

Abstract
Pyridoxal 5′-phosphate (PLP), the active cofactor form of vitamin B6 is required by over 160 PLP-dependent (vitamin B6) enzymes serving diverse biological roles, such as carbohydrates, amino acids, hemes, and neurotransmitters metabolism. Three key enzymes, pyridoxal kinase (PL kinase), pyridoxine 5′-phosphate oxidase (PNPO), and phosphatases metabolize and supply PLP to PLP-dependent enzymes through the salvage pathway. In born errors in the salvage enzymes are known to cause inadequate levels of PLP in the cell, particularly in neuronal cells. The resulting PLP deficiency is known to cause or implicated in several pathologies, most notably seizures. One such disorder, PNPO-dependent neonatal epileptic encephalopathy (NEE) results from natural mutations in PNPO and leads to null or reduced enzymatic activity. NEE does not respond to conventional antiepileptic drugs but may respond to treatment with the B6 vitamers PLP and/or pyridoxine (PN). In born errors that lead to PLP deficiency in cells have also been reported in PL kinase, however, to date none has been associated with epilepsy or seizure. One such pathology is polyneuropathy that responds to PLP therapy. Phosphatase deficiency or hypophosphatasia disorder due to pathogenic mutations in alkaline phosphatase is known to cause seizures that respond to PN therapy. In this article, we review the biochemical features of in born errors pertaining to the salvage enzyme’s deficiency that leads to NEE and other pathologies. We also present perspective on vitamin B6 treatment for these disorders, along with attempts to develop zebrafish model to study the NEE syndrome in vivo.
Keywords: Seizures, Neonatal epileptic encephalopathy, Pyridoxal 5′-phosphate, Pyridoxal kinase, Pyridoxine, Pyridoxine 5′-phosphate oxidase, Phosphatase, Salvage enzyme, Vitamin B6, In born errors, PLP-Dependent enzymes, Hypophosphatasia, Zebrafish
1. Introduction
Humans, unlike bacteria and plants, cannot synthesize pyridoxal 5’-phosphate (PLP) de novo and must rely on a salvage pathway that involve pyridoxal kinase (PL kinase), Pyridoxine 50-phosphate oxidase (PNPO), and phosphatases in metabolizing and supplying adequate amount of PLP to dozens of vitamin B6 or PLP-dependent enzymes [1]. The B6 enzymes serve vital roles in pathways involving glucose, lipid and amino acid metabolism, heme, DNA/RNA and many neurotransmitter syntheses. Not surprisingly, in born errors in the salvage pathway are known to cause or implicated in several pathologies, most notably seizures [2–19]. Neonatal epileptic encephalopathy (NEE) is one of the most wellstudied salvage enzyme in-born error related seizures [3,4,6,8–10,12–19]. Clinically, the phenotypes observed in NEE patients may include fetal distress, anemia, acidosis, asphyxia and hypoglycemia [6,10,12,20]. Early treatment of patients is critical to prevent health deterioration and death [10,15]. Surviving children are dependent on pyridoxine (PN) or PLP to control the disease for life, and in most instances may be developmentally handicapped [3,4,6,8–10,12–16,21]. There are known cases, however, where B6-dependent NEE result in mild to moderate developmental delay [15].
Vitamin B6-dependent NEE or other B6-dependent pathologies deriving from a deficiency of cellular PLP can be caused by inborn errors in the B6 salvage enzymes PL kinase, PNPO, or phosphatases [15,22]. Pathogenic mutations in PNPO has been implicated in several NEE that are referred to as PNPO deficiency-related NEE or PNPO-dependent NEE (PNPO-NEE), while defects in PL kinase are known to cause polyneuropathy but not seizures [23]. Alkaline phosphatase deficiency or hypophosphatasia disorder affects PLP import into the brain and has been implicated in seizure disorders [24–27]. Apart from the three salvage enzymes, inborn errors in other pathways that affect B6 homeostasis have also been implicated in PLP deficiency, resulting in pathologies, including seizures. Mutations in PROSC, a PLP binding protein that is suggested to be involved in homeostasis regulation of PLP and transport of PLP to activate apo-B6 enzyme, have been associated with PLP deficiency and seizures [28]. Pathogenic mutations in ALDH7A1 (α-aminoadipic semialdehyde dehydrogenase; also known as antiquitin), an aldehyde dehydrogenase enzyme leads to accumulation of intermediates that react with PLP, reducing its bioavailability and causing a so-called PN-dependent epilepsy [5,9,29]. Patients with such an array of disorders are able to control their seizures with either PN and/or PLP, usually with lifelong dependency on these B6 vitamers. A recent review by Wilson et al. describes the biochemical and clinical aspects of B6- responsive seizures and their treatments [30]. Beside seizures, other multifactorial neurological pathologies, such as Alzheimer’s, autism, Parkinson’s and schizophrenia etc. have also been implicated with PLP deficiency [31–37].
Vitamin B6 naturally exists as PN, PM, and PL and their phosphorylated analogs, PNP, PMP and PLP, respectively, with PLP, and to a lesser extent PMP being the active cofactor form for B6 enzymes [1,38]. PLP, PNP or PMP is salvaged from food and PLP/PMP-dependent enzymes by PL kinase, PNPO and phosphatases [1,38]. Once synthesized, PLP is utilized for their activity by several B6 enzymes. We and others have been studying cellular PLP homeostasis with respect to PL Kinase and PNPO [1,15,16,22,38,39]. We previously published a review article on how the molecular mechanisms of inborn errors in PNPO affect the cellular PLP levels leading to NEE [15]. Here we revisit the molecular defects of the three salvage enzymes associated with seizures and other pathologies, along with zebrafish model to study the syndrome in vivo, as well as a perspective on vitamin B6 treatment for some of the disorders.
2. B6 vitamers and PLP metabolism
2.1. Chemical structures of B6 vitamers
The chemical structures of all six B6 vitamers, including the primary forms PL, PN, and PM, and their respective phosphorylated derivatives PLP, PNP and PMP are shown in Fig. 1. All six molecules share the 2-methyl-3-hydroxy pyridine moiety and the structural differences among them are due to the substituents on C4 and C5 carbon residues. PLP, the most versatile organic cofactor in biology is utilized as a cofactor by several PLP-dependent enzymes that are involved in amino acid metabolism, neurotransmitter biosynthesis, lipid metabolism, carbohydrate breakdown, heme synthesis, nucleic acid synthesis, among others [40,41].
Fig. 1.
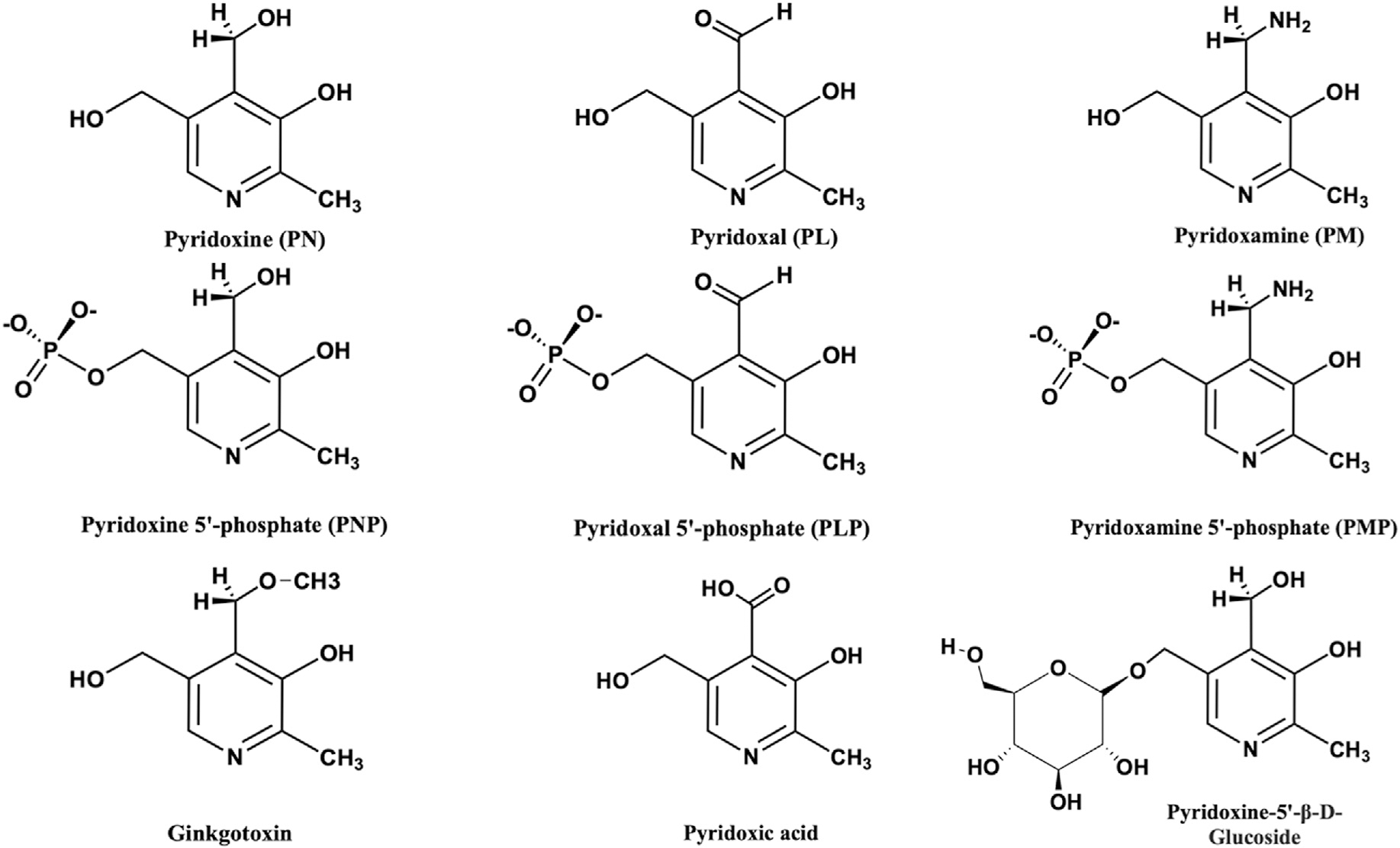
Chemical structure of B6 vitamers and derivatives.
2.2. Salvage enzymes and the supply of cofactor PLP
While plants and microorganisms are capable of de novo PLP synthesis, mammals can only obtain PLP recycled from B6 vitamers ingested with food or from degraded B6 enzymes [1]. These include primary B6 vitamers in food sources [30], PLP and to a lesser extent, PMP that is associated to glycogen phosphorylase in animal derived food sources, and PN and PN-5′-beta-D-glucoside from plant sources (Fig. 1) [42–44]. The commercially available PN hydrochloride is usually the form that is used to fortify foods.
Unlike, the primary vitamers, PLP, PNP and PMP need to be dephosphorylated first into PL, PN and PM, respectively by intestinal phosphatase before absorption into the portal system, and then taken into the liver to be re-phosphorylated by ATP-dependent PL kinase to PMP, PNP and PLP respectively [30,45]. Following, FMN-dependent PNPO oxidizes PNP and PMP to PLP (Fig. 2). PLP is then released to the blood stream in association with albumin for distribution to various tissues. Unlike the primary B6 vitamers, PLP has to be dephosphorylated by phosphatases to gain entry into target cells, e.g. brain and then converted back to PLP by intracellular PL kinase [30,46,47]. Vitamin B6 deficiency is rare in humans on a normal diet. In fact humans require small amounts of vitamin B6 in the diet since the salvage pathway is very efficient in providing enough PLP for enzymatic activity. Nonetheless, a problem in the salvage pathway and/or delivering of PLP to newly formed apo-B6 enzymes, might end up in vitamin B6-associated pathogenesis. The human genes encoding PNPO and PL kinase are located on chromosomes 17q21.2 and 21q22.3, respectively. The expression of mRNA of all three enzymes of the salvage pathway is ubiquitous but is highly regulated at the level of transcription in a tissue-specific manner. While PL kinase (isoforms) is expressed in all tissues [48], the major sites of PNPO expression are liver, skeletal muscle and kidneys [49], with phosphatase expressed in the liver, kidney and bone [25]. PNPO is also present, to a lesser extent, in the brain, in particular in the cerebral cortex [49].
Fig. 2.
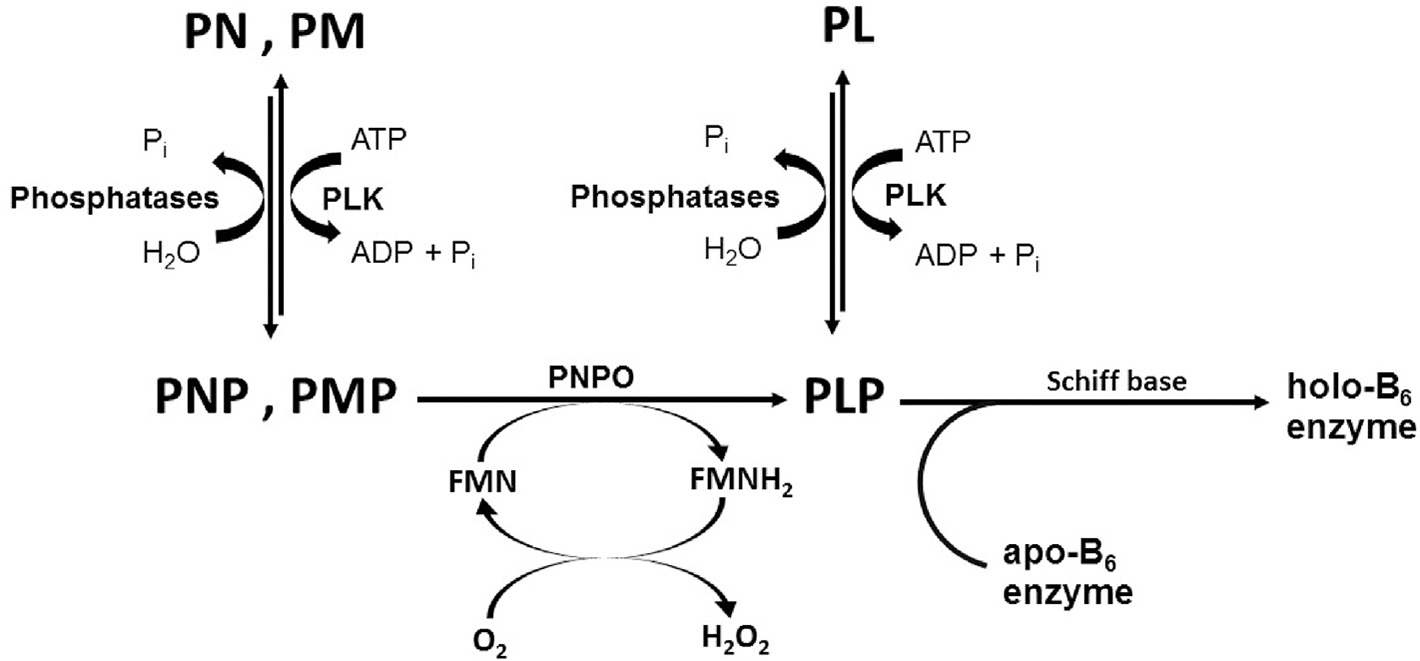
Vitamin B6 salvage pathway.
Due to the potential toxicity of PLP as a result of the reactive aldehyde, its concentration in the cell is maintained at a very low level [50]. High levels of PLP in the cell or excessive consumption has been linked to neurotoxic effect [51]. The concentration of free PLP in eukaryotic cells has been suggested to be as low as 1 μM [52]. PLP is regulated through a PLP feedback inhibition of PL kinase and PNPO [53–57], which has been demonstrated to be allosteric in nature [16,58]. A second regulatory mechanism is the dephosphorylation of PLP to the less toxic PL by phosphatases. Aldehyde oxidases and NAD-dependent dehydrogenases also keep the level of PLP low by converting cellular PL to 4-pyridoxic acid (Fig. 1) [59]. Although much work has been done in the field of PLP-dependent enzymes, we are still yet to fully understand how the reactive and potentially toxic PLP is made available to newly formed apo-B6 enzymes.
The low free PLP concentration in the cell is not enough for activating newly formed apo-B6 enzymes into the holo-forms [15,39,52,60], raising an intriguing question of how PLP is delivered to newly formed apo-B6 enzymes. In vitro studies show physical interactions between the salvage and B6 enzymes for productive transfer of free PLP [39,61], suggesting that in vivo in born errors in the salvage enzymes that disrupt such interactions could prevent B6 activation with dire consequences. PLP binds tightly to PNPO and PL kinase, which is suggested to be the basis of the feedback inhibition of both enzymes activities, and further demonstrated to be allosteric in nature [16,53–58]. The tightly bound PLP does not dissociate from the enzymes during size exclusion chromatography or dialysis [53–57]. Interestingly, in the presence of apo-B6 enzyme, the tightly bound PLP is transferred to activate apo-B6 enzyme into the active holo-enzyme, which has been proposed as a possible role in vivo for the tight-binding PLP, whether in wild-type or pathogenic variant [15,39,57,60–62]. From the foregoing, the importance of vitamin B6 in several cellular processes and in the onset of pathologies as a result of PLP deficiency should also be looked at from the perspective of whether the delivery mechanism is working appropriately.
2.3. Role of PLP-dependent enzymes in brain function
PLP-dependent enzymes are involved in the metabolism of several neurotransmitters, such as L-glutamate, gammaaminobutyric acid (GABA), histamine, dopamine, serotonin, epinephrine, norepinephrine, D-serine, and glycine, and thus their levels could be affected by PLP deficiency [22]. L-glutamate and GABA, are the main excitatory and inhibitory neurotransmitters in the central nervous system (CNS), respectively. The PLP-dependent enzyme, branched-chain aminotransferase catalyzes the synthesis of L-glutamate from α-ketoglutarate and branched-chain amino acids [63]. The PLP-dependent enzyme, L-glutamic acid decarboxylase (GAD) catalyzes the synthesis of GABA from L-glutamate [64]. Whereas the PLP-dependent enzyme, histidine decarboxylase catalyzes the formation of histamine from histidine [65]. Aromatic L-amino acid decarboxylase, also a PLP-dependent enzyme is involved in the formation of dopamine and serotonin [66], the former a precursor of epinephrine and norepinephrine. The PLP-dependent enzyme, serine racemase catalyzes the synthesis of D-serine from L-serine [37]. While glycine is the primary inhibitory neurotransmitter of the spinal cord and brainstem, it has excitatory effects in the cerebral cortex owing to its agonism for the glutamatergic N-methyl-D-aspartic acid (NMDA) receptor. The PLP-dependent enzyme, serine hydroxymethyltransferase (SHMT) catalyzes the synthesis of glycine from L-serine [67]. Several PLP-dependent enzymes are also known to be participate in the kynurenine oxidative pathway of tryptophan degradation that are implicated in physiological tuning of the CNS, as well the etiogenesis and progression of several human neurodegenerative disorders [68,69].
From the foregoing, it is clear that, insufficient PLP levels in the brain could lead to loss or decrease activity of several PLP-dependent enzymes that could impact on the availability of some or several neurotransmitters, potentially manifesting in seizures. The potential of PLP deficiency in the brain is likely to occur if the salvage enzyme activity is compromised in the brain since PLP must be synthesized within the brain tissues from the non-phosphorylated B6 vitamers. Even a modest decrease in PLP availability could have more dire consequences on brain function than would be expected because of the large number of enzymes that require PLP for activation. Some apo-B6 enzymes may compete better for PLP and thus function properly, while others may not, manifesting in various pathologies, as exemplified by DOPA decarboxylase with PLP deficiency [70].
3. PL kinase
PL kinase, a member of the ribokinase superfamily, serves as an essential salvage enzyme in the production of PLP, by catalyzing the ATP-dependent phosphorylation of primary B6 vitamers (Fig. 2). The enzyme has been purified and characterized from various sources [71–74]. Most eukaryotic organisms contain a single PL kinase, encoded by the pdxK gene. Under physiological conditions at pH 7.3, Mg2+ and K+ are the metal ions required for enzyme activity [55,73]. The Km for substrates, PL and MgATP in the presence of Kþ have been estimated to be lower than 10 μM and 25 μM, respectively, with kcat of about 85 min−1. The mechanism of phosphorylation by PL Kinase follows a random sequential substrate addition [53,73,74].
The crystal structure of PL kinase has been determined and show conserved fold across species [53,73–75]. PL kinase is a homodimer, with two active sites that are formed exclusively by each monomer (Fig. 3). The residues making interactions with the ATP and PL are highly conserved. The γ-phosphate of the bound ATP faces the 5′-hydroxymethyl group of primary substrate, PL. A conserved Asp at the active site makes a hydrogen-bond interaction with the 5′-hydroxymethyl group of PL, which has been shown to be the catalytic base [76]. Further, the ability of hPL Kinase to trap a PLP in the presence of the MgATP substrate suggested that PL kinase might use this mechanism to self-regulate its activity [53].
Fig. 3.
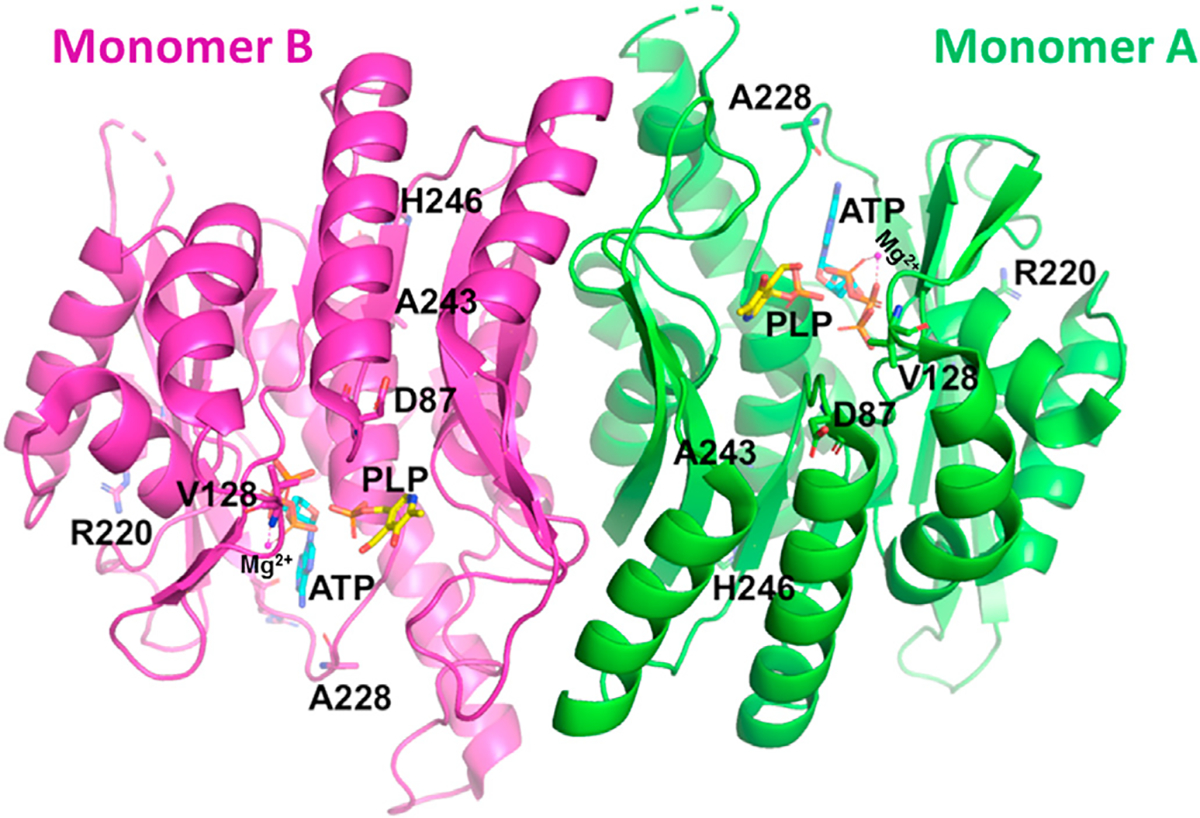
Crystal structure of human PL kinase in complex with ATP (cyan) and PLP (yellow) (PDB code: 3KEU). Distribution of the mutated residues (A228T, R220 N, D87H, V128I, H246Q and A243G) are shown in the structure.
3.1. Pathogenic mutations in PL kinase
To date epilepsy or seizures due to mutations in the gene pdxK that codes for PL Kinase has not been reported. The genetic basis of variation among human erythrocyte PL kinase activity was reported by Flanagan and Beutler in 2006 [77]. Erythrocyte PL kinase activity of African Americans was found to be lower than persons with European or Asian ancestry and was attributed to 8bp pdxK promoter insertion [77]. Flanagan and Beutler also suggested that tissue specific variations in the activity of human PL kinase enzyme could be attributed to polymorphisms in the PdxK gene promoter region [77].
A recent work by Chelban et al. reported two biallelic mutations (p.A228T and p.R220N; Fig. 3) in the pdxK gene of five individual from two unrelated families that led to polyneuropathy and optic atrophy [23]. PLP supplement at 50 mg/day led to clinical improvement after the first 3 months. Nonetheless, individuals become wheelchair-bound and blind if the disorder is left untreated [23]. The authors proposed a conformational rearrangement in the mutant PL kinase around the ATP binding pocket, which they suggested resulted in weak ATP binding with concomitant reduced PL kinase activity [23]. The absence of seizures in the patients was attributed to the fact that PL kinase activity is tissue specific under PLP deficiency, and neuronal PLP supply was not affected [23]. For example, rats with PLP deficiency showed rapid decrease in PL kinase in the peripheries (liver, muscle and plasma) while in the brain, PL kinase activity and B6 supply was maintained [78,79]. Consistently, patients with the defective PL kinase showed normal cognitive function.
In another recent study with Drosophila, mutations in the pdxK gene were shown to cause chromosome aberrations (CABs) as well as increased glucose content in larval hemolymph [80], that were rescued with wild type human pdxK. However, four PL kinase human variants: D87H, V128I, H246Q and A243G (Fig. 3) were unable to completely rescue CABs and glucose content elicited by the pdxK mutations. The four mutant proteins showed reduced catalytic activity and/or reduced affinity for PLP precursors. These results suggest that mutations in pdxK gene in human can result in diseases.
3.2. Inhibition of PL kinase activity by drugs/compounds
Human PL kinase activity is known to be inhibited by several drugs and compounds that are directed at different targets with a concomitant deficiency in PLP, causing unwanted disorders, e.g. epileptic convulsions, leg paralysis, seizures, and loss of consciousness [22,81]. PLP supplementation is known to correct these symptoms. Examples of such inhibitors are progabide, theophylline or ginkgotoxin (4′-O-methylpyridoxine; Fig. 1), the latter from Ginkgo biloba [22,82–84]. Ginkgo biloba is available over the counter and is widely used in the treatment of several conditions, e.g. asthma, irritable bladder, depression, dizziness, and tinnitus. In some instances ginkgotoxin poisoning has led to death, both in humans and cattle [85,86].
Ginkgotoxin is structurally similar to the three primary B6 vitamers (Fig. 1). Our group has elucidated the molecular basis of ginkgotoxin and theophylline PL kinase inhibitory activities [84]. The study showed hPL kinase binds ginkgotoxin or theophylline at the substrate site, in a similar fashion as the natural substrate PL, with Ki of 3 μM, and 50 μM, respectively [84]. Similarly, the phosphorylated product of ginkgotoxin also binds at the active site, and suggested to be an inhibitor of the kinase [84]. Consistently, normal or epileptic patients treated with theophylline show reduced serum concentration of PLP [87]. The active site of human PL kinase appears to be a sink for several compounds, particularly those that share structural characteristics with B6 vitamers. Such compounds if suspected to be neurotoxic, can be investigated for possible PL kinase inhibition, and recommendation of vitamin B6 supplementation would be needed.
4. PNP oxidase (PNPO)
The flavin mononucleotide (FMN)-dependent enzyme, PNPO is the second enzyme involved in the B6 salvage pathway in E. coli and mammalian cells, as well as involved the terminal step in the de novo vitamin B6 biosynthesis pathway in E. coli [1,54–56,88–91]. FMN is a phosphorylated form of vitamin B2 (riboflavin). Our group was the first to elucidate both the atomic structure and catalytic mechanism of PNPO, which involves direct hydride transfer from the 4′ hydroxyl group of PNP or the 4’ amino group of PMP to FMN to form PLP and FMNH2, respectively [91]. The enzyme exhibits a low catalytic rate constant of 0.2–0.8 sec−1 and Km for PNP or PMP is in the low micromolar range [88,89,91]. The product PLP is an inhibitor of the enzyme [16,54,56,58,88–91].
The crystal structure of PNPO from several species, including human have been determined, and show similar dimer structure with two FMN molecules bound non-covalently at the dimer interface (Fig. 4) [88,89,91–93]. The isoalloxazine ring, and the ribityl group of FMN make both hydrophobic and hydrogen-bond interactions with the two subunits. The FMN phosphate makes several hydrophilic interactions with amino acids that are mostlybasic e.g. Arg95, Arg141, Arg161, Ser175, Arg116, Lys117 and Arg229. The crystal structure of PNP or PLP with PNPO shows PNP/PLP to bind at the re-face of FMN, with the pyridine ring of PNP/PLP and the pyrazine and pyrimidine portions of the FMN isoalloxazine ring stack parallel to each other to make extensive van der Waals interactions. The atoms involved in the hydride transfer, C4’ of PLP and N5 of FMN are separated by about 3.4Å (Fig. 5A). The PLP or PNP phosphate moiety makes extensive salt bridges and hydrogen bond interactions to Lys100, Arg161, Arg225, Tyr157 and Ser165. The PLP hydroxyl, aldehyde and pyridine nitrogen make director water-mediated hydrogen bond interactions to His227, Glu77 and Trp206, respectively. These interactions help to stabilize FMN and substrate binding, and also ensure correct orientation of FMN and PNP for catalysis [88,89,91–93]. Several of the residues involved in both FMN and the substrate binding are conserved, and are interestingly involved in majority of the observed pathogenic mutations that cause NEE (Fig. 5).
Fig. 4.
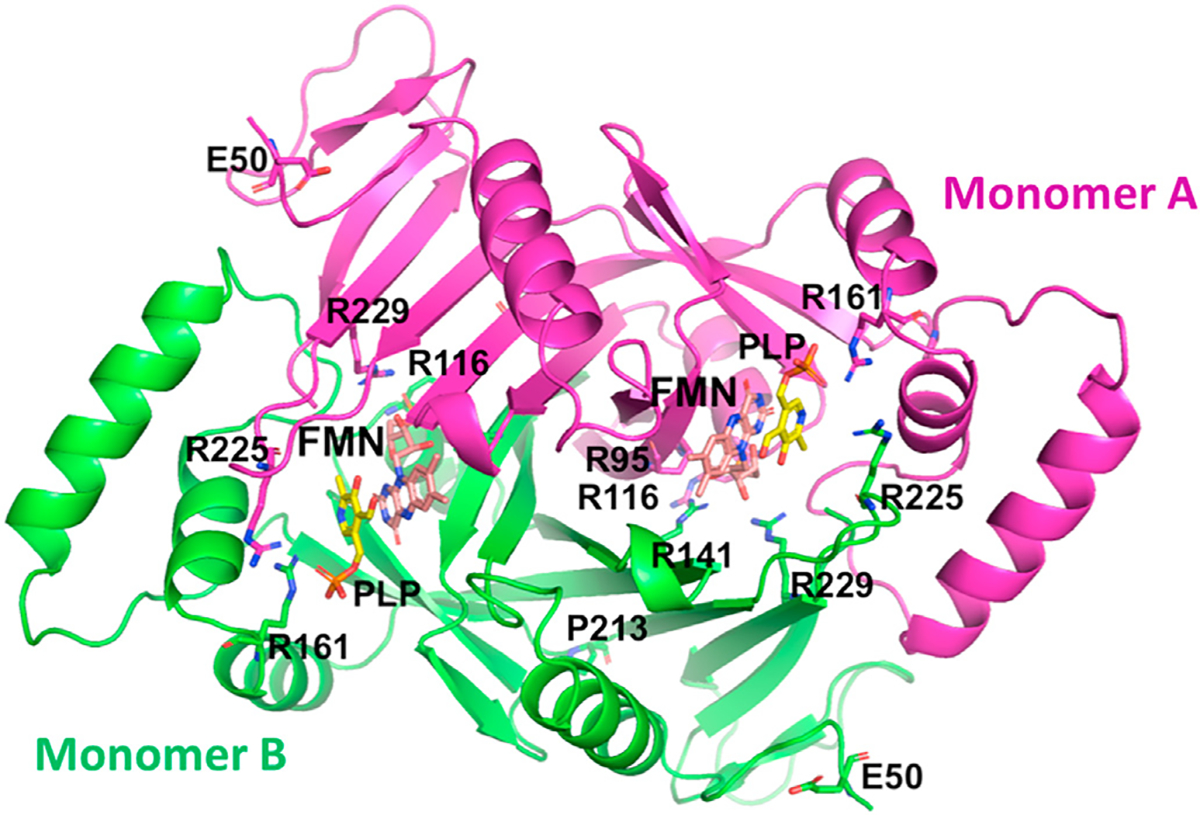
Crystal structure of human PNPO in complex with FMN (cyan) and PLP (yellow) (PDB code: 1NRG). Distribution of the NEE mutation residues in the crystal structure is also shown.
Fig. 5.
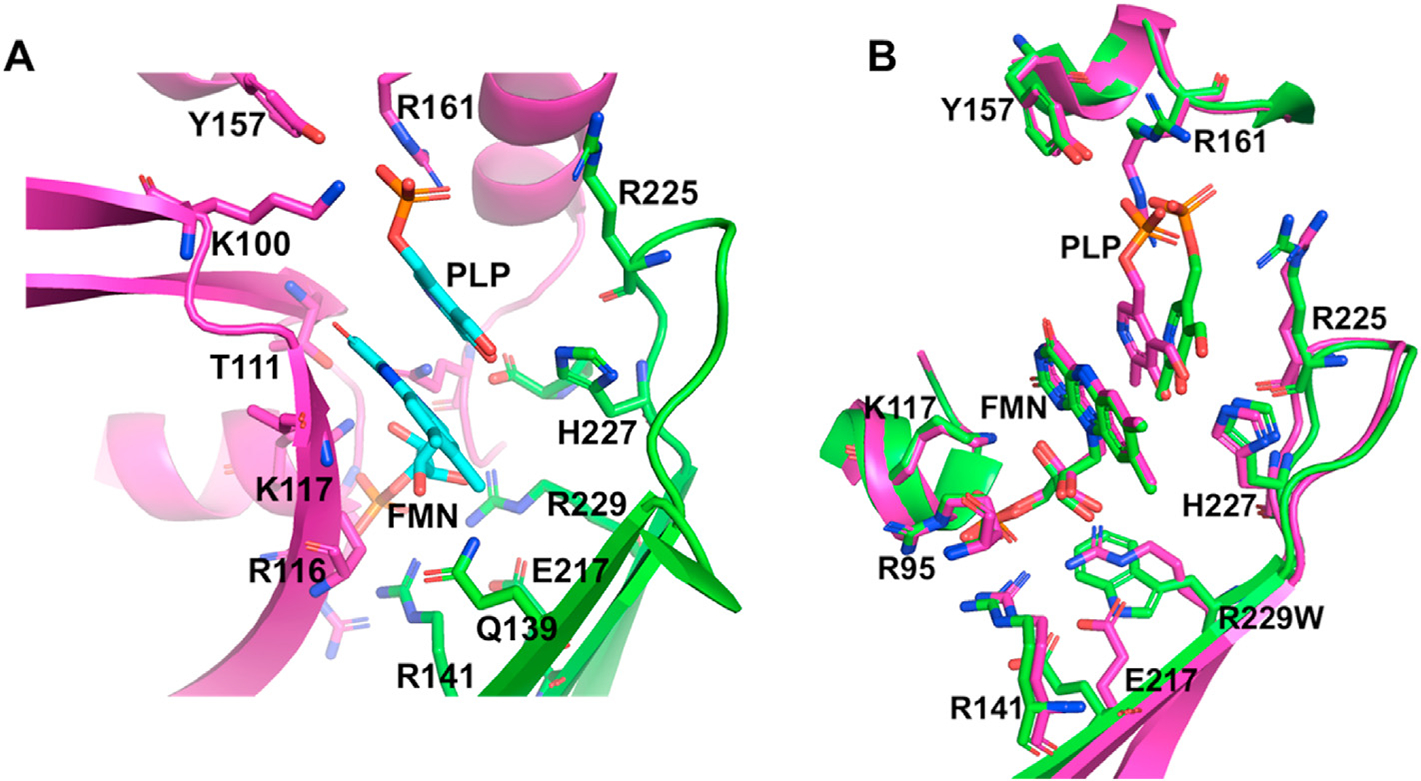
(A). The active site structure of wild-type human PNPO showing bound PLP at the re-face of FMN. (B). Superposition of the active site of the wild type (magenta) and R229W mutant (green) forms of PNPO.
4.1. PNPO deficiency-related NEE
PNPO deficiency-related NEE (PNPO-NEE) was first described by Kuo and Wang as a syndrome leading to severe seizures in new born children [94]. In 2005, mutations in the pdxH gene, which encodes hPNPO, were demonstrated to be a cause for this autosomal recessive condition by Mills et al. [10], and since then several other pathogenic mutations have been identified by other groups [3,4,6,8–10,12–19,95]. Several are missense mutations that involve active site arginine residues, such as R229W, R95C, and R225H. PNPO-NEE patients show distinct clinical features and neurological pathologies. For example, neonates are born premature with features that mimic organic academia, and with seizures [6,10,12,14,20,96]. Patient show elevated glycine and threonine, elevated L-DOPA and 3-methoxytyrosine, and decreased homovanillic acid and 5-hydroxyindoleacetic acid [6,10,12,14,20,96].
Infants with PNPO-NEE do not respond to treatment with antiepileptic drugs but may respond well to PLP, and in some instances to PN [3,4,6,8–10,12–16]. If untreated, the infants may die or have severe neuro-developmental impairments. Most of the mutations involve one amino acid and is located at the active site, impairing enzymes normal catalytic function. The resulting enzyme cannot effectively catalyze PNP or PMP to the product PLP, disrupting the function of many other proteins and enzymes that depend on PLP for activity. The typical dose of PLP to treat NEE is 30–50 mg/kg/day but sometime may exceed 60 mg/kg/day [6]. Although, PLP or PN therapy are shown to be effective in controlling seizures, approximately 56% of PNPO-NEE patients suffer developmental delay/intellectual disability [6]. Early diagnosis and treatment are very important in predicting the disease outcome since untreated patients usually die, while survivors are left with poor neurocognitive outcome [6,97,98].
As pointed out by Mills and co-workers, the clinical spectrum of PNPO deficiency is much broader than earlier reported [4,8]. Patients with PNPO mutations are divided into three categories: 1) those with neonatal onset seizures responding to PLP; 2) a patient with infantile spasms responsive to PLP; and 3) those with seizures onset under three months of age responding to PN. Certain genotypes (R225 H/C and D33V) were suggested to more likely result in seizures that respond to PN, while R116Q, R229W, E50K and X262Q mutations appear to result in infertility, miscarriage and prematurity [8].
The observation that PLP is the normal therapy for PNPO-NEE is understood in the context of the salvage pathway for PLP metabolism [15]. PN or PM is phosphorylated to PNP or PMP by PL kinase, and requires PNPO to change the PNP/PMP into PLP, explaining why PLP is the standard therapy for PNPO-NEE as defective PNPO may not be able to oxidize PNP/PMP to PLP. However, the large doses of PLP used in therapy are known to have toxic side effects [10,15,95]. Thus the dose of PLP used in the treatment of PNPO deficiency-related epilepsy needs to be carefully selected to avoid such toxicity [99–102].
As noted above, seizures generally manifest early after birth, during first few days of life [8,16,19,95]. Variable developmental delays were observed with almost 25% deaths based on seizure onset and time for diagnosis and treatment. Early diagnosis of NEE associated with molecular defects in PNPO and treatment with PLP and/or PN, and as suggested below perhaps in conjunction with riboflavin may be essential for optimal seizure control and favorable developmental outcomes. Emphasis should also be placed on early testing and disease detection, especially in cases of suspected family history of PNPO deficiency.
4.2. Pathogenic mutations in PNPO
The recent review by Alghamdi et al. [95], presented the phenotypic and genotypic spectrum of 87 patients with confirmed PNPO deficiency and a comprehensive review of the clinical, biochemical and molecular observations for the mutations. Based on the type of mutation observed in human PNPO, the variants could be separated into 4 different categories (Table 1).
Table 1.
PNPO mutations affecting enzyme structure and function.
| Category | Variants | Mutations affecting |
|---|---|---|
|
| ||
| I | R95C, R116Q, R141C, R161C, R225 C/L/H and R229W/Q | catalytic site and ligand binding |
| II | E50K, P231S, G118Q/R | fold and protein stability |
| III | Residue deletion (S93S_A94Ldel), pre-mature stop codons (A174X), Loss of stop codons (X262Q) Frame shifts combine with loss of (L83W_fs*17, P150R_fs*27) |
Ligand binding and protein fold |
| IV | D33V | Neither ligand binding nor fold |
From Table 1, it appears that mutations of key amino acid residues can 1) affect folding of the enzyme, 2) influence the binding affinity of substrates, PMP or PNP, 3) reduce the affinity for the cofactor FMN and 4) affect catalytic activity. Distorted FMN binding site points towards a need for riboflavin supplementation along with active form of vitamin B6. Genotypic, as well as phenotypic spectrum of PNPO deficiency is wide with most serious complications appeared to be caused by category I and III variants (arginine missense mutants, and deletion mutants of hPNPO). Our group and others have characterized several of the PNPO variant proteins, including R229W and R95C [39,103], and G118R, R141C, R116Q, R225H, R116Q/R225H and X262Q [16,104].
4.3. PNPO R229W and R95C mutants show reduced enzymatic activity
The molecular basis of how PNPO R229W and R95C mutants lead to reduced PNPO activity and NEE have been elucidated [39,103]. Neither of these two mutations, which are located at the active site, affect the overall structure or thermal stability of the enzyme. Nonetheless, significant structural changes were observed at the active site that led to weak affinity of the cofactor FMN and/or substrate. The strictly conserved Arg229 makes several interactions, including a hydrogen-bond with the FMN phosphate moiety(Fig. 5A), intra- and inter-subunit hydrogen-bond with Arg141, Ser175, Glu217 and Ala260. The R229W variant showed only 0.2% of the wild-type enzyme activity in vitro with ~192-fold and ~4.5-fold decrease in PNP affinity and catalytic rate constant, respectively, while FMN affinity for the R229W mutant is reduced by ~50-fold; effectively preventing catalysis of PNP/PMP to PLP. Structural studies showed that the substituted tryptophan residue (Fig. 5B) as expected was unableto make the salt-bridge interactions with FMN, and several of the conserved R229 protein interactions. The substitution with the bigger tryptophan also resulted in a C[103]. The PNPO R95C mutant also exhibited a ~5-fold decrease in kcat, ~70-fold increase in substrate Km, and ~15% reduction in FMN affinity when compared to the wild-type enzyme. Arg95 is involved in salt-bridge/hydrogen-bond interactions with the FMN phosphate that is absent in the Cys95 mutant, explaining the diminished binding of FMN.
The studies with the R229W and R95C missense mutations explain why PN may not be universally successful in treating NEE as the mutation may prevent the enzyme in oxidizing PNP (or PMP) efficiently to form PLP. Treatment of NEE with PLP or PL maybe the most effective since the wild type PL kinase can convert PL to PLP. Nonetheless, it is clear that not enough PLP is made by PL kinase alone to meet the cell’s requirements; thus, both the salvage pathway enzymes may be required to provide enough PLP for cellular activity. The reduced affinity of FMN in both the R229W and R95C mutants also suggest that a combinatorial therapy of riboflavin (vitamin B2), PN and PLP may offer some improvement over the current treatment protocol of PN or PLP.
An interesting observation was that even though R229W and R95C show decreased or null enzymatic activity, these mutants like the wild-type PNPO bind PLP tightly at a non-catalytic site, which did not release even when dialyzed [39]. However, in the presence of apo-B6 enzyme, the tightly bound PLP is released to activate the apo-B6 enzyme into the catalytic holo-form [39]. Based on the fact that these mutants and the wild-type form specific interactions with several B6 enzymes with dissociation constants ranging from 0.3 to 12.3 μM, and the fact that the transfer of the PLP was not affected by phosphatases, we proposed that the tight-binding PLP is physiologically relevant for apo-B6 activation, whether by wild-type or variant [39]. If this hypothesis is correct, it would suggest that certain variants, e.g. R229W that are capable of binding PLP tightly and transferring it to activate apo-B6 enzyme, may explain why therapeutic PLP works in treating NEE [39]. It also suggests that variants that are both null and cannot bind and transfer PLP will be more lethal.
4.4. Biochemical characterization of other human PNPO mutants
The PNPO variants G118R, R141C, R225H, R116Q, X262Q and R116Q/R225H have been studied by other groups [16,104]. The purified R116Q protein was characterized with respect to quaternary structure, kinetic properties, thermal stability as well as ligand binding [104]. The mutant protein did retain overall structure but showed decreased thermal stability (Tm of 57 °C vs 63 °C for wild-type). The mutant protein also showed reduced affinity for FMN with only slightly reduced PLP affinity.
The five G118R, R141C, R225H, R116Q, X262Q mutants, and the R116Q/R225H double mutant have also been expressed, purified and characterized [16]. G118R and R141C were previously shown to exhibit mild clinical outcome [49]. Replacement of the stop codon with a glutamine residue (X262Q) resulted in a longer protein; with 28 amino acids added at the C-terminal end of the chain [30]. CD spectra of all variants, with the exception of G118R and X262Q showed no significant secondary structural differences compared to the wild-type enzyme, suggesting that the mutations did not result in any significant effect on the overall structure of the enzyme.
The R225H and R116Q/R225H variants showed ~77% saturation with FMN, while the R141C, G118R, and X262Q variants showed 57%, 44% and 22% FMN saturation, respectively. The G118R variant showed lower affinity for FMN (KD of 94 ± 19 nM) when compared to the wild type enzyme (13.1 ± 1.7 nM) [104], while R141C and X262Q variants showed even significantly lower affinity (KD values of 1.3 ± 0.3 μM and 2.4 ± 0.4 μM, respectively). Whether in the presence or absence of FMN, all variants were found to be less stable than the wild type (Tm = 55.4 ± 0.1 °C), except the R225H variant (Tm = 60.5 ± 0.1 °C). All amino acid replacements led to increased KM values and decreased kCAT. The R225H variant showed the most drastic change with a 27-fold lower kCAT and 6-fold higher KM. The activity of the X262Q variant could not be measured even at high (200 μM) FMN concentrations.
5. Hypophosphatasia (HPP)
Hypophosphatasia (HPP) is an inherited metabolic disease caused by pathogenic mutations in the tissue-nonspecific alkaline phosphatase (TNSALP) [25,105–107]. TNSALP is expressed in the liver, kidney and bone [25], and functions to convert PLP to PL in order to cross the blood–brain barrier. HPP is characterized by deficiency of TNSALP leading to excess extracellular PLP. Molecular testing shows defects in TNSALP occur in the ALPL gene located on chromosome 1p36.1-p34 [108]. The gene consists of 12 exons distributed over 50 kb. TNSALP is shown to be involved in the regulation of neurotransmission in the cerebral cortex [109]. Patients with the most severe form present with PN-dependent seizures in the neonatal period that can precede the detection of bone disease [30,106,107,110]. PN is part of the treatment regimen in children with seizures [30,106,107].
Functional TNSALP exists as a homodimer of ~160 kDa, and is anchored by glycosylphosphatidylinositol (GIP) to the outer membrane of the cells [105]. Inborn errors that cause deficiency of the proteins responsible for GPI anchor processing have also been implicated in brain PLP deficiency and B6-responsive seizures [111].
Even though there appears to be several known mutations in ALPL, perhaps significant number being pathogenic, only few HPP patients have been reported in the literature with PN-responsive seizures [110]. HPP is diagnosed by low amount or absence of alkaline phosphatase in serum. Substrates of alkaline phosphatase are elevated, which can be measured in the urine [phosphoethanolamine (PEA)] or serum [PLP and inorganic pyrophosphate (PPi)]. Molecular testing of the TNSALP gene is also used to diagnose the disease. Definite diagnosis of the disease was made by sequencing analysis of the TNSALP gene that showed compound heterozygosity for a mutation in exon 9, c.875_881delCAGGGGAinsT, and a previously reported mutation in exon 12, c.1559delT [112]. Another study also detected a homozygous p.267_268delHF (c.799_804delCACTTC) mutation in the ALPL gene [110]. Recently enzyme replacement therapy (ERT) that treats the bone disease has become available [113].
6. Other pathological conditions that potentially may involve inborn errors in the PLP salvage enzymes
Several disorders, e.g., autism, schizophrenia, attention deficit hyperactive disorder, anxiety disorders, learning disability, Alzheimer’s disease, and Parkinson’s disease have been implicated or suggested to be caused by vitamin B6 deficiency as a result of pathogenic mutations in the salvage enzymes. The link between Autism and vitamin B6 has been explored by several groups with conflicting results. One such study by Adams et al. reported children with autism spectrum to show higher concentrations of primary B6 vitamers and significantly lower levels of PLP as compared with normal subjects [114] that the authors attributed to defective PL kinase. A recent study by Obara et al. also suggested that a certain phenotypic subset of autistic subjects respond to B6 and magnesium supplementation [115], although the authors did not correlate their findings with any defect in the B6 metabolism pathway. Another recent study by Laue [116] that examined the link between gut bacteria and autism spectrum disorder (ASD) found that two functional pathways, L-ornithine and vitamin B6 biosynthesis, were associated with better social skills at 3 years, prompting the authors to suggest that PLP biosynthesis and salvage are relevant to the effectiveness of high-dose vitamin B6 and magnesium supplementation in reducing autistic symptoms.
Individuals with Down syndrome (chromosome 21 trisomy) that are characterized by epileptic episodes have been reported to display a significant alteration of B6 metabolism [117]. The study reported increased PL kinase activity in the patients compared to normal subjects. A recent review by Katz et al. [118] reported that Vitamin B6 has been used successfully to treat the epileptic spasms associated with Down syndrome, although in the majority of cases only when used in combination with other therapies, e.g. Prednisone or ACTH.
Several polymorphisms in the PNPO gene have been associated with schizophrenia in the Japanese population [119]. In a recent case-control study in a large Japanese cohort, the investigators observed decreased serum PL levels in patients with schizophrenia [120]. However, they found no correlation between PL levels and schizophrenia risk using a Mendelian randomization.
Other neurological disorders including seizures, attention deficit hyperactive disorder, anxiety disorders, learning disability, Alzheimer’s disease, and Parkinson’s disease, have also been associated with defect in the B6 metabolic pathway although without any conclusive evidence [121–124].
Although there are no direct evidences that link these neurological disorders to mutations in PL kinase or PNPO or phosphatase, the genes are candidates for mutational analysis in affected patients. Identification of such an inborn error could help understand the role of the mutation in the enzymatic activity and the associated phenotypes, and potential therapy.
7. Zebrafish model
Zebrafish shares about 70% similarity with human genetic, and thus have obvious advantages over rodents for use in both basic medical sciences and clinical research, including target identification, disease modeling and drug development [125,126].
The zebrafish pdxH gene (zpdxH) contains 7 exons located on chromosome 12 that codes for zebrafish PNPO (zPNPO) made up of 267 amino acids [127]. The amino acid sequence and structure of zPNPO and hPNPO are very similar, suggesting that zebrafish is a good animal model for studying PNPO-deficiency NEE [127]. Our group has reported a seizure model in zebrafish larvae using ginkgotoxin, and shown to inhibit the formation of the neurotransmitter, GABA [127]. The ginkgotoxin induced seizure was alleviated by the addition of either PLP or GABA. As mentioned earlier ginkgotoxin is structurally similar to the three primary B6 vitamers, and like the B6 vitamers, is a substrate for PL kinase, which converts it to ginkgotoxin phosphate to inhibit the enzyme [128,129]. The inhibition has been implicated in decreased GABA and epileptic convulsions [127,130].
In another recent study, our group generated knockdown zPNPO by injecting zebrafish embryos with morpholino oligonucleotide (MO) to block protein translation [126]. The knockdown resulted in brain malformation and impaired locomotor activity, as well as a defective circulation system. Zebrafish zpdxH mRNA was found distributed globally in developing embryos and ubiquitously among organs in adult fish, reflecting its importance in embryogenesis, which is further supported by the developmental delay and the anomalies occurring in a wide spectrum of organs/tissues observed in zPNPO morphants [126]. Expectedly, the morphological phenotypes observed in zPNPO morphants recapitulate some of the clinical features observed with PNPO- NEE patients [126].
As expected from clinical observations in humans, providing the morphants with PLP resulted in improved morphological and behavioral anomalies, especially rescuing the eye, heart, and swim bladder [126]. Of interest is that PM, even at significantly lower concentration was able to rescue the malfunction of the brain, eye, and swim bladder, while PL could only rescue the malformation of the swim bladder, with PN infective in rescuing any anomaly. The observation that the rescue effect of PM was significantly at lower concentration than PLP, presents a possible new therapeutic option for PNPO-NEE. It is interesting that PM showed rescue effects, since like PN, PM is unlikely to be metabolized into PLP due to the knockdown of zPNPO expression. Transaminases are the largest family of PLP-dependent enzymes that catalytically convert amino acids into the corresponding α-keto acids. Unlike other PLP-dependent enzymes, transaminases can use PMP to form the holo-enzymes, thus reducing the cell’s need for PLP because of the increased production of PMP by PL kinase from the added PM. PLP and PM, in some instances are able to rescue different anomalies, implying possible tissue specificity for B6 metabolism and/or requirement. This observation suggests that combinatorial use of PM and PLP for PNPO-NEE treatment may reduce dose requirements and potential toxicity of the use of PLP alone.
8. Conclusion
Several inborn errors in the vitamin B6 salvage pathway enzymes, involving PNPO, PL kinase and phosphatases have been identified. These pathogenic mutations in most instances lead to defective metabolism of PLP, either through reduced or null activities of these enzymes, or potentially affect PLP transport. Consequently, not enough PLP is available to activate newly-formed apoB6 enzymes. In majority of cases, the most immediate effect of PLP deficiency is seen in neurological or seizure disorders, e.g. convulsions and epileptic encephalopathy [3,4,6,8–10,12–19], and also suspected in Alzheimer’s, Autism, Parkinson’s and Schizophrenia [31–37]. Only the errors in PNPO and phosphatase have been shown to cause seizures, most notably PNPO-deficient NEE that is responsive to PLP and sometimes to PN, and for phosphatases, pyridoxine-dependent HPP that is responsive to PN. In contrast, pathogenic mutations in PL kinase have not been shown to cause seizures but rather polyneuropathy, which is responsive to PLP. Interestingly, inactivation of PL kinase by drugs or compounds, which is known to lead to PLP deficiency, cause seizures and epileptic convulsion that are responsive to vitamin B6 therapy. There are other instances when inborn errors in certain metabolic pathways, e.g. the antiquitin (ALDH7A1) gene lead to accumulation of intermediates that react with PLP, reducing its availability; inborn errors in certain PLP carrier proteins, e.g. PROSC that disrupt normal intracellular PLP transfer.
While supplementation of vitamin B6 works for several of these pathologies, it is also clear that definitive mechanisms that underlie some of these pathologies have not been well established, and studies on the effect of supplementation of B6 vitamers sometimes show conflicting results. The genes coding for these enzymes are clearly candidates for mutational analysis in affected patients. If a pathogenic inborn error is identified, it could be investigated to determine its role in the enzymatic activity and the associated phenotypes, and potentially lead to a pharmacologic intervention.
Acknowledgements
This work was partially supported in part by NIH/NIMHD grant R01MD009124 (to M. K. S), NIH/NIGMS grant R01GM129793 (to V. de C.-L.). The authors also acknowledge, with thanks, Deanship of Scientific Research (DSR) at King Abdulaziz University, Jeddah, SA under grant no. FP-61-44 for funding and support of this work.
Abbreviations:
- NEE
Neonatal epileptic encephalopathy
- PL
Pyridoxal
- PM
Pyridoxamine
- PN
Pyridoxine
- PLP
Pyridoxal 5′-phosphate
- PMP
Pyridoxamine 5′-phosphate
- PNP
Pyridoxine 5′-phosphate
- PL kinase
Pyridoxal kinase
- PNPO
Pyridoxine 5′-phosphate oxidase
- TNSALP
Tissue nonspecific alkaline phosphatase
- GABA
Gamma amino butyric acid
- HPP
Hypophosphatasia
Footnotes
Additional information
The authors declare no competing interests.
References
- [1].di Salvo ML, Contestabile R, Safo MK, Vitamin B(6) salvage enzymes: mechanism, structure and regulation, Biochim. Biophys. Acta 1814 (2011) 1597–1608, 10.1016/j.bbapap.2010.12.006. [DOI] [PubMed] [Google Scholar]
- [2].Stockler S, Plecko B, Gospe SM, Coulter-Mackie M, Connolly M, van Karnebeek C, Mercimek-Mahmutoglu S, Hartmann H, Scharer G, Struijs E, et al. , Pyridoxine dependent epilepsy and antiquitin deficiency clinical and molecular characteristics and recommendations for diagnosis, treatment and follow-up, Mol.Genet.Metab. (2011), 10.1016/j.ymgme.2011.05.014. [DOI] [PubMed] [Google Scholar]
- [3].Plecko B, Stockler S, Vitamin B6 dependent seizures, Can. J. Neurol. Sci. 36 (Suppl 2) (2009) S73–S77. [PubMed] [Google Scholar]
- [4].Plecko B, Paul K, Mills P, Clayton P, Paschke E, Maier O, Hasselmann O, Schmiedel G, Kanz S, Connolly M, et al. , Pyridoxine responsiveness in novel mutations of the PNPO gene, Neurology 82 (2014) 1425–1433, 10.1212/WNL.0000000000000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Plecko B, Paul K, Paschke E, Stoeckler-Ipsiroglu S, Struys E, Jakobs C, Hartmann H, Luecke T, di Capua M, Korenke C, et al. , Biochemical and molecular characterization of 18 patients with pyridoxine-dependent epilepsy and mutations of the antiquitin (ALDH7A1) gene, Hum. Mutat. 28 (2007) 19–26, 10.1002/humu.20433. [DOI] [PubMed] [Google Scholar]
- [6].Hoffmann GF, Schmitt B, Windfuhr M, Wagner N, Strehl H, Bagci S, Franz AR, Mills PB, Clayton PT, Baumgartner MR, et al. , Pyridoxal 5’-phosphate may Be curative in early-onset epileptic encephalopathy, J. Inherit. Metab. Dis. 30 (2007) 96e99, 10.1007/s10545-006-0508-4. [DOI] [PubMed] [Google Scholar]
- [7].Hartmann H, Fingerhut M, Jakobs C, Plecko B, Status epilepticus in a neonate treated with pyridoxine because of a familial recurrence risk for antiquitin deficiency: pyridoxine toxicity? Dev. Med. Child Neurol. (2011) 10.1111/j.1469-8749.2011.04033.x. [DOI] [PubMed] [Google Scholar]
- [8].Mills PB, Camuzeaux SSM, Footitt EJ, Mills KA, Gissen P, Fisher L, Das KB, Varadkar SM, Zuberi S, McWilliam R, et al. , Epilepsy due to PNPO mutations: genotype, environment and treatment affect presentation and outcome, Brain 137 (2014) 1350e1360, 10.1093/brain/awu051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mills PB, Struys E, Jakobs C, Plecko B, Baxter P, Baumgartner M, Willemsen MAAP, Omran H, Tacke U, Uhlenberg B, et al. , Mutations in antiquitin in individuals with pyridoxine-dependent seizures, Nat. Med. 12 (2006) 307–309, 10.1038/nm1366. [DOI] [PubMed] [Google Scholar]
- [10].Mills PB, Surtees RAH, Champion MP, Beesley CE, Dalton N, Scambler PJ, Heales SJR, Briddon A, Scheimberg I, Hoffmann GF, et al. , Neonatal epileptic encephalopathy caused by mutations in the PNPO gene encoding pyridox(Am)ine 5’-phosphate oxidase, Hum. Mol. Genet. 14 (2005) 1077–1086, 10.1093/hmg/ddi120. [DOI] [PubMed] [Google Scholar]
- [11].Gospe SM, Pyridoxine-dependent seizures: new genetic and biochemical clues to help with diagnosis and treatment, Curr. Opin. Neurol. 19 (2006) 148–153, 10.1097/01.wco.0000218230.81301.12. [DOI] [PubMed] [Google Scholar]
- [12].Gospe SM, Neonatal vitamin-responsive epileptic encephalopathies, Chang Gung Med. J. 33 (2010) 1–12. [PubMed] [Google Scholar]
- [13].Gospe SM, Pyridoxine-dependent epilepsy and pyridoxine phosphate oxidase deficiency: unique clinical symptoms and non-specific EEG characteristics, Dev. Med. Child Neurol. 52 (2010) 602e603, 10.1111/j.1469-8749.2010.03668.x. [DOI] [PubMed] [Google Scholar]
- [14].Ormazabal A, Oppenheim M, Serrano M, García-Cazorla A, Campistol J, Ribes A, Ruiz A, Moreno J, Hyland K, Clayton P, et al. , Pyridoxal 5’-phosphate values in cerebrospinal fluid: reference values and diagnosis of PNPO deficiency in paediatric patients, Mol. Genet. Metabol. 94 (2008) 173–177, 10.1016/j.ymgme.2008.01.004. [DOI] [PubMed] [Google Scholar]
- [15].Ghatge MS, Di Salvo ML, Contestabile R, Eseonu DN, Karve S, Schirch V, Safo MK, Molecular defects of vitamin B6 metabolism associated with neonatal epileptic encephalopathy, in: R. Tanasescu (Ed.), Miscellanea on Encephalopathies - A Second Look, InTech, 2012, p. 267. [Google Scholar]
- [16].Barile A, Nogues I, di Salvo ML, Bunik V, Contestabile R, Tramonti A, Molecular characterization of pyridoxine 5’-phosphate oxidase and its pathogenic forms associated with neonatal epileptic encephalopathy, Sci. Rep. 10 (2020) 13621, 10.1038/s41598-020-70598-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bowling FG, Pyridoxine supply in human development, Semin. Cell Dev. Biol. 22 (2011) 611–618, 10.1016/j.semcdb.2011.05.003. [DOI] [PubMed] [Google Scholar]
- [18].Balasubramaniam S, Bowling F, Carpenter K, Earl J, Chaitow J, Pitt J, Mornet E, Sillence D, Ellaway C, Perinatal hypophosphatasia presenting as neonatal epileptic encephalopathy with abnormal neurotransmitter metabolism secondary to reduced Co-factor pyridoxal-5’-phosphate availability, J. Inherit. Metab. Dis. 33 (Suppl 3) (2010) S25–S33, 10.1007/s10545-009-9012-y. [DOI] [PubMed] [Google Scholar]
- [19].Wilson MP, Footitt EJ, Papandreou A, Uudelepp M-L, Pressler R, Stevenson DC, Gabriel C, McSweeney M, Baggot M, Burke D, et al. , An LC-MS/MS-Based method for the quantification of pyridox(Am)ine 5’-phosphate oxidase activity in dried blood spots from patients with epilepsy, Anal. Chem. 89 (2017) 8892e8900, 10.1021/acs.analchem.7b01358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Batllori M, Molero-Luis M, Ormazabal A, Casado M, Sierra C, García-Cazorla A, Kurian M, Pope S, Heales SJ, Artuch R, Analysis of human cerebrospinal fluid monoamines and their cofactors by HPLC, Nat. Protoc. 12 (2017) 2359–2375, 10.1038/nprot.2017.103. [DOI] [PubMed] [Google Scholar]
- [21].Burd L, Stenehjem A, Franceschini LA, Kerbeshian J, A 15-year follow-up of a boy with pyridoxine (vitamin B6)-dependent seizures with autism, breath holding, and severe mental retardation, J. Child Neurol. 15 (2000) 763–765, 10.1177/088307380001501111. [DOI] [PubMed] [Google Scholar]
- [22].di Salvo ML, Safo MK, Contestabile R, Biomedical aspects of pyridoxal 5’-phosphate availability, Front Biosci (Elite Ed) 4 (2012) 897–913. [DOI] [PubMed] [Google Scholar]
- [23].Chelban V, Wilson MP, Warman Chardon J, Vandrovcova J, Zanetti MN, Zamba-Papanicolaou E, Efthymiou S, Pope S, Conte MR, Abis G, et al. , PDXK mutations cause polyneuropathy responsive to pyridoxal 5’-phosphate supplementation, Ann. Neurol. 86 (2019) 225e240, 10.1002/ana.25524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Seefried L, Kishnani PS, Moseley S, Denker AE, Watsky E, Whyte MP, Dahir KM, Pharmacodynamics of asfotase alfa in adults with pediatric-onset hypophosphatasia, Bone (2020) 115664, 10.1016/j.bone.2020.115664. [DOI] [PubMed] [Google Scholar]
- [25].Salles JP Hypophosphatasia, Biological and clinical aspects, avenues for therapy, Clin. Biochem. Rev. 41 (2020) 13e27, 10.33176/AACB-19-00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Talwar D, Catchpole A, Wadsworth JM, Toole BJ, McMillan DC, The relationship between plasma albumin, alkaline phosphatase and pyridoxal phosphate concentrations in plasma and red cells: implications for assessing vitamin B6 status, Clin. Nutr. 39 (2020) 2824–2831, 10.1016/j.clnu.2019.12.012. [DOI] [PubMed] [Google Scholar]
- [27].Colazo JM, Hu JR, Dahir KM, Simmons JH, Neurological symptoms in hypophosphatasia, Osteoporos. Int. 30 (2019) 469–480, 10.1007/s00198-018-4691-6. [DOI] [PubMed] [Google Scholar]
- [28].Darin N, Reid E, Prunetti L, Samuelsson L, Husain RA, Wilson M, El Yacoubi B, Footitt E, Chong WK, Wilson LC, et al. , Mutations in PROSC disrupt cellular pyridoxal phosphate homeostasis and cause vitamin-B6-dependent epilepsy, Am. J. Hum. Genet. 99 (2016) 1325e1337, 10.1016/j.ajhg.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Scharer G, Brocker C, Vasiliou V, Creadon-Swindell G, Gallagher RC, Spector E, Van Hove JL, The genotypic and phenotypic spectrum of pyridoxine-dependent epilepsy due to mutations in ALDH7A1, J. Inherit. Metab. Dis. 33 (2010) 571–581, 10.1007/s10545-010-9187-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wilson MP, Plecko B, Mills PB, Clayton PT, Disorders affecting vitamin B6 metabolism, J. Inherit. Metab. Dis. 42 (2019) 629–646, 10.1002/jimd.12060. [DOI] [PubMed] [Google Scholar]
- [31].Bohme I, Luddens H, The inhibitory neural circuitry as target of antiepileptic drugs, Curr. Med. Chem. 8 (2001) 1257–1274. [DOI] [PubMed] [Google Scholar]
- [32].Lloyd KG, Munari C, Worms P, Bossi L, Bancaud J, Talairach J, Morselli PL, The role of GABA mediated neurotransmission in convulsive states, Adv. Biochem. Psychopharmacol. 26 (1981) 199–206. [PubMed] [Google Scholar]
- [33].Nishino N, Fujiwara H, Noguchi-Kuno SA, Tanaka C, GABAA receptor but not muscarinic receptor density was decreased in the brain of patients with Parkinson’s disease, Jpn. J. Pharmacol. 48 (1988) 331–339. [DOI] [PubMed] [Google Scholar]
- [34].Aoyagi T, Wada T, Kojima F, Nagai M, Harada S, Takeuchi T, Hirokawa K, Increase in aminobutyrate aminotransferase and cholineacetyltransferase in cerebrum of aged rats, Chem. Pharm. Bull. 38 (1990) 1750–1752. [DOI] [PubMed] [Google Scholar]
- [35].Butterworth J, Yates CM, Simpson J, Phosphate-activated glutaminase in relation to huntington’s disease and agonal state, J. Neurochem. 41 (1983) 440–447. [DOI] [PubMed] [Google Scholar]
- [36].Schrag A, Psychiatric aspects of Parkinson’s disease–an update, J. Neurol. 251 (2004) 795–804, 10.1007/s00415-004-0483-3. [DOI] [PubMed] [Google Scholar]
- [37].Snyder SH, Ferris CD, Novel neurotransmitters and their neuropsychiatric relevance, Am. J. Psychiatr. 157 (2000) 1738e1751, 10.1176/appi.ajp.157.11.1738. [DOI] [PubMed] [Google Scholar]
- [38].McCormick DB, Chen H, Update on interconversions of vitamin B-6 with its coenzyme, J. Nutr. 129 (1999) 325–327, 10.1093/jn/129.2.325. [DOI] [PubMed] [Google Scholar]
- [39].Ghatge MS, Karve SS, David TMS, Ahmed MH, Musayev FN, Cunningham K, Schirch V, Safo MK, Inactive mutants of human pyridoxine 5’-phosphate oxidase: a possible role for a noncatalytic pyridoxal 5’-phosphate tight binding site, FEBS Open Bio 6 (2016) 398–408, 10.1002/2211-5463.12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Percudani R, Peracchi A, The B6 database: a tool for the description and classification of vitamin B6-dependent enzymatic activities and of the corresponding protein families, BMC Bioinf. 10 (2009) 273, 10.1186/1471-2105-10-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Schneider G, Käck H, Lindqvist Y, The manifold of vitamin B6 dependent enzymes, Structure 8 (2000) R1–R6. [DOI] [PubMed] [Google Scholar]
- [42].McCormick DB, Two interconnected B vitamins: riboflavin and pyridoxine, Physiol. Rev. 69 (1989) 1170–1198. [DOI] [PubMed] [Google Scholar]
- [43].Mackey AD, Lieu SO, Carman C, Gregory JF, Hydrolytic activity toward pyridoxine-5’-beta-D-glucoside in rat intestinal mucosa is not increased by vitamin B-6 deficiency: effect of basal diet composition and pyridoxine intake, J. Nutr. 133 (2003) 1362–1367. [DOI] [PubMed] [Google Scholar]
- [44].Armada LJ, Mackey AD, Gregory JF, Intestinal brush border membrane catalyzes hydrolysis of pyridoxine-5’-beta-D-glucoside and exhibits parallel developmental changes of hydrolytic activities toward pyridoxine-5’-beta-D-glucoside and lactose in rats, J. Nutr. 132 (2002) 2695–2699. [DOI] [PubMed] [Google Scholar]
- [45].Said HM, Recent advances in carrier-mediated intestinal absorption of water-soluble vitamins, Annu. Rev. Physiol. 66 (2004) 419–446, 10.1146/annurev.physiol.66.032102.144611. [DOI] [PubMed] [Google Scholar]
- [46].Surtees R, Mills P, Clayton P, Inborn errors affecting vitamin B 6 metabolism, Future Neurol. 1 (2006) 615–620, 10.2217/14796708.1.5.615. [DOI] [Google Scholar]
- [47].Merrill AH, Horiike K, McCormick DB, Evidence for the regulation of pyridoxal 5-phosphate formation in liver by pyridoxamine (pyridoxine) 5-phosphate oxidase, Biochem. Biophys. Res. Commun. 83 (1978) 984–990. [DOI] [PubMed] [Google Scholar]
- [48].Fang X, Zhou ZM, Lu L, Yin LL, Li JM, Zhen Y, Wang H, Sha JH, Expression of a novel pyridoxal kinase MRNA splice variant, PKH-T, in human testis, Asian J. Androl. 6 (2004) 83–91. [PubMed] [Google Scholar]
- [49].Kang JH, Hong ML, Kim DW, Park J, Kang TC, Won MH, Baek NI, Moon BJ, Choi SY, Kwon OS, Genomic organization, tissue distribution and deletion mutation of human pyridoxine 5’-phosphate oxidase, Eur. J. Biochem. 271 (2004) 2452–2461, 10.1111/j.1432-1033.2004.04175.x. [DOI] [PubMed] [Google Scholar]
- [50].Fu TF, di Salvo M, Schirch V, Distribution of B6 vitamers in Escherichia coli as determined by enzymatic assay, Anal. Biochem. 298 (2001) 314–321, 10.1006/abio.2001.5401. [DOI] [PubMed] [Google Scholar]
- [51].Chung J-Y, Choi J-H, Hwang C-Y, Youn H-Y, Pyridoxine induced neuropathy by subcutaneous administration in dogs, J. Vet. Sci. 9 (2008) 127, 10.4142/jvs.2008.9.2.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Li TK, Lumeng L, Veitch RL, Regulation of pyridoxal 5’-phosphate metabolism in liver, Biochem. Biophys. Res. Commun. 61 (1974) 677–684. [DOI] [PubMed] [Google Scholar]
- [53].Safo MK, Musayev FN, di Salvo ML, Hunt S, Claude J-B, Schirch V, Crystal structure of pyridoxal kinase from the Escherichia coli PdxK gene: implications for the classification of pyridoxal kinases, J. Bacteriol. 188 (2006) 4542–4552, 10.1128/JB.00122-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Choi SY, Churchich JE, Zaiden E, Kwok F, Brain pyridoxine-5-phosphate oxidase. Modulation of its catalytic activity by reaction with pyridoxal 5-phosphate and analogs, J. Biol. Chem. 262 (1987) 12013–12017. [PubMed] [Google Scholar]
- [55].Di Salvo M, Yang E, Zhao G, Winkler ME, Schirch V, Expression, purification, and characterization of recombinant Escherichia coli pyridoxine 5’-phosphate oxidase, Protein Expr. Purif. 13 (1998) 349–356, 10.1006/prep.1998.0904. [DOI] [PubMed] [Google Scholar]
- [56].Zhao G, Winkler ME, Kinetic limitation and cellular amount of pyridoxine (pyridoxamine) 5’-phosphate oxidase of Escherichia coli K-12, J. Bacteriol. 177 (1995) 883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Yang ES, Schirch V, Tight binding of pyridoxal 5’-phosphate to recombinant Escherichia coli pyridoxine 5’-phosphate oxidase, Arch. Biochem. Biophys. 377 (2000) 109–114, 10.1006/abbi.2000.1737. [DOI] [PubMed] [Google Scholar]
- [58].Barile A, Tramonti A, di Salvo ML, Nogués I, Nardella C, Malatesta F, Contestabile R, Allosteric feedback inhibition of pyridoxine 5’-phosphate oxidase from Escherichia coli, J. Biol. Chem. 294 (2019) 15593–15603, 10.1074/jbc.RA119.009697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Stanulovic M, Jeremic V, Leskovac V, Chaykin S, New pathway of conversion of pyridoxal to 4-pyridoxic acid, Enzyme 21 (1976) 357–369. [DOI] [PubMed] [Google Scholar]
- [60].Kim YT, Kwok F, Churchich JE, Interactions of pyridoxal kinase and aspartate aminotransferase emission anisotropy and compartmentation studies, J. Biol. Chem. 263 (1988) 13712e13717. [PubMed] [Google Scholar]
- [61].Cheung P-Y, Fong C-C, Ng K-T, Lam W-C, Leung Y-C, Tsang C-W, Yang M, Wong M-S, Interaction between pyridoxal kinase and pyridoxal-5-phosphate-dependent enzymes, J. Biochem. 134 (2003) 731e738. [DOI] [PubMed] [Google Scholar]
- [62].Ghatge MS, Contestabile R, di Salvo ML, Desai JV, Gandhi AK, Camara CM, Florio R, Gonzalez IN, Parroni A, Schirch V, et al. , Pyridoxal 5’-phosphate is a slow tight binding inhibitor of E. Coli pyridoxal kinase, PloS One 7 (2012), e41680, 10.1371/journal.pone.0041680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Yudkoff M, Interactions in the metabolism of glutamate and the branchedchain amino acids and ketoacids in the CNS, Neurochem. Res. 42 (2017) 10e18, 10.1007/s11064-016-2057-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Silverman RB, Design and mechanism of GABA aminotransferase inactivators. Treatments for epilepsies and addictions, Chem. Rev. 118 (2018) 4037–4070, 10.1021/acs.chemrev.8b00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Huang H, Li Y, Liang J, Finkelman FD, Molecular regulation of histamine synthesis, Front. Immunol. 9 (2018), 10.3389/fimmu.2018.01392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Montioli R, Battini R, Paiardini A, Tolve M, Bertoldi M, Carducci C, Leuzzi V, Borri Voltattorni C, A novel compound heterozygous genotype Associated with aromatic amino acid decarboxylase deficiency: clinical aspects and biochemical studies, Mol. Genet. Metabol. 127 (2019) 132–137, 10.1016/j.ymgme.2019.05.004. [DOI] [PubMed] [Google Scholar]
- [67].Zieske LR, Davis L, Decarboxylation of Glycine by serine hydroxymethyltransferase in the presence of lipoic acid, J. Biol. Chem. 258 (1983) 10355–10359. [PubMed] [Google Scholar]
- [68].Stone TW, Mackay GM, Forrest CM, Clark CJ, Darlington LG, Tryptophan metabolites and brain disorders, Clin. Chem. Lab. Med. 41 (2003) 852–859, 10.1515/CCLM.2003.129. [DOI] [PubMed] [Google Scholar]
- [69].Schwarcz R, Pellicciari R, Manipulation of brain kynurenines: glial targets, neuronal effects, and clinical opportunities, J.Pharmacol.Exp.Ther. 303 (2002) 1–10, 10.1124/jpet.102.034439. [DOI] [PubMed] [Google Scholar]
- [70].Rahman MK, Nagatsu T, Sakurai T, Hori S, Abe M, Matsuda M, Effect of pyridoxal phosphate deficiency on aromatic L-amino acid decarboxylase activity with L-DOPA and L-5-hydroxytryptophan as substrates in rats, Jpn. J. Pharmacol. 32 (1982) 803–811. [DOI] [PubMed] [Google Scholar]
- [71].Kwok F, Churchich JE, Brain pyridoxal kinase. Purification, substrate specificities, and sensitized photodestruction of an essential histidine, J. Biol. Chem. 254 (1979) 6489–6495. [PubMed] [Google Scholar]
- [72].Hanna MC, Turner AJ, Kirkness EF, Human pyridoxal kinase. CDNA cloning, expression, and modulation by ligands of the benzodiazepine receptor, J. Biol. Chem. 272 (1997) 10756e10760. [DOI] [PubMed] [Google Scholar]
- [73].Musayev FN, di Salvo ML, Ko T-P, Gandhi AK, Goswami A, Schirch V, Safo MK, Crystal structure of human pyridoxal kinase: structural basis of M(+) and M(2+) activation, Protein Sci. 16 (2007) 2184–2194, 10.1110/ps.073022107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Li M-H, Kwok F, Chang W-R, Lau C-K, Zhang J-P, Lo SCL, Jiang T, Liang D-C, Crystal structure of brain pyridoxal kinase, a novel member of the ribokinase superfamily, J. Biol. Chem. 277 (2002) 46385–46390, 10.1074/jbc.M208600200. [DOI] [PubMed] [Google Scholar]
- [75].Newman JA, Das SK, Sedelnikova SE, Rice DW, The crystal structure of an ADP complex of Bacillus subtilis pyridoxal kinase provides evidence for the parallel emergence of enzyme activity during evolution, J. Mol. Biol. 363 (2006) 520–530, 10.1016/j.jmb.2006.08.013. [DOI] [PubMed] [Google Scholar]
- [76].Gandhi AK, Ghatge MS, Musayev FN, Sease A, Aboagye SO, di Salvo ML, Schirch V, Safo MK, Kinetic and structural studies of the role of the active site residue Asp235 of human pyridoxal kinase, Biochem. Biophys. Res. Commun. 381 (2009) 12–15, 10.1016/j.bbrc.2009.01.170. [DOI] [PubMed] [Google Scholar]
- [77].Flanagan JM, Beutler E, The genetic basis of human erythrocyte pyridoxal kinase activity variation, Haematologica 91 (2006) 801–804. [PubMed] [Google Scholar]
- [78].Meisler NT, Thanassi JW, Pyridoxine kinase, pyridoxine phosphate phosphatase and pyridoxine phosphate oxidase activities in control and B-6-Deficient rat liver and brain, J. Nutr. 110 (1980) 1965e1975, 10.1093/jn/110.10.1965. [DOI] [PubMed] [Google Scholar]
- [79].Lumeng L, Ryan MP, Li T-K, Validation of the diagnostic value of plasma pyridoxal 50-phosphate measurements in vitamin B6 nutrition of the rat, J. Nutr. 108 (1978) 545e553, 10.1093/jn/108.4.545. [DOI] [PubMed] [Google Scholar]
- [80].Mascolo E, Barile A, Mecarelli LS, Amoroso N, Merigliano C, Massimi A, Saggio I, Hansen T, Tramonti A, Di Salvo ML, et al. , The expression of four pyridoxal kinase (PDXK) human variants in Drosophila impacts on genome integrity, Sci. Rep. 9 (2019), 10.1038/s41598-019-50673-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Morocutti C, Colazza GB, Soldati G, D’Alessio C, Damiano M, Casali C, Pierelli F, Charcot-marie-tooth disease in molise, a central-southern region of Italy: an epidemiological study, Neuroepidemiology 21 (2002) 241–245, 10.1159/000065642. [DOI] [PubMed] [Google Scholar]
- [82].Glenn GM, Krober MS, Kelly P, McCarty J, Weir M, Pyridoxine as therapy in theophylline-induced seizures, Vet. Hum. Toxicol. 37 (1995) 342–345. [PubMed] [Google Scholar]
- [83].Hasegawa S, Oda Y, Ichiyama T, Hori Y, Furukawa S, Ginkgo nut intoxication in a 2-year-old male, Pediatr. Neurol. 35 (2006) 275–276, 10.1016/j.pediatrneurol.2006.05.008. [DOI] [PubMed] [Google Scholar]
- [84].Gandhi AK, Desai JV, Ghatge MS, di Salvo ML, Di Biase S, Danso-Danquah R, Musayev FN, Contestabile R, Schirch V, Safo MK, Crystal structures of human pyridoxal kinase in complex with the neurotoxins, ginkgotoxin and theophylline: insights into pyridoxal kinase inhibition, PloS One 7 (2012), e40954, 10.1371/journal.pone.0040954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Kajiyama Y, Fujii K, Takeuchi H, Manabe Y, Ginkgo seed poisoning, Pediatrics 109 (2002) 325–327. [DOI] [PubMed] [Google Scholar]
- [86].Leistner E, Drewke C, Ginkgo biloba and ginkgotoxin, J. Nat. Prod. 73 (2010) 86–92, 10.1021/np9005019. [DOI] [PubMed] [Google Scholar]
- [87].Delport R, Ubbink JB, Serfontein WJ, Becker PJ, Walters L, Vitamin B6 nutritional status in asthma: the effect of theophylline therapy on plasma pyridoxal-5’-phosphate and pyridoxal levels, Int. J. Vitam. Nutr. Res. 58 (1988) 67–72. [PubMed] [Google Scholar]
- [88].di Salvo ML, Safo MK, Musayev FN, Bossa F, Schirch V, Structure and mechanism of Escherichia coli pyridoxine 5’-phosphate oxidase, Biochim. Biophys. Acta 1647 (2003) 76–82. [DOI] [PubMed] [Google Scholar]
- [89].Musayev FN, Di Salvo ML, Ko T-P, Schirch V, Safo MK, Structure and properties of recombinant human pyridoxine 5’-phosphate oxidase, Protein Sci. 12 (2003) 1455–1463, 10.1110/ps.0356203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Kazarinoff MN, McCormick DB, Rabbit liver pyridoxamine (pyridoxine) 5’-phosphate oxidase. Purification and properties, J. Biol. Chem. 250 (1975) 3436–3442. [PubMed] [Google Scholar]
- [91].di Salvo ML, Ko T-P, Musayev FN, Raboni S, Schirch V, Safo MK, Active site structure and stereospecificity of Escherichia coli pyridoxine-5’-phosphate oxidase, J. Mol. Biol. 315 (2002) 385–397, 10.1006/jmbi.2001.5254. [DOI] [PubMed] [Google Scholar]
- [92].Safo MK, Musayev FN, di Salvo ML, Schirch V, X-ray structure of Escherichia coli pyridoxine 5’-phosphate oxidase complexed with pyridoxal 5’-phosphate at 2.0 A resolution, J. Mol. Biol. 310 (2001) 817–826, 10.1006/jmbi.2001.4734. [DOI] [PubMed] [Google Scholar]
- [93].Pedelacq JD, Rho BS, Kim CY, Waldo GS, Lekin TP, Segelke BW, Rupp B, Hung LW, Kim SI, Terwilliger TC, Crystal structure of a putative pyridoxine 5’-phosphate oxidase (Rv2607) from, Mycobacterium Tuberculosis. Proteins 62 (2006) 563–569, 10.1002/prot.20824. [DOI] [PubMed] [Google Scholar]
- [94].Kuo M-F, Wang H-S, Pyridoxal phosphate-responsive epilepsy with resistance to pyridoxine, Pediatr. Neurol. 26 (2002) 146–147, 10.1016/S0887-8994(01)00357-5. [DOI] [PubMed] [Google Scholar]
- [95].Alghamdi M, Bashiri FA, Abdelhakim M, Adly N, Jamjoom DZ, Sumaily KM, Alghanem B, Arold ST, Phenotypic and molecular spectrum of pyridoxamine-50-phosphate oxidase deficiency: a scoping review of 87 cases of pyridoxamine-50-phosphate oxidase deficiency, Clin. Genet. (2020), 10.1111/cge.13843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Footitt EJ, Heales SJ, Mills PB, Allen GFG, Oppenheim M, Clayton PT, Pyridoxal 5’-phosphate in cerebrospinal fluid; factors affecting concentration, J. Inherit. Metab. Dis. 34 (2011) 529–538, 10.1007/s10545-011-9279-7. [DOI] [PubMed] [Google Scholar]
- [97].Bagci S, Zschocke J, Hoffmann GF, Bast T, Klepper J, Muller A, Heep A, Bartmann P, Franz AR, Pyridoxal phosphate-dependent neonatal epileptic encephalopathy, Arch. Dis. Child. Fetal Neonatal Ed. 93 (2008) F151–F152, 10.1136/adc.2006.115162. [DOI] [PubMed] [Google Scholar]
- [98].Ruiz A, García-Villoria J, Ormazabal A, Zschocke J, Fiol M, Navarro-Sastre A, Artuch R, Vilaseca MA, Ribes A, A new fatal case of pyridox(Am)ine 5’-phosphate oxidase (PNPO) deficiency, Mol. Genet. Metabol. 93 (2008) 216–218, 10.1016/j.ymgme.2007.10.003. [DOI] [PubMed] [Google Scholar]
- [99].Schmitt B, Baumgartner M, Mills PB, Clayton PT, Jakobs C, Keller E, Wohlrab G, Seizures and paroxysmal events: symptoms pointing to the diagnosis of pyridoxine-dependent epilepsy and pyridoxine phosphate oxidase deficiency, Dev. Med. Child Neurol. 52 (2010) e133–e142, 10.1111/j.1469-8749.2010.03660.x. [DOI] [PubMed] [Google Scholar]
- [100].Sudarsanam A, Singh H, Wilcken B, Stormon M, Arbuckle S, Schmitt B, Clayton P, Earl J, Webster R, Cirrhosis associated with pyridoxal 5’-phosphate treatment of pyridoxamine 5’-phosphate oxidase deficiency, JIMD Rep 17 (2014) 67–70, 10.1007/8904_2014_338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Coman D, Lewindon P, Clayton P, Riney K, PNPO deficiency and cirrhosis: expanding the clinical phenotype? JIMD Rep 25 (2016) 71–75, 10.1007/8904_2015_456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Porri S, Fluss J, Plecko B, Paschke E, Korff CM, Kern I, Positive outcome following early diagnosis and treatment of pyridoxal-5’-phosphate oxidase deficiency: a case report, Neuropediatrics 45 (2014) 64–68, 10.1055/s-0033-1353489. [DOI] [PubMed] [Google Scholar]
- [103].Musayev FN, Di Salvo ML, Saavedra MA, Contestabile R, Ghatge MS, Haynes A, Schirch V, Safo MK, Molecular basis of reduced pyridoxine 5’-phosphate oxidase catalytic activity in neonatal epileptic encephalopathy disorder, J. Biol. Chem. 284 (2009) 30949–30956, 10.1074/jbc.M109.038372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].di Salvo ML, Mastrangelo M, Nogués I, Tolve M, Paiardini A, Carducci C, Mei D, Montomoli M, Tramonti A, Guerrini R, et al. , Pyridoxine-50-Phosphate oxidase (pnpo) deficiency: clinical and biochemical alterations associated with the C.347g > A (P.·Arg116gln) mutation, Mol. Genet. Metabol. 122 (2017) 135–142, 10.1016/j.ymgme.2017.08.003. [DOI] [PubMed] [Google Scholar]
- [105].Orimo H, Pathophysiology of hypophosphatasia and the potential role of asfotase alfa, Therapeut. Clin. Risk Manag. 777 (2016), 10.2147/TCRM.S87956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Whyte MP, Hypophosphatasia: an overview for 2017, Bone 102 (2017) 15–25, 10.1016/j.bone.2017.02.011. [DOI] [PubMed] [Google Scholar]
- [107].Baumgartner-Sigl S, Haberlandt E, Mumm S, Scholl-Burgi S, Sergi C, Ryan L, Ericson KL, Whyte MP, Hogler W, Pyridoxine-responsive seizures as the first symptom of infantile hypophosphatasia caused by two novel missense mutations (c.677T>C, p.M226T; c.1112C>T, p.T371I) of the tissue-nonspecific alkaline phosphatase gene, Bone 40 (2007) 1655–1661, 10.1016/j.bone.2007.01.020. [DOI] [PubMed] [Google Scholar]
- [108].Khan AA, Josse R, Kannu P, Villeneuve J, Paul T, Van Uum S, Greenberg CR, Hypophosphatasia: Canadian update on diagnosis and management, Osteoporos. Int. 30 (2019) 1713–1722, 10.1007/s00198-019-04921-y. [DOI] [PubMed] [Google Scholar]
- [109].Whyte MP, Physiological role of alkaline phosphatase explored in hypophosphatasia: hypophosphatasia and alkaline phosphatase, Ann. N. Y. Acad. Sci. 1192 (2010) 190–200, 10.1111/j.1749-6632.2010.05387.x. [DOI] [PubMed] [Google Scholar]
- [110].Güzel Nur B, Çelmeli G, Manguoǧlu E, Soyucen E, Bircan İ, Mıhçı E, Pyridoxine-responsive seizures in infantile hypophosphatasia and a novel homozygous mutation in ALPL gene, Journal of Clinical Research in Pediatric Endocrinology 8 (2016) 360–364, 10.4274/jcrpe.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Kuki I, Takahashi Y, Okazaki S, Kawawaki H, Ehara E, Inoue N, Kinoshita T, Murakami Y, Vitamin B6-responsive epilepsy due to inherited GPI deficiency, Neurology 81 (2013) 1467–1469, 10.1212/WNL.0b013e3182a8411a. [DOI] [PubMed] [Google Scholar]
- [112].Orimo H, Hayashi Z, Watanabe A, Hirayama T, Hirayama T, Novel missense and frameshift mutations in the tissue-nonspecific alkaline phosphatase gene in a Japanese patient with hypophosphatasia, Hum. Mol. Genet. 3 (1994) 1683–1684, 10.1093/hmg/3.9.1683. [DOI] [PubMed] [Google Scholar]
- [113].Whyte MP, Hypophosphatasia — aetiology, nosology, pathogenesis, diagnosis and treatment, Nat. Rev. Endocrinol. 12 (2016) 233–246, 10.1038/nrendo.2016.14. [DOI] [PubMed] [Google Scholar]
- [114].Adams JB, George F, Audhya T, Abnormally high plasma levels of vitamin B6 in children with autism not taking supplements compared to controls not taking supplements, J. Alternative Compl. Med. 12 (2006) 59–63, 10.1089/act.2006.12.59. [DOI] [PubMed] [Google Scholar]
- [115].Obara T, Ishikuro M, Tamiya G, Ueki M, Yamanaka C, Mizuno S, Kikuya M, Metoki H, Matsubara H, Nagai M, et al. , Potential identification of vitamin B6 responsiveness in autism spectrum disorder utilizing phenotype variables and machine learning methods, Sci. Rep. 8 (2018), 10.1038/s41598-018-33110-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Laue HE, Korrick SA, Baker ER, Karagas MR, Madan JC, Prospective associations of the infant gut microbiome and microbial function with social behaviors related to autism at age 3 years, Sci. Rep. 10 (2020), 10.1038/s41598-020-72386-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Coburn SP, Mahuren JD, Schaltenbrand WE, Increased activity of pyridoxal kinase in tongue in down’s syndrome, J. Ment. Defic. Res. 35 (Pt 6) (1991) 543–547. [DOI] [PubMed] [Google Scholar]
- [118].Kats DJ, Roche KJ, Skotko BG, Epileptic spasms in individuals with Down syndrome: a review of the current literature, Epilepsia Open 5 (2020) 344–353, 10.1002/epi4.12412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Song H, Ueno S, Numata S, Iga J, Shibuya-Tayoshi S, Nakataki M, Tayoshi S, Yamauchi K, Sumitani S, Tomotake T, et al. , Association between PNPO and schizophrenia in the Japanese population, Schizophr. Res. 97 (2007) 264–270, 10.1016/j.schres.2007.08.004. [DOI] [PubMed] [Google Scholar]
- [120].Tomioka Y, Numata S, Kinoshita M, Umehara H, Watanabe S, Nakataki M, Iwayama Y, Toyota T, Ikeda M, Yamamori H, et al. , Decreased serum pyridoxal levels in schizophrenia: meta-analysis and mendelian randomization analysis, J. Psychiatry Neurosci. 43 (2018) 194–200, 10.1503/jpn.170053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Sandyk R, Pardeshi R, Pyridoxine improves drug-induced parkinsonism and psychosis in a schizophrenic patient, Int. J. Neurosci. 52 (1990) 225–232. [DOI] [PubMed] [Google Scholar]
- [122].Nogovitsina OR, Levitina EV, Effect of MAGNE-B6 on the clinical and biochemical manifestations of the syndrome of attention deficit and hyperactivity in children, Eksp. Klin. Farmakol. 69 (2006) 74–77. [PubMed] [Google Scholar]
- [123].Herrmann W, Lorenzl S, Obeid R, [Review of the role of hyperhomocysteinemia and B-vitamin deficiency in neurological and psychiatric disorders–current evidence and preliminary recommendations], Fortschr. Neurol. Psychiatr. 75 (2007) 515–527, 10.1055/s-2007-980112. [DOI] [PubMed] [Google Scholar]
- [124].Fuso A, Nicolia V, Cavallaro RA, Ricceri L, D’Anselmi F, Coluccia P, Calamandrei G, Scarpa S, B-vitamin deprivation induces hyperhomocysteinemia and brain S-adenosylhomocysteine, depletes brain S-adenosylmethionine, and enhances PS1 and BACE expression and amyloidbeta deposition in mice, Mol. Cell. Neurosci. 37 (2008) 731–746, 10.1016/j.mcn.2007.12.018. [DOI] [PubMed] [Google Scholar]
- [125].Kari G, Rodeck U, Dicker AP, Zebrafish: an emerging model system for human disease and drug discovery, Clin. Pharmacol. Ther. 82 (2007) 70–80, 10.1038/sj.clpt.6100223. [DOI] [PubMed] [Google Scholar]
- [126].Chen P-Y, Tu H-C, Schirch V, Safo MK, Fu T-F, Pyridoxamine supplementation effectively reverses the abnormal phenotypes of zebrafish larvae with PNPO deficiency, Front. Pharmacol. 10 (2019), 10.3389/fphar.2019.01086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Lee G-H, Sung S-Y, Chang W-N, Kao T-T, Du H-C, Hsiao T-H, Safo MK, Fu T-F, Zebrafish larvae exposed to ginkgotoxin exhibit seizure-like behavior that is relieved by pyridoxal-5’-phosphate, GABA and antiepileptic drugs, Dis Model Mech 5 (2012) 785–795, 10.1242/dmm.009449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Kastner U, Hallmen C, Wiese M, Leistner E, Drewke C, The human pyridoxal kinase, a plausible target for ginkgotoxin from Ginkgo biloba, FEBS J. 274 (2007) 1036–1045, 10.1111/j.1742-4658.2007.05654.x. [DOI] [PubMed] [Google Scholar]
- [129].Salamon N, Gurgui C, Leistner E, Drewke C, Influence of antivitamins ginkgotoxin 5’-phosphate and deoxypyridoxine 5’-phosphate on human pyridoxine 5’-phosphate oxidase, Planta Med. 75 (2009) 563e567, 10.1055/s-0029-1185482. [DOI] [PubMed] [Google Scholar]
- [130].Yagi M, Wada K, Sakata M, Kokubo M, Haga M, Studies on the constituents of edible and medicinal plants. IV. Determination of 4-O-methylpyridoxine in serum of the patient with gin-nan food poisoning, Yakugaku Zasshi 113 (1993) 596–599. [DOI] [PubMed] [Google Scholar]