

Abstract
Neuronal communication relies on precisely maintained synaptic vesicle (SV) clusters, which assemble via liquid-liquid phase separation (LLPS). This process requires synapsins, the major synaptic phosphoproteins, which are known to bind actin. The reorganization of SVs, synapsins and actin is a hallmark of synaptic activity, but their interplay is still unclear. Here we combined the reconstitution approaches and super-resolution imaging to dissect the roles of synapsin-SV condensates in the organization of the presynaptic actin cytoskeleton. Our data indicate that LLPS of synapsin initiates actin polymerization, allowing for SV:synapsin:actin assemblies to facilitate the mesoscale organization of SV clusters along axons mimicking the native presynaptic organization in both lamprey and mammalian synapses. Understanding the relationship between the actin network and synapsin-SVs condensates is an essential building block on a roadmap to unravel how coordinated neurotransmission along the axon enables circuit function and behavior.
Introduction
Liquid-liquid phase separation (LLPS) is a major mechanism for organizing macromolecules, particularly proteins with intrinsically disordered regions, in compartments not limited by a membrane or a scaffold (Banani et al, 2017). However, it is currently unclear how such a complex concoction operates to allow for intracellular trafficking, signaling and metabolic processes to occur with high spatio-temporal precision. We recently discovered that the cluster of synaptic vesicles (SVs) at nerve terminals represents a biomolecular condensate, which is packed with membrane-bound organelles, the synaptic vesicles (Milovanovic et al, 2018; Sansevrino et al, 2023; Alfken et al, 2024). While decades of research have pointed out that SVs are clustered by synapsins, a family of phospho-proteins highly abundant at nerve terminals (De Camilli et al, 1990; Rosahl et al, 1995; Pieribone et al, 1995), only recently the intrinsically disordered region (IDR) of synapsin was shown to be central for the assembly of SV condensates (Milovanovic et al, 2018; Hoffmann et al, 2023b). These data corroborate the analysis in living synapses both upon acute and chronic disruption of synapsins. For example, the injection of anti-synapsin antibodies in the giant reticulospinal synapse of the lamprey results in the dispersion of SVs upon depolarization and at rest (Pieribone et al, 1995; Pechstein et al, 2020). Similarly, the chronic depletion of synapsins in mice – i.e., through synapsin gene deletions – results in less vesicles accumulating at the synaptic boutons; the SVs within the bouton are more dispersed than in wildtype synapses (Rosahl et al, 1995; Gitler et al, 2004).
All synapsin proteins contain a well-conserved central region, so-called Domain C (Südhof et al, 1989; Benfenati et al, 1992), which was identified as an actin-binding site (Bähler & Greengard, 1987; Petrucci & Morrow, 1987; Petrucci et al, 1988; Bähler et al, 1989). For several decades, the presence of actin within the synaptic vesicle clusters has been repeatedly demonstrated, from the classical EM assessments (Hirokawa et al, 1989) to three dimensional reconstructions based on multimodal analyses (Wilhelm et al, 2014). Nucleation-dependent processes such as actin polymerization are particularly sensitive to changes in the concentration of G-actin and actin-regulatory proteins (Case et al, 2019). As synapsins both undergo LLPS and bind to actin, SV condensates are poised to act as molecular beacons for the formation of nucleated actin assemblies in an activity-dependent manner. According to the current model of SV cluster/actin interaction, synapsins at low concentrations nucleate actin and, together with SVs, lower the critical concentration of actin required for the assembly of filaments (Bähler & Greengard, 1987; Valtorta et al, 1992). The acute perturbations of actin and synapsin and the analysis upon electrical activity led to a model in which actin forms corrals around SV clusters with filaments (so-called actin tracks) extending to both the active zone and endocytic sites (Shupliakov et al, 2002; Bloom et al, 2003; Sankaranarayanan et al, 2003). Synapsin was shown to closely associate with both vesicles in the cluster and actin networks involved in synaptic vesicle recycling (Cingolani & Goda, 2008; Rust & Maritzen, 2015; Soykan et al, 2017).
However, this model of the actin network at synaptic boutons does not corroborate with several important findings. First, the acute depletion of synapsins both at rest and upon stimulation disrupts the clustering of SVs (Pieribone et al, 1995; Pechstein et al, 2020). Conversely to synapsin perturbations, disruptions of actin at lamprey and mouse hippocampal synapses do not disperse the distal SV cluster (Bourne et al, 2006; Bloom et al, 2003; Sankaranarayanan et al, 2003). Second, while synapsin colocalizes with the SV clusters, imaging data at lamprey and mouse synapses indicate the vast majority of actin is outside the SV cluster at the periactive zone, not precisely overlapping with synapsin signal (Shupliakov et al, 2002; Wilhelm et al, 2014). Third, quantitative mass spectrometry indicates that synapsins are present at three times higher concentration than actin at the synaptic bouton (Wilhelm et al, 2014), arguing against them being mere crosslinkers of actin filaments. Fourth, our recent work on synapsin phase separation implies that proteins including actin can be recruited to the synapsin-SV phase through both specific protein-protein interactions and transient associations (Milovanovic et al, 2018). In light of these new data, how synapsin-driven SV clustering coordinates with the actin network remains elusive.
Here we set out to determine the cause-effect relationship between the formation of synapsin-SV condensates and actin organization at the synaptic boutons. Our data show that LLPS of synapsin condensates initiate actin polymerization, allowing for SV:synapsin:actin assemblies to be coupled with cortical actin and actin rings and to facilitate the mesoscale organization of SVs along axons.
Results
Synapsin 1 condensates sequester actin monomers and stimulate actin polymerization
We first sought to investigate the role of synapsin 1 condensation on actin polymerization. To address this question, we first purified EGFP-tagged full-length synapsin 1 (Syn1-FL) using a mammalian expression system (Supplementary Fig. 1). We next reconstituted Syn1-FL condensates (final concentration - 4 μM) with crowding agent polyethylene glycol [PEG8K, final concentration 3% (w/v)] to mimic the crowded environment of the cytoplasm of synaptic boutons. We observed the Syn1-FL condensates with diameters ranging from 2-4 μm on a glass-bottom dish using a spinning-disk confocal microscope. After 5 minutes, we added ATTO647-labeled G-actin monomers (final concentration 4 μM) to pre-formed Syn1-FL condensates and followed actin polymerization for 35 minutes (Supplementary Fig. 1). We observed robust enrichment of G-actin monomers within Syn1-FL condensates as the fluorescence of actin within Syn1-FL regions increased rapidly over time and plateaued within 20 minutes (Fig. 1A, 2A). After about 20 min, we witnessed polymerized actin within Syn1-FL clusters congregating into radial arrays (Fig. 1C). It is worth highlighting that actin polymerized within Syn1-FL condensates and not within the surrounding solution. Actin polymerization into asters is distinct for Syn1-FL condensates since actin alone did not polymerize into such actin arrays or fibrils in the presence of commercial polymerization buffer (Supplementary Fig. 2A). In contrast to Syn1-FL, actin did not polymerize in the presence of EGFP-protein alone (Supplementary Fig. 2B).
Figure 1: Synapsin 1 condensates are sites for actin polymerization.

A. Representative confocal images from the in-vitro reconstitution of EGFP-Syn1-FL (4 μM, 3% (w/v) PEG 8000) with ATTO-647 labelled G-actin monomers (4 μM) at t = 0 (left) and 35 minutes (right). Buffer used in these experiments, hereafter referred to as ‘reaction buffer’, contained - 25 mM Tris-HCl (pH - 7.4), 150 mM NaCl, 0.5 mM TCEP. Scale bar: 5 μm
B. Confocal images of the EGFP-Syn1-IDR (4 μM, 3% (w/v) PEG 8000) and ATTO-647 labelled G-actin reconstitution (4 μM) in reaction buffer at t = 0 (left) and 35 minutes (right). Scale bar: 5 μm
C. Plot for the quantification of normalized enrichment of actin within EGFP-Syn1-FL phases. Droplets were followed for enrichment within EGFP-Syn1-FL phases for 20 minutes. Error bars represent SD., black line represents the best fit with logistic growth curve: Y=YM*Y0/((YM-Y0)*exp(-k*x) +Y0), YM = 0.9147, Y0 = 0.04611, k = 0.4452, Xint = 2.246, R2 = 0.9355). Data shown here is quantified from 3 independent experiments (N = 3, n = 30).
D. Plot showing the number of Sholl intersections for polymerized actin in presence of EGFP-Syn1-FL, EGFP-Syn1-IDR and EGFP-Syn1-Domain C condensates as a function of distance (in μm). The number of Sholl intersections were counted in 0.5 μm radius increments from the center of the condensates. Each dot represents the mean intersections count for each condition.
E. Plot depicting the quantification of pyrene-actin fluorescence intensity as a function of time. Pyrene-actin (10 μM) polymerization was assessed in presence of EGFP-Syn1-FL or EGFP-Syn1-IDR (10 μM) in 1X actin polymerization buffer (10 mM Tris/HCl, pH = 7.5, 2 mM MgCl2, 50 mM KCl and 0.5 mM ATP). Data shown here is quantified from 3 independent reconstitutions (N = 3).
Figure 2: Actin torus formation around synapsin 1 condensates precedes aster assembly.
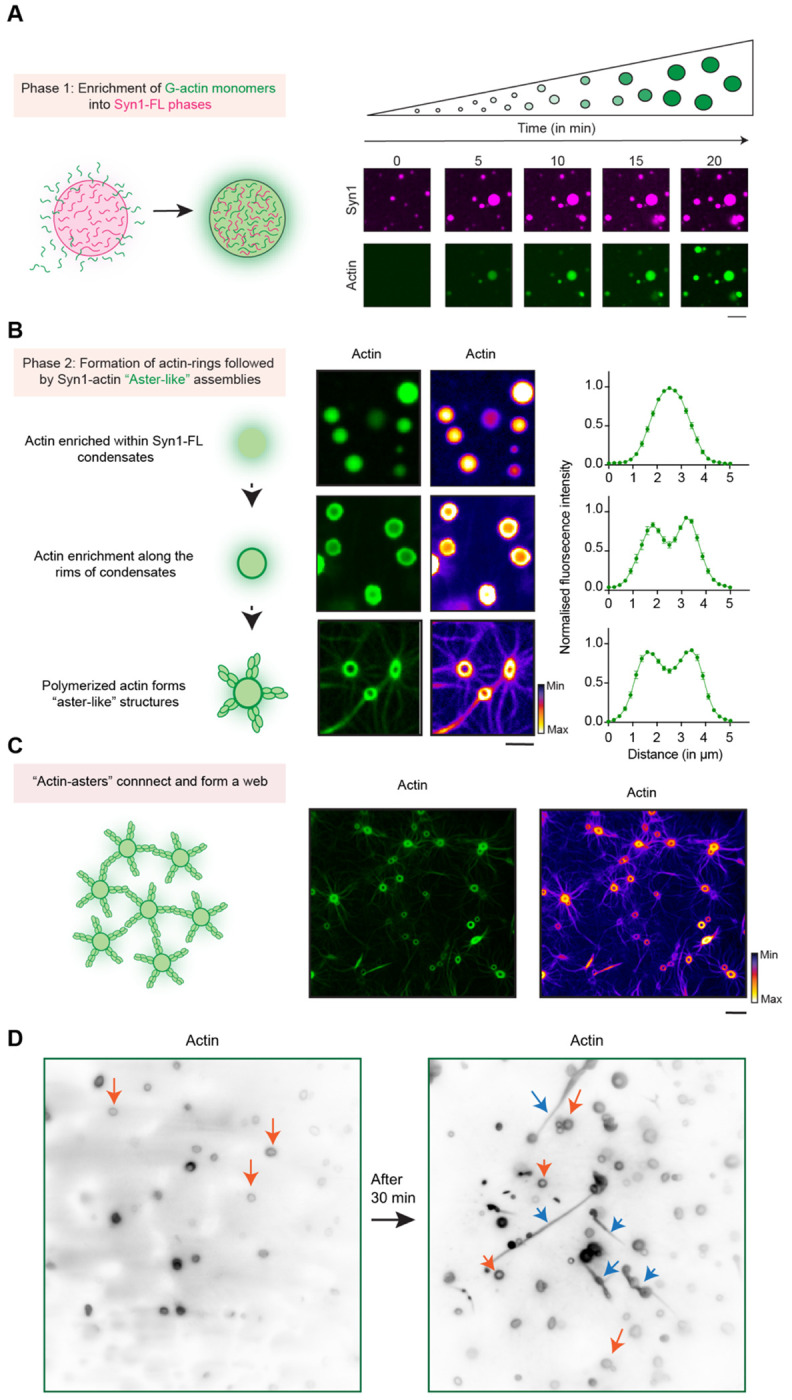
A. Scheme and corresponding representative confocal images from the in-vitro synapsin 1-actin reconstitution assay showing actin enrichment over a period of 20 minutes within EGFP-Syn1-FL condensates. Images were acquired at excitation wavelengths 488 nm and 647 nm for EGFP-Synapsin 1-FL and ATTO647 G-actin respectively. Scale bar, 5 μm.
B. Scheme and corresponding representative confocal images depicting stages of “actin-aster” assembly from EGFP-Syn1-FL condensates; scale bar, 5 μm. Here, the same datasets are shown with two different LUTs (Green and Fire) to highlight ‘torus’ and ‘aster’ structures. Right: line profiles depict enrichment of fluorescently labelled actin within the condensate.
C. Scheme of “actin-asters” connecting and forming “web-like” assembly and corresponding confocal image of polymerized “actin-asters” forming “web-like” structures but with different LUTs (Green and Fire) to highlight ‘torus’ and ‘aster’ structures. Scale bar, 10 μm.
D. Representative images from the EGFP-Syn1-FL and actin reconstitution acquired using TIRF microscopy show “actin-toruses” and “actin-fibers” as indicated with orange and blue arrows respectively. Scale bar, 5 μm.
At early stages, actin was often observed to be homogenously distributed within Syn1-FL condensates, however, it rearranged towards the rims forming so-called “torus-like” structures at later stages followed by dramatic rearrangement into “aster-like” radial assemblies (Fig. 2A, B). The actin-asters emerging from Syn1-FL condensates grew radially and often connected to form networks (Fig. 2C). Interestingly, actin enrichment within the rims persisted even after aster assembly. We found the torus-like distribution of actin within Syn1-FL clusters striking and could also reproduce the same morphology using a different imaging approach - total internal reflection fluorescence (TIRF) microscopy, which allows visualization of structures occurring within <100 nm of the coverslip surface. Beside the above-mentioned morphologies of polymerized actin, we often observed actin-polymerized and elongated fibers frequently accumulating on the glass surface that were positive for both synapsin 1 and actin (Fig. 2D). Actin co-localized with synapsin condensates and had undergone continuous deformations throughout the assembly of these structures. Together, this implies that Syn1-FL condensates are a porous scaffold that permits continuous flux of G-actin monomers, thereby acting as a reaction center for actin polymerization.
Phase separation and actin binding of synapsin both contribute to actin polymerization
We then set out to assess which domain of synapsin 1 is essential for actin polymerization – domain C that can homo and hetero-dimerize (Hosaka & Südhof, 1999) or phase separating IDR region (Milovanovic et al, 2018). We reconstituted actin with synapsin 1 IDR (Syn1-IDR) and synapsin 1-Domain C (Syn1-Dom. C) in the same manner and followed actin polymerization for 35 minutes. We observed that phase-separating Syn1-IDR suffices to polymerize actin. However, the architecture of polymerized actin from Syn1-IDR clusters was completely different from the ones of Syn1-FL condensates, as they appeared more like “networks” rather than “asters” (Fig. 1B). In contrast, Syn1-Dom. C which can homo- and hetero-oligomerize but does not phase separate did not polymerize actin (Supplementary Fig. 3A). Given that there was a difference in the morphology of polymerized actin from Syn1-FL and Syn1-IDR phases, we conducted a Sholl analysis on polymerized actin associated with synapsin condensates and quantified the number of intersections of Sholl spheres with fibrils radiating from the synapsin condensates. Indeed, the count of intersections was higher in the case of actin polymerized from Syn1-FL phases as compared to Syn1-IDR (Fig. 1D). Thus, Syn1-IDR alone can accomplish some actin polymerization, however, other domains–including the actin-binding domain C–are needed to achieve the normal morphologies of the actin structures.
To discern whether any protein that can phase separate and enrich actin into its dense phase meshwork will suffice to trigger actin polymerization (Graham et al, 2023), we repeated these experiments using a stress granule protein G3BP2 (Yang et al, 2020). G3BP2 lacks an actin-binding site like Syn1 has (Jin et al, 2022). Adding actin to the preformed condensates of G3BP2 led to actin accumulation into condensates, as expected given that the dense phase is a dynamic meshwork (Alshareedah et al, 2021), yet it failed to trigger its polymerization (Supplementary Fig. 3B). This clearly indicated that both properties of synapsins - phase separation and the ability to bind actin - are needed to trigger actin polymerization.
We next wanted to dissect whether the difference in the observed network morphology is due to the different polymerization kinetics of Syn1-FL versus Syn-IDR. To assess this, we aimed to quantify differences in kinetics of actin polymerization using a pyrene-actin polymerization assay. For this, we set up reactions of pyrene-labelled actin with Syn1-FL and Syn1-IDR [10 μM pyrene-labelled actin, 10 μM synapsin 1 (either Syn1-FL or Syn1-IDR), 1x actin polymerization buffer with 3% (w/v) PEG 8000)]. We observed no differences in the kinetics of actin polymerization from Syn1-IDR and Syn1-FL phases (Fig. 1E), suggesting that the polymerization activity is preserved but that Domain C is crucial for the stereotypical aster-like morphology. Next, we exploited optical density as a measure to quantify the extent of actin polymerization by Syn1-FL. We set up a reaction of Syn1-FL with actin in polymerizing conditions (4 μM Syn1-FL, 4 μM magnesium-exchanged ATP-actin, 3% (w/v) PEG 8000, 0.5 mM ATP, 140 mM NaCl and 0.01% sodium azide) in a microtiter plate at 37°C and followed turbidity for 48 h at 405 nm. After recording measurements, we visualized the fluorescence of Syn1-FL by microscopy. We noted higher turbidity counts as actin polymerized in the presence of Syn1-FL, while in the absence of either one of them, turbidity stayed minimal (Supplementary Fig. 4). Taken together, our data suggest that Syn1-FL condensates act as reaction centers for the recruitment, nucleation, and polymerization of actin into an elaborate network of interconnected actin-asters. Further, the phase separating IDR region of synapsin 1 is essential for actin polymerization.
Synapsin-SV condensates sequester actin both in reconstituted system and in living synapses
We next examined how synapsin-SVs condensates correspond to actin networks at the synapse. To address this, we reconstituted phases of Syn1-FL (4 μM, 3% (w/v) PEG 8000) with SVs (3 nM, labelled with FM4-64, 1.65 μM) on a glass-bottom dish and imaged using a spinning-disk confocal microscope. After 5 minutes, we added ATTO-647 labelled G-actin monomers (4 μM) to preformed synapsin-SVs condensates and followed actin enrichment within these phases for 35 minutes. Consistently, actin sequestered within synapsin-SV condensates and assembled in actin arrays (Fig. 3A,B). We also reconstituted actin with SVs as a control to test if any SV proteins, including the residual synapsins that are peripherally-associated with the lipid bilayer of SVs, could promote actin polymerization. Yet, we did not observe actin polymerization in the presence of SVs alone (Supplementary Fig. 5A), indicating the central role of synapsins. In line with previous studies (Benfenati et al, 1989; Benfenati et al, 1992), the presence of low concentrations of synapsin 1 enhanced the binding of actin to SVs, as indicated by fluorescence correlation spectroscopy (Supplementary Fig. 5B), but was insufficient to trigger polymerization.
Figure 3: Synapsin-SV condensates sequester actin both in reconstituted system and in living synapses.

A. Left: Representative images of the reconstituted EGFP-Syn1-FL (4 μM, 3% (w/v) PEG 8000) and SVs (3 nM, labelled with FM4-64, 1.65 μM) condensates after adding ATTO-647 labelled G-actin monomers (4 μM) at t = 0 (top) and 35 minutes (bottom). Images were acquired using a spinning disk confocal microscope. Right: Magnified images of polymerized actin-asters formed at t = 35 minutes. Scale bar, 5 μm.
B. Scheme of SV recruitment along the actin fibers.
C. Cartoon illustrating the lamprey spinal cord with giant reticulospinal (RS) axons and microinjection strategy. V, ventral; D, dorsal.
D. Laser-scanning confocal microscopy (LSM) images of a live lamprey reticulospinal axon co-injected with FM4-64 and Alexa Fluor 488-phalloidin for characterizing SVs and actin, respectively. Scale bar, 5 μm. Excitation wavelengths were 488 nm for phalloidin-actin and 560 nm for SVs labelled with FM4-64. Insets 1-3 show clear assembly of actin as a torus around SVs; scale bar, 1 μm.
E. LSM images of immunolabeled lamprey synapses. Synapses were stained for endogenous synapsin 1, endogenous SV2 (an SV marker) and actin (Alexa488-phalloidin). Scale bar, 1 μm.
F. Actin torus in lamprey synapse containing “aster-like” structures indicated with white arrowheads (highlighted regions 1, 2, and 3). Images were acquired using a Zeiss CellDiscoverer7 with LSM900 Airyscan2 (plan apochromat 50x/1.2 objective). Scale bar, 0.5 μm.
Given that actin polymerized into “network-like” structures from Syn1-IDR phases previously, we questioned whether we could reproduce actin sequestering and polymerization from Syn1-IDR clusters in the presence of SVs. Indeed, actin sequestered within Syn1-IDR-SV condensates and polymerized into networks including aster-like structures (Supplementary Fig. 6). This might be either due to synapsin molecules that remain associated with SVs or due to other putative actin binders present at the surface of SVs.
We then aimed to validate our in vitro observations further in vivo using lamprey synapses as a model. Lampreys possess within their spinal cords a subset of giant reticulospinal (RS) axons (20-80 μm) with large en passant glutamatergic synapses (1-2 μm diameter; 1,000-2,000 SVs) along the perimeter of the axolemma, allowing experimental accessibility for exploring the mesoscale organization of presynapse. Consequently, lamprey synapses serve as an excellent model for studying interactions between SVs and actin networks within axons (Shupliakov et al, 2002; Bloom et al, 2003; Bourne et al, 2006). We co-injected Alexa488-phalloidin and FM4-64 for characterizing actin and SVs respectively within the giant axons and imaged using a laser-scanning confocal microscope within 10 minutes (Fig. 3C). We observed that actin assembled as “tori” around the SVs (Fig. 3D), as was previously demonstrated (Shupliakov et al, 2002; Bloom et al, 2003; Bourne et al, 2006). We also performed whole mount immunofluorescence on Alexa-phalloidin injected axons and confirmed the mesoscale organization of the synapsin-SV condensates, surround by the actin toruses (Fig. 3E, Supplementary Fig. 7). Using the advanced confocal airyscan imaging, we were able to capture high-resolution images of actin toruses around SV clusters (Fig. 3E). We ensured that the synapsin 1 antibodies used for the experiments recognize the endogenous lamprey synapsin (Supplementary Fig. 8). Excitingly, we further also observed asters emerging from these “actin-toruses” (Fig. 3F, Supplementary Fig. 7), implying a role of SVs phases in the assembly of actin networks. Altogether, these results consistently imply that synapsin-SV phases sequester and polymerize actin in both minimal reconstitution system and in living lamprey synapses.
The surface of lipid membranes shapes actin polymerization from synapsin condensates
The synaptic bouton is a densely packed environment with numerous membrane-bound organelles and the presynaptic plasma membrane in the vicinity of the SV clusters (Rizzoli & Betz, 2004; Wu et al, 2017). We next employed Giant Unilamellar Vesicle (GUV) as a model system to characterize the effect of membranes on actin polymerization from synapsin condensates. We reconstituted synapsin condensates as previously described and added them to neutrally charged DOPC GUVs (final lipids concentration – 1.5 μM). After 5 minutes, when condensates settled on GUVs surfaces, we added G-actin monomers spiked with ATTO-647 G-actin (final total actin monomers concentration – 4 μM) and added polymerization buffer to the reaction mix. Similar to our previous observations, actin enriched over time within synapsin condensates forming “aster-like” assemblies (Fig. 4A). Interestingly, we observed that many synapsin 1-actin condensates were interconnected with each other by actin filaments, and the length of these connecting filaments were several times longer than the filaments going from condensate to solution without reaching another condensate (Fig. 4B). Interconnected condensates formed elaborate networks, which could span over territories of more than 100 square micrometers (Fig. 4C). The lipid surface of GUVs acted as a template for actin polymerization stemming from the condensates wetting the GUV membrane (Fig. 4D,E). This suggests that synapsin condensates wetting neutrally charged membranes do not inhibit actin polymerization and membranes may serve as a guiding surface for (synapsin-bound) actin filaments. The complex nature of condensates-actin networks and the significant difference in length between “connecting” and “free” actin filaments may indicate a role of synapsin condensates as a “homing beacon/amplifier” for actin polymerization.
Figure 4: Actin aster are stable structures connecting neighboring condensates.

A. Representative confocal image from the reconstituted EGFP-synapsin 1:actin:GUV assemblies. Projection of the maximal intensity, 30 minutes post-incubation. Scale bar, 10 μm.
B. Magnified region from a indicating the long actin fibers connecting the adjacent condensates.
C. Top: Skeletonized actin fibrils from a. Bottom: A single continuous network of synapsin condensates interconnected with a continuous actin network is highlighted in green. Scale bar, 20 μm.
D. Representative confocal image from the reconstituted synapsin 1:actin assemblies spreading over the surface of a GUVs. Projection of the maximal intensity, 30 minutes post-incubation. Scale bar, 3 μm.
E. Schematic representation of the synapsin:actin:GUV association.
F. Representative confocal images of synapsin:actin assemblies before (top) and after (bottom) the addition of 1,6-hexanediol (10% w/v). Treatment of the synapsin:actin assemblies with 1,6-hexanediol partially disperses synapsins without affecting the pre-formed actin asters. Asterisks indicate synapsin condensates without actin, which do disperse upon addition of 1,6-hexanediol, as expected. Scale bar, 10 μm.
G. Quantification of the synapsin 1 and actin partitioning before and after treatment with 1,6-hexanediol. Data from three independent reconstitutions, >1500 condensates analyzed for each condition. ****p<0.0001, ns not significant; Mann-Whitney nonparametric test.
Polymerized actin network stabilizes synapsin condensates
To characterize the architecture of the polymerized actin assemblies in terms of stability and dependance on the persistent existence of synapsin condensates, we further aimed to disperse polymerized “actin-asters” with 10% 1,6-hexanediol (final concentration), an aliphatic alcohol known to disrupt low-affinity hydrophobic interactions (Ribbeck & Görlich, 2002; Kroschwald et al, 2017). To address this aim, we again reconstituted synapsin condensates with DOPC GUVs and G-actin monomers (spiked with ATTO-647 G-actin, as previously described) and after 30 minutes when “actin-asters” were formed, we added 10 % 1,6-hexanediol (final concentration) to the sample. Upon 1,6-hexanediol treatment, synapsin 1-actin condensates that participated in the aster formation had Syn1 partially dispersed, while actin remained unaffected (Fig. 4F). Another aspect of this phenotype is that synapsin remaining after 1,6-hexanediol treatment is attached to the actin core, that is, stabilized. This was confirmed by delta G analysis which showed significantly lowered partitioning of synapsin to dense phase, but no difference in actin partitioning (Fig. 4G). Interestingly, synapsin 1-actin condensates without apparent actin polymerization (i.e., no asters) were completely disrupted (Supplementary Fig. 9). Thus, where actin is polymerized, synapsin1/actin assemblies remain undisrupted with Syn1 enrichment within them, whereas there is a complete dispersion of synapsin 1-actin condensates in the absence of actin polymerization, indicating the stabilizing effect of actin polymerization.
Synapsin condensates are essential for the actin accumulation in synaptic boutons
Previous studies have suggested that perturbation of synapsin function at the active synapse not only led to the drastic reduction of actin at the periphery of the SV cluster but also a corresponding reduction in the number of SVs in the cluster (Pieribone et al, 1995; Bloom et al, 2003); however, neither of the studies examined the effects of synapsin disruptions on synaptic actin. Conversely, synaptic activity and SV recycling critically rely on the local polymerization of actin (Sankaranarayanan et al, 2003; Ogunmowo et al, 2023). We questioned whether synapsin-driven SV condensation was important for actin accumulation at the synaptic boutons. To achieve this, we used mouse hippocampal neurons as a model to assess the role of synapsin phase separation in the context of synapsin-SV interactions with actin. We stained primary hippocampal neurons from WT and synapsin triple knock-out (TKO) mouse with synaptophysin, a well-known SVs integral membrane protein, and phalloidin-StarRed, which allowed the visualization of remarkable axonal actin rings using two-color stimulated depletion emission (STED) microscope (Xu et al, 2013). Furthermore, STED images showed accumulation of actin within synaptic boutons in WT hippocampal neurons as indicated by a strong actin fluorescence signal within SVs clusters (Fig. 5A,C). In contrast, actin failed to accumulate at the synaptic boutons of synapsin TKO hippocampal neurons, which also exhibited dispersed SVs due to the absence of synapsin (Fig. 5B,D). This phenotype was rescued upon transfection with either the full-length EGFP-synapsin 1 and the EGFP-synapsin 1 IDR (Fig. 5E-H,J), which is sufficient to rescue SV clustering (Hoffmann et al, 2023b). Interestingly, the actin rings along the axons remain stable in the absence of synapsins (i.e., in the neurons from synapsin TKO animals; Fig. 5I), suggesting that synapsin/SV condensates are central for the local organization of the actin network in boutons.
Figure 5: Synaptic vesicle condensates are necessary for concentrating actin at the presynaptic boutons.

A, B. Two-color super-resolution STED images of hippocampal neurons (14 DIV) from (a) wild-type and (b) synapsin triple knockout neurons stained with StarRed-phalloidin and anti-synaptophysin (Star580). Scale bar, 1 μm.
C. Magnified region from (a) showing a cohort of synaptic vesicles (anti-synaptophysin) colocalizing with actin (StarRed-phalloidin).
D. Magnified region from (b) indicating a more dispersed signal of SVs (anti-synaptophysin) lacking the colocalization with actin (StarRed-phalloidin). Scale bar, 0.5 μm.
E, F. Representative images of synapsin triple knockout neurons rescued with (e) full-length EGFP-synapsin 1 or (f) EGFP-synapsin 1 IDR and stained with StarRed-phalloidin and anti-synaptophysin (Star580). Scale bar, 1 μm.
G, H. Magnified regions from (e) and (f), respectively, showing colocalization of SVs with actin. Scale bar, 0.5 μm.
I. Autocorrelation of the actin signal along the axons (StarRed-phalloidin; see dotted/while lines in the images in a and b) indicates that actin rings remain unaltered in the absence of synapsins.
J. Actin enrichment in synaptic vesicle cohorts defined as an intensity signal of actin channel within regions positive for synaptophysin (wild-type and synapsin triple knockout) or both synaptophysin and EGFP (in rescue experiments).
Discussions
Our results show that the presynaptic actin network can be reconstituted with a surprisingly small number of components, indicating that synapsin-SV condensates act as reaction centers for organizing actin at the synaptic bouton. Actin monomers are first recruited into synapsin-driven condensates; upon reaching critical concentration within condensates, actin accumulates at the interfaces and polymerizes, without a need for a nucleation factor (Fig. 1,2). Similarly to the asters formed by centrosomes for microtubule organization (Woodruff et al, 2015), the synapsin:actin phase directs SVs along the actin fibers spanning long-range distances of tens of microns (Fig. 4). The IDR region of synapsin, responsible for its phase separation (Milovanovic et al, 2018), suffices to concentrate actin and trigger its polymerization but does not recapitulate the aster-like morphology and long-range fibers. Our in vitro results faithfully recapitulate the actin morphologies observed in vertebrate synapses (Shupliakov et al, 2002; Bloom et al, 2003; Bourne et al, 2006) (Fig. 3), which demonstrate that actin forms a torus around cohorts of synaptic vesicles with actin fibers radiating from these synapsin:SV:actin assemblies.
Condensation as a mechanism for SV organization relies on the surrounding membrane-bound organelles to provide a template for the actin network assembly. Given the long distance and varying topology of the actin network within synaptic boutons (Bingham et al, 2023; Ogunmowo et al, 2023), our model of synapsin:actin assemblies explains that SV trafficking can occur independently of the concentration gradient of SVs, which is particularly important for differential regulation of SV diffusion between the reserve and readily releasable pools of SVs (Joensuu et al, 2016). Indeed, our superresolution STED imaging in murine hippocampal neurons indicates that, there is an absence of actin enrichment in the vicinity of SVs in the absence of synapsins (Fig. 5).
Current thinking regarding the actin involvement in the SV cycle envisions that actin and actin motors would form a range of fixed, distinct structures within a presynaptic compartment, each of which would facilitate a distinct step of the cycle (Cingolani & Goda, 2008; Rust & Maritzen, 2015; Nelson et al, 2013). We propose an alternative although not mutually exclusive possibility in which synapsin-driven SV condensates may act as molecular beacons for locally sequestering actin and directing its polymerization. Together, the minimal machinery for recapitulating synapsin:SV:actin assemblies described here explains the role of actin in maintaining the clusters of SVs and directing the traffic of vesicles both within and between boutons during activity. Understanding the coupling of neighboring SV condensates along the axons is central to understanding the circuit function and the coordination of behavior.
Methods
Cloning
Plasmid encoding for 6xHis-EGFP-rnSynapsin 1 fusion protein (Syn1-FL) is described previously(Milovanovic et al, 2018). For EGFP-Synapsin 1 Domain C and IDR expression plasmids, first, an EGFP A206K mutation was introduced by mutagenesis PCR (#DML0022 and #DML0023) into the Syn1-FL plasmid. Second, the Syn1-FL sequence was exchanged for human synapsin 1 Domain C (Syn1-Dom.C, a.a. 112-420) or human synapsin 1 IDR (Syn1-IDR, a.a. 416-705) sequences using restriction sites BglII and SacI(Hoffmann et al, 2023b). For details of sequences please see Supplementary Table 1.
Protein expression and purification
6xHis-EGFP-rnSynapsin1 (Syn1-FL), 6xHis-EGFP(A206K)-hsSynapsin1-Domain C (112-420) and 6xHis-EGFP(A206K)-hsSynapsin1-IDR (421-705) (Syn1-IDR) were expressed in Expi293F™ cells (ThermoFisher) for three days following induction. Cell lysis was performed by three cycles of freezing in liquid nitrogen and thawing at 37°C in a buffer containing 25 mM Tris-HCl (pH 7.4), 300 mM NaCl, 25 mM imidazole, 0.5 mM TCEP (buffer A) supplemented with EDTA-free Roche Complete protease inhibitors, 10 μg/mL DNaseI and 1 mM MgCl2. The purification process was conducted at 4°C as previously described(Hoffmann et al, 2023b),(Hoffmann et al, 2021). The lysate was further subjected to centrifugation for 1 hour at 20,000xg followed by two-step purification. The first step involved affinity purification of soluble supernatant on a Ni-NTA column (HisTrap™HP, Cytiva, ÄKTA pure 25M). Washing steps were performed with buffer A containing 40 mM imidazole, and elution was performed with buffer A containing 400 mM imidazole. Elution fractions were concentrated (30K MWCO protein concentrator, Pierce) and subjected to size-exclusion chromatography (Superdex™ 200 Increase 10/300, GE Healthcare, ÄKTA pure 25M) in 25 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.5 mM TCEP. Elution fractions were analyzed by SDS-PAGE. All purified proteins were snap-frozen in liquid nitrogen and stored at −80°C until use.
Synaptic vesicle preparation
All isolations were approved by the Institutional Animal Welfare Committees of the State of Lower Saxony, Germany and Max Planck Institute for Multidisciplinary Sciences (Göttingen, DE). Native synaptic vesicles (SVs) from rat brain were prepared following the previously published procedure(Takamori et al, 2006) (Nagy et al, 1976) (Huttner et al, 1983). All isolation steps were performed at 4°C. In brief, brains were isolated from 20 adult rats and homogenized in ice-cold sucrose buffer (5 mM HEPES-KOH pH7.4, 320 mM sucrose supplemented with 0.2 mM PMSF and 1 mg/mL pepstatin A). The homogenate was subjected to centrifugation (10 min at 900gAv), and the supernatant was further centrifuged for 10 min at 12,000gAv. The pellet was washed once with sucrose buffer and subjected to centrifugation for 15 min at 14,500gAv. Synaptosomes were lysed by hypo-osmotic shock, and the lysate was subjected to centrifugation for 20 min at 20,000gAv. Free SVs were collected from the resulting supernatant, which was further ultracentrifuged for 2 h (230,000 gAv) to obtain a crude SV pellet. Synaptic vesicles were further purified by loading the resuspended pellet (40 mM sucrose) on a continuous sucrose density gradient (50–800 mM sucrose) and further centrifugation for 3 h at 110,880gAv. Separation of SVs from residual larger membrane contaminants was achieved by size-exclusion chromatography on controlled pore glass beads (300 nm diameter), equilibrated in glycine buffer (5 mM HEPES-KOH pH 7.40, 300 mM glycine). SV-containing fractions were combined, and synaptic vesicles were collected by centrifugation for 2 h at 230,000gAv and subsequently aliquoted into single-use fractions and snap-frozen in liquid nitrogen.
In vitro reconstitution experiments
All in vitro reconstitutions had a final concentration of 4 μM EGFP-Syn1-variants (FL, IDR, Dom. C), 3% (w/v) PEG 8000, 3 nM SVs (for reactions setups that required SV assessment) and 4 μM ATTO647 actin. Size-exclusion chromatography buffer (hereafter referred to as ‘reaction buffer’, containing - 25 mM Tris-HCl (pH - 7.4), 150 mM NaCl, 0.5 mM TCEP) served as a base for all in vitro reconstitutions. Low-binding pipette tips were used for handling protein and SVs. ATTO647 labelled G-actin monomers from rabbit skeletal muscle (Hypermol, 8158-02) were used for assessing G actin enrichment and polymerization over time within liquid phases of Syn1-variants. The actin mix was freshly prepared during the day of the experiment following the manufacturer’s protocol (Hypermol, 8097-01). Briefly, 10 μM ATTO647 labelled G-actin monomers were ATP and magnesium-exchanged using mono-mix (Hypermol, 5120-01) and ME buffer (Hypermol, 5111-01). The final concentration of actin and ATP in all reaction mixtures was 4 μM and 0.5 mM respectively.
Reconstitutions of Syn1-variants (Syn1-FL, Synl-IDR and Syn1-Dom. C) with G-actin.
For assessing actin enrichment within and polymerization from synapsin 1 phases, 6 μM EGFP-Syn1-variants (FL, IDR, Dom. C) were co-incubated with 3% (w/v) PEG 8000 on a glass bottom dish (Cellvis D35-20-1.5-N). When the condensates became approx. 2-4 μm in size (approx. after 5 minutes), ATP and magnesium-exchanged ATTO647 labelled G-actin monomers were added into these pre-formed condensates ending up at a final concentration of 4 μM each. Actin enrichment and polymerization were followed for 35 minutes.
Reconstitution of Synapsin 1-synapstic vesicle condensates with G-actin.
The effect of SVs on actin enrichment and polymerization was assessed by co-incubating 6 μM EGFP-Syn1-variants (Syn1-FL and Syn1-IDR) with 3% (w/v) PEG 8000 and 3 nM SVs (labelled with 1.65 μM FM4-64) in a tube and placing the mixture on a glass bottom dish (Cellvis D35-20-1.5-N). ATP and magnesium-exchanged ATTO647 labelled G-actin monomers were added into these pre-formed Syn1/SVs condensates approx. after 5 minutes such that the final concentration of actin and EGFP-Syn1-variants in the final reaction mix was 4 μM each. Enrichment of actin within Syn1/SVs phases was followed for 35 minutes.
Reconstitution of Syn1-FL condensates and giant unilamellar vesicles (GUVs) with G-actin.
For assessing the effect of actin polymerization from synapsin 1 phases in the presence of membranes, synapsin 1 condensates were prepared by co-incubating 10 μM Syn1-FL with 7.5% (w/v) PEG 8000 in the reaction buffer for 15 minutes. Subsequently, pre-formed synapsin 1 condensates were added from the top onto GUVs pre-loaded on a 15-well glass slide (ibidi, 81506). Condensates were allowed to settle on the surface of GUVs for approx. 20 minutes. Eventually, ATP and magnesium-exchanged actin (here, unlabeled actin was spiked with ATTO-647 labelled actin; Hypermol, 8101-01 and 8158-02) were added from the top onto the condensate-GUV mix. Thus, the final reaction mix had 3% (w/v) PEG 8000, 4 μM Syn1-FL, 4 μM actin, 74.5 mM NaCl, 0.5 mM ATP, 150 mM sorbitol and 1.5 mM lipids in the form of GUVs. Actin polymerization from synapsin 1 condensates in the presence of GUV was followed after 30 minutes. Reactions involving 1,6-hexanediol were conducted on a glass-bottom dish (Cellvis D35-20-1.5-N). Briefly, after actin polymerization (~ 30 minutes), 1,6 hexanediol (10% final conc.) was added to the reaction mixture followed by a gentle mix and 5 minutes of incubation. Images were acquired both before and after the addition of 1,6 hexanediol at the same locations.
Validation of actin polymerization.
We performed 4 control experiments.
First, G-actin polymerization was validated in the presence of the final purification buffer (i.e., negative control) and in the commercial polymerization buffer, FluMaXx (Hypermol, 5161-01; positive control). In both of the conditions, ATP and magnesium-exchanged ATTO647 labelled G-actin monomers (final conc. − 4 μM) were allowed to polymerize on a glass-bottom dish in respective buffers. Both the reactions were followed for 45 minutes.
Second, we tested actin polymerization in the presence of 6x-His-EGFP protein (4 μM) for 45 minutes to quantify whether the tag affects actin polymerization. Briefly, we prepared a pre-mix of 6x-His-EGFP protein (final conc. - 4 μM) with 3% (w/v) PEG 8,000. ATP and magnesium-exchanged ATTO647 labelled G-actin monomers were added after 5 min to the reaction mixture, and the reaction was followed for 45 minutes.
Third, we tested actin polymerization in the presence of SVs in the reaction buffer (25 mM Tris-HCl (pH - 7.4), 150 mM NaCl, 0.5 mM TCEP) for 45 minutes to validate if any SV-bound proteins could direct actin polymerization. Here, we prepared a pre-mix of 3 nM SVs (labelled with 1.65 μM FM4-64) and 3% (w/v) PEG 8000. Subsequently, ATP and magnesium-exchanged ATTO647 labelled G-actin monomers were added after 5 minutes), and the reaction was followed for 45 minutes.
Fourth, actin polymerization was also examined in the presence of G3BP2 condensates. Here, G3BP2 condensates were formed by mixing G3BP2 (a final concentration of 4 μM) with 3% (w/v) PEG 8000. ATP and magnesium-exchanged ATTO647 labelled G-actin monomers were added to the pre-formed condensates, and the reaction was followed for 45 minutes.
Pyrene actin assay
To deduce the effect of synapsin 1 condensation on the kinetics of actin polymerization, we set up reactions of pyrene-labelled actin (Cytoskeleton, AKL99) with EGFP-Syn-1 variants (Syn1-FL and Syn1-IDR) in absence of crowding agent. Briefly, reaction mixtures containing 10 μM pyrene-labelled actin, 10 μM synapsin 1/variants, 1X actin polymerization buffer (10 mM Tris/HCl, pH = 7.5, 2 mM MgCl2, 50 mM KCl and 0.5 mM ATP) were set up in a 384-well microtiter plate (Greiner Bio-One, 781906). For all the reaction conditions, 1x Monomix buffer was used as a base for adjusting the reaction volume to 30 μl. Several controls were also undertaken for the pyrene actin assay. Pyrene fluorescence was measured at emission wavelength 410 nm (excitation wavelength 365 nm) every 30 seconds for at least 15 minutes at 37°C using Synergy H1 Hybrid Multi-Mode Microplate Reader (BioTek Instruments). Measurements obtained from pyrene-actin assay for each condition were baseline corrected using the average fluorescence value of pyrene-actin in 1x Monomix buffer. Further, data points were plotted using GraphPad Prism software.
Turbidity measurements
To examine the effect of synapsin 1 condensation on actin polymerization, we set up reactions of actin (Hypermol, 8101-01) with EGFP-Syn-1-FL. The changes in turbidity reflect the rate and extent of condensate formation(Hoffmann et al, 2021). Here, we used the further changes in optical density as a proxy of actin polymerization. Reaction setups for polymerizing conditions comprised of 4 μM EGFP-Syn-1-FL, 4 μM ATP and magnesium-exchanged-actin, 3% (w/v) PEG 8000, 0.5 mM ATP, 140 mM NaCl and 0.01% sodium azide. Following control reactions were also undertaken (i) EGFP-Syn-1-FL in 1X Monomix and 3% (w/v) PEG 8000 (ii) Actin and 3% (w/v) PEG 8000. SEC buffer was used as a base for adjusting the reaction volume to 30 μl for all reactions. After mixing, all reactions were transferred into a 384-well microtiter plate (Greiner Bio-One, 781906) and turbidity was measured every 10 minutes for each condition at 405 nm and 37°C in shaking mode for at least 48 h using a Synergy H1 Hybrid Multi-Mode Microplate Reader (BioTek Instruments). Measurements from the microplate reader were baseline corrected (baseline was determined as the turbidity of SEC buffer) and plots were prepared using GraphPad Prism software. After measurements, all conditions were imaged using a Nikon Eclipse TS2-FL microscope, capturing widefield signal and GFP-epifluorescence.
Preparation of Giant Unilamellar Vesicles (GUVs)
Lipids used for GUV preparation were purchased from Avanti Polar Lipids. Neutrally-charged 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) GUVs were prepared by electro formation in Vesicle Prep Pro device (Nanion Technologies GmbH) on the conductive surface of indium tin oxide (ITO)-coated glass slides, as described previously(Hoffmann et al, 2023a). The total lipid concentration was 5 mM. For labelling, lipids were spiked with 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE)-Rhodamin Red (10 μM final concentration). Lipids were first mixed with chloroform for 25 ul total volume and applied in a thin even film of circular shape on a conductive surface of the ITO-coated glass slides. This film was left at room temperature for drying for 20 minutes. Subsequently, a 16 mm O-ring (Nanion Technologies GmbH) was placed around the dried film and the entire assembly was placed inside the Vesicle Prep Pro device. Further, 250 μl of an aqueous solution containing 500 mM D-Sorbitol (MP Biomedicals) was pipetted inside the O-ring. This O-ring was covered with a second ITO-coated glass slide in such a way that the O-ring and solution inside it were sandwiched between conductive surfaces. A program was run on the Vesicle Prep Pro device (amplitude = 3 V; frequency = 5 Hz; temperature = 36°C; time = 2 hours; ramps = 5 min). Afterwards, the upper glass slide was removed from the assembly and the solution inside the O-ring was transferred to a 1,5 ml reaction tube. The vesicle solutions were stored at 4°C and subsequently used for experiments within one week after preparation.
Fluorescence Correlation Spectroscopy
Fluorescence Correlation Spectroscopy (FCS) was conducted using a confocal microscope(Perego et al, 2020). The setup was based on an inverted microscope (Olympus IX73, Olympus Europa SE & CO. KG, Hamburg, Germany). And equipped with two pulsed diode lasers (Cobolt Samba-532 100 mW and Cobolt Calypso-491 25 mW, Cobolt AB, Solna, Sweden), a 60× water immersion objective (UPlanApo, NA = 1.2, Olympus), and two avalanche photodiodes (tau-SPAD, Picoquant GmbH, Berlin, Germany). Additionally, the microscope was equipped for epi-fluorescence microscopy. The SPADs were connected to a digital correlator card (ALV-7004 USB, ALV-Laser Vertriebsgesellschaft mbH, Langen, Germany), and the resulting correlation measurements were analyzed in a post-processing phase using a custom Python script (Python Software Foundation, https://www.python.org).
Synaptic vesicles were patterned as described previously(Perego et al, 2020). Briefly, the vesicles were immobilized on glass coverslips (#1 thickness, Thermo Scientific Technologies Inc., Wilmington, USA) by adding neutravidin (0.05 g/L, Thermo Fisher Scientific, Waltham, MA, USA) and biotinylated anti-synaptotagmin monoclonal antibodies (0.01 g/L, Synaptic Systems GmbH, Gottingen, Germany). The vesicles were labeled with anti-vGLUT1-STAR635P single-domain antibodies (0.05 g/L, Nanotag, Gottingen, Germany) for visualization. For the FCS measurement, approximately 250 μL of either only Alexa Fluo 488-labeled actin (1 mg/L, actin from rabbit muscle, Invitrogen, Carlsbad, CA, USA) or in combination with synapsin (0.1 mg/L), were applied to the patterned vesicle sample. Fluorescence signals were collected using the confocal modality for 15 minutes, divided into 1-minute portions, and averaged to generate the final fluorescence correlation curve. Data portions exhibiting the passage of aggregates or artifacts were excluded from the analysis. The observation volume was calibrated with Atto-488 (AttoTech GmbH, Siegen, Germany) before the fluorescence correlation experiment, resulting in an observation volume of (280 ± 10) nm. All correlation curves were fitted using a Levenberg-Marquardt nonlinear least-square routine.
Microscopy
Spinning disk confocal microscopy.
Imaging of the in vitro reconstitution experiments was performed on a Nikon spinning disk confocal CSU-X (SDC CSU-X) microscope equipped with 2 EMCCD cameras (iXon3 DU-888 Ultra), Andor Revolution SD System (CSU-X)) and a PL APO163 60x/1.4 NA, oil immersion objective. Excitation wavelengths were: 488 nm for EGFP-fusion proteins (Synapsin 1 variants, G3BP2 and EGFP-control), 561 nm for GUVs and SVs (GUVs: (DOPE)-Rhodamine Red, SV: FM4-64) and 647 nm for ATTO647-labeled actin.
Total internal reflection fluorescence (TIRF) microscopy.
TIRF microscopy was performed on a Nikon Ti-E TIRF/STORM setup equipped with a sCMOS camera (Photometrics Prime 95B (95% QE, 1200x1200 pixels, 11 μm pixel size, 41 fps full frame, 82 fps@12-bit)) and a Plan Apo VC 100x/1.49 NA DIC H N2 TIRF oil immersion objective. Excitation wavelength was 647 nm for ATTO647-labeled actin.
Confocal laser scanning microscopy and Airyscan.
Images of fixed lamprey samples were acquired using a Zeiss LSM780 confocal microscope (Plan-Apochromat 40x/1.4 objective). A Zeiss Celldiscoverer 7 with LSM900 Airyscan2 (Plan-Apochromat 50x/1.2 water immersion objective) was used to acquire images with enhanced resolution of actin asters.
Two-color stimulated emission depletion (STED).
Two-color STED microscopy was performed on an Abberior STED system with a 100×/1.4 NA oil UPlanSApo Olympus objective. A single focal plane in the center of neuronal processes was imaged. Pixel size was set to 20 nm and pixel dwell time to 10 μs.
Primary hippocampal neurons
Preparation of hippocampal neurons.
All animal experiments were approved by the Institutional Animal Welfare Committees of the State of Berlin, Germany and Charite University Clinic (Berlin, DE). Hippocampal neurons were prepared following an established procedure(Hoffmann et al, 2023b). Briefly, brains of P0/1 mice of WT (C57BL/6) and synapsin triple-knockout (SYN-TKO: B6; 129-Syn2tm1Pggd Syn3tm1Pggd Syn1tm1Pggd/Mmjax(Gitler et al, 2004) background were dissected followed by hippocampi extraction. The hippocampi were washed with cold supplemented Hank’s balanced salt solution [HBSS-supp, Gibco, 14170112; supplemented with 10 mM HEPES (Gibco, 15630049), 50 units/ml penicillin/streptomycin (Gibco, 15140122), 1 mM sodium pyruvate (Gibco, 11360039) and 6 mM MgCl2 (Roth, KK36.2)]. Subsequently, hippocampi were digested with Papaine (Sigma, P3125) in HBSS-supp for 20 min at 37 °C. The enzymatic reaction was inactivated by washing the cells with plating medium [Neurobasal Medium A (NB-A Gibco; 10888022), supplemented with 5% FBS (Sigma, 12106C), 1% B27 (Gibco, 17504044), 1% Glutamax (Gibco, 35050038), and 50 units/ml penicillin/streptomycin]. The digested tissue was subjected to gentle mechanical dissociation using a P1000 pipette and cells were seeded in a plating medium on PLL-coated glass coverslips (0.1 mg/mL poly-L-lysine, Sigma, P6282). Hippocampal neuronal cultures were maintained at 37°C temperature and 5% CO2 in growth medium (NB-A supplemented with 1% B27, 1% Glutamax, and 50 units/ml penicillin/streptomycin).
Transfection of hippocampal neurons.
Calcium phosphate transfection protocol for neurons was adapted from(Jiang & Chen, 2006). Briefly, a coverslip containing DIV4-6 hippocampal neurons was transferred to a fresh petri dish containing 1 ml of growth medium supplemented with 4 mM kynurenic acid (Sigma, K3375, 20 mM stock solution in NB-A, freshly prepared). DNA mix was composed of 2 μg of the plasmid of interest in 1x TE (10 mM Tris-HCl (pH 7.3), 1 mM EDTA) supplemented with CaCl2 to a final concentration of 250 mM (stock: 10 mM HEPES (pH7.2), 2.5 M CaCl2). To prepare the transfection mix, one-eighth of the DNA mix was added stepwise to 2xHEBS (42 mM HEPES (pH 7.2), 274 mM NaCl, 10 mM KCl, 1.4 mM Na2HPO4, 10 mM glucose) with slow vortexing (~ 600 rpm) for 2-3 seconds between each addition and incubated for 20 min at room temperature. The transfection mix was added to neurons and dishes were incubated at 37 °C, 5% CO2 for 1.5 h. Subsequently, the medium was replaced by 1 mL of NB-A with 4 mM kynurenic acid supplemented with 2.5 mM HCl, followed by an incubation at 37°C, 5% CO2 for 15 min. After the wash step, the coverslip was placed back in the primary feeding culture dish with own conditioned growth medium.
Sample preparation for two-color stimulated emission depletion (STED).
Samples of mouse hippocampal neurons (DIV14) were fixed, immunostained, and imaged as previously described (Rentsch J. et al., 2024). Cells from at least three independent preparations per condition (WT, SYN-TKO, FL-rescue, IDR-rescue) were fixed in 4% PFA (v/v)/PBS for 15 minutes. Samples were quenched in 50 mM NH4Cl/PBS for 30 minutes followed by three brief washes with PBS. A permeabilization/blocking step was performed with 1% BSA (w/v)/0.05 % Saponin (w/v)/4% Horse serum (v/v)/PBS for 45 min. Before antibody staining, the samples were incubated in a drop of Image-iT (ThermoFisher, #R37602) for 30 min. Samples were stained with mouse anti-synaptophysin (Synaptic Systems 101 011, 1:50) overnight at 4°C in 1% BSA (w/v)/0.05 % Saponin (w/v)/4% Horse serum (v/v)/PBS. Samples were washed three times with 1% BSA (w/v)/0.05 % Saponin (w/v)/PBS for 5 min with agitation. Subsequently, samples were incubated with goat anti-mouse-STAR580 (Abberior, ST580-1001-500UG, 1:50) and phalloidin-STARRED (Abberior, STRED-0100-20UG, 1g/L, 1:200) for 1 h in 1% BSA (w/v)/0.05 % Saponin (w/v)/4% Horse serum (v/v)/PBS. After secondary antibody incubation, the samples were washed three times with PBS and analyzed by two-color STED microscopy.
Lamprey experiments
All animal procedures were approved by the Institutional Animal Care and Use Committee at the Marine Biological Laboratory in Woods Hole, MA (USA). After anesthesia, 2-3 cm sections of the lamprey spinal cord were dissected and pinned in a Sylgard-lined dish and submerged in lamprey Ringer, as described in(Walsh et al, 2018). In order to label the actin networks at synapses, giant RS axons were microinjected for 5 minutes with Alexa Fluor 488-conjugated phalloidin (ThermoFisher) diluted in lamprey internal solution (10 mM HEPES pH 7.4, 180 mM KCl). Some axons were also co-injected with FM4-64 (ThermoFisher), which labeled the SV clusters, and then imaged live 10-15 minutes later using a Zeiss LSM780 laser scanning confocal microscope (Plan-Apochromat 40x/1.4 objective, digital zoom 5.2). Other spinal cord preparations were fixed overnight in 4% paraformaldehyde in 0.1M PBS, pH 7.4, and subsequently processed for whole mount immunofluorescence. The primary antibody used for labeling SV clusters was a mouse monoclonal made against synaptic vesicle glycoprotein 2 (SV2; Developmental Hybridoma Studies Bank; 1:100), which was previously characterized in lamprey nervous tissues(Busch & Morgan, 2012)’(Banks et al, 2020)’(Wallace et al, 2024). Synapsin condensates were co-labeled using a rabbit polyclonal antibody made against the IDR of synapsin 1a/b (Synaptic Systems 106 008; 1:100), which was characterized in Supplemental figure 8. Secondary antibodies used were Alexa Fluor plus 647 goat anti-mouse IgG (H+L) and Alexa Fluor plus 594 goat anti-rabbit IgG (H+L), respectively (ThermoFisher; 1:300).
Image processing, data analysis and statistics
All image processing steps such as cropping, brightness adjustments, merging channels, and scale bars were carried out using ImageJ software. Intensities for preparing representative line profiles were noted using ImageJ software. The data obtained post-image analysis was further analyzed using the tools available in the GraphPad Prism software.
Actin enrichment with syn1-FL phases.
For determining actin enrichment within Syn1-FL phases, at first x-y shifts during the acquisition were corrected for individual condensates using the StackRegJ plugin(Slaughter et al, 2013). Further, ROI was created on condensates and their change in intensity over time was computed using the create spectrum jru v1 plugin(Slaughter et al, 2013).
Actin network reconstructions.
For creating actin network reconstructions, images from the actin channel were preprocessed by the difference of Gaussians (Sigmas used were 2.0 and 1.0) to improve fibrils recognition, binarized and skeletonized using ImageJ. Skeletonized images were analyzed and 3D-reconstructed using the SNT plugin(Arshadi et al, 2021).
Sholl analysis.
The SNT plugin was also used for performing Sholl analysis on polymerized actin networks from synapsin 1 phases. Images of actin asters were semi-automatically traced using the plugin and traces were used for Sholl analysis. A series of concentrical spheres (in 0.5 μm radius increments) with centres in synapsin condensates were created and intersections of actin fibrils with these spheres were counted.
Partitioning analysis.
The Delta G analysis was conducted by adapting a previously published pipeline (Baggett DW et al., 2022). Briefly, condensates were segmented using the Otsu algorithm after Gaussian convolution and further mean intensity was determined within and outside the condensates. The partition coefficient (Kp) was calculated as the ratio of mean intensity inside condensates to the mean intensity outside of condensates (see equation 1). Kp was further used for computing Delta G (see equation 2).
| (Eq. 1) |
| (Eq. 2) |
Here, R is the gas constant, its value is 1.9872 and is expressed in units of kcal/mol and T is temperature expressed in Kelvin units(Baggett et al, 2022).
Quantification of two-color STED images.
Analysis was performed using a custom FIJI macro that measures actin intensity inside synaptophysin positive spots. The macro converts the synaptophysin (SV marker) channel into a mask using the built-in FIJI find_mask() function in the WT and SYN-TKO conditions. In the rescue conditions (rescue-FL, rescue-IDR) a shared mask of the GFP (synapsin / IDR) and the synaptophysin channel was created. Thereby, it is ensured that further analysis in the rescue conditions was only performed in synapsin-expressing cells. The mask is then converted into ROIs using the FIJI built-in “Analyze particles” tool. The total intensity of the actin channel is then measured in each ROI (WT n=1592, SYN-TKO n=7095, FL-rescue n=209, IDR-rescue n=460). Normality was tested in OriginPro using Shapiro-Wilk-Test and rejected for the rescue-FL condition (p=7.6E-6 (****)). Mean intensity of all ROIs per condition was compared using Mann-Whitney Test.
Supplementary Material
Acknowledgments.
We thank Advanced Medical Bioimaging Core Facility at Charité for the support. We thank Advanced Medical Bioimaging Core Facility at Charité University Clinic, Berlin, for the support; to Dr. Reinhard Jahn and his entire team (Max Planck Institute for Multidisciplinary Sciences, Göttingen, Germany) for the generous support to use the resources needed for the preparation of native synaptic vesicles. The work is supported by the start-up funds from DZNE, the grants from the German Research Foundation (MI 2104 and SFB1286/B10) and the ERC Grant MemLessInterface (101078172) to DM; the grants from the National Institutes of Health NIA 2RF1 NS078165-12 to JRM and NINDS K99 NS126575-01 to CRV. CH is supported by a fellowship of the Innovative Minds Program of the German Dementia Association.
Footnotes
Competing Interests. The authors declare no competing interests.
Data Availability.
All data generated or analyzed for this study are available within the paper and its associated supplementary information files. All other data presented are available upon reasonable request from the corresponding author.
References
- Alfken J, Neuhaus C, Major A, Taskina A, Hoffmann C, Ganzella M, Petrovic A, Zwicker D, Fernández-Busnadiego R, Jahn R, et al. (2024) Vesicle condensation induced by synapsin: condensate size, geometry, and vesicle shape deformations. Eur Phys J E 47: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alshareedah I, Moosa MM, Pham M, Potoyan DA & Banerjee PR (2021) Programmable viscoelasticity in protein-RNA condensates with disordered sticker-spacer polypeptides. Nat Commun 12: 6620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arshadi C, Gánther U, Eddison M, Harrington KIS & Ferreira TA (2021) SNT: a unifying toolbox for quantification of neuronal anatomy. Nat Methods 18: 374–377 [DOI] [PubMed] [Google Scholar]
- Baggett DW, Medyukhina A, Tripathi S, Shirnekhi HK, Wu H, Pounds SB, Khairy K & Kriwacki R (2022) An Image Analysis Pipeline for Quantifying the Features of Fluorescently-Labeled Biomolecular Condensates in Cells. Front Bioinform 2: 897238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bähler M, Benfenati F, Valtorta F, Czernik AJ & Greengard P (1989) Characterization of synapsin I fragments produced by cysteine-specific cleavage: a study of their interactions with F-actin. J Cell Biol 108: 1841–1849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bähler M & Greengard P (1987) Synapsin I bundles F-actin in a phosphorylation-dependent manner. Nature 326: 704–707 [DOI] [PubMed] [Google Scholar]
- Banani SF, Lee HO, Hyman AA & Rosen MK (2017) Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol 18: 285 298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks SML, Medeiros AT, McQuillan M, Busch DJ, Ibarraran-Viniegra AS, Sousa R, Lafer EM & Morgan JR (2020) Hsc70 Ameliorates the Vesicle Recycling Defects Caused by Excess α-Synuclein at Synapses. eNeuro 7: ENEURO.0448-19.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfenati F, Valtorta F, Bähler M & Greengard P (1989) Synapsin I, a neuron-specific phosphoprotein interacting with small synaptic vesicles and F-actin. Cell Biol Intern Rep 13: 1007 1021 [DOI] [PubMed] [Google Scholar]
- Benfenati F, Valtorta F, Chieregatti E & Greengard P (1992) Interaction of free and synaptic vesicle-bound synapsin I with F-actin. Neuron 8: 377–386 [DOI] [PubMed] [Google Scholar]
- Bingham D, Jakobs CE, Wernert F, Boroni-Rueda F, Jullien N, Schentarra E-M, Friedl K, Moura JDC, Bommel DM van, Caillol G, et al. (2023) Presynapses contain distinct actin nanostructures. J Cell Biol 222: e202208110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom O, Evergren E, Tomilin N, Kjaerulff O, Löw P, Brodin L, Pieribone VA, Greengard P & Shupliakov O (2003) Colocalization of synapsin and actin during synaptic vesicle recycling. J Cell Biol 161: 737–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne J, Morgan JR & Pieribone VA (2006) Actin polymerization regulates clathrin coat maturation during early stages of synaptic vesicle recycling at lamprey synapses. J Comp Neurol 497: 600–609 [DOI] [PubMed] [Google Scholar]
- Busch DJ & Morgan JR (2012) Synuclein accumulation is associated with cell-specific neuronal death after spinal cord injury. J Comp Neurol 520: 1751–1771 [DOI] [PubMed] [Google Scholar]
- Camilli PD, Benfenati F, Valtorta F & Greengard P (1990) The Synapsins. Annu Rev Cell Dev Biol 6: 433. [DOI] [PubMed] [Google Scholar]
- Case LB, Zhang X, Ditlev JA & Rosen MK (2019) Stoichiometry controls activity of phase-separated clusters of actin signaling proteins. Science 363: 1093–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani LA & Goda Y (2008) Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci 9: 344–356 [DOI] [PubMed] [Google Scholar]
- Gitler D, Takagishi Y, Feng J, Ren Y, Rodriguiz RM, Wetsel WC, Greengard P & Augustine GJ (2004) Different presynaptic roles of synapsins at excitatory and inhibitory synapses. J Neurosci 24: 11368 11380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham K, Chandrasekaran A, Wang L, Ladak A, Lafer EM, Rangamani P & Stachowiak JC (2023) Liquid-like VASP condensates drive actin polymerization and dynamic bundling. Nat Phys 19: 574–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Sobue K, Kanda K, Harada A & Yorifuji H (1989) The cytoskeletal architecture of the presynaptic terminal and molecular structure of synapsin 1. J Cell Biol 108: 111–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann C, Murastov G, Tromm JV, Moog J-B, Aslam MA, Matkovic A & Milovanovic D (2023a) Electric Potential at the Interface of Membraneless Organelles Gauged by Graphene. Nano Lett [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann C, Rentsch J, Tsunoyama TA, Chhabra A, Perez GA, Chowdhury R, Trnka F, Korobeinikov AA, Shaib AH, Ganzella M, et al. (2023b) Synapsin condensation controls synaptic vesicle sequestering and dynamics. Nat Commun 14: 6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann C, Sansevrino R, Morabito G, Logan C, Vabulas RM, Ulusoy A, Ganzella M & Milovanovic D (2021) Synapsin Condensates Recruit alpha-Synuclein. J Mol Biol: 166961. [DOI] [PubMed] [Google Scholar]
- Hosaka M & Südhof TC (1999) Homo- and heterodimerization of synapsins. J Biol Chem 274: 16747 16753 [DOI] [PubMed] [Google Scholar]
- Huttner WB, Schiebler W, Greengard P & Camilli PD (1983) Synapsin I (protein I), a nerve terminal-specific phosphoprotein. III. Its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. J Cell Biol 96: 1374–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M & Chen G (2006) High Ca2+-phosphate transfection efficiency in low-density neuronal cultures. Nat Protoc 1: 695–700 [DOI] [PubMed] [Google Scholar]
- Jin G, Zhang Z, Wan J, Wu X, Liu X & Zhang W (2022) G3BP2: Structure and function. Pharmacol Res 186: 106548. [DOI] [PubMed] [Google Scholar]
- Joensuu M, Padmanabhan P, Durisic N, Bademosi ATD, Cooper-Williams E, Morrow IC, Harper CB, Jung W, Parton RG, Goodhill GJ, et al. (2016) Subdiffractional tracking of internalized molecules reveals heterogeneous motion states of synaptic vesicles. J Cell Biol 215: 277 292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroschwald S, Maharana S & Simon A (2017) Hexanediol: a chemical probe to investigate the material properties of membrane-less compartments. Matters [Google Scholar]
- Milovanovic D, Wu Y, Bian X & Camilli PD (2018) A liquid phase of synapsin and lipid vesicles. Science 361: 604 607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy A, Baker RR, Morris SJ & Whittaker VP (1976) The preparation and characterization of synaptic vesicles of high purity. Brain Res 109: 285–309 [DOI] [PubMed] [Google Scholar]
- Nelson JC, Stavoe AKH & Colón-Ramos DA (2013) The actin cytoskeleton in presynaptic assembly. Cell Adhes Migr 7: 379–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogunmowo TH, Jing H, Raychaudhuri S, Kusick GF, Imoto Y, Li S, Itoh K, Ma Y, Jafri H, Dalva MB, et al. (2023) Membrane compression by synaptic vesicle exocytosis triggers ultrafast endocytosis. Nat Commun 14: 2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechstein A, Tomilin N, Fredrich K, Vorontsova O, Sopova E, Evergren E, Haucke V, Brodin L & Shupliakov O (2020) Vesicle Clustering in a Living Synapse Depends on a Synapsin Region that Mediates Phase Separation. Cell Rep 30: 2594–2602.e3 [DOI] [PubMed] [Google Scholar]
- Perego E, Reshetniak S, Lorenz C, Hoffmann C, Milovanović D, Rizzoli SO & Köster S (2020) A minimalist model to measure interactions between proteins and synaptic vesicles. Sci Rep 10: 21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrucci TC, Mooseker MS & Morrow JS (1988) A domain of synapsin I involved with actin bundling shares immunologic cross-reactivity with villin. J Cell Biochem 36: 25–35 [DOI] [PubMed] [Google Scholar]
- Petrucci TC & Morrow JS (1987) Synapsin I: an actin-bundling protein under phosphorylation control. J Cell Biol 105: 1355–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieribone VA, Shupliakov O, Brodin L, Hilfiker-Rothenfluh S, Czernik AJ & Greengard P (1995) Distinct pools of synaptic vesicles in neurotransmitter release. Nature 375: 493 497 [DOI] [PubMed] [Google Scholar]
- Ribbeck K & Görlich D (2002) The permeability barrier of nuclear pore complexes appears to operate via hydrophobic exclusion. EMBO J 21: 2664–2671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzoli SO & Betz WJ (2004) The structural organization of the readily releasable pool of synaptic vesicles. Science 303: 2037 2039 [DOI] [PubMed] [Google Scholar]
- Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, Hammer RE, Malenka RC & Südhof TC (1995) Essential functions of synapsins I and II in synaptic vesicle regulation. Nature 375: 488 493 [DOI] [PubMed] [Google Scholar]
- Rust MB & Maritzen T (2015) Relevance of presynaptic actin dynamics for synapse function and mouse behavior. Exp Cell Res 335: 165–171 [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, Atluri PP & Ryan TA (2003) Actin has a molecular scaffolding, not propulsive, role in presynaptic function. Nat Neurosci 6: 127–135 [DOI] [PubMed] [Google Scholar]
- Sansevrino R, Hoffmann C & Milovanovic D (2023) Condensate biology of synaptic vesicle clusters. Trends Neurosci [DOI] [PubMed] [Google Scholar]
- Shupliakov O, Bloom O, Gustafsson JS, Kjaerulff O, Low P, Tomilin N, Pieribone VA, Greengard P & Brodin L (2002) Impaired recycling of synaptic vesicles after acute perturbation of the presynaptic actin cytoskeleton. Proc Natl Acad Sci USA 99: 14476–14481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaughter BD, Unruh JR, Das A, Smith SE, Rubinstein B & Li R (2013) Non-uniform membrane diffusion enables steady-state cell polarization via vesicular trafficking. Nat Commun 4: 1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soykan T, Kaempf N, Sakaba T, Vollweiter D, Goerdeler F, Puchkov D, Kononenko NL & Haucke V (2017) Synaptic Vesicle Endocytosis Occurs on Multiple Timescales and Is Mediated by Formin-Dependent Actin Assembly. Neuron 93: 854 866.e4 [DOI] [PubMed] [Google Scholar]
- Südhof TC, Czernik AJ, Kao HT, Takei K, Johnston PA, Horiuchi A, Kanazir SD, Wagner MA, Perin MS & Camilli PD (1989) Synapsins: mosaics of shared and individual domains in a family of synaptic vesicle phosphoproteins. Science 245: 1474 1480 [DOI] [PubMed] [Google Scholar]
- Takamori S, Holt M, Stenius K, Lemke EA, Grønborg M, Riedel D, Urlaub H, Schenck S, Brügger B, Ringler P, et al. (2006) Molecular anatomy of a trafficking organelle. Cell 127: 831 846 [DOI] [PubMed] [Google Scholar]
- Valtorta F, Greengard P, Fesce R, Chieregatti E & Benfenati F (1992) Effects of the neuronal phosphoprotein synapsin I on actin polymerization. I. Evidence for a phosphorylation-dependent nucleating effect. J Biol Chem 267: 11281–11288 [PubMed] [Google Scholar]
- Wallace JN, Crockford ZC, Román-Vendrell C, Brady EB, Hoffmann C, Vargas KJ, Potcoava M, Wegman ME, Alford ST, Milovanovic D, et al. (2024) Excess phosphoserine-129 α-synuclein induces synaptic vesicle trafficking and declustering defects at a vertebrate synapse. Mol Biol Cell 35: ar10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh RB, Bloom OE & Morgan JR (2018) Acute Manipulations of Clathrin-Mediated Endocytosis at Presynaptic Nerve Terminals. Methods Mol Biol 1847: 65–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm BG, Mandad S, Truckenbrodt S, Kröhnert K, Schäfer C, Rammner B, Koo SJ, Claßen GA, Krauss M, Haucke V, et al. (2014) Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science 344: 1023 1028 [DOI] [PubMed] [Google Scholar]
- Woodruff JB, Wueseke O, Viscardi V, Mahamid J, Ochoa SD, Bunkenborg J, Widlund PO, Pozniakovsky A, Zanin E, Bahmanyar S, et al. (2015) Centrosomes. Regulated assembly of a supramolecular centrosome scaffold in vitro. Science 348: 808 812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Whiteus C, Xu CS, Hayworth KJ, Weinberg RJ, Hess HF & Camilli PD (2017) Contacts between the endoplasmic reticulum and other membranes in neurons. Proc Natl Acad Sci USA 114: E4859 E4867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Zhong G & Zhuang X (2013) Actin, Spectrin, and Associated Proteins Form a Periodic Cytoskeletal Structure in Axons. Science 339: 452–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P, Mathieu C, Kolaitis R-M, Zhang P, Messing J, Yurtsever U, Yang Z, Wu J, Li Y, Pan Q, et al. (2020) G3BP1 Is a Tunable Switch that Triggers Phase Separation to Assemble Stress Granules. Cell 181: 325–345.e28 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed for this study are available within the paper and its associated supplementary information files. All other data presented are available upon reasonable request from the corresponding author.