

Abstract
Type 2 diabetes mellitus (T2DM) is common and increasing in prevalence worldwide, with devastating public health consequences. While peripheral insulin resistance is a key feature of most forms of T2DM and has been investigated for over a century, research on brain insulin resistance (BIR) has more recently been developed, including in the context of T2DM and non-diabetes states. Recent data support the presence of BIR in the aging brain, even in non-diabetes states, and found that BIR may be a feature in Alzheimer’s disease (AD) and contributes to cognitive impairment. Further, therapies used to treat T2DM are now being investigated in the context of AD treatment and prevention, including insulin. In this review, we offer a definition of BIR, and present evidence for BIR in AD; we discuss the expression, function, and activation of the insulin receptor (INSR) in the brain; how BIR could develop; tools to study BIR; how BIR correlates with current AD hallmarks; and regional/cellular involvement of BIR. We close with a discussion on resilience to both BIR and AD, how current tools can be improved to better understand BIR, and future avenues for research. Overall, this review and position paper highlights BIR as a plausible therapeutic target for the prevention of cognitive decline and dementia due to AD.
Keywords: brain insulin resistance, Alzheimer’s disease, type-2 diabetes mellitus, insulin receptor, cognition
1. Significance: The relation of diabetes, insulin resistance, and cognition
Type-2 diabetes mellitus (T2DM) and cognitive impairment related to Alzheimer’s disease (AD), are among the most prominent, fast-growing, and disabling chronic conditions of aging that negatively impact individuals and societies worldwide. The global prevalence of T2DM and AD were estimated to be 463 million and 57 million respectively in 2019 and are projected to increase to over 700 million and 150 million respectively in 2050 [1, 2]. Furthermore, T2DM has been shown to double the risk of all-cause dementia (clinical syndrome caused by a range of diseases), including dementia attributed to AD (a specific disease with characteristic biomarkers and pathologic features) and other causes [3, 4]. Notably, there are sex differences in T2DM related to cognitive impairment and the risk of developing AD, with women being more affected. Converging lines of evidence suggest that T2DM and dementia are closely associated in terms of risk factors and comorbidities. Several underlying mechanisms are postulated to link T2DM and dementia, and the shared etiology of these multifactorial diseases has been reviewed elsewhere [5]. Notably, T2DM is associated with cerebrovascular disease including stroke, which itself is a major cause of dementia (e.g., vascular contributions to cognitive impairment and dementia [VCID]) [6]. Additionally, inflammation, mitochondrial dysfunction, and other pathways are important features of insulin resistance and have been proposed as plausible links [7, 8]. Another key pathophysiological link is thought to relate to insulin resistance itself [9]. Defined as an impaired biological response of the body to insulin stimulation, peripheral insulin resistance has long been recognized to play a critical role in developing T2DM and in complications related to T2DM, including in the brain. Nevertheless, it was not until recent years that researchers found some aspects of insulin resistance in the brains of individuals with AD who did not have diabetes [10], supporting the idea that AD likely features metabolic disturbances that can present independently of T2DM, and involves insulin resistance in the brain. In addition, a large community-based prospective cohort study also shows that higher blood glucose levels and/or poorly controlled T2DM significantly increase the risk of dementia even in people without clinically-defined diabetes [11]. Moreover, in cognitively normal older adults, peripheral insulin resistance, indicated by high Homeostatic Model Assessment for Insulin Resistance (HOMA-IR) scores [12], has been associated with poorer performance on neurocognitive tests and abnormalities in biomarkers of AD, including increases in phosphorylated tau protein in the cerebrospinal fluid (CSF) and lower global cerebral glucose metabolism on positron emission tomography (PET) scans [13-16]. Given the close associations between T2DM, insulin resistance, dementia, and AD, researchers are currently examining these associations in a range of pre-clinical and clinical studies including testing the clinical benefit of anti-diabetic drugs such as metformin and insulin [17], as well as incretin analogs (e.g., glucagon-like peptide 1 [GLP-1] receptor agonists such as liraglutide, semaglutide) [18], in preventing and treating cognitive decline and dementia [19-23]. For the purpose of this work and future research, we define brain insulin resistance (BIR) as an inadequate response by cells located in the brain, including the cerebral vasculature, to secreted insulin: this inadequate response can be due to a limited CNS availability of insulin in its bioactive form, a limited expression of the insulin receptor (INSR) at the cell surface, a shift in INSR isoform expression, and/or diminished signaling events downstream from the INSR binding.
In the larger context of global health, the significance of the public health, challenge of both diabetes and dementia is further complicated by the Coronavirus Disease 2019 (COVID-19) pandemic caused by the virus SARS-CoV-2. On the one hand, both T2DM and dementia due to AD increase the risk of SARS-CoV-2 infection and the severity of COVID-19 [24, 25]. On the other hand, a diagnosis of COVID-19 has been associated with a higher risk for the development of both conditions, T2DM and dementia due to AD [26-28]. While there is much more research needed on the relation of COVID-19, these shared bi-directional associations further suggest a complex pathophysiology that is common to T2DM and dementia, and research is ongoing to better understand the long-term effects of COVID-19 on metabolism and brain function.
The focus of this review is to: 1) describe our understanding of insulin resistance in the brain and how it relates to, and differs from, peripheral insulin resistance, 2) describe in detail the contributors to BIR, including the role of the insulin receptor (INSR) particularly in the brain, 3) highlight current tools to study BIR, 5) elucidate how BIR may correlate with hallmarks of AD pathology underlying cognitive impairment and decline, and dementia, and 6) define where insulin resistance occurs within the brain and in individual cell types. We discuss pre-clinical, human post-mortem, imaging models in living humans, and human behavioral response to insulin research to interrogate this relationship. We close with a discussion about resilience both to AD and BIR, and ideas on how to improve our definition of BIR so that we may identify it earlier in the hopes of treating it and preventing cognitive decline and dementia.
2. Insulin in the brain
Insulin was discovered in 1921 at the University of Toronto and is one of the most studied hormones over the last 100 years. Largely produced by the pancreatic islets β-cells, insulin is essential to life. Mechanisms of action of insulin include binding to the INSR to induce glucose uptake in many tissues, as well as promoting fatty acid and amino acid uptake. Insulin is utilized as a treatment for diabetes by millions of patients worldwide. In the brain, insulin has several recognized actions, most importantly on metabolism and supporting neuronal function like neuroplasticity, important for cognition [9, 29-31] and emotional behavior [32-34], by modulating brain networks [33, 35, 36] (Fig. 1). However, it should be noted that these roles for insulin in the brain may not extend to all regions. For example, insulin may impact memory directly in the hippocampus, but indirectly in the hypothalamus (i.e., through metabolic pathways). Insulin binds the INSR to induce a wide range of signaling events. Whether or not INSR activation induces changes in cellular glucose uptake in insulin-dependent glucose transporter (GLUT2/4) expressing neurons is still controversial. Reaching a better understanding of how the brain responds to insulin is likely to improve our capacity to possibly treat and prevent dementia, and possibly AD as well.
Figure 1.

Role of insulin in the healthy brain and impairments in brain insulin resistance (BIR) identified in Alzheimer’s disease. Insulin in the brain predominantly originates from the pancreas, yet small amounts of insulin synthesis within the brain and choroid plexus have recently been discovered. Brain insulin is degraded by the insulin degrading enzyme (IDE) to regulate signaling. Under healthy conditions, brain insulin helps regulate mood, glucose metabolism, cognition, food intake, and brain perfusion. In Alzheimer’s disease, when BIR is present, there are decreased levels of brain insulin, decreased levels of IDE leading to increases in amyloid β plaques, increased mood disorders, worsened cognition, impaired glucose metabolism, and decreases in insulin receptor (INSR) activation. Parts of the figure were drawn by using pictures from Servier Medical Art.
The brain is now recognized as an insulin-responsive organ [9, 29, 37] and a few researchers even postulate that there is a small degree of endogenous insulin production in the mammalian brain, including from the human choroid plexus [38, 39]. In the periphery, blood insulin levels increase postprandially, after being released by the pancreas. However, as opposed to peripheral organs, which have access to all insulin the pancreas can produce, the brain must predominantly rely on the blood-brain barrier (BBB) transport of insulin for access [40]. It remains largely unknown how postprandial changes in insulinemia affect the BBB and the brain. Whether insulin concentrations in the brain vary in the same way, given the limited BBB transport, is unlikely but remains unclear. Therefore, access of insulin to the central nervous system (CNS) interstitial fluid could be a regulatory mechanism, which upon failure, contributes to BIR. Even if brain insulin signaling is preserved, lower insulin levels could lead to a decreased response in this pathway. Additionally, the BBB has been shown to be a primary mediator of INSR signaling [41], which can impact insulin signaling in the whole brain [42]. The presence of the BBB contributes a unique source of enhanced regulation of brain insulin signaling. Whether BBB insulin resistance contributes to BIR or vice versa remains to be determined.
As we will discuss in detail in section 3, the degradation of insulin by the insulin-degrading enzyme (IDE) which can also regulate brain insulin availability, similar to what is observed in the periphery. Other events that contribute to BIR are changes in CNS cell type expression of the INSR, issues with the ligand-receptor interactions, and the inability to activate downstream signaling processes through receptor autophosphorylation [40].
While glucose is the preferred brain fuel substrate [43], brain glucose uptake is not regulated by physiological insulin levels in subjects with normal glucose processing. In healthy volunteers, hyperinsulinemia within the normal physiological range does not affect BBB glucose transport or net cerebral glucose metabolism [44, 45], as it is largely regulated by the insulin-independent GLUT1 and GLUT3 [46]. In contrast, insulin may stimulate greater glucose transport to the brain in insulin-resistant patients during hyperglycemia [47]. Additionally, the cognitive response to insulin has also been shown to be independent of glucose. That is, under euglycemic states, hyperinsulinemia appears to improve cognition in subjects with dementia attributed to AD [48]. While memory is also improved under hyperglycemic conditions, memory facilitation is greater with hyperinsulinemia. As an extensive discussion of glucose transport into and within cells of the CNS are beyond the scope of this review, we refer readers to a recent review on brain energy and neurodegeneration [49]. In the following section, we discuss in detail contributors to BIR which can include activation of the INSR and availability of insulin.
3. Contributors to BIR
3.1. Insulin receptor (INSR)
The INSR is produced as a pro-receptor, undergoing proteolytic cleavage to become an active tyrosine kinase receptor. It is composed of two extracellular α chains (ligand binding) and two transmembrane β chains (intracellular signaling), as a homodimer (αβ)2 [50-52] (Fig. 2). During the maturation process, alternative splicing can produce two isoforms of the α-chain: the short A isoform (INSRα-A) truncated by 12 amino acids (exon 11) and the long B isoform (INSRα-B) [51, 53] (Fig. 3). Once insulin binds the extracellular α-chains, in a dose-dependent manner, a conformational change of the tetramer occurs and triggers intrinsic tyrosine phosphorylation within the intracellular β-chain [54-56]. This leads to binding to and phosphorylation of the insulin receptor substrate (IRS), which can ultimately trigger downstream intracellular signaling pathways such as RAS/MAPK (cell growth) and PI3K/Akt/mTOR (metabolism, cell growth), with particular relevance to AD [56-58] (Fig. 2). A decrease in INSR available at the cytoplasmic membrane [41] or a defective coupling to these downstream cellular cascades [10] are postulated as some of the causes of BIR, as well as AD.
Figure 2.
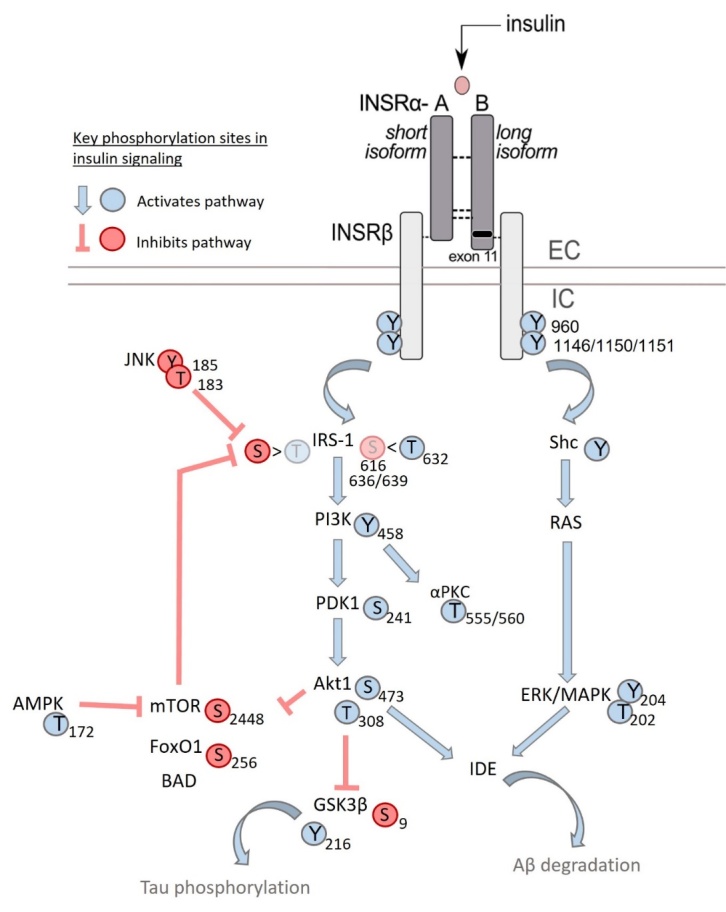
INSR signaling pathway commonly investigated for BIR and phosphorylation sites interrogated. Following insulin binding, the INSR is autophosphorylated at select tyrosine residues, resulting in activation. This triggers activation of the IRS-1 kinase, which can involve either activation on threonine residues or inhibition mediated by serine residues. Threonine phosphorylation of IRS-1 activates the downstream kinases PI3K, PDK1, and Akt1, which all are activated following phosphorylation. Akt activation inhibits GSK3β-mediated tau phosphorylation via tyrosine phosphorylation. Akt1 phosphorylation and subsequent activation can also inhibit mTOR inhibitory phosphorylation. IRS-1 activation can also be inhibited by JNK phosphorylation which limits threonine phosphorylation (activation) and enhances serine phosphorylation (inhibition). The non-canonical INSR signaling pathway involves Shc, Ras, and ERK/MAPK activation. Either Akt or the ERK/MAPK pathway can activate IDE which leads to Aβ degradation. In general, tyrosine and threonine phosphorylation of the kinases present in this pathway elicit activation of the kinase whereas serine phosphorylation inhibits activity of these kinases. Akt, protein kinase B; Aβ, β-amyloid peptide; BAD, Bcl-2-associated death promoter; EC, extracellular; ERK/MAPK, Mitogen-activated protein kinase kinase; FoXO, Forkhead; GSK3β, Glycogen synthase kinase-3 β; IC, intrecellular; INSR, insulin receptor; IRS1, insulin receptor substrates 1; JNK, c-Jun N-terminal kinases; mTor, mammalian target of rapamycin; PI3K, Phosphoinositide 3-kinases; αPKC, atypical protein kinase C.
Figure 3.

Tissue specific expression of INSR isoforms. Alternative splicing results in two isoforms of insulin binding INSRα: A and B. The expression of these isoforms varies between tissues, cell types, and even disease states. Parts of the figure were drawn by using pictures from Servier Medical Art. BBB: blood-brain barrier, BCEC: brain capillary endothelial cell.
3.1a. Location
If BIR is predominantly reliant on the INSR, then it is important to define where the INSR is located in the brain. Autoradiography studies of 125I-insulin binding were one of the primary techniques used to assess the macroscopic localization of the INSR [59, 60]. In rat brain sections, the prominent binding levels of 125I-insulin are in the hippocampal formation [61], choroid plexus [62], hypothalamus [63] as well as the olfactory bulb, cortex, and cerebellum [64, 65]. One of the studies from that time used a cross-linkage strategy to covalently bind 125I-insulin to its receptor to show that most insulin binding sites were retrieved in cerebral microvessels in big animals (pigs and cows) [66].
Early protein analyses showed a wide INSRα distribution throughout the human brain with higher concentrations in the cortex than in the hippocampus and white matter [67]. Immunohistochemistry studies performed on paraformaldehyde-fixed brain sections reported localization of INSRβ in different areas of the rat forebrain (olfactory bulb, hypothalamus, and hippocampus), with staining in cells resembling neurons [68, 69]. The immunoreactivity of antibodies targeting the INSRα also suggested a neuronal localization in the precentral gyrus from human brain sections [67]. The differences between protein and 125I-insulin localization could be due to the specificity of the antibody and non-specific measurement of INSR localization since 125I-insulin binds other receptors.
More recent single-cell RNA transcriptomics analyses in mouse and human whole brain show that INSR mRNAs are largely found in endothelial and glial cells [70-73]. When microvessels are directly compared to parenchyma from the human and mouse brain, the INSR protein was found to be preferentially located on the cerebral vasculature constituting the BBB [41]. Such a high relative concentration of INSR in brain microvessels agrees with a growing number of studies showing that BBB INSR plays a key role in the action of insulin on the brain [41, 42, 74, 75]. The mismatch between cell type localization of INSR mRNA vs protein is likely due to the antibodies available for the INSR. Also, since mRNA levels do not always equate to protein levels, it will be important to pursue global brain proteomics not only on the regional localization of the INSR but also cell type expression.
3.1b. Isoform expression
Further contributing to the complexity of INSR expression throughout the brain, is the question of whether the type of INSRα isoform expressed may also impact insulin signaling. While INSR is ubiquitously expressed, the ratio of INSRα-A/B changes throughout the body [76, 77] (Fig. 3). Insulin-responsive tissues such as the liver, skeletal muscle, adipose tissue, and kidney typically express high levels of INSRα-B [78, 79]. In cells from these organs, INSRα-A is highly expressed during fetal development with a shift toward the B isoform in adults [51, 80-83]. The relative protein abundance of INSR isoforms may result from differences in transcription and maturation processes [51]. The cancer field has provided a large variety of experimental settings to confirm the major impact of the INSRα-A/B ratio on insulin responsivity and downstream cellular response [51]. Not only does the INSRα-A/B ratio influence the relative affinity for ligands, such as insulin-like growth factor (IGF), proinsulin, and insulin, but it also impacts downstream signaling pathways [51, 81]. One specific mechanism is through the ability of (αβ)INSR to heterodimerize into hybrids with (αβ)IGF1-R, resulting in a difference in signaling [51, 56, 84-86]. Higher INSRα-A/B ratios have been observed in the liver of individuals with T2DM [87], as well as in adipocytes from patients with obesity [88] and can be reversed after weight loss induced by bariatric surgery [87, 88]. From this set of data, it has been postulated that a shift toward a higher INSRα-A/B ratio may disrupt the canonical insulin signaling pathway, leading to peripheral insulin resistance [51, 86, 89].
Changes in the INSRα-A/B ratio have recently been implicated in BIR as well. While INSR can be detected in most brain structures, the INSRα-A isoform is expressed chiefly by neurons, in contrast to the INSRα-B isoform which is primarily detected in endothelial cells, astrocytes, and microglia [51, 78, 90-93]. Consistently, higher levels of the B isoform are found in cerebrovascular extracts in both mouse and human brains, compared to parenchymal fractions [41].
In brains of individuals with AD, lower levels of the isoform INSRα-B are present in microvessels, resulting in an increase in the INSRα-A/B ratio [41]. In parenchymal fractions, only the A isoform was detectable, and it remained unchanged with AD pathology [41]. The mechanisms underlying this shift in ratio remain elusive but may involve preferential alternative splicing of INSR, microRNA regulation, and/or post-translational modifications [51]. Clinical relevance stems from the fact that insulin analogs can also be formulated to display higher specificities toward isoforms A and B of INSR [93, 94]. Further studies are needed to examine the relationship between BBB INSR isoform expression, BIR, and AD pathophysiology including amyloid β peptide (Aβ) clearance, Aβ/tau proteostasis, cerebrovascular pathology, and inflammation.
3.2. Availability of bioactive insulin
While changes in INSR or downstream pathways are most often evoked to explain insulin resistance at the molecular level [58, 95], it must also be kept in mind that the bioactivity of insulin itself may vary according to its quaternary structure or soluble component in the surrounding matrix. Human insulin is a classic amyloidogenic protein that aggregates at high temperatures in an acidic solution [96]. While endogenous aggregation of insulin released by the pancreas has not been observed, serum samples from Parkinson’s disease patients display an autoimmune response to insulin oligomers and fibrils, suggesting aggregated insulin may be present in this disease [97]. For example, insulin exists in hexamers or dimers, while only its monomer is expected to bind the INSR [50, 98-100]. The hexamer is the most common form, held together by two zinc ions, and can dissociate into the biologically active monomer for receptor binding. Insulin fibrillation from exogenous administration results in increasing insulin dosing due to a lack of functional insulin availability. The molecular features have been discussed in detail elsewhere [101, 102], and the interacting amino acids between insulin and the INSR have been mapped [99]. Stabilization of the hexameric, dimeric, or monomeric forms of insulin can prevent fibrillation and maintain biological activity.
Alternatively, insulin degradation by the IDE can impact the availability of insulin within the brain. Aβ is also a substrate of IDE [103, 104]. Brain protein expression of IDE is comparable to other major metabolic organs including the liver and pancreas [105]. The muscle is a tissue that expresses the highest amount of IDE protein, along with the stomach and small intestine. Most cell types within the CNS express Ide mRNA [106]. In particular, IDE is also detected at the BBB [41, 107, 108] which not only could be an additional regulator of how much insulin reaches the brain but also a way to control BBB INSR signaling.
As mentioned above, the BBB is also a critical regulator of brain insulin availability [30, 41, 109]. BBB insulin transport is altered under many different pathological conditions, including AD and T2DM [60, 109]. We will discuss later the involvement of the BBB in regulating brain insulin levels and how changes in the transport of insulin into the brain may impact BIR. Now that we have a clearer definition of BIR and how it may arise, we will next discuss ways to interrogate BIR in animal models and humans.
4. Tools to Study BIR
BIR can be interrogated in various ways in mammals from rodents to humans (Fig. 4). Changes in protein levels of the INSR signaling pathway are some of the first endpoints most commonly used to define BIR. Phosphorylation of INSR, of its partner IRS and/or of downstream players such as serine/threonine kinase Akt, glycogen synthase kinase 3 (GSK3), and mTOR [56] are some of the primary mediators (Fig. 2). In most instances, higher tyrosine phosphorylation is a sign of activation whereas serine/threonine phosphorylation is a sign of less active INSR and IRS proteins [56, 58].
Figure 4.

Current tools commonly used to interrogate BIR. We highlight data that utilizes the following tools to investigate BIR: pre-clinical rodent models, human post-mortem in situ insulin stimulation, in vivo neuroimaging especially following insulin stimulation, biomarkers including CNS-derived exosomes, and cognitive response to insulin treatment. Figure created using Biorender.
The most common and reliable technique to measure activation of the signaling cascade remains Western immunoblots as they readily allow the parallel measurement of phosphorylation status in relation to the total number of proteins in the same sample. However, Western immunoblots are semi-quantitative and require a relatively high number of samples. Enzyme-linked immunosorbent assays (ELISAs) are more quantitative and high-throughput but have difficulties detecting phosphorylation sites due to inadequate antibodies. Phosphoproteomic analyses utilizing mass spectrometry have demonstrated tyrosine phosphorylation of a dozen of proteins in response to insulin [110, 111]. While these techniques are promising in uncovering novel mechanisms, they are currently costly and may be less applicable for studies involving several samples, as subsequent confirmation with Western immunoblots or ELISAs is often still necessary. However, just as bulk RNA sequencing has become a popular, genomic tool that has become more affordable over the last decade since it was developed, it is likely phosphoproteomic technology will continue to develop as well [112]. In addition, to obtain further insights on mechanisms, recent techniques utilize human post-mortem tissue, a limited and precious resource to study brain insulin signaling, employ immunohistochemistry and ex vivo stimulation of post-mortem brain tissue with insulin [10]. The electrophysiological response to insulin in rodents or post-mortem tissue can also be used to identify the modulation of neuronal activity [33].
These changes in the INSR signaling pathway can be assessed not only in various rodent models of BIR, including diet-induced, toxin, transgenic, and non-transgenic models but also in human post-mortem tissue. Yet, these latter tissues are not commonly available and methods to collect tissue affect measurement (e.g., need a short postmortem interval to be able to measure response to insulin stimulation). Other tools that are more commonly used as read-outs of BIR in living subjects are neuroimaging and biomarkers (see below).
4.1. BIR assessment in rodent models
Rodent models remain an essential tool for studying insulin response in vivo and inform human research, as well as allow testing hypotheses in experiments based on human data, including common pre-disposing genetic and lifestyle risk factors of BIR and AD like the apolipoprotein E genotype, a high fat diet, and decreased physical activity. In the last few decades, significant efforts have been invested in defining the molecular signature of INSR activation in the brain of rodents. While these cascades have been delineated mostly in various cultured cell models [56, 58], they can be replicated in the muscle and the liver following insulin injection [113-119]. A reduction of this response, assessed with these methods, is then interpreted as evidence of insulin resistance.
However, generating clear proof of INSR activation in the brain triggered by a rise in insulin in the blood circulation has proven challenging, particularly when a small number of animals are used. Table 1 summarizes the range of studies published. From this, we can see that the modes of administration used are varied, associated with different pre-clinical paradigms, brain regions investigated, type or dosage of insulin, dietary status, and euthanasia (method and delay). A first observation is that very few studies directly addressed INSR or IRS-1 phosphorylation, likely due to limited reliable antibodies, and most assessed relative phosphorylation of Akt, GSK3β, or Erk1/2 (Table 1). Intravenously injected insulin at supraphysiological doses is reported to increase pAkt(S473)/Akt ratios in the brain in several but not all studies [115, 117, 120-122]. However, when a hyperinsulinemic clamp is used to fully control blood insulin concentrations, no increases in interstitial fluid levels of insulin or activation of insulin signaling is detected in the hippocampus, contrasting with a clear rise in pAkt in the muscle [123]. Alterations of other kinases are harder to ascertain, and any rise in pAkt(S473)/Akt ratio detected in the brain remains very small compared to the response triggered in the muscle of the same animals [115, 117, 123] (Table 1 Peripheral). By contrast, studies using direct intracerebral or intracerebroventricular (ICV) infusion of insulin consistently show increased activation, mediated by phosphorylation of Akt and GSK3β close to the injection site in the whole brain, hippocampus, or medulla [120, 123-128] (Table 1 ICV). The activation of the cellular cascade downstream of INSR is fast. When detected, the phosphorylation of Akt occurs within 15 minutes after stimulation by insulin in vivo and in vitro [56, 119, 121, 126-129] (Table 1). AD-related factors such as older age or the presence of apolipoprotein E4 (ApoE4) have been found to result in a diminished [120] or enhanced [117] insulin-induced phosphorylation of Akt, respectively (Table 1 Peripheral).
Table 1.
Peripheral and central insulin administration in animal models.
| Ref. | Mouse models | Insulin administration | Brain zone studied (Method) | Insulin administration effects: | ||||
|---|---|---|---|---|---|---|---|---|
| Insulin type, vehicle | fasting information, delay, and death | INSR and IRS-1 activation | Signaling downstream kinases | Others protein activation/phosphorylation | ||||
| Peripheral insulin administration | Intraperitoneal injection | |||||||
| [119] | 5 to 6-month-old C57Bl/6 male | vehicle or insulin (5 IU/kg) no insulin details |
2h fasting Injection 0 to 60 min before death (cervical dislocation) |
Left Cx (WB) | Moderate ↓ of pPI3K(Y458), pAkt(S473), pGSK3β(S9) at 15 min, followed by ↑ until 60 min ↓ pmTor(S2448) (15 min), = pAMPK(T172) and↓ pErk(T202/Y204) (with time 0 -> 60min) |
↓ pTau(S199), (T205), (T212), (S214), (T217), (S262), (S396), and (S404) at 15 min and ↑ after 30 min | ||
| [116] | 4 to 6-month-old 1) male C57Bl/6 ob/ob (OB), lean ob+ (CON) or 2) male, female C57Bl/6 +/- HFD (HFF) | vehicle or insulin (1 IU/kg) no insulin details |
No fasting indication Injection 15min before death (intracardiac perfusion) |
Whole hemisphere (WB) |
CON: ↑ pAkt(S473), pGSK3β(S9), pmTor(S2448) OB or HFF: = pAkt(S473), pGSK3β(S9), pmTor(S2448) (but > CON saline) |
CON: ↑ pαPKC(T555/560), pFoxO1(S256) and pFoxO3a(S253) OB or HFF: = pαPKC(T555/560), pFoxO1(S256) and pFoxO3a(S253) (but > CON saline) |
||
| [118] | 6-month-old inducible liver-specific insulin receptor KO (iLIRKO) 50 or 100% INSR deletion | vehicle or insulin (1 IU/kg) human insulin |
Non-fasted Injection 10 min before death |
Whole Brain (WB) | = INSRβ | 50% deletion: ↓ pAkt(S473), pErk(T202/Y204), pp70S6K(T421/S424) 100% deletion: ø pAkt(S473), pErk(T202/Y204), pp70S6K(T421/S424) |
||
| [342] | 8 to 12-week-old C57BL/6 male mice | saline or insulin (300 IU/kg) bovine pancreas insulin |
Non-fasted Injection 2.5h before death (cervical dislocation, no anesthesia/perfusion) |
Hpc and neocortex (WB) | ↑ pGSK3β(S9)/GSK3β, pJNK(Y185)/JNK = pGSK3β(Y216)/GSK3β, pErk(T202/Y204)/Erk |
|||
| [129] | 6-7-week-old male C57Bl/6 STZ-ip | PBS or insulin (5 IU/kg) bovine pancreas insulin |
o/n fasting Injection 5 to 30 min before death (decapitation) |
Hpc and Cx lysates (WB) | ↑ rapid, transient pAkt (T308, S473), pGSK3β(S9), pGSK3α(S21) (peak at 5 min) = total Akt, GSK3β and GSK3α |
|||
| 6-7 week-old male C57Bl/6 STZ-ip 3 days before ins | Hpc and Cx lysates (WB) | ↓ pAkt (T308, S473) ↑ pGSK3β(S9), pGSK3α(S21) (baseline is high) = total Akt, GSK3β or GSK3α |
||||||
| Intravenous injection | ||||||||
| [117] | 12-month-old APOE3 and APOE4 mice | saline or insulin (33.8 IU/kg) human insulin |
6h fasting Tail vein injection 5 min before death (intracardiac perfusion) |
Microvessels (WB) | = INSRβ, IRS1 | ↓ RAGE, = LRP1 (APOE4 and E3) | ||
| PTCx TBS-soluble (WB) | ↑ pAkt(S473)/Akt (APOE4 > E3) ↑ pGSK3β(S9), pErk(T202/Y204) (APOE4 and E3) = pPDK1(S241)/PDK1, pPI3K(Y458), pERK(T202/Y204), pJNK(T183/Y185)/JNK, pmTor(S2448) (APOE4 and E3) |
↑ pTau(S202) (APOE4 > E3) ↑ pTau (S396/404, T181) = pTau (T231) |
||||||
| = RAGE, LRP1 (APOE4 and E3) | ||||||||
| [120] | C57Bl/6 male young (10-12 weeks) and aged (77-95 weeks) | saline or insulin (1 IU/mouse for 10min) human insulin |
o/n fasting Inferior vena cava injection for 10min (decapitation) |
Whole brain lysate (WB) | ↑ pAkt(S473)/Akt in young mice only = pAkt(S473)/Akt in aged mice |
|||
| [115] | 15-month-old female 3xTg-AD +/- HFD | saline or insulin (33.8 IU/kg) human insulin |
6h fasting Tail vein injection 5 min before death (intracardiac perfusion |
PTCx soluble fraction (WB) |
↑ pAkt(S473) = pGSK3β(S9) (no diet effect) |
= pTau (S202, T181, S396)/Tau (also in Hpc) | ||
| = RAGE, LRP1, IDE (soluble and membrane) | ||||||||
| [121] | 8-week-old C57Bl/6 | saline or insulin (1 mIU or 4 IU) Rapid human recombinant insulin (Actrapid®) |
8h fasting Inferior vena cava injection 5 to 20 min before death (intracardiac perfusion) |
Whole brain (WB) | ↑ pINSR from 5 to 20 min (1 mU) with immunoprecipitated INSR | ↑ pAkt(S473)/Akt (max 10 min), ↑ pGSK3β(S9)/GSK3β with time (max 10 to 20 min), ↑ pERK/ERK (max 10 min) (1 mU) |
↑ pTau(S202) since 5 to 20 min (4 U > 1 mU) = pTau(T231) (4 U and 1 mU) |
|
| [122] | 8-week-old male mice Akt2-/-, Akt3-/- (DKO) | saline or insulin (10 mIU/g) human recombinant insulin |
o/n fasting Inferior vena cava injection 12 min before death |
Whole brain (WB) | = pAkt(S473), pGSK3β(S9) | |||
| Hyperinsulinemic clamps | ||||||||
| [123] | 3- or 12-month-old heterozygous APP/PS1 | 0.1%BSA-PBS, or insulin (4 mIU/kg/min) regular human recombinant insulin (Humulin®R) |
No fasting indication Hyperinsulinemic-euglycemic clamp 60 or 90 min before death |
Hpc and Hyp lysates (ELISA) | = pAkt(S473)/Akt (young and aged APP/PS1) | |||
| [121] | 12-week-old C57Bl/6 male or NIRKO mice | saline + 0.1%BSA, or insulin (200 mIU/kg) regular human insulin (Actrapid®) |
o/n fasting Right internal jugular vein hyperinsulinemic-euglycemic clamp 10 to 60 min before death |
Whole brain (WB) |
↑ pErk/Erk (max 10 min) NIRKO: = pAkt(S473)/Akt, pGSK3β(S9)/GSK3β, pErk/Erk (no variation) |
↑ pTau(S202) since 10 to 60min NIRKO: = pTau(S202) from 0 to 20 min (no variation) |
||
| Central insulin administration | Intracerebroventricular injection | |||||||
| [125] | 8-day-old male layer chicks | saline with 0.1% Evans blue and 43 µM chloric acid, or insulin (100 pmol/chicks) porcine insulin |
3h fasting 9 days injections, death 30 min after the last one (decapitation) |
Medulla (WB) | ↑↑ pAkt/Akt, ↑ pErk/Erk | |||
| [124] | young (4 months) and aged (24 months) male Wistar rats | saline or insulin (1 or 20 mIU) regular human recombinant insulin (Humulin®R) |
No fasting indication 5 days injections, death after the last one |
Right Hpc (WB) | Young rats: = INSRβ Aged rats: ↑ INSRβ |
Young rats: ↑ pGSK3β(S9)/GSK3β, = mTor Aged rats: ↑pGSK3β(S9)/GSK3β, ↑ mTor |
||
| [120] | young (10-12 weeks) and aged (77-95 weeks) C57Bl/6 male mice | saline or insulin (3.75 mIU/5µl 5.41nmol/ml) human insulin (Actrapid®) |
o/n fasting | Whole brain lysate (WB) | ↑ pAkt(S473)/Akt in young and aged mice | |||
| [126] | 2-month-old CF1 (wild-type) mice | saline or insulin (5mIU) no insulin details |
Non-fasted Injection 15min or 24h before death (decapitation) |
Hpc synaptic membrane (WB) | ↑ INSR (15 min > 24h) | |||
| Hpc homogenate (WB) | = INSR ↑ pINSR(Y) (5 mUI 15 min > 24h) |
↑ pAkt(S473) (15 min) | ||||||
| [128] | adult male Sprague-Dawley rats | saline or insulin (6 mU/6µl) human recombinant insulin |
o/n fasting Injection 15min or up to 60 min before death |
Hpc (WB) |
= INSRβ | ↑ pAkt/Akt with time between 15-45min, and ↓ after 60 min | ||
| Intracarotid perfusion | ||||||||
| [343] | 16-month-old Non-transgenic (NTg) or 3xTg-AD (3x) | oxygenated bicarbonate buffered + saline or insulin (350 nM) regular human recombinant insulin (Novolin®ge Toronto) |
Non-fasted Intracarotid perfusion 2 min before death (decapitation) |
Microvessels (WB) | NTg; ↑ pINSRβ(Y1150/1551)/INSRβ*, = pro-INSR, INSRβ, INSRα 3x: = pINSRβ(Y1150/1551)/INSRβ*, = pro-INSR, INSRβ, INSRα |
= BACE1, eNOS, caveolin-1, P-gp, LPR1, RAGE, Neprilysin, IDE (NTg and 3x) | ||
| Intracerebral injection | ||||||||
| [123] | 3- or 12-month-old heterozygous APP/PS1 | artificial CSF +/- insulin (40 or 400nM for 1h) regular human recombinant insulin (Humulin®R) |
No fasting indication Hippocampal reverse microdialysis 1h before death |
Hpc around the injection site (ELISA and WB) | ↑ pAkt(S473)/Akt (400 nM) (young and aged APP/PS1) | |||
| [127] | 1-month-old male Sprague-Dawley rats | artificial ECF +/- insulin (100µU) regular human recombinant insulin (Humulin®R) |
No fasting indication Hippocampal reverse microdialysis 10 min before death |
Hpc around the injection site (WB) | ↑ pAkt | |||
Summary of insulin delivery interventions (peripheral or central) in animal models and their impact on downstream activation of insulin signaling. The majority of the studies have investigated downstream signaling rather than insulin receptor (INSR) and first effector (IRS1) activation. Studies include control animals as well diet-induced metabolic impairment (HFD), type 2 diabetes mellitus (T2DM), or genetic insulin resistance models (LIRKO, ob/ob, STZ-ip), insulin signaling knock-out model (DKO, NIRKO), or in Alzheimer’s disease neuropathological models (APP/PS1, 3xTg-AD). ↑, ↓ or = indicate increased, decreased or equal levels, respectively, following insulin stimulation compared to control. Abbreviations : AD, Alzheimer’s disease; Akt, Protein kinase B; AMPK, AMP-activated protein kinase; BACE1, β-site APP cleaving enzyme 1; BSA, bovine serum albumin; Cx, cortex; ELISA, enzyme-linked immunosorbent assay; ECF, extracellular fluid; eNOS, Endothelial NOS or nitric oxide synthase; Erk, Extracellular signal-regulated kinase; FoxO, forkhead box transcription factors; GSK3β, Glycogen synthase kinase-3β; HFD, high fat diet; Hpc, hippocampus; Hyp, hypothalamus; IB, immunoblot; IDE, Insulin-degrading enzyme; INSR, Insulin receptor; IP, immunoprecipitated; IRS1, insulin receptor substrate 1; IU, international unit; JNK, c-Jun N-terminal kinases; KO, knock-out; LRP1, Low density lipoprotein receptor-related protein 1; mTor, mammalian target of rapamycin; ob/ob, obese mice leptin resistant; P70S6K, Ribosomal protein S6 kinase β-1; PDK, phosphoinositide-dependent protein kinase-1; P-gp, p-glycoprotein; PI3K, Phosphoinositide 3-kinase; PTCx, parieto-temporal cortex; RAGE, Receptor for advanced glycation endproducts; STZ-ip, Streptozotocin-intraperitoneally injection; WB, western blotting; αPKC, protein kinase Cα.
Although less commonly measured, the phosphorylation of INSRβ is the initial activation step specifically following insulin or IGF binding [56]. A more limited number of publications report activation of INSRβ following insulin administration through intravenous, ICV, or intracarotid routes [41, 121, 126] (Table 1). For example, INSRβ phosphorylation can be more readily observed in microvessel extracts after intracarotid administration [41], suggesting a response preferentially located in the endothelium of the brain vasculature.
Within neurons, one major consequence of signaling cascades downstream of the INSR is the modulation of ionic channels which promotes changes in electrical activity. The fact that insulin is able to modulate the excitability or firing rates of neurons has been determined in several brain regions including the hypothalamus, ventral tegmental area (VTA), hippocampus, and dorsal raphe nucleus using patch-clamp electrophysiology. Interestingly, these regions are brain areas involved in functions including feeding behaviors, memory, emotion, sleep, or motivation; functions which are also modulated in response to intranasal delivery of insulin. Thus, in the hypothalamus, insulin has been shown to modulate the electrical activity of the anorexigenic pro-opiomelanocortin neurons [130-133], the orexigenic neuropeptide Y neurons [134, 135], neurons within the ventromedial hypothalamus involved in the control of the counter-regulatory response to hypoglycemia [136], or melanin-concentrating hormone neurons of the lateral hypothalamus [137]. In extra-hypothalamic regions, insulin has been shown to modulate the electrical activity of VTA dopaminergic neurons [138, 139], hippocampus neurons [140], serotonergic neurons [33], or olfactory neurons of the olfactory epithelium [141]. To our knowledge, only two families of ionic channels have been suggested to mediate the effect of insulin including potassium ATP-dependent channel responsible for the hyperpolarizing effects of insulin [134-136, 142], or canonical transient-receptor potential (TRPC) involved in the excitatory effect of insulin [130, 132]. Interestingly, in a mouse model of T2DM, the response to insulin is impaired in several of these neuronal populations [33, 139, 142] supporting evidence for BIR. Of note, in these patch-clamp studies, the direct effect of insulin on neurons suggests BIR is present in the neuron itself. These data are particularly important to aid in differentiating between BIR at the neuronal level versus limited BBB transport of insulin under various conditions in vivo studies.
In sum, these studies show that the detection of INSR activation by circulating insulin is difficult, but perhaps clearer when insulin is injected directly into the brain circumventing BBB transport or when cell populations are independently assessed (i.e., microvessels following carotid insulin injection or neuronal patch clamp studies). Supraphysiological doses or dietary modifications [143] are also often necessary to increase brain levels of insulin. However, supraphysiological doses may not accurately reflect what occurs under normal conditions. First, insulin BBB transport is saturable [144, 145]. Therefore, increasing doses of insulin above the saturable rate will not result in further increases in brain insulin. Second, the activation of Akt/GSK3β/Erk pathway is more frequently reported than the direct assessment of the phosphorylation of INSR/IRS-1. This issue limits data interpretation because while we can reasonably attribute a higher phosphorylation rate of INSR and IRS to the action of insulin, downstream kinases involved in the INSR cascade are shared by a wide variety of cell signaling pathways [146], which makes it more difficult to tightly link such endpoints to a specific response to insulin. Finally, studies are most often performed on brain homogenates, which do not provide cellular localization and may dilute the signal of INSR-activated cells, as will be discussed later. Keeping in mind these challenges, pre-clinical models offer suitable means to investigate the mechanism underlying BIR in vivo. Since blood-derived insulin must interact with the BBB before it can access downstream cell cascades in the CNS, studying brain insulin response within the neurovascular unit may also facilitate the investigation of BIR.
4.1a. Diet-induced models
The most frequently used method to generate insulin resistance in an animal is to induce T2DM following an excessive intake of fat and/or sugar [33, 115, 143, 147-149]. The scientific literature is replete with demonstrations of acquired signs of insulin resistance in the periphery, most particularly in the liver or muscle [113-115, 150].
Combining transgenic induction of AD-like pathology with a high-fat diet (HFD) has been useful to shed light on AD-relevant brain-periphery interactions. Spontaneous changes in glucose response are often described in these models and are greater with aging [123, 143, 151-155] (Fig. 2). Furthermore, in rodent models of AD neuropathology, the intake of a HFD generally aggravates cognitive performance and brain Aβ pathology, while effects on tau and synaptic markers, including synaptophysin and syntaxin-3, are less consistent [115, 149, 153, 156-163]. However, an alternative study has shown transcriptional levels of the INSR in the hippocampus are decreased in an AD mouse model (Tg2576) and feeding a HFD for 10 months to these mice restored levels to controls [164]. Cognition was also improved in the AD HFD mice. Sex-dependent peripheral glucose intolerance and insulin resistance are typically observed as well, with females often affected earlier [157, 160, 165]. A single insulin injection led to a restoration of soluble Aβ levels in cortex and memory function in the 3xTg-AD mouse model on a HFD, possibly associated with greater Aβ clearance through the BBB [115, 166]. How insulin and BIR is able to alter Aβ levels is largely unknown, but it is thought that brain insulin can impact Aβ clearance [167] and/or processing [168].
While a HFD paradigm is efficient to generate insulin resistance in the periphery, signs of BIR have been less studied so far. One of the first studies using HFD-fed Tg2576 mice reported a decrease in basal INSR activation and downstream signaling (see Table 1) in the cortex, compared to a chow-fed control group [169]. AD mice on a HFD had poorer memory, increased brain Aβ neuropathology with higher levels of Aβ40/42 in the hippocampus, an increased Aβ production in the cortex (more APP γ-C-terminal fragments and plaque burden), and lower levels of IDE [169]. This was mirrored in a similar study exploring HFD in another AD mouse model (A7-Tg), where HFD increased both soluble Aβ42, insoluble Aβ40 and Aβ42, and amyloid plaque formation, which was due to altered ISF Aβ42 clearance [143]. In another study, no difference in basal INSRβ activation was observed in the hippocampus of wild-type (WT) or the APP/PS1 mouse model of AD after 6 months of HFD, only a decrease in pAkt(S473), along with, surprisingly, higher relative phosphorylation of other kinases (Table 1) [170]. In addition, 3-month-old HFD-fed APP/PS1 showed no differences in the activation of these kinases in the hippocampus and no change in tau protein phosphorylation of various epitopes [171]. One study reported HFD accelerates the ApoE4 impairment of neuronal insulin signaling by trapping the INSR in endosomes [172]. A limitation of most published animal studies is that they measure basal INSR activation without stimulation by insulin, so the brain’s resistance to insulin remains uncertain. The little available evidence in insulin-injected animals suggests that INSR activation may still be triggered in animal models of AD, even following HFD-induced insulin resistance, similar to the treatment of advanced T2DM [115, 123, 166]. The possibility of a bidirectional relationship between AD and T2DM suggests that improving insulin signaling in the periphery or in the brain could break this cycle and be therapeutic in both diseases [115, 166, 173-175].
In summary, identifying BIR in HFD-fed animal models has proven more difficult than identifying peripheral insulin resistance. Discrepancies between studies stem from differences in the age of exposure to the HFD, as well as models used (AD or otherwise, as some of these models are prone to atherosclerosis), and endpoints assessed, with the lack of information on the exact diets used remaining the most constraining issue. Still, important gene-diet interactions have been uncovered, reinforcing the hypothesis of targeting brain insulin signaling in AD.
4.1b. Toxin models
ICV injection of the diabetogenic toxin streptozotocin (ICV-STZ) is often described as a model of BIR or “sporadic AD”; however, this classification is problematic. On the one hand, some aspects of T2DM and AD are found with this approach. Indeed, ICV-STZ brains present with decreased brain glucose metabolism and impairment in the brain insulin-INSR system [176-179]. This model often displays a rise in neuroinflammatory markers, oxidative stress, reduction in cerebral energy metabolism, and functional changes including learning and memory deficits. While the method of action of STZ within the brain remains unclear, this toxin-based approach can increase Aβ and p-tau neuropathology levels in rodent models of humanized Aβ or tau [180-182]. In AD animal models, STZ-ICV injection was found to worsen neuroinflammation, cognition deficits, and neuropathology [180, 182-184]. However, BIR status has mostly been assessed by evidence of changes in the basal phosphorylation downstream INSR substrates without a direct insulin challenge [179, 182, 185]. In addition, ICV injection of STZ is known to also result in some STZ moving into the blood and inducing insulin deficiency, a model of T1DM, not T2DM, in the periphery [179] by selectively ablating the insulin-producing pancreatic beta cells. In sum, it is difficult with this model to determine whether the AD-relevant effects observed of STZ result from the induction of central (brain) or peripheral insulin resistance and how this model relates to T2DM or AD.
4.1c. Genetic/inducible models
Other mouse models considered tools for investigating BIR include either genetic models of brain INSR deletion or the use of lentiviruses to knock-down or overexpress levels of the INSR in select regions. In 2000, the first brain-specific INSR knock-down mouse was generated [186]. Since then, many other models have been generated not only to investigate the role of INSR in the whole brain but also within select CNS-cell types, including endothelial cells located at the BBB [30, 187-189]. Most recently, these brain INSR-deficient models have been crossed with mouse models of AD to further interrogate the relationship between INSR and AD pathology [190].
One limitation of such approaches is that cells (e.g., neurons, astrocytes, microglia) from the whole brain are targeted. While cell-specific effects will be identified, effects due to sub-populations within these cell types will be lost. However, it is possible to selectively knock-out the INSR in selective cell populations, which was recently shown in a neuronal population. Martin et al. recently developed SeIRKO mice in which the INSR is selectively knocked-out in brain serotonergic neurons [33]. They showed that the anxiolytic effect of insulin is abolished in SeIRKO mice in response to intranasal (INL) insulin. More studies are needed to question the effect of insulin in other selective neuronal networks. Nevertheless, such a selective approach is important to evaluate the effect of insulin on the control of brain functions acutely, past the developmental stage as brain insulin signaling is involved in growth and development [64].
Another alternative approach to investigating the impact of BIR in various mouse models is to manipulate the brain INSR, using lentiviral or adenoviral techniques. This tool is particularly useful to study the impact of the INSR in various brain regions and specific cell types, in a variety of mouse models across the lifespan. Lentiviral-mediated knockdown can be used to reduce INSR levels locally [191]. Using this tool, the role of the INSR in the hypothalamus and hippocampus has been further elucidated in various physiological states [32, 191-194]. Alternatively, efforts to increase brain expression of a constitutively active INSR using an adeno-associated virus targeting neurons, show memory can be enhanced in aging [195]. Limitations to these approaches (both viral and knock-down) are that they do not typically completely eliminate the INSR and often do not target every cell type (i.e. microglia in particular are hard to target using adenoviral or lentiviral approaches). However, these approaches still prove to be useful in identifying cellular contributions for INSR signaling.
The selective INSR antagonist, S961 [196], is also useful for exploring the impact of acute insulin resistance on BIR under various conditions. For example, S961 has been used to identify the role of the INSR at the BBB on BIR and insulin BBB transport [41, 75, 197-199]. Additionally, S961 has been used to better understand the delivery of INL insulin to the brain [200]. S961 has also been used to induce BIR acutely to assess the impact on insulin BBB transport [201]. These tools highlight the pre-clinical models available for interrogating the role of BIR in AD.
4.2. Human post-mortem tissue
Post-mortem investigation of the human brain is another important and useful tool to study the presence of BIR in aging and its association with brain function, cognition, and dementia in particular [9]. Using human brain tissue to study BIR and signaling may provide insights into diseases that are uniquely human, including insights into mechanisms of disease [202]. Indeed, Talbot et al were the first to show in post-mortem human AD brain sections from the University of Pennsylvania and Rush University Medical Center cohorts, that BIR was present, defined by the ex vivo response to insulin stimulation, and that levels of pIRS1(S616, S636/639) negatively correlated with cognition [10] (Fig. 2). Nevertheless, the application of post-mortem investigations of BIR is still relatively limited because the post-mortem brain tissue cannot be easily acquired, especially of persons with and without AD and in whom detailed clinical data (e.g., cognitive data, dementia status, etc.) are available. Therefore, the ability to interrogate the relationship between BIR and genetic or lifestyle risk factors have largely been unexplored using this tool. Another caveat is that these individuals are usually at the later stages of disease progression so studying the development and progression of disease is difficult. Further, there are changes in brain structure, metabolism, and function that occur in the highly variable peri- and post-mortem interval which complicate the experiments and interpretation of results. Indeed, experiments suggest that a short post-mortem interval is important in order to successfully conduct ex vivo stimulation with insulin. Finally, the findings from human post-mortem studies may not be completely clinically translatable, which is a significant limitation in reaching the goal of prevention of cognitive decline in living persons with BIR.
4.3. In vivo neuroimaging
In vivo neuroimaging has the potential to provide measurements of a subset of the various physiological events characterizing BIR, with notable gaps and limitations. One of the most significant gaps in the neuroimaging-based assessment of BIR is the relative lack of techniques for assessing insulin transport across the BBB and binding to receptors in the brain. PET with a radiolabeled insulin ligand would appear to be ideal for this application because dynamic PET techniques, together with compartmental modeling techniques, have the ability to provide separate estimates of BBB exchange and receptor occupancy. However, to our knowledge, data from only one such radiolabeled insulin ligand has been published to date [203], and this data came from mice only. Lacking such insulin PET radioligands, PET radioligands that selectively bind to the insulin-like growth factor 1 (IGF-1) receptor, including radiolabeled IGF-1 itself, could be useful as proxy indicators of INSR binding due to the high level of cross-binding between these molecules and their respective receptors [204-206]. However, there is still relatively little work to date validating such IGF-1 ligands in living humans and post-mortem samples to understand the role they could play in the study of brain insulin action.
The individual biochemical events in the cascade of events that follow INSR binding—including IRS-1 phosphorylation, Akt activation, and downstream modulation of mTOR, GSK3, FOXO, and other pathways—are all difficult to measure directly using in vivo neuroimaging techniques. Glucose uptake into the cell, which is one consequence of these biochemical events in select cell populations, is however commonly measured using PET imaging with 18F-fluorodeoxyglucose (FDG), a glucose analog that is readily taken up by the cell in place of glucose. Because FDG PET imaging is clinically useful for the evaluation of a number of diseases, especially cancer but also AD, FDG PET is readily available at many research centers and now also in academic medical (clinical) centers, and it has been explored in conjunction with either peripheral or central insulin challenges as a proxy measure of BIR. In hyperinsulinemic clamp paradigms, circulating insulin levels are increased via continuous infusions of insulin; the amount of FDG taken up by the brain is compared between high and basal insulin conditions to assess how much additional glucose uptake is provoked by the increase in insulin [44, 47, 207]. Alternatively, somatostatin infusions allow assessment of the sensitivity of brain glucose uptake to reductions in steady-state circulating insulin levels [208]. Finally, naturalistic experiment designs seek to relate neuroimaging-based metabolic measures to modulations of circulating insulin levels that were caused not by the experimenter, but by the participant, via self-administration of insulin for diabetes treatment [209]. The presumed assumption that ties these studies to BIR is that changes in steady-state levels of peripheral insulin correspond tightly to changes in central levels. There is emerging evidence that brain production of insulin at the choroid plexus may follow a distinct regulation process than systemic insulin, driven by serotonin and not glucose signaling [39]. INL insulin challenges attempt to avoid this difficult assumption by administering insulin directly to the brain, with minimal spillover into the periphery [210, 211], but there are surprisingly few studies that relate INL insulin challenges to changes in FDG PET metabolism [212]. One study observed that individuals with AD or mild cognitive impairment (MCI) randomized to chronic INL insulin exhibited attenuated declines in FDG PET uptake over time compared to those randomized to placebo [212]. However, utilizing FDG PET solely as a marker for BIR proves difficult as 1) BIR is not simply tied to brain glucose metabolism and 2) insulin action has different effects on the vasculature (initiating vasodilation) versus the synapse (plasticity), which speaks to its role as a trophic factor or potentially, energy modulator. This highlights the need to improve our methods for assessing BIR or combine tools to further understand BIR in the context of AD.
Magnetic resonance spectroscopy (MRS) provides an alternative means for assessing brain glucose utilization responses to peripheral or central insulin challenges. Infusing a fuel substrate (such as glucose) labeled with 13-carbon (13C), together with the collection of 13C MRS measurements, allows for the simultaneous and dynamic measurement of both the 13C-labeled fuel and the by-products of its metabolism [213]. The glucose metabolic flux estimates resulting from such measurements could potentially be compared between differing insulin states, although human data using 13C MRS with 13C-labeled glucose and insulin manipulation has been limited [214]. Recent advances in 1-hydrogen (1H) MRS allow for measurement of the concentration of endogenous glucose in the brain with no need for a stable isotope infusion [215], although to date it also does not appear that comparisons of these measurements across differing insulin states have been made.
MRS also allows for the measurement of other molecular indicators of cell metabolism beyond glucose utilization. Classical 1-hydrogen (1H) MRS techniques allow for the measurement of neuronal function and integrity markers such as N-acetylaspartate (NAA) and choline-containing compounds (Cho), as well as the regulatory compound myo-inositol and the neurotransmitters glutamate, glutamine, and GABA (sometimes measured all together as a composite variable, Glx), and creatine (Cr) as an indicator of total energy use. Among these indicators, one study suggested that under hyperinsulinemic-euglycemic clamp conditions, the NAA/Cr and NAA/H2O ratios in frontal cortex, and the Glx/Cr and Glx/H2O ratios in frontal and temporal cortices, are elevated compared to basal conditions, while frontal Cho/Cr and temporal Cho/H2O and myo-inositol/H2O ratios are decreased relative to basal conditions [216]. In addition, 31-phosphorus (31P) MRS can provide dynamic measurements of endogenous brain levels of the energetic products adenosine triphosphate (ATP) and phosphocreatine (PCr), both of which appear to increase as soon as ten minutes after an acute INL insulin challenge [217]. However, it should be noted that the number of such MRS studies is very small, likely due to limited accessibility, and that further, the sample size in these studies is also very small, raising the possibility for biased results.
More common are studies that use dynamic magnetic resonance imaging (MRI) techniques to assess the downstream cerebrovascular consequences of altered neural and glial metabolic activity. Blood oxygenation level-dependent (BOLD) functional MRI (fMRI) dynamically measures local relative concentrations of oxygenated hemoglobin, which change with a complex time course in response to local changes in neural and glial metabolic demands [218]. BOLD fMRI accomplishes this using conventional MRI hardware, without the injections or ionizing radiation of FDG PET, and without stable isotope infusions. fMRI, used to image the brain’s default mode network (DMN) which is a network of regions that are more active at rest than during an effortful task, is disrupted in AD patients and those with increased AD risk [219]. Similarly, T2DM patients show differences in DMN functional connectivity compared with controls, and those differences are associated with measures of insulin resistance in selected brain regions [220]. These findings agree with FDG PET measures showing brain glucose hypometabolism in the same areas in T2DM patients [14, 221].
Because of its relative ease of use, changes in BOLD fMRI signal amplitude in response to glucose infusions and peripheral insulin infusion have been assessed. In one study, the BOLD signal amplitude in the resting state increased in a distributed cortical composite region of interest, as well as a hypothalamic region of interest, in response to a 2-minute steady-state 3 mg/kg of body weight infusion of glucose [222]. In another study, BOLD responses to finger tapping, simple reaction time, and four-choice reaction tasks differed in multiple brain locations during a hypoglycemic clamp compared to basal conditions [223]. In addition, changes in the resting-state inter-regional synchrony of the BOLD signal (the so-called “functional connectivity”) in response to INL insulin challenge have also been assessed. One of these studies suggested that functional connectivity between the right hippocampus and the medial prefrontal cortex lowers in response to a 160 IU acute dose of INL insulin [224]. Another study suggested that BOLD fMRI-based functional connectivity between the hippocampus and other regions, including the hypothalamus and prefrontal DMN regions, increased following an acute dose of INL insulin [35]. A third study suggested that the intrinsic local statistical characteristics of the BOLD signal in the hypothalamus and orbitofrontal cortex are modified by INL insulin [225]. Other studies have used a similar technique, perfusion MRI, to measure cerebral blood flow responses to an acute INL insulin challenge [225]. One such study suggested that cerebral blood flow in the bilateral amygdalae reduced significantly compared to basal conditions after an INL insulin challenge [226]. Another study suggested that right putamen cerebral blood flow increased in normal weight adults following INL insulin and that this response was blunted in overweight or obese adults [224]. Perfusion MRI differs from BOLD fMRI in that its measurements isolate cerebral blood flow, while BOLD measurements represent the culmination of cerebral blood flow, cerebral blood volume, and local oxygen metabolism. Note that while in vivo neuroimaging studies of brain responses to insulin challenges have largely focused on measurements from a single imaging modality, multi-modal studies that combine BOLD fMRI, perfusion MRI, and MRS appear to be on the way [227].
4.4. Cognitive response to insulin
Similar to imaging studies assessing the response to an external stimulus, such as insulin administration, an alternative approach assessing cognitive response can be used as a proxy for BIR. Insulin administration either via a hyperinsulinemic clamp [48] or via INL delivery [228-232] may enhance memory in selected populations [233]. Therefore, impaired cognitive responses to insulin administration in this setting may be a result of BIR, more specifically impaired insulin transport across the BBB resulting in insulin deficiency in the brain. Indeed, in individuals within populations known to have BIR, such as in ApoE4 individuals or those with AD or MCI, the response to INL insulin is impaired [212, 234-237]. Previous rodent studies have shown there are no differences in the brain distribution of intranasally-administered insulin, indicating the availability of insulin is not the issue with this administrative route [200]. Furthermore, there is a differential sensitivity to INL insulin between men and women [210, 238] and sex differences in brain insulin signaling have been noted [224]. Cognitive response to external insulin administration is an indirect method for assessing BIR and assumptions must be made. However, it is still a useful tool for elucidating relationships and tracking changes longitudinally. Nonetheless, challenges remain in the production of pharmaceutical formulations for the brain delivery of insulin which continue to make its’ use in research and translation into the clinic difficult [229].
4.5. Biofluid biomarkers
Biofluid-based indicators of the many physiological events involved in brain insulin processing would greatly improve the efficiency of learning about BIR in living human subjects because the current in vivo neuroimaging approaches described above are expensive, technically elaborate, and limited in tracking physiological processes over time. In addition, biofluid-based measures have the potential to assess physiological events that are currently not measurable by in vivo neuroimaging techniques. Markers of IRS-1, with various patterns of phosphorylation, derived from blood-based exosomes enriched for neuronal origin, are currently of intense interest. Phosphorylation of IRS-1 is one of the key biochemical steps leading from INSR binding to downstream metabolic effects. These exosomes are small extracellular vesicles that are released regularly by neurons, cross the BBB into circulation, and carry markers of their neuronal origin to enable their identification. A growing body of work has related the abundance of exosome-derived markers to standard AD-related markers and compared them between groups of differing clinical status [239-241]. CSF-based markers of insulin and insulin-related molecules are alternative biomarkers that avoid the complexities of BBB transport at the expense of a more risky, invasive procedure to obtain the biofluid. CSF-derived insulin concentration has been assessed in relation to AD clinical status and AD-related biomarkers [242, 243], to responses to dietary intervention [244], and to measures of structural brain aging [245]. Concentrations of various biomarkers related to insulin or to peripheral insulin resistance have also been assessed in CSF and blood of subjects from various cohorts, including those with AD (Table 2) [243, 246-248]. Assessment of blood or CSF levels of insulin does not yield consistent results but often have contradictory trends [242, 244, 247, 249-254]. In one study, higher insulin levels in the CSF were associated with worse global cognition and higher p-tau levels, particularly in women and in ApoE4 non-carriers [242]. Although results from studies are not in agreement, several report higher adiponectin and lower leptin levels in blood drawn from subjects with MCI or AD compared to controls (Table 2). More consistently, higher concentrations of IGF-1 [252, 255-257], IGF-2 [256, 258, 259], insulin-like growth factor binding protein (IGFBP)-1 [257, 258], IGFBP-2 [246, 256-262] and IGFBP-3,4,5 [252, 256, 257] have been associated with a diagnosis of AD (Table 2). Overall, despite considerable inter-study variability, biomarker studies depict a complex metabolic signature of AD, possibly including elevated concentrations of IGFs or IGFBPs in both CSF and blood [243, 246-248]. Despite these changes in CSF and blood biomarkers, omics-based signatures of brain insulin signaling from any biofluid, have been slow to develop [263].
Table 2.
Summary of studies on insulin-related biomarkers in Alzheimer’s disease, using various assays in plasma, serum, cerebrospinal fluid, or brain homogenates.
| Cohort (n) | Higher levels in | Body fluid or tissue | Summary of main results | Additional notes | Ref. |
|---|---|---|---|---|---|
| Insulin | |||||
| NCI/MCI,AD (21/19) NINCDS/ADRDA criteria | Ctrl | CSF | NCI > MCI/AD (.005) | [251] | |
| = | Serum | ns | |||
| SCI/aMCI/AD (45/44/49) |
AD (women) |
CSF | aMCI < SCI < AD (.059) Women: Significantly different with MMSE |
Women: higher levels associated with worsen global cognition APOE4 non carriers: correlation with CSF p-tau and t-tau |
[242] |
| Non-elderly/elderly (116/96) with 65years-old as threshold, with MetS MoCA |
Plasma | Non-elderly MetS: correlation with MoCA | [344] | ||
| Ctrl/sporadic AD (60/60) neuropsychological evaluation (DSM) + MRI | AD | Serum | Ctrl < AD (<.0001) | [249] | |
| Ctrl/other dementia/stable MCI /AD (15/13/32/60) | = | Serum | ns | [252] | |
| = | CSF | ns | |||
| Ctrl/AD (12/16) Braak staging |
Ctrl | CSF | Ctrl > AD (.0009) | [247] | |
| Ctrl | Brain homogenate | B0-1 (Ctrl) > B6 (AD) (.05) | |||
| Ctrl like or AD like (29/30) | CSF | CSF Ctrl like: inverse association between WMHs and insulin CSF levels (parieto-occipital region) | [245] | ||
| Ctrl/aMCI (20/29) with 4 weeks high-SFA/glucose (HIGH) diet or a low SFA/glucose (LOW) |
AD | CSF | HIGH diet: Ctrl < MCI/AD LOW diet: Ctrl < MCI/AD |
[244] | |
| AD | Plasma | HIGH diet: Ctrl = MCI/AD LOW diet: Ctrl < MCI/AD |
|||
| Ctrl/AD (24/21) Braak staging |
Ctrl | FCx | B0-1 (Ctrl) > B2-3/B4-5/B6 (<.001) | [345] | |
| Ctrl/AD (26/28) Braak staging |
Ctrl | FCx, Hpc, Hyp | Ctrl > AD (Hyp .01; Hpc .002; ns FCx) | [346] | |
| Ctrl/AD (16/27) | = | CSF | ns | [253] | |
| Ctrl/mild AD/severe AD (25/14/11) Or Ctrl/homozE4/non-homozE4 (14/6/19) |
AD | Plasma | Ctrl, mild AD < moderate/severe AD (.05) APOE4: normal, homoz ApoE4 < non homoz E4 |
[250] | |
| Ctrl | CSF | Ctrl > mild AD > moderate/severe AD (.05) APOE4 : ns |
|||
| Ctrl/AD (26/54) |
AD | CSF | Ctrl < AD (.001) | [254] | |
| AD | Plasma (after OGTT/fasted) | Ctrl < AD (.001)(after OGTT); ns (fasted) |
|||
| Leptin | |||||
| Ctrl with neurological but not degenerative disease/AD (23/26) | = | Plasma | ns | [347] | |
| Cross-sec study 669 participants | Plasma | Associated with cognitive impairment | [348] | ||
| Ctrl/MCI,AD (21/19) NINCDS/ADRDA criteria | = | CSF | ns | [251] | |
| Ctrl | Serum | Ctrl > MCI/AD (.0002) | |||
| Ctrl/AD (25/30) Turkish MMSE, CDR, GDS |
= | Plasma | ns | [349] | |
| Ctrl/MCI with T2DM (63/61) MoCA |
Ctrl | Plasma | Women: Ctrl > MCI | Associated with MoCA (higher levels associated with better cognition) | [350] |
| NCI/MCI/AD (21/8/13) NINCDS/ADRDA criteria | AD | CSF | MCI < Ctrl < AD (.05) Women > Men |
↘ Leptin receptor in AD (CSF and hpc) | [351] |
| Ctrl/ AD (60/60) neuropsycho evaluation (DSM-IV) + MRI | Ctrl | Serum | Ctrl > AD (<.0001) | [249] | |
| Ctrl/AD (12/16) Braak staging |
= | CSF | ns | [247] | |
| AD | Brain homogenate | B0-1/B2-4 < AD (B6) (<.05) | |||
| Ctrl/AD (37/41) NINCDS-ADRDA criteria |
Ctrl | Plasma | Ctrl > AD | [352] | |
| 785 participants 111 incident dementia (89 AD) |
Ctrl | Plasma | Ctrl > dementia/AD | [353] | |
| Adiponectin | |||||
| Ctrl with neurological but not degenerative disease/AD (23/26) | = | Plasma | ns | [347] | |
| 535 non-demented elderly, with neuropsychological tests | Plasma | Women: inverse association with cognitive outcomes | [354] | ||
| Ctrl/sporadic AD (60/60) neuropsychological evaluation (DSM) + MRI | AD | Serum | Ctrl < AD (<.0001) | [249] | |
| NCI/MCI/AD (51/65/41) | MCI/AD | Serum | NCI < MCI, AD (<.001) | [355] | |
| NCI/MCI/AD (28/18/27) NINCDS-ADRD |
MCI/AD | Plasma | NCI < MCI, AD | [356] | |
| MCI | CSF | NCI < MCI (ns AD) | |||
| Ctrl/AD (37/41) NINCDS-ADRDA criteria |
= | Plasma | ns | [352] | |
| Ghrelin | |||||
| Ctrl/MCI,AD (21/19) NINCDS/ADRDA criteria | = | CSF | ns | [251] | |
| AD | Serum | Ctrl < MCI/AD (<.0001) | |||
| NCI/MCI (30/22) neuropsychological tests |
= | Serum | ns (total ghrelin) | [357] | |
| MCI | Serum | NCI < MCI (<.001) (acylated ghrelin) | |||
| Ctrl/AD (12/16) Braak staging |
Ctrl | CSF | Ctrl > AD (.005) | [247] | |
| = | Brain homogenate | ns | |||
| GIP | |||||
| Cross sectional studies, 3001 older people MMSE and AQT |
Serum (2h OGTT/fasted) | Correlation with MMSE (2h OGTT) (higher levels are associated with better cognition); ns (fasted) | [358] | ||
| Ctrl/MCI,AD (21/19) NINCDS/ADRDA criteria | MCI/AD | CSF | Ctrl < MCI/AD (.02) | [251] | |
| = | Serum | ns | |||
| Ctrl/AD (12/16) Braak staging |
= | CSF | ns | [247] | |
| mild NFT pathology | Brain homogenate | B0-1/6 < B3-4 (<.01) | |||
| GLP-1 | |||||
| Cross sectional studies, 3001 older people MMSE and AQT |
Plasma (2h OGTT/fasted) | Correlation with MMSE (2h OGTT) (higher levels are associated with better cognition); ns (fasted) | [358] | ||
| Ctrl/MCI,AD (21/19) NINCDS/ADRDA criteria | = | CSF | ns | [251] | |
| MCI/AD | Serum | Ctrl < MCI/AD (<.0001) | |||
| Ctrl/AD (12/16) Braak staging |
Ctrl | CSF | Ctrl > AD (.012) | [247] | |
| Ctrl | Brain homogenate | B0-1 (Ctrl) > B6 (AD) (.05) | |||
| IGF-1 | |||||
| Ctrl/AD (36/ 40) | CSF | Correlation with CSF t-tau, p-tau | [255] | ||
| Ctrl/other dementia/stable MCI /AD (15/13/32/60) | = | CSF | ns | [252] | |
| AD | Serum | Ctrl < AD (.01) | Inverse correlation with CSF Aβ42 | ||
| Ctrl/AD (41 total) | AD | CSF | Ctrl < AD (.0001) | [257] | |
| AD | Serum | Ctrl < AD (.0001) | |||
| Ctrl/AD (24/21) Braak staging |
Ctrl | FCx | B0-1 (Ctrl) > B4-5/B6 (<.001) | [345] | |
| Ctrl/AD (26/28) Braak staging |
Ctrl | FCx, Hpc, Hyp | Ctrl > AD (Hyp .07; FCx .006; ns Hpc) | [346] | |
| Ctrl/AD (10/10) | = | CSF | ns | [256] | |
| AD | Serum | Ctrl < AD (<.01) | |||
| IGF-2 | |||||
| Ctrl/other dementia/stable MCI/AD (20/15/13/32) | = | Serum | ns | [258] | |
| AD | CSF | Men: Con,MCI < AD | Correlation with t-tau and p-Tau | ||
| Ctrl/AD (72/92) | AD | CSF | Ctrl < AD (.005) | [259] | |
| Ctrl/AD (24/21) Braak staging |
Ctrl | FCx | B0-1 (Ctrl) > B2-3/B4-5/B6 (<.05) | [345] | |
| Ctrl/AD (26/28) Braak staging |
Ctrl | FCx, Hpc, Hyp | Ctrl > AD (Hyp .01; Hpc .04; ns FCx) | [346] | |
| Ctrl/AD (10/10) | AD | CSF | Ctrl < AD (<.01) | [256] | |
| AD | Serum | Ctrl < AD (<.01) | |||
| IGFBP-1 | |||||
| Ctrl/other dementia/stable MCI/AD (20/15/13/32) | = | CSF | ns | [258] | |
| = | serum | ns | |||
| Ctrl/AD (41 total) | AD | CSF | Ctrl < AD (.0001) | [257] | |
| AD | Serum | Ctrl < AD (.0001) | |||
| IGFBP-2 | |||||
| 1596 participants with 131 dementia cases including 98 AD cases | AD | Plasma | Associated with an increased risk of dementia and AD | [260] | |
| NCI/MCI/AD (58/197/99) NINCDS/ADRDA criteria |
= | CSF | ns | [246] | |
| MCI | Plasma | AD, NCI < MCI (<.0001) | Inverse correlation with episodic memory performance Amyloid-negative individuals (CSF Aβ42): Inverse correlation with hpc volume |
||
| Ctrl/MCI/AD (45/134/66) | Correlation with t-tau | [359] | |||
| Ctrl/other dementia/stable MCI/AD (20/15/13/32) | = | Serum | ns | [258] | |
| AD (men) |
CSF | Men: MCI < AD | Correlation with t-tau and p-Tau | ||
| Ctrl/AD (72/92) | AD | CSF | Ctrl < AD (.005) | [259] | |
| NCI/MCI/AD (211/149/331) | AD | Plasma | Associated with cognitive decline and AD diagnosis | [262] | |
| Ctrl/AD from the AIBL cohort (754/207) |
AD | Plasma | Ctrl < AD (<.0001) | [261] | |
| Ctrl/AD (8/8) Braak staging |
Ctrl | TCx | Ctrl > AD (.05) | [360] | |
| Ctrl/AD (41 participants) | AD | CSF | Ctrl < AD (.0001) | [257] | |
| AD | Serum | Ctrl < AD (.0001) | |||
| Ctrl/AD (10/10) | AD | CSF | Ctrl < AD (<.001) | [256] | |
| IGFBP-3 | |||||
| Ctrl/Other dementia/stableMCI /AD (15/13/32/60) | = | CSF | ns | (Johansson et al. 2013) | |
| sMCI/AD | Serum | Ctrl < sMCI, AD (.01) | Inverse correlation with CSF Aβ42 | ||
| Ctrl/AD (41 total) | AD | CSF | Ctrl < AD (.0001) | [257] | |
| IGFBP-4,5 | |||||
| Ctrl/AD (41 total) | AD | CSF | Ctrl < AD (.0001) | [257] | |
| IGFBP-6 | |||||
| Ctrl/AD (41 total) | AD | CSF | Ctrl < AD (.0001) | [257] | |
| Ctrl/AD (10/10) | AD | CSF | Ctrl < AD (<.001) | [256] | |
| FGF21 | |||||
| 569 participants (Ctrl/T2Dwo complications/T2Dw complications/AD (102/92/162/93/120) | = | Plasma | ns Ctrl versus AD (but higher in T2D versus AD) APOE4 status has no impact |
Correlates with age as centenarian has the highest plasma level, and BMI | [361] |
| Ctrl/MCI (39/92) MoCA |
MCI | Plasma | Ctrl < MCI (.004) | [362] | |
| Nonelderly/elderly with 65 years-old as threshold, with MetS (116/96) MoCA |
Plasma | Non-elderly MetS: Inverse correlation with MoCA (lower levels associated with better cognition) | [344] | ||
Background colors: Red cells = Higher levels observed in MCI and/or AD; Purple cells = Brain tissue instead of CSF or blood Abbreviations: (a)MCI, (amnestic) mild cognitive impairment; AD, Alzheimer’s disease; Aβ, β-amyloid; CDR, Clinical Dementia Rating; Ctrl/NCI, controle/no cognitive impairment; CSF, cerebrospinal fluid; DSM, Diagnostic and Statistical Manual of Mental Disorders; E4, apolipoprotein E4; ELISA, enzyme-linked immunosorbent assay; FCx, frontal cortex; FGF21, Fibroblast growth factor 21; GDS, Global Deterioration Scale; GIP, Glucose-dependent insulinotropic polypeptide; GLP-1, Glucagon-like peptide 1; Hpc, hippocampus; Hyp, hypothalamus; IGF, Insulin-like growth factor; IGFBP, Insulin-like growth factor binding protein; LIA, line immunoassay; MetS, metabolic syndrome; MMSE, Mini-Mental State Examination; MoCA, Montreal Cognitive Assessment; MRI, magnetic resonance imaging; NFT, neurofibrillary tangles; NINCDS-ADRD, National Institute of Neurological and Communicative Diseases and Stroke/Alzheimer's Disease and Related Disorders Association; OGTT, Oral glucose tolerance test; RIA, radioimmunoassay; SCI, subjective cognitive decline; SFA, saturated fatty acids; T2D, type 2 diabetes; TCx, temporal cortex; WMHs, white matter hyperintensities.
5. Correlation of BIR with AD hallmarks
5.1. AD Hallmarks
The Jack et al. model of the progress of pathophysiologic processes in AD was first published in 2010 and since expanded upon, describes a hypothetical model of the temporal sequence of AD biomarkers intended to be a framework for in vivo staging of AD [264, 265]. Measures of Aβ deposition and neurodegeneration were the primary markers for AD, with Aβ deposition occurring earlier, then followed by tau-mediated dysfunction, and later by the development of memory impairment. This model was quickly revised in 2012 to include advancements in amyloid imaging, MRI, and FDG PET. Most notably, the current definition of AD is the AT(N) framework, which states that a definitive diagnosis of AD must include Aβ, tau, and some aspect of neurodegeneration at autopsy [266]. A range in cognitive impairment is still last to follow these biomarkers. Since then, many have proposed modifications to the model, including adding a vascular component given most AD is accompanied by vascular abnormalities. Such an addition is pertinent to T2DM and insulin resistance, among other vascular risk factors. While we recognize many other factors may play a role in the development of AD, including neuroinflammation, oxidative stress, and changes in miRNA, we propose BIR also fits into this framework. We do not know where BIR falls in the development of AD. However, recent efforts to identify where in the course of disease progression BIR begins to develop have begun to be explored, and we suspect BIR may develop before the manifestation of cognitive impairment [41]. Further, we do not yet know whether BIR is a cause or a consequence of AD neuropathology, or a more independent factor in brain aging which relates to cognitive impairment. Unfortunately, the molecular pathways involved in BIR and the interactions with AD-related pathology are largely unknown but could provide valuable mechanistic insights that enhance our understanding of the disease process. In efforts to further define this relationship, investigators have begun to interrogate the development of BIR as it relates to AD pathology in both rodents and humans. We discuss in this section how the tools for interrogating BIR described above can aid in correlating directly to hallmarks of AD defined by the Jack et al. model.
5.2. Rodent models
As described above, animal models can be used to directly probe BIR in vivo, in association with AD biomarkers. Rodent models are particularly useful on the one hand to generate a phenotype of obesity and diabetes, and on the other hand to generate brain AD-like neuropathology, to study the causal interactions between both diseases. Mouse models of AD often display glucose impairment and insulin intolerance, exacerbated with HFD [149]. However, the correlation between BIR and AD pathology has not largely been assessed. BBB insulin interactions are impaired in the aged SAMP8 model of AD, after the presence of increased Aβ, compared to young controls [267]. INSRα-B protein levels at the BBB are significantly reduced in another AD mouse model, the 3xTg-AD mice, at 18 months of age compared WT controls, with a significant linear trend from 6 to 18 months of age [41]. Additionally, in the 3xTg-AD mouse model, insulin-induced activation of the INSR at the BBB is blunted, beginning at 6 months of age [41]. In female 3x-Tg-AD mice, pathological development of Aβ deposits, p-tau, and cognitive deficits typically appear by 6 months of age [268]. These data support the link between BIR and AD hallmarks but also suggest that BBB insulin resistance may correlate with AD hallmarks as well.
Other studies have tried to investigate the relationship of BIR with AD pathology. The A7-Tg AD mouse model deposits Aβ at approximately 12 months of age [143]. At 3 months of age, prior to changes in the insoluble levels of Aβ, an experiment placed mice on a HFD. After just 2 months on this diet, the cerebral cortex INSR response to peripherally administered insulin was blunted, but insulin BBB transport was also decreased. At 9 months of age, insoluble Aβ was significantly increased in the cerebral cortex and attributed to the HFD. These data suggest impairments in insulin BBB transport may impact brain insulin signaling and that these effects precede AD pathology. One caveat to many of the mouse models of AD is they do not typically follow the natural progression of the disease, resulting in accelerated AD neuropathology. Indeed, pre-clinical studies may not translate completely to human responses.
5.3. Post-mortem human brain tissue
Complementary investigations have been performed in post-mortem human brain tissue. In an earlier study of the post-mortem brain cortex of 17 older patients with AD, the densities of INSR were significantly increased, compared with age-matched controls [269]. Correlations to the levels of AD hallmarks in the same samples were not performed. A larger study using post-mortem examinations of the brain in AD cases demonstrated that the hippocampal formation and cerebellar cortex of patients with AD compared to age-matched controls exhibited markedly reduced responses to insulin signaling in IRS-1/IRS-2 and PI3K pathways, seemingly irrespective of T2DM status and total INSR protein level [10]. In the same study, biomarkers of brain insulin signaling, in particular IRS-1, were positively associated with increased levels of oligomeric Aβ plaques. Further, phosphorylated IRS-1 was negatively associated with cognition, including episodic memory, the cognitive domain typically affected earliest in the clinical expression of AD [10]. Similarly, leveraging post-mortem human brain tissue, evidence of BIR was also found in patients with amnestic mild cognitive impairment and early-onset AD in Down syndrome [270, 271]. Hyperphosphorylation of mTOR positively correlated with Aβ [270]. INSR and IRS1 levels positively correlated with the synapse markers, syntaxin, and PSD95, and this impairment in INSR signaling occurred early in Down syndrome before the development of AD pathology [271]. In a community-based cohort study of older persons with and without T2DM who came to an autopsy, brain Akt phosphorylation, a downstream event in the insulin signaling pathway, was associated with the level of AD neuropathology and lower cognitive function [202].
In post-mortem human brain microvessels, significant inverse correlations exist between INSRα-B levels and neuritic plaques, but not with parenchymal Aβ40 or Aβ42 [41]. Furthermore, there was a positive association between INSRβ and total soluble tau and an inverse relationship with neurofibrillary tangles. These data continue to support a relation not only between BIR and AD pathology but also between BBB insulin resistance and AD.
5.4. Biomarkers
Several studies have used brain insulin-related measurements from living humans to suggest that these biomarkers differ between cognitively normal older adults and those clinically diagnosed with AD, AD prodromal syndromes, and different neurodegenerative diseases [243, 272]. In addition, some of these studies have further assessed associations between fluid biomarkers of brain insulin signaling and biomarkers associated with AD [240]. Two studies have assessed whether chronic INL insulin administration modified AD biomarkers, with one showing null findings [212] and the other suggesting that the intervention modified CSF tau-P181/Aβ42 ratio [273]. Observational studies have suggested that various brain insulin-related measures extracted from blood or CSF are associated with MRI-based measures of neurodegeneration [240, 246, 274, 275] although the degree to which the spatial pattern of neurodegeneration is suggestive of AD is mixed. Indeed, some authors explicitly suggest that the neurodegeneration associated with brain insulin markers may be non-AD-related because findings were only evident in amyloid-negative individuals [246]. There have been two reports that brain insulin markers are associated with CSF tau burden [242, 274], and seemingly no reports that the brain insulin measures are associated with amyloid burden specifically (except for the aforementioned tau-to-amyloid ratio data in [273]). Yet, methodological heterogeneity in the literature on correlations between brain insulin markers and AD markers is high, with differing papers using CSF or blood to quantify insulin, a variety of insulin-related hormones or proteins, and exosome-derived molecules. Populations under study were also heterogeneous with respect to age, clinical status, and AD biomarker status. In addition, seemingly absent from this literature are neuroimaging-based measures of brain insulin, amyloid, and tau.
6. CNS cell specificity and regional localization
Thus far, research surrounding BIR is predominantly approached from a whole brain or individual region point of view, although advancements in interrogating the impact of insulin signaling in sub-cellular populations are evident [33]. A central question to consider is whether the brain is considered wholly an insulin-sensitive organ or whether insulin sensitivity varies across types of brain cells (e.g., neurons, astrocytes, endothelial cells) and regions, dependent on the age, disease, or stage of disease, and other factors (e.g., exposures). The very definition of BIR could also differ for each type of cell or region in the brain. However, with the recent advances in single-cell data and omics-driven data, researchers are more aware that specific cell populations may be drivers or initiators of a given pathology.
6.1. Neural cell types
As briefly introduced above in Section 4.1c, recent work on the impact of CNS cell-specific characterization of the INSR has been explored in the context of BIR. In mice that lacked the neural INSR, cognition is preserved, but there was also no change in metabolic parameters either including body weight, food intake, and adiposity [186]. This model has a lifelong loss of INSR signaling, so compensation from other signaling pathways is likely present. Additionally, this model was driven by the CaMKII promoter, which is expressed in an array of CNS cell types in the mouse including neurons, oligodendrocytes, and macrophages and is not as specific to neurons in humans as we now know [106].
Lentiviruses targeting the INSR are packaged into the human cytomegalovirus (CMV), which primarily targets neurons [276]. This knock-down model, when delivered to the hippocampus, displays impaired cognition compared to mice treated with the control lentivirus [194]. On the other hand, over-expressing the hippocampal INSR using a constitutively active INSR in aged rats enhanced memory recall [277] and neuronal glucose metabolism [195]. Adeno-associated virus 9 (AAV9) vectors are designed to primarily target neurons using the neuron-specific human synapsin-1 promoter. Neuronal-specific knock-down of the INSR using the same synapsin-1 Cre driver does not affect the mortality of the Tg2576 AD mouse model, unlike the loss of the IGF-1R [278]. The impact of knocking-down or increasing the INSR solely in the hippocampus in the presence of AD pathology remains to be determined.
Genetically targeted knock-down of the INSR can also be useful to pinpoint cell types involved. Using electrophysiological and behavioral approaches, it was recently shown that serotoninergic neurons of the raphe nuclei can display insulin resistance in HFD-fed animals [33]. Neuronal INSR signaling is not the only CNS cell type investigated in its cognitive role. An astrocyte-specific knock-down of the INSR, both constitutive and inducible, has also been generated. Genetic knock-down of the astrocytic INSR in adult mice displayed no change in cognition compared to controls [190]. However, when this genetic model was crossed with a mouse model of AD, loss of the astrocytic INSR resulted in further impaired cognition and enhanced amyloid plaque deposition, indicating a protective role of the astrocytic INSR against AD pathology and cognitive decline. Additionally, the astrocytic INSR is involved in neurovascular coupling, with decreased brain perfusion occurring in older mice [279].
6.2. BBB cell types
In order to better define the cellular localization of BIR, one has to consider the prominent role of the BBB located at the interface between the blood and the brain. Blood flow in the brain is directed through a vast network of capillaries where the BBB plays a key role in the exchange of nutrients and physiological information between the periphery and the brain. The adult human brain has roughly 400 miles of blood vessels, making it the most vascularized organ in the body [280]. This is a large surface through which the periphery interacts with the CNS, where each brain cell is irrigated by a capillary [281]. Transporters and receptors expressed by the BBB can regulate not only the transport of blood-derived compounds but also their downstream signaling.
Since most insulin is produced by the pancreas, then released into circulation, it is logical to assume that most of it will interact first with the BBB before having any impact on cells located deeper within the brain. Emerging work suggests that some insulin gets to the brain by crossing the relatively permeable choroid plexus through the blood-CSF barrier (BCSFB) [282, 283]. However, the surface area of the BBB is manifold larger than the BCSFB [280, 284] and the majority of nutrient exchange occurs at the level of the capillaries. Although considered a barrier due to its specific capacity to block the entry of small polar molecules, the BBB is actually formed by a complex nexus of cells, such as endothelial cells, pericytes, astrocytes, microglia, and neurons, forming a neurovascular unit (NVU) [285-288]. Whereas a neuron-centric perspective has prevailed in the past [65, 69, 289], the role of the BBB and the NVU in brain insulin response is now being increasingly recognized [30, 41, 42, 60, 74, 290]. Since increases in INSR phosphorylation in brain endothelial cells following intracarotid insulin administration is particularly strong compared to the parenchyma [41], it is important to consider the role of the BBB in the response. Previous work has also demonstrated that removing INSR from astrocytes or neurons impacts insulin signaling as well [90, 291-293]. These cells in culture respond to low concentrations of insulin, likely similar to those in the brain ISF [294, 295]. In summary, although a majority of INSR in the brain is found in the NVU, a significant portion is present in other cells within the parenchyma, where they can interact with low levels of insulin.
The regulation of INSR expression or post-transcriptional events has also been proposed as mechanisms leading to insulin resistance. Studies in the liver indicate that the amount of biologically active INSR at the cytoplasmic membrane can be reduced by its cleavage by the β-site amyloid precursor protein cleaving enzyme 1 (BACE1), particularly in T2DM [296]. The use of BACE1 inhibitors to enhance insulin signaling has thus been proposed [296-298]. Within the AD BBB, a strong association between INSRα-B, BACE1, and APPβ-CTF was found in microvessels, suggesting that INSRα-B is reduced along with an increase in BACE1 activity [41]. This suggests that BIR at the BBB may involve BACE1 cleavage of INSR, as was recently shown in the liver and plasma [296-298].
6.3. Regional involvement in BIR
Studies investigating the regional impact of the INSR on memory and cognition in rodents have only recently begun to be explored. Targeted knock-down of the INSR in the hippocampus impaired memory [194] while knock-down in the hypothalamus may have an indirect effect on memory through changes in whole-body metabolism [193]. These data suggest that while the INSR itself may contribute to cognition within the hippocampus, the INSR in other regions, including the hypothalamus, may indirectly be involved in mediating cognition through changes in whole-body metabolism. However, whether some regions are more vulnerable to BIR and why has not been investigated. To do this, a vast descriptive study investigating the development of BIR across brain regions as AD progresses would need to be undertaken. This could be accomplished using any of the tools described in Section 4 but would benefit most by investigating the in situ regional response to insulin stimulation.
Two separate lines of neuroimaging work have addressed the degree that brain responses to insulin, defects in brain responses to insulin, and brain byproducts of such defects, may be region-specific. First, the set of studies assessing neuroimaging responses to peripheral or INL insulin challenges naturally sought to assess whether those responses are region-specific. Among these studies, FDG PET responses to acute insulin challenges were fairly global [47, 207, 208] or null [44]. Functional and perfusion MRI responses to insulin-related challenges have been inconsistent across studies but have featured the prefrontal cortex, hypothalamus, cerebellum, amygdala, hippocampus, and occipital cortex [222, 223, 225, 226, 299]. Second, a group of studies sought to determine whether chronic brain insulin exposures or biomarkers of brain insulin signaling are associated with localized measures of brain volume from structural MRI, to investigate whether greater levels of brain insulin signaling may have long-term beneficial effects for brain tissue survival in specific regions. The bilateral parietal-occipital junction, middle temporal gyri, temporal lobe generally, and hippocampi were mentioned as regions whose volumes correlated with various brain insulin measures [239, 240, 246]; chronic INL insulin was associated with greater preservation of tissue volumes in a broad range of cortical regions including several frontal, temporal, and parietal regions that have been implicated in the pathological progression of AD [273]. Interestingly, in one study the spatial pattern of preserved brain tissue associated with elevated measures of brain insulin signaling was similar to the spatial pattern of IRS-1 expression in a standard brain atlas, consistent with the beneficial effects of brain insulin signaling localized to sites of IRS-1 action.
6.4. Discovery-driven omics approaches
Applying omics approaches for studying BIR phenomenon directly in humans is not straightforward due to the complexity of defining the phenotype. Many of the high-throughput transcriptomics and proteomics studies invariably used mouse or rat models. More recently however, metabolomics have been utilized to understand the global metabolomic changes occurring in the brains and serum of patients with AD, followed up by validation with the APP/PS1 mouse model of AD, as a means to identify new therapeutic targets [300]. The question about BIR and the effect of insulin can be posed in two corresponding ways. One way is to evaluate the differences in gene expression between control and BIR mouse models. An alternative approach is to evaluate the response to insulin treatment, most often via INL delivery. Although these are different questions, they revolve around the same phenomenon of BIR and its rescue with INL insulin. Thus, presumably omics investigations could point to important mediators.
One of the earlier studies investigated the effect of INL insulin using an aged F344 rat model [301]. Although 3-months of INL insulin treatment did not result in significant memory improvement, the effect was noted by a decrease of insulin binding (quantified using a 125I-labled insulin assay) in the thalamus but not the hippocampus. However, according to microarray data, the hippocampal gene expression pattern was altered both by aging and most importantly by INL insulin treatment. Out of the pathways upregulated by INL insulin, of particular interest are anti-inflammatory and synaptic stabilization. In addition, there is also an observation of antiproliferative and tumor suppressor pathways. This counterintuitive observation can be explained by the fact that over the long-term treatment, the hippocampus became less responsive to insulin, potentially as part of negative feedback, which is often not exhibited in the clinical setting [232, 235, 302].
An alternative animal model of BIR is the SAMP8 mouse that is prone to accelerated aging [303]. The improvements in memory of SAMP8 mice upon short-term or repeated INL insulin administration were statistically significant [304]. In the SAMP8 mouse model of AD, a single INL insulin treatment altered the expression of over 300 genes within the hippocampus, just four hours following administration [305]. The majority of these genes were involved in pathways such as T-cell receptor signaling, cell adhesion molecules, cytokine-cytokine receptor interactions, and chemokine signaling. Noteworthy, the pathways related to inflammation were altered similar to the study described above using aged rats, rather than direct changes in the insulin signaling pathway [301].
To answer the complementary question of what gene expression patterns are associated with BIR to begin with, omics approaches can be applied directly to the characterization of the hippocampus or other areas of interest directly to BIR mouse models. For example, transcriptomics analysis of the cortex of the HFD mouse models [306] showed an increased expression of genes related to immune response, such as Trem2 and Tyrobp, known to be part of the innate immune system. The down-regulated genes are related to neuron projections and synaptic transmission. Although, from this study, it isn’t clear if the impact on the neurons is the consequence of inflammation or an independent process. Yet, the theme of increased metabolism-related inflammation involving BIR is consistent with other models.
A similar pattern was observed in SAMP8 mice when compared to cognitively normal SAMR1 mice [307]. Further proteomic analysis in the same study of the hippocampus revealed a number of affected pathways. Specifically, the majority of the differentially expressed proteins are involved in the cytoskeleton and cell motility regulation. Cholinergic components involving the receptors and esterase enzymes showed a downward trend in the case of accelerated aging. However, the inflammatory cytokines were up in SAMP8 mice, consistently with the observations in other models and study designs.
Perhaps the primary reason for the hippocampus getting much attention is because of its role in AD. However, other brain components such as the BBB [30, 305] and choroid plexus, at the junction of the BCSFB [39, 308], have also been crucially implicated in insulin function in the brain, as discussed. Future discovery-type omics studies with a broader scope encompassing the brain components should be informative for understanding the roles of different regions and structures within the brain.
Several follow-up questions on BIR model experiments remain, and include the identification of the specific cell types, and which brain region(s) is playing the key role. One approach to address these questions is through deploying one of the emerging sequencing techniques that is based on isolation and sequencing of the RNA contents of the individual cell nuclei [309, 310]. Alternatively, one can make a hypothesis on the role of a particular cell type and perform an experiment on isolated primary cell culture.
Investigations of the primary murine astrocytes confirmed that to completely blunt the effect of insulin, knock-out of both INSR and IGF1R is required [311]. Regardless of the mediating receptor, the RNA-seq data from this study indicates that insulin stimulation, rather than IGF-1 stimulation, triggers the suppression of autophagy genes. At the same time, stimulation caused upregulation of the ribosomal and heat shock protein, highlighting the regulation of proteostasis. A broader effect also involved cholesterol synthesis and mitochondrial homeostasis.
An informative study using immortalized preadipocytes isolated from genetically engineered mice dissected the effect of different INSR domains on the signaling [312]. Although the source of the cell isn’t from the brain, the connection can be drawn based on the relevance of the insulin signaling between the brain and adipose tissues. The study, using a skillful combination of genetic engineering and liquid chromatography with tandem mass spectrometry (LC-MS/MS) phosphoproteomics, has demonstrated an alternative INSR signaling pathway, that is independent of the ligand and tyrosine-kinase activity, abbreviated as LYK-I. These new signaling actions of the INSR include changes in the phosphorylation of proteins involved in the regulation of extracellular matrix organization, cell cycle, and immune-related pathways. LYK-I of the INSR increases the cellular sensitivity to apoptosis. Whether changes in INSR protein expression in pathophysiological conditions, such as aging or AD, drive the LYK-I response remains to be answered.
Beyond just the study of the overall gene expression changes between control and BIR animal models or in response to INL insulin, omics techniques can play a further role in elucidating the mechanisms of pathology and treatment. As mentioned, single nucleus RNA sequencing (snRNA-seq) will be a valuable tool that can identify a specific cell type involved in pathology and/or response to INL insulin. Once the cell type is determined, the omics techniques aimed at functional interrogation of the signaling pathways or individual proteins will be of particular interest. Ideally, such studies should be conducted on primary cell cultures as this would allow more accurate control of the experiments. Similar to the aforementioned study [312], insulin treatment in combination with LC-MS/MS proteomics should reveal the exact mechanistic differences and effects associated with pathology and treatment. Isolation of individual proteins, such as INSR or IRS-1/2 adaptors, combined with LC-MS/MS would provide an in-depth characterization of the post-translational modification and binding partners dynamics. Finally, integrated multi-post-translational modification characterization approaches have been demonstrated to provide valuable information on the functional state of the proteome [313-315]. The most impactful applications are those designed to focus on a very specific question to avoid the complexity of interpretation with multi-omics studies.
7. Remaining Questions/Concepts
Many questions about insulin resistance in the brain remain. Given the current state of knowledge, we reiterate our definition of BIR: a complex condition that encompasses one or more causes for an inadequate response of the brain and cerebral vasculature INSR to insulin, including as it may relate to CNS insulin availability, CNS INSR and isoform expression, and/or downstream signaling events of the INSR in the brain. So far, data suggest that BIR correlates with cognitive impairment [10]. Why and how BIR arises remains to be elucidated and investigated. And, importantly, the functional and clinical consequences of BIR, including in aging and AD, continue to be explored.
Despite the clear link between BIR and AD, there are potential confounding factors that likely influence this relationship. Indeed, there are many factors that can lead to peripheral insulin resistance and cognitive decline including genetic predisposition, lifestyle factors, and co-morbidities. Importantly, ApoE4, which is a genetic risk factor for AD [317] and for heart disease and stroke [318, 319], has been shown to interact with BIR in the impairment of cognition [316]. Further, lifestyle factors such as exercise and diet can also impact BIR and are involved in cognitive decline, including independently from AD [320]. In a clinical study just last year, it was shown that a sedentary lifestyle, resulting in obesity, leads to BIR, and that exercise could reverse this [321]. Additionally, smoking can cause peripheral insulin resistance [322] and faster cognitive decline [323]; however, the impact on BIR is largely unknown. Other co-morbidities including vascular and sleep disorders that are associated with peripheral insulin resistance and have also been found to worsen cognitive decline. For example, white matter hyperintensities on brain MRI scans are often attributed to underlying cerebral small vessel disease, are known to be associated with cognitive impairment, and found to be increased in persons with peripheral insulin resistance found on brain PET scans [324]. Furthermore, INI reduces white matter hyperintensity progression and improves cognition [231]. Sleep dysfunction is well documented to negatively impact insulin sensitivity and metabolism [325-327]. Sleep dysfunction is also associated with AD, leading to Aβ accumulation [328-331], but the relationship with BIR is unknown. These and other confounding factors highlight the complexity of the BIR-AD relationship. Furthering our understanding on the mechanisms between BIR (and not just peripheral insulin resistance), cognitive decline, and AD pathology will help us more fully identify these interactions.
One important knowledge gap regarding the association between BIR and dementia attributed to AD or otherwise involves resilience, which can be broadly defined as a successful adaptation, adjustment, and compensation process that occurs despite exposure to significant adverse factors including pathology [332]. Indeed, T2DM and insulin resistance can substantially increase the risk of dementia. Yet, there are older people who have T2DM or insulin resistance but do not develop cognitive impairment during their lifetime. In line with this clinical observation, a pre-clinical study using brain/neuron-specific insulin receptor knockout mice (NIRKO) found that rodents with BIR exhibited intact short- and long-term learning and memory despite having significantly increased levels of p-tau protein, a hallmark of neurodegenerative diseases [293]. While in an animal model, this finding suggests that BIR alone may not be sufficient for the development of dementia, and cognitive resilience may be at play in engaging compensatory mechanisms. Although the exact mechanisms are still waiting to be elucidated, emerging studies show that both genetic factors (e.g., a resilience gene along the bile acid metabolism pathway) and non-genetic factors (e.g., physical and cognitive activities and social connectedness, among others) may contribute to cognitive resilience [333-338]. Conversely, it should also be noted that not all people with dementia develop BIR. The underlying mechanisms also largely remain unknown, but may critically involve modifiable lifestyle behaviors such as exercise and a healthy diet, which have been associated with brain insulin sensitivity in animal studies [339, 340]. For example, by combining INL administration of insulin with functional MRI of the brain, a recently published clinical trial further demonstrated that an 8-week exercise intervention could restore brain insulin sensitivity in sedentary adults who are overweight or obese [321]. Further, studies on subjects who have undergone bariatric surgery might also provide some answers. Besides improving insulin resistance, Roux-en-Y gastric bypass was recently shown to decrease cerebral glucose metabolic rate and improve cognition [341]. This finding suggests that BIR is not a fixed trait but is modifiable and potentially a treatable state.
Improving our tools to study BIR in living humans will aid our understanding of how BIR relates to AD. Generating a PET ligand that is sensitive enough to detect changes in insulin BBB transport and visualize regional access to insulin in vivo would be one advancement. Further enhancing the sensitivity of some of the current biomarkers, present in both CSF and blood, including extracellular vesicles displaying markers for BIR, would also aid in tracking the progression of BIR alongside other pathological hallmarks of AD. The advancement of these tools would allow us to identify whether ameliorating BIR has any impact on the clinical progression of AD or impairments in cognition, which has been shown repeatedly in pre-clinical studies. Moreover, using a combination of tools, mentioned here and novel tools being developed, offer the opportunity to understand at a more granular level what processes are involved in BIR. And, studies are needed that use different approaches, from cell culture to animal models and human studies, including human studies of diverse populations that are clinic- and community-based, and observational and interventional (clinical trials). Given the large and growing public health challenge of dementia attributed to AD, research needs to focus on potentially modifiable factors such as BIR. While research has come a long way in this regard, much work remains to identify plausible therapeutic targets in BIR, for the treatment and prevention of cognitive decline and dementia.
Acknowledgements
This work was funded by a Supplement from the United States National Institute of Health (NIH)-funded Diabetes Research Center at the University of Washington P30 DK017047-44 (to E.M.R.), as well as NIH grants RF1 AG059621 and RF1 AG074549 (to Z.A.), and a Canadian CIHR grant PJT 168927 (to F. C.). F.C. was a Fonds de Recherche du Quebec-Sante (FRQ-S) research scholar and M.L. was supported by a scholarship from the FRQ-S. The work was also funded by the LIA OptiNutriBrain (X.F., F.C.) and the RRI Food4BrainHealth (X.F. and F.C.). The APC was covered by X grant (Z.A.).
Parts of the figures were drawn by using pictures from Servier Medical Art as noted in the figure legends. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/). Biorender was also used to create one of the figures.
Funding Statement
This work was funded by a Supplement from the United States National Institute of Health (NIH)-funded Diabetes Research Center at the University of Washington P30 DK017047-44 (to E.M.R.), as well as NIH grants RF1 AG059621 and RF1 AG074549 (to Z.A.), and a Canadian CIHR grant PJT 168927 (to F. C.). F.C. was a Fonds de Recherche du Quebec-Sante (FRQ-S) research scholar and M.L. was supported by a scholarship from the FRQ-S. The work was also funded by the LIA OptiNutriBrain (X.F., F.C.) and the RRI Food4BrainHealth (X.F. and F.C.). The APC was covered by X grant (Z.A.).
References
- [1].Collaborators GBDDF (2022). Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the Global Burden of Disease Study 2019. Lancet Public Health, 7:e105-e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, et al. (2019). Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res Clin Pract, 157:107843. [DOI] [PubMed] [Google Scholar]
- [3].Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA (2004). Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol, 61:661-666. [DOI] [PubMed] [Google Scholar]
- [4].Livingston G, Huntley J, Sommerlad A, Ames D, Ballard C, Banerjee S, et al. (2020). Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet, 396:413-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sato N, Morishita R (2014). Brain alterations and clinical symptoms of dementia in diabetes: abeta/tau-dependent and independent mechanisms. Front Endocrinol (Lausanne), 5:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zheng B, Su B, Udeh-Momoh C, Price G, Tzoulaki I, Vamos EP, et al. (2022). Associations of Cardiovascular and Non-Cardiovascular Comorbidities with Dementia Risk in Patients with Diabetes: Results from a Large UK Cohort Study. J Prev Alzheimers Dis, 9:86-91. [DOI] [PubMed] [Google Scholar]
- [7].Patel VN, Chorawala MR, Shah MB, Shah KC, Dave BP, Shah MP, et al. (2022). Emerging Pathophysiological Mechanisms Linking Diabetes Mellitus and Alzheimer's Disease: An Old Wine in a New Bottle. J Alzheimers Dis Rep, 6:349-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Watson GS, Craft S (2006). Insulin resistance, inflammation, and cognition in Alzheimer's Disease: lessons for multiple sclerosis. J Neurol Sci, 245:21-33. [DOI] [PubMed] [Google Scholar]
- [9].Arnold SE, Arvanitakis Z, Macauley-Rambach SL, Koenig AM, Wang HY, Ahima RS, et al. (2018). Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol, 14:168-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, et al. (2012). Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest, 122:1316-1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Crane PK, Walker R, Hubbard RA, Li G, Nathan DM, Zheng H, et al. (2013). Glucose levels and risk of dementia. N Engl J Med, 369:540-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wallace TM, Levy JC, Matthews DR (2004). Use and abuse of HOMA modeling. Diabetes Care, 27:1487-1495. [DOI] [PubMed] [Google Scholar]
- [13].Laws SM, Gaskin S, Woodfield A, Srikanth V, Bruce D, Fraser PE, et al. (2017). Insulin resistance is associated with reductions in specific cognitive domains and increases in CSF tau in cognitively normal adults. Sci Rep, 7:9766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Willette AA, Bendlin BB, Starks EJ, Birdsill AC, Johnson SC, Christian BT, et al. (2015). Association of Insulin Resistance With Cerebral Glucose Uptake in Late Middle-Aged Adults at Risk for Alzheimer Disease. JAMA Neurol, 72:1013-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Small GW, Ercoli LM, Silverman DH, Huang SC, Komo S, Bookheimer SY, et al. (2000). Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer's disease. Proc Natl Acad Sci U S A, 97:6037-6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Woodfield A, Porter T, Gilani I, Noordin S, Li QX, Collins S, et al. (2022). Insulin resistance, cognition and Alzheimer's disease biomarkers: Evidence that CSF Abeta42 moderates the association between insulin resistance and increased CSF tau levels. Neurobiol Aging, 114:38-48. [DOI] [PubMed] [Google Scholar]
- [17].Craft S, Raman R, Chow TW, Rafii MS, Sun CK, Rissman RA, et al. (2020). Safety, Efficacy, and Feasibility of Intranasal Insulin for the Treatment of Mild Cognitive Impairment and Alzheimer Disease Dementia: A Randomized Clinical Trial. JAMA Neurol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Femminella GD, Frangou E, Love SB, Busza G, Holmes C, Ritchie C, et al. (2019). Evaluating the effects of the novel GLP-1 analogue liraglutide in Alzheimer's disease: study protocol for a randomised controlled trial (ELAD study). Trials, 20:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Moran C, Callisaya ML, Srikanth V, Arvanitakis Z (2019). Diabetes Therapies for Dementia. Curr Neurol Neurosci Rep, 19:58. [DOI] [PubMed] [Google Scholar]
- [20].Luchsinger JA, Perez T, Chang H, Mehta P, Steffener J, Pradabhan G, et al. (2016). Metformin in Amnestic Mild Cognitive Impairment: Results of a Pilot Randomized Placebo Controlled Clinical Trial. J Alzheimers Dis, 51:501-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cummings J, Ortiz A, Castellino J, Kinney J (2022). Diabetes: Risk factor and translational therapeutic implications for Alzheimer's disease. Eur J Neurosci, 56:5727-5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Cummings J, Lee G, Nahed P, Kambar M, Zhong K, Fonseca J, et al. (2022). Alzheimer's disease drug development pipeline: 2022. Alzheimers Dement (N Y), 8:e12295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Nowell J, Blunt E, Edison P (2023). Incretin and insulin signaling as novel therapeutic targets for Alzheimer's and Parkinson's disease. Mol Psychiatry, 28:217-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhou Y, Chi J, Lv W, Wang Y (2021). Obesity and diabetes as high-risk factors for severe coronavirus disease 2019 (Covid-19). Diabetes Metab Res Rev, 37:e3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liu N, Sun J, Wang X, Zhao M, Huang Q, Li H (2020). The Impact of Dementia on the Clinical Outcome of COVID-19: A Systematic Review and Meta-Analysis. J Alzheimers Dis, 78:1775-1782. [DOI] [PubMed] [Google Scholar]
- [26].Ssentongo P, Zhang Y, Witmer L, Chinchilli VM, Ba DM (2022). Association of COVID-19 with diabetes: a systematic review and meta-analysis. Sci Rep, 12:20191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Taquet M, Luciano S, Geddes JR, Harrison PJ (2021). Bidirectional associations between COVID-19 and psychiatric disorder: retrospective cohort studies of 62 354 COVID-19 cases in the USA. Lancet Psychiatry, 8:130-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Rubino F, Amiel SA, Zimmet P, Alberti G, Bornstein S, Eckel RH, et al. (2020). New-Onset Diabetes in Covid-19. N Engl J Med, 383:789-790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chen W, Cai W, Hoover B, Kahn CR (2022). Insulin action in the brain: cell types, circuits, and diseases. Trends Neurosci, 45:384-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Rhea EM, Banks WA (2019). Role of the Blood-Brain Barrier in Central Nervous System Insulin Resistance. Frontiers in Neuroscience, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ferrario CR, Reagan LP (2018). Insulin-mediated synaptic plasticity in the CNS: Anatomical, functional and temporal contexts. Neuropharmacology, 136:182-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Reagan LP, Cowan HB, Woodruff JL, Piroli GG, Erichsen JM, Evans AN, et al. (2021). Hippocampal-specific insulin resistance elicits behavioral despair and hippocampal dendritic atrophy. Neurobiol Stress, 15:100354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Martin H, Bullich S, Martinat M, Chataigner M, Di Miceli M, Simon V, et al. (2022). Insulin modulates emotional behavior through a serotonin-dependent mechanism. Mol Psychiatry. [DOI] [PubMed] [Google Scholar]
- [34].Kleinridders A, Cai W, Cappellucci L, Ghazarian A, Collins WR, Vienberg SG, et al. (2015). Insulin resistance in brain alters dopamine turnover and causes behavioral disorders. Proc Natl Acad Sci U S A, 112:3463-3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kullmann S, Heni M, Veit R, Scheffler K, Machann J, Haring HU, et al. (2017). Intranasal insulin enhances brain functional connectivity mediating the relationship between adiposity and subjective feeling of hunger. Sci Rep, 7:1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kullmann S, Heni M, Fritsche A, Preissl H (2015). Insulin Action in the Human Brain: Evidence from Neuroimaging Studies. Journal of Neuroendocrinology, 27:419-423. [DOI] [PubMed] [Google Scholar]
- [37].Milstein JL, Ferris HA (2021). The brain as an insulin-sensitive metabolic organ. Mol Metab, 52:101234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Devaskar SU, Giddings SJ, Rajakumar PA, Carnaghi LR, Menon RK, Zahm DS (1994). Insulin gene expression and insulin synthesis in mammalian neuronal cells. J Biol Chem, 269:8445-8454. [PubMed] [Google Scholar]
- [39].Mazucanti CH, Liu QR, Lang D, Huang N, O'Connell JF, Camandola S, et al. (2019). Release of insulin produced by the choroid plexis is regulated by serotonergic signaling. JCI Insight, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Rhea EM, Banks WA, Raber J (2022). Insulin Resistance in Peripheral Tissues and the Brain: A Tale of Two Sites. Biomedicines, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Leclerc M, Bourassa P, Tremblay C, Caron V, Sugere C, Emond V, et al. (2022). Cerebrovascular insulin receptors are defective in Alzheimer's disease. Brain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Konishi M, Sakaguchi M, Lockhart SM, Cai W, Li ME, Homan EP, et al. (2017). Endothelial insulin receptors differentially control insulin signaling kinetics in peripheral tissues and brain of mice. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chih CP, Roberts EL Jr. (2003). Energy substrates for neurons during neural activity: a critical review of the astrocyte-neuron lactate shuttle hypothesis. J Cereb Blood Flow Metab, 23:1263-1281. [DOI] [PubMed] [Google Scholar]
- [44].Hasselbalch SG, Knudsen GM, Videbaek C, Pinborg LH, Schmidt JF, Holm S, et al. (1999). No effect of insulin on glucose blood-brain barrier transport and cerebral metabolism in humans. Diabetes, 48:1915-1921. [DOI] [PubMed] [Google Scholar]
- [45].Seaquist ER, Damberg GS, Tkac I, Gruetter R (2001). The effect of insulin on in vivo cerebral glucose concentrations and rates of glucose transport/metabolism in humans. Diabetes, 50:2203-2209. [DOI] [PubMed] [Google Scholar]
- [46].Gruetter R, Ugurbil K, Seaquist ER (1998). Steady-state cerebral glucose concentrations and transport in the human brain. J Neurochem, 70:397-408. [DOI] [PubMed] [Google Scholar]
- [47].Hirvonen J, Virtanen KA, Nummenmaa L, Hannukainen JC, Honka MJ, Bucci M, et al. (2011). Effects of Insulin on Brain Glucose Metabolism in Impaired Glucose Tolerance. Diabetes, 60:443-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Craft S, Newcomer J, Kanne S, Dagogo-Jack S, Cryer P, Sheline Y, et al. (1996). Memory improvement following induced hyperinsulinemia in Alzheimer's disease. Neurobiol Aging, 17:123-130. [DOI] [PubMed] [Google Scholar]
- [49].Cunnane SC, Trushina E, Morland C, Prigione A, Casadesus G, Andrews ZB, et al. (2020). Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nat Rev Drug Discov, 19:609-633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Weiss MA, Lawrence MC (2018). A thing of beauty: Structure and function of insulin's "aromatic triplet". Diabetes Obes Metab, 20 Suppl 2:51-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Belfiore A, Malaguarnera R, Vella V, Lawrence MC, Sciacca L, Frasca F, et al. (2017). Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr Rev, 38:379-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lee J, Pilch PF (1994). The insulin receptor: structure, function, and signaling. Am J Physiol, 266:C319-334. [DOI] [PubMed] [Google Scholar]
- [53].White MF, Kahn CR (2021). Insulin action at a molecular level - 100 years of progress. Mol Metab, 52:101304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Knudsen L, Hansen BF, Jensen P, Pedersen TA, Vestergaard K, Schaffer L, et al. (2012). Agonism and antagonism at the insulin receptor. PLoS One, 7:e51972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ottensmeyer FP, Beniac DR, Luo RZ, Yip CC (2000). Mechanism of transmembrane signaling: insulin binding and the insulin receptor. Biochemistry, 39:12103-12112. [DOI] [PubMed] [Google Scholar]
- [56].Haeusler RA, McGraw TE, Accili D (2018). Biochemical and cellular properties of insulin receptor signalling. Nat Rev Mol Cell Biol, 19:31-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Saltiel AR, Kahn CR (2001). Insulin signalling and the regulation of glucose and lipid metabolism. Nature, 414:799-806. [DOI] [PubMed] [Google Scholar]
- [58].Boucher J, Kleinridders A, Kahn CR (2014). Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ferreira ST (2021). Brain insulin, insulin-like growth factor 1 and glucagon-like peptide 1 signalling in Alzheimer's disease. J Neuroendocrinol, 33:e12959. [DOI] [PubMed] [Google Scholar]
- [60].Rhea EM, Banks WA (2021). A historical perspective on the interactions of insulin at the blood-brain barrier. J Neuroendocrinol, 33:e12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Dore S, Kar S, Rowe W, Quirion R (1997). Distribution and levels of [125I]IGF-I, [125I]IGF-II and [125I]insulin receptor binding sites in the hippocampus of aged memory-unimpaired and -impaired rats. Neuroscience, 80:1033-1040. [DOI] [PubMed] [Google Scholar]
- [62].Baskin DG, Brewitt B, Davidson DA, Corp E, Paquette T, Figlewicz DP, et al. (1986). Quantitative autoradiographic evidence for insulin receptors in the choroid plexus of the rat brain. Diabetes, 35:246-249. [DOI] [PubMed] [Google Scholar]
- [63].Baskin DG, Porte D Jr., Guest K, Dorsa DM (1983). Regional concentrations of insulin in the rat brain. Endocrinology, 112:898-903. [DOI] [PubMed] [Google Scholar]
- [64].Kar S, Chabot JG, Quirion R (1993). Quantitative autoradiographic localization of [125I]insulin-like growth factor I, [125I]insulin-like growth factor II, and [125I]insulin receptor binding sites in developing and adult rat brain. J Comp Neurol, 333:375-397. [DOI] [PubMed] [Google Scholar]
- [65].Heidenreich KA (1991). Insulin in the brain what is its role? Trends Endocrinol Metab, 2:9-12. [DOI] [PubMed] [Google Scholar]
- [66].Haskell JF, Meezan E, Pillion DJ (1985). Identification of the insulin receptor of cerebral microvessels. Am J Physiol, 248:E115-125. [DOI] [PubMed] [Google Scholar]
- [67].Frolich L, Blum-Degen D, Bernstein HG, Engelsberger S, Humrich J, Laufer S, et al. (1998). Brain insulin and insulin receptors in aging and sporadic Alzheimer's disease. J Neural Transm (Vienna), 105:423-438. [DOI] [PubMed] [Google Scholar]
- [68].Unger J, McNeill TH, Moxley RT 3rd, White M, Moss A, Livingston JN (1989). Distribution of insulin receptor-like immunoreactivity in the rat forebrain. Neuroscience, 31:143-157. [DOI] [PubMed] [Google Scholar]
- [69].Moss AM, Unger JW, Moxley RT, Livingston JN (1990). Location of phosphotyrosine-containing proteins by immunocytochemistry in the rat forebrain corresponds to the distribution of the insulin receptor. Proc Natl Acad Sci U S A, 87:4453-4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Zhang W, Liu QY, Haqqani AS, Leclerc S, Liu Z, Fauteux F, et al. (2020). Differential expression of receptors mediating receptor-mediated transcytosis (RMT) in brain microvessels, brain parenchyma and peripheral tissues of the mouse and the human. Fluids and Barriers of the CNS, 17:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Vanlandewijck M, He L, Mae MA, Andrae J, Ando K, Del Gaudio F, et al. (2018). A molecular atlas of cell types and zonation in the brain vasculature. Nature, 554:475-480. [DOI] [PubMed] [Google Scholar]
- [72].Yang AC, Vest RT, Kern F, Lee DP, Agam M, Maat CA, et al. (2022). A human brain vascular atlas reveals diverse mediators of Alzheimer's risk. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].He L, Vanlandewijck M, Mäe MA, Andrae J, Ando K, Del Gaudio F, et al. (2018). Single-cell RNA sequencing of mouse brain and lung vascular and vessel-associated cell types. Sci Data, 5:180160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Vicent D, Ilany J, Kondo T, Naruse K, Fisher SJ, Kisanuki YY, et al. (2003). The role of endothelial insulin signaling in the regulation of vascular tone and insulin resistance. J Clin Invest, 111:1373-1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Rhea EM, Rask-Madsen C, Banks WA (2018). Insulin transport across the blood-brain barrier can occur independently of the insulin receptor. J Physiol, 596:4753-4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Diaz-Castroverde S, Baos S, Luque M, Di Scala M, González-Aseguinolaza G, Gómez-Hernández A, et al. (2016). Prevalent role of the insulin receptor isoform A in the regulation of hepatic glycogen metabolism in hepatocytes and in mice. Diabetologia, 59:2702-2710. [DOI] [PubMed] [Google Scholar]
- [77].Lopez-Pastor AR, Gomez-Hernandez A, Diaz-Castroverde S, Gonzalez-Aseguinolaza G, Gonzalez-Rodriguez A, Garcia G, et al. (2019). Liver-specific insulin receptor isoform A expression enhances hepatic glucose uptake and ameliorates liver steatosis in a mouse model of diet-induced obesity. Dis Model Mech, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Mosthaf L, Grako K, Dull TJ, Coussens L, Ullrich A, McClain DA (1990). Functionally distinct insulin receptors generated by tissue-specific alternative splicing. Embo j, 9:2409-2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Moller DE, Yokota A, Caro JF, Flier JS (1989). Tissue-specific expression of two alternatively spliced insulin receptor mRNAs in man. Mol Endocrinol, 3:1263-1269. [DOI] [PubMed] [Google Scholar]
- [80].Frasca F, Pandini G, Scalia P, Sciacca L, Mineo R, Costantino A, et al. (1999). Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol, 19:3278-3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R (2009). Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev, 30:586-623. [DOI] [PubMed] [Google Scholar]
- [82].Sesti G, Tullio AN, D'Alfonso R, Napolitano ML, Marini MA, Borboni P, et al. (1994). Tissue-specific expression of two alternatively spliced isoforms of the human insulin receptor protein. Acta Diabetol, 31:59-65. [DOI] [PubMed] [Google Scholar]
- [83].Benecke H, Flier JS, Moller DE (1992). Alternatively spliced variants of the insulin receptor protein. Expression in normal and diabetic human tissues. J Clin Invest, 89:2066-2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Pandini G, Frasca F, Mineo R, Sciacca L, Vigneri R, Belfiore A (2002). Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J Biol Chem, 277:39684-39695. [DOI] [PubMed] [Google Scholar]
- [85].Benyoucef S, Surinya KH, Hadaschik D, Siddle K (2007). Characterization of insulin/IGF hybrid receptors: contributions of the insulin receptor L2 and Fn1 domains and the alternatively spliced exon 11 sequence to ligand binding and receptor activation. Biochem J, 403:603-613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Escribano O, Beneit N, Rubio-Longás C, López-Pastor AR, Gómez-Hernández A (2017). The Role of Insulin Receptor Isoforms in Diabetes and Its Metabolic and Vascular Complications. J Diabetes Res, 2017:1403206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Besic V, Shi H, Stubbs RS, Hayes MT (2015). Aberrant liver insulin receptor isoform a expression normalises with remission of type 2 diabetes after gastric bypass surgery. PLoS One, 10:e0119270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Kaminska D, Hamalainen M, Cederberg H, Kakela P, Venesmaa S, Miettinen P, et al. (2014). Adipose tissue INSR splicing in humans associates with fasting insulin level and is regulated by weight loss. Diabetologia, 57:347-351. [DOI] [PubMed] [Google Scholar]
- [89].Hughes TM, Craft S (2016). The role of insulin in the vascular contributions to age-related dementia. Biochim Biophys Acta, 1862:983-991. [DOI] [PubMed] [Google Scholar]
- [90].Garwood CJ, Ratcliffe LE, Morgan SV, Simpson JE, Owens H, Vazquez-Villasenor I, et al. (2015). Insulin and IGF1 signalling pathways in human astrocytes in vitro and in vivo; characterisation, subcellular localisation and modulation of the receptors. Mol Brain, 8:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Spencer B, Rank L, Metcalf J, Desplats P (2018). Identification of Insulin Receptor Splice Variant B in Neurons by in situ Detection in Human Brain Samples. Sci Rep, 8:4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Joost HG (1995). Structural and functional heterogeneity of insulin receptors. Cell Signal, 7:85-91. [DOI] [PubMed] [Google Scholar]
- [93].Vienberg SG, Bouman SD, Sørensen H, Stidsen CE, Kjeldsen T, Glendorf T, et al. (2011). Receptor-isoform-selective insulin analogues give tissue-preferential effects. Biochem J, 440:301-308. [DOI] [PubMed] [Google Scholar]
- [94].Páníková T, Mitrová K, Halamová T, Mrzílková K, Pícha J, Chrudinová M, et al. (2021). Insulin Analogues with Altered Insulin Receptor Isoform Binding Specificities and Enhanced Aggregation Stabilities. J Med Chem, 64:14848-14859. [DOI] [PubMed] [Google Scholar]
- [95].Wilcox G (2005). Insulin and insulin resistance. Clin Biochem Rev, 26:19-39. [PMC free article] [PubMed] [Google Scholar]
- [96].Librizzi F, Rischel C (2005). The kinetic behavior of insulin fibrillation is determined by heterogeneous nucleation pathways. Protein Sci, 14:3129-3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Wilhelm KR, Yanamandra K, Gruden MA, Zamotin V, Malisauskas M, Casaite V, et al. (2007). Immune reactivity towards insulin, its amyloid and protein S100B in blood sera of Parkinson's disease patients. Eur J Neurol, 14:327-334. [DOI] [PubMed] [Google Scholar]
- [98].Menting JG, Yang Y, Chan SJ, Phillips NB, Smith BJ, Whittaker J, et al. (2014). Protective hinge in insulin opens to enable its receptor engagement. Proc Natl Acad Sci U S A, 111:E3395-3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Menting JG, Whittaker J, Margetts MB, Whittaker LJ, Kong GK, Smith BJ, et al. (2013). How insulin engages its primary binding site on the insulin receptor. Nature, 493:241-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Nielsen L, Frokjaer S, Brange J, Uversky VN, Fink AL (2001). Probing the mechanism of insulin fibril formation with insulin mutants. Biochemistry, 40:8397-8409. [DOI] [PubMed] [Google Scholar]
- [101].Das A, Shah M, Saraogi I (2022). Molecular Aspects of Insulin Aggregation and Various Therapeutic Interventions. ACS Bio & Med Chem Au, 2:205-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Zakova L, Kletvikova E, Lepsik M, Collinsova M, Watson CJ, Turkenburg JP, et al. (2014). Human insulin analogues modified at the B26 site reveal a hormone conformation that is undetected in the receptor complex. Acta Crystallogr D Biol Crystallogr, 70:2765-2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Miller BC, Eckman EA, Sambamurti K, Dobbs N, Chow KM, Eckman CB, et al. (2003). Amyloid-beta peptide levels in brain are inversely correlated with insulysin activity levels in vivo. Proc Natl Acad Sci U S A, 100:6221-6226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, et al. (2003). Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A, 100:4162-4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. (2015). Proteomics. Tissue-based map of the human proteome. Science, 347:1260419. [DOI] [PubMed] [Google Scholar]
- [106].Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci, 34:11929-11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Ito S, Ohtsuki S, Murata S, Katsukura Y, Suzuki H, Funaki M, et al. (2014). Involvement of insulin-degrading enzyme in insulin- and atrial natriuretic peptide-sensitive internalization of amyloid-beta peptide in mouse brain capillary endothelial cells. J Alzheimers Dis, 38:185-200. [DOI] [PubMed] [Google Scholar]
- [108].Matsumoto K, Chiba Y, Fujihara R, Kubo H, Sakamoto H, Ueno M (2015). Immunohistochemical analysis of transporters related to clearance of amyloid-beta peptides through blood-cerebrospinal fluid barrier in human brain. Histochem Cell Biol, 144:597-611. [DOI] [PubMed] [Google Scholar]
- [109].Banks WA, Owen JB, Erickson MA (2012). Insulin in the brain: there and back again. Pharmacol Ther, 136:82-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Krüger M, Kratchmarova I, Blagoev B, Tseng YH, Kahn CR, Mann M (2008). Dissection of the insulin signaling pathway via quantitative phosphoproteomics. Proc Natl Acad Sci U S A, 105:2451-2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Humphrey SJ, Azimifar SB, Mann M (2015). High-throughput phosphoproteomics reveals in vivo insulin signaling dynamics. Nat Biotechnol, 33:990-995. [DOI] [PubMed] [Google Scholar]
- [112].Arrington JV, Hsu CC, Elder SG, Andy Tao W (2017). Recent advances in phosphoproteomics and application to neurological diseases. Analyst, 142:4373-4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Tsuchiya K, Accili D (2013). Liver sinusoidal endothelial cells link hyperinsulinemia to hepatic insulin resistance. Diabetes, 62:1478-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Kubota T, Kubota N, Kumagai H, Yamaguchi S, Kozono H, Takahashi T, et al. (2011). Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell Metab, 13:294-307. [DOI] [PubMed] [Google Scholar]
- [115].Vandal M, White PJ, Tremblay C, St-Amour I, Chevrier G, Emond V, et al. (2014). Insulin reverses the high-fat diet-induced increase in brain Abeta and improves memory in an animal model of Alzheimer disease. Diabetes, 63:4291-4301. [DOI] [PubMed] [Google Scholar]
- [116].Sajan M, Hansen B, Ivey R 3rd, Sajan J, Ari C, Song S, et al. (2016). Brain Insulin Signaling Is Increased in Insulin-Resistant States and Decreases in FOXOs and PGC-1α and Increases in Aβ1-40/42 and Phospho-Tau May Abet Alzheimer Development. Diabetes, 65:1892-1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Traversy MT, Vandal M, Tremblay C, Tournissac M, Giguere-Rancourt A, Bennett AD, et al. (2017). Altered cerebral insulin response in transgenic mice expressing the epsilon-4 allele of the human apolipoprotein E gene. Psychoneuroendocrinology, 77:203-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Escribano O, Guillén C, Nevado C, Gómez-Hernández A, Kahn CR, Benito M (2009). Beta-Cell hyperplasia induced by hepatic insulin resistance: role of a liver-pancreas endocrine axis through insulin receptor A isoform. Diabetes, 58:820-828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Jiang Y, Li L, Dai CL, Zhou R, Gong CX, Iqbal K, et al. (2020). Effect of Peripheral Insulin Administration on Phosphorylation of Tau in the Brain. J Alzheimers Dis, 75:1377-1390. [DOI] [PubMed] [Google Scholar]
- [120].Sartorius T, Peter A, Heni M, Maetzler W, Fritsche A, Haring HU, et al. (2015). The brain response to peripheral insulin declines with age: a contribution of the blood-brain barrier? PLoS One, 10:e0126804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Freude S, Plum L, Schnitker J, Leeser U, Udelhoven M, Krone W, et al. (2005). Peripheral Hyperinsulinemia Promotes Tau Phosphorylation In Vivo. Diabetes, 54:3343-3348. [DOI] [PubMed] [Google Scholar]
- [122].Dummler B, Tschopp O, Hynx D, Yang Z-Z, Dirnhofer S, Hemmings BA (2006). Life with a Single Isoform of Akt: Mice Lacking Akt2 and Akt3 Are Viable but Display Impaired Glucose Homeostasis and Growth Deficiencies. Molecular and Cellular Biology, 26:8042-8051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Stanley M, Macauley SL, Caesar EE, Koscal LJ, Moritz W, Robinson GO, et al. (2016). The Effects of Peripheral and Central High Insulin on Brain Insulin Signaling and Amyloid-beta in Young and Old APP/PS1 Mice. J Neurosci, 36:11704-11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Haas CB, Kalinine E, Zimmer ER, Hansel G, Brochier AW, Oses JP, et al. (2016). Brain Insulin Administration Triggers Distinct Cognitive and Neurotrophic Responses in Young and Aged Rats. Mol Neurobiol, 53:5807-5817. [DOI] [PubMed] [Google Scholar]
- [125].Saneyasu T, Ueno M, Nagata K, Kewan A, Honda K, Kamisoyama H (2021). Central administration of insulin and refeeding lead to Akt and ERK phosphorylation in the chicken medulla. Neurosci Lett, 758:136008. [DOI] [PubMed] [Google Scholar]
- [126].Muller AP, Gnoatto J, Moreira JD, Zimmer ER, Haas CB, Lulhier F, et al. (2011). Exercise increases insulin signaling in the hippocampus: physiological effects and pharmacological impact of intracerebroventricular insulin administration in mice. Hippocampus, 21:1082-1092. [DOI] [PubMed] [Google Scholar]
- [127].McNay EC, Ong CT, McCrimmon RJ, Cresswell J, Bogan JS, Sherwin RS (2010). Hippocampal memory processes are modulated by insulin and high-fat-induced insulin resistance. Neurobiol Learn Mem, 93:546-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Grillo CA, Piroli GG, Hendry RM, Reagan LP (2009). Insulin-stimulated translocation of GLUT4 to the plasma membrane in rat hippocampus is PI3-kinase dependent. Brain Res, 1296:35-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Clodfelder-Miller B, De Sarno P, Zmijewska AA, Song L, Jope RS (2005). Physiological and pathological changes in glucose regulate brain Akt and glycogen synthase kinase-3. J Biol Chem, 280:39723-39731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Dodd GT, Michael NJ, Lee-Young RS, Mangiafico SP, Pryor JT, Munder AC, et al. (2018). Insulin regulates POMC neuronal plasticity to control glucose metabolism. Elife, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Williams KW, Margatho LO, Lee CE, Choi M, Lee S, Scott MM, et al. (2010). Segregation of acute leptin and insulin effects in distinct populations of arcuate proopiomelanocortin neurons. J Neurosci, 30:2472-2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Qiu J, Zhang C, Borgquist A, Nestor CC, Smith AW, Bosch MA, et al. (2014). Insulin excites anorexigenic proopiomelanocortin neurons via activation of canonical transient receptor potential channels. Cell Metab, 19:682-693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Benoit SC, Air EL, Coolen LM, Strauss R, Jackman A, Clegg DJ, et al. (2002). The catabolic action of insulin in the brain is mediated by melanocortins. J Neurosci, 22:9048-9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Pocai A, Lam TK, Gutierrez-Juarez R, Obici S, Schwartz GJ, Bryan J, et al. (2005). Hypothalamic K(ATP) channels control hepatic glucose production. Nature, 434:1026-1031. [DOI] [PubMed] [Google Scholar]
- [135].Konner AC, Janoschek R, Plum L, Jordan SD, Rother E, Ma X, et al. (2007). Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab, 5:438-449. [DOI] [PubMed] [Google Scholar]
- [136].Cotero VE, Routh VH (2009). Insulin blunts the response of glucose-excited neurons in the ventrolateral-ventromedial hypothalamic nucleus to decreased glucose. Am J Physiol Endocrinol Metab, 296:E1101-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Hausen AC, Ruud J, Jiang H, Hess S, Varbanov H, Kloppenburg P, et al. (2016). Insulin-Dependent Activation of MCH Neurons Impairs Locomotor Activity and Insulin Sensitivity in Obesity. Cell Rep, 17:2512-2521. [DOI] [PubMed] [Google Scholar]
- [138].Liu S, Labouebe G, Karunakaran S, Clee SM, Borgland SL (2013). Effect of insulin on excitatory synaptic transmission onto dopamine neurons of the ventral tegmental area in a mouse model of hyperinsulinemia. Nutr Diabetes, 3:e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Labouebe G, Liu S, Dias C, Zou H, Wong JC, Karunakaran S, et al. (2013). Insulin induces long-term depression of ventral tegmental area dopamine neurons via endocannabinoids. Nat Neurosci, 16:300-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Hammoud H, Netsyk O, Tafreshiha AS, Korol SV, Jin Z, Li JP, et al. (2021). Insulin differentially modulates GABA signalling in hippocampal neurons and, in an age-dependent manner, normalizes GABA-activated currents in the tg-APPSwe mouse model of Alzheimer's disease. Acta Physiol (Oxf), 232:e13623. [DOI] [PubMed] [Google Scholar]
- [141].Savigner A, Duchamp-Viret P, Grosmaitre X, Chaput M, Garcia S, Ma M, et al. (2009). Modulation of spontaneous and odorant-evoked activity of rat olfactory sensory neurons by two anorectic peptides, insulin and leptin. J Neurophysiol, 101:2898-2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Spanswick D, Smith MA, Mirshamsi S, Routh VH, Ashford ML (2000). Insulin activates ATP-sensitive K+ channels in hypothalamic neurons of lean, but not obese rats. Nat Neurosci, 3:757-758. [DOI] [PubMed] [Google Scholar]
- [143].Wakabayashi T, Yamaguchi K, Matsui K, Sano T, Kubota T, Hashimoto T, et al. (2019). Differential effects of diet- and genetically-induced brain insulin resistance on amyloid pathology in a mouse model of Alzheimer’s disease. Molecular Neurodegeneration, 14:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Baura GD, Foster DM, Porte D Jr., Kahn SE, Bergman RN, Cobelli C, et al. (1993). Saturable transport of insulin from plasma into the central nervous system of dogs in vivo. A mechanism for regulated insulin delivery to the brain. J Clin Invest, 92:1824-1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Banks WA, Jaspan JB, Huang W, Kastin AJ (1997). Transport of insulin across the blood-brain barrier: saturability at euglycemic doses of insulin. Peptides, 18:1423-1429. [DOI] [PubMed] [Google Scholar]
- [146].De Felice FG, Goncalves RA, Ferreira ST (2022). Impaired insulin signalling and allostatic load in Alzheimer disease. Nat Rev Neurosci, 23:215-230. [DOI] [PubMed] [Google Scholar]
- [147].Bosoi CR, Vandal M, Tournissac M, Leclerc M, Fanet H, Mitchell PL, et al. (2021). High-Fat Diet Modulates Hepatic Amyloid β and Cerebrosterol Metabolism in the Triple Transgenic Mouse Model of Alzheimer's Disease. Hepatol Commun, 5:446-460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Jeon BT, Jeong EA, Shin HJ, Lee Y, Lee DH, Kim HJ, et al. (2012). Resveratrol attenuates obesity-associated peripheral and central inflammation and improves memory deficit in mice fed a high-fat diet. Diabetes, 61:1444-1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Amelianchik A, Sweetland-Martin L, Norris EH (2022). The effect of dietary fat consumption on Alzheimer's disease pathogenesis in mouse models. Transl Psychiatry, 12:293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Lalli CA, Pauli JR, Prada PO, Cintra DE, Ropelle ER, Velloso LA, et al. (2008). Statin modulates insulin signaling and insulin resistance in liver and muscle of rats fed a high-fat diet. Metabolism, 57:57-65. [DOI] [PubMed] [Google Scholar]
- [151].Griffith CM, Macklin LN, Cai Y, Sharp AA, Yan XX, Reagan LP, et al. (2019). Impaired Glucose Tolerance and Reduced Plasma Insulin Precede Decreased AKT Phosphorylation and GLUT3 Translocation in the Hippocampus of Old 3xTg-AD Mice. J Alzheimers Dis, 68:809-837. [DOI] [PubMed] [Google Scholar]
- [152].Velazquez R, Tran A, Ishimwe E, Denner L, Dave N, Oddo S, et al. (2017). Central insulin dysregulation and energy dyshomeostasis in two mouse models of Alzheimer's disease. Neurobiol Aging, 58:1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Vandal M, White PJ, Chevrier G, Tremblay C, St-Amour I, Planel E, et al. (2015). Age-dependent impairment of glucose tolerance in the 3xTg-AD mouse model of Alzheimer's disease. FASEB J, 29:4273-4284. [DOI] [PubMed] [Google Scholar]
- [154].Wijesekara N, Gonçalves RA, Ahrens R, Ha K, De Felice FG, Fraser PE (2021). Combination of human tau and islet amyloid polypeptide exacerbates metabolic dysfunction in transgenic mice. [J] Pathol. [DOI] [PubMed] [Google Scholar]
- [155].Wijesekara N, Ahrens R, Sabale M, Wu L, Ha K, Verdile G, et al. (2017). Amyloid-beta and islet amyloid pathologies link Alzheimer's disease and type 2 diabetes in a transgenic model. FASEB J, 31:5409-5418. [DOI] [PubMed] [Google Scholar]
- [156].Sah SK, Lee C, Jang JH, Park GH (2017). Effect of high-fat diet on cognitive impairment in triple-transgenic mice model of Alzheimer's disease. Biochem Biophys Res Commun, 493:731-736. [DOI] [PubMed] [Google Scholar]
- [157].Gannon OJ, Robison LS, Salinero AE, Abi-Ghanem C, Mansour FM, Kelly RD, et al. (2022). High-fat diet exacerbates cognitive decline in mouse models of Alzheimer's disease and mixed dementia in a sex-dependent manner. J Neuroinflammation, 19:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Knight EM, Martins IV, Gumusgoz S, Allan SM, Lawrence CB (2014). High-fat diet-induced memory impairment in triple-transgenic Alzheimer's disease (3xTgAD) mice is independent of changes in amyloid and tau pathology. Neurobiol Aging, 35:1821-1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Julien C, Tremblay C, Phivilay A, Berthiaume L, Emond V, Julien P, et al. (2010). High-fat diet aggravates amyloid-beta and tau pathologies in the 3xTg-AD mouse model. Neurobiol Aging, 31:1516-1531. [DOI] [PubMed] [Google Scholar]
- [160].Barron AM, Rosario ER, Elteriefi R, Pike CJ (2013). Sex-specific effects of high fat diet on indices of metabolic syndrome in 3xTg-AD mice: implications for Alzheimer's disease. PLoS One, 8:e78554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Mancini G, Dias C, Lourenço CF, Laranjinha J, de Bem A, Ledo A (2021). A High Fat/Cholesterol Diet Recapitulates Some Alzheimer's Disease-Like Features in Mice: Focus on Hippocampal Mitochondrial Dysfunction. J Alzheimers Dis, 82:1619-1633. [DOI] [PubMed] [Google Scholar]
- [162].Sanguinetti E, Guzzardi MA, Panetta D, Tripodi M, De Sena V, Quaglierini M, et al. (2019). Combined Effect of Fatty Diet and Cognitive Decline on Brain Metabolism, Food Intake, Body Weight, and Counteraction by Intranasal Insulin Therapy in 3xTg Mice. Front Cell Neurosci, 13:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].King MR, Anderson NJ, Deciu M, Guernsey LS, Cundiff M, Hajizadeh S, et al. (2020). Insulin deficiency, but not resistance, exaggerates cognitive deficits in transgenic mice expressing human amyloid and tau proteins. Reversal by Exendin-4 treatment. J Neurosci Res, 98:2357-2369. [DOI] [PubMed] [Google Scholar]
- [164].Elhaik Goldman S, Goez D, Last D, Naor S, Liraz Zaltsman S, Sharvit-Ginon I, et al. (2018). High-fat diet protects the blood-brain barrier in an Alzheimer's disease mouse model. Aging Cell, 17:e12818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Robison LS, Gannon OJ, Thomas MA, Salinero AE, Abi-Ghanem C, Poitelon Y, et al. (2020). Role of sex and high-fat diet in metabolic and hypothalamic disturbances in the 3xTg-AD mouse model of Alzheimer's disease. J Neuroinflammation, 17:285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Vandal M, Bourassa P, Calon F (2015). Can insulin signaling pathways be targeted to transport Abeta out of the brain? Front Aging Neurosci, 7:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Swaminathan SK, Ahlschwede KM, Sarma V, Curran GL, Omtri RS, Decklever T, et al. (2018). Insulin differentially affects the distribution kinetics of amyloid beta 40 and 42 in plasma and brain. J Cereb Blood Flow Metab, 38:904-918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Wang X, Zheng W, Xie JW, Wang T, Wang SL, Teng WP, et al. (2010). Insulin deficiency exacerbates cerebral amyloidosis and behavioral deficits in an Alzheimer transgenic mouse model. Mol Neurodegener, 5:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Ho L, Qin W, Pompl PN, Xiang Z, Wang J, Zhao Z, et al. (2004). Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer's disease. FASEB J, 18:902-904. [DOI] [PubMed] [Google Scholar]
- [170].Petrov D, Pedros I, Artiach G, Sureda FX, Barroso E, Pallas M, et al. (2015). High-fat diet-induced deregulation of hippocampal insulin signaling and mitochondrial homeostasis deficiences contribute to Alzheimer disease pathology in rodents. Biochim Biophys Acta, 1852:1687-1699. [DOI] [PubMed] [Google Scholar]
- [171].Ettcheto M, Petrov D, Pedros I, Alva N, Carbonell T, Beas-Zarate C, et al. (2016). Evaluation of Neuropathological Effects of a High-Fat Diet in a Presymptomatic Alzheimer's Disease Stage in APP/PS1 Mice. J Alzheimers Dis, 54:233-251. [DOI] [PubMed] [Google Scholar]
- [172].Zhao N, Liu CC, Van Ingelgom AJ, Martens YA, Linares C, Knight JA, et al. (2017). Apolipoprotein E4 Impairs Neuronal Insulin Signaling by Trapping Insulin Receptor in the Endosomes. Neuron, 96:115-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Rojas M, Chávez-Castillo M, Bautista J, Ortega Á, Nava M, Salazar J, et al. (2021). Alzheimer's disease and type 2 diabetes mellitus: Pathophysiologic and pharmacotherapeutics links. World J Diabetes, 12:745-766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Benedict C, Grillo CA (2018). Insulin Resistance as a Therapeutic Target in the Treatment of Alzheimer's Disease: A State-of-the-Art Review. Front Neurosci, 12:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Ferreira LSS, Fernandes CS, Vieira MNN, De Felice FG (2018). Insulin Resistance in Alzheimer's Disease. Front Neurosci, 12:830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Nitsch R, Hoyer S (1991). Local action of the diabetogenic drug, streptozotocin, on glucose and energy metabolism in rat brain cortex. Neurosci Lett, 128:199-202. [DOI] [PubMed] [Google Scholar]
- [177].El Sayed NS, Kandil EA, Ghoneum MH (2021). Enhancement of Insulin/PI3K/Akt Signaling Pathway and Modulation of Gut Microbiome by Probiotics Fermentation Technology, a Kefir Grain Product, in Sporadic Alzheimer's Disease Model in Mice. Front Pharmacol, 12:666502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Kamat PK, Kalani A, Rai S, Tota SK, Kumar A, Ahmad AS (2016). Streptozotocin Intracerebroventricular-Induced Neurotoxicity and Brain Insulin Resistance: a Therapeutic Intervention for Treatment of Sporadic Alzheimer’s Disease (sAD)-Like Pathology. Molecular Neurobiology, 53:4548-4562. [DOI] [PubMed] [Google Scholar]
- [179].Grieb P (2016). Intracerebroventricular Streptozotocin Injections as a Model of Alzheimer's Disease: in Search of a Relevant Mechanism. Mol Neurobiol, 53:1741-1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Plaschke K, Kopitz J, Siegelin M, Schliebs R, Salkovic-Petrisic M, Riederer P, et al. (2010). Insulin-resistant brain state after intracerebroventricular streptozotocin injection exacerbates Alzheimer-like changes in Tg2576 AbetaPP-overexpressing mice. J Alzheimers Dis, 19:691-704. [DOI] [PubMed] [Google Scholar]
- [181].Salkovic-Petrisic M, Knezovic A, Hoyer S, Riederer P (2013). What have we learned from the streptozotocin-induced animal model of sporadic Alzheimer's disease, about the therapeutic strategies in Alzheimer's research. J Neural Transm (Vienna), 120:233-252. [DOI] [PubMed] [Google Scholar]
- [182].Chen Y, Liang Z, Tian Z, Blanchard J, Dai CL, Chalbot S, et al. (2014). Intracerebroventricular streptozotocin exacerbates Alzheimer-like changes of 3xTg-AD mice. Mol Neurobiol, 49:547-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Chen Y, Tian Z, Liang Z, Sun S, Dai CL, Lee MH, et al. (2012). Brain gene expression of a sporadic (icv-STZ Mouse) and a familial mouse model (3xTg-AD mouse) of Alzheimer's disease. PLoS One, 7:e51432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Kelliny S, Lin L, Deng I, Xiong J, Zhou F, Al-Hawwas M, et al. (2021). A New Approach to Model Sporadic Alzheimer’s Disease by Intracerebroventricular Streptozotocin Injection in APP/PS1 Mice. Molecular Neurobiology, 58:3692-3711. [DOI] [PubMed] [Google Scholar]
- [185].Deng Y, Li B, Liu Y, Iqbal K, Grundke-Iqbal I, Gong CX (2009). Dysregulation of insulin signaling, glucose transporters, O-GlcNAcylation, and phosphorylation of tau and neurofilaments in the brain: Implication for Alzheimer's disease. Am J Pathol, 175:2089-2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Bruning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, et al. (2000). Role of brain insulin receptor in control of body weight and reproduction. Science, 289:2122-2125. [DOI] [PubMed] [Google Scholar]
- [187].Lee SH, Zabolotny JM, Huang H, Lee H, Kim YB (2016). Insulin in the nervous system and the mind: Functions in metabolism, memory, and mood. Mol Metab, 5:589-601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Garcia-Caceres C, Quarta C, Varela L, Gao Y, Gruber T, Legutko B, et al. (2016). Astrocytic Insulin Signaling Couples Brain Glucose Uptake with Nutrient Availability. Cell, 166:867-880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Milanova IV, Korpel NL, Correa-da-Silva F, Berends E, Osman S, la Fleur SE, et al. (2022). Loss of Microglial Insulin Receptor Leads to Sex-Dependent Metabolic Disorders in Obese Mice. Int J Mol Sci, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Zegarra-Valdivia J, Fernandez AM, Martinez-Rachadell L, Herrero-Labrador R, Fernandes J, Torres Aleman I (2022). Insulin and insulin-like growth factor-I receptors in astrocytes exert different effects on behavior and Alzheimer s-like pathology. F1000Res, 11:663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Grillo CA, Tamashiro KL, Piroli GG, Melhorn S, Gass JT, Newsom RJ, et al. (2007). Lentivirus-mediated downregulation of hypothalamic insulin receptor expression. Physiol Behav, 92:691-701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Grillo CA, Woodruff JL, Macht VA, Reagan LP (2019). Insulin resistance and hippocampal dysfunction: Disentangling peripheral and brain causes from consequences. Exp Neurol, 318:71-77. [DOI] [PubMed] [Google Scholar]
- [193].Grillo CA, Piroli GG, Kaigler KF, Wilson SP, Wilson MA, Reagan LP (2011). Downregulation of hypothalamic insulin receptor expression elicits depressive-like behaviors in rats. Behav Brain Res, 222:230-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Grillo CA, Piroli GG, Lawrence RC, Wrighten SA, Green AJ, Wilson SP, et al. (2015). Hippocampal Insulin Resistance Impairs Spatial Learning and Synaptic Plasticity. Diabetes, 64:3927-3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Frazier HN, Ghoweri AO, Anderson KL, Lin RL, Popa GJ, Mendenhall MD, et al. (2020). Elevating Insulin Signaling Using a Constitutively Active Insulin Receptor Increases Glucose Metabolism and Expression of GLUT3 in Hippocampal Neurons. Front Neurosci, 14:668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Schaffer L, Brand CL, Hansen BF, Ribel U, Shaw AC, Slaaby R, et al. (2008). A novel high-affinity peptide antagonist to the insulin receptor. Biochem Biophys Res Commun, 376:380-383. [DOI] [PubMed] [Google Scholar]
- [197].Hersom M, Helms HC, Schmalz C, Pedersen TA, Buckley ST, Brodin B (2018). The insulin receptor is expressed and functional in cultured blood-brain barrier endothelial cells but does not mediate insulin entry from blood to brain. Am J Physiol Endocrinol Metab, 315:E531-E542. [DOI] [PubMed] [Google Scholar]
- [198].Gray SM, Aylor KW, Barrett EJ (2017). Unravelling the regulation of insulin transport across the brain endothelial cell. Diabetologia, 60:1512-1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Meijer RI, Gray SM, Aylor KW, Barrett EJ (2016). Pathways for insulin access to the brain: the role of the microvascular endothelial cell. Am J Physiol Heart Circ Physiol, 311:H1132-H1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Rhea EM, Humann SR, Nirkhe S, Farr SA, Morley JE, Banks WA (2017). Intranasal Insulin Transport is Preserved in Aged SAMP8 Mice and is Altered by Albumin and Insulin Receptor Inhibition. J Alzheimers Dis, 57:241-252. [DOI] [PubMed] [Google Scholar]
- [201].Nguyen V, Thomas P, Pemberton S, Babin A, Noonan C, Weaver R, et al. (2023). Central nervous system insulin signaling can influence the rate of insulin influx into brain. Fluids Barriers CNS, 20:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Arvanitakis Z, Wang HY, Capuano AW, Khan A, Taib B, Anokye-Danso F, et al. (2020). Brain Insulin Signaling, Alzheimer Disease Pathology, and Cognitive Function. Ann Neurol, 88:513-525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Taubel JC, Nelson NR, Bansal A, Curran GL, Wang L, Wang Z, et al. (2022). Design, Synthesis, and Preliminary Evaluation of [(68)Ga]Ga-NOTA-Insulin as a PET Probe in an Alzheimer's Disease Mouse Model. Bioconjug Chem, 33:892-906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Bhuiyan M, Kucharski A, Freifelder R, Won L, Kraig R, Chen CT (2020). Semi-automatic radiolabeling of insulin-like growth factor-1 protein for the verification of its nose-to-brain delivery by positron emission tomography imaging. Journal of Nuclear Medicine, 61. [Google Scholar]
- [205].Prabhakaran J, Dewey SL, McClure R, Simpson NR, Tantawy MN, Mann JJ, et al. (2017). In vivo evaluation of IGF1R/IR PET ligand [F-18]BMS-754807 in rodents. Bioorganic & Medicinal Chemistry Letters, 27:941-943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Sai KKS, Prabhakaran J, Sattiraju A, Mann JJ, Mintz A, Kumar JSD (2017). Radiosynthesis and evaluation of IGF1R PET ligand [C-11]GSK1838705A. Bioorganic & Medicinal Chemistry Letters, 27:2895-2897. [DOI] [PubMed] [Google Scholar]
- [207].Rebelos E, Bucci M, Karjalainen T, Oikonen V, Bertoldo A, Hannukainen JC, et al. (2021). Insulin Resistance Is Associated With Enhanced Brain Glucose Uptake During Euglycemic Hyperinsulinemia: A Large-Scale PET Cohort. Diabetes Care, 44:788-794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Bingham EM, Hopkins D, Smith D, Pernet A, Hallett W, Reed L, et al. (2002). The role of insulin in human brain glucose metabolism - An (18)fluoro-deoxyglucose positron emission tomography study. Diabetes, 51:3384-3390. [DOI] [PubMed] [Google Scholar]
- [209].Chabriat H, Sachon C, Levasseur M, Grimaldi A, Pappata S, Rougemont D, et al. (1994). Brain Metabolism after Recurrent Insulin-Induced Hypoglycemic Episodes - a Pet Study. Journal of Neurology Neurosurgery and Psychiatry, 57:1360-1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Benedict C, Kern W, Schultes B, Born J, Hallschmid M (2008). Differential sensitivity of men and women to anorexigenic and memory-improving effects of intranasal insulin. J Clin Endocrinol Metab, 93:1339-1344. [DOI] [PubMed] [Google Scholar]
- [211].Hallschmid M (2021). Intranasal insulin. J Neuroendocrinol, 33:e12934. [DOI] [PubMed] [Google Scholar]
- [212].Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, et al. (2012). Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol, 69:29-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].McDougal DH, Darpolor MM, DuVall MA, Sutton EF, Morrison CD, Gadde KM, et al. (2018). Glial acetate metabolism is increased following a 72-h fast in metabolically healthy men and correlates with susceptibility to hypoglycemia. Acta Diabetol, 55:1029-1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Lapidot A, Haber S (2000). Effect of acute insulin-induced hypoglycemia on fetal versus adult brain fuel utilization, assessed by (13)C MRS isotopomer analysis of [U-(13)C]glucose metabolites. Dev Neurosci, 22:444-455. [DOI] [PubMed] [Google Scholar]
- [215].Wijtenburg SA, Kapogiannis D, Korenic SA, Mullins RJ, Tran J, Gaston FE, et al. (2019). Brain insulin resistance and altered brain glucose are related to memory impairments in schizophrenia. Schizophr Res, 208:324-330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Karczewska-Kupczewska M, Tarasow E, Nikolajuk A, Stefanowicz M, Matulewicz N, Otziomek E, et al. (2013). The effect of insulin infusion on the metabolites in cerebral tissues assessed with proton magnetic resonance spectroscopy in young healthy subjects with high and low insulin sensitivity. Diabetes Care, 36:2787-2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Jauch-Chara K, Friedrich A, Rezmer M, Melchert UH, H GS-E, Hallschmid M, et al. (2012). Intranasal insulin suppresses food intake via enhancement of brain energy levels in humans. Diabetes, 61:2261-2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Sheth SA, Nemoto M, Guiou M, Walker M, Pouratian N, Toga AW (2004). Linear and nonlinear relationships between neuronal activity, oxygen metabolism, and hemodynamic responses. Neuron, 42:347-355. [DOI] [PubMed] [Google Scholar]
- [219].Krajcovicova L, Marecek R, Mikl M, Rektorova I (2014). Disruption of resting functional connectivity in Alzheimer's patients and at-risk subjects. Curr Neurol Neurosci Rep, 14:491. [DOI] [PubMed] [Google Scholar]
- [220].Musen G, Jacobson AM, Bolo NR, Simonson DC, Shenton ME, McCartney RL, et al. (2012). Resting-state brain functional connectivity is altered in type 2 diabetes. Diabetes, 61:2375-2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Baker LD, Cross DJ, Minoshima S, Belongia D, Watson GS, Craft S (2011). Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch Neurol, 68:51-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Purnell JQ, Klopfenstein BA, Stevens AA, Havel PJ, Adams SH, Dunn TN, et al. (2011). Brain functional magnetic resonance imaging response to glucose and fructose infusions in humans. Diabetes Obes Metab, 13:229-234. [DOI] [PubMed] [Google Scholar]
- [223].Rosenthal JM, Amiel SA, Yaguez L, Bullmore E, Hopkins D, Evans M, et al. (2001). The effect of acute hypoglycemia on brain function and activation: a functional magnetic resonance imaging study. Diabetes, 50:1618-1626. [DOI] [PubMed] [Google Scholar]
- [224].Wagner L, Veit R, Fritsche L, Haring HU, Fritsche A, Birkenfeld AL, et al. (2022). Sex differences in central insulin action: Effect of intranasal insulin on neural food cue reactivity in adults with normal weight and overweight. Int J Obes (Lond), 46:1662-1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Kullmann S, Frank S, Heni M, Ketterer C, Veit R, Haring HU, et al. (2013). Intranasal insulin modulates intrinsic reward and prefrontal circuitry of the human brain in lean women. Neuroendocrinology, 97:176-182. [DOI] [PubMed] [Google Scholar]
- [226].Wingrove J, Swedrowska M, Scherliess R, Parry M, Ramjeeawon M, Taylor D, et al. (2019). Characterisation of nasal devices for delivery of insulin to the brain and evaluation in humans using functional magnetic resonance imaging. J Control Release, 302:140-147. [DOI] [PubMed] [Google Scholar]
- [227].Stogios N, Hamel L, Smith E, Sanches M, Remington G, Voineskos A, et al. (2022). Investigating the effects of antipsychotics on brain insulin action: Study protocol for a multi-modality magnetic resonance imaging (MRI) study in healthy controls. PLoS One, 17:e0277211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Benedict C, Hallschmid M, Hatke A, Schultes B, Fehm HL, Born J, et al. (2004). Intranasal insulin improves memory in humans. Psychoneuroendocrinology, 29:1326-1334. [DOI] [PubMed] [Google Scholar]
- [229].Kellar D, Craft S (2020). Brain insulin resistance in Alzheimer's disease and related disorders: mechanisms and therapeutic approaches. Lancet Neurol, 19:758-766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Kellar D, Register T, Lockhart SN, Aisen P, Raman R, Rissman RA, et al. (2022). Intranasal insulin modulates cerebrospinal fluid markers of neuroinflammation in mild cognitive impairment and Alzheimer's disease: a randomized trial. Sci Rep, 12:1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Kellar D, Lockhart SN, Aisen P, Raman R, Rissman RA, Brewer J, et al. (2021). Intranasal Insulin Reduces White Matter Hyperintensity Progression in Association with Improvements in Cognition and CSF Biomarker Profiles in Mild Cognitive Impairment and Alzheimer's Disease. J Prev Alzheimers Dis, 8:240-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Claxton A, Baker LD, Hanson A, Trittschuh EH, Cholerton B, Morgan A, et al. (2015). Long Acting Intranasal Insulin Detemir Improves Cognition for Adults with Mild Cognitive Impairment or Early-Stage Alzheimer's Disease Dementia. J Alzheimers Dis, 45:1269-1270. [DOI] [PubMed] [Google Scholar]
- [233].Morris JK, Burns JM (2012). Insulin: an emerging treatment for Alzheimer's disease dementia? Curr Neurol Neurosci Rep, 12:520-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Reger MA, Watson GS, Frey WH 2nd, Baker LD, Cholerton B, Keeling ML, et al. (2006). Effects of intranasal insulin on cognition in memory-impaired older adults: modulation by APOE genotype. Neurobiol Aging, 27:451-458. [DOI] [PubMed] [Google Scholar]
- [235].Reger MA, Watson GS, Green PS, Baker LD, Cholerton B, Fishel MA, et al. (2008). Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J Alzheimers Dis, 13:323-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Claxton A, Baker LD, Wilkinson CW, Trittschuh EH, Chapman D, Watson GS, et al. (2013). Sex and ApoE genotype differences in treatment response to two doses of intranasal insulin in adults with mild cognitive impairment or Alzheimer's disease. J Alzheimers Dis, 35:789-797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Novak V, Milberg W, Hao Y, Munshi M, Novak P, Galica A, et al. (2014). Enhancement of vasoreactivity and cognition by intranasal insulin in type 2 diabetes. Diabetes Care, 37:751-759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Hallschmid M, Benedict C, Schultes B, Born J, Kern W (2008). Obese men respond to cognitive but not to catabolic brain insulin signaling. Int J Obes (Lond), 32:275-282. [DOI] [PubMed] [Google Scholar]
- [239].Walker KA, Chawla S, Nogueras-Ortiz C, Coresh J, Sharrett AR, Wong DF, et al. (2021). Neuronal insulin signaling and brain structure in nondemented older adults: the Atherosclerosis Risk in Communities Study. Neurobiol Aging, 97:65-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Mullins RJ, Mustapic M, Goetzl EJ, Kapogiannis D (2017). Exosomal biomarkers of brain insulin resistance associated with regional atrophy in Alzheimer's disease. Hum Brain Mapp, 38:1933-1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Mansur RB, Delgado-Peraza F, Subramaniapillai M, Lee Y, Iacobucci M, Nasri F, et al. (2021). Exploring brain insulin resistance in adults with bipolar depression using extracellular vesicles of neuronal origin. J Psychiatr Res, 133:82-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Geijselaers SLC, Aalten P, Ramakers I, De Deyn PP, Heijboer AC, Koek HL, et al. (2018). Association of Cerebrospinal Fluid (CSF) Insulin with Cognitive Performance and CSF Biomarkers of Alzheimer's Disease. J Alzheimers Dis, 61:309-320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Sagues-Sese E, Rioja J, Garzon-Maldonado FJ, Narvaez M, Garcia-Arnes JA, Garcia-Casares N (2022). Insulin-Related Biomarkers in Cerebrospinal Fluid in Mild Cognitive Impairment and Alzheimer's Disease: A Systematic Review. J Alzheimers Dis, 90:1-13. [DOI] [PubMed] [Google Scholar]
- [244].Bayer-Carter JL, Green PS, Montine TJ, VanFossen B, Baker LD, Watson GS, et al. (2011). Diet intervention and cerebrospinal fluid biomarkers in amnestic mild cognitive impairment. Arch Neurol, 68:743-752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Besga A, Cedazo-Minguez A, Kareholt I, Solomon A, Bjorkhem I, Winblad B, et al. (2012). Differences in brain cholesterol metabolism and insulin in two subgroups of patients with different CSF biomarkers but similar white matter lesions suggest different pathogenic mechanisms. Neurosci Lett, 510:121-126. [DOI] [PubMed] [Google Scholar]
- [246].Lane EM, Hohman TJ, Jefferson AL, Alzheimer's Disease Neuroimaging I (2017). Insulin-like growth factor binding protein-2 interactions with Alzheimer's disease biomarkers. Brain Imaging Behav, 11:1779-1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Lee S, Tong M, Hang S, Deochand C, de la Monte S (2013). CSF and Brain Indices of Insulin Resistance, Oxidative Stress and Neuro-Inflammation in Early versus Late Alzheimer's Disease. J Alzheimers Dis Parkinsonism, 3:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Tournissac M, Leclerc M, Valentin-Escalera J, Vandal M, Bosoi CR, Planel E, et al. (2021). Metabolic determinants of Alzheimer's disease: A focus on thermoregulation. Ageing Res Rev, 72:101462. [DOI] [PubMed] [Google Scholar]
- [249].Khemka VK, Bagchi D, Bandyopadhyay K, Bir A, Chattopadhyay M, Biswas A, et al. (2014). Altered serum levels of adipokines and insulin in probable Alzheimer's disease. J Alzheimers Dis, 41:525-533. [DOI] [PubMed] [Google Scholar]
- [250].Craft S, Peskind E, Schwartz MW, Schellenberg GD, Raskind M, Porte D Jr. (1998). Cerebrospinal fluid and plasma insulin levels in Alzheimer's disease: relationship to severity of dementia and apolipoprotein E genotype. Neurology, 50:164-168. [DOI] [PubMed] [Google Scholar]
- [251].de la Monte SM, Tong M, Daiello LA, Ott BR (2019). Early-Stage Alzheimer’s Disease Is Associated with Simultaneous Systemic and Central Nervous System Dysregulation of Insulin-Linked Metabolic Pathways. Journal of Alzheimer's Disease, 68:657-668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Johansson P, Åberg D, Johansson JO, Mattsson N, Hansson O, Ahrén B, et al. (2013). Serum but not cerebrospinal fluid levels of insulin-like growth factor-I (IGF-I) and IGF-binding protein-3 (IGFBP-3) are increased in Alzheimer's disease. Psychoneuroendocrinology, 38:1729-1737. [DOI] [PubMed] [Google Scholar]
- [253].Molina JA, Jimenez-Jimenez FJ, Vargas C, Gomez P, de Bustos F, Gomez-Escalonilla C, et al. (2002). Cerebrospinal fluid levels of insulin in patients with Alzheimer's disease. Acta Neurol Scand, 106:347-350. [DOI] [PubMed] [Google Scholar]
- [254].Fujisawa Y, Sasaki K, Akiyama K (1991). Increased insulin levels after OGTT load in peripheral blood and cerebrospinal fluid of patients with dementia of Alzheimer type. Biol Psychiatry, 30:1219-1228. [DOI] [PubMed] [Google Scholar]
- [255].Horvath A, Salman Z, Quinlan P, Wallin A, Svensson J (2020). Patients with Alzheimer's Disease Have Increased Levels of Insulin-like Growth Factor-I in Serum but not in Cerebrospinal Fluid. J Alzheimers Dis, 75:289-298. [DOI] [PubMed] [Google Scholar]
- [256].Tham A, Nordberg A, Grissom FE, Carlsson-Skwirut C, Viitanen M, Sara VR (1993). Insulin-like growth factors and insulin-like growth factor binding proteins in cerebrospinal fluid and serum of patients with dementia of the Alzheimer type. J Neural Transm Park Dis Dement Sect, 5:165-176. [DOI] [PubMed] [Google Scholar]
- [257].Salehi Z, Mashayekhi F, Naji M (2008). Insulin like growth factor-1 and insulin like growth factor binding proteins in the cerebrospinal fluid and serum from patients with Alzheimer's disease. Biofactors, 33:99-106. [DOI] [PubMed] [Google Scholar]
- [258].Aberg D, Johansson P, Isgaard J, Wallin A, Johansson JO, Andreasson U, et al. (2015). Increased Cerebrospinal Fluid Level of Insulin-like Growth Factor-II in Male Patients with Alzheimer's Disease. J Alzheimers Dis, 48:637-646. [DOI] [PubMed] [Google Scholar]
- [259].Hertze J, Nägga K, Minthon L, Hansson O (2014). Changes in cerebrospinal fluid and blood plasma levels of IGF-II and its binding proteins in Alzheimer's disease: an observational study. BMC Neurol, 14:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].McGrath ER, Himali JJ, Levy D, Conner SC, DeCarli CS, Pase MP, et al. (2019). Circulating IGFBP-2: a novel biomarker for incident dementia. Ann Clin Transl Neurol, 6:1659-1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [261].Doecke JD, Laws SM, Faux NG, Wilson W, Burnham SC, Lam C-P, et al. (2012). Blood-Based Protein Biomarkers for Diagnosis of Alzheimer Disease. Archives of Neurology, 69:1318-1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Sattlecker M, Kiddle SJ, Newhouse S, Proitsi P, Nelson S, Williams S, et al. (2014). Alzheimer's disease biomarker discovery using SOMAscan multiplexed protein technology. Alzheimer's & Dementia, 10:724-734. [DOI] [PubMed] [Google Scholar]
- [263].Cogswell JP, Ward J, Taylor IA, Waters M, Shi Y, Cannon B, et al. (2008). Identification of miRNA changes in Alzheimer's disease brain and CSF yields putative biomarkers and insights into disease pathways. J Alzheimers Dis, 14:27-41. [DOI] [PubMed] [Google Scholar]
- [264].Jack CR Jr., Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, et al. (2010). Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol, 9:119-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [265].Jack CR Jr., Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. (2013). Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol, 12:207-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [266].Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. (2018). NIA-AA Research Framework: Toward a biological definition of Alzheimer's disease. Alzheimers Dement, 14:535-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [267].Banks WA, Farr SA, Morley JE (2000). Permeability of the blood-brain barrier to albumin and insulin in the young and aged SAMP8 mouse. J Gerontol A Biol Sci Med Sci, 55:B601-606. [DOI] [PubMed] [Google Scholar]
- [268].Belfiore R, Rodin A, Ferreira E, Velazquez R, Branca C, Caccamo A, et al. (2019). Temporal and regional progression of Alzheimer's disease-like pathology in 3xTg-AD mice. Aging Cell, 18:e12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [269].Frolich L, Blum-Degen D, Riederer P, Hoyer S (1999). A disturbance in the neuronal insulin receptor signal transduction in sporadic Alzheimer's disease. Ann N Y Acad Sci, 893:290-293. [DOI] [PubMed] [Google Scholar]
- [270].Tramutola A, Triplett JC, Di Domenico F, Niedowicz DM, Murphy MP, Coccia R, et al. (2015). Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J Neurochem, 133:739-749. [DOI] [PubMed] [Google Scholar]
- [271].Tramutola A, Lanzillotta C, Di Domenico F, Head E, Butterfield DA, Perluigi M, et al. (2020). Brain insulin resistance triggers early onset Alzheimer disease in Down syndrome. Neurobiol Dis, 137:104772. [DOI] [PubMed] [Google Scholar]
- [272].Kapogiannis D, Boxer A, Schwartz JB, Abner EL, Biragyn A, Masharani U, et al. (2015). Dysfunctionally phosphorylated type 1 insulin receptor substrate in neural-derived blood exosomes of preclinical Alzheimer's disease. FASEB J, 29:589-596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [273].Craft S, Claxton A, Baker LD, Hanson AJ, Cholerton B, Trittschuh EH, et al. (2017). Effects of Regular and Long-Acting Insulin on Cognition and Alzheimer's Disease Biomarkers: A Pilot Clinical Trial. J Alzheimers Dis, 57:1325-1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [274].Bonham LW, Geier EG, Steele NZR, Holland D, Miller BL, Dale AM, et al. (2018). Insulin-Like Growth Factor Binding Protein 2 Is Associated With Biomarkers of Alzheimer's Disease Pathology and Shows Differential Expression in Transgenic Mice. Front Neurosci, 12:476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [275].Wittfeld K, Raman MR, Conner SC, Aslam A, Teumer A, Nauck M, et al. (2022). Insulin-Like Growth Factor, Inflammation, and MRI Markers of Alzheimer's Disease in Predominantly Middle-Aged Adults. J Alzheimers Dis, 88:311-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Blomer U, Naldini L, Kafri T, Trono D, Verma IM, Gage FH (1997). Highly efficient and sustained gene transfer in adult neurons with a lentivirus vector. J Virol, 71:6641-6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [277].Frazier HN, Anderson KL, Ghoweri AO, Lin RL, Hawkinson TR, Popa GJ, et al. (2020). Molecular elevation of insulin receptor signaling improves memory recall in aged Fischer 344 rats. Aging Cell, 19:e13220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [278].Freude S, Hettich MM, Schumann C, Stohr O, Koch L, Kohler C, et al. (2009). Neuronal IGF-1 resistance reduces Abeta accumulation and protects against premature death in a model of Alzheimer's disease. FASEB J, 23:3315-3324. [DOI] [PubMed] [Google Scholar]
- [279].Fernandez AM, Martinez-Rachadell L, Navarrete M, Pose-Utrilla J, Davila JC, Pignatelli J, et al. (2022). Insulin regulates neurovascular coupling through astrocytes. Proc Natl Acad Sci U S A, 119:e2204527119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [280].Thorne RG, Lange ECMd, Hammarlund-Udenaes M. 2022. Appendix: Central Nervous System Anatomy and Physiology: Structure-Function Relationships, Blood Supply, Ventricles and Brain Fluids.: Springer. 763-790 pp. [Google Scholar]
- [281].Pardridge WM (2012). Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab, 32:1959-1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [282].Gray SM, Meijer RI, Barrett EJ (2014). Insulin regulates brain function, but how does it get there? Diabetes, 63:3992-3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [283].Schwartz MW, Bergman RN, Kahn SE, Taborsky GJ Jr., Fisher LD, Sipols AJ, et al. (1991). Evidence for entry of plasma insulin into cerebrospinal fluid through an intermediate compartment in dogs. Quantitative aspects and implications for transport. J Clin Invest, 88:1272-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [284].Pardridge WM (2016). CSF, blood-brain barrier, and brain drug delivery. Expert Opin Drug Deliv, 13:963-975. [DOI] [PubMed] [Google Scholar]
- [285].Presa JL, Saravia F, Bagi Z, Filosa JA (2020). Vasculo-Neuronal Coupling and Neurovascular Coupling at the Neurovascular Unit: Impact of Hypertension. Front Physiol, 11:584135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [286].Iadecola C (2017). The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron, 96:17-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [287].Storck SE, Hartz AMS, Pietrzik CU (2021). The Blood-Brain Barrier in Alzheimer's Disease. Handb Exp Pharmacol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [288].Soto-Rojas LO, Pacheco-Herrero M, Martínez-Gómez PA, Campa-Córdoba BB, Apátiga-Pérez R, Villegas-Rojas MM, et al. (2021). The Neurovascular Unit Dysfunction in Alzheimer's Disease. Int J Mol Sci, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [289].Weyhenmeyer JA, Reiner AM, Reynolds I, Killian A (1985). Light and electron microscopic analysis of insulin binding sites on neurons in dissociated brain cell cultures. Brain Res Bull, 14:415-421. [DOI] [PubMed] [Google Scholar]
- [290].Banks WA (2019). The blood-brain barrier as an endocrine tissue. Nat Rev Endocrinol, 15:444-455. [DOI] [PubMed] [Google Scholar]
- [291].García-Cáceres C, Quarta C, Varela L, Gao Y, Gruber T, Legutko B, et al. (2016). Astrocytic Insulin Signaling Couples Brain Glucose Uptake with Nutrient Availability. Cell, 166:867-880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [292].Cai W, Xue C, Sakaguchi M, Konishi M, Shirazian A, Ferris HA, et al. (2018). Insulin regulates astrocyte gliotransmission and modulates behavior. The Journal of Clinical Investigation, 128:2914-2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [293].Schubert M, Gautam D, Surjo D, Ueki K, Baudler S, Schubert D, et al. (2004). Role for neuronal insulin resistance in neurodegenerative diseases. Proc Natl Acad Sci U S A, 101:3100-3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [294].Jin Z, Jin Y, Kumar-Mendu S, Degerman E, Groop L, Birnir B (2011). Insulin reduces neuronal excitability by turning on GABA(A) channels that generate tonic current. PLoS One, 6:e16188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [295].Gralle M, Labrecque S, Salesse C, De Koninck P (2021). Spatial dynamics of the insulin receptor in living neurons. J Neurochem, 156:88-105. [DOI] [PubMed] [Google Scholar]
- [296].Meakin PJ, Mezzapesa A, Benabou E, Haas ME, Bonardo B, Grino M, et al. (2018). The beta secretase BACE1 regulates the expression of insulin receptor in the liver. Nat Commun, 9:1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [297].Bao H, Liu Y, Zhang M, Chen Z, Zhang W, Ge Y, et al. (2021). Increased beta-site APP cleaving enzyme 1-mediated insulin receptor cleavage in type 2 diabetes mellitus with cognitive impairment. Alzheimers Dement, 17:1097-1108. [DOI] [PubMed] [Google Scholar]
- [298].Gaborit B, Govers R, Altié A, Brunel JM, Morange P, Peiretti F (2021). The aminosterol Claramine inhibits β-secretase 1-mediated insulin receptor cleavage. Journal of Biological Chemistry: 100818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [299].Kullmann S, Heni M, Veit R, Scheffler K, Machann J, Haring HU, et al. (2015). Selective insulin resistance in homeostatic and cognitive control brain areas in overweight and obese adults. Diabetes Care, 38:1044-1050. [DOI] [PubMed] [Google Scholar]
- [300].Baloni P, Arnold M, Buitrago L, Nho K, Moreno H, Huynh K, et al. (2022). Multi-Omic analyses characterize the ceramide/sphingomyelin pathway as a therapeutic target in Alzheimer's disease. Commun Biol, 5:1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [301].Frazier HN, Ghoweri AO, Sudkamp E, Johnson ES, Anderson KL, Fox G, et al. (2019). Long-term intranasal insulin aspart: a profile of gene expression, memory, and insulin receptors in aged F344 rats. J Gerontol A Biol Sci Med Sci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [302].Reger MA, Watson GS, Green PS, Wilkinson CW, Baker LD, Cholerton B, et al. (2008). Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology, 70:440-448. [DOI] [PubMed] [Google Scholar]
- [303].Rhea EM, Banks WA (2017). The SAMP8 mouse for investigating memory and the role of insulin in the brain. Exp Gerontol, 94:64-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [304].Salameh TS, Bullock KM, Hujoel IA, Niehoff ML, Wolden-Hanson T, Kim J, et al. (2015). Central Nervous System Delivery of Intranasal Insulin: Mechanisms of Uptake and Effects on Cognition. J Alzheimers Dis, 47:715-728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [305].Rhea EM, Nirkhe S, Nguyen S, Pemberton S, Bammler TK, Beyer R, et al. (2019). Molecular Mechanisms of Intranasal Insulin in SAMP8 Mice. J Alzheimers Dis, 71:1361-1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [306].Nam KN, Mounier A, Wolfe CM, Fitz NF, Carter AY, Castranio EL, et al. (2017). Effect of high fat diet on phenotype, brain transcriptome and lipidome in Alzheimer's model mice. Sci Rep, 7:4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [307].Reale M, Costantini E, Aielli L, Di Giuseppe F, Angelucci S, Kamal MA, et al. (2022). Proteomic Signature and mRNA Expression in Hippocampus of SAMP8 and SAMR1 Mice during Aging. Int J Mol Sci, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [308].Gray SM, Barrett EJ (2018). Insulin transport into the brain. Am J Physiol Cell Physiol, 315:C125-C136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [309].Olah M, Menon V, Habib N, Taga MF, Ma Y, Yung CJ, et al. (2020). Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer's disease. Nat Commun, 11:6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [310].Habib N, Avraham-Davidi I, Basu A, Burks T, Shekhar K, Hofree M, et al. (2017). Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat Methods, 14:955-958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [311].Geffken SJ, Moon S, Smith CO, Tang S, Lee HH, Lewis K, et al. (2022). Insulin and IGF-1 elicit robust transcriptional regulation to modulate autophagy in astrocytes. Mol Metab, 66:101647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [312].Nagao H, Jayavelu AK, Cai W, Pan H, Dreyfuss JM, Batista TM, et al. (2023). Unique ligand and kinase-independent roles of the insulin receptor in regulation of cell cycle, senescence and apoptosis. Nat Commun, 14:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [313].Wang LB, Karpova A, Gritsenko MA, Kyle JE, Cao S, Li Y, et al. (2021). Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell, 39:509-528 e520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [314].Satpathy S, Krug K, Jean Beltran PM, Savage SR, Petralia F, Kumar-Sinha C, et al. (2021). A proteogenomic portrait of lung squamous cell carcinoma. Cell, 184:4348-4371 e4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [315].Cao L, Huang C, Cui Zhou D, Hu Y, Lih TM, Savage SR, et al. (2021). Proteogenomic characterization of pancreatic ductal adenocarcinoma. Cell, 184:5031-5052 e5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [316].Johnson LA, Torres ER, Impey S, Stevens JF, Raber J (2017). Apolipoprotein E4 and Insulin Resistance Interact to Impair Cognition and Alter the Epigenome and Metabolome. Sci Rep, 7:43701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [317].Kim J, Basak JM, Holtzman DM (2009). The role of apolipoprotein E in Alzheimer's disease. Neuron, 63:287-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [318].Konialis C, Spengos K, Iliopoulos P, Karapanou S, Gialafos E, Hagnefelt B, et al. (2016). The APOE E4 Allele Confers Increased Risk of Ischemic Stroke Among Greek Carriers. Adv Clin Exp Med, 25:471-478. [DOI] [PubMed] [Google Scholar]
- [319].Zlokovic BV (2013). Cerebrovascular effects of apolipoprotein E: implications for Alzheimer disease. JAMA Neurol, 70:440-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [320].Yan S, Fu W, Wang C, Mao J, Liu B, Zou L, et al. (2020). Association between sedentary behavior and the risk of dementia: a systematic review and meta-analysis. Transl Psychiatry, 10:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [321].Kullmann S, Goj T, Veit R, Fritsche L, Wagner L, Schneeweiss P, et al. (2022). Exercise restores brain insulin sensitivity in sedentary adults who are overweight and obese. JCI Insight, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [322].Artese A, Stamford BA, Moffatt RJ (2019). Cigarette Smoking: An Accessory to the Development of Insulin Resistance. Am J Lifestyle Med, 13:602-605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [323].Sabia S, Elbaz A, Dugravot A, Head J, Shipley M, Hagger-Johnson G, et al. (2012). Impact of smoking on cognitive decline in early old age: the Whitehall II cohort study. Arch Gen Psychiatry, 69:627-635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [324].Sun J, Xu B, Zhang X, He Z, Liu Z, Liu R, et al. (2020). The Mechanisms of Type 2 Diabetes-Related White Matter Intensities: A Review. Front Public Health, 8:498056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [325].Buxton OM, Cain SW, O'Connor SP, Porter JH, Duffy JF, Wang W, et al. (2012). Adverse metabolic consequences in humans of prolonged sleep restriction combined with circadian disruption. Sci Transl Med, 4:129ra143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [326].Buxton OM, Pavlova M, Reid EW, Wang W, Simonson DC, Adler GK (2010). Sleep restriction for 1 week reduces insulin sensitivity in healthy men. Diabetes, 59:2126-2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [327].Spiegel K, Leproult R, Van Cauter E (1999). Impact of sleep debt on metabolic and endocrine function. Lancet, 354:1435-1439. [DOI] [PubMed] [Google Scholar]
- [328].Sprecher KE, Bendlin BB, Racine AM, Okonkwo OC, Christian BT, Koscik RL, et al. (2015). Amyloid burden is associated with self-reported sleep in nondemented late middle-aged adults. Neurobiol Aging, 36:2568-2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [329].Spira AP, Gamaldo AA, An Y, Wu MN, Simonsick EM, Bilgel M, et al. (2013). Self-reported sleep and beta-amyloid deposition in community-dwelling older adults. JAMA Neurol, 70:1537-1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [330].Blattner MS, Panigrahi SK, Toedebusch CD, Hicks TJ, McLeland JS, Banks IR, et al. (2020). Increased Cerebrospinal Fluid Amyloid-beta During Sleep Deprivation in Healthy Middle-Aged Adults Is Not Due to Stress or Circadian Disruption. J Alzheimers Dis, 75:471-482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [331].Shokri-Kojori E, Wang GJ, Wiers CE, Demiral SB, Guo M, Kim SW, et al. (2018). beta-Amyloid accumulation in the human brain after one night of sleep deprivation. Proc Natl Acad Sci U S A, 115:4483-4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [332].Aburn G, Gott M, Hoare K (2016). What is resilience? An Integrative Review of the empirical literature. J Adv Nurs, 72:980-1000. [DOI] [PubMed] [Google Scholar]
- [333].Perry BL, McConnell WR, Coleman ME, Roth AR, Peng S, Apostolova LG (2022). Why the cognitive "fountain of youth" may be upstream: Pathways to dementia risk and resilience through social connectedness. Alzheimers Dement, 18:934-941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [334].Ossenkoppele R, Lyoo CH, Jester-Broms J, Sudre CH, Cho H, Ryu YH, et al. (2020). Assessment of Demographic, Genetic, and Imaging Variables Associated With Brain Resilience and Cognitive Resilience to Pathological Tau in Patients With Alzheimer Disease. JAMA Neurol, 77:632-642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [335].Dumitrescu L, Mahoney ER, Mukherjee S, Lee ML, Bush WS, Engelman CD, et al. (2020). Genetic variants and functional pathways associated with resilience to Alzheimer's disease. Brain, 143:2561-2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [336].Casaletto KB, Renteria MA, Pa J, Tom SE, Harrati A, Armstrong NM, et al. (2020). Late-Life Physical and Cognitive Activities Independently Contribute to Brain and Cognitive Resilience. J Alzheimers Dis, 74:363-376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [337].Perez-Gonzalez M, Mendioroz M, Badesso S, Sucunza D, Roldan M, Espelosin M, et al. (2020). PLA2G4E, a candidate gene for resilience in Alzheimer s disease and a new target for dementia treatment. Prog Neurobiol, 191:101818. [DOI] [PubMed] [Google Scholar]
- [338].Wilson RS, Mendes De Leon CF, Barnes LL, Schneider JA, Bienias JL, Evans DA, et al. (2002). Participation in cognitively stimulating activities and risk of incident Alzheimer disease. JAMA, 287:742-748. [DOI] [PubMed] [Google Scholar]
- [339].Kothari V, Luo Y, Tornabene T, O'Neill AM, Greene MW, Geetha T, et al. (2017). High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim Biophys Acta Mol Basis Dis, 1863:499-508. [DOI] [PubMed] [Google Scholar]
- [340].Malin SK, Stewart NR, Ude AA, Alderman BL (2022). Brain insulin resistance and cognitive function: influence of exercise. J Appl Physiol (1985), 133:1368-1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [341].Dardano A, Aghakhanyan G, Moretto C, Ciccarone A, Bellini R, Sancho Bornez V, et al. (2022). Brain effect of bariatric surgery in people with obesity. Int J Obes (Lond), 46:1671-1677. [DOI] [PubMed] [Google Scholar]