

Summary
Recent technological developments such as cryogenic electron microscopy (Cryo-EM) and X-ray free electron lasers (XFEL) have significantly expanded the available toolkit to visualize large, complex noncoding RNAs and their complexes. Consequently, the quality of the RNA sample, as measured by its chemical monodispersity and conformational homogeneity, has become the bottleneck that frequently precludes effective structural analyses. Here we describe a general RNA sample preparation protocol that combines co-transcriptional RNA folding and RNA-RNA complex assembly, followed by native purification of stoichiometric complexes. We illustrate and discuss the utility of this versatile method in overcoming RNA misfolding and enabling the structural and mechanistic elucidations of the T-box riboswitch-tRNA complexes.
Keywords: Transcription, co-transcriptional folding, RNA-RNA interactions, riboswitches, tRNA, T-box
1. Introduction
Like their protein counterparts, many large, complex noncoding RNAs rely on their three-dimensional structures to execute a wide range of cellular functions such as gene regulation (e.g., riboswitches [1, 2]), RNA processing (e.g., ribozymes [3–5]), and protein synthesis (e.g., ribosomes [6, 7]). The inherently dynamic, programmable conformations that RNA can readily and predictably access make them excellent platforms to host regulatory devices [8, 9]. However, the same conformational flexibility also creates structural heterogeneities that render them challenging targets for high-resolution analyses [10–12]. With the recent advent of direct electron detectors for Cryo-EM and other technical advances in structural biology, the rate-limiting step for visualizing large noncoding RNAs has shifted to sample preparation.
Typically, RNAs samples under structural analyses are synthesized in vitro by high-yield, non-cognate RNA polymerases (RNAPs) such as the T7 RNAP. These enzymes are dramatically faster than the cognate multi-subunit RNAPs that normally transcribe and vectorially fold the RNAs of interest in cells [13, 14], and generally ignore pause signals that can be essential for hierarchical folding of complex structured RNA, especially those that harbor long-range interactions [15]. The challenge of correctly folding complex RNAs in vitro is only exacerbated by the difficulty to detect misfolded RNAs after the fact. Unlike proteins, misfolded RNAs do not usually form aggregates due to their high hydrophilicity and extensive negative charges that discourage inter-particle interactions. As a result, it can be very difficult to detect RNA misfolding, unless an in vitro functional assay is readily available, such as for ribozymes like the RNase P [16]. Finally, refolding complex structured RNAs after initial denaturation is not always successful, akin to refolding proteins from inclusion bodies. For example, the full-length B. subtilis glyQS T-box riboswitch RNA is prone to misfolding, likely due to the absence of a cognate tRNA partner that is normally present during co-transcriptional folding and complex formation in cells. Interestingly, a number of long noncoding RNAs such as HOTAIR exhibit strong tendencies to misfold and form large heterogeneous aggregates in vitro [12].
To help overcome these significant challenges in RNA misfolding and heterogeneity, we illustrate a general protocol below that proved essential to the preparation of RNA samples of sufficient quality, which enabled structural analyses of T-box riboswitch-tRNA complexes using X-ray crystallography, Cryo-EM, and Small-angle X-ray Scattering (SAXS) [17–19]. The method starts with co-transcriptional folding and assembly of the mRNA-tRNA complex, where the misfolding-prone T-box RNA is transcribed in the presence of its cognate tRNA partner, thus facilitating the formation of functionally relevant complexes. Then the method diverges depending on the size and stability of the complexes. With the smaller (~ 46 kD) and less stable T-box discriminator-tRNA complex (Kd ~ 2 μM), size-exclusion chromatography (SEC) is effective in isolating the 1:1 complex without dissociating it. With the larger (~ 78 kD) and highly stable full-length T-box-tRNA complex (Kd < 100 nM), strong anion exchange chromatography on a Mono Q column was key to remove misfolded T-box RNA as well as oligomeric RNAs to obtain highly homogeneous samples for Cryo-EM analysis.
2. Equipment and Materials
DNA oligonucleotides for PCR amplification (Integrated DNA Technologies).
Diethyl pyrocarbonate (DEPC, Sigma) - treated water.
Taq DNA polymerase, 5 U/μL. (New England Biolabs)
Deoxynucleotide triphosphates mix containing dATP, dGTP, dCTP and dTTP, 25 mM each (Thermo Scientific).
Programmable thermal cycler (Bio-Rad).
T7 RNA polymerase, 50 U/μL (New England Biolabs).
10x T7 transcription buffer: 300 mM Tris pH 8.1, 250 mM MgCl2, 100 mM DTT, 20 mM spermidine and 0.01% Triton X-100.
ATP, GTP, GMP, CTP and UTP, 100 mM solution each in DEPC water (Millipore-Sigma).
Inorganic Pyrophosphatase (Millipore-Sigma)
RNA binding buffer: 50 mM HEPES-KOH, pH 7.0, 100 mM KCl, 20 mM MgCl2, 5 mM tris (2-carboxyethyl) phosphine (TCEP).
SYBR Gold Nucleic Acid Gel Stain (Life technologies)
Ethidium Bromide (EtBr) Dye (Millipore-Sigma)
Nanodrop 2000c spectrophotometer (Thermo Scientific)
Benchtop centrifuge: Eppendorf 5424 and 5430R with F-35-6-30 rotor.
Amicon 10K MWCO centrifugal filters (15 ml and 0.5 ml, Millipore-Sigma)
Ultrafree-MC Centrifugal Filter Units (Millipore-Sigma)
Vertical gel electrophoresis apparatus (C.B.S. Scientific)
40% acrylamide solution (acrylamide: bis-acrylamide 29:1, Thermo Scientific)
Ammonium persulfate (APS, Sigma)
PowerPac Hv power supply (Bio-rad)
Gel-imaging system such as an ImageQuant 800 (Cytiva) or a Typhoon scanner (Cytiva).
AKTA Pure 25 FPLC system with F9-C fraction collector (Cytiva)
Superdex 200 Increase 10/300 GL column (Cytiva)
MonoQ 5/50 GL column (Cytiva)
All other chemicals from Sigma-Aldrich unless otherwise specified.
3. Methods
3.1. Choice of T7 promoter and desired RNA 5′ phosphorylation state.
T7 RNA polymerase (RNAP) initiates transcription de novo on T7 promoters. Two classes of T7 promoters namely ϕ2.5 (class II) and ϕ6.5 (class III) are commonly used. The ϕ6.5 class-III promoter has a sequence 5’-TAATACGACGACATATAG, where the final G is the first nucleotide of the synthesized RNA transcript. The ϕ2.5 class-II promoter initiates transcription with ATP and has a typical sequence of 5’-TAATACGACGACATATTA, where the final A is the transcription start site [20]. Both ϕ6.5 and ϕ2.5 class promoters yield higher amount of RNA when the initiator nucleotide (GTP for ϕ6.5 and ATP for ϕ2.5) is followed by one to two Gs. The ϕ6.5 promoter is used as the B. subtilis glyQS T-box riboswitch and its cognate ligand B. subtilis tRNAGly both have a G at the +1 position.
RNA transcripts synthesized by in vitro transcription bear a 5′ triphosphate (PPP) group. The fact that T7 RNAP does not recognize structural features of the initiating nucleotide beyond the GMP/AMP moiety allows convenient 5′ labeling of RNA with fluorophores, biotin, or cofactors like NAD, FAD and CoA using a GMP/AMP derivative [21]. To mimic natural tRNAs which bear a 5′-monophosphate, GMP priming is used to generate tRNAGly, where a GMP:GTP ratio of 10:1 is used, producing a ~90% 5′-monophosphoroylated tRNA sample. If a 5′-OH group is desired on the transcript, a cis-cleaving Hammerhead ribozyme is appended 5′ to the RNA of interest to produce a non-phosphorylated RNA 5′-end (see the section below).
T7 RNAP occasionally produces 5′ heterogeneities and frequently generates 3′ heterogeneities in the form of N+1, N+2, or even longer transcripts [22]. In the next section, we describe a previously reported method that utilizes a cis-cleaving Hammerhead ribozyme and a trans-cleaving Varkud satellite (VS) ribozyme to overcome the 5′ and 3′ heterogeneities that can hamper structural analyses [23].
3.2. (Optional) Homogenization of RNA termini by dual ribozyme processing.
The use of a cis-acting Hammerhead ribozyme and a trans-acting VS ribozyme allows facile synthesis of chemically homogenous RNA transcripts with defined 5′ and 3′ ends. These optional transcript-processing cassettes that flank the RNA of interest can be included in initial gene synthesis design so that they are available if needed. Cleavage by the 5′-appended Hammerhead ribozyme leaves a 5′-OH group on the downstream target RNA, whereas cleavage and removal of the 3′-appended VS substrate RNA (~24 nucleotides, or nts) by the trans-acting VS ribozyme leaves a 2′,3′-cyclic phosphate on the 3′ end of the target RNA. If needed, the cyclic phosphate can be converted to a linear 3′-phosphate by hydrochloric acid (HCl) treatment [18, 24]. Here, the VS ribozyme is provided either in the form of a restriction enzyme-linearized plasmid that encodes a T7 promoter-driven VS gene near its downstream open end, PCR products of a similar segment, or pre-transcribed VS RNA.
As an alternative to the VS ribozyme cleavage approach, RNA transcripts with homogenous 3′-ends can generally be produced using DNA templates that bear two consecutive 2′-O-methyl modifications at the 5′-terminus of the DNA template strand [9, 22, 25]. When PCR amplification is used to generate DNA templates, these 2′-O-methyl modifications can be provided by the reverse primer.
3.3. Streamlined DNA template preparation and co-transcriptional assembly of RNA-RNA complexes.
Assemble a 1 mL PCR reaction by mixing 100 μL 10x Taq buffer, forward and reverse primers to 5 μM final concentration, 100 μL 2.5 mM each dNTP, 5 μL Taq DNA Polymerase (DNAP, 25 units) and 1 μg plasmid DNA. 10x Taq buffer: 100 mM Tris-Cl pH 8.0, 500 mM KCl, 15 mM MgCl2. Verify the expected length of PCR products by agarose gel electrophoresis. Testing showed that the PCR reaction can be used directly as template for in vitro transcription in its entirety without further purification. Removal of the Taq DNAP by phenol/chloroform extraction or PCR product purification is unnecessary.
With appropriate DNA templates in hand, which can include several PCR products or linearized plasmids that encode the target RNA and any applicable trans-acting ribozymes, proceed to analytical in vitro transcription trials. When multiple DNA templates are included in the same reaction, they compete for the same pool of T7 RNAP and exhibit different efficiencies in T7 utilization and RNA yield. To balance the relative yield of the transcripts, such as the target RNA bearing the VS substrate appendage relative to the VS ribozyme, or the tRNA relative to the T-box RNA, several ratios of the DNA templates are generally tested to optimize the production of end-processed target RNA, or stoichiometric RNA-RNA complex formation.
Assemble several small-scale (50 μL) in vitro transcription reactions to optimize complex assembly. Scale up to 1–10 mL for preparative transcription reactions. 1 mg of purified RNA can be reasonably obtained from a 1 mL transcription reaction. For a standard 10 mL reaction, mix 1 mL 10x T7 transcription buffer, 25 μL 2 M MgCl2 (to 5 mM), 500 μL each of 100 mM ATP, CTP, GTP, and UTP (to 5 mM), up to 4 mL (40% v/v) of PCR reaction, appropriate amount of T7 RNAP and inorganic pyrophosphatase, and use DEPC water to bring up to volume. The optimal quantities of the two enzymes are usually empirically determined by titration series that monitor RNA yield using Urea-PAGE. 10x T7 transcription buffer: 300 mM Tris-HCl pH 8.1, 250 mM MgCl2, 100 mM dithiothreitol (DTT), 0.1% Triton X-100, and 20 mM spermine in DEPC water. Incubate at 37 °C for 2–12 hours in a water bath or PCR machine.
Prepare a non-denaturing (or native) gel solution and cast the analytical gel while waiting for the transcription reaction. To make 40 mL 10% non-denaturing polyacrylamide gel solution, mix 8 mL 5x THE (Tris-HEPES-EDTA) buffer, 0.2 mL 2 M MgCl2,10 mL 40% acrylamide solution (acrylamide: bis-acrylamide 29:1), 160 μL 25% Ammonium persulfate (APS), 40 μL TEMED and 22 mL MilliQ water. To prepare 2 L of 5x THE buffer, dissolve in DEPC water 40 g Tris Base, 157 g HEPES and 2 mL 0.5 M EDTA (pH 8.0). Bring to 2 L volume and filter to sterilize.
Combine 5 μL of transcription reaction with 5 μL 2x native gel loading buffer. 2x native gel loading buffer: 100 mM Tris-Cl pH 7.0, 20% (v/v) glycerol, 0.025% (w/v) Bromophenol Blue, 0.025% (w/v) Xylene Cyanol. Load 1–5 μL per lane. Run the native gel in 1x THEM buffer at 25 W (constant power) until the bromophenol blue dye migrates close to the bottom of the gel. 1x THEM buffer (1 L): 200 mL 5x THE buffer, 5 mL 2 M MgCl2 (to 10 mM) and 795 mL MilliQ water. Keep the temperature of the native gels below 30 °C using fans and metal plates affixed to gel plates.
Following electrophoresis, stain the gel in Ethidium Bromide (EtBr) or SYBR Gold staining solution for 5 minutes and destain in deionized MilliQ water for another 5 minutes. Change water 2 times to reduce background staining. Image the gel using an appropriate gel-imaging system such as an ImageQuant 800 or Typhoon scanner.
The transcription reaction may generate some residual precipitation of Magnesium pyrophosphate (Mg2P2O7) despite the presence of inorganic pyrophosphatase in the reaction mixture, producing a cloudy appearance. Remove any precipitate by centrifugation at 9000 g for 5 min.
Use 10K molecular weight cut-off (MWCO) concentrators to concentrate and desalt the supernatant. Centrifuge at 5500 × g for 30 min at 4 °C to reduce the volume by 80–90%. Dilute the sample back to the original volume with a low-salt buffer and repeat the centrifugation.
Recover the RNA sample from the concentrator and filter through a 0.2-micron filter to remove any residual precipitate in preparation for chromatographic separation. Avoid freeze-thaw cycles that may dissociate the RNA-RNA complex.
3.4. Purification of large RNA-RNA complexes by size-exclusion chromatography (SEC).
After desalting and filtration, load the sample onto a size-exclusion column to purify the complex. RNA-RNA complex will be separated from the individual transcripts, abortive transcripts, RNase-cleaved RNA, DNA templates, protein enzymes including Taq DNAP, T7 RNAP and inorganic pyrophosphatase, NTPs and other small molecules (Fig. 1). A Cytiva Superdex 200 Increase 10/300 GL (analytical) or HiLoad Superdex 200 16/60 (preparative) column is effective to separate the B. subtilis glyQS T-box discriminator-tRNAGly complex (144 nts, ~46 kD, Kd ~ 2 μM) from free tRNA (75 nts, ~ 24 kD) (Fig. 1b). Excess tRNA is used to saturate the discriminator, as the free discriminator does not separate well from the complex.
Equilibrate the column by running 2 column volumes (CV) of SEC buffer at a flow rate of 1 mL/min. SEC buffer: 10 mM Tris-HCl pH 7.4, 100 mM KCl and 20 mM MgCl2.
Inject a concentrated RNA complex sample onto the column using an ÄKTA pure system at the flow rate of 1 mL/min and continue with isocratic elution. Reducing sample injection volume increases complex stability and improves separation resolution.
Collect the eluted samples in 0.5 – 1 mL fractions using a fraction collector into 96-well deep-well plates. Analyze relevant fractions by native and denaturing gel electrophoresis and pool appropriate fractions based on the chromatogram and analytical gels (Fig. 1c-d). The sample is now ready for X-ray crystallographic and SAXS analyses (Fig. 3).
Fig. 1. Size-exclusion chromatography (SEC) purification of a T-box discriminator – tRNAGly complex.
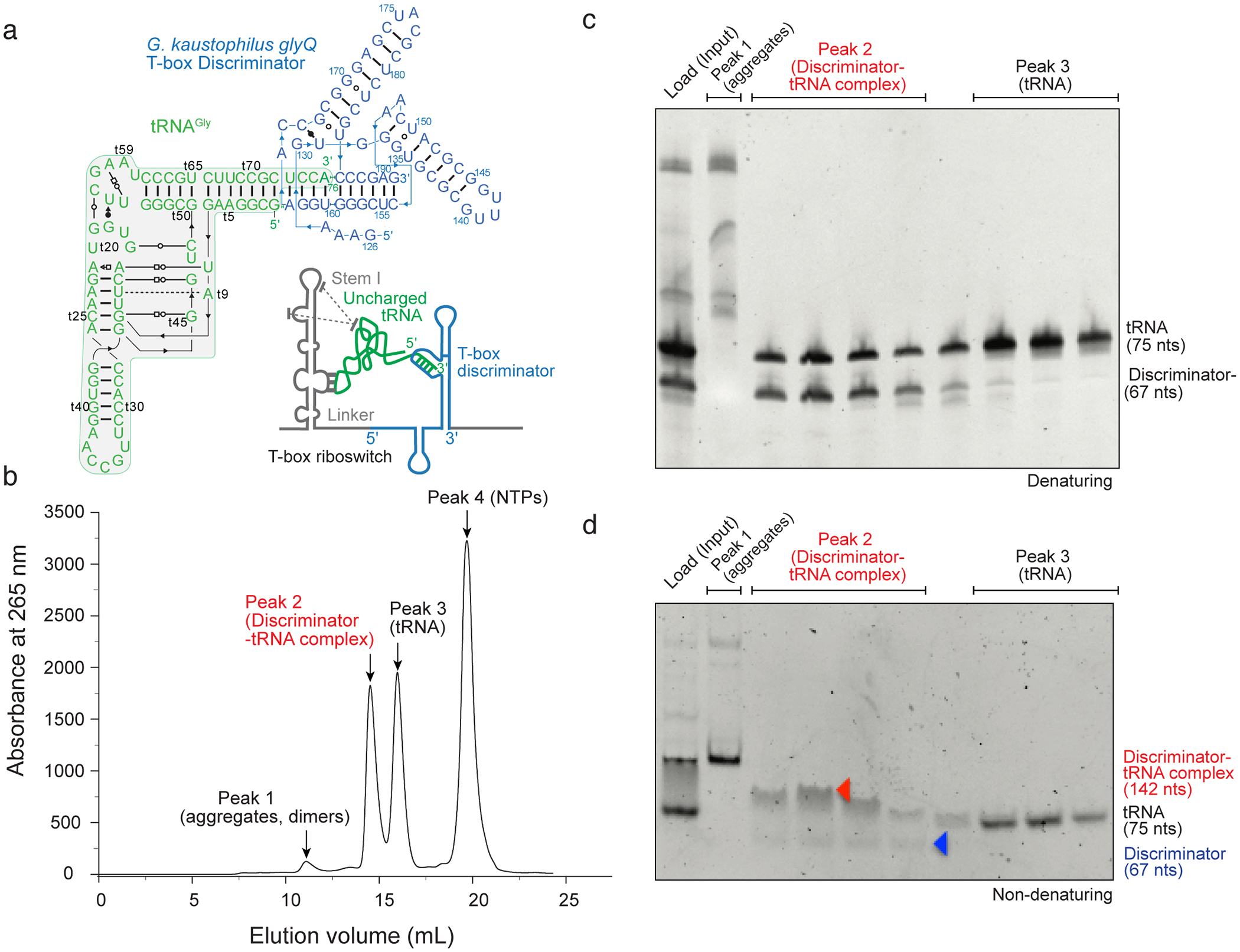
(a) Secondary structure of Geobacillus kaustophilus glyQ T-box Discriminator – tRNAGly complex informed by the co-crystal structure (PDB: 6PMO). Inset: cartoon scheme of a typical full-length glycine-specific T-box riboswitch bound to its cognate tRNA substrate. Dotted lines indicate stacking interactions between the apical region of the Stem I and tRNA elbow. (b) SEC profile of native, co-transcriptionally folded and assembled T-box Discriminator – tRNAGly complex. Main peaks are labeled, and their contents indicated. (c) Denaturing gel (10%) analysis of peak fractions of (b). Bands are annotated. (d) Non-denaturing gel (10%) analysis of the same peak fractions in (b) & (c).
Fig. 3. Crystal and Cryo-EM structures solved using the protocol and corresponding method details.
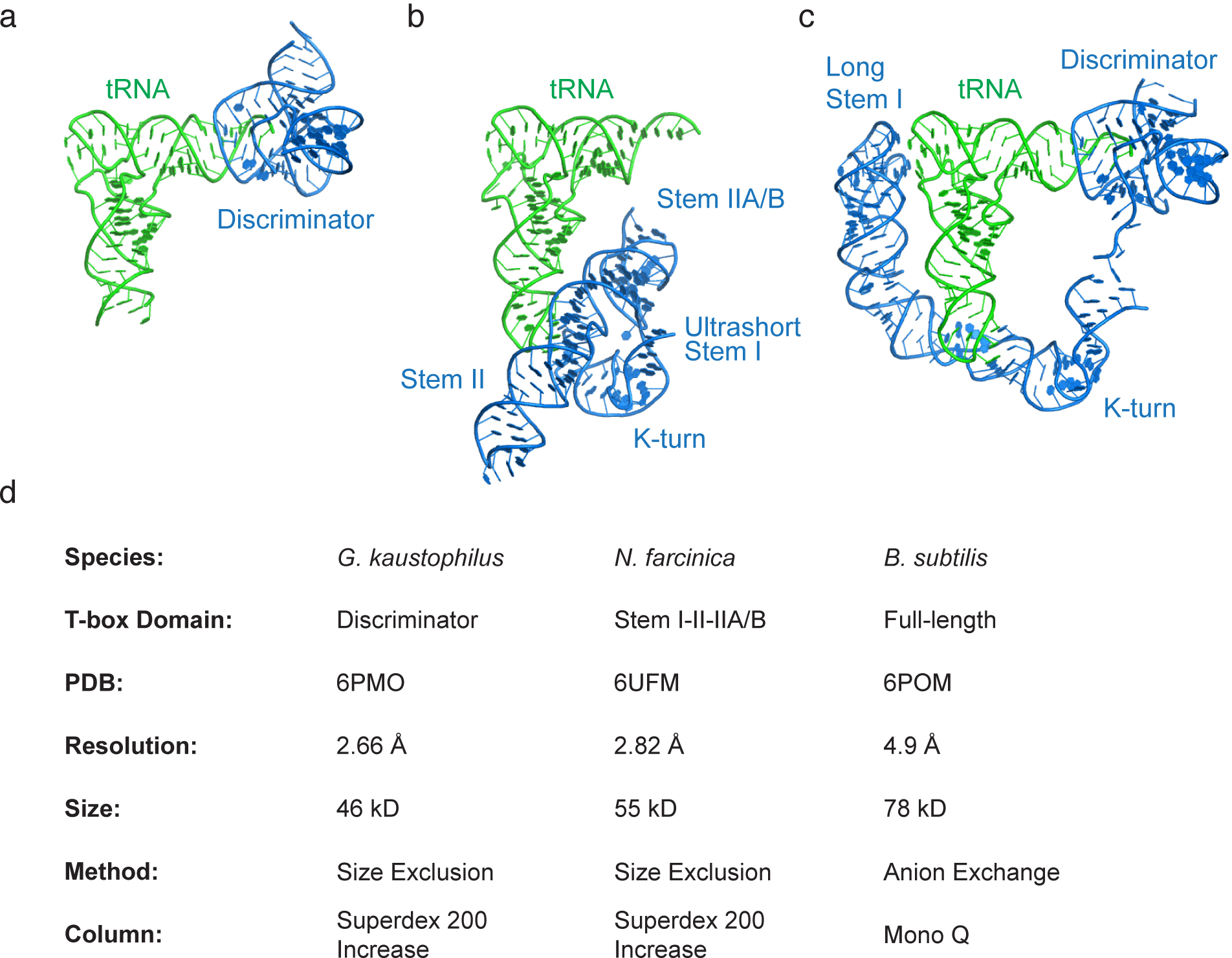
(a) Co-crystal structure of the Geobacillus kaustophilus glyQ T-box Discriminator – tRNAGly complex (PDB: 6PMO). (b) Co-crystal structure of the Nocardia farcinica ileS T-box Stem I-Stem II domains in complex with the cognate tRNAIle (PDB: 6UFM). (c) Cryo-EM structure of the Bacillus subtilis glyQS T-box full-length RNA (anti-terminated conformation) in complex with its cognate tRNAGly (PDB:6POM). (d) Characteristics of RNA samples and method parameters employed to solve the structures in (a-c).
3.5. Purification of large RNA-RNA complexes by anion exchange chromatography.
Anion exchange chromatography on a strong anion exchanger such as the Mono Q achieves superior resolutions than SEC. However, due to the extensive negative charge of larger RNAs, elution generally requires substantial ionic strength (up to 800 mM NaCl) which may dissociate some RNA-RNA complexes under study. The full-length T-box riboswitch and tRNA forms extraordinarily stable complexes due to coordinated, multivalent base-pairing and coaxial stacking interactions (Fig. 2) [18]. This complex can withstand high ionic strengths and enabled high-resolution separation of the 1:1 complex from the free T-box, which is not achievable using SEC as previously reported [26].
Assemble a 5 mL co-transcription reaction by mixing 500 μL 10x T7 transcription buffer, 12.5 μL 2 M MgCl2, 250 μL 100 mM each NTP, 67 μL T7 RNAP (prepared in house), 10 μL inorganic pyrophosphatase (1 Unit/μL), 1 mL full-length B. subtilis glyQS T-box PCR mixture, and 1 mL tRNAGly PCR mixture. Adjust final volume to 5 mL with DEPC water and incubate the reaction at 37 °C for 4–8 h. Desalt and filter the sample as described in Section 3.3.
Equilibrate a Mono Q column by running 5–10 column volumes of binding buffer at a flow rate of 0.5 mL/min. Binding buffer: 10 mM Tris-HCl pH 7.4, 100 mM KCl and 20 mM MgCl2.
Inject the RNA complex sample onto the Mono Q column. Unlike SEC, sample volumes don’t significantly impact resolution on ion exchange columns. Resolution is largely determined by the slope of the eluting salt gradient and the differences in molecular mass, shape, and total surface-exposed charge (Fig. 2b).
Elute the bound RNAs by applying a linear KCl gradient of 100 mM-1 M over 50–100 CV. While the tRNA elutes early at ~ 350–450 mM KCl, stoichiometric full-length T-box-tRNA elutes at ~ 600–700 mM KCl (Fig. 2b). Misfolded or aggregated T-box RNA and other oligomers elute later at > 700 mM KCl, presumably due to higher overall negative charges that stem from more extended conformations, exposed phosphate backbones, or RNA oligomerization or aggregation.
Assess the contents of the fractions by native and denaturing gel electrophoresis (Fig. 2c-d). Pool appropriate fractions that contain the stoichiometric T-box-tRNA complex, desalt and concentrate using Amicon spin concentrators, filter through UltraFree filter units, concentrate further, and measure the concentration using a Nanodrop spectrophotometer prior to storage. The highly purified RNA complex sample is now ready for SAXS, Cryo-EM, or X-ray crystallographic analyses (Fig. 3).
Wash the column with 2 column volumes (CV) of 2 M NaCl, 4 CV of 0.25 M NaOH, 3 CV of 2 M NaCl and 4 CV of DEPC water to remove all the bound molecules. Fill the column with 4 CV of 20% Ethanol for storage.
Fig. 2. Anion-exchange chromatography purification of a full-length T-box – tRNAGly complex.
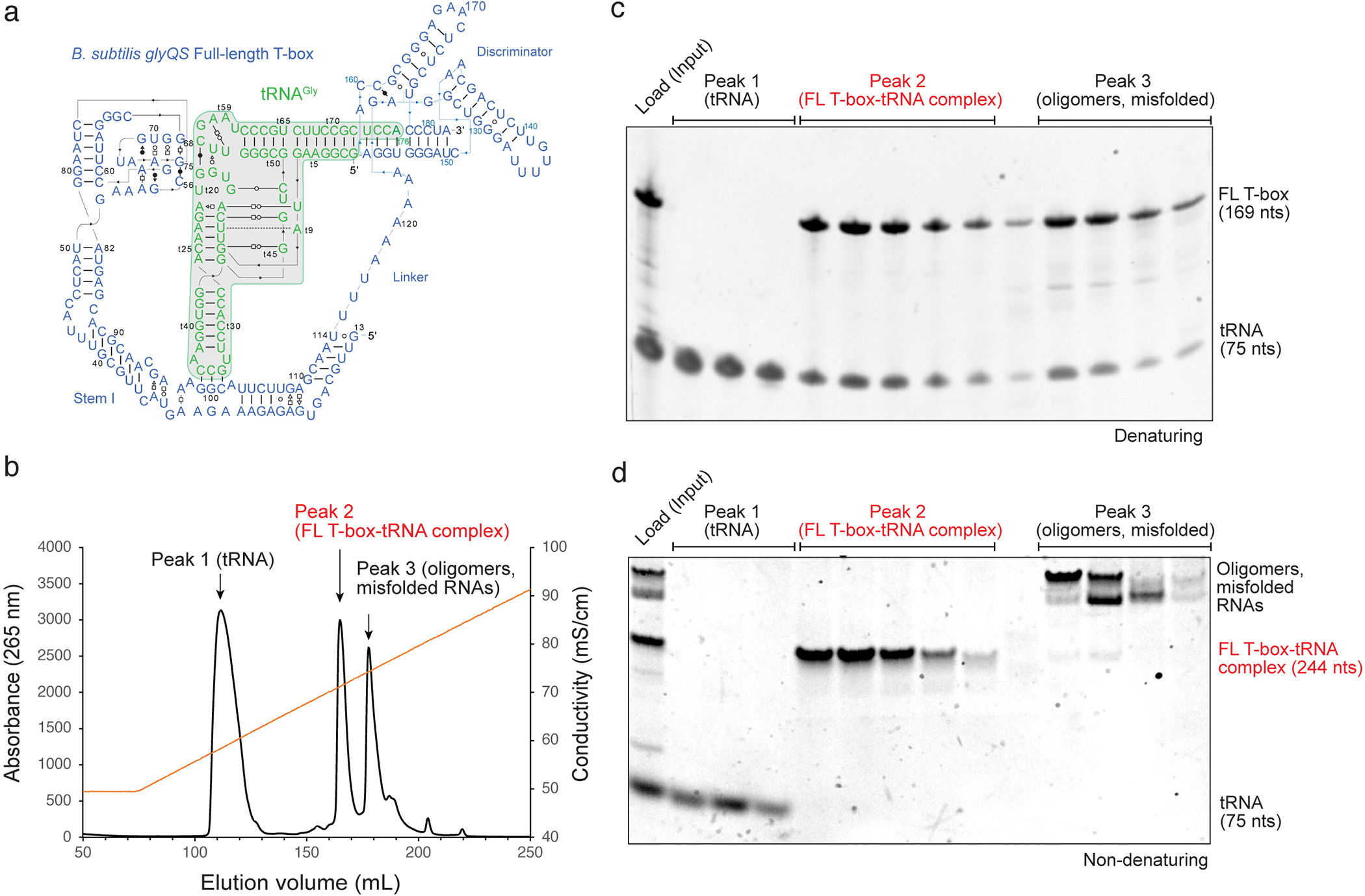
(a) Secondary structure of Bacillus subtilis glyQS full-length T-box– tRNAGly complex informed by two co-crystal structures (PDB: 6PMO; 4LCK) and cryo-EM structure (PDB: 6POM). (b) Mono Q profile of native, co-transcriptionally folded and assembled full-length T-box– tRNAGly complex. Main peaks are labeled, and their contents indicated. The conductivity trace representing the salt gradient is shown in orange. (c) Denaturing gel (10%) analysis of peak fractions of (b). Bands are annotated. (d) Non-denaturing gel (10%) analysis of the same peak fractions in (b) & (c).
4. Notes
tRNA acts as a folding chaperone for the T-box discriminator and full-length T-box. tRNA can be prepared either in advance as purified, refolded RNA added at the start of the in vitro transcription reaction, or be transcribed and folded simultaneously with the T-box RNA.
Due to the extensive negative charge of RNA backbone phosphates, RNAs bear much larger, multi-layered hydration shells. As a result, RNA elutes much earlier from SEC columns, exhibiting apparent molecular weights that are 3–5 times larger than globular proteins.
Similar to proteins, the shape and flexibilities of RNAs impact their behavior on columns such as SEC in additive to their sizes. Correctly folded, functionally relevant RNA-RNA complexes are generally much more compact than their misfolded counterparts and are simultaneously larger in size. As a result, they frequently elute close to more extended individual RNA components. Thus, careful analysis of the stoichiometry of component RNAs in the complex peaks by biochemical analyses can be required to ensure sample homogeneity. Comparison of chromatograms of individual RNAs with their complexes are usually diagnostic of such phenomena. For example, free full-length T-boxes elutes from SEC columns at approximately the same volume as its tRNA complex, giving the false impression of a pure 1:1 complex [26].
For RNA-RNA complexes that depend on Mg2+ for stability, such as the T-box-tRNA complexes, it is important to avoid depriving the sample of Mg2+ at any step of the protocol. Once dissociated, some individual RNAs will misfold and can no longer reform the complex.
ACKNOWLEDGEMENTS
We thank G. Piszczek and D. Wu for support with biophysical analyses, and A. R. Ferré-D’Amaré, K. Suddala and C. Bou-Nader for discussions. This work was supported by the Intramural Research Program of the NIH, The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (ZIADK075136 to J.Z.), and a NIH Deputy Director for Intramural Research (DDIR) Challenge Award to J.Z. The authors declare no conflicts of interests.
References
- 1.Harris KA and Breaker RR, (2018) Large Noncoding RNAs in Bacteria. Microbiol Spectr. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peselis A, Gao A, and Serganov A, (2015) Cooperativity, allostery and synergism in ligand binding to riboswitches. Biochimie. 117:100–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reiter NJ, et al. , (2010) Structure of a bacterial ribonuclease P holoenzyme in complex with tRNA. Nature. 468:784–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suslov NB, et al. , (2015) Crystal structure of the Varkud satellite ribozyme. Nat Chem Biol. 11:840–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferré-D’Amaré AR and Scott WG, Small self-cleaving ribozymes, in The RNA World, Fourth Edition, Gesteland RF, Cech TR, and Atkins JF, Editors. 2010, Cold Spring Harbor Laboratory Press: Cold Spring Harbor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Korostelev A, et al. , (2006) Crystal structure of a 70S ribosome-tRNA complex reveals functional interactions and rearrangements. Cell. 126:1065–77. [DOI] [PubMed] [Google Scholar]
- 7.Ramakrishnan V, (2002) Ribosome structure and the mechanism of translation. Cell. 108:557–572. [DOI] [PubMed] [Google Scholar]
- 8.Bou-Nader C and Zhang J, (2020) Structural Insights into RNA Dimerization: Motifs, Interfaces and Functions. Molecules. 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hood IV, et al. , (2019) Crystal structure of an adenovirus virus-associated RNA. Nat Commun. 10:2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J and Ferre-D’Amare AR, (2014) Dramatic improvement of crystals of large RNAs by cation replacement and dehydration. Structure. 22:1363–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stagno JR, et al. , (2017) Structures of riboswitch RNA reaction states by mix-and-inject XFEL serial crystallography. Nature. 541:242–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Somarowthu S, et al. , (2015) HOTAIR forms an intricate and modular secondary structure. Mol Cell. 58:353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wickiser JK, et al. , (2005) The speed of RNA transcription and metabolite binding kinetics operate an FMN riboswitch. Mol Cell. 18:49–60. [DOI] [PubMed] [Google Scholar]
- 14.Zhang J, Lau MW, and Ferré-D’Amaré AR, (2010) Ribozymes and riboswitches: modulation of RNA function by small molecules. Biochemistry. 49:9123–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang J and Landick R, (2016) A Two-Way Street: Regulatory Interplay between RNA Polymerase and Nascent RNA Structure. Trends Biochem Sci. 41:293–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong TN, Sosnick TR, and Pan T, (2007) Folding of noncoding RNAs during transcription facilitated by pausing-induced nonnative structures. Proc Natl Acad Sci U S A. 104:17995–8000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J, (2020) Unboxing the T-box riboswitches-A glimpse into multivalent and multimodal RNA-RNA interactions. Wiley Interdiscip Rev RNA. 11:e1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li S, et al. , (2019) Structural basis of amino acid surveillance by higher-order tRNA-mRNA interactions. Nat Struct Mol Biol. 26:1094–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suddala KC and Zhang J, (2019) High-affinity recognition of specific tRNAs by an mRNA anticodon-binding groove. Nat Struct Mol Biol. 26:1114–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coleman TM, Wang G, and Huang F, (2004) Superior 5’ homogeneity of RNA from ATP-initiated transcription under the T7 phi 2.5 promoter. Nucleic Acids Res. 32:e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin CT and Coleman JE, (1989) T7 RNA polymerase does not interact with the 5’-phosphate of the initiating nucleotide. Biochemistry. 28:2760–2. [DOI] [PubMed] [Google Scholar]
- 22.Vasilyev N and Serganov A, (2016) Preparation of Short 5’-Triphosphorylated Oligoribonucleotides for Crystallographic and Biochemical Studies. Methods Mol Biol. 1320:11–20. [DOI] [PubMed] [Google Scholar]
- 23.Ferré-D’Amaré AR and Doudna JA, (1996) Use of cis- and trans-ribozymes to remove 5’ and 3’ heterogeneities from milligrams of in vitro transcribed RNA. Nucleic Acids Res. 24:977–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiao H, et al. , (2008) Structural basis of specific tRNA aminoacylation by a small in vitro selected ribozyme. Nature. 454:358–361. [DOI] [PubMed] [Google Scholar]
- 25.Kao C, Zheng M, and Rudisser S, (1999) A simple and efficient method to reduce nontemplated nucleotide addition at the 3 terminus of RNAs transcribed by T7 RNA polymerase. RNA. 5:1268–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang J and Ferré-D’Amaré AR, (2014) Direct evaluation of tRNA aminoacylation status by the T-box riboswitch using tRNA-mRNA stacking and steric readout. Mol Cell. 55:148–55. [DOI] [PMC free article] [PubMed] [Google Scholar]