficits. Recent structural imaging suggests that ALS-FTD with behavioral/cognitive involvement might be a phenotypic variant of ALS rather than a feature of worsening disease.65,66
Limbic-Predominant Age-Related TDP-43 Encephalopathy
TDP-43 neuropathology is strongly associated with late-life cognitive decline67 in adults >80 years of age and is recognized as a unique disease entity named limbic-predominant age-related TDP-43 encephalopathy (LATE).68 LATE neuropathological change (LATE-NC) is associated with amnestic cognitive impairment mimicking Alzheimer’s disease (AD)68 and co-occurs with hippocampal sclerosis in ~40% of cases,69 AD neuropathology in 25%−37% of cases,70,71 or other mixed pathologies (e.g., Lewy body disease), and more rarely in isolation (~6%). A considerable proportion of cognitively normal older adults (11%−36%) have also been found to have TDP-43 proteinopathy, with increasing prevalence with older age.21,72,73 While LATE-NC is a unique disease entity for clinical and research purposes,74 clinical identification of LATE-NC is challenging in the absence of TDP-43 biomarkers.
Others
Other primary TDP-43 proteinopathies include multisystem proteinopathy and the dementia Parkinsonism-ALS complex of Guam. Concomitant TDP-43 pathology has also been observed in AD,75,76 Parkinson’s disease (PD), dementia with Lewy bodies, Huntington’s disease, CBS, and PSP.49
PSYCHIATRIC PRESENTATIONS IN TDP-43 PROTEINOPATHY
FTD and FTLD
NPS are prevalent in FTD and are important to recognize in psychiatric practice as they may be the earliest manifestation of the disease. This prevalence has been derived from autopsy-confirmed cohorts (including both sporadic and genetic cases) and large genetic observational studies (only genetic cases). The frequency of psychotic symptoms (e.g., delusions and hallucinations) in neuropathologically confirmed FTLD ranges from 10% to 32%, with differences based on underlying neuropathology subtype, genetic variant, and clinical syndrome.77,78 Recently, Naasan et al.79 examined patterns of NPS in a large cohort of patients with autopsy-confirmed neurodegenerative pathology, including FTLD-TDP inclusions, FTLD-tau (including Pick’s disease, PSP, CBS), AD, and Lewy body disease. Patterns of NPS differed not only across major neuropathology types, but also by underlying FTLD-TDP subtypes. Patients with FTLD-TDP types A and B were more likely to have delusions (32%−35%) compared with patients with AD (16%) or FTLD-tau (2%−15%) pathology.79 Among patients with FTLD-TDP, psychosis was most prevalent among patients with FTLD-TDP type A (53%), followed by type B (42%) and type C (32%). Patients with FTLD-TDP types A and B more frequently reported hallucinations (24%−30%) than those with FTLD-TDP type C (5%). Patients with FTLD-TDP types A and B were more likely to have delusions in the early stages of disease (i.e., in the first 3 years after disease onset) compared with other diagnostic groups.79 In another study, patients with FTLD-TDP pathology were more likely to report paranoid and self-elevating delusions (e.g., grandiosity, erotomania) than those with any other pathology types. Another pathologically confirmed cohort study found that the presence of hallucinations was a differentiating clinical feature suggesting FTLD-TDP pathology, given that it was absent in FTLD-tau or FTLD-FUS pathology.80 This investigation also found that perseverative/compulsive behavior was significantly more prevalent in TDP-43 types B (93%) and C (77%) compared with other TDP-43 subtypes.80 Collectively, these studies suggest that NPS are particularly prevalent in TDP-43-related diseases, even in sporadic cases, and that the emergence of those symptoms in older adults should trigger an evaluation for other features of TDP-43-related diseases.
Longitudinal cohort studies on genetic FTD (such as the Genetic Frontotemporal Dementia Initiative [GENFI]) have revealed associations between the major FTD genetic variants and NPS across the disease trajectory. C9orf72 expansion carriers typically show higher rates of NPS, including psychosis and somatic delusions, and more severe psychosis compared with noncarriers.5,81–83 The prevalence of psychotic symptoms among C9orf72 mutation carriers ranges from 21% to 56%, and psychotic symptoms frequently precede behavioral and personality changes characteristic of bvFTD by up to 5 years.5,78 A similar finding of more frequent psychotic symptoms among C9orf72 expansion carriers is also observed across the ALS-FTD continuum.84
While not as predominant as in C9orf72 carriers, NPS are also common among GRN carriers. A GENFI cohort analysis found that the most frequent NPS among GRN carriers were depression and anxiety, particularly in the early (43%−56%) and late (40%−100%) stages of disease, compared with hallucinations (0%−32%) and delusions (0%−40%).82 Other studies of symptomatic GRN mutation carriers report similar rates of hallucinations (6%−25%) and delusions (6%−33%).85–87 Overall, C9orf72 and GRN carriers appear to share more similar behavioral and NPS trajectories compared to MAPT carriers (i.e., the common FTD mutation associated with tau pathology), perhaps resulting from their shared TDF-43 pathology.82 These results suggest that while late-onset psychotic symptoms are most indicative of a TDP-43 proteinopathy, these patients can also present with significant anxiety and depressive symptoms. These variations in NPS profile across TDP-43 pathological subtypes might relate to differences in the affected neuronal network (e.g., more posterior parietal involvement in GRN carriers vs temporal poles for TDP-43 type C).88
ALS
In addition to shared genetic and pathological features, ALS has significant clinical overlap with FTD, in what has been termed the frontotemporal spectrum disorder of ALS (ALS-FTD).64 Over 50% of patients with ALS exhibit some form of neuropsychological impairment based on the original Strong criteria.64,89,90 The recently revised Strong consensus criteria classify cognitive/behavioral impairment along a clinical spectrum, including ALS cognitively normal (ALS-cn), with cognitive impairment (ALS-d), with behavioral impairment (ALS-bi), with combined cognitive and behavioral impairment (ALS-cbi), and ALS-FTD.64 In a recent study, patients with ALS-bi, ALS-cbi, ALS-FTD, and bvFTD were all found to have similar patterns of NPS severity, supporting the ALS-FTD spectrum.91
A recent investigation of psychotic symptoms across the ALS-FTD spectrum revealed a high prevalence of psychotic features in patients with ALS (18%), ALS-cbi (22%), bvFTD (39%), and ALS-FTD (55%).84 Common symptoms across subtypes included thought broadcasting, thought repetition, and hallucinations.92 Further, across the ALS-FTD spectrum, C9orj72 carriers had much higher rates of psychotic symptoms (63%) than noncarriers (22%). Collectively, these studies suggest that psychotic symptoms are common across the ALS-FTD spectrum and, while they are more common in patients with ALS-FTD, they may be under-recognized in ALS without associated cognitive impairment.
Interestingly, patients with ALS also have an increased risk of psychiatric disorders before an ALS diagnosis compared with controls.93,94 Using a large national record linkage database, hospitalization for a diagnosis of schizophrenia (SCZ), bipolar disorder (BD), depression, or anxiety was associated with a higher risk of subsequent diagnosis of ALS within the following year.94 This association weakened when the ALS diagnosis occurred more than a year after hospitalization, supporting the view that psychiatric symptoms are prodromal in ALS. Corroborating these findings, another nationwide registry study found an increased risk of psychiatric disorder preceding ALS diagnosis, which peaked in the year prior to diagnosis.93 These findings underscore the importance of evaluating patients with late-onset NPS for motor signs and symptoms, which can be prodromal manifestations of neurodegenerative disease.
LATE
NPS have been reported in some patients with LATE-NC but do not appear to be a common feature. In one study using data from the National Alzheimer’s Coordinating Center (NACC), LATE-NC participants with severe impairment (i.e., Clinical Dementia Rating [CDR®] global score 2–3) were more likely to show symptoms of psychosis than FTLD-TDP participants with severe impairment.74 Among LATE-NC participants with severe impairment, the prevalence of visual hallucinations and delusions were 18% and 25%, respectively.74 LATE-NC has been compared more extensively to AD. Using the UK Brains for Dementia Research cohort, Liu et al.95 found that comorbid AD and LATE-NC was not associated with greater NPS burden than AD alone. Overall, it appears that clinically significant NPS can be a feature of advanced stage LATE; however, little is known about the role and prevalence of NPS in the prodromal stage of this disease.
ROLE OF TDP-43 IN NONDEGENERATTVE MAJOR PSYCHIATRIC DISORDERS
Given the high prevalence of NPS in TDP-43-related disease, investigators have started exploring whether TDP-43 perturbations could also play a direct role in the context of major psychiatric disorders. Recent postmortem reports in psychiatry have noted TDP-43 neuronal inclusions in the hippocampus in a small set of BD cases96 and in patients with late-onset psychosis, including SCZ and BD.97 However, a prior study examining postmortem TDP-43 pathology in major psychiatric disorders observed no difference between older adults who had severe mental illness, primarily SCZ, and controls in the frequency, degree, or morphology of TDP-43 pathology.21 A postmortem investigation of TDP-43 proteinopathy in cognitively normal older adults also found no difference in NPS between adults with TDP-43 proteinopathy and those without.73
A larger body of work has investigated the genetic links between non-degenerative PPD and TDP-43 proteinopathies. Relatives of patients with ALS have an increased risk of psychiatric disorders, including SCZ (three-to fourfold higher risk), psychosis, suicide, and autism spectrum disorder.93,98,99 C9orf72 expansion did not fully account for the increased risk of psychiatric disorders among ALS kindreds.99 Further, in a study of family members of patients with FTD and ALS, kindreds of C9orf72 expansion carriers had higher rates of PPD, including SCZ and mood disorders, compared with relatives of C9orf72 noncarriers.100 Although the precise genetic association remains unclear, these findings support the concept of a neuropsychiatric endophenotype in ALS/FTD kindreds.99
Of note, several association studies have explored GRN variability, plasma progranulin protein (PGRN) levels, and the risk for developing BD. In German and Italian cohorts, Galimberti et al101,102 have reported significantly lower plasma PGRN levels in patients with BD compared with controls, a finding which has been replicated.103 Medication was a potential confounder in BD patients, with the replication analysis showing that lithium influenced PGRN levels. Lithium-treated patients had significantly lower plasma PGRN levels compared with nonlithium-treated patients, although nonlithium-treated patients still showed significantly lower plasma PGRN levels compared with controls.103
At the mechanistic level, TDP-43 inclusions in astrocyte cytoplasm can activate astrocytes and induce inflammation and the secretion of pro-inflammatory factors that contribute to neurodegeneration104 and inflammation.105 In mood disorders, there is some postmortem evidence of abnormal glial pathology; young and mixed age group major depressive disorder patients appear to have less glial fibrillary acidic protein (GFAP) immunoreactive astrocyte density than control patients, while in late-onset depression there is increased density of GFAP immunoreactive astrocytes.106 Taken together, this evidence suggests that TDP-43 plays a critical role in glial cell homeostasis and glial regulation of neuronal function, which might theoretically contribute to psychiatric disturbances; however, neuropathological findings do not support a strong link between TDP-43 accumulation and major psychiatric syndromes.
DIAGNOSIS OF DISEASES DUE TO TDP-43 IN PSYCHIATRY
FTD
Patients with unidentified FTLD-TDP and LATE-NC can be encountered in general psychiatric practice. The most significant limitation in clinical practice is the absence of valid TDP-43-specific fluid or imaging biomarkers, except for genetic screening to determine if FTD is caused by GRN, C9orj72, or MAPT gene mutations.10,107,108 Therefore, diagnosis largely depends on the clinical assessment and a probabilistic method to identify likely TDP-43-related diseases.6
Clinical strategies for distinguishing bvFTD from PPD in cases of late-onset behavioral changes are similar for all pathological subtypes; however, the presence of delusions and hallucinations is particularly concerning for both the sporadic and genetic forms of TDP-43-related disease.6,79 Table 1 lists some of the red flags to look for during clinical assessment.
TABLE 1.
Factors Suggestive of TDP-43 Proteinopathies in Patients With Late-onset NPS and Clinical Action Points
| Red Flags for Possible TDP-43 Proteinopathy | Clinical Action Points |
|---|---|
| Late-onset hallucinations and/or delusions | Explore other indicators, cognitive testing, neuroimaging |
| Nonpsychotic late-onset NPS | Explore other indicators – investigate only if other factors present |
| Family history of FTD, ALS, or early-onset dementia | Collateral history from family +/− genetic testing and referral to genetic counselor if positive cases |
| bvFTD features, including hyperorality and stereotypies | Referral to specialty clinics, neuroimaging, neuropsychology, social cognition tests |
| Language disturbances | Neuropsychological +/− speech therapy assessment, neuroimaging |
| Cognitive deficits | Neuropsychological testing |
| Motor weakness | Neurological exam +/− EMG |
Notes: ALS: amyotrophic lateral sclerosis; bvFTD: behavioral variant frontotemporal dementia; EMG: electromyography; FTD: frontotemporal dementia; NPS: neuropsychiatrie symptoms; TDP-43: transactive response DNA-binding protein 43.
While there is consensus that neuroimaging should be performed in the diagnostic investigation of bvFTD, the sensitivity and specificity of standard magnetic resonance imaging (MRI) and positron emission tomography (PET) tracers for differential diagnosis of bvFTD vs PPD are insufficient in the early, ambiguous stages.7,109 Recent clinical practice recommendations to guide the differential diagnosis of bvFTD vs PPD suggest various approaches to improve diagnostic accuracy, such as the inclusion of at least 1 structured test of social cognition (e.g., Ekman 60 Faces Test, Social Cognition and Emotional Assessment [SEA], or Mini-SEA) to the standard neuropsychological battery for bvFTD,6 and increasingly, the use of neurofilament light chain as a differential marker between PPD and neurodegenerative diseases 110–112 In Edition, the Frontotemporal Dementia versus Primary Psychiatric Disorder (FTD versus PPD) Checklist is a recently developed bedside tool designed to help distinguish between bvFTD and PPD, although further validation is needed.113 While a significant proportion of patients with svPPA have NPS,114 the svPPA diagnosis is more straightforward as neuropsychological and speech assessments have strong utility in recognizing semantic deficits. Genetic testing may be warranted in any patient with unexplained late-onset behavioral disturbance (particularly psychosis) who has a first-degree relative with ALS or FTD.6,115
LATE
LATE-NC presents in older patients with short-term recall deficits and is frequently mixed with other pathologies, sometimes accompanied by NPS-like psychosis as well as hippocampal sclerosis on MRI. Clinically, it is nearly impossible to accurately identify patients with LATE-NC as opposed to AD or mixed dementia. When performed, the absence of amyloid on molecular markers in an elderly patient with short-term memory deficits can be a due to the presence of LATE-NC changes; however, the prevalence of amyloid plaques in older adults in the age range of LATE-NC is very high.
TDP-43 Biomarkers
Efforts are underway to develop sensitive fluid and neuroimaging biomarkers for FTD and TDP-43 proteinopathy. Until reliable FTD biomarkers can be validated and implemented in the clinic, it is important to use all available tools to increase diagnostic accuracy in patients with late-onset behavioral changes. Accurate and early diagnosis of FTD is paramount to facilitate appropriate treatment and enable ongoing and future clinical trials of investigative disease-modifying therapies.
TREATMENT DEVELOPMENTS FOR TDP-43 PROTEINOPATHIES
Investigative therapies specific to FTD and ALS are being vigorously pursued to address the urgent unmet need for treatments. Currently, there are no FDA-approved treatments for FTD. Managing NPS in FTD is limited to nonpharmacological interventions (e.g., behavioral programs)116,117 and off-label psychiatric medications (e.g., selective serotonin reuptake inhibitors [SSRIs]), although evidence supporting the efficacy of these medications in FTD is minimal and certain medications, such as antipsychotics, carry an increased risk of adverse effects for elderly patients with dementia-related psychosis.118,119 Edaravone and riluzole are approved for use in ALS and demonstrate modest increases in survival, while the combination drug sodium phenylbutyrate/taurursodiol appears to confer a larger survival benefit by comparison.120 Novel drug candidates targeting C9orf72 expansions and PGEN haploinsuffidency are in various stages of clinical trial development (see Boxer et al121 for review). The 2 greatest challenges of conducting clinical trials on these therapeutics in FTD are clinical trial recruitment and disease heterogeneity.121
Therapeutic Strategies for TDP-43
It is unclear whether targeting a specific aspect of TDP-43 pathogenesis, such as cytoplasmic mislocalization, post-translational modification, or aggregation, will offer the greatest therapeutic potential.122 Several antibody-based interventions directly targeting TDP-43 are being investigated in cell lines and animal models of TDP-43 proteinopathy.123 Recently, a full-length monoclonal antibody targeting the RR1 domain of TDP-43 was shown to bind specifically to cytoplasmic TDP-43 in the brain and spinal cord tissues of postmortem FTD/ALS patients, and to reduce cytoplasmic TDP-43 in murine spinal cord neurons.124 The translational feasibility of these compounds has yet to be determined.
Therapeutic Strategies for FTD-GRN
A growing body of evidence indicates that restoring PGRN may be an effective therapeutic strategy for FTD-GRN as well as other neurodegenerative diseases (reviewed elsewhere125). In addition to the PGRN haploinsuffidency disease mechanism in FTD-GRN, PGRN has neuroprotective and neurotrophic properties,126,128 and PGRN deficiency is a common feature of neurodegenerative diseases.125 Several approaches are in preclinical and clinical development to augment PGRN levels in FTD-GRN, including blocking the degradation pathway of PGRN, protein replacement therapy, gene therapy, and small molecule histone deacetylase (HDAC) inhibitors (Fig. 2).125
FIGURE 2.
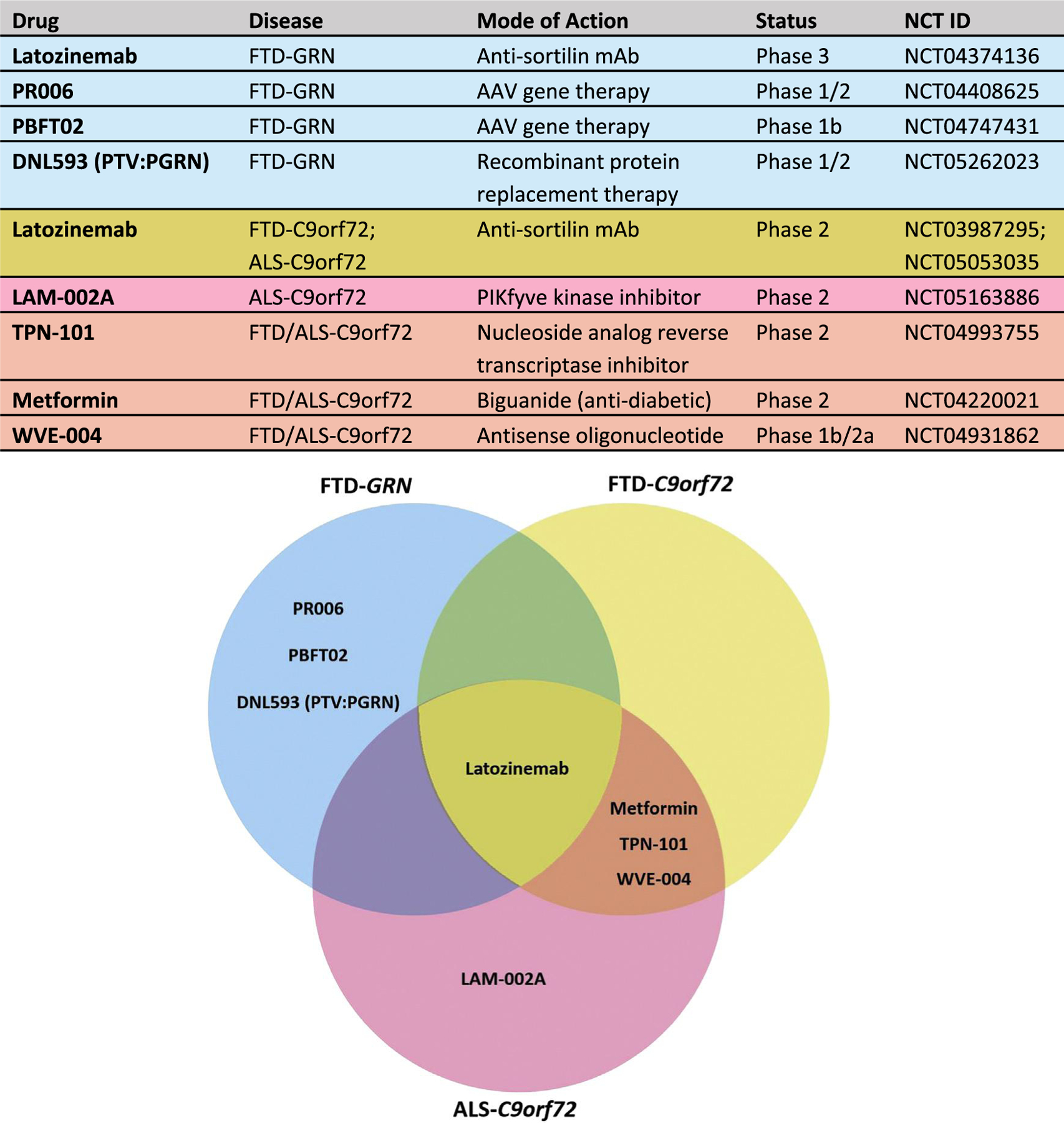
Clinical trials for FTD-GRN and FTD)/ALS-C9orf72. The table lists the drug, target disease, mechanism of action, status, and registration number. The Venn diagram at the bottom provides a visual representation of the drug distributions based on target disease and mutation. AAV, adeno-associated virus; ALS, amyotrophic lateral sclerosis; FTD, frontotemporal dementia; mAb, monoclonal antibody.
In the first approach, a monoclonal antisortilin human antibody, latozinemab (formerly AL001), was developed to block the sortilin-PGRN interaction and prevent PGRN degradation while retaining the ability of PGRN to have functional interactions through alternate trafficking pathways.129 The completed phase 1 study showed that latozinemab increased plasma and CSF PGRN to healthy control levels in GRN mutation carriers.130 Preliminary 12-month results from the phase 2 open-label study (NCT03987295) in patients with FTD-GRN suggest that latozinemab treatment improves multiple biomarkers of disease activity and may slow clinical progression relative to a GENFI2-matched control cohort as measured by the CDR® plus National Alzheimer’s Coordinating Center FTLD Behavior and Language Domains Sum of Boxes (CDR® plus NACC FTLD-SB) scale.131 A pivotal phase 3 trial for latozinemab is ongoing to evaluate its safety and efficacy in patients with FTD-GRN (NCT04374136).
Gene therapy has shown promise for treating rare monogenic diseases and may offer another strategy for elevating PGRN levels in patients with GRN LOF mutations. In this approach, DNA encoding GRN is delivered through an adeno-assodated virus (AAV) vector into the dstema magna. AAV-mediated GRN gene delivery has demonstrated proof of concept in murine models of PGRN deficiency,132,133 although safety concerns over GRN overexpression in the central nervous system were raised when a different AAV vector and intraventricular gene delivery were assodated with hippocampal neurodegeneration in one study.134 Translational safety and feasibility of AAV-mediated GRN gene therapy was demonstrated in nonhuman primates,133 and phase 1/2 trials are underway evaluating 2 different AAV vectors in patients with FTD-GRN (NCT04408625; NCT04747431).
Another therapeutic strategy under investigation for FTD-GRN is intravenous PGRN protein replacement therapy, in which recombinant PGRN protein is fused to an engineered antibody segment that binds to the transferrin receptor to increase blood-brain barrier transport and brain penetrance. A phase 1/2 trial is ongoing for PTV:PGRN (NCT05262023), a protein transport vehicle fused to PGRN, Lastly, HDAC inhibition was shown to enhance PGRN transcription in preclinical studies, although a phase 2 trial of a similar HDAC inhibitor did not find an increase in PGRN levels in participants with prodromal-to-moderate FTD-GRN.137
Therapeutic Strategies for FTD- and ALS-C9orf72
Therapeutic strategies for treating FTD- and ALS-C9orf72 have primarily focused on immunotherapy, antibody-based interventions, and gene therapy approaches (eg., RNA interference, CRISPR-based genome editing, AAV-mediated gene silencing, and AAV-mediated gene delivery including trophic factors).123,138,139 One strategy to counteract the gain-of-toxic function disease mechanism uses antisense oligonucleotides (ASOs) to selectively target repeat-containing RNAs for degradation while preserving CSorf/2 mRNA levels. ASO-mediated therapy has demonstrated proof of concept in reducing nuclear RNA fod and dipeptide repeat proteins in preclinical studies.140,141 Recently, a phase 1 clinical trial of the ASO BIIB078 (NCT03626012) for ALS-C9orf72 patiente did not demonstrate clinical benefit and the open-label extension was terminated (NCT04288856). Unfortunately, a phase lb/2a trial of another ASO targeting the C9orf72 expansion transcript in FTD and ALS, WVE-004, was recently canceled due to lack of clinical benefit despite target engagement (NCT04931862).
Several additional clinical trials are studying new and pre-existing drugs in FTD/ALS-C9orf72. An open-label phase 2 trial is evaluating the safety, pharmacokinetics, and pharmacodynamics of latozinemab, an anti-sortilin antibody designed to block degradation of PGRN, in patients with FTD-C9orf72 (NCT03987295). PGRN overexpression has been shown to reduce TDP-43 aggregation and improve survival in a mouse model of TDP-43 proteinopathy.142 Additional phase 2 trials are investigating pre-existing drugs in FTD/ALS-C9orf72, including: the widely-used antidiabetic agent metformin (NCT04220021; ALS-C9orf72); TPN-101, a nucleoside analog reverse transcriptase inhibitor originally developed for the treatment of HIV (NCT04993755; FTD/ALS-C9orf72); and LAM-002A, a PIKfyve kinase inhibitor that activates transcription factor EB and has been investigated in Thl 7-mediated inflammatory diseases (e.g., psoriasis) as well as B-cell non-Hodgkin lymphoma (NCT05163886; ALS-C9orf72).143
CONCLUSIONS
Late-onset behavioral changes are frequently encountered in general and geriatric psychiatry and present a challenging differential diagnosis. TDP-43 proteinopathies such as FTD and LATE-NC can cause NPS in some patients and pose distinct diagnostic challenges due to symptomatic overlap with PPD, heterogeneous clinical presentation, and the lack of reliable biomarkers. Although much remains to be elucidated, the genetic, neuropathological, and clinical associations reviewed here suggest that NPS are related to the underlying neuropathology and could provide cues for better diagnostic recognition. Indeed, the presence of late-onset NPS, in particular psychosis, can be the initial manifestation of both sporadic and genetic TDP-43-related diseases, and psychiatrists play a key role in the identification and investigations of those patients. While an exploration of the prognostic significance of NPS in TDP-43-related disease is still needed, NPS are associated with faster disease progression and earlier death in patients with AD.144,146 Greater awareness of FTD and other TDP-43 proteinopathies is needed to further refine diagnostic recognition, prevent the use of ineffective treatments with potential negative side effects, and identify carriers of disease-causing mutations who may be eligible for clinical trials of investigational drugs or other appropriate treatment modalities. Improving diagnostic identification among psychiatrists could greatly contribute to the recruitment of eligible participants for research and enable adequately powered clinical trials, thereby accelerating the availability of disease-modifying therapies for TDP-43 proteinopathies.
Highlights.
• What is the primary question addressed by this study?
Neuropsychiatrie symptoms are often early signs of neurodegenerative diseases, contributing to misdiagnosis of these conditions. Among neurodegenerative diseases, TDP-43 proteinopathies are a common cause of neuropsychiatric symptoms and are under-recognized by clinicians, including geriatric psychiatrists. A better understanding of the relationship between neuropsychiatric symptoms and underlying TDP-43 neuropathology may improve recognition, diagnosis, and disease management.
• What is the main finding of this study?
Psychiatric symptoms are prevalent in frontotemporal dementia (FTD) and related disorders and can vary based on TDP-43 neuropathology subtype. Depression and anxiety are often early presentations in FTD caused by GRN gene mutations, while FTD cases caused by repeat expansions in the C9orf72 gene demonstrate a higher rate of psychosis and somatic delusions.
• What is the meaning of the finding?
Late-onset neuropsychiatric symptoms can be the initial manifestation of sporadic and genetic TDP-43-related diseases, including FTD and amyotrophic lateral sclerosis (ALS), and TDP-43 pathology is often comorbid in Alzheimer’s disease. Psychiatrists must play a key role in the differential diagnosis of these patients, who require complex biopsychosocial interventions and may be eligible for emerging clinical trial opportunities for potentially disease-modifying therapies.
ACKNOWLEDGMENTS
Medical writing support and publication assistance were provided by SCIENT Healthcare Communications (Cedar Knolls, NJ, USA).
DISCLOSURES
Medical writing support was funded by Alector, Inc. Academic authors did not receive compensation from Alector for this work. SD has received: research funding support from Alnylam Pharmaceuticals, Biogen, Janssen, Novo Nordisk, and Wave Life Sciences; consultation foes from AI Therapeutics, Eisai, Prevail Therapeutics/Eli Lilly, and QuRALIS; and support for meeting attendance/travel from Eisai SD has participated in advisory boards/DSMBs for IntelGenX. YP has nothing to disclose. JDR has received consulting fees paid to his institution from AC Immune SA, Astex Pharmaceuticals, Biogen, Eisai, Takeda, and UCB. JDR has participated in advisory boards/DSMBs for Alector, Arkuda Therapeutics, Prevail Therapeutics, and Wave Lifo Sciences. EH has received support from NIH grants R01MH120794 and R01AG062268. EF has received research support from Canadian Institutes of Health Research, the Physicians’ Services Incorporated Foundation, and the Weston Foundation and has participated in scientific advisory boards and received honoraria from Denali Therapeutics and Vigil Neuro, and has received honoraria for serving as an Annual Meeting Course Director with the American Academy of Neurology. NT is an employee of Alector, LLC, may have an equity interest in Alector, Inc, and is an unpaid board member for CurePSP.
DATA STATEMENT
The data has not been previously presented orally or by poster at scientific meetings.
References
- 1.Monastero R, Mangiaiasche F, Camarda C, et al. : A systematic review of neuropsychiatrie symptoms in mild cognitive impairment. J Alzheimers Dis 2009; 18:11–30 [DOI] [PubMed] [Google Scholar]
- 2.Santacruz Escudero JM, Beltran J, Palacios A, et al. : Neuropsychiatrie symptoms as predictors of clinical course in neurodegen eration. A longitudinal study. Front Aging Neurosci 2019; 11:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Devanand DP, Lee S, Huey ED, Goldberg TE: Associations between neuropsychiatrie symptoms and neuropathological diagnoses of alzhdmer disease and related dementias. JAMA Psychiatry 2022; 79:359–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Da Silva TBL, Ordonez TN, Bregola AG, et al. : Neuropsychiatrie symptoms in behavioral variant frontotemporal dementia and Alzheimer’s disease: a 12-month follow-up study. Front Neurol 2021; 12:728108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ducharme S, Bajestan S, Dickerson BC, et al. : Psychiatric presentations of C9orf72 mutation: what are the diagnostic implications for clinicians? J Neuropsychiatry din Neurosci 2017; 29:195–205 [DOI] [PubMed] [Google Scholar]
- 6.Ducharme S, Dois A, Laforce R, et al. : Recommendations to difrtinguish behavioural variant frontotemporal dementia from psychiatric disorders. Brain 2020; 143:1632–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krudop WA, Dois A Kerssens CJ, et al. : The pitfall of behavioral variant frontotemporal dementia mimics despite multidisciplinary application of the FTDC criteria. J Alzheimers Dis 2017; 60:959–975 [DOI] [PubMed] [Google Scholar]
- 8.Besser LM, Galvin JE: Diagnostic experience reported by care-givers of patients with frontotemporal degeneration. Neurol Clin Pract 2020; 10:298–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woolley JD, Khan BK, Murthy NK, et al. : The diagnostic chat lenge of psychiatric symptoms in neurodegenerative disease: rates of and risk factors for prior psychiatric diagnosis in patients with early neurodegenerative disease. J din Psychiatry 2011;72:126–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swift IJ, Sogorb-Esteve A, Heller C, et al. : Fluid biomarkers in frontotemporal dementia: past, present and future. J Neurol Neurosurg Psychiatry 2021; 92:204–215 [DOI] [PubMed] [Google Scholar]
- 11.Gomo-Tempini ML, Hiliis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology 2011;76:1006–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olney NT, Spina S, Miller BL: Frontotemporal dementia. Neurol din 2017; 35:339–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rabinovici GD, Miller BL: Frontotemporal lobar degeneration: epidemiology, pathophysiology, diagnosis and management. CNS Drugs 2010; 24:375–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rusina R, Vandenberghe R, Bruffaerts R: Cognitive and behavioral manifestations in ALS: beyond motor system involvement. Diagnostics (Basel) 2021; 11:624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peterson KA, Patterson K, Rowe JB: Language impairment in progressive supranuclear palsy and corticobasal syndrome. J Neurol 2021; 268:796–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Riku Y, Iwasaki Y, Ishigaki S, et al. : Motor neuron TDP-43 proteinopathy in progressive supranuclear palsy and corticobasal degeneration. Brain 2022; 145:2769–2784 [DOI] [PubMed] [Google Scholar]
- 17.Mol MO, Miedema SSM, van Swieten JC, et al. : Molecular pathways involved to frontotemporal lobar degeneration with TDP-43 protetoopathy: what can we learn from proteomics? Int J Mol Sci 2021; 22:10298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neumann M, Sampathu DM, Kwong LK, et al. : Ubiquitinated TDP-43 to frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006; 314:130–133 [DOI] [PubMed] [Google Scholar]
- 19.Irwin DJ, Cairns NJ, Grossman M, et al. : Frontotemporal lobar degeneration: defining phenotypic diversity through personalized medicine. Acta Neuropathol 2015; 129:469–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mackenzie IR, Neumann M: Molecular neuropathology of frontotemporal dementia: insights into disease mechanisms from postmortem studies. J Neutochem 2016; 138(suppl l):54–70 [DOI] [PubMed] [Google Scholar]
- 21.Geser F, Robinson JL, Malunda JA, et al. : Pathological 43-kDa transactivation response DNA-binding protein to older adults with and without severe mental illness. Arch Neurol 2010. 67:1238–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Darby MM, Yolken RH, Sabundyan S: Consistently altered expression of gene sets in postmortem brains of individuals with major psychiatric disorders. Transi Psychiatry 2016; 6: e890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santamaria N, Aihothali M, Alfonso MH, et al. : Intrinsic disorder in proteins involved in amyotrophic lateral sclerosis. Cell Mol Life Sci 2017; 74:1297–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Che MX, Jiang LL, Ii HY, et al. TDP-35 sequesters TDP-43 into cytoplasmic inclusions through binding with RNA. FEBS Lett 2015; 589:1920–1928 [DOI] [PubMed] [Google Scholar]
- 25.Prasad A, Bharathi V, Sivalingam V, et al. : Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front Mol Neurosci 2019; 12:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bjork RT, Mortimore NP, Loganathan S, et al. : Dysregulation of translation in TDP-43 protdnopathies: deficits in the RNA supply chain and local protein production. Front Neurosci 2022; 16:840357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ayala YM, Zago P, D’Ambrogio A, et al. : Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci 2008; 121:3778–3785 [DOI] [PubMed] [Google Scholar]
- 28.Buratti E, Baralle FE: TDP-43: gumming up neurons through protein protein and protein RNA interactions. Trends Biochem Sci 2012; 37:237–247 [DOI] [PubMed] [Google Scholar]
- 29.Ayala YM, De Conti L, Avendano-Vazquez SE, et al. : TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J 2011; 30:277–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aulas A, Vande Velde C: Alterations in stress granule dynamics driven by TDP-43 and FUS: a link to pathological inclusions in ALS? Front Cell Neurosci 2015; 9:423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peng AYT, Agrawal I, Ho WY, et al. : Loss of TDP-43 in astrocytes leads to motor deficits by triggering Allike reactive phenotype and triglial dysfunction. Proc Natl Acad Sci U S A 2020; 117:29101–29112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhopatkar AA, Uversky VN, Rangachari V: Granulins modulate liquid-liquid phase separation and aggregation of the prion-Uke C-terminal domain of the neurodegeneration-assodated protein TDP-43. J Biol Chem 2020; 295:2506–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mackenzie IR, Munoz DG, Kusaka H, et al. : Distinct pathological subtypes of FTLD-FUS. Acta Neuropathol 2011; 121:207–218 [DOI] [PubMed] [Google Scholar]
- 34.Lee EB, Porta S, Michael Baer G, et al. : Expansion of the classification of FTLD-TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol 2017; 134:65–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.SpinelH EG, Mandelli ML, Miller ZA, et al. : Typical and atypical pathology in primary progressive aphasia variants. Ann Neurol 2017; 81:430–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Satadno D, Ferrieux S, NoguesLassiaiDe M, et al. : Primary Progressive Aphasia associated with GRN mutations: new insights into the nonamyloid logopenic variant. Neurology 2021; 97:e88–el02 [DOI] [PubMed] [Google Scholar]
- 37.Van Deerlin VM, Leverenz JB, Bekris LM, et al. : TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol 2008; 7:409–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kabashi E, Valdmanis PN, Dion P, et al. : TARDBP mutations in individuals with sporadic and familial amyotrophic lateral scte rosis. Nat Genet 2008; 40:572–574 [DOI] [PubMed] [Google Scholar]
- 39.van Langenhove T, van der Zee J, van Broeckhoven C: The molecular basis of the frontotemporal lobar degeneration amyotrophic lateral sclerosis spectrum. Ann Med 2012; 44:817–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meneses A, Koga S, O’Leary J, et al. : TDP-43 pathology in Alzheimer’s disease. Mol Neurodegener 2021; 16:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang J, Li C, Shang H: Astrocytes in neurodegeneration : inspiration from genetics. Front Neurosci 2022; 16:882316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Valori CF, Neumann M: Contribution of RNA/DNA binding protein dysfunction in oligodendrocytes in the pathogenesis of the amyotrophic lateral sclerosis/ftontotempotal lobar degeneration spectrum diseases. Front Neurosci 2021; 15:724891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao J, Wang L, Huntley ML, et al. Pathomechanisms of TDP-43 in neurodegeneration. J Neurochem 2018; 146:7–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cascella R, Capitini C, Fani G, et al. Quantification of the relative contributions of loss-of-function and gain-of-function mechanisms in TAR DNA-binding protein 43 (TDP-43) proteinopathies. J Biol Chem 2016; 291:19437–19448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scotter EL, Chen HJ, Shaw CE: TDP-43 proteinopathy and ALS: insights into disease mechanisms and therapeutic targets. Neurotherapeutics 2015; 12:352–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arai T, Hasegawa M, Akiyama H, et al. TDP-43 is a component of ubiquitin positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 2006; 351:602–611 [DOI] [PubMed] [Google Scholar]
- 47.Colombrita C, Onesto E, Megiomi F, et al. : TDP-43 and FUS RNA-binding proteins bind distinct sets of cytoplasmic messenger RNAs and differently regulate their post-transcriptional fete in motoneuron-like cells. J Biol Chem 2012; 287:15635–15647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Buffington SA, Huang W, Costa-Mattioli M Translational control in synaptic plasticity and cognitive dysfunction. Annu Rev Neurosci 2014; 37:17–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.de Boer EMJ, Orie VK, Williams T, et al. : TDP-43 proteinopathies: a new wave of neurodegenerative diseases. J Neurol Neurosurg Psychiatry 2020; 92:86–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011; 134:2456–2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leroy M, Bertoux M, Skrobala E, et al. : Characteristics and progression of patients with frontotemporal dementia in a regional memory clinic network. Alzheimers Res Ther 2021; 13:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boeve BF, Boxer AL, Kumfor F, et al. : Advances and controversies in frontotemporal dementia: diagnosis, biomarkers, and therapeutic considerations. Lancet Neurol 2022; 21:258–272 [DOI] [PubMed] [Google Scholar]
- 53.Mesulam MM: Primary progressive aphasia. Ann Neurol 2001; 49:425–432 [PubMed] [Google Scholar]
- 54.Perry DC, Brown JA, Possin KL, et al. : Clinicopathological correlations in behavioural variant frontotemporal dementia. Brain 2017; 140:3329–3345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kumfor F, Zhen A, Hodges JR, et al. : Apathy in Alzheimer’s disease and frontotemporal dementia: Distinct clinical profiles and neural correlates. Cortex 2018; 103:350–359 [DOI] [PubMed] [Google Scholar]
- 56.Burrell JR, Kiernan MC, Vucic S, et al. : Motor neuron dysfunction in frontotemporal dementia. Brain 2011; 134:2582–2594 [DOI] [PubMed] [Google Scholar]
- 57.Greaves CV, Rohrer JD: An update on genetic frontotemporal dementia. J Neurol 2019; 266:2075–2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zarei S, Carr K, Reiley L, et al. : A comprehensive review of amyotrophic lateral sclerosis. Surg Neurol Int 2015; 6:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mackenzie IR-The neuropathology and clinical phenotype of FTD with progranulin mutations. Acta Neuropathol 2007; 114:49–54 [DOI] [PubMed] [Google Scholar]
- 60.Vance C, Rogdj B, Hortobagyi T, et al. : Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009; 323:1208–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chio A, Calvo A, Mazzini L, et al. : Extensive genetics of ALS: a population-based study in Italy. Neurology 2012; 79:1983–1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mejzini R, Flynn LL, Pitout EL, et al. : ALS genetics, mechanisms, and therapeutics: where are we now? Front Neurosci 2019; 13:1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Longinetti E, Fang F: Epidemiology of amyotrophic lateral sclerosis: an update of recent literature. Curr Opin Neurol 2019; 32:771–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Strong MJ, Abrahams S, Goldstein LH, et al. ; Amyotrophic lateral sclerosis - frontotemporal spectrum disorder (ALS-FTSD): revised diagnostic criteria. Amyotroph Lateral Scler Frontotemporal Degener 2017; 18:153–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Long Z, Irish M, Foxe D, et al. : Heterogeneity of behavioural and language deficits in FTD-MND. J Neurol 2021; 268:2876–2889 [DOI] [PubMed] [Google Scholar]
- 66.Cividini C, Basaia S, Spinelli EG, et al. : Amyotrophic lateral sclerosis-frontotemporal dementia: shared and divergent neural correlates across the clinical spectrum. Neurology 2021; 98: e402–e415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wilson RS, Yu L, Trojanowski JQ, et al. : TDP-43 pathology, cognitive decline, and dementia in old age. JAMA Neurol 2013; 70:1418–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nelson PT, Dickson DW, Trojanowski JQ, et al. : Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 2019; 142:1503–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Besser LM, Teylan MA, Nelson PT: limbic predominant age related TDP-43 encephalopathy (LATE): clinical and neuropath ological associations. J Neuropathol Exp Neurol 2020; 79.305–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.James BD, Wilson RS, Boyle FA, et al. : TDP-43 stage, mixed pathologies, and clinical Alzheimer’s-type dementia. Brain 2016; 139:2983–2993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kapasi A, Yu L, Boyle PA, et al. : Limbic-predominant age-related TDP-43 encephalopathy, ADNC pathology, and cognitive decline in aging. Neurology 2020,95:el951–el962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Arnold SJ, Dugger BN, Beach TG: TDP-43 deposition in prospectively followed, cognitively normal elderly individuals: correlation with argyrophilic grains but not other concomitant pathologies. Acta Neuropathol 2013; 126:51–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nascimento C, Suemoto CK, Rodriguez RD, et al. : Higher prevalence of TDP-43 proteinopathy in cognitively normal Asians: a chn icopathological study on a multiethnic sample. Brain Pathol 2016; 26:177–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Teylan MA, Mock C, Gauthreaux K, et al. Differences in symptomatic presentation and cognitive performance among participants with LATE-NC compared to FTLD-TDP. J Neuropathol Exp Neurol 2021; 80:1024–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.AmadorOrtiz C, Lin WL, Ahmed Z, et al. : TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol 2007; 61:435–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Uryu K, Nakashima-Yasuda H, Forman MS, et al. : Concomitant TAR-DNA binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopa- thies. J Neuropathol Exp Neurol 2008; 67:555–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Landqvist Waldo M, Gustafson L, Passant U, et al. : Psychotic symptoms in frontotemporal dementia: a diagnostic dilemma? Int Psychogeriatr 2015; 27:531–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shinagawa S, Nakajima S, PKtman E, et al. : Psychosis in frontotemporal dementia. J Alzheimers Dis 2014; 42:485–499 [DOI] [PubMed] [Google Scholar]
- 79.Naasan G, Shdo SM, Rodriguez EM, et al. : Psychosis in neurodegenerative disease: differential patterns of hallucination and delusion symptoms. Brain 2021; 144:999–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Scarioni M,Gami-Patel P,Timar Y, et al. : Frontotemporal dementia: correlations between psychiatric symptoms and pathology. Ann Neurol 2020; 87:950–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Snowden JS, Rollinson S, Thompson JC, et al. : Distinct clinical and pathological characteristics of frontotemporal dementia associated with C90RF72 mutations. Brain 2012; 135:693–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Benussi A, Premi E, Gazzina S, et al. : Progression of behavioral disturbances and neuropsychiatric symptoms in patients with genetic frontotemporal dementia. JAMA Netw Open 2021; 4: e2030194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Takada LT, Sha SJ: Neuropsychiatrie features of C9orf72-associated behavioral variant frontotemporal dementia and frontotemporal dementia with motor neuron disease. Alzheimers Res Ther 2012; 4:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Devenney EM, Tu S, Caga J, et al. : Neural mechanisms of psychosis vulnerability and perceptual abnormalities in the ALS-FTD spectrum Ann Clin Transi Neurol 2021; 8:1576–1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Le Ber I, Camuzat A, Hannequin D, et al. : Phenotype variability in progranulin mutation carriers: a clinical, neuropsychological, imaging and genetic study. Brain 2008; 131:732–746 [DOI] [PubMed] [Google Scholar]
- 86.Sellami L, Bocchetta M, Masellis M, et al. : Distinct neuroanatomical correlates of neuropsychiatric symptoms in the three main forms of genetic frontotemporal dementia in the GENFI cohort. J Alzheimers Dis 2018; 65:147–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Snowden JS, Adams J, Harris J, et al. : Distinct clinical and pathological phenotypes in frontotemporal dementia associated with MAPT, PGRN and C9orf72 mutations. Amyotroph Lateral Sder Frontotemporal Degener 2015; 16:497–505 [DOI] [PubMed] [Google Scholar]
- 88.Whitwell JL: FTD spectrum: neuroimaging across the FTD spectrum Prog Mol Biol Transi Sci 2019; 165:187–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Elamin M, Phukan J, Bede P, et al. : Executive dysfunction is a negative prognostic indicator in patients with ALS without dementia. Neurology 2011; 76:1263–1269 [DOI] [PubMed] [Google Scholar]
- 90.Montuschi A, Iazzolino B, Calvo A, et al. : Cognitive correlates in amyotrophic lateral sclerosis: a population-based study in Italy. J Neurol Neurosurg Psychiatry 2015; 86:168–173 [DOI] [PubMed] [Google Scholar]
- 91.Devenney EM, McErlean K, Tse NY, et al. Factors that influence non-motor impairment across the ALS-FTD spectrum: impact of phenotype, sex, age, onset and disease stage. Front Neurol 2021; 12:743688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zucchi E, Ticozzi N, Mandrioli J: Psychiatric symptoms in amyotrophic lateral sclerosis: beyond a motor neuron disorder. Front Neutosci 2019,13:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Longinetti E, Mariosa D, Larsson H, et al. : Neurodegenerative and psychiatric diseases among families with amyotrophic lateral sclerosis. Neurology 2017; 89:578–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Turner MR, Goldacre R, Talbot K, et al. : Psychiatric disorders prior to amyotrophic lateral sclerosis. Ann Neurol 2016; 80:935–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu KY, Reeves S, McAleese KE, et al. : Neuropsychiatrie symptoms in limbic-predominant age-related TDP-43 encephalopathy and Alzheimer’s disease. Brain 2020; 143:3842–3849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nascimento C, Nunes PV, Kim HK, et al. : Increased levels of TAR DNA-binding protein 43 in the hippocampus of subjects with bipolar disorder: a postmortem study. J Neural Ttansm (Vienna) 2022; 129:95–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cao B, Passos IC, Mwangi B, et al. : Hippocampal subfield volumes in mood disorders. Mol Psychiatry 2017; 22:1352–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Byrne S, Heverin M, Elamin M, et al. : Aggregation of neurologic and neuropsychiatric disease in amyotrophic lateral sclerosis kindreds: a population-based case-control cohort study of familial and sporadic amyotrophic lateral sclerosis. Ann Neurol 2013; 74:699–708 [DOI] [PubMed] [Google Scholar]
- 99.O’Brien M, Burke T, Heverin M, et al. : Clustering of neuropsychiatrie disease in fitstdegree and second-Jegree relatives of patients with amyotrophic lateral sclerosis. JAMA Neurol 2017; 74:1425–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Devenney EM, Ahmed RM, Halliday G, et al. : Psychiatric disorders in C9orf72 kindreds: study of 1,414 family members. Neurology 2018; 91:el498–el507 [DOI] [PubMed] [Google Scholar]
- 101.Galimberti D, Dell’Osso B, Fenoglio C, et al. : Progranulin gene variability and plasma levels in bipolar disorder and schizophrenia. PLoS One 2012; 7:e32l64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Galimberti D, Prunas C, Paoli RA, et al. : Ptogranulin gene vari ability influences the risk for bipolar I disorder, but not bipolar n disorder. Bipolar Disord 2014; 16:769–772 [DOI] [PubMed] [Google Scholar]
- 103.Kittel Schneider S, Weigl J, Volkert J, et al. : Further evidence for plasma progranuiin as a biomarker in bipolar disorder. J Affect Disord 2014; 157:87–91 [DOI] [PubMed] [Google Scholar]
- 104.Lee S, Kim S, Kang HY, et al. : The overexpression of TDP-43 in astrocytes causes neurodegeneration via a FTPlB-mediated inflammatory response. J Neuroinflammation 2020; 17:299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kim JH, Rahman MH, Park D, et al. : Identification of genetic modifiers of TDP-43: inflammatory activation of astrocytes for neuroinflammation Cells 2021; 10:676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhou X, Xiao Q, Xie L, et al. : Astrocyte, a promising target for mood disorder interventions. Front Mol Neurosci 2019,12:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Feneberg E, Gray E, Ansorge O, et al. : Towards a TDP-43-based biomarker for ALS and FTLD. Mol Neurobiol 2018; 55:7789–7801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Meeter LH, Kaat LD, Rohrer JD, et al. Imaging and fluid biomarkers in frontotempotal dementia. Nat Rev Neurol 2017; 13:406–419 [DOI] [PubMed] [Google Scholar]
- 109.Vijverberg EG, Wattjes MP, Dols A, et al. Diagnostic accuracy of MRI and additional [18F]FDG-PET for behavioral variant frontotempotal dementia in patients with late onset behavioral changes. J Alzheimers Dis 2016; 53:1287–1297 [DOI] [PubMed] [Google Scholar]
- 110.Al Shweiki MR, Steinacker P, Oeckl P, et al. : Neurofilament light chain as a blood biomarker to differentiate psychiatric disorders from behavioural variant frontotempotal dementia. J Psychiatr Res 2019; 113:137–140 [DOI] [PubMed] [Google Scholar]
- 111.Karantali E, Kazis D, Chatzikonstantinou S, et al. : The role of neurofilament light chain in frontotempotal dementia: a meta-analysis. Aging CHn Exp Res 2021; 33:869–881 [DOI] [PubMed] [Google Scholar]
- 112.Katisko K, Cajanus A, Jääskeläinen O, et al. : Serum neurofilament light chain is a discriminative biomarker between frontotempotal lobar degeneration and primary psychiatric disorders. J Neurol 2020, 267:162–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ducharme S, Pearl-Dowler L, Gossink F, et al. : The frontotempotal dementia versus primary psychiatric disorder (FTD versus PPD) checklist: a bedside clinical tool to identify behavioral variant FTD in patients with late-Onset behavioral changes. J Alz heimers Dis 2019; 67:113–124 [DOI] [PubMed] [Google Scholar]
- 114.de la Sablonniere J, Tastevin M, Lavoie M, et al. : Longitudinal changes in cognition, behaviours, and functional abilities in the three main variants of primary progressive Aphasia: a literature review. Brain Sd 2021; 11:1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Turner MR, Al-chalabi A, Chio A, et al. : Genetic screening in sporadic ALS and FTD. J Neurol Neurosurg Psychiatry 2017; 88:1042–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Barton C, Ketelle R, Merrilees J, et al. : Non-pharmacological management of behavioral symptoms in frontotemporal and other dementias. Curr Neurol Neurosci Rep 2016; 16:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Onyike CU: Psychiatric aspects of dementia. Continuum (Min neap Minn) 2016; 22:600–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tsai RM, Boxer AL: Treatment of frontotemporal dementia. Curr Treat Options Neurol 2014; 16:319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Khoury R, Liu Y, Sheheryar Q, et al. : Pharmacotherapy for frontotemporal dementia. CNS Drugs 2021; 35:425–438 [DOI] [PubMed] [Google Scholar]
- 120.Johnson SA, Fang T, De Marchi F, et al. Pharmacotherapy for amyotrophic lateral sclerosis: a review of approved and upcoming agents. Drugs 2022; 82:1367–1388 [DOI] [PubMed] [Google Scholar]
- 121.Boxer AL, Gold M, Feldman H, et al. : New directions in clinical trials for frontotempotal lobar degeneration: methods and outcome measures. Alzheimers Dement 2020; 16:131–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Buratti E: Targeting TDP-43 proteinopathy with drugs and drug-like small molecules. Br J Pharmacol 2021; 178:1298–1315 [DOI] [PubMed] [Google Scholar]
- 123.Poulto-Briere A, Rezaei E, Pozzi S: Antibody-based therapeutic interventions for amyotrophic lateral sclerosis: a systematic literature review. Front Neurosci 2021; 15:790114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pozzi S, Codron P, Soucy G, et al. : Monoclonal full-length antibody against TAR DNA binding protein 43 reduces related proteinopathy in neurons. JCI Insight 2020; 5:el4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rhinn H, Tatton N, McCaughey S, Kumellas M, Rosenthal A Progranufin as a therapeutic target in neurodegenexative diseases. Trends Pharmacol Sci 2022; 43:641–652 [DOI] [PubMed] [Google Scholar]
- 126.De Muynck L, Herdewyn S, Beel S, et al. : The neurotrophic properties of progranuiin depend on the granuHn E domain but do not require sortilin binding. Neurobiol Aging 2013; 34:2541–2547 [DOI] [PubMed] [Google Scholar]
- 127.Gass J, Lee WC, Cook C, et al. : Progranuiin regulates neuronal outgrowth independent of sortilin. Mol Neurodegener 2012; 7:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Van Damme P, Van Hoecke A, Lambrechts D, et al. Progranuiin fonctions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J Cell Biol 2008; 181:37–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhou X, Sun L, Bastos de Oliveira F, et al. : Prosaposin facilitates sortilin independent lysosomal trafficking of progranuiin. J Cell Biol 2015; 210:991–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Paul R, Long H, Tu G, et al. : AL001 blocks the Sortilin/PGRN interaction and is a potential therapy for FTD-GRN (4422). Neurology 2021; 96(15 Suppl):4422 [Google Scholar]
- 131.Huang JY, Paul R, Yeh F, et al. A phase 2 study of AL001 in frontotempotal dementia patients carrying a granulin mutation (P5–3.005). Neurology 2022; 98(18 suppl):3173 [Google Scholar]
- 132.Arrant AE, Onyilo VC, Unger DE, et al. : Progranuiin gene therapy improves lysosomal dysfunction and microglial pathology associated with frontotempotal dementia and neuronal ceroid lipofuscinosis. J Neurosci 2018; 38:2341–2358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hinderer C, Miller R, Dyer C, et al. : Adeno-associated virus sero-type 1-based gene therapy for FTD caused by GRN mutations. Ann Cto Transi Neurol 2020; 7:1843–1853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Amado DA, Riedets JM, Diatta F, et al. : AAV-mediated progranulin delivery to a mouse model of progranuiin deficiency causes T cell-mediated toxicity. Mol Ther 2019, 27:465–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Logan TDS, Simon MJ, Davis S, et al. : A brain penetrant progranuiin biotherapeutic rescues lysosomal and inflammatory phenotypes to the brain of GRN knockout mice. Alzheimer’s Dement 2020; 16:e25 [Google Scholar]
- 136.Cenik B, Sephton CF, Dewey CM, et al. SuberoylaniHde hydroxamic acid (vorinostat) up-regulates progranuiin transcription: rational therapeutic approach to frontotempotal dementia. J Biol Chem 2011; 286:16101–16108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ljubenkov PA, Edwards L, Iaccartoo L, et al. : Effect of the histone deacetylase inhibitor FRM-0334 on progranuiin levels to patients with progranuiin gene haplotosuffidency: a randomized clinical trial. JAMA Netw Open 2021; 4: e2125584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hautbergue GM, Cleary JD, Guo S, et al. : Therapeutic strategies for C9orf72 amyotrophic lateral sclerosis and frontotempotal dementia. Curr Opto Neutol 2021; 34:748–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Amado DA, Davidson BL: Gene therapy for ALS: a review. Mol Ther 2021; 29:3345–3358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jiang J, Zhu Q, Gendron TF, et al. : Gain of toxicity from ALS/FTD-Linked repeat expansions to C90RF72 is alleviated by anti-sense oligonucleotides targeting GGGGCC-containtog RNAs. Neuron 2016; 90:535–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lagier-Tourenne C, Baughn M, Rigo F, et al. : Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A 2013; 110:E4530–E4539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Beel S, Herdewyn S, Fazal R, et al. : FrogranuHn reduces insoluble TDP-43 levels, slows down axonal degeneration and prolongs survival in mutant TDP-43 mice. Mol Neurodegener 2018; 13:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gayle S, Landrette S, Beeharry N, et al. Identification of apilimod as a first-in-class PIKfyve kinase inhibitor for treatment of B-ceil non-Hodgkin lymphoma. Blood 2017; 129:1768–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Peters ME, Schwartz S, Han D, et al. : Neuropsychiatrie symptoms as predictors of progression to severe Alzheimer’s dementia and death: the Cache County dementia Progression Study. Am J Psychiatry 2015; 172:460–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Connors MH, Ames D, Woodward M, et al. : Psychosis and clinical outcomes in Alzheimer disease: a longitudinal study. Am J Geriatr Psychiatry 2018; 26:304–313 [DOI] [PubMed] [Google Scholar]
- 146.Rabins PV, Schwartz S, Hack BS, et al. : Predictors of progression to severe Alzheimer’s disease in an incidence sample. AIzheimers Dement 2013; 9:204–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data has not been previously presented orally or by poster at scientific meetings.