

Abstract
Aging leads to progressive deterioration of the structure and function of arteries, which eventually contributes to the development of vascular aging-related diseases. N6-methyladenosine (m6A) is the most prevalent modification in eukaryotic RNAs. This reversible m6A RNA modification is dynamically regulated by writers, erasers, and readers, playing a critical role in various physiological and pathological conditions by affecting almost all stages of the RNA life cycle. Recent studies have highlighted the involvement of m6A in vascular aging and related diseases, shedding light on its potential clinical significance. In this paper, we comprehensively discuss the current understanding of m6A in vascular aging and its clinical implications. We discuss the molecular insights into m6A and its association with clinical realities, emphasizing its significance in unraveling the mechanisms underlying vascular aging. Furthermore, we explore the possibility of m6A and its regulators as clinical indicators for early diagnosis and prognosis prediction and investigate the therapeutic potential of m6A-associated anti-aging approaches. We also examine the challenges and future directions in this field and highlight the necessity of integrating m6A knowledge into patient-centered care. Finally, we emphasize the need for multidisciplinary collaboration to advance the field of m6A research and its clinical application.
Keywords: disease, vascular aging, N6-methyladenosine, RNA modification, senescence
1. Introduction
Aging leads to progressive deterioration of the structure and function of arteries. More specifically, aging induces intrinsic and extrinsic cellular changes that affect the phenotypes and behavior of building cells of the vascular wall, especially endothelial cells (ECs) and vascular smooth muscle cells (VSMCs). Macroscopically, aging vasculature presents dilated lumen, thickened wall, diffused stiffness, and impaired angiogenesis. These alterations seriously compromise proper tissue function and eventually contribute to the development of vascular diseases [1, 2]. Age-related macrovascular diseases, such as cardiovascular diseases (CVDs), are the leading cause of death and a major driver of disability in the elderly worldwide [3]. Aging-induced structural and functional changes in microvessels exert a pivotal role in the aging process of many organs and are one of the common denominators of various aging-related diseases, such as Alzheimer's disease (AD), vascular cognitive impairment, and kidney disease [4]. Understanding vascular aging and its molecular mechanisms is crucial for therapeutic approaches to age-related diseases.
N6-methyladenosine (m6A) methylation, an emerging frontier in epigenetic research, involves the methylation at the N6 position of adenine in RNA. It has been found to play a significant role in vascular aging and related diseases [5-7]. To better learn about the discovery and research history of m6A in these diseases, we review the timeline of m6A (Fig. 1). RNA modifications have gained attention comparable to DNA and histone modifications in the field of epigenetics. Since the 1950s, over one hundred distinct RNA chemical modification types have been identified [8]. The discovery of m6A modification dates back to 1974 [9, 10]. m6A was identified as the most prevalent internal modification on messenger RNAs (mRNAs) in a diverse spectrum of eukaryotes (eg. yeast [11], plants [12], insects [13], mammals [9, 10, 14]), multiple viruses [15], and bacteria [16]. Nonetheless, due to the lack of powerful detective techniques to support in-depth studies on the distribution and function of m6A modification, limited progress was made during the following decades. In the 1990s, m6A methyltransferase was purified and characterized as a multicomponent protein complex along with the identification of methyltransferase-like 3 (METTL3) as one of its components [17, 18]. In 2011, fat mass and obesity-associated protein (FTO) was identified as the first m6A demethylase, revealing a reversible regulatory mechanism [19]. Hereafter, a series of other regulatory machineries of writers, erasers, and readers, have been identified [20]. The landscape of m6A modification at a transcriptome-wide level was first compiled by m6A-seq or methylated RNA immunoprecipitation sequencing (MeRIP-seq) in 2012 [21, 22]. m6A sites are non-randomly distributed in the transcriptome and are adjacent to consensus motif RRACH (R=A or G; H=A, C or U), enriched in the 3' untranslated region (3' UTR), near the stop codons, and within long internal exons [21, 22]. m6A is deposited in both coding and non-coding RNA (ncRNA) by m6A methyltransferases (writers), removed by demethylases (erasers), and recognized by binding proteins (readers) to affect almost all stages of RNA life cycle [20]. The robust technical advancements supporting extensive research on m6A and its function have catalyzed the emergence of a novel field of study known as “epitranscriptomics”. Thus far, an overwhelming number of reports have demonstrated the multifaceted effects of m6A on almost all major biological processes and human diseases (including vascular aging and related diseases) via expression regulation of various RNAs [23, 24]. Promisingly, recent studies start to focus on revealing novel therapies targeting m6A modification and their preclinical efficacy, heralding an era of translational research in this field [25].
Figure 1.

Timeline of the discovery and research history of m6A modification. Major findings in the m6A field are highlighted. This figure was created with the aid of Biorender (https://biorender.com/).
In this paper, we will compile emerging evidence on the role of m6A methylation in vascular aging and highlight its potential clinical application in vascular aging-related diseases.
2. m6A Modification: Bridging Molecular Insights and Clinical Realities
2.1. Biological effects of m6A regulators
As mentioned above, m6A modification is a reversible process where m6A is installed by writers, removed by erasers, and recognized by readers (Fig. 2). Moreover, m6A can regulate RNA fate and metabolism (e.g., splicing, maturation, nuclear export, degradation, stabilization, and translation), which in turn, underlies their critical role in a myriad of physiological and pathological processes [26-28].
Figure 2.
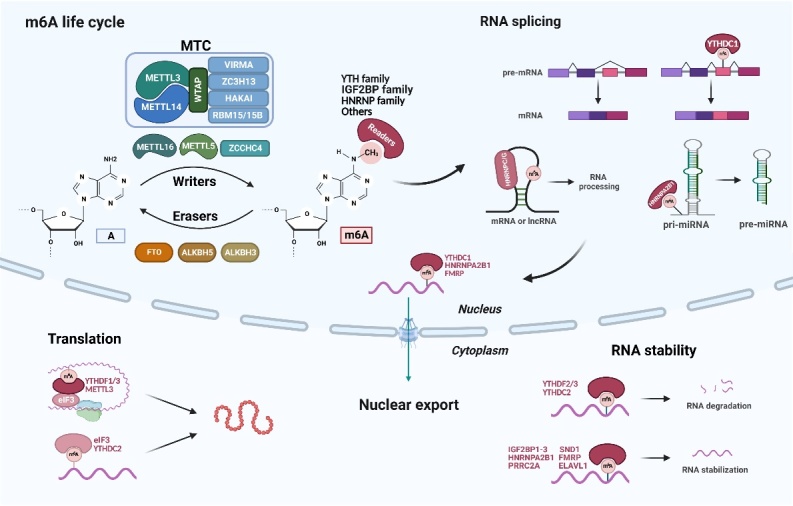
The composition and function of m6A modification. m6A methylation is a dynamic and reversible epigenetic modification that is installed by writers, removed by erasers, and recognized by readers. RNA m6A methylation can affect almost all stages of the RNA life cycle, including RNA splicing, maturation, nuclear export, degradation, stabilization, and translation. This figure was created with the aid of Biorender (https://biorender.com/).
2.1.1. Writers
m6A methylation is catalyzed by m6A methyltransferase complex (MTC) [17] that consists of a key component, METTL3/METTL14 dimer, and cofactors such as Wilms tumor 1 associated protein (WTAP), vir-like m6A methyltransferase-associated protein (VIRMA, also called KIAA29), RNA-binding motif protein 15/15B (RBM15/15B), zinc finger CCCH-type containing 13 (ZC3H13), and HAKAI (also called CBLL1) [29-34]. METTL3, the first-identified MTC component, is the only subunit with catalytic activity in MTC that catalyzes the transfer of methyl groups from S-adenosylmethionine (SAM) to adenine in RNA [18, 35-37]. METTL14 is the other core part in MTC that unites with METTL3 forming a stable heterodimer [29]. Unlike METTL3, METTL14 contains a degenerate active site and presents non-catalytic effects on structurally supporting the catalytic function of METTL3 as well as substrate recognition [35-37]. WTAP serves as a regulatory subunit that interacts with METTL3 and METTL14 to recruit them into nuclear speckles, enhancing the catalytic activity of m6A methyltransferase. In addition, WTAP and METTL3 regulate genes associated with transcription and RNA processing [30]. VIRMA directs METTL3/METTL14/ WTAP to mediate region-selective mRNA methylation in 3'UTR and near stop codon [38]. RBM15/RBM15B recruits MTC to specific RNA sites, which leads to the methylation of adenosine in neighboring consensus motifs [32]. ZC3H13 can bridge RBM15 to WTAP [34] and is necessary for the nuclear localization of MTC [33]. HAKAI, a recently identified MTC component, is needed for stabilizing other constituents of MTC through its ubiquitination domain [39].
Despite large research efforts on the role of MTC in mRNA m6A modification, there is evidence revealing other m6A writers (METTL16, METTL5, and ZCCHC4) to catalyze m6A methylation of a subset of RNAs [40-42]. METTL16 is reported to mediate m6A modification in U6 small nuclear RNA (snRNA) and MAT2A mRNA [42, 43]. ZCCHC4 is the enzyme responsible for m6A modification of 28S ribosomal RNA (rRNA) and some mRNAs [40, 41]. METTL5 exerts m6A methyltransferase function on 18S rRNA by forming a heterodimer with methyltransferase activator TRMT112 [41].
2.1.2. Erasers
The demethylation of m6A modification in RNA is mediated by erasers including FTO, AlkB homolog 5 (ALKBH5), and ALKBH3. FTO is the first identified m6A demethylase [19]. In addition to m6A demethylation, FTO has been proven to catalyze other patterns of RNA modifications, such as N6,2'-O-dimethyladenosine (m6Am) and N1-methyladenosine (m1A) [44-46]. ALKBH5 is another established m6A eraser. Unlike FTO, ALKBH5 appears a specific role in m6A [47]. Moreover, recent studies have discovered a new m6A demethylase, ALKBH3, which participates in the demethylation of tRNA [48]. The enzymatic activity of these erasers relies on their oxidative function in a Fe(II)/2-oxoglutarate (2-OG)-dependent manner [49].
2.1.3. Readers
Writers and erasers mediate the deposition and removal of m6A, respectively. However, it is readers that recognize m6A and determine different fates of RNA after modification, including but not limited to YT521-B homology (YTH) domain family, splicing factor heterogeneous nuclear ribonucleoproteins (HNRNP) family, insulin-like growth factor 2 mRNA binding protein (IGF2BP) family.
Different readers present different biological roles [50]. The YTH domain family contains YTH domain family protein 1-3 (YTHDF1-3) and YTH domain containing protein 1-2 (YTHDC1-2). The canonical model considers that YTHDF1 enhances mRNA translation [51]; YTHDF2 accelerates mRNA decay [52]. YTHDF3 exerts dual functions to promote translation and strengthen degradation through cooperation with YTHDF1 and YTHDF2, respectively [53, 54]. However, contradictory results have been observed that the functions of YTHDFs are redundant [55]. YTHDC1 regulates mRNA splicing [56] and nuclear export [57]. YTHDC2 has been revealed to elevate translation efficiency and reduce abundance of target mRNA [58]. HNRNP family includes HNRNPA2B1, HNRNPC, HNRNPG. HNRNPA2B1 is an m6A reader regulating multiple processes of RNA metabolism such as primary microRNA (pri-miRNA) processing, alternative splicing [59], nucleocytoplasmic trafficking [60], and stabilization [61]. m6A leads to RNA allostery to increase the accessibility of HNRNPC and HNRNPG, a phenomenon termed the "m6A switch", which can affect mRNA expression and alternative splicing [62, 63]. IGF2BP family members, IGF2BP1-3, have been reported to enhance the stability of target mRNA through recognition of m6A [64].
In addition to the three aforementioned well-studied protein families, several other readers have been reported. Fragile X mental-retardation protein (FMRP) has been demonstrated to modulate m6A-dependent mRNA nuclear export [65] and stability [66]. Recent studies have identified other readers that stabilize mRNA through m6A modification, including proline-rich coiled-coil 2A (PRRC2A) [67], ELAV-like RNA binding protein 1 (ELAVL1) [68, 69], and staphylococcal nuclease and tudor domain containing 1 (SND1) [70]. Another m6A reader, eukaryotic translation initiation factor 3 (eIF3), can promote translation by binding 5’-UTR m6A [71]. METTL3 located in nuclear speckles serves as methyltransferase as mentioned before. Interestingly, in contrast to its nuclear existence as an m6A writer, cytoplasmic METTL3 acts as a potential m6A reader instead. Mechanistically, METTL3 promotes mRNA translation through interaction with eIF3h to form RNA looping [72].
2.2. Potential regulatory mechanisms of m6A modification in vascular aging
Vascular aging is a complicated biological process driven by intertwined cellular and molecular mechanisms (such as oxidative stress, chronic inflammation, mitochondrial dysfunction, autophagy, DNA damage, and cellular senescence) contributing to various aging-related diseases in major organs [4]. m6A methylation is well-documented to modulate the expression of target genes involved in these mechanisms and therefore may apply pleiotropic activities on vascular aging [73-77]. The interplay between m6A modification and key pathways implicated in vascular aging bridges molecular insights with clinical realities, highlighting its significance in understanding disease pathogenesis (Fig. 3).
Figure 3.

Potential regulatory mechanisms of m6A modification in vascular aging. The aberrant m6A modification participates in various molecular and cellular mechanisms that lead to vascular aging, including oxidative stress, chronic inflammation, mitochondrial dysfunction, autophagy, DNA damage, and cellular senescence. This figure was created with the aid of Biorender (https://biorender.com/). ROS: reactive oxygen species; SG: stress granule; EC: endothelial cell; VSMC: vascular smooth muscle cell.
2.2.1. Oxidative stress
Oxidative stress is an imbalance between pro-oxidation and anti-oxidation systems, resulting in the overproduction of reactive oxygen species (ROS), which can contribute to various diseases by damaging cells and tissues [78]. Oxidative stress is considered one of the main determinants in vascular aging and related diseases [79, 80]. Aging vasculature produces excessive ROS, primarily generated by reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, mitochondrial respiratory chain, xanthine oxidase, and uncoupled endothelial nitric oxide (NO) synthase [81]. The overproduction of ROS leads to endothelial dysfunction and large artery stenosis [82-84]. Adequate production of NO by the vascular endothelium exerts pivotal roles in regulating vasodilation and tissue perfusion [85], while compromised bioavailability of endothelium-derived NO is critical for mediating the effects of oxidative stress in blood vessel function with aging [82, 86]. Additionally, the weakening antioxidant response mediated by nuclear factor erythrocyte 2-related factor 2 (NRF2) and the inactivation of mitochondrial manganese superoxide dismutase (SOD2) also result in chronic oxidative stress in the aging vasculature [87, 88]. It has demonstrated that through regulating Kelch-like ECH-associated protein 1 (KEAP1)/NRF2 signaling, METTL3/m6A/miR-873-5p axis mitigates oxidative stress and cell apoptosis [89].
ROS induces cell senescence along with altered m6A level and expression profiles of m6A modification regulators and their target genes [90]. Further, METTL3/METTL14-catalyzed m6A has been revealed to promote P21 expression, thus aggravating oxidative stress-induced cellular senescence [91]. These studies indicate the potential role of m6A in age-related pathophysiological changes induced by oxidative stress.
Under oxidative stress, mRNAs are additionally m6A-modified in the 5' UTR, facilitating mRNA triaging to stress granules (SGs), which are membraneless granules mainly containing untranslated mRNAs and RNA-binding proteins in cells in response to stress conditions [92]. YTHDF proteins, especially YTHDF1/3 are crucial in regulating SG formation [92, 93]. These results suggest a novel function of m6A modification in regulating the oxidative stress response.
2.2.2. Chronic inflammation
Inflammation is a protective response to various internal and external stimuli such as injury, infection, and tissue stress that may challenge homeostasis; however, persistent, or excessive inflammation can cause tissue injury and diseases [94]. “Inflamm-aging” is a result of an imbalance between inflammatory and anti-inflammatory networks, and represents the chronic, low-grade, non-infective inflammation that develops with age [95]. Importantly, this sterile inflammation has been regarded as a dominator of vascular aging and a central player in most vascular aging-related diseases [96-98]. Several anti-inflammatory interventions such as canakinumab and colchicine have been proven effective in the context of some vascular aging-related afflictions [99]. Emerging evidence has unraveled the role of m6A modification in the modulation of inflammation [100].
Increased abundance and changed polarization of macrophages are important hallmarks of aging [101]. Recent studies have established that m6A modification alters when stimulated by oxidized low-density lipoprotein (oxLDL) [102, 103], lipopolysaccharide (LPS) [104], or interferon-γ (IFN-γ) [105], or IL-4 [106] in macrophages, and therefore dynamically regulating macrophage inflammation and polarization. Literature has described that m6A participates in the modulation of macrophage polarization through targeting signal transducer and activator of transcription 1 (STAT1) [105, 107], and myeloid differentiation primary response 88 (MYD88) [104]. Emerging evidence has established that m6A regulators, METTL3 [107], METTL14 [104], FTO [108], IGF2BP2 [106], and YTHDF2 [105] are involved in the modulation of macrophage polarization. Specifically, METTL3 [107] and METTL14 [104] promote macrophage M1 polarization, while IGF2BP2 is a positive regulator of M2 activation [106]. Downregulation of FTO suppresses both M1 and M2 macrophage polarization [108].
In addition to the regulation of macrophage inflammation, m6A also plays an important role in EC and VSMC inflammation. For example, METTL3- and METTL14-mediated m6A modification promotes EC inflammation through regulating NOD-like receptor protein 1 (NLRP1), Krüppel-like factor 4 (KLF4), or forkhead box O1 (FOXO1) mRNA [109, 110]. Moreover, downregulation of FTO expression suppresses VSMC inflammation by targeting nuclear receptor subfamily 4, group A, member3 (NR4A3) [111].
An in-depth exploration of the interplay between m6A and inflammation is of great importance to identify more pathogenic pathways and develop promising therapeutic approaches to inflammation-associated diseases.
2.2.3. Mitochondrial dysfunction
Dysfunctional mitochondria underly the pathological mechanism of vascular aging [83]. Structural and functional alterations in mitochondria potentially result in EC and VSMC senescence, including dysfunctional mitochondrial dynamics, mitochondrial energy metabolism, mitophagy, and consequent mitochondrial DNA mutations [112-114]. Particularly, excessive ROS produced by dysfunctional mitochondria exert a pivotal role in vascular aging as described above. Additionally, numerous interplays exist between mitochondrial dysfunction and other age-related molecular and cellular mechanisms, such as low-grade inflammation and telomere attrition, which further exacerbate vascular aging [114].
Emerging evidence suggests the regulatory function of m6A modification in mitochondrial homeostasis. Specifically, METTL3 coordinates with YTHDF2 to enhance oxLDL-induced mitochondrial dysfunction and inflammation in monocytes by promoting m6A-mediated mRNA degradation of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), a key regulator of mitochondrial biogenesis [115]. METTL3 and YTHDF2 synergistically downregulate electron transport chain proteins cytochrome c (CYCS) and NADH: ubiquinone oxidoreductase subunit C2 (NDUFC2), and diminish ATP generation and oxygen consumption, and thus elevate ROS production [115]. These results provide a potential mechanism for vascular aging-related diseases.
2.2.4. Autophagy
Autophagy serves as a highly conserved intracellular degradation and recycling mechanism of senescent or malfunctioning organelles to maintain cellular renovation and homeostasis. Notably, it is believed that autophagy is a fundamental mechanism for the maintenance of cardiovascular homeostasis during aging [116]. Experiments demonstrated that autophagy-deficient EC appears increased oxidative stress, impaired NO bioavailability, and increased inflammatory mediators. Autophagic activity is decreased in vascular tissues of aged humans and mice [117]. Dysregulation of autophagic processes is associated with vascular aging and related diseases [118]. The aspects of arterial aging can be reversed with the administration of autophagy enhancers such as spermidine [119] and nicotinamide mononucleotide [120].
Recent studies have shown that m6A modification plays important roles in autophagy regulatory networks by regulating autophagy-related genes, such as transcription factor EB (TFEB) [121], unc-51-like kinase 1 (ULK1) [122], and autophagy-related (ATG) genes [123, 124], which serves as a potential mechanism in vascular aging. For example, METTL3 and ALKBH5 reversely regulate m6A modification of TFEB to affect autophagic flux [121]. FTO potentiates the initiation of autophagy through increasing ULK1 mRNA stability by demethylation [122]. Notably, the m6A writer METTL3 has been identified as a regulator of autophagy to affect VSMC behavior, serving as a potential mechanism of vascular aging. Specifically, METTL3 promotes autophagosome formation by upregulating the expression of ATG5 and ATG7, and thus inhibits VSMC proliferation and prevents VSMCs from switching to synthetic phenotype [124]. Further studies are needed to fully elucidate the mechanisms underlying the role of m6A modification in autophagy and its potential implications for vascular aging-related diseases.
2.2.5. DNA damage
DNA damage refers to the structural alteration of DNA caused by various endogenous and exogenous factors such as oxidation, replication errors, radiation, and chemical compounds, which can affect its normal function [125]. DNA damage has been implicated in the pathogenesis of vascular aging. It is reported that mice with genomic instability present increased vascular stiffness, vasodilator dysfunction, and blood pressure, suggesting the association between variations in human DNA repair genes and vascular aging [126]. Moreover, accumulating evidence indicates that DNA damage gives rise to endothelial dysfunction, inflammation, and cellular senescence, which all are hallmarks of vascular aging [4, 127].
Recent studies clarified the correlation between m6A and DNA damage and repair [128, 129]. Dysregulation of m6A modification has been implicated in the development of genomic instability, which can contribute to the onset and progression of various diseases, including aging-related disorders [76]. Recent studies have shown that m6A modification and its regulators play a critical role in the DNA damage response (DDR) pathway. Specifically, m6A regulators, such as METTL3 [130-132], METTL14 [132, 133], FTO [132], ALKBH5 [134], YTHDC1 [131], regulate DDR pathway through m6A in response to different stimuli such as ROS [134], ultraviolet [132], radiation [131], and various chemicals [130, 131, 133]. For example, METTL3-catalyzed m6A modification promotes DDR by stabilizing mRNAs of DNA damage repair factors, RAD51 recombinase (RAD51) and X-ray repair cross-complementing 5 (XRCC5), thereby maintaining cell survival [130]. Moreover, DNA damage repair has recently been verified to be closely related to m6A-modified retrotransposable element (RTE) RNAs, especially intronic long interspersed element-1 (LINE-1), which often inhibit hosting gene transcription and preferentially reside in host genes with vital functions in DNA damage repair [135]. METTL3-m6A-YTHDC1 axis facilitates homologous recombination-mediated repair by promoting the accumulation of DNA-RNA hybrids at double-strand break sites [131].
These findings suggest that m6A modification plays a crucial role in regulating DNA damage and repair processes, contributing to the development of aging and age-related diseases. However, further investigation is needed to explore the role and mechanisms of m6A modification in vascular aging through DNA damage.
2.2.6. Cellular senescence
Cellular senescence is a state of irreversible growth arrest that occurs after a finite number of divisions or in response to diverse stimuli, where cells present distinctive phenotypic switching [136]. Senescent cells accumulate in tissues during aging and contribute to the development of age-related diseases, including vascular aging. Evidence suggests that chronic senolytic treatment targeting senescent cells can attenuate aspects of vascular aging [137]. Senescent cells secrete senescence-associated secretory phenotype (SASP), including pro-inflammatory cytokines, chemokines, growth factors, and proteases. These SASP components drive autocrine and paracrine signaling, impairing vascular function, and promoting the development of vascular aging-related diseases such as atherosclerosis (AS) [76, 138].
Several studies have reported that alterations in m6A levels or in the expression of m6A regulators are associated with cellular senescence [91, 139, 140]. Emerging evidence further demonstrated that m6A regulators play a key role in cell senescence [77]. For example, METTL3/14 levels gradually decrease in cells experiencing replicative senescence, while the overexpression of METTL14 mitigates both replicative and premature senescence [140]. Similarly, METTL3 overexpression reverses the premature senescence of human mesenchymal stem cells (MSC) through m6A-mediated MIS12 mRNA stabilization in cooperation with IGF2BP2 [141]. However, in another study, METTL3 was found to promote autophagy-regulated senescence in fibroblast-like synoviocytes [123]. Notably, researchers unraveled that METTL14 and FTO affect cell cycle to regulate senescence through m6A methylation. METTL14 regulates m6A-mediated maturation of miR-34a-5p, which promotes cell cycle arrest and senescence by targeting Sirtuin-1 (SIRT1) [142]. Another study revealed that FTO depletion increases m6A-mediated degradation of Cyclin D1 mRNA, contributing to impaired cell cycle progression [143].
Recently, FTO and YTHDF2 have been found to be related to EC senescence, suggesting a potential mechanism of vascular aging. Li et al. reported the role of FTO in promoting EC senescence [144]. The results suggest that targeting m6A regulators or their target genes may represent a promising strategy for preventing or treating cellular senescence and its associated pathologies. Further studies are needed to fully elucidate the mechanisms underlying the role of m6A modification in cellular senescence and its potential implications for therapeutic interventions in vascular aging.
3. Unraveling Vascular Aging: Clinical Relevance
Vascular aging significantly impacts human health, leading to an increased risk of diseases. Identifying clinical manifestations associated with vascular aging allows for early detection and intervention. Accumulating evidence suggests that m6A presents pleiotropic effects on these diseases through gene expression regulation (Table 1). In this section, the role of m6A modification in different vascular aging-related diseases is explored, including CVDs, encephalopathy, and chronic kidney disease (CKD) (Fig. 4).
Table 1.
The role of m6A in vascular aging-related diseases.
| Disease | m6A regulator | Target genes | Function | Ref |
|---|---|---|---|---|
| Vascular diseases | ||||
| AS | METTL3 | NLRP1↑, KLF4↓ | Mediates proatherogenic inflammatory responses in ECs induced by disturbed blood flow | [109] |
| METTL3 | NPC1L1↑ | Promotes EC dysfunction and AS development | [152] | |
| METTL3 | JAK2↑ | Promotes oxLDL-stimulated EC dysfunction and AS progression | [153] | |
| METTL3 | STAT1↑ | Enhances oxLDL-induced inflammation in macrophages | [102] | |
| METTL3 | BRAF↑ | Facilitates macrophage inflammatory response and AS | [158] | |
| METTL3 | pri-miR-375-3p↓ | Promotes AS progression and destabilizes AS plaques by facilitating oxLDL-induced phenotypic transformation of VSMCs | [160] | |
| METTL3 | EGFR↓ | Attenuates endothelial atherogenic progression | [156] | |
| METTL14 | FOXO1↑ | Induces endothelial inflammation and contributes to AS progression | [110] | |
| METTL14 | pri-miR-19a↓ | Promotes the proliferation and invasion of atherosclerotic vascular ECs | [154] | |
| METTL14 | P65↑ | Decreases EC viability and enhances EC apoptosis stimulated by oxLDL | [155] | |
| METTL14 | MYD88↑ | Mediates macrophage inflammation and development of AS plaques | [104] | |
| METTL14 | UCHL5↑ | Exacerbates AS and VSMC phenotypic switching | [161] | |
| Hypertension | FTO | NR4A3↑ | DHA attenuates AngII-induced VSMC proliferation and inflammation by downregulating FTO expression | [111] |
| AAD | METTL3 | pri-miR-34a↓ | Promotes the formation of aortic aneurysm in mice | [175] |
| METTL3 | SLC7A11↓, FSP1↓ | Promotes ferroptosis of VSMCs | [177] | |
| METTL3-METTL14 complex | RIP3↑ | Promotes necroptosis and inflammation of VSMCs and aortic aneurysms progression | [179] | |
| KIAA1429, ALKBH5 | pri-miR-143-3p↓ | Oppositely affect aortic dissection progression through modulating VSMC proliferation | [180] | |
| FTO | KLF5↑ | Mediates AngII-induced VSMC proliferation and migration | [176] | |
| Heart diseases | ||||
| MI | METTL3 | circ_0029589↓ | Mediates IRF-1-induced macrophage pyroptosis and inflammation | [187] |
| METTL3 | pri-miR-503↓ | Evokes miR-503 biogenesis in ECs; exosomal miR-503 triggers mitochondrial dysfunction and cardiomyocyte death | [188] | |
| METTL3 | pri-let-7e↓, pri-miR-17-92↓ |
Improves post-ischemic neovascularization in MI mice | [191] | |
| METTL3 | SMAD2/3↑ | Aggravates MI-induced cardiac fibrosis through the activation of cardiac fibroblasts | [193] | |
| ALKBH5 | YTHDF1↑ | Reduces infarct size, improves cardiac function, and enhances cardiomyocyte proliferation | [189] | |
| ALKBH5 | SPHK1↑ | Maintains EC angiogenesis during acute ischemic stress | [190] | |
| HF | METTL3 | MAP3K6↑, MAP4K5↑, MAPK14↑ | Controls cardiac homeostasis and hypertrophy | [183] |
| FTO | SERCA2a↑ | Alleviates ischemia-induced decrease in cardiac function | [182] | |
| FTO | - | Mice with cardiomyocyte-specific knockout of FTO presents worsened cardiac function | [197] | |
| FTO | PGAM2↑ | Mitigates cardiac dysfunction in HF mice through modulating glycolysis | [199] | |
| FTO | - | FTO overexpression counteracts exercise benefits in HFpEF mice by inducing myocyte apoptosis, myocardial fibrosis, and myocyte hypertrophy | [201] | |
| Encephalopathy | ||||
| Stroke | METTL3 | pri-miR-335↓ | Induces stress granule formation and attenuates the apoptosis of injury neuronal cells | [209] |
| FTO | - | Minimizes poststroke brain damage and neurobehavioral deficits | [211] | |
| FTO | PLPP3↑ | Mediates circSCMH1-promoted vascular repair after stroke | [212] | |
| YTHDC1 | PTEN↓ | Attenuates post-ischemic brain injury | [210] | |
| YTHDF1 | P65↑ | miR-421-3p presents anti-inflammatory effects in cerebral IRI by targeting YTHDF1 | [213] | |
| AD | METTL3 | Cyclin D2↓ | METTL3 knockout in the hippocampus leads to memory deficits, synaptic loss, and neuronal death | [220] |
| METTL3 | STUB1↑ | Facilitates autophagic clearance of p-Tau in AD cell model | [221] | |
| METTL3 | ARC↑ | Rescues Aβ-stimulated decrease in ARC expression | [223] | |
| METTL3 | DNMT3A↑ | METTL3 ablation in monocyte-derived macrophages attenuates AD pathology | [224] | |
| FTO | - | Conditional knockout of FTO in the neurons decreases cognitive deficits in AD mice | [227] | |
| HNRNPA2B1 | - | Serves as a connector between oTau and m6A-modified RNAs, subsequently regulating stress response and mediating the development of tauopathy | [228] | |
| IGF2BP2 | - | IGF2BP2-related gene modules are significantly enriched in AD-associated biological processes | [229] | |
| Kidney diseases | ||||
| CKD | METTL3 | pri-miR-21↓ | Promotes renal fibrosis by regulating inflammation | [235] |
| METTL3 | lncRNA MALAT1↑ | Promotes TGF-β1-induced renal fibrosis | [236] | |
| METTL14 | Klotho↓ | Motivates VSMC osteogenic conversion stimulated by indoxyl sulfate | [242] | |
| WTAP | NLRP3↑ | Induces cell pyroptosis and inflammation in DN models | [238] | |
| FTO | lncRNA GAS5↓ | Promotes renal epithelial-mesenchymal transition and inflammation response | [237] | |
| FTO | SOCS1↑ | Overexpression of FTO attenuates inflammation response and kidney injury of DN | [239] |
The upward arrow represents “upregulated”; the downward arrow represents “downregulated”.
Figure 4.

Dysregulation of m6A modifiers in vascular aging-related diseases. Red-colored modifiers indicate a pathogenic role, green-colored modifiers indicate a protective role, while yellow-colored ones have controversial roles reported, in the specific disease type. This figure was created with the aid of Biorender (https://biorender.com/). AD: Alzheimer's disease; HF: heart failure; MI: myocardial infarction; CKD: chronic kidney disease; AAD: aortic aneurysm/dissection; AS: atherosclerosis.
3.1. The roles of m6A in aging-related vascular diseases
Aging is the major risk factor for vascular diseases [145]. Marked aging changes in the vasculature make arteries more susceptible to vascular diseases. Recent research highlights the importance of m6A modification in modulating gene expression and maintaining vascular health during aging, while the dysregulation of m6A methylation may contribute to vascular dysfunction and age-related vascular diseases, such as AS, hypertension, and aortic aneurysm/dissection (AAD).
3.1.1. m6A in AS
AS, a primary cause of CVDs, is an inflammatory disease that occurs in the large arteries [146]. Atherosclerotic plaques are characterized by accumulated and transformed lipids, macrophages, VSMCs, and necrotic cell debris in the subendothelial space just underneath the EC layer in the artery wall [101]. Vascular aging promotes the occurrence and development of AS, which in turn accelerates the process of vascular aging. Interestingly, altered levels of m6A and its regulators are identified in AS tissues, which indicates the potential interplay between m6A and AS [147, 148]. Notably, accumulating data have revealed the regulatory roles of m6A modification in the initiation and progression of AS through regulating EC dysfunction and inflammation, macrophage inflammation, and VSMC phenotypic transformation [149, 150].
EC dysfunction and inflammation are key pathological features in AS [151]. It is reported that METTL3-catalyzed m6A modification mediates proatherogenic inflammatory responses in ECs induced by disturbed blood flow through upregulating NLRP1 and downregulating KLF4 expression [109]. Further, METTL3-dependent m6A methylation of Niemann-Pick C1-Like 1 (NPC1L1) promotes EC dysfunction and AS development, possibly through regulating the mitogen-activated protein kinase (MAPK) pathway [152]. On the contrary, silencing METTL3 attenuates oxLDL-stimulated EC dysfunction and impedes AS progression in vivo by repressing the JAK2/STAT3 pathway via m6A/IGF2BP1-dependent regulatory mechanisms [153]. METTL14-mediated m6A modification also regulates the pathogenesis of AS by targeting ECs. Specifically, METTL14 facilitates the translation of FOXO1 mRNA through YTHDF1 recognition in an m6A-dependent manner, which elevates adhesion molecule expression, induces endothelial inflammation, and ultimately contributes to the progression of AS [110]. Besides, METTL14 promotes the proliferation and invasion of atherosclerotic vascular ECs, probably through m6A-mediated pri-miR-19a processing [154]. METTL14 decreases EC viability and enhances EC apoptosis stimulated by oxLDL through m6A modification of P65; while knockdown of METTL14 can suppress AS progression in vivo [155]. On the other hand, m6A can also exert protective effects on AS. Mechanistically, METTL3 attenuates endothelial atherogenic progression through m6A-dependent mRNA decay of epidermal growth factor receptor (EGFR), a molecule related to EC dysfunction [156].
In addition to ECs, macrophages are pivotal players in the vascular inflammatory process of AS [157]. METTL3 and METTL14-mediated m6A modification affects AS process by regulating macrophage inflammation. Specifically, METTL3 facilitates the m6A modification of STAT1 and v-Raf murine sarcoma viral oncogene homolog B (BRAF) to enhance oxLDL-induced inflammation in macrophages, thus promoting AS progression [102, 158]. METTL14 mediates macrophage inflammation and development of AS plaques via m6A-modified mRNA stabilization of MYD88 that regulates the nuclear factor kappa B (NF-κB)/IL-6 signaling [104]. The RNA binding protein Matrin-3 (MATR3) inhibits oxLDL-induced macrophage inflammation and attenuates AS development by promoting the formation of METTL3-METTL14 complex and m6A-mediated mRNA decay of MAPK [103].
It is well-documented that the phenotypic transformation of VSMCs to proliferative synthetic cells contributes to AS development [159]. Recent evidence has revealed the role of m6A in this process. Specifically, silencing METTL3 attenuates AS progression and stabilizes AS plaques by mitigating oxLDL-induced phenotypic transformation of VSMCs partly through m6A-modified pri-miR-375 processing [160]. Moreover, METTL14 recruits YTHDF1 to enhance m6A and expression level of Ubiquitin C-terminal hydrolase L5 (UCHL5), which exacerbates AS and VSMC phenotypic switching by stabilizing the NLRP3 inflammasome [161].
3.1.2. m6A in hypertension
Vascular aging is a crucial player in the pathogenesis of hypertension. Hypertension has become a crucial risk factor for mortality and CVDs [162, 163], which develops as a result of disorders of the renin-angiotensin-aldosterone system (RAAS), the sympathetic nervous system, and the immune system [164].
Research on the function of m6A modification in hypertension is still in its infancy. A study has unraveled that FTO rs9939609 is negatively associated with mean and diastolic blood pressure in male hypertension patients [165]. In another study, 1236 m6A-associated single nucleotide polymorphisms (SNPs) are nominally associated with blood pressure, and among them, rs7398833 in CUX2 and rs13096477 in SLC4A7 are the most significant [166], highlighting the potential effects of m6A in blood pressure regulation. Further, m6A RNA methylomes are altered and the abundance of m6A methylation is reduced in pericytes of spontaneously hypertensive rats [164]. Later on, another study described that the downregulation of FTO expression suppresses angiotensin II (AngII)-induced VSMC proliferation and inflammation by regulating the m6A methylation of NR4A3 [111]. These results suggest the potential roles of m6A in hypertension-related vascular complications. Further exploration is warranted for how m6A modulates gene expression to trigger hypertension progression.
3.1.3. m6A in AAD
Changed mechanical properties of the vessel wall with aging increase the fragility of artery and make it prone to aneurysm. Aortic aneurysm is the second most common disorder involving the aorta after AS, and its global burden remains high [167]. It is identified as a localized dilation of aorta attributed to acute factors (eg. trauma) or diseases (eg. hypertension). The large majority of aortic aneurysms are asymptomatic. However, progressive enlargement of aortic aneurysm increases the risk for aortic dissection, which can be life-threatening [168].
The m6A abundance remarkably increases in AAD in comparison with healthy aorta tissues and acts as a risk factor for aortic aneurysm rupture [169, 170]. Recent data from bioinformatic analyses suggest the potential roles of m6A in AAD [170-173]. VSMC dysfunction is a major contributor to the development and progression of aortic aneurysm and dissection [174]. Further evidence has revealed the possible involvement of m6A methylation in these diseases via regulating VSMC function [175-177]. Specifically, METTL3 promotes the formation of aortic aneurysm in mice through m6A-promoted miR-34a maturation and consequent downregulation of SIRT1 in VSMCs [175]. Importantly, miR-34a has been found to stimulate VSMC senescence by SIRT1 and enhance the expression of pro-inflammatory senescence-associated secretory phenotype [178]. METTL3-METTL14 complex promotes necroptosis and inflammation of VSMCs and progression of abdominal aortic aneurysms by mediating m6A modification of receptor-interacting protein 3 (RIP3) [179]. Moreover, the expression of METTL3 and FTO is increased in human aortic dissection tissues [176, 177]. METTL3 promotes ferroptosis of VSMCs by downregulating key ferroptosis regulatory proteins, solute carrier family 7 member 11 (SLC7A11) and ferroptosis suppressor protein 1 (FSP1); while the specific inhibitor of ferroptosis can alleviate the development and rupture of aortic dissection in vivo [177]. In another study, FTO mediates AngII-induced VSMC proliferation and migration, probably through m6A demethylation of KLF5 [176]. Additionally, KIAA1429 and ALKBH5 oppositely affect aortic dissection progression through modulating VSMC proliferation by m6A-regulated pri-miR-143-3p maturation [180].
Overall, m6A methylation plays a great role in AAD by regulating the proliferation, migration, senescence, and programmed cell death of VSMCs. Future work on revealing the underlying mechanisms of the interplay between m6A and AAD are needed.
3.2. The roles of m6A in vascular aging-related heart diseases
Structural and functional alterations of the heart occur with aging, such as increased stiffness, myocardial hypertrophy, and cardiac dysfunction [181]. The role of m6A in the maintenance of cardiac homeostasis has been established [182, 183]. Emerging literature highlights the significance of m6A modification in regulating vascular aging-related heart diseases such as myocardial infarction (MI) and heart failure (HF) [6, 24].
3.2.1. m6A in MI
The interplay between vascular aging and AS leads to the occurrence of MI. MI is a life-threatening disorder characterized by an abrupt drop in coronary blood flow, contributing to ischemia and, eventually, the loss of myocardium [184]. Recent studies have indicated that m6A presents multifaceted effects on MI and approaches targeting m6A exhibit potential for treatment of MI [24, 185].
m6A is an important player during the progression of MI. It is well-established that inflammation and cell death are key pathological alterations during the progression of MI [186]. It is reported that the expression and m6A level of hsa_circ_0029589 is decreased, while METTL3 expression is increased in macrophages from patients with acute coronary syndrome. Of note, IFN regulatory factor-1 (IRF-1) triggers macrophage pyroptosis and inflammation by inhibiting circ_0029589 via METTL3-mediated m6A modification [187]. Further, m6A-installed molecules can be transferred from ECs to cardiomyocytes and thus induce cardiac injury in MI. Mechanistically, hypoxia-induced METTL3 overexpression promotes m6A-mediated miR-503 maturation in ECs, which is transported to cardiomyocytes, eventually leading to mitochondrial dysfunction and cardiomyocyte death after MI [188].
m6A plays important roles in post-MI repair and regeneration by regulating cardiomyocyte proliferation, EC angiogenesis, and cardiac fibrosis. For instance, improving the expression of ALKBH5 remarkably decreases the infarct size, improves cardiac function, and enhances cardiomyocyte proliferation after MI in mice. This effect is mediated through ALKBH5-promoted YTHDF1 mRNA stability in an m6A-dependent manner in cardiomyocytes, which eventually contributes to increased Yes-associated protein (YAP) translation [189]. Moreover, ALKBH5 maintains EC angiogenesis during acute ischemic stress, which may be mediated by the demethylation of sphingosine kinase-1 (SPHK1) [190]. Similarly, implantation of METTL3-overexpression ECs improves post-ischemic neovascularization in MI mice by regulating the maturation of let-7e-5p and miR-18a-5p [191]. A recent MeRIP-seq analysis of heart samples from MI and control rats identified m6A-modified hub mRNAs and found their association with angiogenesis and apoptosis [192]. However, the role of these transcripts in MI pathologies needs to be further validated. In terms of cardiac fibrosis, METTL3 exhibits a pro-fibrotic role in the myocardium after MI. METTL3 silencing attenuates MI-induced cardiac fibrosis in vivo through the inactivation of cardiac fibroblasts mediated by m6A methylation of fibrosis-related genes such as small mothers against decapentaplegic homolog (SMAD) 2/3 [193].
3.2.2. m6A in HF
HF is defined as a clinical syndrome consisting of symptoms and/or signs raised by structural or functional abnormality of ventricular filling or cardiac ejection [194]. Vascular aging-related reduction of coronary blood flow, AS, and hypertension are important mechanisms underlying HF pathogenesis. This disorder is the leading cause of hospitalizations in the elderly with high morbidity and mortality [195, 196]. HF is associated with deregulated epigenetic processes and abnormal gene expression [197]. Especially, RNA m6A methylation has been well-documented to participate in the development of HF [185, 198].
m6A levels in human, pig, and mouse failing hearts are increased compared to those in normal controls [182]. Also, transcriptome profiling of m6A is changed in the HF mouse model and failing human hearts, and differentially methylated transcripts code for proteins mainly associated with cardiac muscle differentiation and metabolic processes [197]. FTO is downregulated in failing mammalian hearts and hypoxic cardiomyocytes, and FTO overexpression in failing mouse hearts alleviates ischemia-induced decrease in cardiac function by selectively demethylating cardiac contractile transcripts such as sarcoplasmic reticulum calcium ATPase 2a (SERCA2a) [182]; while mice with cardiomyocyte-specific knockout of FTO presents worsened cardiac function compared to control mice [197], suggesting the necessity of FTO for maintaining cardiac homeostasis. Further studies revealed the underlying regulatory mechanism of FTO on HF progression. Specifically, FTO mitigates cardiac dysfunction in pressure overload-induced HF mice through modulating glycolysis at least partially by removing phosphoglycerate mutase 2 (PGAM2) m6A and also modulating glucose uptake probably by regulating the protein kinase B (AKT)-glucose transporter type 4 (GLUT4) pathway [199].
In addition to FTO, METTL3 also participates in the process of HF. It is reported that overexpression of METTL3 facilitates cardiomyocyte hypertrophy both in vitro and in vivo largely through m6A methylation of mitogen-activated protein kinases, whereas cardiac-specific METTL3 knockout mice present structural and functional characteristics of HF with aging and stress [183].
HF with preserved ejection fraction (HFpEF) is a large subset of HF. Whereas, fundamental biological processes involved in this disturbance remain largely elusive. A recent study found that m6A regulators (e.g., METTL3, METTL4, KIAA1429, FTO, and YTHDF2) are differentially expressed and m6A landscape is changed in HFpEF patients and/or mice, suggesting that m6A may act on the development of HFpEF [200]. Further, exercise training has been shown to alleviate myocardial phenotypes in high-fat diet (HFD)-induced HFpEF mice with altered m6A patterns and decreased FTO expression. However, overexpression of FTO counteracts the beneficial effects of exercise in HFpEF mice by inducing myocyte apoptosis, myocardial fibrosis, and myocyte hypertrophy [201].
Collectively, these studies suggest that METTL3 and FTO control cardiac homeostasis in an m6A-dependent manner, while their dysregulation is involved in HF progression. Further efforts to probe the underlying mechanisms of m6A and its regulators on the development of HF will help to provide new insight into biomarker identification and therapy exploration for HF.
3.3. The roles of m6A in vascular aging-related encephalopathy
Similar to the mechanism of vascular aging-related kidney diseases, elevated pulse pressure induced by vascular aging promotes the structural and functional abnormality of cerebral microvessels. These alterations can ultimately lead to cerebrovascular diseases and cognitive impairment [202].
3.3.1. m6A in stroke
Vascular aging is an important player in the pathogenesis of stroke [203]. Stroke remains the second leading cause of death worldwide. The incidence and fatality of stroke increased heavily in the past three decades, particularly among the elderly [204]. Increasing data suggest the role of m6A in the pathogenesis of stroke [205].
Mo et al. identified 310 m6A-SNPs that were nominally associated with ischemic stroke [206]. Further data suggest that stroke changes the m6A profile. After cerebral ischemia, m6A abundance is increased and m6A epitranscriptome is changed significantly in mouse cortex [207, 208]. Further, METTL3-catalyzed m6A methylation promotes the maturation of miR-335, which induces stress granule formation and attenuates the apoptosis of injury neuronal cells by targeting eukaryotic translation termination factor (eRF1) in the early stage of acute ischemic stroke [209]. Several other m6A regulators have also been found to play a role in stroke. For example, YTHDC1 attenuates post-ischemic brain injury by promoting PTEN mRNA degradation to increase AKT phosphorylation [210]. Interestingly, exogenous FTO substantially minimizes poststroke brain damage and neurobehavioral deficits in vivo [211]. circSCMH1 promotes vascular repair after stroke through FTO-regulated m6A methylation of phospholipid phosphatase 3 (PLPP3) [212]. Moreover, miR-421-3p presents anti-inflammatory effects in cerebral ischemia/reperfusion injury by targeting YTHDF1 which mediates m6A modification of P65 mRNA to regulate its translation [213].
3.3.2. m6A in AD
AD is a complex neurodegenerative disease and is the leading cause of dementia in the elderly. The pathological hallmarks of AD are amyloid plaques consisting of β-amyloid (Aβ) peptides and neurofibrillary tangles composed of hyperphosphorylated tau in the brain. Age-related vascular alteration is a possible pathogenic factor in AD progression [214].
Evidence suggests that m6A modification is associated with AD [215]. Decreased m6A levels are detected in brain tissues of aged mice and AD patients [216, 217]. Of note, plentiful AD-associated transcripts present dysregulated m6A methylation in the AD mouse models and subsequently affect their protein levels, which indicates the potential pathogenic role of m6A in AD progression [216-218].
Reduced neuronal m6A abundance and METTL3 expression are found in human AD brains [219, 220]. METTL3 in the insoluble fractions is positively correlated with the abundance of insoluble tau protein in human AD samples [219]. Of note, METTL3 knockout in the hippocampus leads to memory deficits, synaptic loss, and neuronal death, which might be mediated by m6A dysregulation of the cell cycle gene, Cyclin D2 [220]. A recent study reported that METTL3 facilitates autophagic clearance of p-Tau in the Aβ-induced cell model of AD via m6A-mediated stabilization of STIP1 homology and U-box containing protein 1 (STUB1) [221]. The expression of activity-regulated cytoskeleton-associated protein (ARC) is reduced in AD patients and cell models, which functions as a key factor for AD [222]. Interestingly, METTL3 rescues Aβ-stimulated decrease in ARC expression through YTHDF1-recognized m6A methylation [223]. However, METTL3 ablation in monocyte-derived macrophages attenuates AD pathology in a mouse model induced by Aβ-injection [224]. Further experiments revealed that METTL3 deficiency decreases m6A modification of DNA methyltransferase 3A (DNMT3A) that subsequently affects alpha-tubulin acetyltransferase 1 (ATAT1) expression [224].
An early prospective cohort study suggests that the FTO AA genotype is associated with a higher AD risk [225]. Consistently, another study also identified genetic variations in the FTO gene that might contribute to AD risk, and detected reduced FTO expression in human AD brains, indicating the potential function of FTO in AD pathology [226]. A more recent study revealed that conditional knockout of FTO in the neurons decreases cognitive deficits in AD mice. Mechanistic experiments suggested that FTO induces tau phosphorylation through activating tuberous sclerosis complex 1 (TSC1)-mTOR signaling [227]. Whereas few studies focus on the role of FTO-mediated m6A demethylation in AD progression.
Emerging evidence revealed the association of HNRNPA2B1 with tau that mediates the development of tauopathy [228]. Mechanistically, HNRNPA2B1 serves as a connector between oligomeric tau (oTau) and m6A-modified RNAs under AD conditions, subsequently regulating stress response and protein synthesis. While knockdown of HNRNPA2B1 reduces tau-induced neurodegeneration by dampening the association of oTau with m6A transcripts [228].
Bioinformatic analysis found that IGF2BP2 is highly expressed in human AD brain tissues. Besides, gene modules related to IGF2BP2 are significantly enriched in AD-associated biological processes, such as cytokine-cytokine receptor interaction and the TGF-β signaling pathway. Importantly, a diagnostic model for AD based on IGF2BP2-related genes has been constructed and validated [229]. These results indicate the potential of IGF2BP2 and associated m6A methylation in AD diagnosis and therapy.
Although several researchers have reported the role of m6A regulators in AD, especially METTL3, FTO, HNRNPA2B1, and IGF2BP2, the specific underlying molecular mechanisms remain largely unknown. Thus, in-depth mechanistic studies are warranted to provide a better understanding of the function of m6A in AD [230].
3.4. The roles of m6A in vascular aging-related kidney diseases
3.4.1. m6A in CKD
Chronic kidney disease (CKD) refers to chronic abnormalities of renal function and/or structure, leading to a marked global disease burden [231]. Renal function progressively declines with aging. Vascular aging is significantly associated with CKD development [232]. Vascular aging-related artery stiffness can increase pulse pressure; its wave penetrates deeper into susceptible renal microvasculature, contributing to renal microvascular damage that may ultimately promote renal dysfunction and the development of end-stage renal disease [233].
Accumulating data indicate that m6A plays an important role in the pathogenesis of CKD. Renal fibrosis is a hallmark and common outcome in a variety of progressive CKD [234]. m6A may modulate renal fibrosis by targeting ncRNAs. For instance, METTL3-catalyzed m6A modification promotes renal fibrosis by promoting miR-21-5p maturation. Further experiments recovered that miR-21-5p promotes inflammation probably through the activation of Sprouty RTK signaling antagonist 1 (SPRY1)/extracellular signal-regulated kinase (ERK)/NF-κB pathway [235]. Another study reported that METTL3 modifies m6A methylation on long-non-coding RNA (lncRNA) MALAT1 to upregulate MALAT1 expression, which thereby promotes transforming growth factor β1 (TGF-β1)-induced renal fibrosis through miR-145/FAK signaling [236]. Moreover, Li et al. found that FTO promotes renal epithelial-mesenchymal transition (EMT) and inflammation response, which may be mediated by regulating m6A demethylation of lncRNA GAS5 [237]. These results provide a better understanding of novel mechanisms of m6A in CKD, which helps drug development for renal fibrosis.
Emerging evidence has also uncovered the important role of m6A in diabetic nephropathy (DN) progression. Lan et al. reported that WTAP induces NLRP3 m6A methylation to mediate NLRP3 inflammasome activation in an IGF2BP1-dependent manner, further regulating cell pyroptosis and inflammation in DN models [238]. A recent study demonstrated that FTO expression is decreased in DN patients. Overexpression of FTO exerts protective effects during the pathogenesis of DN by increasing the expression of suppressors of cytokine signaling 1 (SOCS1) to attenuate inflammation response and kidney injury [239].
Vascular calcification is recognized as a common complication and marker of higher mortality and cardiovascular events in CKD patients [240, 241]. Previous research has established the role of m6A in regulating vascular calcification. Specifically, METTL14-dependent m6A motivates VSMC osteogenic conversion stimulated by indoxyl sulfate, possibly through methylating Klotho and inducing its degradation [242].
In summary, m6A methylation might affect the development of CKD by regulating the expression of genes involved in inflammation, EMT, fibrosis, and vascular calcification.
4. m6A as a Clinical Indicator
Vascular aging is an important cause of organ aging and a common pathogenesis of various chronic diseases. Its related diseases are highly harmful and have become a global challenge [243]. Currently, there is a lack of effective treatment options to reverse the progression of vascular aging. Therefore, further research is urgent to develop optimal clinical indicators.
Biomarkers refer to biological indicators that are objectively measured for the detection or evaluation of physiological processes, pathological processes, and the effectiveness of therapeutic approaches [244]. Although significant advances have been achieved in research on vascular aging mechanisms as stated above, effective methods for early identification of vascular aging remain limited. There is a growing need to develop novel biomarkers of vascular aging with simple operation, high sensitivity, strong specificity, and low cost. Biomarkers measurable in biofluids are potentially efficient, sensitive, and easily accessible indicators for the early detection of vascular aging-related diseases. Inflammatory factors such as C-reactive protein (CRP), interleukin-6 (IL-6), IL-1 receptor antagonist (IL-1Ra), and oxLDL, along with other circulating biomolecules like fibroblast growth factor 21 (FGF21), Fibulin-1, and miRNAs, have been reported as potential biomarkers of vascular aging. Additionally, the number and senescence of endothelial progenitor cells (EPCs) and senescence of immune cells such as macrophages, T cells, and B cells have been identified as circulating biomarkers of vascular aging as well [181]. Emerging evidence has revealed that epigenetic alterations including m6A modification during aging are closely linked with vascular aging [181, 245]. A fast response to environmental stimuli underlies the potential of m6A as the biomarker for the diagnosis of vascular aging and its related diseases. Some literature has reported the diagnostic and prognostic potential of m6A and its regulators in aspects of these conditions including coronary artery disease (CAD), HF, stroke, and kidney disease.
Decreased m6A abundance was measured in peripheral blood mononuclear cells (PBMCs) obtained from CAD patients compared to controls, where differentially methylated genes were revealed to participate in the pathogenesis of AS [246]. Of note, a diagnostic model of acute MI was established based on m6A-related genes, including FTO, WTAP, YTHDC1, IGFBP3, and CBLL1 [247]. Moreover, a genome-wide association study suggests that WTAP SNP is significantly associated with MI progression [248].
Reduced m6A abundance was observed in blood samples obtained during reperfusion in MI patients who developed HF four months after MI compared to those without HF [249]. Several m6A regulators (METTL3, METTL4, KIAA1429, FTO, and YTHDF2) were upregulated in PBMCs derived from HFpEF patients compared with the health group [200]. More efforts should be made to explore the feasibility of m6A as a novel biomarker in HF.
Additionally, the link between m6A-SNPs and the risk of ischemic stroke has been identified [250]. Eighty-four local genes (containing 87 m6A-SNPs) were detected to be differentially expressed in the peripheral blood from ischemic stroke patients, suggesting these m6A-SNPs as functional polymorphisms and new genetic biomarkers for ischemic stroke susceptibility [250].
Aside from circulating m6A, urine m6A has also been identified as a candidate biomarker in vascular aging-related kidney diseases. DN patients exhibit a marked reduction in urine m6A abundance compared to type 2 diabetes and normal glucose-tolerant cohorts. In particular, the levels of urine m6A decrease progressively as the disease worsens [251].
Further experimental and clinical research is required to confirm the utility of m6A methylation as a clinical indicator for vascular aging-related diseases. Therefore, future exploration should emphasize the evaluation of the relation between m6A and traditional clinical indicators of these disorders such as myocardial enzyme in MI, and N-terminal pro-brain natriuretic peptide in HF [252].
5. Therapeutic Potentials: Targeting m6A for Clinical Benefit
Healthy lifestyles (e.g., physical exercise, smoking cessation, certain diet regimens) are considered the easiest and most fundamental strategy in preventing vascular aging-related diseases. However, drugs targeting key aging-related molecular and cellular changes are still promising clinical therapies for these diseases. Existing clinical studies have observed the potential preventing effects of some clinically available drugs (e.g., statins, renin-angiotensin system inhibitors, and metformin) on vascular aging phenotypes during their administration for other medical conditions [4, 83]. Nonetheless, there are no medications developed specifically for the prevention and treatment of vascular aging. In recent years, multiple therapeutic approaches based on epigenetic alterations during aging have been developed for the aging process and age-related diseases [245]. Of note, aberrant m6A modification has been implicated in various molecular and cellular processes involved in vascular aging by regulating gene expression, contributing to the progression of related diseases. Therefore, targeting dysregulated m6A and its regulatory proteins provides potential therapeutic methods for various vascular aging-related diseases. Promising results have been obtained through research in this area (Fig. 5).
Figure 5.
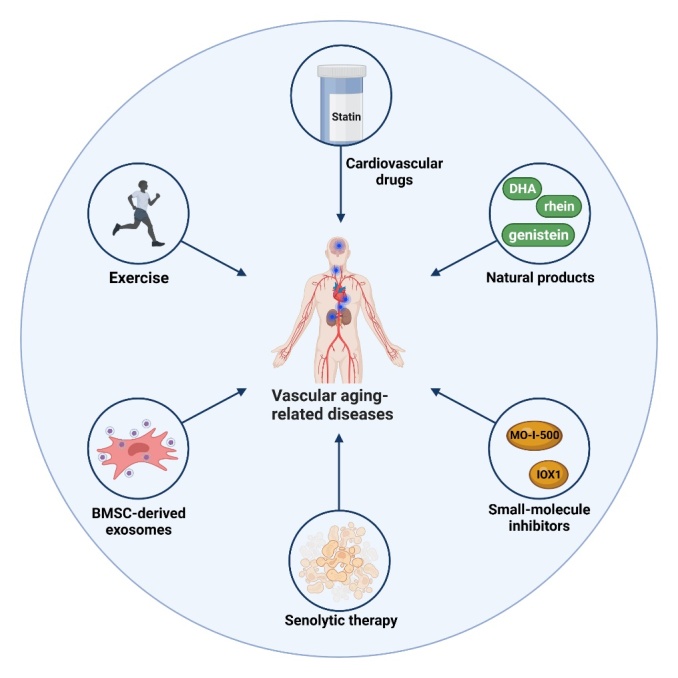
m6A-associated therapies for vascular aging-related diseases. m6A-associated anti-aging approaches present protective effects in vascular aging-related diseases, including exercise, senolytic therapy, BMSC-derived exosomes, cardiovascular drugs, natural products, and small-molecule inhibitors targeting m6A regulatory proteins. This figure was created with the aid of Biorender (https://biorender.com/). DHA: dihydroartemisinin; BMSC: bone marrow mesenchymal stem cell.
Recent data have described the protective effects of exercise on vascular aging-related diseases. Yang et al. reported that exercise attenuates endothelial pyroptosis and AS by downregulating METTL14 and m6A methylation of lncRNA NEAT1 [253]. Additionally, exercise training can alleviate HFpEF phenotypes by altering m6A modification patterns in mice [201].
Bone marrow MSC (BMSC)-derived exosomes exhibit promising therapeutic roles in various human diseases [254]. Emerging evidence suggests that BMSC-derived exosomal KLF4 promotes lncRNA-ZFAS1 expression to inhibit m6A methylation of dynamin-related protein 1 (DRP1) by targeting FTO, therefore mitigating mitochondrial dysfunction and ischemic stroke [255].
Moreover, a recent study reported that senolytic therapy of the combination of dasatinib and quercetin can attenuate LPS-induced EC senescence by upregulating YTHDF2 which destabilizes mitogen-activated protein kinase kinase 4 (MAP2K4) and mitogen-activated protein kinase kinase kinase kinase 4 (MAP4K4) mRNAs [256].
Traditional cardiovascular drugs such as classic lipid-lowering drugs, statins, can reduce FTO protein level and exerts protective effects on ECs by attenuating inflammation and increasing NO production. FTO attenuated statin-mediated effects on ECs by targeting KLF2 and endothelial NOS (eNOS) through m6A demethylation and YTHDF3-mediated stabilization [257].
More than that, natural products from traditional medicine could be used as a chemical library for m6A-targeting anti-aging drug discovery. Recent evidence has identified some natural products that exhibit activating or inhibitory effects on m6A regulatory proteins (e.g., FTO and ALKBH5) and have potential therapeutic effects in various vascular aging-related diseases. For example, rhein, an anthraquinone concentrated in Rheumrhabarbarum, has been identified as a reversible and competitive inhibitor of FTO [20]. Inhibition of m6A demethylation through rhein treatment has partially rescued neurodegenerative changes induced by METTL3 knockdown [220]. Dihydroartemisinin, a first-line antimalarial drug originated from the natural small-molecule compound artemisinin, has also been shown to block FTO expression, leading to inhibiting AngII-regulated VSMC proliferation and inflammation by increasing NR4A3 m6A methylation [111]. These results indicate the potential therapeutic role of dihydroartemisinin in hypertension-related vascular complications. Moreover, genistein, an isoflavone in soybean products widely used as a dietary supplement, has been found to elevate ALKBH5 expression and m6A levels. Genistein alleviates renal fibrosis by restoring ALKBH5 to regulate epithelial-to-mesenchymal transition [258].
Further, some pre-clinical experiments suggest that small-molecule inhibitors targeting dysregulated m6A regulators (e.g., FTO and ALKBH5) have potential therapeutic benefits in several vascular aging-related disorders. 5-carboxy-8-hydroxyquinoline (IOX1), a broad-spectrum inhibitor of most 2-OG oxygenases, can significantly repress ALKBH5 activity in a cofactor 2-OG competitive manner [259]. Since ALKBH5 plays a critical regulatory role in acute MI, IOX1 has been investigated for its therapeutic potential in treating acute MI. Notably, IOX1 was loaded onto bioengineered ferritin nanocage that can selectively target dying cells in the infarct territory. When administrated to the acute MI model, this nanocage improves cardiac function and minimizes the infarct area [260]. Given that disrupted m6A signaling is believed to play a role in AD pathogenesis, MO-I-500, a newly developed pharmacological inhibitor of FTO, has been revealed to promote cell survival and inhibit mitochondrial dysfunction in streptozotocin-treated astrocytes [261]. These findings suggest the therapeutic potential of MO-I-500 in AD.
The above findings suggest the potential therapeutic value of m6A-associated strategies for vascular aging-related diseases. These strategies include exercise, senolytic therapy, BMSC-derived exosomes, as well as targeting m6A regulatory proteins through cardiovascular drugs, natural products, and small-molecule inhibitors. Further research is required to explore the underlying mechanisms and translate these findings into clinical practice.
6. Patient-Centered Considerations
Patient-centered considerations are needed to improve outcomes, with a particular focus on the elderly population who often exhibit more complex care needs compared to younger counterparts [262, 263]. In-depth understanding of the intricate mechanisms that underlie diseases is crucial for clinician to customize interventions within a patient-centered framework [262].
In the context of patient-centered considerations, incorporating the knowledge of m6A modification into the management of vascular aging-related diseases can provide valuable insights. First, understanding the role of m6A in the pathophysiology of vascular aging and related diseases is helpful to strengthen clinical reasoning. By clarifying the underlying molecular mechanisms, clinicians can deepen their understanding of the disease process and identify potential therapeutic targets. This helps guide treatment decisions and tailor interventions for individual patients. Second, detecting the m6A landscape can guide patient subgrouping to obtain more targeted interventions. The distinct m6A patterns among individuals may be related to disease progression and treatment response differences [185, 264]. By stratifying patients according to the m6A landscape, clinicians can identify subgroups that may benefit from certain treatments, allowing them to develop personalized interventions [265]. Finally, m6A-associated biomarkers can be useful for personalized clinical diagnosis and treatment. By integrating technologies such as high-throughput technologies and bioinformatics analysis, clinicians can identify m6A-modified transcripts or m6A regulators as potential biomarkers to achieve early diagnosis, efficacy monitoring, and prognosis prediction [247, 248].
The integration of the knowledge of m6A into patient-centered care needs further research and validation in vascular aging-related diseases. However, a deeper understanding will help clinicians enhance their clinical reasoning, customize interventions through patient subgrouping, and apply m6A-related biomarkers to clinical diagnosis and treatment. These measures may contribute to a more individualized approach to the management of vascular aging-related diseases with a view to improving patient outcomes.
7. Challenges and Future Directions
Despite some advances and meaningful insights have been made in the investigation of m6A in vascular aging, challenges and exciting avenues for future research and clinical application remain. Further research is required to establish the precise function and potential mechanisms of m6A in this context, as well as its potential for clinical translation in the diagnosis and treatment of related diseases. Specifically, in-depth mechanistic studies on how m6A regulators coordinate to affect RNA fate and function and how specific m6A site is involved in different cellular signal pathways may bring a better understanding of its role in vascular aging. The development of more efficient and accurate methods to profile m6A RNA methylomes in various disease models can help to delineate condition-specific m6A patterns and identify precise methylation sites during vascular aging. Especially, ncRNA is also an important component of epigenetics in vascular aging [203]; however, studies are limited on its interplay with m6A modification during vascular aging progression. Further exploration is needed to enrich the knowledge in this area. Moreover, through integrative analysis of multi-omics data, a comprehensive analysis of the mechanisms underlying endothelial dysfunction and VSMC phenotypic transformation during vascular aging can be conducted. This approach is particularly useful for identifying novel potential targets for interventions in vascular aging. Currently, many small molecule inhibitors and activators of m6A regulatory factors have been developed, however, they are mostly explored in oncological disorders with poor specificity, efficacy, and safety [20]. Future work should be made to develop m6A inhibitors and activators with improved specificity, efficacy, and safety, and evaluate their function in vascular aging phenotypes. Epitranscriptome editing, similar to genome editing, has the potential to restore or remove functional m6A sites that are dysregulated or mutated in human diseases [23]. This approach might someday be applied to clinical practice in vascular aging-related diseases. In the future, clinical trials validating the diagnostic and therapeutic efficacy of m6A-related biomarkers and targeting therapy in vascular aging and related diseases could pave the way for new strategies for early detection and therapy.
8. A Multidisciplinary Approach: Collaborating for Progress
The complexity of vascular aging and related diseases is evident in the multifactorial influences (genetic factors, environmental factors), intertwined mechanisms, and multisystem involvement. Advancing the field of m6A research and its clinical application in these disorders necessitates a multidisciplinary approach. Collaboration among professionals such as clinicians, geneticists, molecular biologists, bioinformaticians, and pharmacologists enables to bridge the knowledge gap among different health care professionals, which helps to fully understand the effects of m6A on vascular aging. Collaboration is essential for revealing the epigenetic role in the disease process and provides a scientific basis, technical support, and possibility of novel diagnostic methods and targeted therapies.
9. Conclusion: Shaping the Future of Vascular Aging Care
m6A modification, as a key part of epitranscriptomics, has developed vigorously in the past decade. In this paper, we summarized the role of m6A methylation in vascular aging and related diseases and discussed its clinical prospect. The interaction between m6A modification and key pathways related to vascular aging bridges molecular insights and clinical realities. m6A exerts pleiotropic functions in various vascular aging-related diseases. m6A and its regulators detected in biofluid present diagnostic and prognostic potential in these conditions. m6A-associated anti-aging approaches exhibit potential protective effects in multiple vascular aging-related disorders. Moreover, integrating the knowledge of m6A into patient-centred care may help to improve clinical outcomes. While some progress has been made, there are still challenges ahead that demand further effort. Multidisciplinary collaboration is required to tackle these challenges and advance the field. With the continuous improvement and development of technology, the exploration of m6A can help to reveal new mechanistic insights into vascular aging process and may provide clinical diagnostic tools and therapeutic modalities for related diseases.
Authors' contributions
Chen Li wrote the manuscript and generated the figures. Le Liu summarized the table and revised the manuscript. You-Shuo Liu and Shuang Li conceived the idea, guided the writing process, and supervised the manuscript. All authors have reviewed and approved the final manuscript.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 82071593, 81974223, and 82101663).
Funding Statement
This work was supported by the National Natural Science Foundation of China (No. 82071593, 81974223, and 82101663).
Footnotes
Competing interests
The authors declare that they have no competing interests.
References
- [1].Mistriotis P, Andreadis ST (2017). Vascular aging: Molecular mechanisms and potential treatments for vascular rejuvenation. Ageing Res Rev, 37:94-116. [DOI] [PubMed] [Google Scholar]
- [2].Zhao Y, Liu Y-S (2021). Longevity Factor FOXO3: A Key Regulator in Aging-Related Vascular Diseases. Front Cardiovasc Med, 8:778674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Collaborators GDaI (2020). Global burden of 369 diseases and injuries in 204 countries and territories, 1990-2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet, 396:1204-1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ungvari Z, Tarantini S, Sorond F, Merkely B, Csiszar A (2020). Mechanisms of Vascular Aging, A Geroscience Perspective: JACC Focus Seminar. J Am Coll Cardiol, 75:931-941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Liu H, Huang Y, Lu S, Yuan D, Liu J (2023). Global Trends of Lipid Metabolism Research in Epigenetics Field: A Bibliometric Analysis from 2012-2021. Int J Environ Res Public Health, 20:2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sikorski V, Vento A, Kankuri E (2022). Emerging roles of the RNA modifications N6-methyladenosine and adenosine-to-inosine in cardiovascular diseases. Mol Ther Nucleic Acids, 29:426-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jiapaer Z, Su D, Hua L, Lehmann HI, Gokulnath P, Vulugundam G, et al. (2022). Regulation and roles of RNA modifications in aging-related diseases. Aging Cell, 21:e13657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Boccaletto P, Stefaniak F, Ray A, Cappannini A, Mukherjee S, Purta E, et al. (2022). MODOMICS: a database of RNA modification pathways. 2021 update. Nucleic Acids Res, 50:D231-D235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Desrosiers R, Friderici K, Rottman F (1974). Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A, 71:3971-3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Perry RP, Kelley DE (1974). Existence of methylated messenger RNA in mouse L cells. Cell, 1:37-42. [Google Scholar]
- [11].Clancy MJ, Shambaugh ME, Timpte CS, Bokar JA (2002). Induction of sporulation in Saccharomyces cerevisiae leads to the formation of N6-methyladenosine in mRNA: a potential mechanism for the activity of the IME4 gene. Nucleic Acids Res, 30:4509-4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kennedy TD, Lane BG (1979). Wheat embryo ribonucleates. XIII. Methyl-substituted nucleoside constituents and 5'-terminal dinucleotide sequences in bulk poly(AR)-rich RNA from imbibing wheat embryos. Can J Biochem, 57:927-931. [DOI] [PubMed] [Google Scholar]
- [13].Zhang G, Xu Y, Wang X, Zhu Y, Wang L, Zhang W, et al. (2022). Dynamic FMR1 granule phase switch instructed by m6A modification contributes to maternal RNA decay. Nat Commun, 13:859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Adams JM, Cory S (1975). Modified nucleosides and bizarre 5'-termini in mouse myeloma mRNA. Nature, 255:28-33. [DOI] [PubMed] [Google Scholar]
- [15].Krug RM, Morgan MA, Shatkin AJ (1976). Influenza viral mRNA contains internal N6-methyladenosine and 5'-terminal 7-methylguanosine in cap structures. J Virol, 20:45-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Schmidt W, Arnold HH, Kersten H (1975). Biosynthetic pathway of ribothymidine in B. subtilis and M. lysodeikticus involving different coenzymes for transfer RNA and ribosomal RNA. Nucleic Acids Res, 2:1043-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bokar JA, Rath-Shambaugh ME, Ludwiczak R, Narayan P, Rottman F (1994). Characterization and partial purification of mRNA N6-adenosine methyltransferase from HeLa cell nuclei. Internal mRNA methylation requires a multisubunit complex. J Biol Chem, 269:17697-17704. [PubMed] [Google Scholar]
- [18].Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM (1997). Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA, 3:1233-1247. [PMC free article] [PubMed] [Google Scholar]
- [19].Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. (2011). N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol, 7:885-887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Deng L-J, Deng W-Q, Fan S-R, Chen M-F, Qi M, Lyu W-Y, et al. (2022). m6A modification: recent advances, anticancer targeted drug discovery and beyond. Mol Cancer, 21:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. (2012). Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature, 485:201-206. [DOI] [PubMed] [Google Scholar]
- [22].Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR (2012). Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell, 149:1635-1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Huang H, Weng H, Chen J (2020). m6A Modification in Coding and Non-coding RNAs: Roles and Therapeutic Implications in Cancer. Cancer Cell, 37:270-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kumari R, Ranjan P, Suleiman ZG, Goswami SK, Li J, Prasad R, et al. (2022). mRNA modifications in cardiovascular biology and disease: with a focus on m6A modification. Cardiovasc Res, 118:1680-1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cully M (2019). Chemical inhibitors make their RNA epigenetic mark. Nat Rev Drug Discov, 18:892-894. [DOI] [PubMed] [Google Scholar]
- [26].An Y, Duan H (2022). The role of m6A RNA methylation in cancer metabolism. Mol Cancer, 21:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Li Y, Meng L, Zhao B (2022). The roles of N6-methyladenosine methylation in the regulation of bone development, bone remodeling and osteoporosis. Pharmacol Ther, 238:108174. [DOI] [PubMed] [Google Scholar]
- [28].Boulias K, Greer EL (2023). Biological roles of adenine methylation in RNA. Nat Rev Genet, 24:143-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, et al. (2014). A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol, 10:93-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ping X-L, Sun B-F, Wang L, Xiao W, Yang X, Wang W-J, et al. (2014). Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res, 24:177-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, et al. (2014). Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5' sites. Cell Rep, 8:284-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Patil DP, Chen C-K, Pickering BF, Chow A, Jackson C, Guttman M, et al. (2016). m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature, 537:369-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wen J, Lv R, Ma H, Shen H, He C, Wang J, et al. (2018). Zc3h13 Regulates Nuclear RNA m6A Methylation and Mouse Embryonic Stem Cell Self-Renewal. Mol Cell, 69:1028-1038.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Knuckles P, Lence T, Haussmann IU, Jacob D, Kreim N, Carl SH, et al. (2018). Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m6A machinery component Wtap/Fl(2)d. Genes Dev, 32:415-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, et al. (2016). Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature, 534:575-578. [DOI] [PubMed] [Google Scholar]
- [36].Wang P, Doxtader KA, Nam Y (2016). Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol Cell, 63:306-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Śledź P, Jinek M (2016). Structural insights into the molecular mechanism of the m(6)A writer complex. Elife, 5:e18434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z, et al. (2018). VIRMA mediates preferential m6A mRNA methylation in 3'UTR and near stop codon and associates with alternative polyadenylation. Cell Discov, 4:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bawankar P, Lence T, Paolantoni C, Haussmann IU, Kazlauskiene M, Jacob D, et al. (2021). Hakai is required for stabilization of core components of the m(6)A mRNA methylation machinery. Nat Commun, 12:3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ma H, Wang X, Cai J, Dai Q, Natchiar SK, Lv R, et al. (2019). N6-Methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation. Nat Chem Biol, 15:88-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].van Tran N, Ernst FGM, Hawley BR, Zorbas C, Ulryck N, Hackert P, et al. (2019). The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res, 47:7719-7733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, et al. (2017). The U6 snRNA m6A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell, 169:824-835.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Shima H, Matsumoto M, Ishigami Y, Ebina M, Muto A, Sato Y, et al. (2017). S-Adenosylmethionine Synthesis Is Regulated by Selective N-Adenosine Methylation and mRNA Degradation Involving METTL16 and YTHDC1. Cell Rep, 21:3354-3363. [DOI] [PubMed] [Google Scholar]
- [44].Gerken T, Girard CA, Tung Y-CL, Webby CJ, Saudek V, Hewitson KS, et al. (2007). The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science, 318:1469-1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, et al. (2017). Reversible methylation of m6Am in the 5' cap controls mRNA stability. Nature, 541:371-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wei J, Liu F, Lu Z, Fei Q, Ai Y, He PC, et al. (2018). Differential m(6)A, m(6)A(m), and m(1)A Demethylation Mediated by FTO in the Cell Nucleus and Cytoplasm. Mol Cell, 71:973-985.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. (2013). ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell, 49:18-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ueda Y, Ooshio I, Fusamae Y, Kitae K, Kawaguchi M, Jingushi K, et al. (2017). AlkB homolog 3-mediated tRNA demethylation promotes protein synthesis in cancer cells. Sci Rep, 7:42271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wang T, Kong S, Tao M, Ju S (2020). The potential role of RNA N6-methyladenosine in Cancer progression. Mol Cancer, 19:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Jiang X, Liu B, Nie Z, Duan L, Xiong Q, Jin Z, et al. (2021). The role of m6A modification in the biological functions and diseases. Signal Transduct Target Ther, 6:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. (2015). N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell, 161:1388-1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. (2014). N6-methyladenosine-dependent regulation of messenger RNA stability. Nature, 505:117-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, et al. (2017). YTHDF3 facilitates translation and decay of N-methyladenosine-modified RNA. Cell Res, 27:315-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Li A, Chen Y-S, Ping X-L, Yang X, Xiao W, Yang Y, et al. (2017). Cytoplasmic m6A reader YTHDF3 promotes mRNA translation. Cell Res, 27:444-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zaccara S, Jaffrey SR (2020). A Unified Model for the Function of YTHDF Proteins in Regulating m6A-Modified mRNA. Cell, 181:1582-1595.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Xiao W, Adhikari S, Dahal U, Chen Y-S, Hao Y-J, Sun B-F, et al. (2016). Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol Cell, 61:507-519. [DOI] [PubMed] [Google Scholar]
- [57].Roundtree IA, Luo GZ, Zhang Z, Wang X, Zhou T, Cui Y, et al. (2017). YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. Elife, 6:e31311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hsu PJ, Zhu Y, Ma H, Guo Y, Shi X, Liu Y, et al. (2017). Ythdc2 is an N-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res, 27:1115-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Alarcón CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF (2015). HNRNPA2B1 Is a Mediator of m(6)A-Dependent Nuclear RNA Processing Events. Cell, 162:1299-1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Wang L, Wen M, Cao X (2019). Nuclear hnRNPA2B1 initiates and amplifies the innate immune response to DNA viruses. Science, 365:eaav0758. [DOI] [PubMed] [Google Scholar]
- [61].Jiang F, Tang X, Tang C, Hua Z, Ke M, Wang C, et al. (2021). HNRNPA2B1 promotes multiple myeloma progression by increasing AKT3 expression via m6A-dependent stabilization of ILF3 mRNA. J Hematol Oncol, 14:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T (2015). N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature, 518:560-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Liu N, Zhou KI, Parisien M, Dai Q, Diatchenko L, Pan T (2017). N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res, 45:6051-6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. (2018). Recognition of RNA N-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol, 20:285-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Edens BM, Vissers C, Su J, Arumugam S, Xu Z, Shi H, et al. (2019). FMRP Modulates Neural Differentiation through m6A-Dependent mRNA Nuclear Export. Cell Rep, 28:845-854.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Zhang F, Kang Y, Wang M, Li Y, Xu T, Yang W, et al. (2018). Fragile X mental retardation protein modulates the stability of its m6A-marked messenger RNA targets. Hum Mol Genet, 27:3936-3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Wu R, Li A, Sun B, Sun J-G, Zhang J, Zhang T, et al. (2019). A novel m6A reader Prrc2a controls oligodendroglial specification and myelination. Cell Res, 29:23-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Yue B, Song C, Yang L, Cui R, Cheng X, Zhang Z, et al. (2019). METTL3-mediated N6-methyladenosine modification is critical for epithelial-mesenchymal transition and metastasis of gastric cancer. Mol Cancer, 18:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Visvanathan A, Patil V, Arora A, Hegde AS, Arivazhagan A, Santosh V, et al. (2018). Essential role of METTL3-mediated m6A modification in glioma stem-like cells maintenance and radioresistance. Oncogene, 37:522-533. [DOI] [PubMed] [Google Scholar]
- [70].Baquero-Perez B, Antanaviciute A, Yonchev ID, Carr IM, Wilson SA, Whitehouse A (2019). The Tudor SND1 protein is an m6A RNA reader essential for replication of Kaposi's sarcoma-associated herpesvirus. Elife, 8:e47261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, et al. (2015). 5' UTR m(6)A Promotes Cap-Independent Translation. Cell, 163:999-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Choe J, Lin S, Zhang W, Liu Q, Wang L, Ramirez-Moya J, et al. (2018). mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature, 561:556-560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Chen X, Wang J, Tahir M, Zhang F, Ran Y, Liu Z, et al. (2021). Current insights into the implications of m6A RNA methylation and autophagy interaction in human diseases. Cell Biosci, 11:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Wilkinson E, Cui Y-H, He Y-Y (2021). Context-Dependent Roles of RNA Modifications in Stress Responses and Diseases. Int J Mol Sci, 22:1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Xu Z, Lv B, Qin Y, Zhang B (2022). Emerging Roles and Mechanism of m6A Methylation in Cardiometabolic Diseases. Cells, 11:1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Sun J, Cheng B, Su Y, Li M, Ma S, Zhang Y, et al. (2022). The Potential Role of m6A RNA Methylation in the Aging Process and Aging-Associated Diseases. Front Genet, 13:869950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Zhang L, Xia J (2023). N6-Methyladenosine Methylation of mRNA in Cell Senescence. Cell Mol Neurobiol, 43:27-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Farías JG, Molina VM, Carrasco RA, Zepeda AB, Figueroa E, Letelier P, et al. (2017). Antioxidant Therapeutic Strategies for Cardiovascular Conditions Associated with Oxidative Stress. Nutrients, 9:966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].El Assar M, Angulo J, Rodríguez-Mañas L (2013). Oxidative stress and vascular inflammation in aging. Free Radic Biol Med, 65:380-401. [DOI] [PubMed] [Google Scholar]
- [80].Luo J, Mills K, le Cessie S, Noordam R, van Heemst D (2020). Ageing, age-related diseases and oxidative stress: What to do next? Ageing Res Rev, 57:100982. [DOI] [PubMed] [Google Scholar]
- [81].Förstermann U, Xia N, Li H (2017). Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ Res, 120:713-735. [DOI] [PubMed] [Google Scholar]
- [82].Csiszar A, Ungvari Z, Edwards JG, Kaminski P, Wolin MS, Koller A, et al. (2002). Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ Res, 90:1159-1166. [DOI] [PubMed] [Google Scholar]
- [83].Ungvari Z, Tarantini S, Donato AJ, Galvan V, Csiszar A (2018). Mechanisms of Vascular Aging. Circ Res, 123:849-867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Canugovi C, Stevenson MD, Vendrov AE, Hayami T, Robidoux J, Xiao H, et al. (2019). Increased mitochondrial NADPH oxidase 4 (NOX4) expression in aging is a causative factor in aortic stiffening. Redox Biol, 26:101288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Tejero J, Shiva S, Gladwin MT (2019). Sources of Vascular Nitric Oxide and Reactive Oxygen Species and Their Regulation. Physiol Rev, 99:311-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].van der Loo B, Labugger R, Skepper JN, Bachschmid M, Kilo J, Powell JM, et al. (2000). Enhanced peroxynitrite formation is associated with vascular aging. J Exp Med, 192:1731-1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Ungvari Z, Bailey-Downs L, Gautam T, Sosnowska D, Wang M, Monticone RE, et al. (2011). Age-associated vascular oxidative stress, Nrf2 dysfunction, and NF-{kappa}B activation in the nonhuman primate Macaca mulatta. J Gerontol A Biol Sci Med Sci, 66:866-875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Ungvari Z, Bailey-Downs L, Sosnowska D, Gautam T, Koncz P, Losonczy G, et al. (2011). Vascular oxidative stress in aging: a homeostatic failure due to dysregulation of NRF2-mediated antioxidant response. Am J Physiol Heart Circ Physiol, 301:H363-H372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Wang J, Ishfaq M, Xu L, Xia C, Chen C, Li J (2019). METTL3/m6A/miRNA-873-5p Attenuated Oxidative Stress and Apoptosis in Colistin-Induced Kidney Injury by Modulating Keap1/Nrf2 Pathway. Front Pharmacol, 10:517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Wu F, Zhang L, Lai C, Peng X, Yu S, Zhou C, et al. (2022). Dynamic Alteration Profile and New Role of RNA m6A Methylation in Replicative and HO-Induced Premature Senescence of Human Embryonic Lung Fibroblasts. Int J Mol Sci, 23:9271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Li Q, Li X, Tang H, Jiang B, Dou Y, Gorospe M, et al. (2017). NSUN2-Mediated m5C Methylation and METTL3/METTL14-Mediated m6A Methylation Cooperatively Enhance p21 Translation. J Cell Biochem, 118:2587-2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Anders M, Chelysheva I, Goebel I, Trenkner T, Zhou J, Mao Y, et al. (2018). Dynamic m6A methylation facilitates mRNA triaging to stress granules. Life Sci Alliance, 1:e201800113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Fu Y, Zhuang X (2020). m6A-binding YTHDF proteins promote stress granule formation. Nat Chem Biol, 16:955-963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Medzhitov R (2021). The spectrum of inflammatory responses. Science, 374:1070-1075. [DOI] [PubMed] [Google Scholar]
- [95].Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, et al. (2007). Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev, 128:92-105. [DOI] [PubMed] [Google Scholar]
- [96].Liberale L, Badimon L, Montecucco F, Lüscher TF, Libby P, Camici GG (2022). Inflammation, Aging, and Cardiovascular Disease: JACC Review Topic of the Week. J Am Coll Cardiol, 79:837-847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Liu L, Ni YQ, Zhan JK, Liu YS (2021). The Role of SGLT2 Inhibitors in Vascular Aging. Aging Dis, 12:1323-1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Cui X-Y, Zhan J-K, Liu Y-S (2021). Roles and functions of antisense lncRNA in vascular aging. Ageing Res Rev, 72:101480. [DOI] [PubMed] [Google Scholar]
- [99].Liberale L, Montecucco F, Schwarz L, Lüscher TF, Camici GG (2021). Inflammation and cardiovascular diseases: lessons from seminal clinical trials. Cardiovasc Res, 117:411-422. [DOI] [PubMed] [Google Scholar]
- [100].Zhu X, Tang H, Yang M, Yin K (2023). N6-methyladenosine in macrophage function: a novel target for metabolic diseases. Trends Endocrinol Metab, 34:66-84. [DOI] [PubMed] [Google Scholar]
- [101].Björkegren JLM, Lusis AJ (2022). Atherosclerosis: Recent developments. Cell, 185:1630-1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Li Z, Xu Q, Huangfu N, Chen X, Zhu J (2022). Mettl3 promotes oxLDL-mediated inflammation through activating STAT1 signaling. J Clin Lab Anal, 36:e24019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Sun Z, Chen W, Wang Z, Wang S, Zan J, Zheng L, et al. (2022). Matr3 reshapes m6A modification complex to alleviate macrophage inflammation during atherosclerosis. Clin Immunol, 245:109176. [DOI] [PubMed] [Google Scholar]
- [104].Zheng Y, Li Y, Ran X, Wang D, Zheng X, Zhang M, et al. (2022). Mettl14 mediates the inflammatory response of macrophages in atherosclerosis through the NF-κB/IL-6 signaling pathway. Cell Mol Life Sci, 79:311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Huangfu N, Zheng W, Xu Z, Wang S, Wang Y, Cheng J, et al. (2020). RBM4 regulates M1 macrophages polarization through targeting STAT1-mediated glycolysis. Int Immunopharmacol, 83:106432. [DOI] [PubMed] [Google Scholar]
- [106].Wang X, Ji Y, Feng P, Liu R, Li G, Zheng J, et al. (2021). The m6A Reader IGF2BP2 Regulates Macrophage Phenotypic Activation and Inflammatory Diseases by Stabilizing TSC1 and PPARγ. Adv Sci (Weinh), 8:2100209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Liu Y, Liu Z, Tang H, Shen Y, Gong Z, Xie N, et al. (2019). The N6-methyladenosine (m6A)-forming enzyme METTL3 facilitates M1 macrophage polarization through the methylation of STAT1 mRNA. Am J Physiol Cell Physiol, 317:C762-C775. [DOI] [PubMed] [Google Scholar]
- [108].Gu X, Zhang Y, Li D, Cai H, Cai L, Xu Q (2020). N6-methyladenosine demethylase FTO promotes M1 and M2 macrophage activation. Cell Signal, 69:109553. [DOI] [PubMed] [Google Scholar]
- [109].Chien C-S, Li JY-S, Chien Y, Wang M-L, Yarmishyn AA, Tsai P-H, et al. (2021). METTL3-dependent N-methyladenosine RNA modification mediates the atherogenic inflammatory cascades in vascular endothelium. Proc Natl Acad Sci U S A, 118:e2025070118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Jian D, Wang Y, Jian L, Tang H, Rao L, Chen K, et al. (2020). METTL14 aggravates endothelial inflammation and atherosclerosis by increasing FOXO1 N6-methyladeosine modifications. Theranostics, 10:8939-8956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Huo Y-B, Gao X, Peng Q, Nie Q, Bi W (2022). Dihydroartemisinin alleviates AngII-induced vascular smooth muscle cell proliferation and inflammatory response by blocking the FTO/NR4A3 axis. Inflamm Res, 71:243-253. [DOI] [PubMed] [Google Scholar]
- [112].Gioscia-Ryan RA, LaRocca TJ, Sindler AL, Zigler MC, Murphy MP, Seals DR (2014). Mitochondria-targeted antioxidant (MitoQ) ameliorates age-related arterial endothelial dysfunction in mice. J Physiol, 592:2549-2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Ma D, Zheng B, Liu H-L, Zhao Y-B, Liu X, Zhang X-H, et al. (2020). Klf5 down-regulation induces vascular senescence through eIF5a depletion and mitochondrial fission. PLoS Biol, 18:e3000808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Li Y-J, Jin X, Li D, Lu J, Zhang X-N, Yang S-J, et al. (2022). New insights into vascular aging: Emerging role of mitochondria function. Biomed Pharmacother, 156:113954. [DOI] [PubMed] [Google Scholar]
- [115].Zhang X, Li X, Jia H, An G, Ni J (2021). The m6A methyltransferase METTL3 modifies PGC-1α mRNA promoting mitochondrial dysfunction and oxLDL-induced inflammation in monocytes. J Biol Chem, 297:101058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Abdellatif M, Sedej S, Carmona-Gutierrez D, Madeo F, Kroemer G (2018). Autophagy in Cardiovascular Aging. Circ Res, 123:803-824. [DOI] [PubMed] [Google Scholar]
- [117].LaRocca TJ, Henson GD, Thorburn A, Sindler AL, Pierce GL, Seals DR (2012). Translational evidence that impaired autophagy contributes to arterial ageing. J Physiol, 590:3305-3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Mizushima N, Levine B (2020). Autophagy in Human Diseases. N Engl J Med, 383:1564-1576. [DOI] [PubMed] [Google Scholar]
- [119].LaRocca TJ, Gioscia-Ryan RA, Hearon CM, Seals DR (2013). The autophagy enhancer spermidine reverses arterial aging. Mech Ageing Dev, 134:314-320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].de Picciotto NE, Gano LB, Johnson LC, Martens CR, Sindler AL, Mills KF, et al. (2016). Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell, 15:522-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Song H, Feng X, Zhang H, Luo Y, Huang J, Lin M, et al. (2019). METTL3 and ALKBH5 oppositely regulate m6A modification of TFEB mRNA, which dictates the fate of hypoxia/reoxygenation-treated cardiomyocytes. Autophagy, 15:1419-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Jin S, Zhang X, Miao Y, Liang P, Zhu K, She Y, et al. (2018). m6A RNA modification controls autophagy through upregulating ULK1 protein abundance. Cell Res, 28:955-957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Chen X, Gong W, Shao X, Shi T, Zhang L, Dong J, et al. (2022). METTL3-mediated m6A modification of ATG7 regulates autophagy-GATA4 axis to promote cellular senescence and osteoarthritis progression. Ann Rheum Dis, 81:87-99. [DOI] [PubMed] [Google Scholar]
- [124].Fang Z-M, Zhang S-M, Luo H, Jiang D-S, Huo B, Zhong X, et al. (2023). Methyltransferase-like 3 suppresses phenotypic switching of vascular smooth muscle cells by activating autophagosome formation. Cell Prolif, 56:e13386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Katerji M, Duerksen-Hughes PJ (2021). DNA damage in cancer development: special implications in viral oncogenesis. Am J Cancer Res, 11:3956-3979. [PMC free article] [PubMed] [Google Scholar]
- [126].Durik M, Kavousi M, van der Pluijm I, Isaacs A, Cheng C, Verdonk K, et al. (2012). Nucleotide excision DNA repair is associated with age-related vascular dysfunction. Circulation, 126:468-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Di Micco R, Krizhanovsky V, Baker D, d'Adda di Fagagna F (2021). Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol, 22:75-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Wilkinson E, Cui Y-H, He Y-Y (2022). Roles of RNA Modifications in Diverse Cellular Functions. Front Cell Dev Biol, 10:828683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Zhou M, Liu W, Zhang J, Sun N (2021). RNA m6A Modification in Immunocytes and DNA Repair: The Biological Functions and Prospects in Clinical Application. Front Cell Dev Biol, 9:794754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Fu H, Zhu X, Di Q, Sun J, Jiang Q, Xu Q (2023). m6A contributes to a pro-survival state in GC-2 cells by facilitating DNA damage repair: Novel perspectives on the mechanism underlying DEHP genotoxicity in male germ cells. Sci Total Environ, 859:160432. [DOI] [PubMed] [Google Scholar]
- [131].Zhang C, Chen L, Peng D, Jiang A, He Y, Zeng Y, et al. (2020). METTL3 and N6-Methyladenosine Promote Homologous Recombination-Mediated Repair of DSBs by Modulating DNA-RNA Hybrid Accumulation. Mol Cell, 79:425-442.e7. [DOI] [PubMed] [Google Scholar]
- [132].Xiang Y, Laurent B, Hsu C-H, Nachtergaele S, Lu Z, Sheng W, et al. (2017). RNA m6A methylation regulates the ultraviolet-induced DNA damage response. Nature, 543:573-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Wang Z, Pan Z, Adhikari S, Harada BT, Shen L, Yuan W, et al. (2021). m6 A deposition is regulated by PRMT1-mediated arginine methylation of METTL14 in its disordered C-terminal region. EMBO J, 40:e106309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Yu F, Wei J, Cui X, Yu C, Ni W, Bungert J, et al. (2021). Post-translational modification of RNA m6A demethylase ALKBH5 regulates ROS-induced DNA damage response. Nucleic Acids Res, 49:5779-5797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Xiong F, Wang R, Lee J-H, Li S, Chen S-F, Liao Z, et al. (2021). RNA m6A modification orchestrates a LINE-1-host interaction that facilitates retrotransposition and contributes to long gene vulnerability. Cell Res, 31:861-885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Mathon NF, Lloyd AC (2001). Cell senescence and cancer. Nat Rev Cancer, 1:203-213. [DOI] [PubMed] [Google Scholar]
- [137].Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, et al. (2016). Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell, 15:973-977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Regnault V, Challande P, Pinet F, Li Z, Lacolley P (2021). Cell senescence: basic mechanisms and the need for computational networks in vascular ageing. Cardiovasc Res, 117:1841-1858. [DOI] [PubMed] [Google Scholar]
- [139].Min K-W, Zealy RW, Davila S, Fomin M, Cummings JC, Makowsky D, et al. (2018). Profiling of m6A RNA modifications identified an age-associated regulation of AGO2 mRNA stability. Aging Cell, 17:e12753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Zhang J, Ao Y, Zhang Z, Mo Y, Peng L, Jiang Y, et al. (2020). Lamin A safeguards the m(6) A methylase METTL14 nuclear speckle reservoir to prevent cellular senescence. Aging Cell, 19:e13215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Wu Z, Shi Y, Lu M, Song M, Yu Z, Wang J, et al. (2020). METTL3 counteracts premature aging via m6A-dependent stabilization of MIS12 mRNA. Nucleic Acids Res, 48:11083-11096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Zhu H, Sun B, Zhu L, Zou G, Shen Q (2021). N6-Methyladenosine Induced miR-34a-5p Promotes TNF-α-Induced Nucleus Pulposus Cell Senescence by Targeting SIRT1. Front Cell Dev Biol, 9:642437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Hirayama M, Wei F-Y, Chujo T, Oki S, Yakita M, Kobayashi D, et al. (2020). FTO Demethylates Cyclin D1 mRNA and Controls Cell-Cycle Progression. Cell Rep, 31:107464. [DOI] [PubMed] [Google Scholar]
- [144].Li N, Luo R, Zhang W, Wu Y, Hu C, Liu M, et al. (2023). IL-17A promotes endothelial cell senescence by up-regulating the expression of FTO through activating JNK signal pathway. Biogerontology, 24:99-110. [DOI] [PubMed] [Google Scholar]
- [145].Lakatta EG, Levy D (2003). Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part I: aging arteries: a "set up" for vascular disease. Circulation, 107:139-146. [DOI] [PubMed] [Google Scholar]
- [146].Saigusa R, Winkels H, Ley K (2020). T cell subsets and functions in atherosclerosis. Nat Rev Cardiol, 17:387-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Quiles-Jiménez A, Gregersen I, Mittelstedt Leal de Sousa M, Abbas A, Kong XY, Alseth I, et al. (2020). N6-methyladenosine in RNA of atherosclerotic plaques: An epitranscriptomic signature of human carotid atherosclerosis. Biochem Biophys Res Commun, 533:631-637. [DOI] [PubMed] [Google Scholar]
- [148].Liu M, Xu K, Saaoud F, Shao Y, Zhang R, Lu Y, et al. (2022). 29 m6A-RNA Methylation (Epitranscriptomic) Regulators Are Regulated in 41 Diseases including Atherosclerosis and Tumors Potentially via ROS Regulation - 102 Transcriptomic Dataset Analyses. J Immunol Res, 2022:1433323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Wu Y, Zhan S, Xu Y, Gao X (2021). RNA modifications in cardiovascular diseases, the potential therapeutic targets. Life Sci, 278:119565. [DOI] [PubMed] [Google Scholar]
- [150].Fu J, Cui X, Zhang X, Cheng M, Li X, Guo Z, et al. (2021). The Role of m6A Ribonucleic Acid Modification in the Occurrence of Atherosclerosis. Front Genet, 12:733871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Gimbrone MA, García-Cardeña G (2016). Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ Res, 118:620-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Zhang G, Li X, Huang X (2023). m6A-related bioinformatics analysis and functional characterization reveals that METTL3-mediated NPC1L1 mRNA hypermethylation facilitates progression of atherosclerosis via inactivation of the MAPK pathway. Inflamm Res, 72:429-442. [DOI] [PubMed] [Google Scholar]
- [153].Dong G, Yu J, Shan G, Su L, Yu N, Yang S (2021). N6-Methyladenosine Methyltransferase METTL3 Promotes Angiogenesis and Atherosclerosis by Upregulating the JAK2/STAT3 Pathway via m6A Reader IGF2BP1. Front Cell Dev Biol, 9:731810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Zhang BY, Han L, Tang YF, Zhang GX, Fan XL, Zhang JJ, et al. (2020). METTL14 regulates M6A methylation-modified primary miR-19a to promote cardiovascular endothelial cell proliferation and invasion. Eur Rev Med Pharmacol Sci, 24:7015-7023. [DOI] [PubMed] [Google Scholar]
- [155].Liu Y, Luo G, Tang Q, Song Y, Liu D, Wang H, et al. (2022). Methyltransferase-like 14 silencing relieves the development of atherosclerosis via m6A modification of p65 mRNA. Bioengineered, 13:11832-11843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Li B, Zhang T, Liu M, Cui Z, Zhang Y, Liu M, et al. (2022). RNA -methyladenosine modulates endothelial atherogenic responses to disturbed flow in mice. Elife, 11:e69906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Orecchioni M, Kobiyama K, Winkels H, Ghosheh Y, McArdle S, Mikulski Z, et al. (2022). Olfactory receptor 2 in vascular macrophages drives atherosclerosis by NLRP3-dependent IL-1 production. Science, 375:214-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Li Q, Yu L, Gao A, Ren R, Zhang J, Cao L, et al. (2023). METTL3 (Methyltransferase Like 3)-Dependent N6-Methyladenosine Modification on Braf mRNA Promotes Macrophage Inflammatory Response and Atherosclerosis in Mice. Arterioscler Thromb Vasc Biol, 43:755-773. [DOI] [PubMed] [Google Scholar]
- [159].Miano JM, Fisher EA, Majesky MW (2021). Fate and State of Vascular Smooth Muscle Cells in Atherosclerosis. Circulation, 143:2110-2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Chen J, Lai K, Yong X, Yin H, Chen Z, Wang H, et al. (2023). Silencing METTL3 Stabilizes Atherosclerotic Plaques by Regulating the Phenotypic Transformation of Vascular Smooth Muscle Cells via the miR-375-3p/PDK1 Axis. Cardiovasc Drugs Ther, 37:471-486. [DOI] [PubMed] [Google Scholar]
- [161].Yang X, Wang C, Zhu G, Guo Z, Fan L (2023). METTL14/YTHDF1 axis-modified UCHL5 aggravates atherosclerosis by activating the NLRP3 inflammasome. Experimental cell research, 427:113587. [DOI] [PubMed] [Google Scholar]
- [162].Collaborators GRF (2020). Global burden of 87 risk factors in 204 countries and territories, 1990-2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet, 396:1223-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Brunström M, Carlberg B (2018). Association of Blood Pressure Lowering With Mortality and Cardiovascular Disease Across Blood Pressure Levels: A Systematic Review and Meta-analysis. JAMA Intern Med, 178:28-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Wu Q, Yuan X, Han R, Zhang H, Xiu R (2019). Epitranscriptomic mechanisms of N6-methyladenosine methylation regulating mammalian hypertension development by determined spontaneously hypertensive rats pericytes. Epigenomics, 11:1359-1370. [DOI] [PubMed] [Google Scholar]
- [165].Marcadenti A, Fuchs FD, Matte U, Sperb F, Moreira LB, Fuchs SC (2013). Effects of FTO RS9939906 and MC4R RS17782313 on obesity, type 2 diabetes mellitus and blood pressure in patients with hypertension. Cardiovasc Diabetol, 12:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Mo X-B, Lei S-F, Zhang Y-H, Zhang H (2019). Examination of the associations between m6A-associated single-nucleotide polymorphisms and blood pressure. Hypertens Res, 42:1582-1589. [DOI] [PubMed] [Google Scholar]
- [167].Bossone E, Eagle KA (2021). Epidemiology and management of aortic disease: aortic aneurysms and acute aortic syndromes. Nat Rev Cardiol, 18:331-348. [DOI] [PubMed] [Google Scholar]
- [168].Milewicz DM, Ramirez F (2019). Therapies for Thoracic Aortic Aneurysms and Acute Aortic Dissections. Arterioscler Thromb Vasc Biol, 39:126-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].He Y, Xing J, Wang S, Xin S, Han Y, Zhang J (2019). Increased m6A methylation level is associated with the progression of human abdominal aortic aneurysm. Ann Transl Med, 7:797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Zhou X, Chen Z, Zhou J, Liu Y, Fan R, Sun T (2021). Transcriptome and N6-Methyladenosine RNA Methylome Analyses in Aortic Dissection and Normal Human Aorta. Front Cardiovasc Med, 8:627380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Li T, Wang T, Jing J, Sun L (2021). Expression Pattern and Clinical Value of Key m6A RNA Modification Regulators in Abdominal Aortic Aneurysm. J Inflamm Res, 14:4245-4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Fu C, Feng L, Zhang J, Sun D (2022). Bioinformatic analyses of the role of m6A RNA methylation regulators in abdominal aortic aneurysm. Ann Transl Med, 10:547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Yin F, Zhang H, Guo P, Wu Y, Zhao X, Li F, et al. (2022). Comprehensive Analysis of Key m6A Modification Related Genes and Immune Infiltrates in Human Aortic Dissection. Front Cardiovasc Med, 9:831561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Rombouts KB, van Merrienboer TAR, Ket JCF, Bogunovic N, van der Velden J, Yeung KK (2022). The role of vascular smooth muscle cells in the development of aortic aneurysms and dissections. Eur J Clin Invest, 52:e13697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Zhong L, He X, Song H, Sun Y, Chen G, Si X, et al. (2020). METTL3 Induces AAA Development and Progression by Modulating N6-Methyladenosine-Dependent Primary miR34a Processing. Mol Ther Nucleic Acids, 21:394-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Ma D, Liu X, Zhang JJ, Zhao JJ, Xiong YJ, Chang Q, et al. (2020). Vascular Smooth Muscle FTO Promotes Aortic Dissecting Aneurysms via m6A Modification of Klf5. Front Cardiovasc Med, 7:592550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Li N, Yi X, He Y, Huo B, Chen Y, Zhang Z, et al. (2022). Targeting Ferroptosis as a Novel Approach to Alleviate Aortic Dissection. Int J Biol Sci, 18:4118-4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Badi I, Burba I, Ruggeri C, Zeni F, Bertolotti M, Scopece A, et al. (2015). MicroRNA-34a Induces Vascular Smooth Muscle Cells Senescence by SIRT1 Downregulation and Promotes the Expression of Age-Associated Pro-inflammatory Secretory Factors. J Gerontol A Biol Sci Med Sci, 70:1304-1311. [DOI] [PubMed] [Google Scholar]
- [179].Li K, Zhang D, Zhai S, Wu H, Liu H (2023). METTL3-METTL14 complex induces necroptosis and inflammation of vascular smooth muscle cells via promoting N6 methyladenosine mRNA methylation of receptor-interacting protein 3 in abdominal aortic aneurysms. J Cell Commun Signal, 17:897-914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Wang P, Wang Z, Zhang M, Wu Q, Shi F, Yuan S (2021). KIAA1429 and ALKBH5 Oppositely Influence Aortic Dissection Progression Regulating the Maturation of Pri-miR-143-3p in an m6A-Dependent Manner. Front Cell Dev Biol, 9:668377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Bao H, Cao J, Chen M, Chen M, Chen W, Chen X, et al. (2023). Biomarkers of aging. Sci China Life Sci, 66:893-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Mathiyalagan P, Adamiak M, Mayourian J, Sassi Y, Liang Y, Agarwal N, et al. (2019). FTO-Dependent N6-Methyladenosine Regulates Cardiac Function During Remodeling and Repair. Circulation, 139:518-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Dorn LE, Lasman L, Chen J, Xu X, Hund TJ, Medvedovic M, et al. (2019). The N6-Methyladenosine mRNA Methylase METTL3 Controls Cardiac Homeostasis and Hypertrophy. Circulation, 139:533-545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Bhatt DL, Lopes RD, Harrington RA (2022). Diagnosis and Treatment of Acute Coronary Syndromes: A Review. JAMA, 327:662-675. [DOI] [PubMed] [Google Scholar]
- [185].Liu C, Gu L, Deng W, Meng Q, Li N, Dai G, et al. (2022). N6-Methyladenosine RNA Methylation in Cardiovascular Diseases. Front Cardiovasc Med, 9:887838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Hadas Y, Vincek AS, Youssef E, Żak MM, Chepurko E, Sultana N, et al. (2020). Altering Sphingolipid Metabolism Attenuates Cell Death and Inflammatory Response After Myocardial Infarction. Circulation, 141:916-930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Guo M, Yan R, Ji Q, Yao H, Sun M, Duan L, et al. (2020). IFN regulatory Factor-1 induced macrophage pyroptosis by modulating m6A modification of circ_0029589 in patients with acute coronary syndrome. Int Immunopharmacol, 86:106800. [DOI] [PubMed] [Google Scholar]
- [188].Sun P, Wang C, Mang G, Xu X, Fu S, Chen J, et al. (2022). Extracellular vesicle-packaged mitochondrial disturbing miRNA exacerbates cardiac injury during acute myocardial infarction. Clin Transl Med, 12:e779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Han Z, Wang X, Xu Z, Cao Y, Gong R, Yu Y, et al. (2021). ALKBH5 regulates cardiomyocyte proliferation and heart regeneration by demethylating the mRNA of YTHDF1. Theranostics, 11:3000-3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Kumari R, Dutta R, Ranjan P, Suleiman ZG, Goswami SK, Li J, et al. (2021). ALKBH5 Regulates SPHK1-Dependent Endothelial Cell Angiogenesis Following Ischemic Stress. Front Cardiovasc Med, 8:817304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Chamorro-Jorganes A, Sweaad WK, Katare R, Besnier M, Anwar M, Beazley-Long N, et al. (2021). METTL3 Regulates Angiogenesis by Modulating let-7e-5p and miRNA-18a-5p Expression in Endothelial Cells. Arterioscler Thromb Vasc Biol, 41:e325-e337. [DOI] [PubMed] [Google Scholar]
- [192].Zhang Y, Hua W, Dang Y, Cheng Y, Wang J, Zhang X, et al. (2021). Validated Impacts of N6-Methyladenosine Methylated mRNAs on Apoptosis and Angiogenesis in Myocardial Infarction Based on MeRIP-Seq Analysis. Front Mol Biosci, 8:789923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Li T, Zhuang Y, Yang W, Xie Y, Shang W, Su S, et al. (2021). Silencing of METTL3 attenuates cardiac fibrosis induced by myocardial infarction via inhibiting the activation of cardiac fibroblasts. FASEB J, 35:e21162. [DOI] [PubMed] [Google Scholar]
- [194].Bozkurt B, Coats AJS, Tsutsui H, Abdelhamid CM, Adamopoulos S, Albert N, et al. (2021). Universal definition and classification of heart failure: a report of the Heart Failure Society of America, Heart Failure Association of the European Society of Cardiology, Japanese Heart Failure Society and Writing Committee of the Universal Definition of Heart Failure: Endorsed by the Canadian Heart Failure Society, Heart Failure Association of India, Cardiac Society of Australia and New Zealand, and Chinese Heart Failure Association. Eur J Heart Fail, 23:352-380. [DOI] [PubMed] [Google Scholar]
- [195].Heidenreich PA, Bozkurt B, Aguilar D, Allen LA, Byun JJ, Colvin MM, et al. (2022). 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: Executive Summary: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol, 79:1757-1780. [DOI] [PubMed] [Google Scholar]
- [196].Savarese G, Becher PM, Lund LH, Seferovic P, Rosano GMC, Coats AJS (2023). Global burden of heart failure: a comprehensive and updated review of epidemiology. Cardiovasc Res, 118:3272-3287. [DOI] [PubMed] [Google Scholar]
- [197].Berulava T, Buchholz E, Elerdashvili V, Pena T, Islam MR, Lbik D, et al. (2020). Changes in m6A RNA methylation contribute to heart failure progression by modulating translation. Eur J Heart Fail, 22:54-66. [DOI] [PubMed] [Google Scholar]
- [198].Fan S, Hu Y (2022). Role of m6A Methylation in the Occurrence and Development of Heart Failure. Front Cardiovasc Med, 9:892113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Zhang B, Jiang H, Wu J, Cai Y, Dong Z, Zhao Y, et al. (2021). m6A demethylase FTO attenuates cardiac dysfunction by regulating glucose uptake and glycolysis in mice with pressure overload-induced heart failure. Signal Transduct Target Ther, 6:377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Zhang B, Xu Y, Cui X, Jiang H, Luo W, Weng X, et al. (2021). Alteration of m6A RNA Methylation in Heart Failure With Preserved Ejection Fraction. Front Cardiovasc Med, 8:647806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Liu K, Ju W, Ouyang S, Liu Z, He F, Hao J, et al. (2022). Exercise training ameliorates myocardial phenotypes in heart failure with preserved ejection fraction by changing N6-methyladenosine modification in mice model. Front Cell Dev Biol, 10:954769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Thorin-Trescases N, de Montgolfier O, Pinçon A, Raignault A, Caland L, Labbé P, et al. (2018). Impact of pulse pressure on cerebrovascular events leading to age-related cognitive decline. Am J Physiol Heart Circ Physiol, 314:H1214-H1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Ni Y-Q, Lin X, Zhan J-K, Liu Y-S (2020). Roles and Functions of Exosomal Non-coding RNAs in Vascular Aging. Aging Dis, 11:164-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Collaborators GS (2021). Global, regional, and national burden of stroke and its risk factors, 1990-2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol, 20:795-820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Chang H, Yang J, Wang Q, Zhao J, Zhu R (2022). Role of N6-methyladenosine modification in pathogenesis of ischemic stroke. Expert Rev Mol Diagn, 22:295-303. [DOI] [PubMed] [Google Scholar]
- [206].Mo X-B, Lei S-F, Zhang Y-H, Zhang H (2019). Integrative Analysis Identified and as Potential Causal Genes for Ischemic Stroke. Front Neurol, 10:517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Chokkalla AK, Mehta SL, Kim T, Chelluboina B, Kim J, Vemuganti R (2019). Transient Focal Ischemia Significantly Alters the m6A Epitranscriptomic Tagging of RNAs in the Brain. Stroke, 50:2912-2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Li Y, Li H, Luo Y, Li X, Chen Z, Zhang W, et al. (2022). The Alteration Profiles of m6A-Tagged circRNAs in the Peri-Infarct Cortex After Cerebral Ischemia in Mice. Front Neurosci, 16:869081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Si W, Li Y, Ye S, Li Z, Liu Y, Kuang W, et al. (2020). Methyltransferase 3 Mediated miRNA m6A Methylation Promotes Stress Granule Formation in the Early Stage of Acute Ischemic Stroke. Front Mol Neurosci, 13:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Zhang Z, Wang Q, Zhao X, Shao L, Liu G, Zheng X, et al. (2020). YTHDC1 mitigates ischemic stroke by promoting Akt phosphorylation through destabilizing PTEN mRNA. Cell Death Dis, 11:977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Chokkalla AK, Jeong S, Mehta SL, Davis CK, Morris-Blanco KC, Bathula S, et al. (2023). Cerebroprotective Role of N6-Methyladenosine Demethylase FTO (Fat Mass and Obesity-Associated Protein) After Experimental Stroke. Stroke, 54:245-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Li B, Xi W, Bai Y, Liu X, Zhang Y, Li L, et al. (2023). FTO-dependent m6A modification of Plpp3 in circSCMH1-regulated vascular repair and functional recovery following stroke. Nat Commun, 14:489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Zheng L, Tang X, Lu M, Sun S, Xie S, Cai J, et al. (2020). microRNA-421-3p prevents inflammatory response in cerebral ischemia/reperfusion injury through targeting m6A Reader YTHDF1 to inhibit p65 mRNA translation. Int Immunopharmacol, 88:106937. [DOI] [PubMed] [Google Scholar]
- [214].Cortes-Canteli M, Iadecola C (2020). Alzheimer's Disease and Vascular Aging: JACC Focus Seminar. J Am Coll Cardiol, 75:942-951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Shafik AM, Allen EG, Jin P (2022). Epitranscriptomic dynamics in brain development and disease. Mol Psychiatry, 27:3633-3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Castro-Hernández R, Berulava T, Metelova M, Epple R, Peña Centeno T, Richter J, et al. (2023). Conserved reduction of m6A RNA modifications during aging and neurodegeneration is linked to changes in synaptic transcripts. Proc Natl Acad Sci U S A, 120:e2204933120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Shafik AM, Zhang F, Guo Z, Dai Q, Pajdzik K, Li Y, et al. (2021). N6-methyladenosine dynamics in neurodevelopment and aging, and its potential role in Alzheimer's disease. Genome Biol, 22:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Han M, Liu Z, Xu Y, Liu X, Wang D, Li F, et al. (2020). Abnormality of m6A mRNA Methylation Is Involved in Alzheimer's Disease. Front Neurosci, 14:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Huang H, Camats-Perna J, Medeiros R, Anggono V, Widagdo J (2020). Altered Expression of the m6A Methyltransferase METTL3 in Alzheimer's Disease. eNeuro, 7:ENEURO.0125-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Zhao F, Xu Y, Gao S, Qin L, Austria Q, Siedlak SL, et al. (2021). METTL3-dependent RNA m6A dysregulation contributes to neurodegeneration in Alzheimer's disease through aberrant cell cycle events. Mol Neurodegener, 16:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Tang Z, Cao J, Yao J, Fan X, Zhao J, Zhao M, et al. (2023). KDM1A-mediated upregulation of METTL3 ameliorates Alzheimer's disease via enhancing autophagic clearance of p-Tau through m6A-dependent regulation of STUB1. Free Radic Biol Med, 195:343-358. [DOI] [PubMed] [Google Scholar]
- [222].Bi R, Kong L-L, Xu M, Li G-D, Zhang D-F, Li T, et al. (2018). The Arc Gene Confers Genetic Susceptibility to Alzheimer's Disease in Han Chinese. Mol Neurobiol, 55:1217-1226. [DOI] [PubMed] [Google Scholar]
- [223].Xu C, Huang H, Zhang M, Zhang P, Li Z, Liu X, et al. (2022). Methyltransferase-Like 3 Rescues the Amyloid-beta protein-Induced Reduction of Activity-Regulated Cytoskeleton Associated Protein Expression YTHDF1-Dependent N6-Methyladenosine Modification. Front Aging Neurosci, 14:890134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Yin H, Ju Z, Zheng M, Zhang X, Zuo W, Wang Y, et al. (2023). Loss of the m6A methyltransferase METTL3 in monocyte-derived macrophages ameliorates Alzheimer's disease pathology in mice. PLoS Biol, 21:e3002017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Keller L, Xu W, Wang H-X, Winblad B, Fratiglioni L, Graff C (2011). The obesity related gene, FTO, interacts with APOE, and is associated with Alzheimer's disease risk: a prospective cohort study. J Alzheimers Dis, 23:461-469. [DOI] [PubMed] [Google Scholar]
- [226].Reitz C, Tosto G, Mayeux R, Luchsinger JA (2012). Genetic variants in the Fat and Obesity Associated (FTO) gene and risk of Alzheimer's disease. PLoS One, 7:e50354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Li H, Ren Y, Mao K, Hua F, Yang Y, Wei N, et al. (2018). FTO is involved in Alzheimer's disease by targeting TSC1-mTOR-Tau signaling. Biochem Biophys Res Commun, 498:234-239. [DOI] [PubMed] [Google Scholar]
- [228].Jiang L, Lin W, Zhang C, Ash PEA, Verma M, Kwan J, et al. (2021). Interaction of tau with HNRNPA2B1 and N-methyladenosine RNA mediates the progression of tauopathy. Mol Cell, 81:4209-4227.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Deng Y, Zhu H, Xiao L, Liu C, Liu Y-L, Gao W (2021). Identification of the function and mechanism of m6A reader IGF2BP2 in Alzheimer's disease. Aging, 13:24086-24100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Zhang N, Ding C, Zuo Y, Peng Y, Zuo L (2022). N6-methyladenosine and Neurological Diseases. Mol Neurobiol, 59:1925-1937. [DOI] [PubMed] [Google Scholar]
- [231].Shlipak MG, Tummalapalli SL, Boulware LE, Grams ME, Ix JH, Jha V, et al. (2021). The case for early identification and intervention of chronic kidney disease: conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int, 99:34-47. [DOI] [PubMed] [Google Scholar]
- [232].Kim ED, Tanaka H, Ballew SH, Sang Y, Heiss G, Coresh J, et al. (2018). Associations Between Kidney Disease Measures and Regional Pulse Wave Velocity in a Large Community-Based Cohort: The Atherosclerosis Risk in Communities (ARIC) Study. Am J Kidney Dis, 72:682-690. [DOI] [PubMed] [Google Scholar]
- [233].Woodard T, Sigurdsson S, Gotal JD, Torjesen AA, Inker LA, Aspelund T, et al. (2015). Mediation analysis of aortic stiffness and renal microvascular function. J Am Soc Nephrol, 26:1181-1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Zhou D, Liu Y (2016). Renal fibrosis in 2015: Understanding the mechanisms of kidney fibrosis. Nat Rev Nephrol, 12:68-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Liu E, Lv L, Zhan Y, Ma Y, Feng J, He Y, et al. (2021). METTL3/N6-methyladenosine/ miR-21-5p promotes obstructive renal fibrosis by regulating inflammation through SPRY1/ERK/NF-κB pathway activation. J Cell Mol Med, 25:7660-7674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Liu P, Zhang B, Chen Z, He Y, Du Y, Liu Y, et al. (2020). m(6)A-induced lncRNA MALAT1 aggravates renal fibrogenesis in obstructive nephropathy through the miR-145/FAK pathway. Aging, 12:5280-5299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Li X, Li Y, Wang Y, He X (2022). The m6A demethylase FTO promotes renal epithelial-mesenchymal transition by reducing the m6A modification of lncRNA GAS5. Cytokine, 159:156000. [DOI] [PubMed] [Google Scholar]
- [238].Lan J, Xu B, Shi X, Pan Q, Tao Q (2022). WTAP-mediated N6-methyladenosine modification of NLRP3 mRNA in kidney injury of diabetic nephropathy. Cell Mol Biol Lett, 27:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Sun Q, Geng H, Zhao M, Li Y, Chen X, Sha Q, et al. (2022). FTO-mediated m6 A modification of SOCS1 mRNA promotes the progression of diabetic kidney disease. Clin Transl Med, 12:e942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Lanzer P, Hannan FM, Lanzer JD, Janzen J, Raggi P, Furniss D, et al. (2021). Medial Arterial Calcification: JACC State-of-the-Art Review. J Am Coll Cardiol, 78:1145-1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Rennenberg RJMW, Kessels AGH, Schurgers LJ, van Engelshoven JMA, de Leeuw PW, Kroon AA (2009). Vascular calcifications as a marker of increased cardiovascular risk: a meta-analysis. Vasc Health Risk Manag, 5:185-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Chen J, Ning Y, Zhang H, Song N, Gu Y, Shi Y, et al. (2019). METTL14-dependent m6A regulates vascular calcification induced by indoxyl sulfate. Life Sci, 239:117034. [DOI] [PubMed] [Google Scholar]
- [243].Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, et al. (2020). Global Burden of Cardiovascular Diseases and Risk Factors, 1990-2019: Update From the GBD 2019 Study. J Am Coll Cardiol, 76:2982-3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].BDW Group (2001). Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther, 69:89-95. [DOI] [PubMed] [Google Scholar]
- [245].Wang K, Liu H, Hu Q, Wang L, Liu J, Zheng Z, et al. (2022). Epigenetic regulation of aging: implications for interventions of aging and diseases. Signal Transduct Target Ther, 7:374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Deng K, Ning X, Ren X, Yang B, Li J, Cao J, et al. (2021). Transcriptome-wide N6-methyladenosine methylation landscape of coronary artery disease. Epigenomics, 13:793-808. [DOI] [PubMed] [Google Scholar]
- [247].Liang C, Wang S, Zhang M, Li T (2022). Diagnosis, clustering, and immune cell infiltration analysis of m6A-related genes in patients with acute myocardial infarction-a bioinformatics analysis. J Thorac Dis, 14:1607-1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Shi X, Cao Y, Zhang X, Gu C, Liang F, Xue J, et al. (2021). Comprehensive Analysis of N6-Methyladenosine RNA Methylation Regulators Expression Identify Distinct Molecular Subtypes of Myocardial Infarction. Front Cell Dev Biol, 9:756483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Vausort M, Niedolistek M, Lumley AI, Oknińska M, Paterek A, Mączewski M, et al. (2022). Regulation of N6-Methyladenosine after Myocardial Infarction. Cells, 11:2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Zhu R, Tian D, Zhao Y, Zhang C, Liu X (2021). Genome-Wide Detection of m6A-Associated Genetic Polymorphisms Associated with Ischemic Stroke. J Mol Neurosci, 71:2107-2115. [DOI] [PubMed] [Google Scholar]
- [251].Wan S-J, Hua Q, Xing Y-J, Cheng Y, Zhou S-M, Sun Y, et al. (2022). Decreased Urine N6-methyladenosine level is closely associated with the presence of diabetic nephropathy in type 2 diabetes mellitus. Front Endocrinol (Lausanne), 13:986419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Zhang B, Jiang H, Dong Z, Sun A, Ge J (2021). The critical roles of m6A modification in metabolic abnormality and cardiovascular diseases. Genes Dis, 8:746-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Yang Q, Chen S, Wang X, Yang X, Chen L, Huang T, et al. (2023). Exercise Mitigates Endothelial Pyroptosis and Atherosclerosis by Downregulating NEAT1 Through N6-Methyladenosine Modifications. Arterioscler Thromb Vasc Biol, 43:910-926. [DOI] [PubMed] [Google Scholar]
- [254].Li C, Ni Y-Q, Xu H, Xiang Q-Y, Zhao Y, Zhan J-K, et al. (2021). Roles and mechanisms of exosomal non-coding RNAs in human health and diseases. Signal Transduct Target Ther, 6:383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Wang Q-S, Xiao R-J, Peng J, Yu Z-T, Fu J-Q, Xia Y (2023). Bone Marrow Mesenchymal Stem Cell-Derived Exosomal KLF4 Alleviated Ischemic Stroke Through Inhibiting N6-Methyladenosine Modification Level of Drp1 by Targeting lncRNA-ZFAS1. Mol Neurobiol, 60:3945-3962. [DOI] [PubMed] [Google Scholar]
- [256].Fan T, Du Y, Zhang M, Zhu AR, Zhang J (2022). Senolytics Cocktail Dasatinib and Quercetin Alleviate Human Umbilical Vein Endothelial Cell Senescence via the TRAF6-MAPK-NF-κB Axis in a YTHDF2-Dependent Manner. Gerontology, 68:920-934. [DOI] [PubMed] [Google Scholar]
- [257].Mo W, Chen Z, Zhang X, Dai G, Ma D, Pan J, et al. (2022). N6-Methyladenosine Demethylase FTO (Fat Mass and Obesity-Associated Protein) as a Novel Mediator of Statin Effects in Human Endothelial Cells. Arterioscler Thromb Vasc Biol, 42:644-658. [DOI] [PubMed] [Google Scholar]
- [258].Ning Y, Chen J, Shi Y, Song N, Yu X, Fang Y, et al. (2020). Genistein Ameliorates Renal Fibrosis Through Regulation Snail via m6A RNA Demethylase ALKBH5. Front Pharmacol, 11:579265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [259].Li F, Kennedy S, Hajian T, Gibson E, Seitova A, Xu C, et al. (2016). A Radioactivity-Based Assay for Screening Human m6A-RNA Methyltransferase, METTL3-METTL14 Complex, and Demethylase ALKBH5. J Biomol Screen, 21:290-297. [DOI] [PubMed] [Google Scholar]
- [260].Cheng P, Han H, Chen F, Cheng L, Ma C, Huang H, et al. (2022). Amelioration of acute myocardial infarction injury through targeted ferritin nanocages loaded with an ALKBH5 inhibitor. Acta Biomater, 140:481-491. [DOI] [PubMed] [Google Scholar]
- [261].Cockova Z, Honc O, Telensky P, Olsen MJ, Novotny J (2021). Streptozotocin-Induced Astrocyte Mitochondrial Dysfunction Is Ameliorated by FTO Inhibitor MO-I-500. ACS Chem Neurosci, 12:3818-3828. [DOI] [PubMed] [Google Scholar]
- [262].Polli A, Nijs J, Ickmans K, Velkeniers B, Godderis L (2019). Linking Lifestyle Factors to Complex Pain States: 3 Reasons Why Understanding Epigenetics May Improve the Delivery of Patient-Centered Care. J Orthop Sports Phys Ther, 49:683-687. [DOI] [PubMed] [Google Scholar]
- [263].Kogan AC, Wilber K, Mosqueda L (2016). Person-Centered Care for Older Adults with Chronic Conditions and Functional Impairment: A Systematic Literature Review. J Am Geriatr Soc, 64:e1-7. [DOI] [PubMed] [Google Scholar]
- [264].Liu Z, Zou H, Dang Q, Xu H, Liu L, Zhang Y, et al. (2022). Biological and pharmacological roles of m(6)A modifications in cancer drug resistance. Mol Cancer, 21:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [265].Chong W, Shang L, Liu J, Fang Z, Du F, Wu H, et al. (2021). m(6)A regulator-based methylation modification patterns characterized by distinct tumor microenvironment immune profiles in colon cancer. Theranostics, 11:2201-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]