

Abstract

In the last quarter-century, the field of molecular dynamics (MD) has undergone a remarkable transformation, propelled by substantial enhancements in software, hardware, and underlying methodologies. In this Perspective, we contemplate the future trajectory of MD simulations and their possible look at the year 2050. We spotlight the pivotal role of artificial intelligence (AI) in shaping the future of MD and the broader field of computational physical chemistry. We outline critical strategies and initiatives that are essential for the seamless integration of such technologies. Our discussion delves into topics like multiscale modeling, adept management of ever-increasing data deluge, the establishment of centralized simulation databases, and the autonomous refinement, cross-validation, and self-expansion of these repositories. The successful implementation of these advancements requires scientific transparency, a cautiously optimistic approach to interpreting AI-driven simulations and their analysis, and a mindset that prioritizes knowledge-motivated research alongside AI-enhanced big data exploration. While history reminds us that the trajectory of technological progress can be unpredictable, this Perspective offers guidance on preparedness and proactive measures, aiming to steer future advancements in the most beneficial and successful direction.
Keywords: molecular dynamics, multiscale modeling, artificial intelligence, simulation databases, computational physical chemistry, computational biophysics
Introduction
In just 26 years, we will reach the year 2050. Reflecting back to 1998, 26 years ago, the field of molecular dynamics has dramatically transformed from its pioneering days of simulating mainly hundreds of atoms over several nanoseconds.1,2 Currently, we operate with intricate and multiscale models involving millions of atoms/particles simulated over the microsecond time scale.3 Such remarkable progress, marking a significant leap in computational physical chemistry, has been fueled by substantial technological, hardware, and methodological advancements. This evolution is poised to continue and even accelerate with the integration of artificial intelligence (AI) concepts, optimizing human efforts in the preparation, execution, analysis, and even review of MD simulations.
However, this progression has its challenges. The number of simulation studies and, thereby, the volume of data generated by the expanding scientific community are constantly growing, Figure 1, risking the possibility of valuable insights being lost in an overwhelming amount of information. The complexity of models is also increasing, heightening the risk of “garbage in–garbage out” scenarios due to potential mistakes in the preparation phase. With the continuously growing impact of MD simulations in, for instance, biomedical applications,4,5 the potentially incorrect or misleading results can have increasingly severe consequences. Therefore, a key challenge for computational physical chemists (and also scientific publishers) by 2050 will be effectively utilizing, managing, and critically reviewing this wealth of data.6
Figure 1.

Molecular dynamics simulations and artificial
intelligence in scientific
publishing. Data collected from Scopus as of December 21, 2023. The
search was based on finding occurrences of “molecular dynamics”
either alone (“MD”) or in combination with “artificial
intelligence”, “machine learning”, “neural
network”, and “deep learning” phrases (“MD
+ AI”) within the title, abstract, or keywords of publications
indexed in the Scopus database. The collected data were fitted and
extrapolated to the year 2050. The projected values,  , are shown in the inlet rectangles and
compared with publication data from the years 1998 and 2023 (N1998 and N2023,
respectively). The utilized Scopus data set is available on Zenodo
under DOI: 10.5281/zenodo.10673694.
, are shown in the inlet rectangles and
compared with publication data from the years 1998 and 2023 (N1998 and N2023,
respectively). The utilized Scopus data set is available on Zenodo
under DOI: 10.5281/zenodo.10673694.
The role of AI in shaping the future of many fields, including computational physical chemistry, also becomes apparent. AI is already often coupled to MD simulations: the noticeable growth of combined MD and AI studies started less than ten years ago, and further AI’s involvement is likely to surge dramatically, Figure 1. While this combination of methods is promising to expedite groundbreaking discoveries, the integration of AI is set to further amplify the volume of data generated, potentially reaching scales unmanageable by human researchers alone. This scenario suggests a future in which AI-driven simulations become so advanced and numerous that their review and analysis may be feasible only with the assistance of, or entirely by, equally sophisticated AI systems. Moreover, such AI-driven analysis could become essential in formulating initial hypotheses and recommending simpler simulations to verify or refute them.
In this Perspective, we envision a future in 2050 (elaborating on current trends and necessary actions to take) where every MD simulation can be traced through global open-access databases integrated with sophisticated AI models. These databases, ideally encompassing every conceivable MD simulation, would link to peer-reviewed publications or open-access platforms housing detailed simulation data and additional necessary information. They would also accommodate nonpublished, failure, and even “wrong” data, thus reducing redundant efforts and encouraging a more comprehensive scientific dialogue. Advanced AI models in this envisioned future would not only organize these data but also evaluate, refine, and improve simulation force fields by aligning them with experimental data and cross-validating various model resolutions, such as all-atom vs coarse-grained simulations. Furthermore, these models would autonomously conduct new MD simulations, identify inconsistencies, suggest enhanced parameters or even new phenomena, and enable the modeling of increasingly complex and multiscale systems. However, realizing this vision requires a monumental collaborative effort involving back-tracking—or rather regenerating according to Moore’s law—likely millions of existing simulation trajectories and fostering open scientific collaboration worldwide—a necessary “entrance fee to 2050” for securing a bright future in computational physical chemistry.
For conciseness and focus, this Perspective primarily discusses molecular simulations within biological systems that bridge the fields of physical chemistry and biophysics. Biological systems, with their diverse sizes and time scales, provide a rich context for discussing computational advancements. For instance, compare the first ever one-microsecond-long simulation capturing the folding of a 36-residue-long peptide in 19987 and the 500 ps long simulations of a 64-dipalmitoylphosphatidylcholine bilayer a year earlier8 with today’s simulations where a 1 μs duration is often the minimum benchmark for force field comparison.9 Moreover, modern simulations frequently can involve much larger proteins embedded in multicomponent lipid bilayers containing hundreds of lipids.10,11 Current biological simulations set even more ambitious milestones, both in terms of simulation length (ranging from microseconds to seconds) and system size and complexity—from realistic cellular membranes12−15 to an entire ribosome,16 minimal cell,17 virus capsid,18,19 or envelope structure of SARS-CoV-2.20 These and many other examples provide a valuable lens through which to view the evolution and future of MD simulations.
This Perspective does not delve into the technical details or listing available AI models, and we refer instead to a large number of recent works, overviews, and special issues exclusively dedicated to this topic, see, e.g., refs (21−33) and references therein. We rather focus on the practical applications of AI concepts, presuming that by 2050, AI models will achieve unprecedented levels of sophistication, and we need just to choose which ones to use or allow AI to make these decisions for us. We briefly discuss the current state of molecular dynamics simulations, the emerging challenges in data storage, the potential application of AI, and what steps should be undertaken by 2050 or even right now. For clarity, in this Perspective, the term “AI” typically refers to a variety of terms, including “machine learning”, “neural networks”, “large language models”, and others. Though not technically precise, this simplifies our discussion without compromising the overall understanding.
It is also important to recognize the uncertainties inherent in forecasting over such an extended time frame. As we stand in 2024, our vision for 2050 is shaped by the developments we observe today. The unpredictable nature of technological evolution, especially in a field as dynamic as AI, means that the future may significantly deviate from our current expectations. Nevertheless, certain trends and necessary actions are already evident, and here, we aim to underscore the importance of adaptability in navigating the constantly evolving scientific landscape.
Current Prospects and the Need of Artificial Intelligence
We live in an era marked by the continuous and likely exponential growth of information including scientific data. Individual scientists or even large research teams cannot feasibly process it all. Even now, it is close to, if not impossible, for a scientist to read or browse through all of the new relevant articles in the field of interest, let alone examine the publicly available MD simulations. Therefore, developing tools that enable researchers to stay up to date becomes a practical necessity.
While preferences for such tools are subjective, the successful existing tools for similar purposes are typically intuitive, straightforward to use, and visually appealing. Most likely, precisely these characteristics contributed to the widespread popularity of AI-managed user-chat interfaces like ChatGPT (https://chat.openai.com), Bard with its most recent Gemini AI model (https://bard.google.com/), or Copilot (https://copilot.microsoft.com/). Adopting and advancing a similar idea for specific scientific purposes seems to be an optimal solution for the near future. The chat-like approach could revolutionize the initial interaction with scientific information or even performing MD simulations, see Figure 2 for a possible future example, making AI the primary tool for administration and search functions. The final data may still be presented in traditional formats, such as tables or diagrams, to meet conventional expectations for scientific data display.
Figure 2.
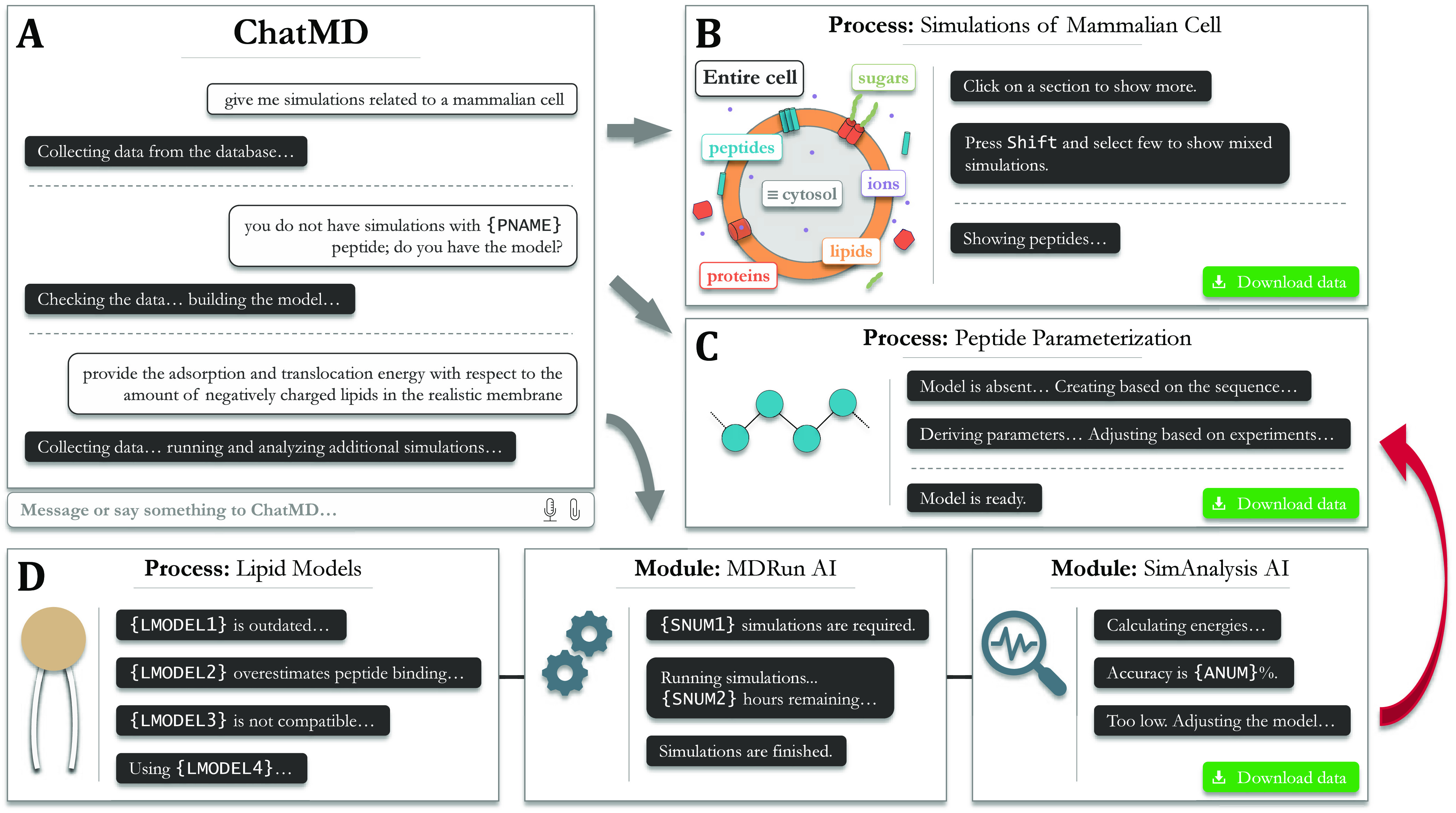
Possible schematic interface of a futuristic molecular dynamics AI-maintained engine capable of setting up, running, and analyzing simulations. (A) Initial user-interaction interface. (B) Dashboard of molecular dynamics simulations of mammalian cells. (C) New molecule parametrization interface: adding an unlisted peptide. (D) Interface of a membrane model selection compatible with a new peptide model followed by simulation and analysis interfaces.
While the prospect of AI models managing MD simulations in the coming years is feasible, verifying their consistency and accuracy is a more profound challenge. The adage “garbage in, garbage out” is particularly pertinent. It is crucial to acknowledge that simulation results are affected not just by the chosen model (called “force field” in classical MD) but also by subtle variations in simulation protocols, software choices, and data analysis methods.34,35 Minor discrepancies might be inconsequential on a smaller scale, but an AI model trained on extensive data sets could amplify these errors. This could result in “hallucinating simulations or force fields” that appear to generate at first glance realistic but ultimately inaccurate results.
A less acknowledged issue is whether the converse concept—good in, good out—always holds true. The answer is not necessarily affirmative since a successful combination of a model and simulation protocol for one system does not guarantee success for even a slightly altered system.36,37 An intriguing consideration, for example, is whether current atomistic force fields can maintain the stability of secondary and tertiary protein structures over millisecond time scales.38 It is possible that some, or perhaps all, of the force fields may not be adequate for the next era of simulations. Similar concerns apply to MD simulation codes: extended time scales of larger systems are already revealing numerical inaccuracies and bugs that are usually not apparent in smaller systems and shorter simulations.39
Overall, the key is to carefully select a model that is appropriate for the specific application.40 Currently, most large-scale simulations rely on established models extensively validated by the scientific community. These simulations yield groundbreaking insights despite the inherent model limitations; even all-atom force fields are essentially a coarse-grained representation of quantum reality. Typically, the model limitations are identified, and potential solutions are proposed.41−43 However, these solutions could be system-dependent or even not widely known and not incorporated into the standard protocols associated with the original model. Such circumstances lead to a complex sequence of updates and inconsistencies between simulations that can be difficult to track. This problem can be mitigated by implementing open-science practices aimed to collect simulations into dedicated repositories for cross-evaluation44−46 (see section Challenges in Data Management for representative examples such as NMRlipids46). In all these scenarios, AI can play an invaluable role in model comparison and validation, offering insights and guidance that might otherwise be unattainable even when performing replica simulations to prevent “false positive” conclusions.47
Beyond MD models and simulations per se, related tools like AlphaFold48 are also expected to evolve and advance further. Apart from known strengths and limitations of the current AlphaFold2,49−51 the main fundamental concern is that the single structure does not represent functional motions of a protein.52 Therefore, future tools might be capable of predicting the dynamics of molecules, offering more than a static snapshot of their structures (one might suggest a title like “AlphaDynamics”). The involvement of MD concepts in such tools is very promising, and the first steps have already been made, see, e.g., ref (53), where multiple Boltzmann-ranked ensembles from a single protein sequence can be generated and then propagated by MD. However, the quality of these tools will again rely on the quality of reference data that might be more ambiguous due to the different models and resolutions used and time scales explored.
This leads us to envision global AI-administrated databases as self-expanding and self-correcting entities, not merely repositories awaiting human input. Integrated AI within these databases should identify errors and inconsistencies. AI could also try to autonomously fix them by performing missing simulations or asking for human intervention to distinguish possible errors from yet-unknown phenomena. Without AI functionalities, the substantial efforts invested in creating these databases may not be fully exploited, especially given the continuous refinement of the models and the generation of new simulations. In the end, we could be equipped with AI tools that organize data, allow “communication” with them, verify and double-check accuracy, and fill in missing gaps without explicit requests. The human role would then be as a moderator who applies sanity checks, makes major decisions and critical adjustments, and distills the key insights, essentially trying to make sense of AI-proposed data interpretations. In modern terms, a scientist should be able to effectively “prompt” the AI to provide or generate information or simulation and interact with it until a desirable and meaningful outcome is achieved. Realizing this vision requires thoughtful and careful implementation of technologies to ensure widespread adoption and success.
Coupling Molecular Dynamics and Artificial Intelligence
To grasp how AI can enhance MD simulations, it is helpful to characterize the current state and typical applications. Below, we discuss MD simulations along several critical points, which, looking ahead to 2050, are anticipated to evolve dramatically with the help of AI.
First of all, the model resolution is a crucial factor determining the underlying accuracy and accessible scales of MD simulations.54 It can be categorized into the following: (i) ab initio molecular dynamics, where the nuclei are treated as classical particles, while the electronic structure and interatomic forces are calculated from first-principles;55 (ii) all-atom molecular dynamics, where each atom (or a ghost/virtual particle) is modeled as a sphere with a constant partial charge, while interatomic interactions are derived from (semi)empirical potentials; (iii) coarse-grained molecular dynamics, where multiple atoms are united into larger units called “beads” and further used to construct small biological entities such as amino acids, nucleotides, or saccharides (see, e.g., popular Martini,56 SPICA,57 or SIRAH58 force fields); interactions in coarse-grained models are often described using potentials similar to those in all-atom simulations; (iv) ultra coarse-grained models with implicit solvent, representing large biological entities with arbitrary building blocks,59,60 while interactions, including their form, are specifically tailored for particular purposes or systems of interest.
Selecting the appropriate model resolution is typically the first step in the design of a research plan. This choice determines the simulation’s capabilities, namely, which system’s properties can be accurately described, if at all. Each of the listed approaches has significantly contributed to the development of MD simulations, as illustrated by various examples spanning from 1998 until now; see Table 1. These examples essentially represent “(among) first-of-their-kind” events, potentially viewed as significant milestones within the field, in terms of scientific discoveries or methodological innovations.
Table 1. Examples of Notable Molecular Dynamics Studies.
| method | system and simulated time | landmark | reference |
|---|---|---|---|
| 1998 | |||
| Car–Parrinello ab initio MD | glucose in 58 waters, several ps | first ab initio MD simulation of a solvated glucose | (61) |
| all-atom force field MD | HP-36 peptide in ∼3000 waters, 1 μs | first μs-long simulation of a protein folding | (7) |
| all-atom force field MD | N/A | presentation of CHARMM22 all-atom force field for proteins | (62) |
| coarse-grained MD | a few hundreds CG particles, up to 5 μsa | pioneering CG simulation of a lipid bilayer self-assembly | (63) |
| 2022–2023 | |||
| Born–Oppenheimer ab initio MD | chignolin in water, 10,000 × 225 fs | fully explored conformational space of a 10-residue protein at ab initio MD level | (64) |
| all-atom force field MD | N/A | description of Anton 3 supercomputer enabling ms-long biosimulations | (65) |
| all-atom MD | ∼44 million atoms, several ns | atomistic simulation of HIV capsid using machine-learning potential | (66) |
| coarse-grained MD | ∼37 million CG particles, 1 μs | a coarse-grained simulation of the entire SARS-CoV-2 envelope | (20) |
| coarse-grained MD | ∼550 million CG particles, not simulated | first coarse-grained Martini model of an entire cell | (17) |
| 2050 | |||
| Multiscale ms-long simulations of an entire cell? | |||
Approximate time scale.
While the progress over the last years is evident, simulations incorporating multiple resolutions within a single system generally advance more slowly due to their methodological and technical complexity. In the future, we may witness the emergence of advanced hybrid methodologies like AI-accelerated multiscale simulations. These simulations might adopt varying resolution levels for different system parts (for example, a quantum description of active sites while the remainder is described classically), dynamically adjust them, and even attempt to extrapolate system behavior to extend the time scale. In current terms, this possible approach could be termed as the automatically generated and regulated on-the-fly version of quantum mechanics/molecular mechanics, or simply the future iteration of it.67,68 Such simulations would potentially facilitate the modeling of processes, where both the quantum description and associated conformational changes are crucial, e.g., enzyme catalysis69 or electron transfer in proteins.70 Similarly, coupling AA and CG resolutions in a single simulation would improve the accessible time scales while maintaining atomistic accuracy where necessary. Although AA/CG models already exist,71−73 they again lack the capability for on-the-fly adjustment of resolution partitioning. However, this challenge is primarily technical and appears to be surmountable with the integration of AI algorithms.
The fusion of simulations with experiments is also expected to become more seamless, with AI playing a pivotal role in bridging these two realms such as suggesting new simulations or experiments for improved correlation. The cornerstone of molecular simulations is gaining the information not accessible by the available experimental methods, e.g., the simulation is actually a stand-alone research aimed at elucidating mechanisms of various phenomena or making predictive analyses. However, simulations can also play a synergistic role in conjunction with experimental studies, i.e., the information gathered by experiments could focus simulations and vice versa.74 For instance, MD simulations can be performed imposing restraints from NMR or cryogenic electron microscopy experiments.75,76 To enhance that further, we anticipate a broader integration of high-performance computing and AI, fostering a more complex interplay. This integration may be particularly fascinating as simulations and experiments typically drift toward each other: simulations strive to characterize larger biological assemblies over extended time scales, whereas the most recent experimental techniques increase the resolution to capture events occurring over shorter periods. Together, these approaches should aim to replicate in vivo, i.e., biologically relevant systems and conditions as closely as possible.
The model and method development is also a vital part of computational physical chemistry, which can greatly benefit from AI concepts. The (semi)automatized or/and AI-driven model/force field generation is already under extensive development77−91 but yet to be tested beyond the current “comfort zone” of a limited range of systems. While methodological studies frequently incorporate experimental data, more often than not, they repurpose already available data from well-understood systems. A classic example is the alanine dipeptide—a small, thoroughly studied system where a biomolecule exhibits rare events in solution at room temperature. It is typically used for benchmarking various enhanced sampling methods92 and lately, the quality of machine learning potentials.93,94 Therefore, generating more reference (“training”) data is essential for the development and validation of novel force field parameters. The ever-increasing computational power, including promising advances in quantum computing,95 is anticipated to become increasingly important in producing such data, e.g., through accelerated and AI-enhanced first-principles calculations.
However, the reference data must always be carefully evaluated. As simulations scale up in size and complexity, they naturally become more prone to errors arising from either human oversights or previously unexplored regions of conformational space. Consequently, ensuring the transferability and transparency of simulation data is crucial despite technical hurdles. Overall, simulations can be classified into three categories based on their complexity and shareability: (i) simulations that are straightforward to perform, reproduce, and share; (ii) simulations that are challenging but feasible to perform, reproduce, and share; (iii) simulations that are often impossible for a given researcher to perform or reproduce and are similarly difficult to share. The last category thus demands the main attention in the upcoming years.
The need for transparency is even more important, given that the field of MD simulations has significantly lowered the entry barrier, making them increasingly routine and readily utilized as a “black box”. Tools like CHARMM-GUI96 and Martinize97 exemplify how simulations can be set up and run within mere minutes or hours, which is a stark and positive change from just a few years ago. Similarly, the accessibility of force field parameters in various simulation packages has been improved. For instance, CHARMM-GUI supports the generation of simulation files for major force fields like CHARMM98 and AMBER99 compatible with not only “home” MD engines100,101 but also open-source codes such as Gromacs,102 NAMD,103 OpenMM,104 or Desmond.105 The existence of web-based databases sharing molecular topologies and force field parameters also contributes positively.106,107 However, there is a potential downside to this accessibility: the widespread availability of simulations could lead to a decline in their quality. The development of multiple user-friendly and open-source codes (which, undoubtedly, is a great step forward) enables the execution of molecular simulations with little knowledge of the underlying physics and MD algorithms. The incorporation of AI may further simplify employing MD, reducing it to merely instructing an AI tool to execute a simulation without the user needing to select a specific force field, software, or simulation parameters. While this could lead to unbiased AI-driven decision-making, it places considerable “responsibility” on the AI tools, including cross-validation and searching for errors.
Nevertheless, human involvement remains an integral part of the development of AI tools. These tools are defined by the model architecture, the data used for training, the objectives they aim to achieve, evaluation metrics, the process of fine-tuning, and subsequent validation. At every stage, human decisions may play a critical role, simultaneously guiding the direction of AI and introducing a degree of subjective bias into the outcomes it produces. For example, selecting a force field for the MD simulation reflects a personal decision influenced by numerous factors. These considerations, whether intentional or subconscious, may also influence the design of AI tools tasked with identifying “the best” force field. Thus, although AI tools are called to automate processes and analyze data volumes beyond human capability, their development and oversight still fundamentally depend on human expertise and judgment. Recognizing the potential diversity of AI tools developed by various teams, one will be expected to test a broad array of those to ensure the consistency and robustness of the results they yield.
Finally, all of these potential AI-driven advancements will also lead to a substantial increase in data generation, with its own set of challenges and implications. Larger, more complex simulations will produce vast amounts of data, demanding storage and analysis. Thereby, this data surge will place significant pressure on the quality of training data for AI tools of various types.108 To mitigate the risks associated with the potential data explosion and facilitate efficient error detection, simulations should be subjected to rigorous cross-validation. Such validation becomes feasible only if the training data are collected in shared vast databases, underscoring the need for a unified approach to data management and transparency.
Challenges in Data Management
Science, driven by both successes and mistakes, thrives on open communication and data sharing. In the realm of MD simulations, managing and accessing vast data volumes is essential yet challenging.109 This becomes increasingly critical when considering the need for well-managed and meticulously verified training data for AI applications. While the format of simulation raw data can be easily standardized, managing large data sets at institutional, national, or global levels is increasingly demanding, both technically and financially.110−112 Even with standardized formats, storing simulation files proves more complex than storing files such as .pdb, which essentially represent the end result of protein structure determination. Consequently, efficient indexing and data storage, imperative for leveraging years of accumulated research, emerges as a critical and mandatory objective to be achieved well before 2050. The data indexing should not be limited to technical aspects but should also account for scientific context, e.g., by highlighting the connections between data sets that are not related from a digital perspective. Although there has been steady progress in this direction over the past years, monumental efforts are still required for significant advancements.
Typically, currently existing databases focus on specific research or methodological areas,113−119 like Protein Data Bank,113 Uniprot,118 or NMR global databank,119 to mention only a few. The existence of such databases was central in developing predictive tools such as AlphaFold48 or RoseTTAFold.120 The idea of databases dedicated to MD models and simulations has been around since at least 1999,44,45,121−128 but progress has been limited due to the constraints of resources among researchers, who typically maintain these databases as a secondary task.
In addition to prominent platforms like GitHub (https://github.com/) or GitLab (https://gitlab.com/), repositories such as Zenodo (https://zenodo.org/), Figshare (https://figshare.com/), Open Science Framework (https://osf.io/), Dryad (https://datadryad.org/stash), or Mendeley Data (https://data.mendeley.com/) are increasingly used to store simulation data and codes. However, these repositories are primarily intended for hosting supplementary materials for peer-reviewed publications, such as simulation input files. Storage of hundreds of gigabytes of simulation trajectories presents a nontrivial challenge for these platforms. For example, Zenodo’s upload limit stands at 50 GB in January 2024, which might be insufficient for the trajectories and simulation data of many publications. Although it is theoretically possible to divide the data across multiple repositories, one must upload dozens of segmented parts when dealing with exceptionally large and complexly fragmented data.
Nevertheless, there are already valuable examples that demonstrate how to store and further analyze large amounts of simulation data, Figure 3. Tiemann et al.129 recently conducted a systematic analysis of the MD data accumulated in Zenodo, Figshare, and Open Science Framework repositories. They introduced a designated prototype application, https://mdverse.streamlit.app/, that efficiently organizes the collected MD data, Figure 3A. Building on their findings, they offered practical data management tips, such as storing original files uncompressed, providing detailed metadata, and linking to related resources. These recommendations highlight the critical role of well-indexed data; without proper indexing, even accessible data can become useless.130
Figure 3.

Examples of databases and applications that store and organize molecular dynamics data. (A) MDverse data explorer (https://mdverse.streamlit.app/): an online application navigating through the simulation data collected from Zenodo (https://zenodo.org/), Figshare (https://figshare.com/), and Open Science Framework (https://osf.io/) repositories. (B) NMRlipids databank (https://databank.nmrlipids.fi/): a catalog containing atomistic MD simulations of biologically relevant lipid membranes. The search result for “POPC” is shown. (C) BioExcel-CV19 (https://bioexcel-cv19.bsc.es/): a platform designed to provide web access to atomistic MD trajectories for macromolecules involved in the COVID-19 disease.
The NMRlipids project,46,131https://nmrlipids.blogspot.com/, is, in turn, a promising example of an MD databank, https://databank.nmrlipids.fi/, focused on a well-defined topic. It contains various lipid-membrane simulation data spanning different compositions and modeled conditions, generated using various force fields and MD codes and compared to experiments when available. The interface of this databank is straightforward and intuitive, Figure 3B, suitable for the relatively simple nature of the data. However, more complex data sets would require more flexible and efficient interfaces for effective data navigation. In the biological context, the variability of modeled systems and their composition are apparent, while lipid simulations benefit from the countable and categorizable nature of lipid types. It is thereby more advantageous to offer programmatic access via the API — application programming interface. The NMRlipids project,46 alongside other initiatives,132 has already made significant progress in this area, allowing a smoother training of related AI models.
Finally, the COVID-19 pandemic showcased how global crises can catalyze the development of centralized databases, as seen with the creation of the pandemic-dedicated database, https://covid.bioexcel.eu/, with related molecular simulations,132−134https://bioexcel-cv19.bsc.es/, Figure 3C. This event underscored the value and potential of such databases. Probably, as a consequence, in 2023, a highly ambitious and promising initiative, Molecular Dynamics Data Bank (MDDB, https://mddbr.eu/), received funding, aiming to establish a comprehensive European database for MD data. This initiative seeks to develop an infrastructure for efficient data storage, exchange, analysis, and integration, enhancing the utility and accessibility of MD data.
These positive trends in data storage practices and the rise of larger initiatives are encouraging. However, adhering to FAIR principles135−137—ensuring data are findable, accessible, interoperable, and reusable—remains a challenge in practical implementation. Additionally, sharing “failure data”, which could prevent redundant efforts among research groups, is still largely overlooked. Such data are rarely published or made publicly available. To effectively leverage data from centralized databases, it is also essential to establish and implement various controls and “moderators” to oversee and manage these comprehensive repositories. Fortunately, these challenges are mainly well-known, and we possess the necessary knowledge and skills to address them.
Pathways to 2050
The path of computational physical chemistry toward 2050 relies on several pillars. First, we should rigorously gather, index, organize, and verify the simulation data. Second, an effective search interface is needed to navigate the vast quantities of data, since any database lacking robust search functionality would fall short of its potential. Third, we need to maximize data utilization and foster self-expansion, ensuring that the collected data contribute to advancing the field rather than merely exist as a static digital library. Finally, incorporating control mechanisms into AI-driven research is crucial to avert the accumulation of extensive but flawed data.
Data sharing and accessibility are central to this vision, since transparency is crucial for AI model training and related force field development as it reduces uncertainties in AI applications. The scientific community should be more open with their findings, and publishers should enforce data accessibility. Currently, data availability is often labeled as “available upon reasonable request”, which is not always the case.138 The practices of raw data sharing, particularly simulations, have already been implemented by several publishers.139,140 More transparent data accessibility should be a standard publishing requirement well earlier than by 2050, of course, taking into account the protection of intellectual property and proprietary data.
Another key aspect is to ensure the accessibility of peer-reviewed articles associated with simulation data to prevent limited accessibility of the necessary data to AI codes. While data sets should have thorough indexing and metadata, related manuscripts often contain vital information not found in the data sets themselves. The current trend toward open-access policies141 is expected to result in all scientific journals being open-access by 2050, greatly enhancing data accessibility. Generally, AI should enhance the scientific dialogue and information exchange, e.g., by making scientific tools more widely available, suggesting potential collaborators for specific research projects or transcending possible language barriers.
From another point of view, while AI integration is inevitable, it must be approached with caution. AI can retrieve patterns beyond human perception, but distinguishing real insights from noise is vital. A major concern is a possible amplification of minor AI errors, leading to “false positive” scientific claims. The human bias in AI development could also exacerbate this issue, highlighting the need for skepticism and vigilance. Additionally, scientific data and publications naturally contain errors,142 and although they can be found, it is not that simple to quickly fix them.143,144 A record number of retracted papers in 2023 is particularly alarming.145,146 Thus, AI should be capable of autonomously filtering this information to avoid being misled by it while also being able to discern potential errors from novel insights that lie outside of its training data.
We also should not replace the “knowledge first” concept with the “big amount of data” that AI provides. For instance, while AI algorithms can already streamline the predictions of protein–ligand binding free energies147,148 or free energy of permeation through the membrane,149 it may struggle with seemingly next-step tasks, such as screening of antimicrobial peptides. Even if we collect all amino-acid sequences known to be effective in a single database and then apply AI to fill in the gaps, the results are far from being guaranteed to be successful. The efficiency of such peptides heavily depends on multiple factors, many of them counter-acting each other, e.g., mechanism of action, selectivity, peptide’s structure, net charge, and amphiphilicity, target microorganism, minimum inhibitory concentration, or possible side effects.150,151 Even if AI successfully predicts certain sequences, more is needed to elucidate their functional mechanisms and operational conditions. Thus, a deep understanding of the underlying processes remains more important than blind screening through vast pools of possible combinations (however, it is fair to recognize that if a drug developed by AI proves effective, it represents a huge achievement regardless).
In other words, AI principles should serve to augment and enhance physics-based simulations rather than to replace them. Although AI algorithms can be used to extrapolate the system’s behavior using potentials of arbitrary form, the breakdown and analysis of contributing physical forces, e.g., electrostatics or dispersion interactions, remains crucial. Furthermore, while AI-generated force fields and neural network (“’black box”) potentials show promise,21 their application in large-scale simulations needs further exploration. The active development of “black-box” potentials logically necessitates the creation of AI tools that moderate the products of other AI codes.152
Attention should also be paid to various biological subtopics. For instance, while proteins and lipids have received significant focus, carbohydrate research is gaining momentum only now153−156 (including the development of designated databases157), partly due to the important role of glycans in the conformational dynamics and shielding of COVID-19.158 The adequately distributed efforts across various fields can foster interdisciplinary communication and facilitate unraveling fundamental scientific questions from multiple perspectives.
In conclusion, as we approach 2050, the integration of AI in computational physical chemistry and biophysics holds tremendous potential for revolutionizing molecular dynamics simulations and data analysis. However, achieving the necessary progress mandates a responsible approach to data management, ensuring a future foundation built on accuracy and integrity. The future computational physical chemist will need to proficiently develop and manage diverse AI tools, addressing various objectives while being mindful of the inherent human biases in AI creation. The potential emergence of artificial general intelligence (AGI)159 could further transform not only physical chemistry but also the broader scientific landscape and society. However, the actual definition of AGI and its practical implementation and contribution to physical chemistry remain a topic for future discussions.
Final Remarks
Molecular dynamics simulations are in constant evolution, and making predictions about their future is an exciting exercise. It is becoming clear that artificial intelligence will significantly influence computational physical chemistry among other fields. By 2050, computational scientists will likely be armed with an array of AI tools. These tools will enable them to navigate through existing data, develop new models, refine current ones, initiate simulations, or even delegate these tasks to AI, including creatively analyzing the outcomes. A critical skill will likely be the ability to effectively communicate with or “prompt” future AI systems. Sharping this skill will presumably require specialized training, including educational programs designed for students. The development of AI tools would also benefit from and perhaps even require robust databases of meticulously accumulated and organized knowledge. These databases provide the indispensable “raw” material for training diverse AI applications and searching intriguing scientific questions emerging from inconsistencies often present in these data.
On a final note, the future is inherently unpredictable. While the emergence of tools like AlphaFold and ChatGPT could be anticipated, their actual impact has been staggering, reshaping not only science but also everyday life. Thus, while our forecasts for future developments might seem reasonable at the moment, new discoveries often lead to even more rapid and unexpected advancements. This accelerated evolution demands from scientists both creativity and adaptability. One such creative approach might be to ask AI itself regarding its vision for 2050. ChatGPT-4’s response to the question “Answer in one sentence how will molecular dynamics and computational physical chemistry look like in 2050” was the following (https://chat.openai.com/share/6025fea1-edc9-493c-8f9c-8a8908f343b2): “By 2050, molecular dynamics and computational physical chemistry will likely be highly advanced, utilizing quantum computing and AI to simulate complex systems with unprecedented speed and accuracy.” Therefore, we can approach the future with optimism, unless ChatGPT-4 was hallucinating.
Acknowledgments
This work was supported by the Czech Science Foundation (grant 24-11274S), the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement No. 101001470), and the project “National Institute of Virology and Bacteriology (Program EXCELES, ID Project No LX22NPO5103)—Funded by the European Union—Next Generation EU”. The authors thank the members of groups of Robert Vácha, Pavel Jungwirth, and Hector Martinez-Seara for the endless discussions about molecular simulations—their past, current, and future—many outcomes of which entered the unconscious and crystallized in this Perspective. The authors acknowledge the supervised usage of ChatGPT and Grammarly for improving the readability of the manuscript.
Author Contributions
CRediT: Denys Biriukov conceptualization, formal analysis, funding acquisition, investigation, methodology, project administration, visualization, writing-original draft, writing-review & editing; Robert Vacha conceptualization, funding acquisition, project administration, writing-review & editing.
The authors declare no competing financial interest.
Special Issue
Published as part of ACS Physical Chemistry Auvirtual special issue “Visions for the Future of Physical Chemistry in 2050”.
References
- Ciccotti G.; Dellago C.; Ferrario M.; Hernández E. R.; Tuckerman M. E. Molecular simulations: past, present, and future (a Topical Issue in EPJB). Eur. Phys. J. B 2022, 95, 1–12. 10.1140/epjb/s10051-021-00249-x. [DOI] [Google Scholar]
- Frenkel D.; Smit B.. Underst. Mol. Simul. from Algorithms to Appl.. Third Ed.; Elsevier, 2023; pp 1–728, DOI: 10.1016/C2009-0-63921-0. [DOI] [Google Scholar]
- Gupta C.; Sarkar D.; Tieleman D. P.; Singharoy A. The ugly, bad, and good stories of large-scale biomolecular simulations. Curr. Opin. Struct. Biol. 2022, 73, 102338. 10.1016/j.sbi.2022.102338. [DOI] [PubMed] [Google Scholar]
- Hollingsworth S. A.; Dror R. O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. 10.1016/j.neuron.2018.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed M.; Maldonado A. M.; Durrant J. D. From Byte to Bench to Bedside: Molecular Dynamics Simulations and Drug Discovery. BMC Biol. 2023, 21, 1–4. 10.1186/s12915-023-01791-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sever R. We need a plan D. Nat. Methods 2023, 20, 473–474. 10.1038/s41592-023-01817-y. [DOI] [PubMed] [Google Scholar]
- Duan Y.; Kollman P. A. Pathways to a protein folding intermediate observed in a 1-microsecond simulation in aqueous solution. Science 1998, 282, 740–744. 10.1126/science.282.5389.740. [DOI] [PubMed] [Google Scholar]
- Berger O.; Edholm O.; Jähnig F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 1997, 72, 2002–2013. 10.1016/S0006-3495(97)78845-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javanainen M.; Heftberger P.; Madsen J. J.; Miettinen M. S.; Pabst G.; Ollila O. H. Quantitative Comparison against Experiments Reveals Imperfections in Force Fields’ Descriptions of POPC–Cholesterol Interactions. J. Chem. Theory Comput. 2023, 19, 6342–6352. 10.1021/acs.jctc.3c00648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karki S.; Javanainen M.; Rehan S.; Tranter D.; Kellosalo J.; Huiskonen J. T.; Happonen L.; Paavilainen V. Molecular view of ER membrane remodeling by the Sec61/TRAP translocon. EMBO Rep 2023, 24, e57910 10.15252/embr.202357910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javanainen M.; Karki S.; Tranter D.; Biriukov D.; Paavilainen V. O.. The Sec61/TRAP Translocon Scrambles Lipids. bioRxiv, November 23, 2023, ver. 1. 10.1101/2023.11.23.568215 (accessed 2024-03-04). [DOI]
- Ingólfsson H. I.; Melo M. N.; Van Eerden F. J.; Arnarez C.; Lopez C. A.; Wassenaar T. A.; Periole X.; De Vries A. H.; Tieleman D. P.; Marrink S. J. Lipid organization of the plasma membrane. J. Am. Chem. Soc. 2014, 136, 14554–14559. 10.1021/ja507832e. [DOI] [PubMed] [Google Scholar]
- Lorent J. H.; Levental K. R.; Ganesan L.; Rivera-Longsworth G.; Sezgin E.; Doktorova M.; Lyman E.; Levental I. Plasma membranes are asymmetric in lipid unsaturation, packing and protein shape. Nat. Chem. Biol. 2020, 16, 644–652. 10.1038/s41589-020-0529-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doktorova M.; Symons J. L.; Zhang X.; Wang H.-Y.; Schlegel J.; Lorent J. H.; Heberle F. A.; Sezgin E.; Lyman E.; Levental K. R.; Levental I.. Cell Membranes Sustain Phospholipid Imbalance Via Cholesterol Asymmetry. bioRxiv, July 31, 2023, ver. 1. 10.1101/2023.07.30.551157 (accessed 2024-03-04). [DOI]
- Biriukov D.; Javanainen M. Efficient Simulations of Solvent Asymmetry Across Lipid Membranes Using Flat-Bottom Restraints. J. Chem. Theory Comput. 2023, 19, 6332–6341. 10.1021/acs.jctc.3c00614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock L. V.; Gabrielli S.; Kolár̃ M. H.; Grubmüller H. Simulation of Complex Biomolecular Systems: The Ribosome Challenge. Annu. Rev. Biophys. 2023, 52, 361–390. 10.1146/annurev-biophys-111622-091147. [DOI] [PubMed] [Google Scholar]
- Stevens J. A.; Grünewald F.; van Tilburg P. A.; König M.; Gilbert B. R.; Brier T. A.; Thornburg Z. R.; Luthey-Schulten Z.; Marrink S. J. Molecular dynamics simulation of an entire cell. Front. Chem. 2023, 11, 1106495. 10.3389/fchem.2023.1106495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perilla J. R.; Schulten K. Physical properties of the HIV-1 capsid from all-atom molecular dynamics simulations. Nat. Commun. 2017, 8, 1–10. 10.1038/ncomms15959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasova E.; Nerukh D. All-Atom Molecular Dynamics Simulations of Whole Viruses. J. Phys. Chem. Lett. 2018, 9, 5805–5809. 10.1021/acs.jpclett.8b02298. [DOI] [PubMed] [Google Scholar]
- Pezeshkian W.; Grünewald F.; Narykov O.; Lu S.; Arkhipova V.; Solodovnikov A.; Wassenaar T. A.; Marrink S. J.; Korkin D. Molecular architecture and dynamics of SARS-CoV-2 envelope by integrative modeling. Structure 2023, 31, 492–503.e7. 10.1016/j.str.2023.02.006. [DOI] [PubMed] [Google Scholar]
- Behler J.; Parrinello M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 2007, 98, 146401. 10.1103/PhysRevLett.98.146401. [DOI] [PubMed] [Google Scholar]
- Smith J. S.; Nebgen B.; Lubbers N.; Isayev O.; Roitberg A. E. Less is more: Sampling chemical space with active learning. J. Chem. Phys. 2018, 148, 241733. 10.1063/1.5023802. [DOI] [PubMed] [Google Scholar]
- Schütt K. T.; Kessel P.; Gastegger M.; Nicoli K. A.; Tkatchenko A.; Müller K. R. SchNetPack: A Deep Learning Toolbox for Atomistic Systems. J. Chem. Theory Comput. 2019, 15, 448–455. 10.1021/acs.jctc.8b00908. [DOI] [PubMed] [Google Scholar]
- Gao X.; Ramezanghorbani F.; Isayev O.; Smith J. S.; Roitberg A. E. TorchANI: A Free and Open Source PyTorch-Based Deep Learning Implementation of the ANI Neural Network Potentials. J. Chem. Inf. Model. 2020, 60, 3408–3415. 10.1021/acs.jcim.0c00451. [DOI] [PubMed] [Google Scholar]
- Noé F.; Tkatchenko A.; Müller K. R.; Clementi C. Machine learning for molecular simulation. Annu. Rev. Phys. Chem. 2020, 71, 361–390. 10.1146/annurev-physchem-042018-052331. [DOI] [PubMed] [Google Scholar]
- Doerr S.; Majewski M.; Pérez A.; Krämer A.; Clementi C.; Noe F.; Giorgino T.; De Fabritiis G. TorchMD: A Deep Learning Framework for Molecular Simulations. J. Chem. Theory Comput. 2021, 17, 2355–2363. 10.1021/acs.jctc.0c01343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glielmo A.; Husic B. E.; Rodriguez A.; Clementi C.; Noé F.; Laio A. Unsupervised Learning Methods for Molecular Simulation Data. Chem. Rev. 2021, 121, 9722–9758. 10.1021/acs.chemrev.0c01195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmud M.; Kaiser M. S.; McGinnity T. M.; Hussain A. Deep Learning in Mining Biological Data. Cognit. Comput. 2021, 13, 1–33. 10.1007/s12559-020-09773-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan F. J.; Shi Y. Effects of data quality and quantity on deep learning for protein-ligand binding affinity prediction. Bioorg. Med. Chem. 2022, 72, 117003. 10.1016/j.bmc.2022.117003. [DOI] [PubMed] [Google Scholar]
- Ketkaew R.; Luber S. DeepCV: A Deep Learning Framework for Blind Search of Collective Variables in Expanded Configurational Space. J. Chem. Inf. Model. 2022, 62, 6352–6364. 10.1021/acs.jcim.2c00883. [DOI] [PubMed] [Google Scholar]
- Unsleber J. P.; Grimmel S. A.; Reiher M. Chemoton 2.0: Autonomous Exploration of Chemical Reaction Networks. J. Chem. Theory Comput. 2022, 18, 5393–5409. 10.1021/acs.jctc.2c00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson N. E.; Savoie B. M.; Statt A.; Webb M. A. Introduction to Machine Learning for Molecular Simulation. J. Chem. Theory Comput. 2023, 19, 4335–4337. 10.1021/acs.jctc.3c00735. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Chen D.; Xia Y.; Huang Y. P.; Lin X.; Han X.; Ni N.; Wang Z.; Yu F.; Yang L.; Yang Y. I.; Gao Y. Q. Artificial Intelligence Enhanced Molecular Simulations. J. Chem. Theory Comput. 2023, 19, 4338–4350. 10.1021/acs.jctc.3c00214. [DOI] [PubMed] [Google Scholar]
- Lee J.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. 10.1021/acs.jctc.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thallmair S.; Javanainen M.; Fábián B.; Martinez-Seara H.; Marrink S. J. Nonconverged Constraints Cause Artificial Temperature Gradients in Lipid Bilayer Simulations. J. Phys. Chem. B 2021, 125, 9537–9546. 10.1021/acs.jpcb.1c03665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph S.; Aluru N. R. Pumping of confined water in carbon nanotubes by rotation-translation coupling. Phys. Rev. Lett. 2008, 101, 064502. 10.1103/PhysRevLett.101.064502. [DOI] [PubMed] [Google Scholar]
- Bonthuis D. J.; Falk K.; Kaplan C. N.; Horinek D.; Berker A. N.; Bocquet L.; Netz R. R. Comment on ”Pumping of confined water in carbon nanotubes by rotation-translation coupling. Phys. Rev. Lett. 2010, 105, 064502. 10.1103/PhysRevLett.105.209401. [DOI] [PubMed] [Google Scholar]
- Shaw D. E.; et al. Anton 3: Twenty Microseconds of Molecular Dynamics Simulation before Lunch. Int. Conf. High Perform. Comput. Networking, Storage Anal. SC 2021, 1–11. 10.1145/3458817.3487397. [DOI] [Google Scholar]
- Kim H.; Fábián B.; Hummer G. Neighbor List Artifacts in Molecular Dynamics Simulations. J. Chem. Theory Comput. 2023, 19, 8919–8929. 10.1021/acs.jctc.3c00777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biriukov D.; Wang H. W.; Rampal N.; Tempra C.; Kula P.; Neuefeind J. C.; Stack A. G.; Předota M. The ”good,” the ”bad,” and the ”hidden” in neutron scattering and molecular dynamics of ionic aqueous solutions. J. Chem. Phys. 2022, 156, 194505. 10.1063/5.0093643. [DOI] [PubMed] [Google Scholar]
- Sandoval-Perez A.; Pluhackova K.; Böckmann R. A. Critical Comparison of Biomembrane Force Fields: Protein-Lipid Interactions at the Membrane Interface. J. Chem. Theory Comput. 2017, 13, 2310–2321. 10.1021/acs.jctc.7b00001. [DOI] [PubMed] [Google Scholar]
- Javanainen M.; Martinez-Seara H.; Vattulainen I. Excessive aggregation of membrane proteins in the Martini model. PLoS One 2017, 12, e0187936 10.1371/journal.pone.0187936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo J.; Aksimentiev A. New tricks for old dogs: Improving the accuracy of biomolecular force fields by pair-specific corrections to non-bonded interactions. Phys. Chem. Chem. Phys. 2018, 20, 8432–8449. 10.1039/C7CP08185E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer T.; D’Abramo M.; Hospital A.; Rueda M.; Ferrer-Costa C.; Pérez A.; Carrillo O.; Camps J.; Fenollosa C.; Repchevsky D.; Gelpí J. L.; Orozco M. MoDEL (Molecular Dynamics Extended Library): A Database of Atomistic Molecular Dynamics Trajectories. Structure 2010, 18, 1399–1409. 10.1016/j.str.2010.07.013. [DOI] [PubMed] [Google Scholar]
- Hospital A.; Andrio P.; Cugnasco C.; Codo L.; Becerra Y.; Dans P. D.; Battistini F.; Torres J.; Gõni R.; Orozco M.; Gelpí J. L. BIGNASim: A NoSQL database structure and analysis portal for nucleic acids simulation data. Nucleic Acids Res. 2016, 44, D272–D278. 10.1093/nar/gkv1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiirikki A. M.; et al. Overlay databank unlocks data-driven analyses of biomolecules for all. Nat. Commun. 2024, 15, 1136. 10.1038/s41467-024-45189-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp B.; Ospina L.; Deane C. M. Avoiding False Positive Conclusions in Molecular Simulation: The Importance of Replicas. J. Chem. Theory Comput. 2018, 14, 6127–6138. 10.1021/acs.jctc.8b00391. [DOI] [PubMed] [Google Scholar]
- Jumper J.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride J. M.; Polev K.; Abdirasulov A.; Reinharz V.; Grzybowski B. A.; Tlusty T. AlphaFold2 can predict single-mutation effects on structure and phenotype. Phys. Rev. Lett. 2023, 131, 218401. 10.1103/PhysRevLett.131.218401. [DOI] [PubMed] [Google Scholar]
- Chakravarty D.; Porter L. L. AlphaFold2 fails to predict protein fold switching. Protein Sci. 2022, 31, e4353 10.1002/pro.4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger T. C.; Liebschner D.; Croll T. I.; Williams C. J.; McCoy A. J.; Poon B. K.; Afonine P. V.; Oeffner R. D.; Richardson J. S.; Read R. J.; Adams P. D. AlphaFold predictions are valuable hypotheses and accelerate but do not replace experimental structure determination. Nat. Methods 2024, 21, 110–116. 10.1038/s41592-023-02087-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane T. J. Protein structure prediction has reached the single-structure frontier. Nat. Methods 2023, 20, 170–173. 10.1038/s41592-022-01760-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vani B. P.; Aranganathan A.; Wang D.; Tiwary P. AlphaFold2-RAVE: From Sequence to Boltzmann Ranking. J. Chem. Theory Comput. 2023, 19, 4351–4354. 10.1021/acs.jctc.3c00290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kmiecik S.; Gront D.; Kolinski M.; Wieteska L.; Dawid A. E.; Kolinski A. Coarse-Grained Protein Models and Their Applications. Chem. Rev. 2016, 116, 7898–7936. 10.1021/acs.chemrev.6b00163. [DOI] [PubMed] [Google Scholar]
- Marx D.; Hutter J.. Ab Initio Molecular Dynamics Basic Theory and Advanced Methods; Cambridge University Press, 2009; pp 1–567. 10.1017/CBO9780511609633 [DOI] [Google Scholar]
- Souza P. C.; et al. Martini 3: a general purpose force field for coarse-grained molecular dynamics. Nat. Methods 2021, 18, 382–388. 10.1038/s41592-021-01098-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T.; Miyazaki Y.; Harada S.; Kumar A.; Vanni S.; Shinoda W. Improved Protein Model in SPICA Force Field. J. Chem. Theory Comput. 2023, 19, 8967–8977. 10.1021/acs.jctc.3c01016. [DOI] [PubMed] [Google Scholar]
- Klein F.; Soñora M.; Helene Santos L.; Nazareno Frigini E.; Ballesteros-Casallas A.; Rodrigo Machado M.; Pantano S. The SIRAH force field: A suite for simulations of complex biological systems at the coarse-grained and multiscale levels. J. Struct. Biol. 2023, 215, 107985. 10.1016/j.jsb.2023.107985. [DOI] [PubMed] [Google Scholar]
- Vácha R.; Frenkel D. Relation between molecular shape and the morphology of self-assembling aggregates: A simulation study. Biophys. J. 2011, 101, 1432–1439. 10.1016/j.bpj.2011.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukeník L.; Mukhamedova L.; Procházková M.; Škubník K.; Plevka P.; Vácha R. Cargo Release from Nonenveloped Viruses and Virus-like Nanoparticles: Capsid Rupture or Pore Formation. ACS Nano 2021, 15, 19233–19243. 10.1021/acsnano.1c04814. [DOI] [PubMed] [Google Scholar]
- Molteni C.; Parrinello M. Glucose in aqueous solution by first principles molecular dynamics. J. Am. Chem. Soc. 1998, 120, 2168–2171. 10.1021/ja973008q. [DOI] [Google Scholar]
- MacKerell A. D.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- Goetz R.; Lipowsky R. Computer simulations of bilayer membranes: Self-assembly and interfacial tension. J. Chem. Phys. 1998, 108, 7397–7409. 10.1063/1.476160. [DOI] [Google Scholar]
- Wang T.; He X.; Li M.; Shao B.; Liu T. Y. AIMD-Chig: Exploring the conformational space of a 166-atom protein Chignolin with ab initio molecular dynamics. Sci. Data 2023, 10, 1–12. 10.1038/s41597-023-02465-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim K. S.; Greskamp B.; Towles B.; Edwards B.; Grossman J. P.; Shaw D. E. The Specialized High-Performance Network on Anton 3. Proc. - Int. Symp. High-Performance Comput. Archit. 2022, 1211–1223. 10.1109/HPCA53966.2022.00092. [DOI] [Google Scholar]
- Kozinsky B.; Musaelian A.; Johansson A.; Batzner S. Scaling the Leading Accuracy of Deep Equivariant Models to Biomolecular Simulations of Realistic Size. Proc. Int. Conf. High Perform. Comput. Networking, Storage Anal. SC 2023 2023, 1–12. 10.1145/3581784.3627041. [DOI] [Google Scholar]
- Csizi K. S.; Reiher M. Universal QM/MM approaches for general nanoscale applications. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2023, 13, e1656 10.1002/wcms.1656. [DOI] [Google Scholar]
- Raghavan B.; Paulikat M.; Ahmad K.; Callea L.; Rizzi A.; Ippoliti E.; Mandelli D.; Bonati L.; De Vivo M.; Carloni P. Drug Design in the Exascale Era: A Perspective from Massively Parallel QM/MM Simulations. J. Chem. Inf. Model. 2023, 63, 3647–3658. 10.1021/acs.jcim.3c00557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamerlin S. C.; Warshel A. At the dawn of the 21st century: Is dynamics the missing link for understanding enzyme catalysis. Proteins Struct. Funct. Bioinforma. 2010, 78, 1339–1375. 10.1002/prot.22654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontkanen O. V.; Biriukov D.; Futera Z. Reorganization free energy of copper proteins in solution, in vacuum, and on metal surfaces. J. Chem. Phys. 2022, 156, 175101. 10.1063/5.0085141. [DOI] [PubMed] [Google Scholar]
- Rzepiela A. J.; Louhivuori M.; Peter C.; Marrink S. J. Hybrid simulations: Combining atomistic and coarse-grained force fields using virtual sites. Phys. Chem. Chem. Phys. 2011, 13, 10437–10448. 10.1039/c0cp02981e. [DOI] [PubMed] [Google Scholar]
- Kar P.; Feig M. Hybrid All-Atom/Coarse-Grained Simulations of Proteins by Direct Coupling of CHARMM and PRIMO Force Fields. J. Chem. Theory Comput. 2017, 13, 5753–5765. 10.1021/acs.jctc.7b00840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; De Vries A. H.; Barnoud J.; Pezeshkian W.; Melcr J.; Marrink S. J. Dual Resolution Membrane Simulations Using Virtual Sites. J. Phys. Chem. B 2020, 124, 3944–3953. 10.1021/acs.jpcb.0c01842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolicato F.; et al. Disulfide bridge-dependent dimerization triggers FGF2 membrane translocation into the extracellular space. eLife 2024, 12, RP88579. 10.7554/eLife.88579.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B.; Scheek R. M. Orientation restraints in molecular dynamics simulations using time and ensemble averaging. J. Magn. Reson. 2003, 164, 19–27. 10.1016/S1090-7807(03)00178-2. [DOI] [PubMed] [Google Scholar]
- Igaev M.; Kutzner C.; Bock L. V.; Vaiana A. C.; Grubmüller H. Automated cryo-EM structure refinement using correlation-driven molecular dynamics. eLife 2019, 8, e43542 10.7554/eLife.43542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reith D.; Meyer H.; Müller-Plathe F. CG-OPT: A software package for automatic force field design. Comput. Phys. Commun. 2002, 148, 299–313. 10.1016/S0010-4655(02)00562-3. [DOI] [Google Scholar]
- Krämer A.; Hülsmann M.; Köddermann T.; Reith D. Automated parameterization of intermolecular pair potentials using global optimization techniques. Comput. Phys. Commun. 2014, 185, 3228–3239. 10.1016/j.cpc.2014.08.022. [DOI] [Google Scholar]
- Wang L. P.; Martinez T. J.; Pande V. S. Building force fields: An automatic, systematic, and reproducible approach. J. Phys. Chem. Lett. 2014, 5, 1885–1891. 10.1021/jz500737m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz R. M.; Walker R. C. Paramfit: Automated optimization of force field parameters for molecular dynamics simulations. J. Comput. Chem. 2015, 36, 79–87. 10.1002/jcc.23775. [DOI] [PubMed] [Google Scholar]
- Zahariev F.; De Silva N.; Gordon M. S.; Windus T. L.; Pérez García M. ParFit: A Python-Based Object-Oriented Program for Fitting Molecular Mechanics Parameters to ab Initio Data. J. Chem. Inf. Model. 2017, 57, 391–396. 10.1021/acs.jcim.6b00654. [DOI] [PubMed] [Google Scholar]
- Sauceda H. E.; Gastegger M.; Chmiela S.; Müller K. R.; Tkatchenko A. Molecular force fields with gradient-domain machine learning (GDML): Comparison and synergies with classical force fields. J. Chem. Phys. 2020, 153, 124109. 10.1063/5.0023005. [DOI] [PubMed] [Google Scholar]
- Befort B. J.; Defever R. S.; Tow G. M.; Dowling A. W.; Maginn E. J. Machine Learning Directed Optimization of Classical Molecular Modeling Force Fields. J. Chem. Inf. Model. 2021, 61, 4400–4414. 10.1021/acs.jcim.1c00448. [DOI] [PubMed] [Google Scholar]
- Unke O. T.; Chmiela S.; Sauceda H. E.; Gastegger M.; Poltavsky I.; Schütt K. T.; Tkatchenko A.; Müller K. R. Machine Learning Force Fields. Chem. Rev. 2021, 121, 10142–10186. 10.1021/acs.chemrev.0c01111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unke O. T.; Chmiela S.; Gastegger M.; Schütt K. T.; Sauceda H. E.; Müller K. R. SpookyNet: Learning force fields with electronic degrees of freedom and nonlocal effects. Nat. Commun. 2021, 12, 1–14. 10.1038/s41467-021-27504-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y.; Krämer A.; Venable R. M.; Simmonett A. C.; Mackerell A. D.; Klauda J. B.; Pastor R. W.; Brooks B. R. Semi-automated Optimization of the CHARMM36 Lipid Force Field to Include Explicit Treatment of Long-Range Dispersion. J. Chem. Theory Comput. 2021, 17, 1562–1580. 10.1021/acs.jctc.0c01326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Empereur-Mot C.; Pedersen K. B.; Capelli R.; Crippa M.; Caruso C.; Perrone M.; Souza P. C. T.; Marrink S. J.; Pavan G. M. Automatic Optimization of Lipid Models in the Martini Force Field Using SwarmCG. J. Chem. Inf. Model. 2023, 63, 3827. 10.1021/acs.jcim.3c00530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmiela S.; Vassilev-Galindo V.; Unke O. T.; Kabylda A.; Sauceda H. E.; Tkatchenko A.; Müller K. R. Accurate global machine learning force fields for molecules with hundreds of atoms. Sci. Adv. 2023, 9, eadf087 10.1126/sciadv.adf0873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illarionov A.; et al. Combining Force Fields and Neural Networks for an Accurate Representation of Chemically Diverse Molecular Interactions. J. Am. Chem. Soc. 2023, 145, 23620–23629. 10.1021/jacs.3c07628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.; Yang X.; Zhao X.; Li Z.; Lu M.; Xie X.; Yan J. Applications and Advances in Machine Learning Force Fields. J. Chem. Inf. Model. 2023, 63, 6972–6985. 10.1021/acs.jcim.3c00889. [DOI] [PubMed] [Google Scholar]
- Chen G.; Inizan T. J.; Plé T.; Lagardère L.; Piquemal J.-P.; Maday Y.. Advancing Force Fields Parameterization: A Directed Graph Attention Networks Approach. ChemRxiv, December 22, 2023, ver. 1. 10.26434/chemrxiv-2023-nz8hc (accessed 2024-03-04). [DOI] [PubMed]
- Bonomi M.; Barducci A.; Parrinello M. Reconstructing the equilibrium boltzmann distribution from well-tempered metadynamics. J. Comput. Chem. 2009, 30, 1615–1621. 10.1002/jcc.21305. [DOI] [PubMed] [Google Scholar]
- Kikutsuji T.; Mori Y.; Okazaki K. I.; Mori T.; Kim K.; Matubayasi N. Explaining reaction coordinates of alanine dipeptide isomerization obtained from deep neural networks using Explainable Artificial Intelligence (XAI). J. Chem. Phys. 2022, 156, 154108. 10.1063/5.0087310. [DOI] [PubMed] [Google Scholar]
- Yao S.; Van R.; Pan X.; Park J. H.; Mao Y.; Pu J.; Mei Y.; Shao Y. Machine learning based implicit solvent model for aqueous-solution alanine dipeptide molecular dynamics simulations. RSC Adv. 2023, 13, 4565–4577. 10.1039/D2RA08180F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baiardi A.; Christandl M.; Reiher M. Quantum Computing for Molecular Biology. ChemBioChem. 2023, 24, e202300120 10.1002/cbic.202300120. [DOI] [PubMed] [Google Scholar]
- Feng S.; Park S.; Choi Y. K.; Im W. CHARMM-GUI Membrane Builder: Past, Current, and Future Developments and Applications. J. Chem. Theory Comput. 2023, 19, 2161–2185. 10.1021/acs.jctc.2c01246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroon P. C.; Grunewald F.; Barnoud J.; van Tilburg M.; Souza P. C. T.; Wassenaar T. A.; Marrink S. J. Martinize2 and Vermouth: Unified Framework for Topology Generation. eLife 2023, 12, RP90627. 10.7554/eLife.90627.1. [DOI] [Google Scholar]
- Huang J.; Rauscher S.; Nawrocki G.; Ran T.; Feig M.; De Groot B. L.; Grubmüller H.; MacKerell A. D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian C.; Kasavajhala K.; Belfon K. A.; Raguette L.; Huang H.; Migues A. N.; Bickel J.; Wang Y.; Pincay J.; Wu Q.; Simmerling C. Ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. 10.1021/acs.jctc.9b00591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon-Ferrer R.; Case D. A.; Walker R. C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. 10.1002/wcms.1121. [DOI] [Google Scholar]
- Brooks B. R.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Páll S.; Smith J. C.; Hess B.; Lindahl E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Phillips J. C.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 44130. 10.1063/5.0014475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman P.; Swails J.; Chodera J. D.; McGibbon R. T.; Zhao Y.; Beauchamp K. A.; Wang L. P.; Simmonett A. C.; Harrigan M. P.; Stern C. D.; Wiewiora R. P.; Brooks B. R.; Pande V. S. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017, 13, e1005659 10.1371/journal.pcbi.1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers K. J.; Chow D. E.; Xu H.; Dror R. O.; Eastwood M. P.; Gregersen B. A.; Klepeis J. L.; Kolossvary I.; Moraes M. A.; Sacerdoti F. D.; Salmon J. K.; Shan Y.; Shaw D. E.. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, New York, 2007, 43–43. 10.1109/sc.2006.54 [DOI]
- Hilpert C.; Beranger L.; Souza P. C.; Vainikka P. A.; Nieto V.; Marrink S. J.; Monticelli L.; Launay G. Facilitating CG Simulations with MAD: The MArtini Database Server. J. Chem. Inf. Model. 2023, 63, 702–710. 10.1021/acs.jcim.2c01375. [DOI] [PubMed] [Google Scholar]
- Schmitt S.; Kanagalingam G.; Fleckenstein F.; Froescher D.; Hasse H.; Stephan S. Extension of the MolMod Database to Transferable Force Fields. J. Chem. Inf. Model. 2023, 63, 7148–7158. 10.1021/acs.jcim.3c01484. [DOI] [PubMed] [Google Scholar]
- Boothroyd S.; Wang L. P.; Mobley D. L.; Chodera J. D.; Shirts M. R. Open Force Field Evaluator: An Automated, Efficient, and Scalable Framework for the Estimation of Physical Properties from Molecular Simulation. J. Chem. Theory Comput. 2022, 18, 3566–3576. 10.1021/acs.jctc.1c01111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hospital A.; Battistini F.; Soliva R.; Gelpí J. L.; Orozco M. Surviving the deluge of biosimulation data. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2020, 10, e1449 10.1002/wcms.1449. [DOI] [Google Scholar]
- Marx V. The big challenges of big data. Nature 2013, 498, 255–260. 10.1038/498255a. [DOI] [PubMed] [Google Scholar]
- Stephens Z. D.; Lee S. Y.; Faghri F.; Campbell R. H.; Zhai C.; Efron M. J.; Iyer R.; Schatz M. C.; Sinha S.; Robinson G. E. Big data: Astronomical or genomical?. PLoS Biol. 2015, 13, e1002195 10.1371/journal.pbio.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson S. L.; Way G. P.; Bittremieux W.; Armache J. P.; Haendel M. A.; Hoffman M. M. Sharing biological data: why, when, and how. FEBS Lett. 2021, 595, 847–863. 10.1002/1873-3468.14067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H.; Henrick K.; Nakamura H. Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 2003, 10, 980. 10.1038/nsb1203-980. [DOI] [PubMed] [Google Scholar]
- Domański J.; Stansfeld P. J.; Sansom M. S.; Beckstein O. Lipidbook: A public repository for force-field parameters used in membrane simulations. J. Membr. Biol. 2010, 236, 255–258. 10.1007/s00232-010-9296-8. [DOI] [PubMed] [Google Scholar]
- Newport T. D.; Sansom M. S.; Stansfeld P. J. The MemProtMD database: A resource for membrane-embedded protein structures and their lipid interactions. Nucleic Acids Res. 2019, 47, D390–D397. 10.1093/nar/gky1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Espigares I.; et al. GPCRmd uncovers the dynamics of the 3D-GPCRome. Nat. Methods 2020, 17, 777–787. 10.1038/s41592-020-0884-y. [DOI] [PubMed] [Google Scholar]
- Suarez-Leston F.; Calvelo M.; Tolufashe G. F.; Muñoz A.; Veleiro U.; Porto C.; Bastos M.; Piñeiro Á.; Garcia-Fandino R. SuPepMem: A database of innate immune system peptides and their cell membrane interactions. Comput. Struct. Biotechnol. J. 2022, 20, 874–881. 10.1016/j.csbj.2022.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A.; et al. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. 10.1093/nar/gkac1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoch J. C.; et al. Biological Magnetic Resonance Data Bank. Nucleic Acids Res. 2023, 51, D368–D376. 10.1093/nar/gkac1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek M.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. 10.1126/science.abj8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig M.; Abdullah M.; Johnsson L.; Pettitt B. M. Large scale distributed data repository: Design of a molecular dynamics trajectory database. Futur. Gener. Comput. Syst. 1999, 16, 101–110. 10.1016/S0167-739X(99)00039-4. [DOI] [Google Scholar]
- Tai K.; Murdock S.; Wu B.; Ng M. H.; Johnston S.; Fangohr H.; Cox S. J.; Jeffreys P.; Essex J. W.; Sansom M. S. BioSimGrid: Towards a worldwide repository for biomolecular simulations. Org. Biomol. Chem. 2004, 2, 3219–3221. 10.1039/b411352g. [DOI] [PubMed] [Google Scholar]
- van der Kamp M. W.; Schaeffer R. D.; Jonsson A. L.; Scouras A. D.; Simms A. M.; Toofanny R. D.; Benson N. C.; Anderson P. C.; Merkley E. D.; Rysavy S.; Bromley D.; Beck D. A.; Daggett V. Dynameomics: A Comprehensive Database of Protein Dynamics. Structure 2010, 18, 423–435. 10.1016/j.str.2010.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham M. J.; et al. Sharing Data from Molecular Simulations. J. Chem. Inf. Model. 2019, 59, 4093–4099. 10.1021/acs.jcim.9b00665. [DOI] [PubMed] [Google Scholar]
- Hildebrand P. W.; Rose A. S.; Tiemann J. K. Bringing Molecular Dynamics Simulation Data into View. Trends Biochem. Sci. 2019, 44, 902–913. 10.1016/j.tibs.2019.06.004. [DOI] [PubMed] [Google Scholar]
- Abriata L. A.; Lepore R.; Dal Peraro M. About the need to make computational models of biological macromolecules available and discoverable. Bioinformatics 2020, 36, 2952–2954. 10.1093/bioinformatics/btaa086. [DOI] [PubMed] [Google Scholar]
- Bekker G. J.; Kawabata T.; Kurisu G. The Biological Structure Model Archive (BSM-Arc): an archive for in silico models and simulations. Biophys. Rev. 2020, 12, 371–375. 10.1007/s12551-020-00632-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antila H. S.; Kav B.; Miettinen M. S.; Martinez-Seara H.; Jungwirth P.; Ollila O. H. Emerging Era of Biomolecular Membrane Simulations: Automated Physically-Justified Force Field Development and Quality-Evaluated Databanks. J. Phys. Chem. B 2022, 126, 4169–4183. 10.1021/acs.jpcb.2c01954. [DOI] [Google Scholar]
- Tiemann J. K. S.; Szczuka M.; Bouarroudj L.; Oussaren M.; Garcia S.; Howard R. J.; Delemotte L.; Lindahl E.; Baaden M.; Lindorff-Larsen K.; Chavent M.; Poulain P. MDverse: Shedding Light on the Dark Matter of Molecular Dynamics Simulations. eLife 2023, 12, RP90061. 10.7554/eLife.90061.1. [DOI] [Google Scholar]
- Musen M. A. Without appropriate metadata, data-sharing mandates are pointless. Nature 2022, 609, 222. 10.1038/d41586-022-02820-7. [DOI] [PubMed] [Google Scholar]
- Antila H. S.; M. Ferreira T.; Ollila O. H.; Miettinen M. S. Using Open Data to Rapidly Benchmark Biomolecular Simulations: Phospholipid Conformational Dynamics. J. Chem. Inf. Model. 2021, 61, 938–949. 10.1021/acs.jcim.0c01299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrán D.; Hospital A.; Gelpí J. L.; Orozco M. A new paradigm for molecular dynamics databases: the COVID-19 database, the legacy of a titanic community effort. Nucleic Acids Res. 2024, 52, D393. 10.1093/nar/gkad991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaro R. E.; Mulholland A. J. A Community Letter Regarding Sharing Biomolecular Simulation Data for COVID-19. J. Chem. Inf. Model. 2020, 60, 2653–2656. 10.1021/acs.jcim.0c00319. [DOI] [PubMed] [Google Scholar]
- Mulholland A. J.; Amaro R. E. COVID19 - Computational Chemists Meet the Moment. J. Chem. Inf. Model. 2020, 60, 5724–5726. 10.1021/acs.jcim.0c01395. [DOI] [PubMed] [Google Scholar]
- Wilkinson M. D.; et al. Comment: The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 1–9. 10.1038/sdata.2016.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonomi M.; et al. Promoting transparency and reproducibility in enhanced molecular simulations. Nat. Methods 2019, 16, 670–673. 10.1038/s41592-019-0506-8. [DOI] [PubMed] [Google Scholar]
- Porubsky V. L.; Goldberg A. P.; Rampadarath A. K.; Nickerson D. P.; Karr J. R.; Sauro H. M. Best Practices for Making Reproducible Biochemical Models. Cell Syst. 2020, 11, 109–120. 10.1016/j.cels.2020.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabelica M.; Bojčić R.; Puljak L. Many researchers were not compliant with their published data sharing statement: a mixed-methods study. J. Clin. Epidemiol. 2022, 150, 33–41. 10.1016/j.jclinepi.2022.05.019. [DOI] [PubMed] [Google Scholar]
- Elofsson A.; Hess B.; Lindahl E.; Onufriev A.; van der Spoel D.; Wallqvist A. Ten simple rules on how to create open access and reproducible molecular simulations of biological systems. PLoS Comput. Biol. 2019, 15, e1006649 10.1371/journal.pcbi.1006649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merz K. M.; Amaro R.; Cournia Z.; Rarey M.; Soares T.; Tropsha A.; Wahab H. A.; Wang R. Editorial: Method and Data Sharing and Reproducibility of Scientific Results. J. Chem. Inf. Model. 2020, 60, 5868–5869. 10.1021/acs.jcim.0c01389. [DOI] [PubMed] [Google Scholar]
- Solomon G. C.; Zhang J. Z.; Cuk T. ACS Physical Chemistry Au: A Journal Celebrating Open Science across the Broad Horizons of Physical Chemistry. ACS Phys. Chem. Au 2021, 1, 1–2. 10.1021/acsphyschemau.1c00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong-ekkabut J.; Karttunen M. The good, the bad and the user in soft matter simulations. Biochim. Biophys. Acta - Biomembr. 2016, 1858, 2529–2538. 10.1016/j.bbamem.2016.02.004. [DOI] [PubMed] [Google Scholar]
- Linton J. D. Research: All journals need to correct errors. Nature 2013, 504, 33. 10.1038/504033d. [DOI] [PubMed] [Google Scholar]
- Allison D. B.; Brown A. W.; George B. J.; Kaiser K. A. Reproducibility: A tragedy of errors. Nature 2016, 530, 27–29. 10.1038/530027a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Noorden R. More than 10,000 research papers were retracted in 2023 - a new record. Nature 2023, 624, 479–481. 10.1038/d41586-023-03974-8. [DOI] [PubMed] [Google Scholar]
- Liverpool L. AI intensifies fight against ’paper mills’ that churn out fake research. Nature 2023, 618, 222–223. 10.1038/d41586-023-01780-w. [DOI] [PubMed] [Google Scholar]
- Gapsys V.; Hahn D. F.; Tresadern G.; Mobley D. L.; Rampp M.; De Groot B. L. Pre-Exascale Computing of Protein-Ligand Binding Free Energies with Open Source Software for Drug Design. J. Chem. Inf. Model. 2022, 62, 1172–1177. 10.1021/acs.jcim.1c01445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyro G. W.; Brent R. I.; Batista V. S. HAC-Net: A Hybrid Attention-Based Convolutional Neural Network for Highly Accurate Protein-Ligand Binding Affinity Prediction. J. Chem. Inf. Model. 2023, 63, 1947–1960. 10.1021/acs.jcim.3c00251. [DOI] [PubMed] [Google Scholar]
- Dutta P.; Jain D.; Gupta R.; Rai B. Deep learning models for the estimation of free energy of permeation of small molecules across lipid membranes. Digit. Discovery 2023, 2, 189–201. 10.1039/D2DD00119E. [DOI] [Google Scholar]
- Kabelka I.; Vácha R. Advances in Molecular Understanding of α-Helical Membrane-Active Peptides. Acc. Chem. Res. 2021, 54, 2196–2204. 10.1021/acs.accounts.1c00047. [DOI] [PubMed] [Google Scholar]
- Deb R.; Kabelka I.; PÅibyl J.; Vácha R.. De novo design of peptides that form transmembrane barrel pores killing antibiotic resistant bacteria. bioRxiv, May 9, 2022, ver. 1. 10.1101/2022.05.09.491086 (accessed 2024-03-04). [DOI]
- Fonseca G.; Poltavsky I.; Tkatchenko A. Force Field Analysis Software and Tools (FFAST): Assessing Machine Learning Force Fields Under the Microscope. J. Chem. Theory Comput. 2023, 19, 8706–8717. 10.1021/acs.jctc.3c00985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez S.; et al. Glycosaminoglycans: What Remains To Be Deciphered?. JACS Au 2023, 3, 628–656. 10.1021/jacsau.2c00569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallet S. D.; Berthollier C.; Ricard-Blum S. The glycosaminoglycan interactome 2.0. Am. J. Physiol. - Cell Physiol. 2022, 322, C1271–C1278. 10.1152/ajpcell.00095.2022. [DOI] [PubMed] [Google Scholar]
- Riopedre-Fernandez M.; Biriukov D.; Dračínský M.; Martinez-Seara H. Hyaluronan-arginine enhanced and dynamic interaction emerges from distinctive molecular signature due to electrostatics and side-chain specificity. Carbohydr. Polym. 2024, 325, 121568. 10.1016/j.carbpol.2023.121568. [DOI] [PubMed] [Google Scholar]
- Ives C. M.; Singh O.; D’Andrea S.; Fogarty C. A.; Harbison A. M.; Satheesan A.; Tropea B.; Fadda E.. Restoring Protein Glycosylation with GlycoShape. bioRxiv, December 11, 2023. 10.1101/2023.12.11.571101 (accessed 2024-03-04). [DOI]
- Ricard-Blum S.; Perez S. Glycosaminoglycan interaction networks and databases. Curr. Opin. Struct. Biol. 2022, 74, 102355. 10.1016/j.sbi.2022.102355. [DOI] [PubMed] [Google Scholar]
- Casalino L.; Gaieb Z.; Goldsmith J. A.; Hjorth C. K.; Dommer A. C.; Harbison A. M.; Fogarty C. A.; Barros E. P.; Taylor B. C.; Mclellan J. S.; Fadda E.; Amaro R. E. Beyond shielding: The roles of glycans in the SARS-CoV-2 spike protein. ACS Cent. Sci. 2020, 6, 1722–1734. 10.1021/acscentsci.0c01056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubeck S.; Chandrasekaran V.; Eldan R.; Gehrke J.; Horvitz E.; Kamar E.; Lee P.; Lee Y. T.; Li Y.; Lundberg S.; Nori H.; Palangi H.; Ribeiro M. T.; Zhang Y.. Sparks of Artificial General Intelligence: Early experiments with GPT-4. arXiv, April 13, 2023, ver. 5. 10.48550/arXiv.2303.12712 (accessed 2024-03-04). [DOI]