

Abstract
Copper (Cu), an essential micronutrient, participates in several physiological processes, including cell proliferation and development. Notably, the disturbance of Cu homeostasis promotes tumor progression through the generation of oxidative stress. Chronic or excessive accumulation of reactive oxygen species (ROS) causes lipid peroxidation, protein denaturation, and enzyme inactivation, which leads to a breakdown of intracellular homeostasis and exacerbates tumor progression. The disruption of the ROS scavenging mechanism also reduces resistance to oxidative stress, leading to further deterioration in a disease state, and maintenance of redox homeostasis is thought to inhibit the onset and progression of various diseases. Superoxide dismutase 3 (SOD3), a Cu-containing secretory antioxidative enzyme, plays a key role in extracellular redox regulation, and the significant reduction in SOD3 facilitates tumor progression. Furthermore, the significant induction of SOD3 participates in tumor metastasis. This review focuses on the role of Cu homeostasis and antioxidative enzymes, including SOD3, in tumor progression, to help clarify the role of redox regulation.
Keywords: superoxide dismutase 3, copper, tumor, reactive oxygen species, glutathione
Introduction
Reactive oxygen species (ROS) have important physiological functions such as bactericidal action in the immune system and action as signaling molecules.(1–3) However, excessive ROS accumulation due to the failure of ROS scavenging mechanisms causes oxidative modification of DNA, lipids, and proteins, and plays a significant role in the development and progression of various diseases such as tumors and atherosclerosis.(4,5) To protect the cells and tissues from the damaging effects of oxidative stress, mammals have several antioxidative enzymes, including superoxide dismutase (SOD), and catalase and antioxidative compounds, including glutathione (GSH).(6,7) SOD catalyzes the dismutation of superoxide into hydrogen peroxide and oxygen. Mammalian cells contain three types of SOD isozymes, namely SOD1, SOD2, and SOD3, which cooperatively protect tissues from oxidative stress by differentiating their intracellular and extracellular localization.(8) SOD1 and SOD3 have copper (Cu) ions in their catalytic domains, but SOD2 has manganese ions. Furthermore, significant Cu accumulation has been observed in several tumor tissues, including breast, lung, colon, and neuroblastoma tissues, which occurs through the dysregulation of Cu transporters and their chaperone proteins.(9–11) Accordingly, Cu dysregulation in tumor tissues may be involved in the dysfunction of SOD enzymes and other Cu-containing enzymes, thus exacerbating tumor progression. SOD1 expression is highly expressed in most lung cancer cells and plays a key role in its progression through the regulation of redox mediated signaling, and its pharmacological inhibition induces non-small lung cancer cell death.(12–14) On the other hand, compared to SOD1, there have been few reports on the clinical significance and role of SOD3 in tumor progression. This review focuses first on the role of SOD3 in tumor progression and then discusses Cu dysregulation in tumor progression. Hopefully, this review will contribute to a better understanding the relationship between Cu- and SOD3-mediated extracellular redox regulation and tumor progression.
Role of SOD3 in Tumor Progression
SOD3 is the only extracellularly localized SOD isozyme and is distributed by binding to heparan sulfate expressed on the cell surface, using the C-terminal heparin-binding domain.(15,16) Although SOD3 levels are relatively low compared with the levels of SOD1 and SOD2, decreased SOD3 activity has been observed in mouse models of diabetes mellitus.(17,18) In addition, a decrease in infarct size after coronary ischemia has been observed in animals overexpressing SOD3, whereas an increase in infarct size has been reported in SOD3 knockout mice.(19,20) In tumor tissues, endogenous administration of recombinant SOD3 was shown to decrease the malignant potential of cancer cells.(21) Accordingly, SOD3 is considered to have antioxidative and antitumor properties. Moreover, it is important to strictly control SOD3 expression in cancer cells because SOD3 overexpression has two aspects: an overexpression of SOD3 acts in a cancer-suppressive manner, but a moderate overexpression acts in a cancer-promoting manner.(22) SOD3 expression was found to be higher in metastatic MDA-MB-231 cells than in non-metastatic MCF7 cells, suggesting that SOD3 is involved in breast cancer cell progression.(23) Moderate and sustained overexpression of SOD3 may promote tumor progression in advanced undifferentiated cancer cells. Furthermore, SOD3 activates small GTPase signaling, which is involved in cell proliferation; thus, SOD3 is considered to have both antitumor and tumor-promoting properties.(24) Overall, maintaining adequate SOD3 expression is important in suppressing oxidative stress-mediated diseases and tumors. However, the molecular mechanisms underlying SOD3 regulation in tumor tissues have not been fully elucidated.
Regulation of SOD3 Expression and Function
Epigenetic regulation of SOD3 expression
In contrast to SOD1 and SOD2, which are ubiquitously expressed, SOD3 expression is cell- and tissue-specific, and several epigenetic modifications were reported to play a critical role in the regulation of SOD3. Epigenetics usually refers to changes in gene expression that are inherited through mitosis, without changes in the DNA sequence, and are involved in tumor development and progression.(25) DNA methylation, a major epigenetic factor, occurs at the 5' position of cytosine within CpG, and hypermethylation within target gene promoters is involved in tissue- and cell-specific gene reduction.(26) DNA methylation within the SOD3 promoter region plays a critical role in its reduction in human lung cancer A549 cells and human monocytic THP-1 cells, which is accompanied by the marked reduction in DNA demethylase ten-eleven-translocation 1 (TET1).(27–30) Considering that TET1 requires oxygen for the DNA demethylation process, DNA demethylation is likely dysregulated in hypoxic tumor tissues, and SOD3 expression may be reduced. However, some DNA methylation inhibitors, such as 5-azacytidine, fail to induce SOD3 expression in MCF7 cells, suggesting that there is another mechanism governing SOD3 expression.
Histone modifications of the N-terminal tail, including acetylation and methylation at lysine and arginine residues, are also involved in gene regulation. Histone acetylase, p300, and myocyte enhancer factor 2 (MEF2) participate in SOD3 expression. In addition, histone deacetylase 1 (HDAC1) plays a key role in SOD3 reduction in tumor-associated cells. Some epigenetic reagents, including the inhibitors of HDAC and DNA methyltransferases, contribute to maintaining redox homeostasis.(31) It was also demonstrated that natural product-derived 4-hydroperoxy-2-decenoic acid ethyl ester and exenatide induce SOD3 expression through histone acetylation and DNA demethylation, respectively.(32) These compounds may control extracellular redox homeostasis, which ultimately inhibits tumor progression.
Transcriptional regulation of SOD3 expression
NF-E2-related factor 2 (Nrf2), a transcription factor, plays a critical role in the regulation of redox homeostasis through the induction of several antioxidative enzymes, including SOD2, heme oxygenase 1 (HO-1), and the cystine/glutamate antiporter SLC7A11.(33) Notably, the Nrf2-mediated induction of antioxidative properties is involved not only in the antioxidative defense against oxidative stress-induced injury but also in antitumor therapy. SLC7A11 regulates intracellular redox homeostasis by facilitating GSH synthesis and increasing the activity of glutathione peroxidase 4, which is a selenoenzyme that reduces membrane phospholipid hydroperoxides.(34) Recent studies revealed that Nrf2 activation and increased SLC7A11 are closely associated with tumor progression. Thus, reagents that inhibit SLC7A11 function may induce ferroptosis, a form of controlled cell death induced by ferrous ions and excessive lipid peroxidation. Some reports suggested that SOD3 expression is also regulated by Nrf2, but the molecular mechanisms underlying SOD3 expression remain unclear.(35) In the process of elucidating the molecular mechanisms for SOD3 regulation, forkhead box O1 (FOXO1), a transcription factor that binds to the promoter regions of several genes involved in glucose and lipid metabolism, was found to regulate SOD3 expression in MDA-MB-231 cells.(23) FOXO1 was considered an antitumor transcription factor; however, recent studies revealed that FOXO1 also functions as a pro-metastatic transcription factor.(36,37) Another report suggested that FOXO1 functions as a tumor promoter and partially facilitates MDA-MB-231 cell metastasis through the induction of SOD3. Furthermore, Jumonji domain-containing protein-3 (JMJD3), a histone demethylase that regulates trimethylation of histone H3 on lysine 27, plays an important role in SOD3 regulation in cooperation with FOXO1. Considering that the combined action of FOXO1 and MEF2 is required for myocardin regulation, FOXO1/MEF2/JMJD3 may function cooperatively to induce SOD3.(38) Further studies are necessary to clarify the exact molecular mechanisms governing SOD3 regulation in tumor cells.
Regulation of SOD3 secretion and function
N-glycosylation on asparagine residues functions as a key posttranscriptional modification that determines the secretion and activity of glycosylated proteins.(39) Notably, SOD3 has a conserved and putative N-glycosylation site (N-X-S/T). It was reported that glycosylated SOD3 was secreted into extracellular spaces, but its amino acid mutation, N89Q, suppresses its secretion. In addition, non-glycosylated or mutated SOD3 mainly resides in the endoplasmic reticulum, indicating that their secretion is not blocked by abnormal intracellular trafficking to a particular cellular compartment such as the Golgi apparatus.(40) Recent studies have revealed that fucose addition on SOD3 mediated by α1,6-fucosyltransferase (FUT8) plays an important role in its secretion and antitumor property in non-small cell lung cancer A549 cells (Fig. 1).(41) FUT8 is frequently upregulated in tumor cells and plays a critical role in immune evasion through the regulation of transforming growth factor-β, epidermal growth factor, and E-cadherin, which are involved in tumor progression.(32–44) Accordingly, FUT8 may exhibit antitumor and pro-tumor properties. However, as noted above, SOD3 expression is epigenetically silenced in A549 cells; thus, some epigenetic inhibitors may exhibit antitumor effects, in part, through the induction and functional secretion of SOD3.
Fig. 1.

Involvement of N-glycosylation in SOD3 secretion. SOD3 receives Cu from ATP7A in the trans-Golgi network. N-glycosylation, especially FUT8-mediated fucosylation, plays a critical role in SOD3 secretion to extracellular spaces. In addition, non-fucosylated SOD3 does not show antioxidative properties. Cleaved- and R213G mutated-SOD3 are secreted in extracellular spaces, but they cannot bind to heparan sulfate because they do not have the conserved heparin-binding domain. Accordingly, their serum concentrations drastically increase compared with uncleaved or wildtype SOD3.
It has been well recognized that a genetic mutation in the C-terminal heparin-binding domain (R213G) reduces the heparin-binding affinity and increases its serum level, leading to an increased risk of cardiovascular diseases.(45,46) Furin-like protease and unknown carboxypeptidase are involved in the cleavage of SOD3, leading to the reduction in its heparin-binding affinity.(47) Furin is the first identified proprotein convertase and is highly expressed in tumor tissues. Since a wide range of proproteins are involved in the various physiological and pathological processes in tumor progression, furin has been proposed as a potential therapeutic target for tumors. In addition, the terminal sialic acid residues on SOD3 are closely related to its enzymatic cleavage and secretion, suggesting that regulation of the cleavage and secretion by N-glycan may be associated with tumor progression.(48) However, the molecular mechanism and relevance of SOD3 desorption and N-glycosylation have not been fully elucidated, as well as the relationship between SOD3 function and cancer.
Cu in Tumor Progression
Cu, an essential micronutrient, plays a pivotal role in several physiological processes.(49–51) The disturbance of Cu homeostasis is involved in neurological and cardiac diseases, as well as tumor progression. The bioavailability of Cu is strictly controlled by Cu transporters, including Cu transporter 1 (CTR1), Cu chaperone for SOD1 (CCS), antioxidant-1 (Atox-1), and Cu-transporting P-type ATPase α and β (ATP7A and ATP7B).(52,53) These copper transporters supply Cu ions to Cu-containing enzymes that are involved in a variety of metabolic processes, including aerobic respiration, superoxide dismutation, and extracellular matrix synthesis.(54) Studies suggest that Cu levels generally increase in tumor tissues, including breast, liver, lung, and oral tissues, thus promoting tumor progression.(9–11)
CTR1 plays a key role in Cu (I) uptake, and excess Cu accumulation in tumor tissues is due to the significant induction of CTR1. In addition, the six-transmembrane epithelial antigen of the prostate (STEAP) family proteins (STEAP1-6) are highly expressed in tumor tissues.(55) Hence, STEAP/CTR1-mediated Cu uptake is considered to facilitate tumor progression. Cu taken up through CTR1 is supplied to SOD1 and SOD3 through CCS or Atox-1 and ATP7A, respectively. As excess Cu generates oxidative stress, cells need to deliver Cu to its chaperon proteins without toxic reactions, which is facilitated by the abundance of thiol compounds, mainly GSH.(56) The level of GSH depends on the cell type and is relatively high in several tumor cells. Thus, tumor cells use high concentrations of GSH for Cu-mediated growth and resistance to oxidative stress. A recent study reported that intracellular Cu induces the expression of SLC7A11, leading to an increase in intracellular cysteine to produce GSH (Fig. 2).(57) In addition, Cu induces programmed cell death ligand 1 (PD-L1), a key protein in the immune checkpoint, in neuroblastoma and glioblastoma, suggesting that abnormal Cu homeostasis is involved in antitumor immune responses. Concerning Cu-mediated immune responses, GSH may also play a pivotal role in the intracellular trafficking of Cu; thus, redox homeostasis may be a potential therapeutic target for tumor progression.
Fig. 2.
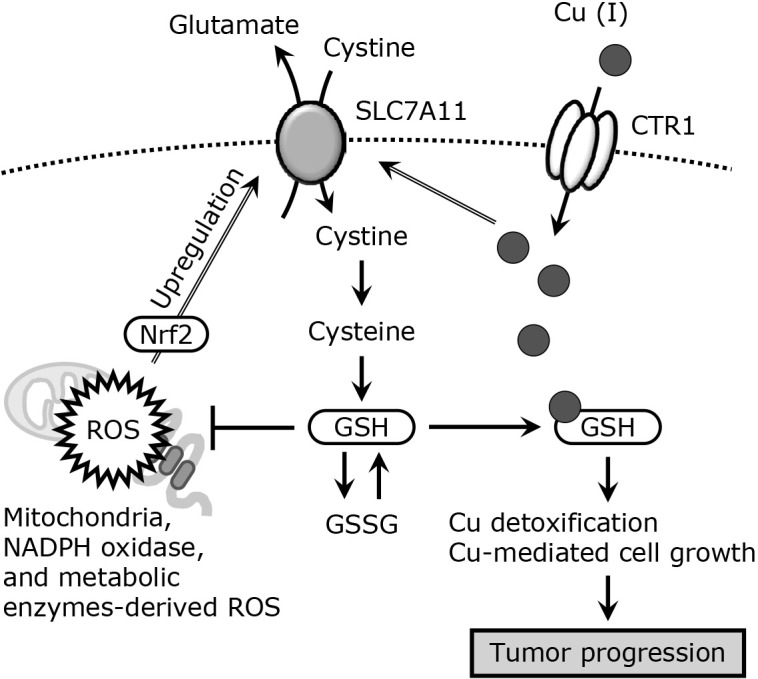
GSH facilitates Cu-mediated cell growth. Mitochondrial, NADPH oxidase, or metabolic enzyme-mediated ROS accumulation induces SLC7A11 expression through the activation of Nrf2.tif. SLC7A11 increases intracellular GSH by promoting cystine uptake. Excess accumulation of Cu is observed in several tumor cells, and Cu also induces SLC7A11 expression. Increased GSH protects the cells from the damaging effects of ROS and participates in Cu detoxification by binding to Cu, which leads to Cu-mediated cell growth and tumor progression. GSSG, oxidized-GSH.
The expression of Atox-1, a Cu chaperone for ATP7A, is also upregulated in tumors, and Atox-1 is considered to function as a tumor promoter.(58) Recent studies have shown that genetic deletion of Atox-1 reduces the rate and direction of single-cell migration.(59) In addition to being a Cu chaperone, Atox-1 functions as a Cu-dependent transcription factor. Atox-1 has a conserved lysine-rich domain (KKTGK) at its C-terminal region, which functions in its nuclear localization, and its nuclear localization is highly observed in metastatic tumor cells.(60) It has been reported that Atox-1 regulates SOD3 expression in tumor-associated macrophages, suggesting that Atox-1 may be involved in tumor progression through the induction of SOD3.(61) Atox-1 also upregulates some tumor-associated genes such as cyclin D1 and MDM2.(62) In addition, a recent study revealed that tumor necrosis factor-α (TNF-α)-mediated Atox-1 nuclear translocation is accompanied by TNF receptor-associated factor 4 (TRAF4) in a Cu-dependent manner.(63) TRAF4 promotes lung cancer aggressiveness and high-grade ovarian cancer progression. Thus, inhibition of the combined action of TRAF4 and Atox-1 could be a novel therapeutic target.
The Cu-containing secretory enzymes, including SOD3 and lysyl oxidases, acquire Cu from ATP7A, a Cu-exporting P-type ATPase, in the trans-Golgi network.(64) This function of ATP7A was determined by gene silencing, which caused a reduction in SOD3 expression and activity. In addition, ATP7A influences the cellular concentration of Cu by facilitating Cu egress, which helps protect cells from Cu-mediated oxidative stress. A significant reduction in ATP7A increased the intracellular Cu levels and ROS accumulation in neuroblastoma cells.(65) Recently, cuproptosis has been identified as a novel form of Cu-mediated programmed cell death.(66) This occurs by the addition of a Cu ionophore, elesclomol, which facilitates Cu (II) uptake in a CTR1-independent manner and Cu accumulation in mitochondria.(67) Ferredoxin 1-mediated reduction in Cu ions and the loss of iron-sulfur clusters, as well as the aggregation of lipoylated proteins, are thought to lead to cell death. Notably, decreased expression of ATP7A and SLC7A11 is also involved in cuproptosis, which hinders physiological Cu utilization.(68,69) Many details are unknown because this concept has only been discovered recently, but clarifying this link between cell death and cancer may lead to new targets for cancer therapy.
Conclusion
Cu plays an essential role in several physiological processes, but its dysregulation is involved in tumor progression. SOD3 also exhibits antitumor and pro-tumor effects. Therefore, it is necessary to understand the exact molecular mechanisms underlying the cell-specific and tissue-specific expression of SOD3, secretion, and function in tumor cells, which contribute to the elucidation of the pathological biochemistry, especially in redox homeostasis, in tumor progression.
Acknowledgments
This study was supported in part by grants from JSPS KAKENHI Grant Numbers 23790190, 26460090, and 17K08277, a Sasakawa Scientific Research Grant from The Japan Science Society (2018-4007), and a grant for the encouragement of young scientists from Gifu Pharmaceutical University. TK was awarded the SFRR Japan Award of Scientific Excellent 2023.
Abbreviations
- Atox-1
antioxidant-1
- ATP7A
Cu-transporting P-type ATPase α
- ATP7B
Cu-transporting P-type ATPase β
- CCS
Cu chaperone for superoxide dismutase 1
- CTR1
Cu transporter 1
- Cu
copper
- FOXO1
forkhead box O1
- FUT8
α1,6-fucosyltransferase
- GSH
glutathione
- GSSG
oxidized-GSH
- HDAC1
histone deacetylase 1
- HO-1
heme oxygenase 1
- JMJD3
Jumonji domain-containing protein-3
- MEF2
myocyte enhancer factor 2
- Nrf2
NF-E2-related factor 2
- PD-L1
programmed cell death ligand 1
- SOD
superoxide dismutase
- ROS
reactive oxygen species
- STEAP
six-transmembrane epithelial antigen of the prostate
- TET1
ten-eleven-translocation 1
- TNF-α
tumor necrosis factor-α
- TRAF4
TNF receptor-associated factor 4
Conflict of Interest
No potential conflicts of interest were disclosed.
References
- 1.Akaike T, Nishida M, Fujii S. Regulation of redox signalling by an electrophilic cyclic nucleotide. J Biochem 2013; 153: 131–138. [DOI] [PubMed] [Google Scholar]
- 2.Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006; 312: 1882–1883. [DOI] [PubMed] [Google Scholar]
- 3.D'Autréaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol 2007; 8: 813–824. [DOI] [PubMed] [Google Scholar]
- 4.Kamata H, Hirata H. Redox regulation of cellular signalling. Cell Signal 1999; 11: 1–14. [DOI] [PubMed] [Google Scholar]
- 5.Faraci FM. Vascular protection. Stroke 2003; 34: 327–329. [DOI] [PubMed] [Google Scholar]
- 6.Furukawa S, Fujita T, Shimabukuro M, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 2004; 114: 1752–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts CK, Barnard RJ, Sindhu RK, Jurczak M, Ehdaie A, Vaziri ND. Oxidative stress and dysregulation of NAD(P)H oxidase and antioxidant enzymes in diet-induced metabolic syndrome. Metabolism 2006; 55: 928–934. [DOI] [PubMed] [Google Scholar]
- 8.Zelko IN, Mariani TJ, Folz RJ. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med 2002; 33: 337–349. [DOI] [PubMed] [Google Scholar]
- 9.Jouybari L, Kiani F, Islami F, et al. Copper concentrations in breast cancer: a systematic review and meta-analysis. Curr Med Chem 2020; 27: 6373–6383. [DOI] [PubMed] [Google Scholar]
- 10.Zabłocka-Słowińska K, Prescha A, Płaczkowska S, et al. Serum and whole blood Cu and Zn status in predicting mortality in lung cancer patients. Nutrients 2020; 13: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stepien M, Jenab M, Freisling H, et al. Pre-diagnostic copper and zinc biomarkers and colorectal cancer risk in the European prospective investigation into cancer and nutrition cohort. Carcinogenesis 2017; 38: 699–707. [DOI] [PubMed] [Google Scholar]
- 12.Liu S, Li B, Xu J, et al. SOD1 promotes cell proliferation and metastasis in non-small cell lung cancer via an miR-409-3p/SOD1/SETDB1 epigenetic regulatory feedforward loop. Front Cell Dev Biol 2020; 8: 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glasauer A, Sena LA, Diebold LP, Mazar AP, Chandel NS. Targeting SOD1 reduces experimental non-small-cell lung cancer. J Clin Invest 2014; 124: 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Somwar R, Erdjument-Bromage H, Larsson E, et al. Superoxide dismutase 1 (SOD1) is a target for a small molecule identified in a screen for inhibitors of the growth of lung adenocarcinoma cell lines. Proc Natl Acad Sci U S A 2011; 108: 16375–16380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marklund SL. Human copper-containing superoxide dismutase of high molecular weight. Proc Natl Acad Sci U S A 1982; 79: 7634–7638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ookawara T, Imazeki N, Matsubara O, et al. Tissue distribution of immunoreactive mouse extracellular superoxide dismutase. Am J Physiol 1998; 275: C840–C847. [DOI] [PubMed] [Google Scholar]
- 17.Svensk AM, Soini Y, Pääkkö P, Hiravikoski P, Kinnula VL. Differential expression of superoxide dismutases in lung cancer. Am J Clin Pathol 2004; 122: 395–404. [DOI] [PubMed] [Google Scholar]
- 18.Teoh ML, Fitzgerald MP, Oberley LW, Domann FE. Overexpression of extracellular superoxide dismutase attenuates heparanase expression and inhibits breast carcinoma cell growth and invasion. Cancer Res 2009; 69: 6355–6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Q, Bolli R, Qiu Y, Tang XL, Guo Y, French BA. Gene therapy with extracellular superoxide dismutase protects conscious rabbits against myocardial infarction. Circulation 2001; 103: 1893–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu Z, Xu X, Hu X, et al. Extracellular superoxide dismutase deficiency exacerbates pressure overload-induced left ventricular hypertrophy and dysfunction. Hypertension 2008; 51: 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mira E, Carmona-Rodríguez L, Pérez-Villamil B, et al. SOD3 improves the tumor response to chemotherapy by stabilizing endothelial HIF-2α. Nat Commun 2018; 9: 575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laukkanen MO. Extracellular superoxide dismutase: growth promoter or tumor suppressor? Oxid Med Cell Longev 2016; 2016: 3612589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kamiya T, Yamaguchi Y, Oka M, Hara H. Combined action of FOXO1 and superoxide dismutase 3 promotes MDA-MB-231 cell migration. Free Radic Res 2022; 56: 106–114. [DOI] [PubMed] [Google Scholar]
- 24.Laukkanen MO, Cammarota F, Esposito T, Salvatore M, Castellone MD. Extracellular superoxide dismutase regulates the expression of small GTPase regulatory proteins GEFs, GAPs, and GDI. PLoS One 2015; 9: e0121441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kristensen LS, Nielsen HM, Hansen LL. Epigenetics and cancer treatment. Eur J Pharmacol 2009; 625: 131–142. [DOI] [PubMed] [Google Scholar]
- 26.Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 2007; 447: 425–432. [DOI] [PubMed] [Google Scholar]
- 27.Zelko IN, Mueller MR, Folz RJ. CpG methylation attenuates Sp1 and Sp3 binding to the human extracellular superoxide dismutase promoter and regulates its cell-specific expression. Free Radic Biol Med 2010; 48: 895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yasuda H, Mizukami K, Hayashi M, Kamiya T, Hara H, Adachi T. Exendin-4 promotes extracellular-superoxide dismutase expression in A549 cells through DNA demethylation. J Clin Biochem Nutr 2016; 58: 34–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kamiya T, Machiura M, Makino J, Hara H, Hozumi I, Adachi T. Epigenetic regulation of extracellular-superoxide dismutase in human monocytes. Free Radic Biol Med 2013; 61: 197–205. [DOI] [PubMed] [Google Scholar]
- 30.Kamiya T, Nakahara R, Mori N, Hara H, Adachi T. Ten-eleven translocation 1 functions as a mediator of SOD3 expression in human lung cancer A549 cells. Free Radic Res 2017; 51: 329–336. [DOI] [PubMed] [Google Scholar]
- 31.Ichihara M, Kamiya T, Hara H, Adachi T. The MEF2A and MEF2D function as scaffold proteins that interact with HDAC1 or p300 in SOD3 expression in THP-1 cells. Free Radic Res 2018; 52: 799–807. [DOI] [PubMed] [Google Scholar]
- 32.Makino J, Ogasawara R, Kamiya T, et al. Royal jelly constituents increase the expression of extracellular superoxide dismutase through histone acetylation in monocytic THP-1 cells. J Nat Prod 2016; 79: 1137–1143. [DOI] [PubMed] [Google Scholar]
- 33.Yamamoto M, Kensler TW, Motohashi H. The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol Rev 2018; 98: 1169–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu X, Shen S, Qin J, et al. High co-expression of SLC7A11 and GPX4 as a predictor of platinum resistance and poor prognosis in patients with epithelial ovarian cancer. BJOG 2022; 129 Suppl 2: 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singh B, Bhat HK. Superoxide dismutase 3 is induced by antioxidants, inhibits oxidative DNA damage and is associated with inhibition of estrogen-induced breast cancer. Carcinogenesis 2012; 33: 2601–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eijkelenboom A, Burgering BM. FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol 2013; 14: 83–97. [DOI] [PubMed] [Google Scholar]
- 37.Han CY, Cho KB, Choi HS, Han HK, Kang KW. Role of FoxO1 activation in MDR1 expression in adriamycin-resistant breast cancer cells. Carcinogenesis 2008; 29: 1837–1844. [DOI] [PubMed] [Google Scholar]
- 38.Creemers EE, Sutherland LB, McAnally J, Richardson JA, Olson EN. Myocardin is a direct transcriptional target of Mef2, Tead and Foxo proteins during cardiovascular development. Development 2006; 133: 4245–4256. [DOI] [PubMed] [Google Scholar]
- 39.Goto Y, Niwa Y, Suzuki T, Uematsu S, Dohmae N, Simizu S. N-glycosylation is required for secretion and enzymatic activity of human hyaluronidase1. FEBS Open Bio 2014; 4: 554–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ota F, Kizuka Y, Kitazume S, Adachi T, Taniguchi N. N-glycosylation is essential for the secretion of extracellular superoxide dismutase. FEBS Lett 2016; 590: 3357–3367. [DOI] [PubMed] [Google Scholar]
- 41.Ohkawa Y, Kitano M, Maeda K, et al. Core fucosylation is required for the secretion of and the enzymatic activity of SOD3 in nonsmall-cell lung cancer cells. Antioxid Redox Signal 2023; 38: 1201–1211. [DOI] [PubMed] [Google Scholar]
- 42.Tu CF, Wu MY, Lin YC, Kannagi R, Yang RB. FUT8 promotes breast cancer cell invasiveness by remodeling TGF-β receptor core fucosylation. Breast Cancer Res 2017; 19: 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kelel M, Yang RB, Tsai TF, et al. FUT8 remodeling of EGFR regulates epidermal keratinocyte proliferation during psoriasis development. J Invest Dermatol 2021; 141: 512–522. [DOI] [PubMed] [Google Scholar]
- 44.Hu P, Shi B, Geng F, Zhang C, Wu W, Wu XZ. E-cadherin core fucosylation regulates nuclear β-catenin accumulation in lung cancer cells. Glycoconj J 2008; 25: 843–850. [DOI] [PubMed] [Google Scholar]
- 45.Adachi T, Morihara N, Yamazaki N, et al. An arginine-213 to glycine mutation in human extracellular-superoxide dismutase reduces susceptibility to trypsin-like proteinases. J Biochem 1996; 120: 184–188. [DOI] [PubMed] [Google Scholar]
- 46.Kwon MJ, Lee KY, Ham WG, Tak LJ, Agrahari G, Kim TY. Pathologic properties of SOD3 variant R213G in the cardiovascular system through the altered neutrophils function. PLoS One 2020; 15: e0227449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bowler RP, Nicks M, Olsen DA, et al. Furin proteolytically processes the heparin-binding region of extracellular superoxide dismutase. J Biol Chem 2002; 277: 16505–16511. [DOI] [PubMed] [Google Scholar]
- 48.Ota F, Kizuka Y, Nakano M, et al. Sialylation of extracellular superoxide dismutase (EC-SOD) enhances furin-mediated cleavage and secretion. Glycobiology 2017; 27: 1081–1088. [DOI] [PubMed] [Google Scholar]
- 49.Harris ED. Copper transport: an overview. Proc Soc Exp Biol Med 1991; 196: 130–140. [DOI] [PubMed] [Google Scholar]
- 50.Bull PC, Cox DW. Wilson disease and Menkes disease: new handles on heavy-metal transport. Trends Genet 1994; 10: 246–252. [DOI] [PubMed] [Google Scholar]
- 51.Rodríguez JP, Ríos S, González M. Modulation of the proliferation and differentiation of human mesenchymal stem cells by copper. J Cell Biochem 2002; 85: 92–100. [PubMed] [Google Scholar]
- 52.Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY. Function and regulation of human copper-transporting ATPases. Physiol Rev 2007; 87: 1011–1046. [DOI] [PubMed] [Google Scholar]
- 53.Kim BE, Nevitt T, Thiele DJ. Mechanisms for copper acquisition, distribution and regulation. Nat Chem Biol 2008; 4: 176–185. [DOI] [PubMed] [Google Scholar]
- 54.Lelièvre P, Sancey L, Coll JL, Deniaud A, Busser B. The multifaceted roles of copper in cancer: a trace metal element with dysregulated metabolism, but also a target or a bullet for therapy. Cancers (Basel) 2020; 12: 3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen WJ, Wu HT, Li CL, et al. Regulatory roles of six-transmembrane epithelial antigen of the prostate family members in the occurrence and development of malignant tumors. Front Cell Dev Biol 2021; 9: 752426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hatori Y, Clasen S, Hasan NM, Barry AN, Lutsenko S. Functional partnership of the copper export machinery and glutathione balance in human cells. J Biol Chem 2012; 287: 26678–26687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Voli F, Valli E, Lerra L, et al. Intratumoral copper modulates PD-L1 expression and influences tumor immune evasion. Cancer Res 2020; 80: 4129–4144. [DOI] [PubMed] [Google Scholar]
- 58.Blockhuys S, Celauro E, Hildesjö C, et al. Defining the human copper proteome and analysis of its expression variation in cancers. Metallomics 2017; 9: 112–123. [DOI] [PubMed] [Google Scholar]
- 59.Blockhuys S, Wittung-Stafshede P. Copper chaperone Atox1 plays role in breast cancer cell migration. Biochem Biophys Res Commun 2017; 483: 301–304. [DOI] [PubMed] [Google Scholar]
- 60.Itoh S, Kim HW, Nakagawa O, et al. Novel role of antioxidant-1 (Atox1) as a copper-dependent transcription factor involved in cell proliferation. J Biol Chem 2008; 283: 9157–9167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kamiya T, Takeuchi K, Fukudome S, Hara H, Adachi T. Copper chaperone antioxidant-1, Atox-1, is involved in the induction of SOD3 in THP-1 cells. Biometals 2018; 31: 61–68. [DOI] [PubMed] [Google Scholar]
- 62.Sudhahar V, Shi Y, Kaplan JH, Ushio-Fukai M, Fukai T. Whole-transcriptome sequencing analyses of nuclear antixoxidant-1 in endothelial cells: role in inflammation and atherosclerosis. Cells 2022; 11: 2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Das A, Sudhahar V, Ushio-Fukai M, Fukai T. Novel interaction of antioxidant-1 with TRAF4: role in inflammatory responses in endothelial cells. Am J Physiol Cell Physiol 2019; 317: C1161–C1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shanbhag V, Jasmer-McDonald K, Zhu S, et al. ATP7A delivers copper to the lysyl oxidase family of enzymes and promotes tumorigenesis and metastasis. Proc Natl Acad Sci U S A 2019; 116: 6836–6841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kondo M, Hara H, Kamijo F, Kamiya T, Adachi T. 6-Hydroxydopamine disrupts cellular copper homeostasis in human neuroblastoma SH-SY5Y cells. Metallomics 2021; 13: mfab041. [DOI] [PubMed] [Google Scholar]
- 66.Tsvetkov P, Coy S, Petrova B, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022; 375: 1254–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zheng P, Zhou C, Lu L, Liu B, Ding Y. Elesclomol: a copper ionophore targeting mitochondrial metabolism for cancer therapy. J Exp Clin Cancer Res 2022; 41: 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang W, Lu K, Jiang X, et al. Ferroptosis inducers enhanced cuproptosis induced by copper ionophores in primary liver cancer. J Exp Clin Cancer Res 2023; 42: 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gao W, Huang Z, Duan J, Nice EC, Lin J, Huang C. Elesclomol induces copper-dependent ferroptosis in colorectal cancer cells via degradation of ATP7A. Mol Oncol 2021; 15: 3527–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]