

Abstract
Virulent infectious agents such as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and Methicillin Resistant Staphylococcus Aureus (MRSA) induce tissue damage that recruits neutrophils, monocyte and macrophages, leading to T cell exhaustion, fibrosis, vascular leak, epithelial cell depletion, and fatal organ damage. Neutrophils, monocytes, and macrophages recruited to pathogen-infected lungs, including SARS-CoV-2 infected lungs, express phosphatidylinositol 3-kinase gamma (PI3Kγ), a signaling protein that coordinates both granulocyte and monocyte trafficking to diseased tissues and immune suppressive, pro-fibrotic transcription in myeloid cells. PI3Kγ deletion and inhibition with the clinical PI3Kγ inhibitor eganelisib promoted survival in models of infectious diseases, including SARS-CoV-2 and MRSA, by suppressing inflammation, vascular leak, organ damage, and cytokine storm. These results demonstrate essential roles for PI3Kγ in inflammatory lung disease and support the potential use of PI3Kγ inhibitors to suppress inflammation in severe infectious diseases.
One sentence summary:
PI3Kγ inhibition suppresses lethal inflammation associated with severe infectious diseases, such as SARS-CoV-2.
Introduction
SARS-CoV-2, SARS-CoV-1 and Middle Eastern respiratory syndrome coronavirus (MERS-CoV) are viruses that cause lethal infections associated with aberrant pulmonary and systemic inflammation, vascular leak, coagulation, and organ damage (1). These and other viral and bacterial infections induce acute respiratory distress syndrome (ARDS), a disorder characterized by breakdown of lung capillary walls and epithelial integrity, leading to leakage of interstitial fluid, plasma proteins, and leukocytes into alveoli that can cause fatal organ damage (2). Extensive recruitment of granulocytes, monocytes, and macrophages to the lung as a consequence of infection promotes organ damage and scarring as well as cytokine storm, a lethal systemic release of pro-inflammatory cytokines (2). In patients that survive ARDS, extensive lung fibrosis, compromised lung capacity, and systemic inflammation impair recovery from pulmonary and systemic viral infection (2). Single cell sequencing has shown that SARS-CoV-2 infections induce loss of alveolar type I (ATI) and II (ATII) epithelial cells, vascular damage, and inflammation, activation of fibroblasts, and co-infection with pneumonia-causing viruses (3,4,5). In children, SARS-CoV-2 infections generate a rapid interferon (IFN) response that ameliorates disease, whereas in adults, delayed IFN responses to MERS-CoV, SARS-CoV-1, and SARS-CoV-2 can fail to suppress viral infections and exacerbate the damage associated with severe inflammation (6,7).
To identify therapeutic strategies that can reduce inflammatory damage induced by lethal infectious agents such as SARS-CoV-2, we compared immune responses to infections in lung and in bronchoalveolar lavage (BAL) specimens from humans, hamsters, and mice using multiplex immunohistochemistry, fixed tissue RNA sequencing, and spatial transcriptomics. These strategies together identified key roles for immature inflammatory neutrophils and monocyte/macrophages in mediating acute and chronic lung tissue damage during infections.
Research in our laboratory previously identified key roles for the phosphatidylinositol-3 kinase (PI3K) isoform, PI3Kγ, in myeloid cell trafficking and immune suppressive, wound healing, and pro-fibrotic macrophage polarization in cancer and chronic inflammatory diseases (8,9,10,11,12,13,14). We found here that PI3Kγ plays a central role in lung damage in inflammatory diseases such as coronavirus disease 2019 (COVID-19), the respiratory disease caused by SARS-CoV-2 infections. Targeted inhibition of PI3Kγ prevents myeloid cell trafficking, vascular leak, and damage to lung epithelium, providing therapeutic benefit in models of severe lung and systemic infection. These findings support the clinical evaluation of PI3Kγ inhibitors for the treatment of infection-associated inflammation and disease progression.
Results
CD163+ wound-healing macrophages predominate in severe COVID-19
To identify therapeutic strategies that could suppress the lethal inflammation associated with pulmonary disorders such as COVID-19, we compared immune responses in SARS-CoV-2 and other infections in humans, hamsters, and mice using patient lung tissue and animal models of disease. Under institutional review board (IRB) approvals, we characterized immune cell infiltrates in formalin-fixed, paraffin-embedded (FFPE) lung tissue from recently deceased COVID-19 patients to normal human lung tissue obtained during lung surgeries at UCSD, and bronchoalveolar lavage (BAL) specimens from hospitalized, uninfected (normal) and SARS-CoV-2-infected patients (tables S1 to S2) using multiplex immunohistochemistry (mIHC), RNA sequencing of FFPE tissue and spatial transcriptomics. All BAL specimens were collected from hospitalized, living patients (table S1), whereas COVID-19 patient tissues were obtained from rapid autopsies of patients who died from SARS-CoV-2 infections early in the pandemic before vaccines or anti-viral therapies were developed (table S2). The average interval between illness onset and death was 19.9 ± 13.1 days for COVID-19 patients in this study. Patients who were virus positive at the time of death died 5± 2 days after illness onset, whereas virus negative patients died at 28.5 ± 7 days after illness onset, suggesting that most patients died as a consequence of long-term, rather than acute, damage to the lung induced by the viral infection.
Lungs from SARS-CoV-2-infected patients exhibited congested blood vessels, enlarged interstitial septa, and alveoli congested with large mononuclear cells, red blood cells, and protein deposits in contrast to open airways with thin-walled interstitial septa separating alveoli and thin capillaries in normal lung tissue (Fig. 1A; fig. S1A). Sparsely distributed alveolar CD68+ macrophages in normal lungs were replaced by dense accumulations of CD68+ monocytes and macrophages (black arrows) filling alveolar, interstitial, and perivascular spaces in COVID-19 lungs (Fig. 1B; fig. S1A). Myeloperoxidase (MPO)+ neutrophils also were increased in COVID-19 lung tissue (Fig. 1C). Monocyte/macrophages and neutrophils were each significantly (p=0.0024 and p=0.0023, respectively) increased more than ten-fold and were more broadly dispersed in diseased versus normal lungs (Fig. 1D and E); monocyte/macrophages were more abundant than neutrophils. Lungs from COVID-19 patients also exhibited significantly (p=0.0028) increased collagen deposition, identified as blue fibrils in Mason’s trichrome stained tissue (Fig. 1F and G). This extensive macrophage and neutrophil infiltration as well as collagen deposition suggested that the lungs of most deceased COVID-19 patients were undergoing macrophage-driven inflammation and fibrosis.
Figure 1: CD163+ myeloid cells dominate the immune microenvironment of SARS-CoV-2 - infected lungs.
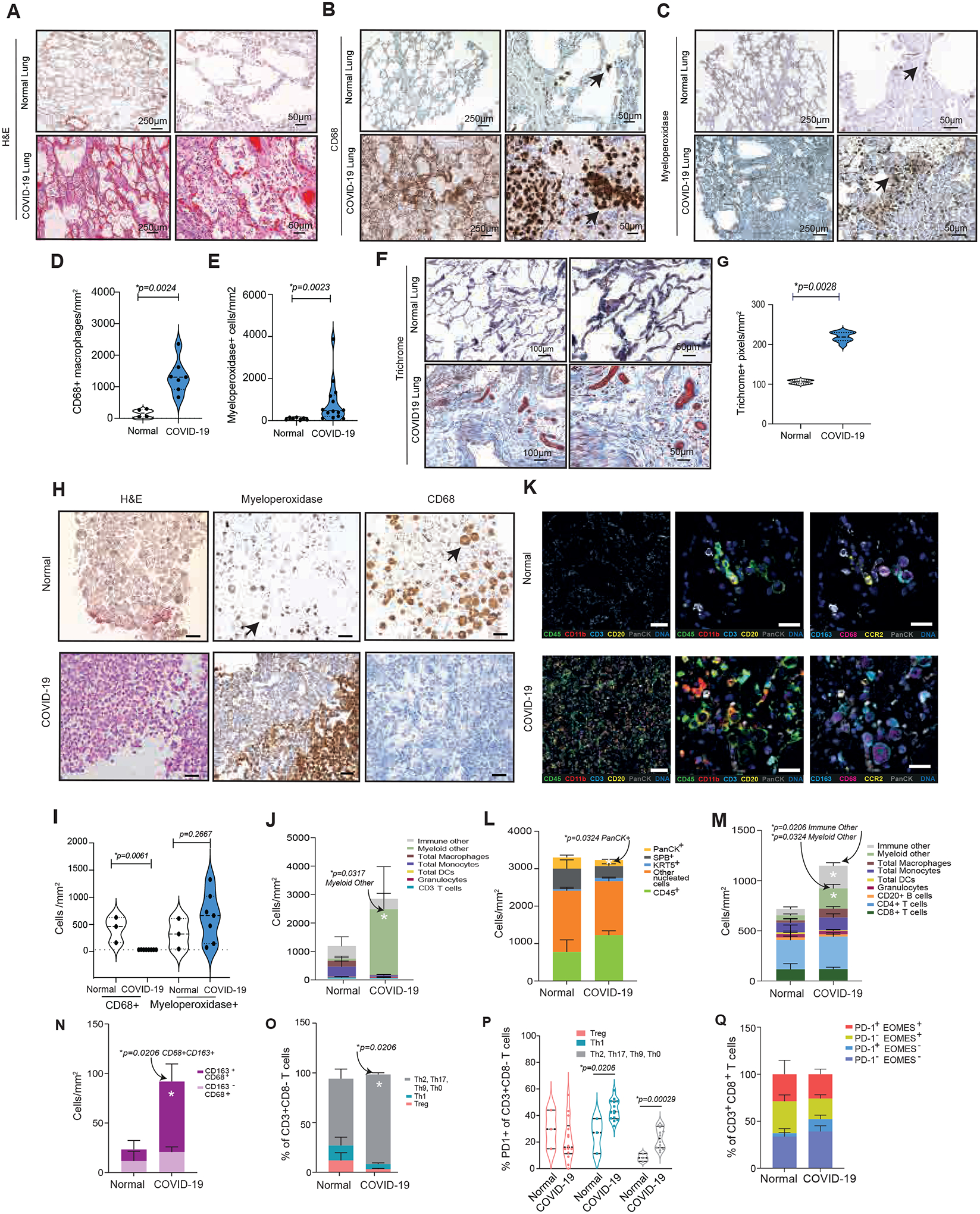
(A to C) H&E (A), anti-CD68 (B), and anti-myeloperoxidase (MPO) (C) stained images of lung tissue from patients that were uninfected (normal) or infected with SARS-CoV-2 (COVID-19); scale bar, 250 or 50 μm; arrows indicate CD68+ macrophages or MPO+ granulocytes. (D-E) Graphs of CD68+ macrophages/mm2 (D) and MPO+ granulocytes/mm2 (E) in normal (n=5) and COVID-19 patient lung tissues (n=14). (F) Images of trichrome-stained lung tissue from normal and COVID-19 patients. Scale bars, 100 or 50 μm. (G) Graphs of trichrome+ pixels/mm2 in lung tissue from normal (n=6) and COVID-19 patients (n=8). (H) Images of H&E, anti-CD68, and anti-MPO-stained sections of cell pellets from BAL from uninfected and COVID-19 infected patients. Arrows indicate granulocytes and macrophages. (I) Graphs of CD68+ macrophages/mm2 and MPO+ granulocytes/mm2 in sections of BAL cells from normal (n=3) and COVID-19 (n=7) patients. (J) Graphs of immune cell proportions within BAL from COVID-19 (n=5) and uninfected normal patients (n=4). (K, left) Pseudo-colored images of multiplex IHC-stained lung tissue from COVID-19 and normal uninfected patients: total immune cells (CD45, green), myeloid cells (CD11b, red), total T cells (CD3, cyan), total CD20+ B cells (CD20, yellow), epithelium (PanCK, white) and DNA (blue). (K, middle) Higher magnification of a subregion from the left column. K, right) Pseudo-colored images for CD68 (magenta); CCR2 (yellow), CD163 (cyan), and PanCK (white); scale bar indicates 50 μm. (L to P) Quantification graphs of multiplex immune markers in lung tissue from COVID-19 (n=14) vs normal (n=3) patients: (L) PanCK epithelial and immune cells, (M) CD45+ cell subsets, (N) CD163+ macrophages, (O) CD3+CD8− cell subsets, (P) CD3+CD8− cells expressing PD-1 and (Q) CD3+CD8+ cells expressing PD-1 or EOMES. For violin plots, dashed lines indicate mean and interquartile range; for stacked and single bar graphs, bars indicate mean ± SEM. For statistical testing, T tests (D, G) or Mann Whitney tests (E, I, J, L, M, N, O, P, Q) were utilized.
Hematoxylin and Eosin (H&E) and immunohistochemical (IHC) analysis of BAL specimens of hospitalized patients (n=3) who were not infected (normal) revealed the presence of large CD68+ alveolar macrophages yet few MPO+ cells. In contrast, BAL specimens from COVID-19 patients (n=7) were characterized by loss of alveolar macrophages and variable changes in MPO+ granulocytes (Fig. 1H and I). We further analyzed BAL specimens of uninfected normal (n=4) and hospitalized COVID-19 patients (n=5) (table S1) by multiplex immune profiling (14) with validated panels of myeloid lineage-identifying antibodies (table S3) followed by quantitative analyses (fig. S1B to D, see plots with red-labelled axes). BAL from COVID-19 patients exhibited a significantly (p=0.0317) increased population of myeloid cells (Fig. 1J) that lacked CCR2 or CD66b expression (here named “myeloid-other”) and are consistent with prior reports of extensive immature neutrophil and monocyte infiltration in COVID-19 patient BAL (4,5).
We investigated biomarkers of immune cell functional states in lung tissue from uninfected and SARS-CoV-2-infected patients with validated panels of lineage-identifying antibodies (table S4). Increased CD45+ (green), CD11b+ cells (red) (Fig. 1K, left and middle panels) and CD163+CD68+ macrophages (purple with blue membrane, Fig 1K, right panel) were observed in pseudo-colored images of lungs from COVID-19 patients. No obvious changes in lymphoid cell (CD45+CD3+ or CD45+CD20+) density were noted (Fig. 1K), but lungs from COVID-19 patients exhibited significantly (p=0.0324) decreased pan-cytokeratin-positive epithelial cells (Fig. 1L)(16). Immature monocytes and neutrophils (myeloid-other, p=0.0206) as well as NK cells, γδ T cells, plasma cells, and other cells, p=0.0324) were increased in COVID-19 lungs (Fig. 1M), as previously reported (2, 3, 4, 5, 6, 17, 18,19). CD68+ macrophages were positive for CD163 (Fig. 1N), a biomarker expressed by immune-suppressive, wound-healing-type macrophages (17,18).
Three COVID-19 patients who died within 7 days of hospitalization were virus-positive at the time of death (table S2). In their tissues, Surfactant Protein B positive (SPB+) ATII cells were decreased, whereas Programmed Cell Death Protein Ligand 1 (PD-L1)+ and Ki67+SPB+ cells were increased (fig. S2A to C), consistent with ongoing lung tissue repair processes. Active viral infection was associated with increased immature myeloid cells (myeloid other), CD163+CD68+ macrophages (fig. S2D and E), as well as increased PD-L1 on dendritic cells and granulocytes (fig. S2F and G).
Although total T cell abundance was unchanged, CD4+ T cells, particularly Foxp3−Tbet−CD4+Th2 cells, were significantly (p=0.0206) increased in lungs from COVID-19 patients (Fig. 1O). Th2 subsets have known roles in resolution of viral infection, wound-healing, and fibrosis, are often associated with CD163+ macrophages, and are increased in COVID-19 patients (19). Increased expression of Programmed Cell Death Protein 1 (PD-1), but not Ki67, (Fig. 1P; fig. S2H and I), suggests the presence of exhausted T cells in the lungs of COVID-19 patients (21, 20). Although high PD-1 and EOMES expression together can signify T cell activation, no increases in the CD8+ PD-1+EOMES+ cells were observed (Fig. 1Q), suggesting an absence of activated CD8+ T cells in COVID-19 lungs. Expression of PD-L1 and PD-1 was also noted on CD20+ B cells in virus-positive COVID-19 lungs (fig. S2J and K) (21). Together, these results suggest a dysregulated immune microenvironment characterized by infiltration of immature granulocytes and monocytes, CD163+ wound-healing type macrophages, exhausted T cells and fibrosis in lungs of patients with lethal COVID-19 disease.
Early inflammation is followed by wound-healing signatures in COVID-19 lungs.
To extend our understanding of the immune microenvironment in airways of COVID-19 patients, we performed RNA sequencing of normal and SARS-CoV-2-infected, formalin-fixed, paraffin-embedded BAL and lung tissue. For bulk RNA sequencing, we utilized a low input ligation-based targeted whole transcriptome expression profiling assay, TempO-Seq, to characterize gene expression signatures in FFPE tissue (22, 23). Transcriptome analysis of BAL specimens (normal, n=5; COVID-19, n=8) revealed that COVID-19 BAL was primarily characterized by increased expression of genes associated with immature neutrophil recruitment (CCL3, CXCL1, CXCL2, CXCL8, S100A8, SELL, CSF3R), pro-inflammatory response pathways(CXCL8/IL8, CCL3, CCL4, IL1B, S100A9, S100A8, OSM), and mRNA editing (APOBEC3A), but also decreased MHC class II (HLA-DMB, HLA-DRA) (Fig. 2A and B). COVID-19 BAL transcriptomes exhibited similarity to inflammatory and infectious disease responses (Fig. 2C; fig. S3A). Together, these data indicate a florid myeloid immune response in COVID-19 lungs.
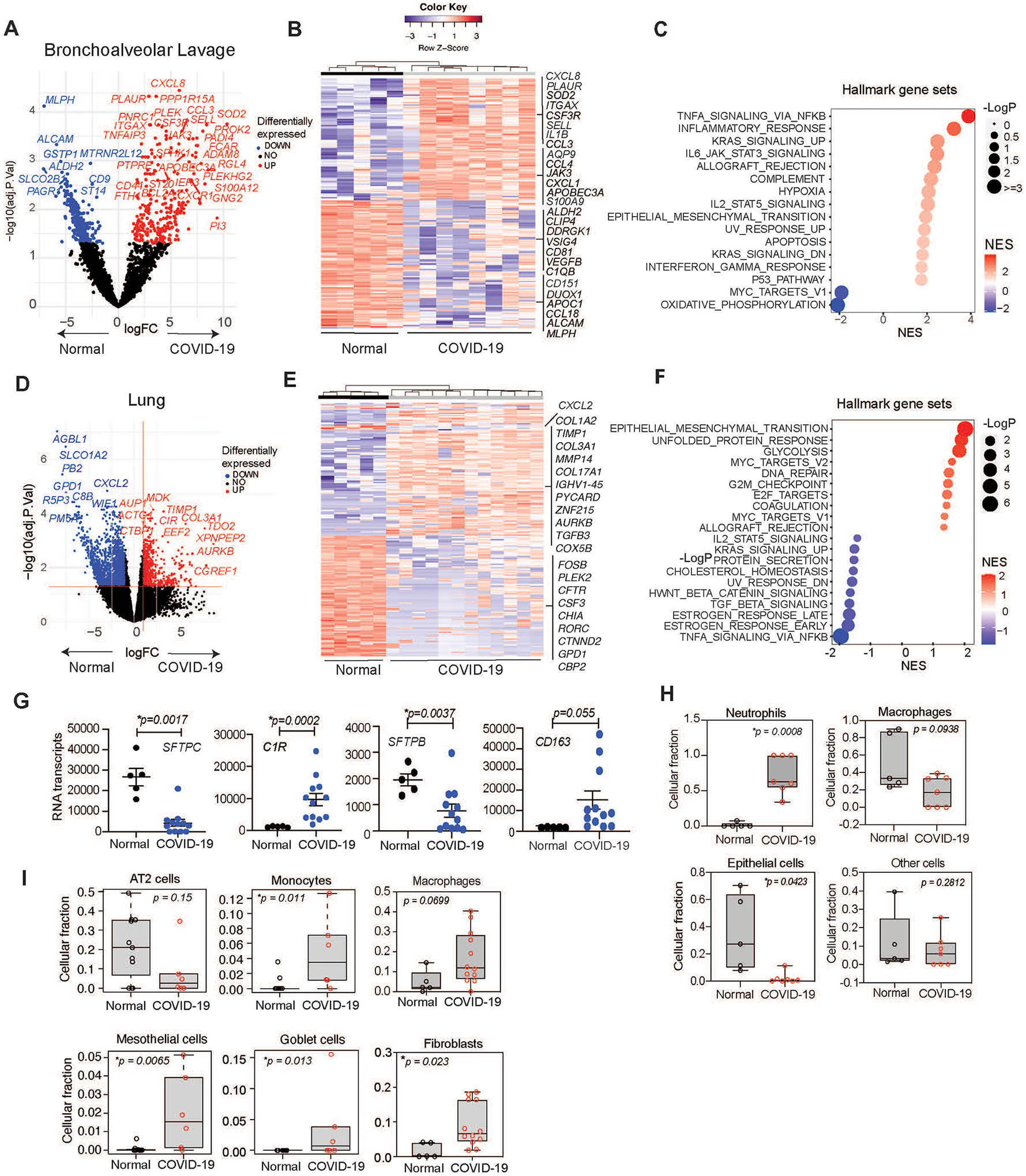
Figure 2. Transcriptomics identify a myeloid cell-rich microenvironment in SARS-CoV-2 infected lung tissue.
(A)Volcano plot of differential mRNA expression in BAL cells from uninfected “normal” (n=5) and COVID-19 infected (n=8) patients. Significant gene expression differences are identified in blue (down-regulated in COVID-19) or red (up-regulated in COVID-19). (B) Heatmap depicting differentially expressed genes in BAL cells from normal uninfected (n=5) and infected patients (n=8). (C) Plot of up- or down-regulated Hallmark pathways in BAL from infected (n=8) versus uninfected patients (n=5) graphed according to normalized expression score (NES) and log adj.p. (D) Volcano plot of differential mRNA expression in lung tissue from normal (n=5) and COVID-19 (n=12) patients. (E) Heatmap depicting differentially expressed genes in lung tissue from normal (n=5) and COVID-19 (n=12) patients. (F) Plot of differentially expressed hallmark pathways in lung tissue from normal and COVID-19 patients graphed according to NES and Log adj. P. (G) Graphs of SFTPC, C1R, SFTPB, and CD163 transcripts in COVID-19 (n=12) versus normal (n=5) patient lung tissue (mean ± SEM), (H-I) Box plots of mean ± SEM cell type deconvolution of (H) BAL from normal (n=5) and COVID-19 (n=8) patients and (I) lung tissue from normal (n=5) and COVID-19 (n=12) patients. p values were determined by limma-vroom.
We performed RNA sequencing of FFPE lung tissue specimens from 5 normal and 12 SARS-CoV-2-infected lung tissues. Specimens from upper and lower lung of two patients each were also sequenced. Transcriptome analysis indicated that lungs from COVID-19 patients exhibited more than 2500 significantly (p adj<0.05) downregulated genes and 813 significantly (p adj<0.05) upregulated genes compared to lungs from uninfected, normal patients (Fig. 2D and E). Volcano plot (Fig. 2D) and heatmaps of differentially-expressed genes, as well as gene set enrichment analysis (GSEA) (Fig. 2E–F) demonstrate upregulated genes associated with tissue remodeling/fibrosis (e.g., TIMP1, TGFB, COL1A1, COL3A1, COL5A1, LOX, MMP14, TGFB3), but decreased neutrophil recruitment genes (e.g., IL1A, CXCL2, CXCL1, CCL8, CSF3R), and lung epithelial function (e.g., CFTR, SFTPB, SFTPC, MUC15) in lungs from COVID-19 patients. GSEA also indicated upregulated genes associated with glycolysis, the unfolded protein response, epithelial to mesenchymal transition, coagulation, the G2M checkpoint, cellular response to stress and neutrophil extracellular trap formation (Fig. 2F; fig. S3B to E). Lung tissue from COVID-19 patients was associated with downregulated genes in pro-inflammatory and cholesterol homeostasis pathways, among others. BAL from COVID-19 patients was more pro-inflammatory and neutrophilic than lung tissue, whereas lung tissue was characterized by more monocyte/macrophages and an overall epithelial-to-mesenchymal transition/fibrosis signature. Diseased lungs exhibited significantly increased gene expression of CD163 (p=0.0006) and C1R (p=0.0003), indicating the presence of wound-healing, immune suppressive-type macrophages. Significant loss of genes associated with normal airway functions, including surfactant proteins (SFTPB p=0.0136, SFTPC p=0.0003) (Fig. 2G; fig. S3F) was observed in diseased lungs, supporting prior descriptions that COVID-19 is associated with the influx of wound-healing macrophages and loss of ATII cells (2, 3, 4, 5). These results are consistent with the presence of a neutrophil rich lung environment in the early stages of disease that is replaced by a macrophage-rich environment and associated fibrosis and progressive COVID-19 disease.
To correlate RNA sequencing and IHC analyses, we applied cell type deconvolution analysis to normalized expression values in BAL and lungs from normal and COVID-19 patients (Fig. 2H and I) (24, 25). This analysis confirmed that normal BAL exhibits a high macrophage/low neutrophil signature, whereas COVID-19 BAL exhibits high neutrophil/low macrophage signature, similar to results from our IHC studies (Fig. 2H). Cell type deconvolution of bulk RNA sequencing data revealed increased monocytes, macrophages, fibroblasts, mesothelial cells and goblet cells and decreased ATII cells in lungs from COVID-19 versus normal patients (Fig. 2I). An increase in goblet cells has been previously associated with chronic obstructive pulmonary disease, a chronic fibrotic disorder (26). These results reveal that lethal COVID-19 is characterized by ATII cell depletion and a monocyte/macrophage/fibroblast-rich, wound-healing, immune-suppressive environment, suggesting impaired lung function as previously shown (27).
We examined the impact of ongoing viral infection at the time of death on gene expression profiles. Tissues of virus-positive patients exhibited signatures of IFN-α and IFN-γ pathways, illustrated by expression of interferon-inducible genes, such as CXCL10, CXCL11, CCL8 and OAS3 (fig. S4A to C). In contrast, virus-negative COVID-19 patient tissues exhibited higher signatures of transforming growth factor (TGF)-β signaling and coagulation (fig. S4C). Virus-positive patients exhibited signatures of cellular stress, cytokine expression, inflammation and loss of normal cellular physiology when compared to lungs of normal, uninfected patients (fig. S4D). Two virus-positive patients at the time death were in treatment for multiple myeloma, a known risk factor for lethal COVID-19 due to the absence of antibody-mediated immune responses in these patients (28). As virus-positive patients died from COVID-19 an average of 5 days after illness onset but virus-negative patients died 28.5 days after illness onset, these data support that death from COVID-19 is associated with either an early insurmountable viral infection or from long-term wound-healing/fibrosis processes that damage normal lung function and architecture.
Macrophages in COVID-19 lungs express a wound-healing gene expression signature
Our studies indicate that increased macrophage content in COVID-19 lungs correlates with fibrosis and death. To explore whether lung myeloid cells promote the wound-healing, pro-fibrotic environment in COVID-19 diseased lungs, we performed spatial profiling of lung tissue using the nanoString GeoMx ImmuneOncology plus COVID-19 platform on CD68+ macrophages, MPO+ granulocytes, and panCK+ epithelium in normal and COVID-19 lung tissue specimens. Volcano plots, heat maps, and GSEA analysis of differentially expressed genes within macrophages demonstrate that COVID-19 lung macrophages exhibit alternatively activated, wound-healing, immune-suppressive gene expression signatures characterized by increased expression of CD163 and IDO, as well as heat shock proteins (HSPA1A), complement/coagulation (C1QA, C1QB, C1R), and tissue remodeling (COL3A1, COL1A1, COL1A2, FN1, CTSS) pathways; with down-regulation of genes in immune response pathways (HLA-DRB5) (fig. S5A to C). Macrophages in virus-positive tissues expressed IFN-associated genes, including IRF8, OAS1, OAS2, CXCL8 (fig. S5A to C). Spatial profiling of granulocytes revealed IFN, oxidative phosphorylation, apoptosis, and complement expression signatures in COVID-19 patients (fig. S5D to F). Lung epithelial cells also expressed signatures of inflammation and TGF-β signaling (fig. S5G to I). These signatures support the hypothesis that during SARS-CoV-2 infection, myeloid cells initially mount an IFN-driven anti-viral response that is superseded by wound-healing responses associated with fibrosis and death, much as has been observed after infection with SARS-CoV-1 and MERS-CoV and in animal models of betacoronavirus infections (6).
PI3Kγ promotes inflammation in models of SARS-CoV-2 infection
These results suggest that strategies that inhibit myeloid cell recruitment to tissues could provide therapeutic benefit in COVID-19 and other severe infectious diseases. One potential target for therapeutic intervention of myeloid cell trafficking in inflammatory diseases is the signaling protein PI3Kγ. PI3Kγ promotes neutrophil and monocyte trafficking to tumors by activating integrin α4β1 expressed on circulating myeloid cells (8); PI3Kγ also promotes immune suppressive, pro-fibrotic transcription in macrophages (9,10,11). Genetic or pharmacological blockade of PI3Kγ suppressed recruitment of myeloid cells to tumor tissues, repolarized macrophages and dendritic cells to promote anti-tumor T cell responses and prevented fibrosis (9, 10, 11). Other studies showed that PI3Kγ inhibition also blocked neutrophil recruitment and degranulation (12) and prevented vascular leak (29,30). Inhibition of PI3Kγ also suppressed fibrosis in association with pancreatic cancer and pancreatitis through the reduction of macrophage expression of pro-fibrotic factors, including TGFB, PDGFA, PDGFB, PLAU and extracellular matrix genes (10).
To investigate whether PI3Kγ inhibition might also ameliorate lung damage associated with SARS-CoV-2 and other infections, we first evaluated PI3Kγ expression in lung tissue from normal and COVID-19 patients. PIK3CG mRNA was expressed both in COVID-19 patient lung tissue and in normal lung (Fig. 3A). Immunohistochemical staining for PI3Kγ and CD68 in serial sections of COVID-19 tissue revealed that PI3Kγ expression overlapped extensively with CD68+ macrophages (Fig. 3B). Fluorescent confocal microscopy of COVID-19 lungs for CD68 (green) and PI3Kγ (red) expression showed considerable overlap of expression (yellow, arrows) (Fig. 3C upper panels). Stochastic optical reconstruction microscopy (STORM) of single macrophages demonstrated that PI3Kγ colocalizes extensively with CD68 in clusters at the cell membrane in COVID-19 infected lungs (Fig. 3C lower panels). Increased macrophage content and macrophage PI3Kγ expression was also observed in lung tissue from patients with other inflammatory lung disorders (fig. S6A and B). Specificity of the antibody for PI3Kγ used in immunohistochemistry studies was confirmed by manufacture’s specifications and Western blotting of immortalized WT, mock transfected and PI3Kγ Crispr-mediated knockout murine macrophages (fig. S6C).
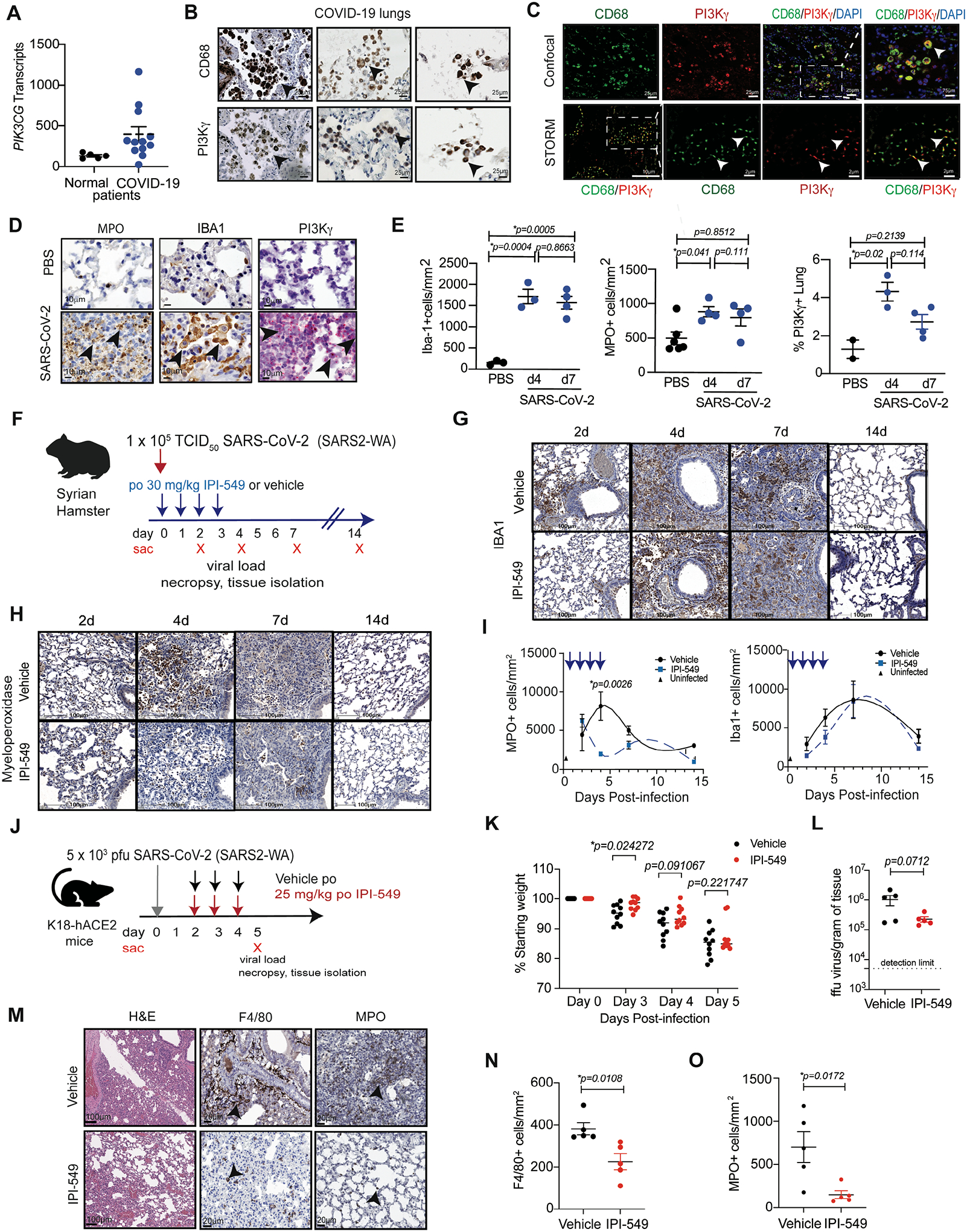
Figure 3. PI3Kγ is promotes inflammation in response to SARS-CoV-2 infection.
(A) Graph of PI3KCG gene transcripts determined by RNA sequencing of FFPE lung tissue from normal (n=5) and COVID-19 patients (n=14). (B) Images of CD68 and PI3Kγ expression in lung tissue from COVID-19 patients. Scale bar indicates 25 μm. Arrowheads indicate CD68+ and PI3Kγ+ cells in serial 5 μm sections. (C) PI3Kγ (red), CD68 (green), and DAPI (blue) confocal (upper panels) and stochastic optical reconstruction microscopy (STORM, lower panels) images of macrophages in COVID-19 infected lungs. Scale bars indicate 25 μm or 75 μm (upper) and 2 μm or 10 μm (lower). Insets indicate fields magnified in subsequent panels. Arrowheads identify CD68+PI3Kγ+ stained cells. (D) Immunostaining for MPO+ granulocytes, IBA1+ macrophages and RNAscope detection of PI3Kγ mRNA in lung tissue from uninfected (PBS) and SARS-CoV-2 infected Syrian golden hamsters. Scale bar indicates 10μm. Arrowheads identify stained cells. (E) Graphs of mean ± SEM IBA1+ macrophages, MPO+ granulocytes and PI3Kγ mRNA+ cells in lung tissue from uninfected (PBS, n=2–5) and SARS-CoV-2 infected (COVID, n=3–4) hamsters. (F) Schematic of hamster infection with SARS-CoV-2 and treatment with vehicle (n=4) or IPI-549 (n=4). (G and H) Images of IBA1+ macrophages (G) and MPO+ granulocytes (H) in lungs from (F). (I) Graphs of mean ± SEM MPO+ neutrophils and IBA1+ macrophages in hamster lungs over time. Arrows indicate days of treatment. (J) Schematic of K18-ACE2 transgenic mice infected with SARS-CoV-2 and treated with vehicle or IPI-549. (K) Graph of median weight loss over time for mice infected with SARS-CoV-2 and treated with vehicle (n=10) or IPI-549 (n=10). (L) Graph of mean ± SEM viral load expressed as focus-forming units (ffu)/gm lung tissue in mice (n=10 per group) from (J). (M) Images of H&E, MPO+ and F4/80+ staining in endpoint tissues from (K). Scale bar indicates 20 μm. Arrowheads indicate F4/80+ macrophages and MPO+ granulocytes. (N and O) Graphs of mean ± SEM F4/80+ macrophages/mm2 (N) and MPO+ granulocytes/mm2 (O) in lung tissue from mice infected with SARS-CoV-2 and treated with vehicle (n=5) or IPI-549 (n=5). Statistical analysis was performed by limma-vroom (A), unpaired T tests (K, L, N, O), or one-way ANOVA with Tukey’s post hoc multiple comparisons test (E, I).
We next evaluated PI3Kγ expression in an animal model of SARS-CoV-2 infection (31). SARS-CoV-2 induces non-lethal respiratory disease in hamsters, with a duration of 10–14 days. When Syrian golden hamsters were infected intranasally with SARS-CoV-2, infiltration of myeloperoxidase+ neutrophil and IBA1+ macrophages into lungs was rapidly induced. Neutrophil infiltration peaked at 4 days after infection, whereas IBA1+ macrophage infiltration peaked 7 days after infection (Fig. 3D and E). SARS-CoV-2 infection of Syrian golden hamsters increased PI3Kγ mRNA expression in these cells, as detected by RNAscope fluorescence microscopy (Fig. 3D and E). Taken together, these data indicate that PI3Kγ is expressed in COVID-19-associated myeloid cells. On the basis of its roles in myeloid cell trafficking in cancer and its tissue specific expression patterns, it is possible that PI3Kγ plays a key role in promoting COVID-19.
To investigate whether PI3Kγ inhibition could serve as a therapeutic strategy for COVID-19, we tested the PI3Kγ inhibitor IPI-549 (eganelisib), a highly selective PI3Kγ inhibitor that is currently in development as a cancer therapeutic and has shown safety and activity in clinical trials (13,14), in a hamster model of SARS-CoV-2 infection (Fig. 3F) (31). Hamsters were inoculated with SARS-CoV-2 and treated with a 4-day course of IPI-549 or vehicle from day 0 to day 4 (Fig. 3F). Neutrophil accumulation was substantially suppressed by IPI-549 treatment, whereas macrophage accumulation was not inhibited, possibly because macrophage accumulation peaks at day 7 post-inoculation (Fig. 3G to I); treatment did not substantially affect weight loss in this non-lethal model of SARS-CoV-2 infection (fig. S7A). These results indicate that PI3Kγ inhibition can suppress neutrophil recruitment induced by SARS-CoV-2 infection and suggest that continuous treatment with IPI-549 might be warranted to further protect lungs from inflammation associated with infection.
We then tested the effect of PI3Kγ inhibition in murine models of SARS-CoV-2 infection. K18-ACE2TG C57BL/6 mice (32) were infected with SARS-CoV-2 and treated two days later with IPI-549 or vehicle (Fig. 3J). IPI-549 treatment of SARS-CoV-2 infected hACE2-TG mice significantly delayed weight loss (Fig. 3K, p=0.02472) and delayed signs of illness, including reduced activity, reduced appetite, and social withdrawal, but did not significantly impact viral load (p=0.0712) or survival (Fig. 3L, fig. S7B). IPI-549 treatment significantly reduced monocyte/macrophage (p=0.0108) and granulocyte (p=0.0172) infiltration, as detected by immunohistochemistry (Fig. 3M to O) or by flow cytometry in lung tissue collected at 5 dpi (fig. S7C to E).
Transcriptomics reveal PI3Kγ inhibition reduces inflammation in mice infected with SARS-CoV-2
To investigate the impact of PI3Kγ inhibition on SARS-CoV-2 infected mouse lungs further, we performed RNA sequencing of lungs from hACE2Tg mice infected intranasally with SARS-CoV-2 and treated 2–5 days post infection (dpi) later with vehicle or IPI-549, as shown in Fig. 3J. Differential expression analysis revealed that just 3 days of IPI-549 treatment down-regulated expression of pro-inflammatory cytokines and transcription factors (e.g., Irf8, Irf5, Tlr9, Tnfa, Osm, S100a4, Ltf) as well as neutrophil, monocyte, and macrophage markers (Adgre1, Cd68, Trem2, Itgam, Apoe, C1qa, Ly6c2), and markers of fibrosis (Col1a1, Col3a1, Col5a2, Fn1, Mmp2, Mmp8) (Fig. 4A). In contrast, genes associated with normal organ development (e.g.,Tgfbr3) or protection from oxidative stress (e.g., Cbr2, Ahr) were upregulated in IPI-549-treated lungs (Fig. 4A). Signatures of macrophage activation, response to infection, inflammation and interferon gamma signaling stimulated by SARS-CoV-2 infection were downregulated in IPI-549 treated lungs, and signatures of epithelial and vascular development were upregulated in IPI-549 treated lungs (Fig. 4B). To determine if differences between experimental conditions were associated with unique cell-type signatures, we assessed the list of differentially expressed genes for enrichment for gene expression markers used to define cell types, as defined in PanglaoDB (33), a single cell sequencing resource for gene expression data collected and integrated from multiple studies. Vehicle-treated, SARS-CoV-2-infected lungs exhibited gene expression signatures of macrophage, monocyte, and myeloid-derived suppressor cells, whereas IPI-549-treated lungs exhibited signatures of lung epithelial cells, including Clara cells, ATI and ATII cells, and ionocytes (Fig. 4C). Examples of downregulated macrophage genes in IPI-549-treated lungs shown in the heatmap in fig. S8 and in graphs of transcripts per million for monocyte/macrophages genes include Adgre1, Itgam, Cd68 and Ly6c2 (Fig. 4D). Pik3cg, the gene for PI3Kγ, was also downregulated upon IPI-549 treatment, indicating a reduction in myeloid cell content. We identified 102 genes that are upregulated in both SARS-CoV-2 infected murine and human lungs but are downregulated in IPI-549 treated lungs (Fig. 4E and F). These genes were associated with signatures of inflammation, neutrophil degranulation, leukocyte migration, SARS-CoV-2 signaling, extracellular matrix disassembly, and interferon gamma responses (Fig. 4E). Select genes that were upregulated in SARS-CoV-2 infected human and mouse lungs and downregulated in IPI-549 treated lungs, including interferon response genes (CGAS, IFIT130), fibrosis-associated genes (COL1A2, COL3A1, FN1) and immune exhaustion genes (LAG3, LILRB4), are shown in Fig. 4F. Taken together, these data indicate that IPI-549 treatment prevents epithelial tissue damage and fibrosis.
Figure 4: PI3Kγ inhibition suppresses gene expression signatures of inflammation and tissue damage during SARSCoV-2 infection.
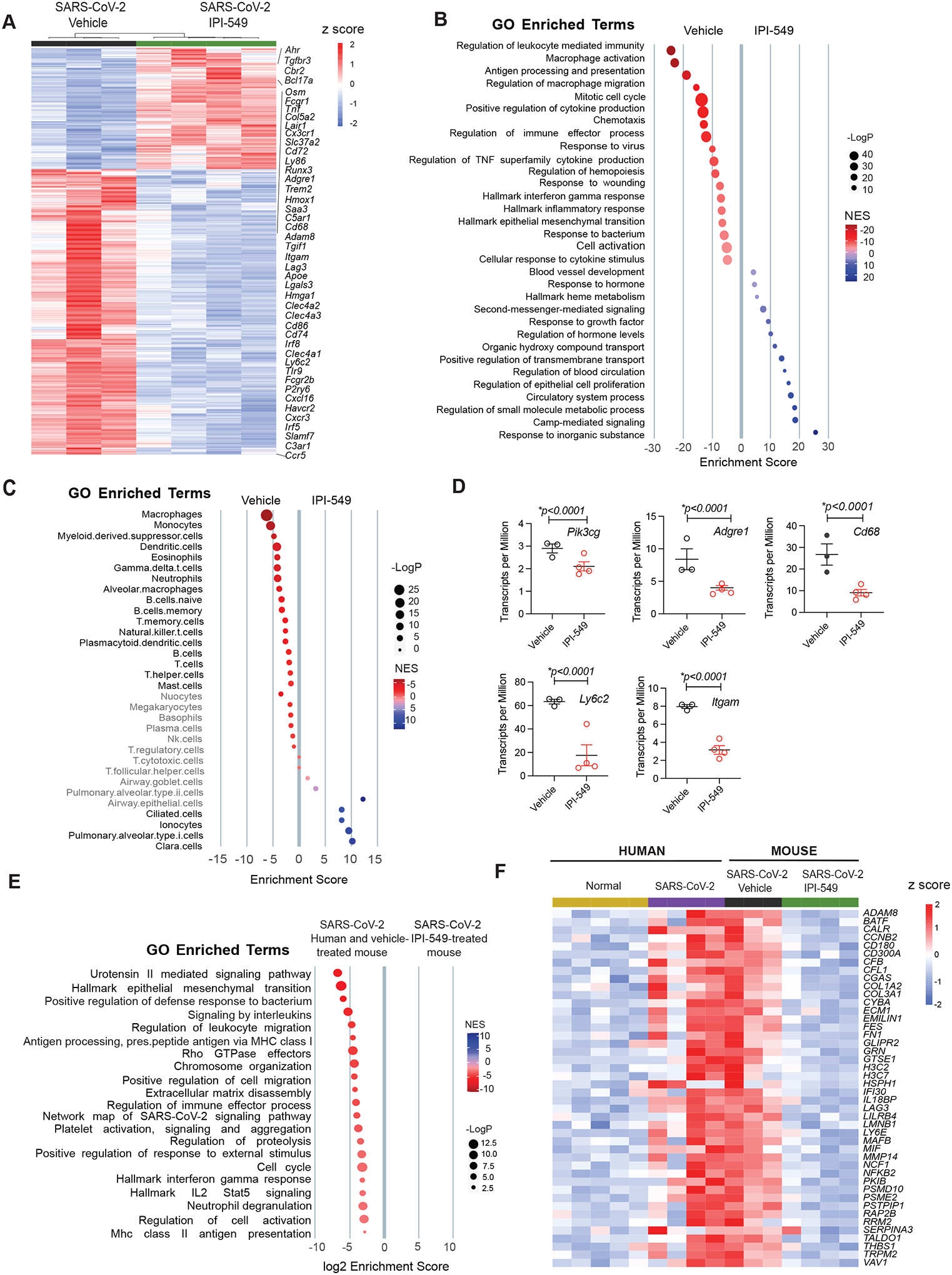
(A and B) Heatmap (A) and gene set enrichment signatures (B) of differentially expressed genes and pathways in SARS-CoV-2 infected, vehicle- or IPI-549-treated mouse lungs at day 5 post-infection. Select gene names are shown. (C) Cell type enrichment signatures expressed in lungs from (A). Cell types shown in grey are not significantly changed in lungs from IPI-549 treated animals. (D) Transcripts per million of myeloid cell genes Pik3cg Adgre1, Cd68, Ly6c2 and Itgam) are shown for lungs from (A). (E) Gene enrichment signatures common to mouse and human SARS-CoV-2 infected lungs that are down-regulated in IPI-549-treated lungs. (F) Heatmap of differentially expressed genes that are upregulated in SARS-CoV-2-infected, IPI-549-treated mouse lungs versus SARS-CoV-2-infected human and mouse lung tissues. Statistical analysis was performed by limma-vroom.
PI3Kγ inhibition reduces inflammation and tissue damage in aged mice infected with SARS-CoV-2
We next examined the effect of PI3Kγ inhibition on mouse-adapted (ma)SARS-CoV-2 infection in aged mice in three separate cohorts. 10-month-old female BALB/c mice were infected intranasally with maSARS-CoV-2 (34) and were treated with vehicle (n=5) or with IPI-549 (n=10) from 0 days post-infection (dpi) until 2 dpi (cohort 1) (Fig. 5A). Tissue and blood were collected from cohort 1 mice on day 2 post-infection for viral load, histological, and other assessments. Additional cohorts of mice were infected and treated with vehicle (n=5) or IPI-549 (n=10) from 0–4 dpi (cohort 2), or with IPI-549 (n=10) from 2–4 dpi (cohort 3) or were mock-infected and treated with vehicle (sham, n=4). IPI-549 significantly (p=0.0133) extended survival of mice infected with SARS-CoV-2, whether treated from 0–4 dpi (cohort 2) or 2–4 dpi (cohort 3) (Fig. 5B). Tissue and blood were collected from mice that were surviving on 6 dpi from cohort 3 (n=2) and sham-infected mice (n=4) for viral load, histological and other assessments. No tissue was collected from mice in cohorts 2 and 3 that died before day 6.
Figure 5: PI3Kγ inhibition suppresses inflammation and promotes survival from maSARS-CoV-2 infection.
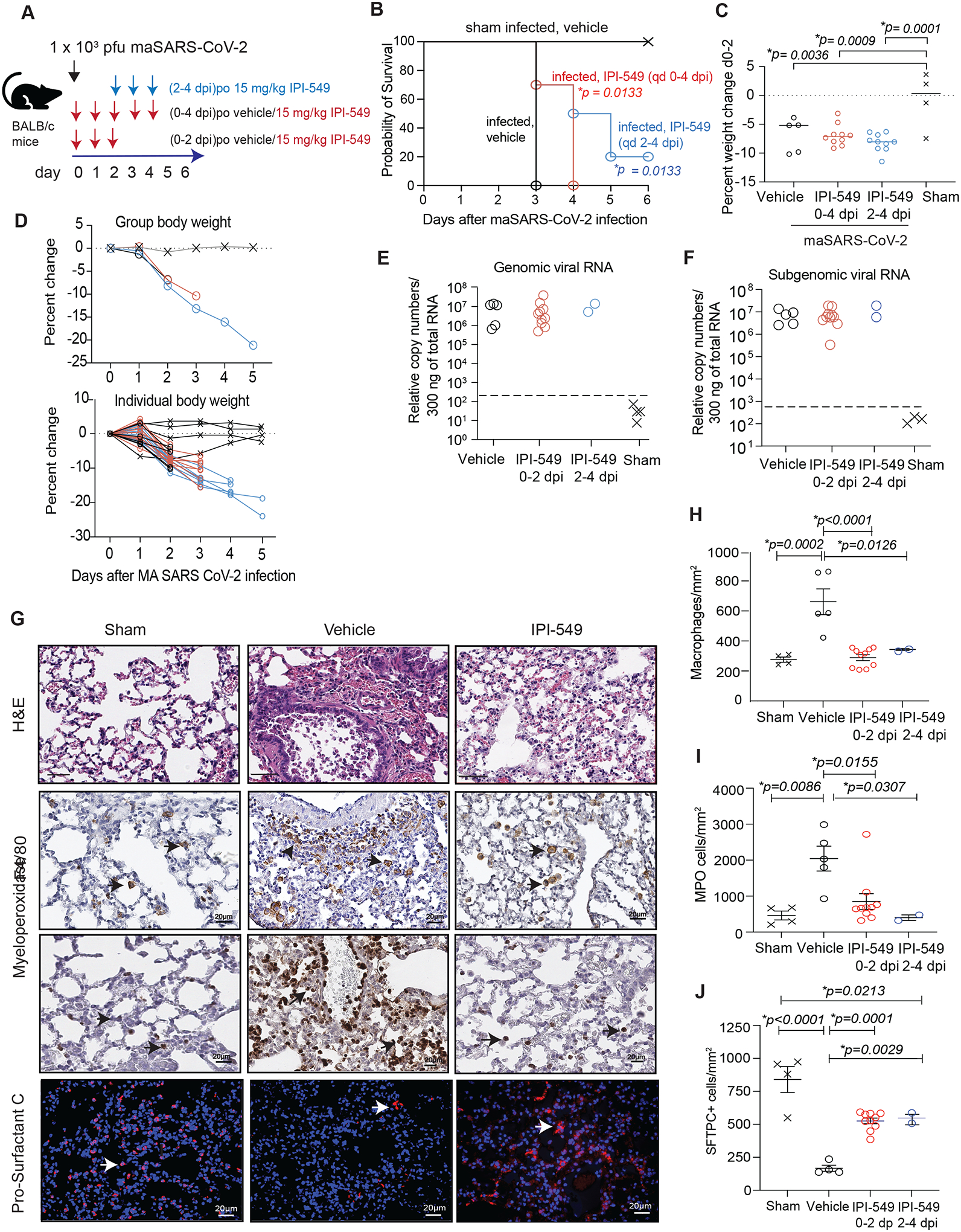
(A) Schematic of maSARS-CoV-2 infection in 10-month-old mice. Cohorts of mice were inoculated with virus and treated with vehicle or IPI-549 from 0 dpi or 2 dpi. Tissue was collected at 2 dpi or 6 dpi (B) Probability of survival graph of mice treated with vehicle alone (black x, sham infected, n=4) or inoculated with virus and treated with vehicle from 0–4 dpi (black open circles, n=5), with IPI-549 from 0–4 dpi (red circles, n=10) or with IPI-549 from 2–4 dpi (blue circles, n=10). (C) Graph of median weight changes at 2 dpi of mice from each group in (B). (D) Graph of group and individual body weight changes over time in mice from (B). (E and F) Graph of genomic (E) and subgenomic (F) viral RNA relative to total RNA in tissues from mice treated 0–2 dpi with vehicle (n=5) or IPI-549 (n=10) (cohort 1) and from surviving mice at day 6 that had been treated 2–4 dpi with IPI-549 (n=2) or sham-infected (n=4). Dashed line indicates lower limit of virus detection. (G) Images of H&E as well as anti-F4/80, anti-MPO, and anti-ProSurfactant C staining of lung tissues from (A). Arrowheads identify CD68+ macrophages, MPO+ granulocytes, and Pro-Surfactant C+ ATII cells. (H to J) Graphs of mean ± SEM of F4/80+ macrophages/mm2 (H), MPO+ granulocytes/mm2 (I), and Pro-Surfactant C+ cells (J) in tissues from (A). Significance determined by Log-rank (Mantel-Cox) test (B), one-way ANOVA with Dunnett’s multiple comparisons test (C) or Tukey’s multiple comparisons test (H-J).
Although PI3Kγ inhibitor treatment did not reduce weight loss induced by infection and did not affect viral load during this assay window in cohorts 1, 2 and 3 (Fig. 5C to F), IPI-549 treatment did reduce lung inflammation in infected mice. We compared lung tissues collected at 2 dpi (cohort 1) and at 6 dpi (cohort 3 and sham infected) for immune cell and epithelial cell content. In contrast to lungs of vehicle-treated, SARS-CoV-2 infected mice, which exhibited substantial macrophage and granulocyte content, lungs of 0–4 dpi (cohort 1) and 2–4 dpi (cohort 3) IPI-549-treated mice exhibited near normal frequencies of macrophages (p<0.0001) and granulocytes (p=0.0155) (Fig. 5G to I). These results indicate that PI3Kγ inhibition suppressed SARS-CoV-2 induced lung inflammation in both treatment regimens. PI3Kγ blockade by IPI-549 also reduced virus-induced lung damage, as ATII loss detected by Pro-Surfactant C staining, septal wall thickening, vascular congestion and hemorrhage were equally suppressed in IPI-549-treated mice from cohorts 1 and 3 (Fig. 5G and J). PI3Kγ inhibition suppressed inflammatory cytokine gene and protein expression, as shown for Tnfa and Cxcl10 mRNA in lung tissue and circulating CXCL10 serum proteins (fig. S9A and B). These results indicate that inhibition of PI3Kγ can suppress the damaging inflammation and cytokine surge that accompanies SARS-CoV-2 infection.
We sequenced mRNA isolated from lung tissue of cohorts 1 and 3 and sham-infected mice. Transcriptome analysis revealed that maSARS-CoV-2 infection downregulated expression of genes associated with normal lung epithelium and alveolar macrophage function (Sftpc, Siglecf, Muc2, Muc4, Muc6) and T cell function (Cd3g, Cd8b1, Rag1, Trdc), and stimulated expression of genes associated with pro-inflammatory response pathways, including cytokine/chemokines (Cxcl9, Cxcl10, Ccl2, Cxcl1, Cxcl2, Il6), interferon response genes (Oasl, Isg15, Irf7, Ifit1, Ifit3, Gbp5, Gbp8, Gbp9) and immune exhaustion (Cd274, Lag3) (fig. S9C). Accordingly, maSARS-CoV-2 infection of mouse lungs was associated with upregulation of gene expression pathways related to interferon gamma responses, inflammation and epithelial-to-mesenchymal transition and downregulation of pathways associated with normal function, including oxidative phosphorylation, DNA repair, and fatty acid metabolism, consistent with SARS-CoV-2 infection in humans (fig. S9D).
To determine the effect of PI3Kγ inhibition on lung function and immune responses, we compared transcriptomes from normal lungs (sham) and maSARS-CoV-2-infected lungs that were treated with vehicle or IPI-549. Heatmaps illustrate that IPI-549 treatment of virus-infected animals from dpi0-2 (cohort 1) only modestly affected gene expression compared with treatment with vehicle (fig. S9E). In contrast, IPI-549 treatment from 2–4 dpi (cohort 3) significantly (p adj <0.05) altered gene expression patterns by reducing immune response gene (ISG) and inflammatory gene expression signatures and increasing expression of lung alveolar macrophage genes, such as Siglecf, Mrc1, Csf1r, Trem2, and Apoe, to that found in sham-treated lung (fig. S9E). Gene signature and pathway analyses revealed that IPI-549 treatment stimulated blood vessel and epithelial morphogenesis signatures characterized by expression of key endothelial cell regulators (Vwf, Fn1, Itga5, Itgb3), alveolar macrophage markers (Trem2, Apoe) and other repair genes (fig. S9F to H). These results are similar to those shown in Fig. 4 for hACE2Tg mice. Taken together, results from these two mouse models suggest that IPI-549 treatment restored some normal alveolar macrophage and epithelial tissue markers and reduced expression of recruited pro-inflammatory macrophage markers, while also upregulating markers of tissue repair.
When gene expression signatures between SARS-CoV-2-infected murine and human lungs were compared, overlap was observed primarily in interferon gamma and viral response signatures; expression of these interferon response signatures was strongly suppressed in IPI-549-treated mouse lungs (fig S9I and J). Importantly, IPI-549 treatment was associated with a reduction in gene expression signatures found in other infectious diseases, including hepatitis A, B and C, pneumonitis, and tuberculosis (fig. S9K). It is notable that PI3Kγ inhibition similarly reduced inflammatory response gene signatures and increased signatures of wound repair and vascularization in both young and old mice. These results indicate that PI3Kγ inhibition can reduce damaging inflammation in the lung and protect normal lung function during infectious lung disease.
PI3Kγ inhibition reduces inflammation, vascular leak, and cytokine storm in mouse models of acute respiratory distress syndrome
To explore whether PI3Kγ inhibition could provide therapeutic benefit in animal models of other pulmonary diseases and identify mechanisms of therapeutic benefit, we inoculated wild-type (WT) and Pik3cg−/− mice with methicillin-resistant Staphylococcus aureus (MRSA) or multi-drug resistant Escherichia coli O157:H7 (Fig. 6A; fig. S10A). Whereas all WT infected mice succumbed to MRSA infection over a period of 24 hours, 40% of Pik3cg−/− mice survived infection (Fig. 6A, p=0.0092). A reduction in serum IL-1β, a key biomarker in systemic inflammation, was observed in Pik3cg−/− animals, although no significant differences in IL-6, bacterial load, or the abilities of WT and Pik3cg−/− macrophages to mediate killing of bacteria in vitro by phagocytosis were noted (fig. S10B to D). These results indicate that PI3Kγ inhibition can provide therapeutic benefits in acute infections even without directly affecting pathogen load. We also examined the effects of PI3Kγ inhibition during sterile inflammation models utilizing both Pik3cg−/− mice and the PI3Kγ inhibitor IPI-549. In a model of sterile inflammation, peritoneal administration of thioglycolate, a bacterial nutrient, stimulated substantial macrophage recruitment to the peritoneum that was suppressed by both PI3Kγ deletion and IPI-549-treatment (Fig. 6B).
Figure 6: PI3Kγ inhibition suppresses inflammation and promotes survival in mouse models of acute respiratory distress syndrome and sepsis.
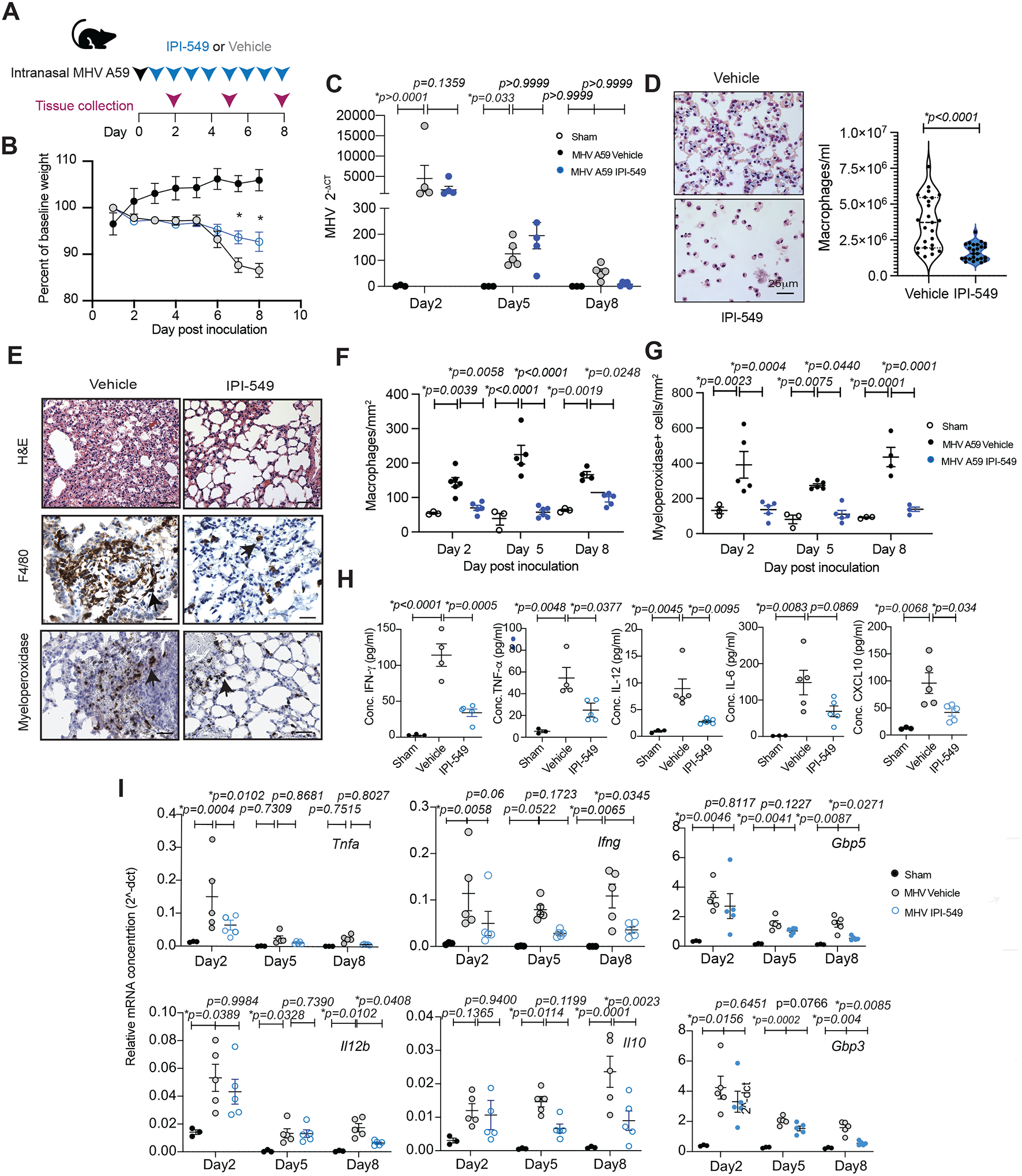
(A) Kaplan-Meier survival plots of WT (n=8) and Pik3cg−/− mice (n=8) inoculated with methicillin-resistant Staphylococcus aureus (MRSA). B) Left, graph of mean ± SEM macrophages in thioglycolate-stimulated peritoneal exudates of WT and Pik3cg−/− (n=4) and in vehicle vs IPI-549 treated mice (n=4). Right, flow cytometry profiles of F4/80+ macrophages in peritoneal exudates from WT and Pik3cg−/− animals with and without thioglycolate treatment. (C) Schematic of animal models of acute respiratory distress syndrome and outcome measures. (D) Kaplan-Meier survival plots of female and male WT (n=10) and Pik3cg−/− (n=10) mice treated once with 10 mg/kg (acute) LPS. (E) Representative images of Giemsa-stained cells in BAL from WT or Pik3cg−/− animals stimulated with 12 mg/kg (acute) LPS or with 3 mg/kg (chronic) LPS or poly I:C and treated with vehicle or IPI-549. Arrowheads indicate macrophages. (F) Graph of macrophages/ml (mean ± SEM) in BAL from WT (n=5) versus Pik3cg−/− (n=5) and vehicle (n=5) versus IPI-549-treated (n=5) animals after treatment with acute LPS, chronic LPS or chronic poly I:C (G) H&E and anti-F4/80-stained images of lungs isolated from WT versus Pik3cg−/− and vehicle versus IPI-549-treated animals 96h after stimulation with chronic LPS. Arrowheads indicate macrophages. (H) Violin plots of macrophages/mm2 (mean and interquartile range indicated by dotted lines) in lungs from WT (n=5) versus Pik3cg−/− (n=5) and vehicle (n=5) versus IPI-549-treated (n=5) animals. (I) Schematic depicting vascular leak assay. (J) Graph of Evans blue concentration (mean μg/ml ± SEM) in lungs of naïve mice (n=7) or mice treated with chronic LPS (n=5), Poly I:C (n=5), or acute LPS (n=9–10) and vehicle or IPI-549-treatment. (K) Graph of mean ± SEM pg/ml TNF-α in serum from naïve (n=5) and chronic LPS-stimulated, vehicle- (n=5) or IPI-549-treated (n=5) animals. (L) Graph of cytokine mRNA expression (mean 2−ΔCT ± SEM) in CD11b+ cells from lungs of chronic LPS-stimulated animals treated with vehicle or IPI-549 (n=3). Statistical significance was determined by Log-rank Mantel-Cox test (A, D), one-way ANOVA with Tukey’s multiple comparisons test (J, K) or t test (B, F, H, L).
To determine whether PI3Kγ controls inflammation in bacterial and viral infection, we investigated mouse models of acute respiratory distress syndrome (ARDS), in which acute or chronic Poly I:C or lipopolysaccharide (LPS) administration induces sterile lung inflammation, vascular leak and local and systemic cytokine elevation (35) (Fig. 6C). Significantly (p=0.0214 female; p=0.0237 male) increased survival from lethal LPS administration was observed in Pik3cg−/− animals (Fig. 6D). Analysis of BAL of treated mice demonstrated that each inflammatory stimulus induced significant (p<0.0001) macrophage recruitment to airways that was suppressed at all time points in both Pik3cg−/− mice and IPI-549-treated mice (Fig. 6E and F; fig. S11A and B). H&E analysis also revealed that PI3Kγ inhibition reduced inflammatory changes in the lung (fig. S11B). In contrast to clear airway spaces in PI3Kγ−/− and IPI-549-treated animals, WT and vehicle-treated animal lungs exhibit thickened septa, congested vessels, and hemorrhage (Fig. 6G; fig. S11B). PI3Kγ inhibition decreased F4/80+ macrophage recruitment to the lungs in all inflammation models, as substantially fewer macrophages were observed in Pik3cg−/− and IPI-549 treated animals than in WT and vehicle treated animals (Fig. 6G and H; fig. S11B).
Vascular leak is a major complicating factor in sepsis, ARDS and viral pneumonia that is induced by histamine and vascular endothelial growth factor secreted by myeloid cells, as well as other cells in injured tissues. We evaluated the effect of PI3Kγ inhibition on vascular leak in the ARDS models using the Miles assays, in which the quantification of intravascular Evans blue dye leakage into tissues serves as a measure of vascular leak (Fig. 6I and J). Both PI3Kγ deletion and IPI-549 treatment suppressed vascular leak in the lungs and other organs in chronic and acute ARDS models, although with some variability between models and tissues (Fig. 6J, fig. S12A to E). Importantly, PI3Kγ inhibition directly suppresses vascular leak induced by VEGF-A (fig. S12F), suggesting added therapeutic benefit to PI3Kγ inhibition in sepsis and infectious diseases characterized by vascular leak.
To determine the role of PI3Kγ on cytokine storm associated with severe inflammation, we examined the effect of inhibition PI3Kγ on inflammatory cytokine expression in serum and lung myeloid cells. Systemic, acute administration of LPS promoted a transient rise in serum cytokines and chemokines, including TNF-α, IL-1β, and IL-6, that peaked at 24 hours and returned to normal within 48 hours (fig.S13A). PI3Kγ inhibition transiently elevated the expression of some inflammatory cytokines upon acute LPS administration (fig. S13B). In contrast, PI3Kγ inhibition suppressed chronic LPS-induced cytokine release, as shown for TNF-α in serum, bronchioalveolar lavage and peritoneal lavage (Fig. 6K; fig. S13C and D). Chronic LPS stimulated RNA expression of inflammatory cytokines in lung CD11b+ cells but not lung CD11b− cells; PI3Kγ inhibition substantially reduced expression of inflammatory cytokines in lung myeloid cells (Fig. 6L; fig. S13E). Together, these data indicate that PI3Kγ inhibition can reduce inflammation and associated inflammatory sequalae such as vascular leak, cytokine storm and lethality in infectious disease models.
PI3Kγ inhibition promotes recovery in murine hepatitis virus infection
To evaluate the impact of dampening myeloid cell infiltration in viral pneumonia, mice that were intranasally inoculated with the betacoronavirus, murine hepatitis virus (MHV-A59) (41), were treated with the PI3Kγ inhibitor IPI-549 or vehicle and compared to sham-infected animals (Fig. 7A). MHV administered intranasally induces non-lethal pneumonia that lasts approximately 10 days (36). Treatment with IPI-549, but not vehicle, partially attenuated virus-induced weight loss without affecting viral load from 5 dpi (Fig. 7B and C). Treatment with IPI-549 substantially inhibited airway inflammation; fewer macrophages were present in BAL collected from IPI-549-treated versus vehicle-treated animals 5 dpi (Fig. 7D). PI3Kγ blockade also reduced lung septal wall thickening, vascular congestion, and hemorrhage (Fig. 7E; fig. S14), while substantially reducing macrophage and granulocyte infiltration of lungs at all time points after viral infection (Fig. 7E to G; fig. S14).
Figure 7: The PI3Kγ inhibitor IPI-549 reduces inflammation and promotes recovery in a mouse model of MHV infection.
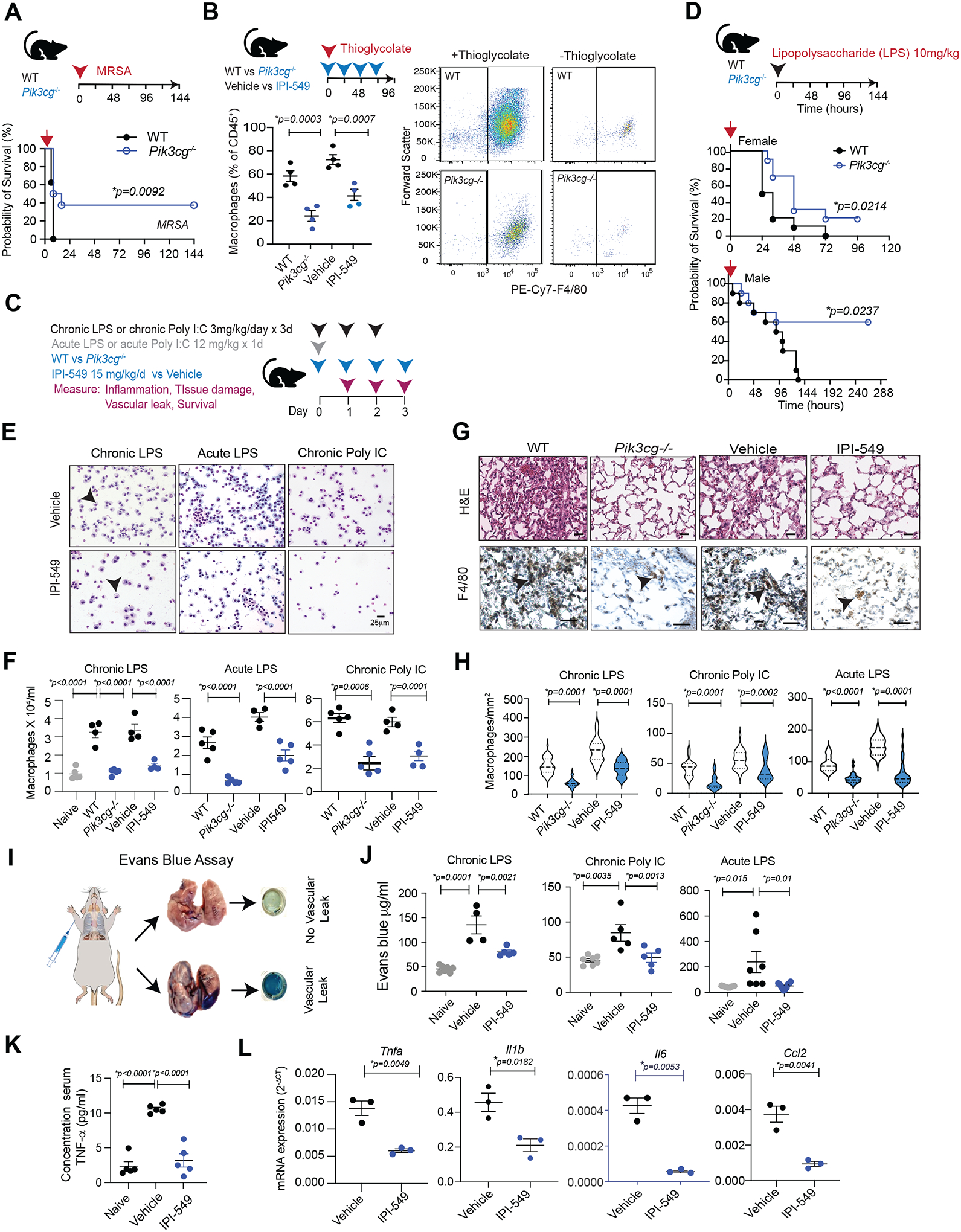
(A) Schematic of MHV A59 pulmonary infection, treatment, and tissue collection. (B) Time course graph of mean ± SEM weight changes in sham-infected (n=5) or MHV-infected mice treated with vehicle (n=5) or IPI-549 (n=5). (C) Graph of mean ± SEM MHV nucleocapsid N gene transcripts (2−ΔCT) determined by quantitative RT-PCR normalized to L32 ribosomal protein housekeeping gene in lungs from sham-infected (n=3) and MHV-infected mice treated with vehicle (n=5 per time point) or IPI-549 (n=5 per time point). (D) Images of Giemsa-stained cells and graphs of macrophages/ml quantification in BAL from MHV-infected animals at 5 dpi after treatment with vehicle or IPI-549. (E) Images of H&E, anti-F4/80-, and anti-MPO-stained lung tissues from (B). Arrowheads indicate macrophages and granulocytes. (F and G) Graphs of mean ± SEM F4/80+ macrophages/mm2 (F) and MPO+ granulocytes/mm2 (G) from (E). (H) Graph of cytokine concentration ± SEM in serum of mice treated from A. (I) Graph of mRNA expression (2−ΔCT) of inflammatory factors relative to Gapdh in lung lysates from A. Statistical significance was determined by t-test (B, D) or by one-way ANOVA with Tukey’s multiple comparisons test (C, F, G, H, I).
Our previous results indicated that PI3Kγ inhibition may reduce virus-induced cytokine storm; therefore, we quantified cytokine concentrations in serum and mRNA expression of cytokines in lung tissue from sham-infected and MHV-infected mice treated with vehicle or IPI-549. IPI-549 treatment reduced TNF-α, IFN-γ, IL-12, IL-6, and CXCL10 in serum (Fig. 7H). Viral infection increased expression of genes encoding pro-inflammatory cytokines, inflammatory signaling factors, and T cell biomarkers in the lung (Fig. 8I). In contrast, PI3Kγ inhibition reduced expression of these factors, particularly at later time points (Fig. 7I). As elevated serum cytokines are associated with severe or lethal coronavirus infection (1,2), these results suggest that PI3Kγ inhibition can suppress myeloid cell accumulation and expression of inflammatory cytokines that contribute to cytokine storm.
Discussion
Infectious diseases like SARS-CoV-2 continue to kill millions of individuals worldwide (37). Despite increased attention to disease prevention and management, new therapeutic approaches are still needed to manage this and other aggressive pulmonary infections, which are characterized by aberrant pulmonary and systemic inflammation leading to acute respiratory distress syndrome, vascular leak, coagulation and fatal organ damage (1,2). Here, we showed using animal models of disease that PI3Kγ inhibition with the clinical inhibitor IPI-549 (eganelisib) can protect against acute respiratory distress syndrome, vascular damage and, in some cases, lethality, induced by severe infectious agents such as MRSA and SARS-CoV-2.
SARS-CoV-2 and other viral and bacterial infections in mouse, hamster, and human are characterized by extensive recruitment of PI3Kγ-expressing wound-healing type, pro-fibrotic monocyte/macrophages and granulocytes to the lungs, concomitant with loss of surfactant C-expressing alveolar cells. Several studies have revealed neutrophilia in blood and extensive neutrophil infiltration of airways in COVID-19 patients (35). Neutrophils can damage tissues in part through NETosis, the release of intracellular proteinases and DNA that can create a physical trap for bacteria but can also induce thrombosis and damage organs (38). Because they self-destruct in tissues, neutrophils can be difficult to study. Our use of spatial transcriptomics and fixed tissue RNA sequencing provided an opportunity to characterize neutrophil and macrophages roles in lungs from individuals with COVID-19. Recent single cell sequencing studies demonstrated increased macrophages and fibroblasts in lungs from COVID-19 patients, consistent with our findings (3).
Our studies uniquely compare and contrast inflammation and transcriptomics in the response to COVID-19 in both BAL specimens and lung tissues from COVID-19 patients. These studies show that BAL reveals a subset of the inflammation and damage done to lungs during diseases like COVID-19 and demonstrates the recruitment of immature monocytes and neutrophils to the site of infection. Analysis of lung tissue, however, reveals that the alveolar spaces of the infected lung are replaced by extensive macrophage recruitment, interstitial inflammation and remodeling of tissue extracellular matrix, features that cannot be revealed in BAL specimens. Extensive areas of the infected lung are compromised by fibrosis and lung collapse and thus would not be accessible to lavage. Ongoing viral infections were associated with greater abundance of immature myeloid cells (myeloid other) and CD163+CD68+ macrophages as well as increased expression of PD-L1 on dendritic cells and granulocytes. Virus negative patients exhibited much greater fibroblast content.
We further showed that PI3Kγ is expressed in myeloid cells from SARS-CoV-2 infected human and animal tissues, suggesting a potential therapeutic target for reduction of inflammation, vascular leak, cytokine storm, and mortality from infectious lung diseases. We then demonstrated that inhibition of myeloid cell recruitment to infected lungs with the clinical PI3Kγ inhibitor IPI-549 promoted survival and recovery in animal models of sterile inflammation and infectious disease. IPI-549, also called eganelisib, is currently in cancer immune therapy Phase 2 clinical trials. Antagonism of PI3Kγreduced myeloid cell recruitment to lungs in several models of inflammation and infection. RNA expression analysis showed that lungs of COVID-19 patients and SARS-CoV-2 infected animals exhibited inflammation as well as a Th2-like, immune suppressed, wound-healing profile, which was suppressed prevented in animals by PI3Kγ inhibition. Our studies thus indicated that PI3Kγ inhibition can reduce inflammatory disease by suppressing organ-damaging sequelae, such as fibrosis, vascular leak, and cytokine storm.
Our study has some limitations. Whereas PI3Kγ inhibition reduces myeloid cell recruitment, weight loss and lung damage associated with SARS-CoV-2 and other infections, our studies did not show a curative benefit in animal models of SARS-CoV-2 infection. PI3Kγ inhibition did extend the survival of aging animals infected with SARS-CoV-2 in two separate cohorts of one study, although these studies need to be repeated and extended. Additionally, our analyses of biological materials from SARS-CoV-2 infected patients and animals were largely limited to fixed tissues for biosafety reasons. Further analyses of the mechanisms by which myeloid cells and PI3Kγ promote SARS-CoV-2-mediated inflammation and disease progression are needed to learn how to modulate viral diseases such as SARS-CoV-2 and the inflammation that is associated with them.
Taken together, these results indicate that PI3Kγ inhibition can reduce recruitment of inflammatory neutrophils, monocytes, and macrophages to the lung, which in turn lessens damage to the lung and distal organs by preventing cytokine surge and vascular leak. These results indicate that IPI-549 might provide benefit to patients with infectious lung diseases including and beyond COVID-19.
MATERIALS AND METHODS
Study Design
In this study, immune cell composition and state were investigated in lungs and BAL specimens from normal patients and patients with COVID-19, using IHC, mIHC RNA sequencing, and spatial transcriptomics. The effect of PI3Kγ inhibition on disease progression was evaluated in Pik3cg−/− mice and mice treated with the PI3Kγ antagonist, IPI-549. Animal models included infection with SARS-CoV-2, mouse hepatitis virus (MHV), methicillin-resistant Staphylococcus aureus (MRSA), and antibiotic-resistant E.coli as well as acute respiratory distress syndrome (ARDS) induced by lipopolysaccharide (LPS) and poly-inosine:cytosine (PolyI:C) administration. Survival, weight loss, inflammation and vascular leak were measured as an index of disease progression.
Human tissue analyses were conducted on de-identified tissue under guidelines established by the Institutional Review Board (IRB) for human subject research of the University of California, San Diego (UCSD). De-identified lung tissue was obtained upon rapid autopsy of deceased COVID-19 patients or from patients with consent during lung cancer surgery at the Moores Cancer Center, UCSD. BAL cells were obtained from patients infected with SARS-CoV-2 and from hospitalized patients without infections with consent.
MHV and ARDS animal studies were performed with approval of the Institutional Animal Care and Use Committee (IACUC) and Institutional Biosafety Committee (IBC) of UCSD. MHV studies were conducted in Animal Biosafety Level 2 (ABSL2) facilities. SARS-CoV-2 infections in K18-hACE2 mice were performed in ABSL3 facilities at The Scripps Research Institute (TSRI) with approval of TSRI IACUC and IBC. MaSARS-CoV-2 models were conducted in ABSL3 facilities at The Institute for Antiviral Research, Animal, Dairy and Veterinary Science, Utah State University, Logan, UT, with the approval of the IACUC and IBC of Utah State University. SARS-CoV-2 hamster studies were performed in ABSL3 facilities at the Department of Infectious Diseases and Global Health, Tufts University Cummings School of Veterinary Medicine with the approval of the IACUC and IBC of Tufts University.
A sample size of at least 10 mice per group provided 80% power to detect mean difference of 2.25 standard deviations (SD) between two groups (based on a two-sample t-test with two-sided 5% significance level). Mice were randomized prior to treatment with inhibitors or controls. Prior to statistical analyses, data were examined for normal distribution and outliers. No data points were omitted. Animal studies were ended when mouse weight loss was greater than 20% of starting body weight or at pre-designated endpoints. All animal and histological studies were performed with investigators blinded as to study group identity. Animal experiments were performed at least 2 times with n=5 to 15 per group, except the maSARS-CoV-2 study was performed once, with n=5 to 20 mice per group.
Statistical Analysis
Results were analyzed using one-way ANOVA with Tukey’s or Dunnett’s post-hoc test for multiple group analyses, Student’s t-test for parametric two sample analysis, Mann Whitney test for non-parametric two sample analysis, and Log-rank (Mantel-Cox) test for survival analysis using Graph Pad Prism version 9.1.0. Data with p≤0.05 were considered statistically significant. Statistical significance in differential gene expression analysis was performed by limma-voom method.
Supplementary Material
Acknowledgements:
The authors thank Thames Pickett and Ann Eakin, NIAID, for access to SARS-CoV-2 animal models through the NIH-ACTIV program, Venkatraman Siddharthan (USU), Shannon O’ Connor (Tufts) and Giovanney Gonzalez (OHSU), for technical assistance, and David Smith (UCSD) for valuable advice.
Funding:
This study was supported by National Institutes of Health grants(R01CA226909, R01CA167426 and R01DE027325 to J.A.V., R01CA226909 subaward to LCM, R01AI176554 to V.N.), the California Office of the President Emergency COVID-19 Research Seed Funding (R00RG2398 to J.A.V.), the Cowlin Family Foundation (to S.P.P.), Fast Grants, Emergent Ventures, Mercatus Center at George Mason University (to A.J.M.), and Cancer Research Institute/Irvington postdoctoral fellowships (J.Z. and A.G.) This work was also supported by NIH UL1TR001442 and NIH S10 OD026929 to the UCSD Center for Computational Biology & Bioinformatics Shared Resource and Genomics Center), by NIH P30CA23100 to UCSD Moores Comprehensive Cancer Center, and Department of Health and Human Services Contract HHSN272201700041I/75N93021F00002 to J.M.
Competing interests:
R.A., B.O.’C., and T.A.M. were employees of and J.A.V. and A.J.M. were consultants for Infinity Pharmaceuticals. D.A.C. and J.A.V. are shareholders in Impact Biosciences and AlphaBeta Therapeutics. L.C.A. is a consultant for Regeneron. V.N. is a consultant for Cellics Therapeutics, I2 Pure, Inc., Clarametyx BioTherapeutics, Iogen, Inc., Staurus Therapeutics and Aequor, Inc. and received research funding from Micronbrane, Inc. and Vaxcyte, Inc. A.J.M. was a consultant for HistoWiz, Inc. C.B. is currently an employee of, and holds equity in, Akoya Biosciences, Inc. J. D. is a current employee of Ionis Pharmaceuticals in Carlsbad, CA, and was a consultant for Lexeo Therapeutics. The other authors declare no competing interests.
Footnotes
Data, code and materials availability:
All data associated with this study are in the paper or the Supplementary Materials. Data and code are available at DOI: 10.5061/dryad.sf7m0cgbm. All raw and processed RNA sequence data are publicly available at Gene Expression Omnibus (GEO) under accession numbers GSE190494, GSE190496, GSE229570, and GSE255211. mIHC protocols are available at dx.doi.org/10.17504/protocols.io.n92ldmmznl5b/v2 with custom color deconvolution macro available at doi: 10.5281/zenodo.11287476. IPI-549 was provided to the University of California, San Diego, The Scripps Research Institute, and Tufts University under Material Transfer Agreements and is commercially available. Resources are available upon reasonable request to J.A.V.
References
- 1.Liu J, Zheng X, Tong Q, Li W, Wang B, Sutter K, Trilling M, Lu M, Dittmer U, Yang D. Overlapping and discrete aspects of the pathology and pathogenesis of the emerging human pathogenic coronaviruses SARS-CoV, MERS-CoV, and 2019-nCoV. J Med Virol 92, 491–494 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giamarellos-Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos N, Damoraki G, Gkavogianni T, Adami ME, Katsaounou P, Ntaganou M, Kyriakopoulou M, Dimopoulos G, Koutsodimitropoulos I, Velissaris D, Koufargyris P, Karageorgos A, Katrini K, Lekakis V, Lupse M, Kotsaki A, Renieris G, Theodoulou D, Panou V, Koukaki E, Koulouris N, Gogos C, Koutsoukou A. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe 27, 992–1000.e3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Melms JC, Biermann J, Huang H, Wang Y, Nair A, Tagore S, Katsyv I, Rendeiro AF, Amin AD, Schapiro D, Frangieh CJ, Luoma AM, Filliol A, Fang Y, Ravichandran H, Clausi MG, Alba GA, Rogava M, Chen SW, Ho P, Montoro DT, Kornberg AE, Han AS, Bakhoum MF, Anandasabapathy N, Suárez-Fariñas M, Bakhoum SF, Bram Y, Borczuk A, Guo XV, Lefkowitch JH, Marboe C, Lagana SM, Del Portillo A, Tsai EJ, Zorn E, Markowitz GS, Schwabe RF, Schwartz RE, Elemento O, Saqi A, Hibshoosh H, Que J, Izar B. A molecular single-cell lung atlas of lethal COVID-19. Nature 595, 114–119 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen ST, Park MD, Del Valle DM, Buckup M, Tabachnikova A, Thompson RC, Simons NW, Mouskas K, Lee B, Geanon D, D’Souza D, Dawson T, Marvin R, Nie K, Zhao Z, LeBerichel J, Chang C, Jamal H, Akturk G, Chaddha U, Mathews K, Acquah S, Brown SA, Reiss M, Harkin T, Feldmann M, Powell CA, Hook JL, Kim-Schulze S, Rahman AH, Brown BD; Mount Sinai COVID-19 Biobank Team; N.D. Beckmann, S. Gnjatic, E. Kenigsberg, A.W. Charney, M. Merad. A shift in lung macrophage composition is associated with COVID-19 severity and recovery. Science Translational Medicine 14, eabn5168 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rendeiro AF, Ravichandran H, Bram Y, Chandar V, Kim J, Meydan C, Park J, Foox J, Hether T, Warren S, Kim Y, Reeves J, Salvatore S, Mason CE, Swanson EC, Borczuk AC, Elemento O, Schwartz RE. The spatial landscape of lung pathology during COVID-19 progression. Nature 593, 564–569 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Channappanavar R, Fehr AR, Zheng J, Wohlford-Lenane C, Abrahante JE, Mack M, Sompallae R, McCray PB Jr, Meyerholz DK, Perlman S. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J Clin Invest 129, 3625–3639 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoshida M, Worlock KB, Huang N, Lindeboom RGH, Butler CR, Kumasaka N, Conde CD, Mamanova L, Bolt L, Richardson L, Polanski K, Madissoon E, Barnes JL, Allen-Hyttinen J, Kilich E, Jones BC, de Wilton A, Wilbrey-Clark A, Sungnak W, Pett JP, Weller J, Prigmore E, Yung H, Mehta P, Saleh A, Saigal A, Chu V, Cohen JM, Cane C, Iordanidou A, Shibuya S, Reuschl AK, Herczeg IT, Argento AC, Wunderink RG, Smith SB, Poor TA, Gao CA, Dematte JE; NU SCRIPT Study Investigators, Reynolds G, Haniffa M, Bowyer GS, Coates M, Clatworthy MR, Calero-Nieto FJ, Göttgens B, O’Callaghan C, Sebire NJ, Jolly C, De Coppi P, Smith CM, Misharin AV, Janes SM, Teichmann SA, Nikolić MZ, Meyer KB. Local and systemic responses to SARS-CoV-2 infection in children and adults. Nature 602, 321–327 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmid MC, Avraamides CJ, Dippold HC, Franco I, Foubert P, Ellies LG, Acevedo LM, Manglicmot JR, Song X, Wrasidlo W, Blair SL, Ginsberg MH, Cheresh DA, Hirsch E, Field SJ, Varner JA. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kgamma, a single convergent point promoting tumor inflammation and progression. Cancer Cell 19, 715–727 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, Woo G, Nguyen AV, Figueiredo CC, Foubert P, Schmid MC, Pink M, Winkler DG, Rausch M, Palombella VJ, Kutok J, McGovern K, Frazer KA, Wu X, Karin M, Sasik R, Cohen EE, Varner JA. PI3Kgamma is a molecular switch that controls immune suppression. Nature 539, 437–442 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaneda MM, Cappello P, Nguyen AV, Ralainirina N, Hardamon CR, Foubert P, Schmid MC, Sun P, Mose E, Bouvet M, Lowy AM, Valasek MA, Sasik R, Novelli F, Hirsch E, Varner JA. Macrophage PI3Kgamma Drives Pancreatic Ductal Adenocarcinoma Progression. Cancer Discov 6, 870–885 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin EL, Souza DG, Fagundes CT, Amaral FA, Assenzio B, Puntorieri V, Del Sorbo L, Fanelli V, Bosco M, Delsedime L, Pinho JF, Lemos VS, Souto FO, Alves-Filho JC, Cunha FQ, Slutsky AS, Ruckle T, Hirsch E, Teixeira MM, Ranieri VM. Phosphoinositide-3 kinase gamma activity contributes to sepsis and organ damage by altering neutrophil recruitment. Am J Respir Crit Care Med 182, 762–773 (2010). [DOI] [PubMed] [Google Scholar]
- 12.De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, Budhu S, Ghosh A, Pink M, Tchaicha J, Douglas M, Tibbitts T, Sharma S, Proctor J, Kosmider N, White K, Stern H, Soglia J, Adams J, Palombella VJ, McGovern K, Kutok JL, Wolchok JD, Merghoub T. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature 539, 443–447 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chmielowski B, Sullivan R, Postow M, Patnaik A, Shapiro G, Cohen EEW, Gutierrez M, Steuer C, Ribas A, Lee L, O’Connell B, Kutok J, Roberts J, Mahabhashyam S, Fjallskog M-L, Wolchok JD, Hong D. The First Clinical/Translational Data from the Expansion Cohorts of a Ph1/1b Study of IPI-549, a Tumor Macrophage-Reprogramming Small Molecule, in Combination with Nivolumab in Advanced Solid Tumors. JITC 6, A264–265 (2018). [Google Scholar]
- 14.Hong DS, Postow M, Chmielowski B, Sullivan R, Patnaik A, Cohen EEW, Shapiro G, Steuer C, Gutierrez M, Yeckes-Rodin H, Ilaria R, O’Connell B, Peng J, Peng G, Zizlsperger N, Tolcher A, Wolchok JD. Eganelisib, a First-in-Class PI3K-γ Inhibitor, in Patients with Advanced Solid Tumors: Results of the Phase 1/1b MARIO-1 Trial. Clin Cancer Res. 22, 3313 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsujikawa T, Kumar S, Borkar RN, Azimi V, Thibault G, Chang YH, Balter A, Kawashima R, Choe G, Sauer D, El Rassi E, Clayburgh DR, Kulesz-Martin MF, Lutz ER, Zheng L, Jaffee EM, Leyshock P, Margolin AA, Mori M, Gray JW, Flint PW, Coussens LM. Quantitative Multiplex Immunohistochemistry Reveals Myeloid-Inflamed Tumor-Immune Complexity Associated with Poor Prognosis. Cell Rep. 19, 203–217 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ni W, Yang X, Yang D, Bao J, Li R, Xiao Y, Hou C, Wang H, Liu J, Yang D, Xu Y, Cao Z, Gao Z. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit Care 24, 422 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barros MH, Hauck F, Dreyer JH, Kempkes B, Niedobitek G. Macrophage polarisation: an immunohistochemical approach for identifying M1 and M2 macrophages. PLoS One 8, e80908 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Skytthe MK, Graversen JH, Moestrup SK. Targeting of CD163(+) Macrophages in Inflammatory and Malignant Diseases. Int J Mol Sci 21, 5497 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gil-Etayo FJ, Suàrez-Fernández P, Cabrera-Marante O, Arroyo D, Garcinuño S, Naranjo L, Pleguezuelo DE, Allende LM, Mancebo E, Lalueza A, Díaz-Simón R, Paz-Artal E, Serrano A. T-Helper Cell Subset Response Is a Determining Factor in COVID-19 Progression. Front Cell Infect Microbiol 11, 624483 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, Chen L, Li M, Liu Y, Wang G, Yuan Z, Feng Z, Zhang Y, Wu Y, Chen Y. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front Immunol 11, 827 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X, Wang G, Wang Z, Liu B, Han N, Li J, Lu C, Liu X, Zhang Q, Yang Q, Wang G. PD-1-expressing B cells suppress CD4(+) and CD8(+) T cells via PD-1/PD-L1-dependent pathway. Mol Immunol 109, 20–26 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Trejo CL, Babić M, Imler E, Gonzalez M, Bibikov SI, Shepard PJ, VanSteenhouse HC, Yeakley JM, Seligmann BE. Extraction-free whole transcriptome gene expression analysis of FFPE sections and histology-directed subareas of tissue. PLoS One 14, e0212031 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turnbull AK, Selli C, Martinez-Perez C, Fernando A, Renshaw L, Keys J, Figueroa JD, He X, Tanioka M, Munro AF, Murphy L, Fawkes A, Clark R, Coutts A, Perou CM, Carey LA, Dixon JM, Sims AH. Unlocking the transcriptomic potential of formalin-fixed paraffin embedded clinical tissues: comparison of gene expression profiling approaches. BMC Bioinformatics 21, 30 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Danaher P, Kim Y, Nelson B, Griswold M, Yang Z, Piazza E, and Beechem JM. Advances in mixed cell deconvolution enable quantification of cell types in spatial transcriptomic data. Nature Commun. 13, 385 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shaykhiev R. Emerging biology of persistent mucous cell hyperplasia in COPD. Thorax 74, 4–6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Szabo PA, Dogra P, Gray JI, Wells SB, Connors TJ, Weisberg SP, Krupska I, Matsumoto R, Poon MML, Idzikowski E, Morris SE, Pasin C, Yates AJ, Ku A, Chait M, Davis-Porada J, Guo XV, Zhou J, Steinle M, Mackay S, Saqi A, Baldwin MR, Sims PA, Farber DL. Longitudinal profiling of respiratory and systemic immune responses reveals myeloid cell-driven lung inflammation in severe COVID-19. Immunity 54, 797–814 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stampfer SD, Goldwater MS, Bujarski S, Regidor B, Zhang W, Feinstein AJ, Swift R, Eshaghian S, Vail E, Berenson JR. Severe breakthrough COVID-19 with a heavily mutated variant in a multiple myeloma patient 10 weeks after vaccination. Clin Infect Pract 13, 100130 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palanki MS, Dneprovskaia E, Doukas J, Fine RM, Hood J, Kang X, Lohse D, Martin M, Noronha G, Soll RM, Wrasidlo W, Yee S, Zhu H. Discovery of 3,3’-(2,4-diaminopteridine-6,7-diyl)diphenol as an isozyme-selective inhibitor of PI3K for the treatment of ischemia reperfusion injury associated with myocardial infarction. J Med Chem 50, 4279–4294 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Serban D, Leng J, Cheresh D. H-ras regulates angiogenesis and vascular permeability by activation of distinct downstream effectors. Circ Res 102, 1350–1358 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tostanoski LH, Wegmann F, Martinot AJ, Loos C, McMahan K, Mercado NB, Yu J, Chan CN, Bondoc S, Starke CE, Nekorchuk M, Busman-Sahay K, Piedra-Mora C, Wrijil LM, Ducat S, Custers J, Atyeo C, Fischinger S, Burke JS, Feldman J, Hauser BM, Caradonna TM, Bondzie EA, Dagotto G, Gebre MS, Jacob-Dolan C, Lin Z, Mahrokhian SH, Nampanya F, Nityanandam R, Pessaint L, Porto M, Ali V, Benetiene D, Tevi K, Andersen H, Lewis MG, Schmidt AG, Lauffenburger DA, Alter G, Estes JD, Schuitemaker H, Zahn R, Barouch DH. Ad26 vaccine protects against SARS-CoV-2 severe clinical disease in hamsters. Nat Med 26, 1694–1700 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCray PB, Pewe L, Wohlford-Lenane C, Hickey M, Manzel L, Shi L, Netland J, Jia HP, Halabi C, Sigmund CD, Meyerholz DK, Kirby P, Look DC, Perlman S. Lethal infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus. J Virol. 81, 813–821 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Franzén O, Gan L-M, Björkegren JLM. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database 2019, baz046 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leist SR, Dinnon KH 3rd, Schäfer A, Tse LV, Okuda K, Hou YJ, West A, Edwards CE, Sanders W, Fritch EJ, Gully KL, Scobey T, Brown AJ, Sheahan TP, Moorman NJ, Boucher RC, Gralinski LE, Montgomery SA, Baric RS. A Mouse-Adapted SARS-CoV-2 Induces Acute Lung Injury and Mortality in Standard Laboratory Mice. Cell 183, 1070–1085 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bastarache JA, Blackwell TS. Development of animal models for the acute respiratory distress syndrome. Dis Model Mech 2, 218–223 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Z, Du J, Chen G, Zhao J, Yang X, Su L, Cheng G, Tang H. Coronavirus MHV-A59 infects the lung and causes severe pneumonia in C57BL/6 mice. Virol Sin 29, 393–402 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hendrickson KW, Peltan ID, Brown SM. The Epidemiology of Acute Respiratory Distress Syndrome Before and After Coronavirus Disease 2019. Crit Care Clin 37, 703–716 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Middleton EA, He XY, Denorme F, Campbell RA, Ng D, Salvatore SP, Mostyka M, Baxter-Stoltzfus A, Borczuk AC, Loda M, Cody MJ, Manne BK, Portier I, Harris ES, Petrey AC, Beswick EJ, Caulin AF, Iovino A, Abegglen LM, Weyrich AS, Rondina MT, Egeblad M, Schiffman JD, Yost CC. Neutrophil extracellular traps (NETs) contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 36, 1169–1179 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jureka AS, Basler CF. Propagation and Quantification of SARS-CoV-2. Methods Mol Biol. 2452,111–129 (2022). [DOI] [PubMed] [Google Scholar]
- 40.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7, R100 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 15, R29 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11, R25 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society: Series B (Methodological) 57, 289–300 (1995). [Google Scholar]
- 46.Tarca AL, Draghici S, Khatri P, Hassan SS, Mittal P, Kim JS, Kim CJ, Kusanovic JP, Romero R. A novel signaling pathway impact analysis. Bioinformatics 25, 75–82 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, Vilo J. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Research 47, W191–W198 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, Sergushichev A. Fast gene set enrichment analysis. bioRxiv doi: 10.1101/060012 (2021). [DOI] [Google Scholar]
- 49.Aid M, Busman-Sahay K, Vidal SJ, Maliga Z, Bondoc S, Starke C, Terry M, Jacobson CA, Wrijil L, Ducat S, Brook OR, Miller AD, Porto M, Pellegrini KL, Pino M, Hoang TN, Chandrashekar A, Patel S, Stephenson K, Bosinger SE, Andersen H, Lewis MG, Hecht JL, Sorger PK, Martinot AJ, Estes JD, Barouch DH. Vascular Disease and Thrombosis in SARS-CoV-2- Infected Rhesus Macaques. Cell 183, 1354–1366 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aid M, Vidal SJ, Piedra-Mora C, Ducat S, Chan CN, Bondoc S, Colarusso A, Starke CE, Nekorchuk M, Busman-Sahay K, Estes JD, Martinot AJ, Barouch DH. Ad26.COV2.S prevents upregulation of SARS-CoV-2 induced pathways of inflammation and thrombosis in hamsters and rhesus macaques. Plos Pathog 18, e1009990 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wong LR, Li K, Sun J, Zhuang Z, Zhao J, McCray PB Jr, Perlman S. Sensitization of non-permissive laboratory mice to SARS-CoV-2 with a replication-deficient adenovirus expressing human ACE2. STAR Protoc. 1, 100169 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bewley KR, Coombes NS, Gagnon L, McInroy L, Baker N, Shaik I, St-Jean JR, St-Amant N, Buttigieg KR, Humphries HE, Godwin KJ, Brunt E, Allen L, Leung S, Brown PJ, Penn EJ, Thomas K, Kulnis G, Hallis B, Carroll M, Funnell S, Charlton S. Quantification of SARS-CoV-2 neutralizing antibody by wild-type plaque reduction neutralization, microneutralization and pseudotyped virus neutralization assays. Nat Protoc. 16, 3114–3140 (2021). [DOI] [PubMed] [Google Scholar]
- 53.Dinnon KH 3rd, Leist SR, Schäfer A, Edwards CE, Martinez DR, Montgomery SA, West A, Yount BL Jr, Hou YJ, Adams LE, Gully KL, Brown AJ, Huang E, Bryant MD, Choong IC, Glenn JS, Gralinski LE, Sheahan TP, Baric RS. A mouse-adapted model of SARS-CoV-2 to test COVID-19 countermeasures. Nature 586, 560–566 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dagotto G, Mercado NB, Martinez DR, Hou YJ, Nkolola JP, Carnahan RH, Crowe JE Jr., Baric RS, Barouch DH. Comparison of Subgenomic and Total RNA in SARS-CoV-2 Challenged Rhesus Macaques. J Virol. 95, e02370–20 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ishige T, Murata S, Taniguchi T, Miyabe A, Kitamura K, Kawasaki K, Nishimura M, Igari H, Matsushita K. Highly sensitive detection of SARS-CoV-2 RNA by multiplex rRT-PCR for molecular diagnosis of COVID-19 by clinical laboratories. Clin Chim Acta 507, 139–142 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kumaki Y, Salazar AM, Wandersee MK, Barnard DL. Prophylactic and therapeutic intranasal administration with an immunomodulator, Hiltonol((R)) (Poly IC:LC), in a lethal SARS-CoV-infected BALB/c mouse model. Antiviral Res. 139, 1–12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are in the paper or the Supplementary Materials. Data and code are available at DOI: 10.5061/dryad.sf7m0cgbm. All raw and processed RNA sequence data are publicly available at Gene Expression Omnibus (GEO) under accession numbers GSE190494, GSE190496, GSE229570, and GSE255211. mIHC protocols are available at dx.doi.org/10.17504/protocols.io.n92ldmmznl5b/v2 with custom color deconvolution macro available at doi: 10.5281/zenodo.11287476. IPI-549 was provided to the University of California, San Diego, The Scripps Research Institute, and Tufts University under Material Transfer Agreements and is commercially available. Resources are available upon reasonable request to J.A.V.