

Abstract
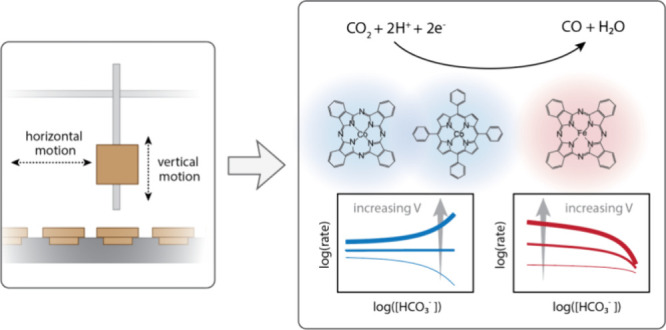
In electrocatalysis, mechanistic analysis of reaction rate data often relies on the linearization of relatively simple rate equations; this is the basis for typical Tafel and reactant order dependence analyses. However, for more complex reaction phenomena, such as surface coverage effects or mixed control, these common linearization strategies will yield incomplete or uninterpretable results. Cohesive kinetic analysis, which is often used in thermocatalysis and involves quantitative model fitting for data collected over a wide range of reaction conditions, requires more data but also provides a more robust strategy for interrogating reaction mechanisms. In this work, we report a robotic system that improves the experimental workflow for collecting electrochemical rate data by automating sequential testing of up to 10 electrochemical cells, where each cell can have a different electrode, electrolyte, gas-phase reactant composition, and applied voltage. We used this system to investigate the mechanism of carbon dioxide electroreduction to carbon monoxide at several immobilized metal tetrapyrroles. Specifically, at cobalt phthalocyanine (CoPc), cobalt tetraphenylporphyrin (CoTPP), and iron phthalocyanine (FePc), we see signatures of complex reaction mechanisms, where observed bicarbonate and CO2 order dependences change with applied potential. We illustrate how phenomena such as electrolyte poisoning and potential-dependent degrees of rate control can explain the observed kinetic behaviors. Our mechanistic analysis suggests that CoPc and CoTPP share a similar reaction mechanism, akin to one previously proposed, whereas the mechanism for FePc likely involves a species later in the catalytic cycle as the most abundant reactive intermediate. Our study illustrates that complex reaction mechanisms that are not amenable to common Tafel and order dependence analyses may be quite prevalent across this class of immobilized metal tetrapyrrole electrocatalysts.
Short abstract
Reaction rate data collected with an automated robot show that several well-studied, heterogeneous CO2 electroreduction catalysts exhibit significant voltage-dependent changes in reaction mechanisms.
1. Introduction
Reaction kinetics analysis in electrocatalysis has traditionally relied on interpretations of linearized rate data. In these analyses, one assumes that, within the rate equation, dependences on different experimental handles (e.g., voltage and concentration) can be factored separately and linearized on log–log or semilog graphs. This is the basis for typical Tafel and reactant order dependence analyses. Although linearization can provide valuable mechanistic insight with relatively light experimental data collection, it requires highly simplifying assumptions. Coverage effects, mixed rate control, and side reactions can all lead to rate behavior that is difficult to analyze using linearization.1−4 Given the substantial effect of voltage on reaction energy landscapes, as well as the general complexity of electrified interphases, it is reasonable to expect that many electrocatalytic reactions follow complex, nonlinearizable reaction mechanisms. For example, since many elementary charge transfer steps display an order of magnitude change in equilibrium constant with only 59 mV of voltage difference (i.e., 59 mV/dec scaling), surface coverages of reaction intermediates could drastically change even within a potential window of just a few hundred millivolts. Additionally, effects from solvent or electrolyte displacement, which are general to solid–liquid interfaces5 but may also have additional potential dependences,6−8 can further complicate reaction rate data.
On the other hand, cohesive kinetic analyses allow for consideration of the aforementioned complexities. Such analyses interrogate possible correlations between variables by using large volumes of data collected over wide ranges of reaction conditions. These data can be fit to general and complex rate equations using quantitative statistical methods.9 Although cohesive kinetic analyses are more common in heterogeneous thermocatalysis,10−13 in electrocatalysis, these analyses, as well as other analyses beyond linearization, have already helped to highlight some of the complex kinetic phenomena14 that underpin the CO2 reduction reaction (CO2RR). For example, a meta-analysis of reported Tafel data across many classes of CO2RR catalysts showed that Tafel slopes do not show a strong preference for commonly interpretable values (e.g., 120, 60, and 40 mV/dec).15 This pointed to the likely prevalence of additional mechanistic phenomena that are not captured with a traditional Tafel analysis. Additionally, cohesive kinetic analysis of complex reaction rate data provided evidence of mechanistic complexities such as competitive electrolyte adsorption and mixed control for CO2RR to CO on cobalt phthalocyanine.16 Finally, kinetic modeling coupled with continuum transport modeling illustrated the importance of mass transport and competing reactions for explaining complex kinetic behavior of CO2RR to CO at Ag.17 Thus, the wider use of cohesive kinetic analysis in electrocatalysis could shed light on complex kinetic behavior that has been unexplained or even missed by typical linearization-based kinetic analyses.
One barrier for implementing cohesive kinetic analysis in electrocatalysis is its heavy data requirement. Collecting the data manually can entail months of intensive yet tedious experimentation, and the data can be subject to human error or variability. On the other hand, automation can provide a tool for accelerating and standardizing such workflows. However, to date, most strategies for automation in electrocatalysis are tailored toward high-throughput catalyst or condition screening.18,19 For example, one automation strategy involves miniaturization, where small (order microliter) electrolyte volumes are employed for rapid materials screening within scanning droplet cells.20−22 However, with such small working volumes, it can be difficult to quantify reaction products; thus, these rapid screening techniques cannot be used to automate the kinetic analysis of electrocatalytic reactions that do not have 100% Faradaic efficiency. Parallelization is another strategy, where arrays of different metal compositions or electrolyte conditions are tested simultaneously.23−25 Parallel setups often do allow for product quantification, but quantification is typically performed either by aggregating effluents from multiple reactors or by manual workup after electrolysis. An automated kinetic analysis workflow would ideally involve online product quantification of individual electrochemical cells, which is possible but likely to be expensive in a parallel configuration. Thus, a strategy involving sequential electrochemical testing and online product quantification of well-controlled reaction conditions within geometrically well-defined reactors would be ideal for automating kinetic analysis. This automation strategy has been used to screen catalysts for the (photo)electrochemical CO2 reduction reaction.26−29 We demonstrate that a similar automation concept can be tailored to automate electrochemical kinetic analysis workflows.
In this work, we introduce a robotic platform designed to automate the collection of electrochemical reaction rate data collection. Our robotic system automatically performs queued electrochemical experiments that, unlike previous approaches, can accommodate different electrode, electrolyte, and gas-phase reactant composition for each individual experiment. This extent of operational versatility is not necessary for high-throughput catalyst screening but is essential for the kinetic analysis we sought to automate, which involves testing fresh electrocatalysts under a wide range of different operating conditions. We used this system to investigate the mechanism of CO2RR to carbon monoxide (CO) at several immobilized metal tetrapyrroles including cobalt phthalocyanine (CoPc), cobalt tetraphenyl porphyrin (CoTPP), and iron phthalocyanine (FePc). The mechanism of CO2RR at these catalysts remains a topic of debate,16,30−42 despite the fact that they are commonly studied and present relatively well-defined active site structures. We show that across these catalysts, nonidealities in reaction rate data are prevalent, specifically evidenced by bicarbonate and CO2 order dependencies that, in addition to being nonlinear and/or noninteger, also change with different applied potentials. We discuss how several reaction phenomena not commonly considered for these catalysts, such as electrolyte poisoning, mixed control, and coverage effects, can explain the trends. Our work demonstrates how automation can assist with cohesive kinetic analysis and sets forth general mechanistic considerations for interpreting complex rate data.
2. Results and Discussion
2.1. Overall Robot Design
The overall robot design consists of a single cell body that houses the reference and counter electrodes, complemented by a line of 10 cell pan pairs that each house a carbon paper working electrode. The cell body moves horizontally to sequentially perform electrochemistry on each cell pan and vertically to open and close itself onto any given cell pan (Figure 1A).
Figure 1.

Robot design for the automated experiments. (A) Schematic of operation, where vertical motion opens and closes a cell body on top of a pair of cell pans and horizontal motion moves the cell body to different cell pans. Together these motions allow automatic execution of a different experiment at each cell pan. (B) Schematic of cell geometry, where the cell body houses ports for working and counter electrodes as well as ports for electrolyte/gas flow in/out of the cell. The top and bottom cell pan sandwich a working electrode, which is pressed into a conducting pin. Blue, red, and gray dots indicate connections to the potentiostat. Seals between cell body and pans are made by o-rings, depicted as black circles. (C) CAD depiction of the overall robot design. (D) Photograph of the robot during operation, while an electrochemical experiment is being executed at one of the cell pans. (E) Schematic illustrating the operational workflow of the automated system.
For any single electrolysis within a given cell pan, the robot closes the cell body onto the cell pan and then pumps the electrolyte liquid through an “electrolyte in” port on the side of the cell body (Figure 1B). There are two possible configurations for the electrolyte source: each cell pan can have its own individual electrolyte that is stored locally in a nearby vial (Figure S2A), or the electrolyte source can be connected to one common reservoir (Figure S2B), so that a given set of electrolyses uses the same electrolyte formulation. In this work, the second option was employed, so that the electrolyte reservoir could be freshly bubbled with CO2. After electrolyte is flowed into the cell, the robot initiates gas flow through the “gas in” tubing (Figure 1B) and then applies voltage and initiates online product quantification. A gas chromatograph connected to the “gas out” port confers the online detection of CO and H2 (Figure 1B). Electrolyses were run for 40 min, and reported rates are averages of data collected between 20 and 40 min of electrolysis. We confirmed no systematic increases or decreases in the reaction rate within this time window (Figure S5). Finally, when the electrolysis is complete, electrolyte is pumped out of the cell and into a waste vial or container via the “electrolyte out” tubing and the cell is opened, completing the run.
Cell body cleaning is achieved by moving and closing the cell body onto a wash station consisting of an empty cell pan pair and then pumping milli-Q water in and out for several cycles (Figure S3). In this work, with the common reservoir electrolyte configuration, the cell body was rinsed after the completion of a full set of 10 electrolyses. We expected that this would not cause contamination issues because the same electrolyte composition was used within any given set of electrolyses and all products were gas-phase.
2.2. Discussion of Cell Design and Data Robustness
The cell assembly is analogous to a three-electrode, undivided compartment cell (Figure 1B). A carbon paper disk working electrode is clamped between a top and bottom cell pan, which forces it into electrical contact with a conductive rod and exposes a well-defined 10 mm diameter circle to the electrolyte. A leak-free Ag/AgCl reference electrode (Innovative Instruments) is inserted on the side of the cell body, and a 0.25 mm diameter platinum wire counter electrode is inserted on the opposite side of the cell body. We opted for this cell design because it uses the same electrode form factor as conventional electrochemical sandwich cell testing and the lack of membrane allowed for more facile liquid and gas management.
We note that as a one-compartment, rather than a two- or three- compartment cell, our setup comes with some considerations. First, without a membrane to separate the anode and cathode, there can be crosstalk between working and counter electrodes. For example, oxygen evolved at the counter electrode can cross over to the working electrode and lead to a parasitic oxygen reduction reaction (ORR) current. We did in fact observe incomplete Faradaic efficiency (FE) closure, where across all runs, an average of only ca. 60% was attributable to CO and H2 formation, and the remaining Faradaic efficiency was presumably lost to oxygen reduction (Figure S6). Additionally, we opted to use a Pt, rather than carbon-based, counter electrode to preclude any possibility of attributing adventitious anode oxidation products to CO2RR. Metal crossover from Pt anodes onto copper CO2RR cathodes has been observed, even in divided cells, to enhance hydrogen evolution.43 We did in fact observe relatively high FEs toward HER, typically around 30% to 60% (Figure S12). However, for dispersed molecular catalysts with relatively low surface coverage, we expected this effect to have a minimal impact on the kinetic interpretation of the partial CO current, which occurs on metal tetrapyrrole sites and should thus be relatively independent of any adventitious HER sites caused by Pt deposition. Finally, while many electrochemical CO2RR cell designs feed CO2 directly to or through the working electrode for improved mass transport, we bubbled CO2 into the electrolyte. We note that the currents in this study are below the theoretical mass transport limited current density (Supplemental Discussion 3.2) and that we did not observe an increase in partial CO current when increasing the flow rate beyond the 20 sccm that was used in this work (Figure S8). However, we acknowledge that flow configuration can have unexpected influence over measured kinetics, even in ostensibly non-transport-limited regimes.44
Given the above considerations, we first evaluated data robustness by using the automated setup to collect data that we could compare with literature precedent. We collected kinetic data for immobilized CoPc and compared it against previously reported results16 that were manually collected in a more classic three-compartment cell. We found that the newly measured rates toward CO production were up to 10 times lower than previously reported (Figure S10) and that Faradaic efficiencies toward CO were lower than expected, with values typically under 30% (Figure S12A). Nonetheless, we did find that previously reported kinetic trends in CO production were preserved (Figure S9) and that the newly collected data were well-correlated with those of the previous work (Figure S10). Importantly, hallmarks of nonideal reaction mechanisms, such as curvature and potential-dependent changes in the bicarbonate order dependence, were clearly evident in the data. Thus, we proceeded to analyze CoTPP and FePc to determine whether signatures of nonideal reaction mechanisms could be more broadly observed at other immobilized tetrapyrroles. Aggregated rates (Figure S11) and selectivities (Figure S12) toward CO and H2 for all three catalysts are provided in the Supporting Information.
2.3. Complex Kinetic Behaviors across CoPc, CoTPP, and FePc
We analyzed the partial currents toward CO production at different applied voltages, CO2 partial pressures, and bicarbonate concentrations. For each of these variables, signatures of mechanistic complexity, such as nonlinear and/or condition-dependent trends, were apparent in the corresponding canonical Tafel, CO2, and bicarbonate dependences.
The three tested catalysts present two general trends in the kinetic data, with the cobalt-based tetrapyrroles (CoPc and CoTPP) displaying qualitatively distinct features from the iron-based tetrapyrrole (FePc). For the bicarbonate dependence, all three catalysts display voltage-dependent bicarbonate inhibition. However, for CoPc and CoTPP, greater bicarbonate inhibition occurs at less reductive potentials (Figure 2B and 2E), whereas for FePc, the opposite is true (Figure 3B). For the CO2 dependence, FePc displays attenuation of the apparent CO2 order (nCO2 = 0.2 ± 0.1) at less reductive potentials, whereas CoPc and CoTPP do not display significant deviations from apparent CO2 orders of 1.
Figure 2.

Kinetic analysis of Co-based tetrapyrroles. (A, D) Structures of catalysts studied: cobalt phthalocyanine (CoPc, A) and cobalt tetraphenyl porphyrin (CoTPP, D). (B, E) Apparent bicarbonate order dependencies at different voltages for CoPc (B) and CoTPP (E). (C, F) Apparent CO2 order dependencies at different voltages for CoPc (C) and CoTPP (F). Points represent experimental data, and lines represent model fits. Error bars represent standard deviation with n ≥ 3. For all experiments, voltages reported vs SHE, total ionic strength held constant using NaClO4, and CO2 at 1 atm. (G) Mechanistic framework that can explain the kinetic observations. Salient phenomena at less reductive voltages are displayed in purple, and salient phenomena at more reductive voltages are displayed in yellow.
Figure 3.

Kinetic analysis of Fe-based tetrapyrrole. (A) Structure of catalyst studied: iron phthalocyanine (FePc, A). (B) Apparent bicarbonate order dependencies at different voltages. (C) Apparent CO2 order dependencies at different voltages. Points represent experimental data, and lines represent model fits. Error bars represent standard deviation with n ≥ 3. For all experiments, voltages reported vs SHE, total ionic strength held constant using NaClO4, and CO2 at 1 atm. (D) Mechanistic framework that can explain the kinetic observations. Salient phenomena at less reductive voltages are colored purple, and salient phenomena at more reductive voltages are displayed in yellow.
Such kinetic signatures have, to our knowledge, only been reported in one previous work at CoPc16 and are unexplainable with mechanisms typically invoked for CO2RR at immobilized tetrapyrroles.30,32,34,37−39,42,45−49 Below, we discuss the mechanistic features that likely underpin the observed trends. For the sake of clarity, the following discussions center around specific mechanistic proposals. However, we note that these mechanisms only qualitatively capture the observed trends but do not quantitatively, via statistical goodness-of-fit metrics, fit all of the experimental data. Thus, rather than proposing specific reaction mechanisms, the following discussion is mainly intended to (1) highlight the unambiguous existence of mechanistic complexity in the rate data at CoPc, CoTPP, and FePc and (2) describe the likely roles of phenomena such as mixed rate control, surface coverage effects, and catalyst poisoning in explaining the complex experimental behavior. Rate law derivations for the presented models are provided in SI Section 5.2, and examples of alternative models that were considered are provided in SI Section 5.1.
2.4. Mechanistic Features Proposed for CoPc and CoTPP
For CoPc and CoTPP, nonideality in the reaction rate data is most apparent for the bicarbonate dependence, where bicarbonate inhibits the reaction at less reductive potentials (low overpotential, or η) but displays a positive dependence at more reductive potentials (high η).
These trends can be explained by a mechanism that invokes two key features: (1) voltage-dependent bicarbonate poisoning and (2) voltage-dependent mixed control between bicarbonate-dependent and bicarbonate-independent reaction pathways (Figure 2G). Such a model has previously been reported to quantitatively describe the kinetic behavior at CoPc16 and can qualitatively capture observed trends for CoPc and CoTPP in this work, where the solid curves in Figure 2B, 2C, 2E, and 2F represent model fits.16
The first feature, bicarbonate poisoning, is modeled with a positive electrosorption valency, meaning that the negative charge on bicarbonate becomes more repelled from (or less attracted to) the surface as the electrode becomes more negatively (or less positively) charged during reductive polarization. Thus, at more reductive potentials (high η values), the local poisoning equilibrium shifts away from θHCO3, which decreases the extent of bicarbonate inhibition.
The second feature, mixed control, is modeled as two concurrent reaction pathways involving a concerted proton–electron transfer (CPET) and sequential proton–electron transfer (SPET). At low η, the CPET with water as a proton donor is dominant, and at high η, the SPET with bicarbonate and water as proton donors becomes more dominant. Thus, at high η, bicarbonate becomes a more kinetically important proton donor, which paired with a decrease in bicarbonate inhibition (vide supra), leads to a positive apparent order in bicarbonate.
We note that the proposed mechanism can also account for apparent attenuation in CO2 order dependence at high η, which has previously been observed at CoPc.16 This phenomenon occurs because of coverage effects, where high η leads to accumulation of the surface species θCOO–. However, for the conditions tested in this work, the apparent orders in CO2 for CoPc and CoTPP remain within error of 1.
2.5. Mechanistic Features Proposed for FePc
For FePc, nonideality is apparent in both the bicarbonate and CO2 dependencies. For the bicarbonate dependence, inhibition increases at more reductive potentials (high η), and for the CO2 dependence, the apparent CO2 order becomes attenuated at less reductive potentials (low η).
These trends can be explained by a mechanism that invokes two key features: (1) voltage-dependent bicarbonate poisoning involving the interplay of two elementary steps and (2) voltage-dependent mixed control between CO desorption and CO2 activation (Figure 3D). One major difference between the model for FePc and that for CoPc and CoTPP is that the most abundant reactive intermediate (MARI), or resting state, of the catalyst is now a CO-adsorbed catalyst site (θCO+) rather than an empty catalyst site (θO). This agrees with the chemical intuition that Fe tends to bind CO more tightly than Co does. The model qualitatively captures observed trends for FePc, where the solid curves in Figure 3B and 3C represent model fits.
The first feature, bicarbonate poisoning, must still be modeled with a positive electrosorption valency, meaning that locally, the equilibrium between θHCO3 and θO disfavors θHCO3 at high η. While this local picture is in ostensible contrast to what is observed experimentally, the contradiction is resolved by accounting for the fact that MARI is a CO-adsorbed site (θCO+). Thus, the coverage of θHCO3 is governed by the interplay between two elementary steps: the local bicarbonate adsorption mentioned above and the voltage-dependent desorption of CO to convert θCO+ to θO. Since the latter step is favored by high η and has a stronger voltage dependence, the overall effect is to increase the coverage of θHCO3 and observe stronger bicarbonate inhibition at high η.
The second feature, mixed control, is modeled as two concurrent pathways, where the voltage-independent desorption of CO is rate limiting at low η and mixed control between voltage-dependent CO desorption and CO2 adsorption are rate limiting at high η. Thus, at low η, because CO desorption is CO2-independent, the rate shows an attenuated dependence in CO2. Then, at high η, CO desorption becomes favored by reductive polarization, and CO2 activation becomes more rate limiting, causing the apparent dependence in CO2 to become first order.
This model also provides intuition for why the CO2 order dependence curves at low and high η intersect, which, alternatively stated, means that at low CO2 partial pressures, voltage inhibits the reaction. In the model, voltage directs reaction flux toward the accumulation of θO, which is an unproductive pathway at low CO2 partial pressures because the subsequent CO2-dependent elementary step that converts θO to θCO2 is sluggish. However, at higher CO2 partial pressures, the CO2-dependent pathway becomes faster, which causes the pathway to become productive and alleviates the inhibitory effect of voltage.
3. Conclusions
In this work, we report a robotic system that enables flexible and automated data collection for heterogeneous electrocatalysis. The resulting experimental workflow was more time- and labor-efficient, enabling faster and more facile rate data collection over a wide range of reaction conditions for several common CO2RR electrocatalysts. Analysis of these data using cohesive kinetic analysis highlighted hallmarks of mechanistic complexity not interpretable via common Tafel or order dependence analyses.
Specifically, across immobilized CoPc, CoTPP, and FePc catalysts, we report CO2 and bicarbonate order dependences that are nonlinear and/or change depending on the applied voltage. We describe how these behaviors can be explained by models that invoke bicarbonate poisoning and a potential-dependent mixed control. We note that beyond an initial discussion of these phenomena at CoPc,16 such kinetic behaviors are not discussed for CO2RR at immobilized metal tetrapyrroles. In the existing mechanistic debates surrounding this class of electrocatalysts, only steps such as CO2 adsorption accompanied30,32,33 or preceded34,35 by electron transfer, CO2 adsorption accompanied by concerted proton electron transfer,36,37,41 CO* formation,50 CO* desorption,39,40,46,51 and discrete metal redox events38−40,52 are among those typically asserted and/or debated. Although the likelihood of voltage-dependent mechanistic changes is acknowledged,34,41,42 it is seldom experimentally interrogated. Our work highlights that kinetic phenomena such as electrolyte poisoning, coverage effects, and mixed control are important considerations for the CO2RR at metal tetrapyrroles. Additionally, our work suggests that ostensibly opposing kinetic observations in the literature might be reconciled by differences in operating conditions, where kinetic measurements reported in a limited range of operating conditions only “see” one part of a much more complicated whole.
Finally, future work extending this initial robotic concept could explore incorporation of a membrane to separate the anode and cathode, as well as alternative electrolyte and gas flow configurations. Additionally, software development to automatically enumerate mechanisms and suggest the most informative experimental conditions could further standardize and accelerate the workflow. We anticipate that, with further engineering efforts, extensions of this automated system may not only improve the throughput and reduce the labor requirements of data collection but also improve reliability and consistency of the data itself. Our work provides an initial demonstration of how the convergence of automation with cohesive kinetic analysis provides an informative experimental strategy for dissecting electrocatalytic reaction complexity.
4. Methods
4.1. Electrode and Electrolyte Preparation
CoPc (Sigma-Aldrich) catalysts were dissolved in N,N-dimethylformamide (DMF) and drop-casted onto calcined Toray060 carbon paper (Fuel Cell Store) electrodes at loadings of 5.98 × 10–11 mol/cm2. CoTPP (Frontier Scientific) and FePc (Sigma-Aldrich) catalysts were dissolved in carbon black-containing DMF solutions and drop-casted at loadings of 5.11 × 10–10 and 8.04 × 10–10 mol/cm2, respectively. Carbon black was used for the less active catalysts to help increase catalyst loading while also avoiding aggregation.53 Thus, all catalysts were tested at low loadings where aggregation was not expected to be an issue.54
Electrolytes were made by preparing stock solutions of 0.5 mol/L Na2CO3 (Sigma-Aldrich) and 1 mol/L NaClO4 (Sigma-Aldrich). Upon preparation, these stock solutions were bubbled overnight with CO2 and stored for further use. Electrolyte solutions with overall 1 mol/L ionic strength were then prepared by mixing the stock solutions in appropriate volumetric ratios to achieve desired bicarbonate concentrations.
4.2. Robot Operation
Typically, electrolyses were queued in batches of 10, with all 10 electrolyses using the same electrolyte composition. Working electrodes were loaded into cell pans, and the electrolyte reservoir was filled with electrolyte and bubbled with CO2. The Pt wire counter electrode resided in the cell body and was not often removed. The leak-free Ag/AgCl reference electrode (Innovative Instruments) was calibrated against a saturated calomel electrode every day and was swapped out every 1–3 days. After queueing and running the 10 electrolyses, the cell body washing sequence would be executed and the cell pans would be washed manually.
4.3. Data Handling
All data files, including electrolyses and reference calibrations, were automatically uploaded to an SQL database. A Python script was used to access files with relevant run conditions. It then output average current, partial CO current, partial H2 current, and operating conditions to an Excel document.
Throughout this work, no unexpected or unusually high safety hazards were encountered.
Acknowledgments
This work was supported by the National Science Foundation under Grant 2204757. K.M. acknowledges funding from the Sloan Foundation. J.S.Z. acknowledges a fellowship from the MIT Energy Initiative, supported by Chevron, as well as an MIT Mathworks fellowship. V.P. acknowledges funding from Fonterra Co-operative Group Limited. This manuscript is adapted from the thesis of J.S.Z.
Glossary
ABBREVIATIONS
- CO2RR
carbon dioxide reduction reaction
- CoPc
cobalt phthalocyanine
- CoTPP
cobalt tetraphenyl porphyrin
- FePc
iron phthalocyanine
- FE
Faradaic efficiency
- HER
hydrogen evolution reaction
- ORR
oxygen reduction reaction
- TOF
turnover frequency
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.3c01295.
Author Contributions
# J.S.Z. and V.P. contributed equally. J.S.Z. and V.P. conceptualized the paper. V.P. designed the robotic device. V.P., J.S.Z., J.H.M., and A.H.L. constructed the device. A.M.L. and J.S.Z. coded the software interface. J.S.Z. and G.Y.C. carried out the electrochemical investigation. J.S.Z. wrote the original draft of the manuscript, and V.P., M.A.Y., I.W.H., and K.M. reviewed and edited its contents. K.M. supervised the work. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Shinagawa T.; Garcia-Esparza A. T.; Takanabe K.. Insight on Tafel slopes from a microkinetic analysis of aqueous electrocatalysis for energy conversion. Sci. Rep. 2015, 5, 10.1038/srep13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynggaard H.; Andreasen A.; Stegelmann C.; Stoltze P. Analysis of simple kinetic models in heterogeneous catalysis. Prog. Surf. Sci. 2004, 77, 71–137. 10.1016/j.progsurf.2004.09.001. [DOI] [Google Scholar]
- Gennero De; Chialvo M. R.; Chialvo A. C. Kinetics of hydrogen evolution reaction with Frumkin adsorption: Re-examination of the Volmer-Heyrovsky and Volmer-Tafel routes. Electrochim. Acta 1998, 44, 841–851. 10.1016/S0013-4686(98)00233-3. [DOI] [Google Scholar]
- Holewinski A.; Linic S. Elementary Mechanisms in Electrocatalysis: Revisiting the ORR Tafel Slope. J. Electrochem. Soc. 2012, 159, H864–H870. 10.1149/2.022211jes. [DOI] [Google Scholar]
- Akinola J.; Barth I.; Goldsmith B. R.; Singh N. Adsorption Energies of Oxygenated Aromatics and Organics on Rhodium and Platinum in Aqueous Phase. ACS Catal. 2020, 10, 4929–4941. 10.1021/acscatal.0c00803. [DOI] [Google Scholar]
- Schultze J. W.; Vetter K. J. Experimental determination and interpretation of the electrosorption valency γ. J. Electroanal. Chem. 1973, 44, 63–81. 10.1016/S0022-0728(73)80515-7. [DOI] [Google Scholar]
- Herrero E.; Mostany J.; Feliu J. M.; Lipkowski J. Thermodynamic studies of anion adsorption at the Pt(111) electrode surface in sulfuric acid solutions. J. Electroanal. Chem. 2002, 534, 79–89. 10.1016/S0022-0728(02)01101-4. [DOI] [Google Scholar]
- Foresti M. L.; Innocenti M.; Hamelin A. Adsorption Behavior of n-Hexanol on Ag(lll) from Aqueous 0.05 M KCIO4. Langmuir 1995, 11, 498–505. 10.1021/la00002a023. [DOI] [Google Scholar]
- Stewart W. E.; Caracotsios M.. Computer-Aided Modeling of Reactive Systems; Wiley-Interscience, 2008. [Google Scholar]
- Miller J. H.; Bui L.; Bhan A. Pathways, mechanisms, and kinetics: A strategy to examine byproduct selectivity in partial oxidation catalytic transformations on reducible oxides. Reaction Chemistry and Engineering 2019, 4, 784–805. 10.1039/C8RE00285A. [DOI] [Google Scholar]
- Miller J. H.; Bhan A. Kinetic Modeling of Acrolein Oxidation Over a Promoted Mo–V Oxide Catalyst. ChemCatChem. 2018, 10, 5511–5522. 10.1002/cctc.201801029. [DOI] [Google Scholar]
- Herrmann S.; Iglesia E. Selective conversion of acetone to isobutene and acetic acid on aluminosilicates: Kinetic coupling between acid-catalyzed and radical-mediated pathways. J. Catal. 2018, 360, 66–80. 10.1016/j.jcat.2018.01.032. [DOI] [Google Scholar]
- Che-Galicia G.; Quintana-Solórzano R.; Ruiz-Martínez R. S.; Valente J. S.; Castillo-Araiza C. O. Kinetic modeling of the oxidative dehydrogenation of ethane to ethylene over a MoVTeNbO catalytic system. Chemical Engineering Journal 2014, 252, 75–88. 10.1016/j.cej.2014.04.042. [DOI] [Google Scholar]
- Lee C. W.; et al. New challenges of electrokinetic studies in investigating the reaction mechanism of electrochemical CO2 reduction. J. Mater. Chem. A Mater. 2018, 6, 14043–14057. 10.1039/C8TA03480J. [DOI] [Google Scholar]
- Limaye A. M.; Zeng J. S.; Willard A. P.; Manthiram K. Bayesian data analysis reveals no preference for cardinal Tafel slopes in CO2 reduction electrocatalysis. Nature Communications 2021 12:1 2021, 12, 1–10. 10.1038/s41467-021-20924-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng J. S.; Corbin N.; Williams K.; Manthiram K. Kinetic Analysis on the Role of Bicarbonate in Carbon Dioxide Electroreduction at Immobilized Cobalt Phthalocyanine. ACS Catal. 2020, 10, 4326–4336. 10.1021/acscatal.9b05272. [DOI] [Google Scholar]
- Rae K.; et al. Beyond Tafel Analysis for Electrochemical CO2 Reduction. (2022) 10.26434/CHEMRXIV-2022-9RX0M. [DOI]
- Woodhouse M.; Parkinson B. A. Combinatorial approaches for the identification and optimization of oxide semiconductors for efficient solar photoelectrolysis. Chem. Soc. Rev. 2009, 38, 197–210. 10.1039/B719545C. [DOI] [PubMed] [Google Scholar]
- Muster T. H.; et al. A review of high throughput and combinatorial electrochemistry. Electrochim. Acta 2011, 56, 9679–9699. 10.1016/j.electacta.2011.09.003. [DOI] [Google Scholar]
- Guevarra D.; et al. High Throughput Discovery of Complex Metal Oxide Electrocatalysts for the Oxygen Reduction Reaction. Electrocatalysis 2022, 13, 1–10. 10.1007/s12678-021-00694-3. [DOI] [Google Scholar]
- Lai Z.; et al. An Automated Test Platform for High-Throughput Micro-Electrochemical Characterization of Metallic Materials and Its Application on a Fe–Cr–Ni Combinatorial Materials Chip. J. Electrochem. Soc. 2021, 168, 091501 10.1149/1945-7111/ac24bc. [DOI] [Google Scholar]
- Gregoire J. M.; Xiang C.; Liu X.; Marcin M.; Jin J. Scanning droplet cell for high throughput electrochemical and photoelectrochemical measurements. Rev. Sci. Instrum. 2013, 84, 024102 10.1063/1.4790419. [DOI] [PubMed] [Google Scholar]
- Neyerlin K. C.; Bugosh G.; Forgie R.; Liu Z.; Strasser P. Combinatorial Study of High-Surface-Area Binary and Ternary Electrocatalysts for the Oxygen Evolution Reaction. J. Electrochem. Soc. 2009, 156, B363. 10.1149/1.3049820. [DOI] [Google Scholar]
- Kolen M.; et al. Combinatorial Screening of Bimetallic Electrocatalysts for Nitrogen Reduction to Ammonia Using a High-Throughput Gas Diffusion Electrode Cell Design. J. Electrochem. Soc. 2022, 169, 124506 10.1149/1945-7111/aca6a7. [DOI] [Google Scholar]
- Rein J.; et al. Unlocking the Potential of High-Throughput Experimentation for Electrochemistry with a Standardized Microscale Reactor. ACS Cent Sci. 2021, 7, 1347–1355. 10.1021/acscentsci.1c00328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M.; et al. Fast Screening for Copper-Based Bimetallic Electrocatalysts: Efficient Electrocatalytic Reduction of CO2 to C2+ Products on Magnesium-Modified Copper. Angew. Chem. 2022, 134, e202213423 10.1002/ange.202213423. [DOI] [PubMed] [Google Scholar]
- Jones R. J. R.; Wang Y.; Lai Y.; Shinde A.; Gregoire J. M. Reactor design and integration with product detection to accelerate screening of electrocatalysts for carbon dioxide reduction. Rev. Sci. Instrum. 2018, 89, 124102 10.1063/1.5049704. [DOI] [PubMed] [Google Scholar]
- Lai Y.; et al. Breaking Scaling Relationships in CO2Reduction on Copper Alloys with Organic Additives. ACS Cent Sci. 2021, 7, 1756–1762. 10.1021/acscentsci.1c00860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y.; et al. Molecular Coatings Improve the Selectivity and Durability of CO2 Reduction Chalcogenide Photocathodes. ACS Energy Lett. 2022, 7, 1195–1201. 10.1021/acsenergylett.1c02762. [DOI] [Google Scholar]
- Choi J.; et al. Steric Modification of a Cobalt Phthalocyanine/Graphene Catalyst to Give Enhanced and Stable Electrochemical CO 2 Reduction to CO. ACS Energy Lett. 2019, 4, 666–672. 10.1021/acsenergylett.8b02355. [DOI] [Google Scholar]
- Kortlever R.; Shen J.; Schouten K. J. P.; Calle-Vallejo F.; Koper M. T. M. Catalysts and Reaction Pathways for the Electrochemical Reduction of Carbon Dioxide. J. Phys. Chem. Lett. 2015, 6, 4073–4082. 10.1021/acs.jpclett.5b01559. [DOI] [PubMed] [Google Scholar]
- Shen J.; et al. Electrocatalytic reduction of carbon dioxide to carbon monoxide and methane at an immobilized cobalt protoporphyrin. Nat. Commun. 2015, 6, 10.1038/ncomms9177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M.; et al. Covalently Grafting Cobalt Porphyrin onto Carbon Nanotubes for Efficient CO 2 Electroreduction. Angewandte Chemie - International Edition 2019, 58, 6595–6599. 10.1002/anie.201900499. [DOI] [PubMed] [Google Scholar]
- Liu Y.; McCrory C. C. L.. Modulating the mechanism of electrocatalytic CO2 reduction by cobalt phthalocyanine through polymer coordination and encapsulation. Nat. Commun. 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman P. M.; McCrory C. C. L.; Rivera Cruz K. E.; Liu Y.; Soucy T. L. Increasing the CO2 reduction activity of cobalt phthalocyanine by modulating the σ-donor strength of axially coordinating ligands. ACS Catal. 2021, 11, 13203–13216. 10.1021/acscatal.1c02379. [DOI] [Google Scholar]
- Miyamoto K.; Asahi R. Water Facilitated Electrochemical Reduction of CO 2 on Cobalt-Porphyrin Catalysts. J. Phys. Chem. C 2019, 123, 9944–9948. 10.1021/acs.jpcc.9b01195. [DOI] [Google Scholar]
- Zhu M.; et al. Electronic Tuning of Cobalt Porphyrins Immobilized on Nitrogen-Doped Graphene for CO2 Reduction. ACS Appl. Energy Mater. 2019, 2, 2435–2440. 10.1021/acsaem.9b00368. [DOI] [Google Scholar]
- Morlanés N.; Takanabe K.; Rodionov V. Simultaneous Reduction of CO2 and Splitting of H2O by a Single Immobilized Cobalt Phthalocyanine Electrocatalyst. ACS Catal. 2016, 6, 3092–3095. 10.1021/acscatal.6b00543. [DOI] [Google Scholar]
- Wu Y.; Liang Y.; Wang H. Heterogeneous Molecular Catalysts of Metal Phthalocyanines for Electrochemical CO2Reduction Reactions. Acc. Chem. Res. 2021, 54, 3149–3159. 10.1021/acs.accounts.1c00200. [DOI] [PubMed] [Google Scholar]
- Zhang X.; et al. Highly selective and active CO2reduction electrocatalysts based on cobalt phthalocyanine/carbon nanotube hybrid structures. Nat. Commun. 2017, 8, 1–8. 10.1038/ncomms14675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Zhu M.; Li J.; Xu J.; Han Y. F. Structure-Activity Relationship of the Polymerized Cobalt Phthalocyanines for Electrocatalytic Carbon Dioxide Reduction. J. Phys. Chem. C 2020, 124, 16501–16507. 10.1021/acs.jpcc.0c04741. [DOI] [Google Scholar]
- Göttle A. J.; Koper M. T. M. Proton-coupled electron transfer in the electrocatalysis of CO2 reduction: prediction of sequential vs. concerted pathways using DFT. Chem. Sci. 2017, 8, 458–465. 10.1039/C6SC02984A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark E. L.; et al. Standards and Protocols for Data Acquisition and Reporting for Studies of the Electrochemical Reduction of Carbon Dioxide. ACS Catal. 2018, 8, 6560–6570. 10.1021/acscatal.8b01340. [DOI] [Google Scholar]
- Watkins N. B.; et al. Hydrodynamics Change Tafel Slopes in Electrochemical CO2 Reduction on Copper. ACS Energy Lett. 2023, 8, 2185–2192. 10.1021/acsenergylett.3c00442. [DOI] [Google Scholar]
- Soucy T. L.; et al. The Influence of pH and Electrolyte Concentration on Fractional Protonation and CO2 Reduction Activity in Polymer-Encapsulated Cobalt Phthalocyanine. J. Phys. Chem. C 2023, 127, 14041. 10.1021/acs.jpcc.3c01490. [DOI] [Google Scholar]
- Zhang X.; et al. Molecular engineering of dispersed nickel phthalocyanines on carbon nanotubes for selective CO2 reduction. Nat. Energy 2020, 5, 684–692. 10.1038/s41560-020-0667-9. [DOI] [Google Scholar]
- Lin S.; et al. Covalent organic frameworks comprising cobalt porphyrins for catalytic CO2 reduction in water. Science (1979) 2015, 349, 1208–1213. 10.1126/science.aac8343. [DOI] [PubMed] [Google Scholar]
- Shen J.; Kolb M. J.; Göttle A. J.; Koper M. T. M. DFT Study on the Mechanism of the Electrochemical Reduction of CO 2 Catalyzed by Cobalt Porphyrins. J. Phys. Chem. C 2016, 120, 15714–15721. 10.1021/acs.jpcc.5b10763. [DOI] [Google Scholar]
- Leung K.; Nielsen I. M. B.; Sai N.; Medforth C.; Shelnutt J. A. Cobalt-porphyrin catalyzed electrochemical reduction of carbon dioxide in water. 2. Mechanism from first principles. J. Phys. Chem. A 2010, 114, 10174–10184. 10.1021/jp1012335. [DOI] [PubMed] [Google Scholar]
- Abe T.; et al. Electrochemical CO2 reduction catalyzed by cobalt octacyanophthalocyanine and its mechanism. J. Porphyr Phthalocyanines 1997, 1, 315–321. . [DOI] [Google Scholar]
- Sinha S.; Zhang R.; Warren J. J. Low Overpotential CO2Activation by a Graphite-Adsorbed Cobalt Porphyrin. ACS Catal. 2020, 10, 12284–12291. 10.1021/acscatal.0c01367. [DOI] [Google Scholar]
- Kaminsky C. J.; Weng S.; Wright J.; Surendranath Y. Adsorbed cobalt porphyrins act like metal surfaces in electrocatalysis. Nature Catalysis 2022 5:5 2022, 5, 430–442. 10.1038/s41929-022-00791-6. [DOI] [Google Scholar]
- Zhu M.; et al. Inductive and electrostatic effects on cobalt porphyrins for heterogeneous electrocatalytic carbon dioxide reduction. Catal. Sci. Technol. 2019, 9, 974–980. 10.1039/C9CY00102F. [DOI] [Google Scholar]
- Zhu M.; Ye R.; Jin K.; Lazouski N.; Manthiram K. Elucidating the Reactivity and Mechanism of CO 2 Electroreduction at Highly Dispersed Cobalt Phthalocyanine. ACS Energy Lett. 2018, 3, 1381–1386. 10.1021/acsenergylett.8b00519. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.