

Abstract
The coronavirus mouse hepatitis virus (MHV) translates its replicase gene (gene 1) into two co-amino-terminal polyproteins, polyprotein 1a and polyprotein 1ab. The gene 1 polyproteins are processed by viral proteinases to yield at least 15 mature products, including a putative RNA helicase from polyprotein 1ab that is presumed to be involved in viral RNA synthesis. Antibodies directed against polypeptides encoded by open reading frame 1b were used to characterize the expression and processing of the MHV helicase and to define the relationship of helicase to the viral nucleocapsid protein (N) and to sites of viral RNA synthesis in MHV-infected cells. The antihelicase antibodies detected a 67-kDa protein in MHV-infected cells that was translated and processed throughout the virus life cycle. Processing of the 67-kDa helicase from polyprotein 1ab was abolished by E64d, a known inhibitor of the MHV 3C-like proteinase. When infected cells were probed for helicase by immunofluorescence laser confocal microscopy, the protein was detected in patterns that varied from punctate perinuclear complexes to large structures that occupied much of the cell cytoplasm. Dual-labeling studies of infected cells for helicase and bromo-UTP-labeled RNA demonstrated that the vast majority of helicase-containing complexes were active in viral RNA synthesis. Dual-labeling studies for helicase and the MHV N protein showed that the two proteins almost completely colocalized, indicating that N was associated with the helicase-containing complexes. This study demonstrates that the putative RNA helicase is closely associated with MHV RNA synthesis and suggests that complexes containing helicase, N, and new viral RNA are the viral replication complexes.
The coronavirus mouse hepatitis virus (MHV) is a member of the recently established order Nidovirales, which also includes the arteriviruses and toroviruses. The member families of this order contain genomes of positive-strand RNA and express their replicase genes as two co-amino-terminal polyproteins. The unique organization and replication strategies of the members of the Nidovirales suggest that they may encode proteins with novel functions in cytoplasmic RNA transcription and replication (10, 25, 45). Much has been learned of the organization, gene expression, and transcription of the coronaviruses and arteriviruses, yet there are many questions that remain to be answered concerning the processing and localization of the viral proteins responsible for replication complex formation and viral RNA transcription and replication.
The genome of mouse hepatitis virus strain A59 (MHV-A59) is a 32-kb single-stranded, positive-sense RNA molecule. Replication and transcription activities of MHV are thought to be mediated by proteins translated from gene 1 of the input genome RNA. Gene 1 comprises the 5′-most two-thirds (22 kb) of the genome and is composed of two open reading frames, ORF 1a and ORF 1b, that overlap but are in different reading frames (4, 5, 27, 35). Translation of ORF 1b occurs following a ribosomal frameshifting event at the 3′ end of ORF 1a (6), and thus translation of gene 1 results in two co-amino-terminal polyproteins of 495 kDa (polyprotein 1a) and 803 kDa (polyprotein 1ab). Since cotranslational processing of the polyproteins occurs, neither pp1a nor pp1ab has been detected in MHV-infected cells or during in vitro translation of purified genome RNA (8, 9).
ORF 1a encodes two experimentally confirmed proteinases that are likely responsible for all of the processing of the MHV gene 1 polyproteins (14, 15, 27, 31, 32), while regions within the ORF 1b portion of the 1ab polyprotein have been predicted to possess RNA-dependent RNA polymerase and helicase activities (13, 16, 24, 27). The polymerase and helicase domains are conserved between the coronaviruses in their location and core amino acid motifs (13). The proteins processed from these regions of the polyprotein have been identified in cells infected with the human coronavirus 229E (229E) and the avian infectious bronchitis virus (17, 28), and the 229E “helicase” (Hel) has been demonstrated to possess ATPase activity in in vitro assays (21). The Hel has not been identified for any other coronavirus. It is presumed that the Hel of coronaviruses is intimately involved in processes of viral RNA transcription and replication, but there has been no experimental determination of the functions of the Hel or of its interaction with viral RNA.
It is postulated that coronavirus RNA synthesis occurs on membrane-attached viral replication complexes that also contain replicase proteins such as polymerase and Hel. Though there has been no experimental identification of active viral replication complexes for any coronavirus, recent work with the arteriviruses has demonstrated that the putative polymerase and Hel proteins localize to perinuclear foci consistent with membranous complexes (36, 48). It also has been shown previously that proteins from polyprotein 1a of equine arteritis virus localize to membranous complexes that are active in RNA synthesis (47). Since the arteriviruses and coronaviruses share many features of genome organization and RNA transcription, it has been presumed that coronaviruses similarly will form replication complexes on cytoplasmic membranes. Gene 1 proteins of coronaviruses have been shown to localize to perinuclear complexes, but nothing is known of the localization of the gene 1 proteins such as Hel presumed to be involved in viral RNA synthesis, or if complexes containing Hel are active in RNA synthesis (3, 20, 26).
In this study, we have defined the expression, processing, and localization of the putative gene 1-encoded Hel in MHV-infected cells. The Hel was detected as a 67-kDa protein that was translated and proteolytically processed throughout the infectious cycle. Hel was detected in MHV-infected cells in cytoplasmic complexes that were initially perinuclear but became more abundant and formed extensive complexes at late times of infection. Hel-containing complexes also were sites of nucleocapsid (N) accumulation and were active in MHV RNA synthesis, suggesting that these membranous structures were the active viral replication complexes and that Hel was directly involved in the process of viral RNA synthesis.
MATERIALS AND METHODS
Viruses, cells, and infection.
MHV-A59 was used throughout this study. Virus was grown and titers were determined in DBT cells (22). DBT cells were used for all experiments, including radiolabeling and immunofluorescence.
Induction and use of antibodies directed against the Hel domain of polyprotein 1ab.
Two different rabbit polyclonal antisera were used in parallel in studies of expression and localization of the putative Hel. All nucleotide sequence numbers are based on the sequence of Lee et al. (27) as modified by Bonilla et al. (4). Amino acid sequences were similarly derived, with ORF 1b amino acid numbers based on presumed fusion of F4460 (ORF 1a) to the sequence FKRI4464 (ORF1b). For the antiserum B1, a reverse transcription-PCR-amplified fragment of gene 1 encompassing the Hel domain was digested with StuI and HindIII and inserted into the SmaI and HindIII sites of pQE-31 (Qiagen), resulting in a cloned fragment from nucleotide (nt) 16787 to 18337. The entire fusion protein included 21 amino acids encoded by the vector at the amino terminus including a six-histidine tag and the portion of the gene 1 polyprotein beginning with the amino acid sequence F5527KQCYA and ending with the amino acid sequence SLMGFKL6043 (see Fig. 1). The fusion protein extended 69 amino acids beyond the predicted carboxy-terminal cleavage site of the putative Hel Q_S5982-3. The B1 fusion protein was expressed in Escherichia coli, purified on Ni-nitrilotriacetic acid resin, concentrated by dialysis, and used to induce antibodies in rabbits at Cocalico Inc. The rabbit polyclonal antiserum MHV-96.8 was raised against the synthetic peptide 5965SLNFTTLTLDKINNPRLQ5982, at the C-terminal end of the putative Hel.
FIG. 1.

Gene 1 structure and anti-Hel antibodies. The organization of the MHV genome is shown, with the location of gene 1, the organization of ORF 1a and ORF 1b, and the polymerase and Hel domains shown to scale. The region of the B1 antiserum directed against the B1 fusion protein (white box) and the 96.8 antiserum (black bar) are shown. The entire sequence of the 96.8 peptide is shown, and the boundaries of the B1 fusion protein are indicated, as described in Materials and Methods.
Immunoprecipitation, radiolabeling, and proteinase inhibition.
Infections, radiolabeling, pulse-labeling, and pulse-chase experiments with MHV-A59 in DBT cells were performed as previously described (7, 9, 30). Briefly, confluent monolayers of DBT cells were infected with MHV-A59 in Dulbecco modified Eagle medium–2% fetal calf serum for 5 h at 37°C. For detection of protein, cells were incubated with medium containing actinomycin D (5 μg/ml) for 1 h, followed by addition of medium containing [35S]methionine for times as indicated for individual experiments. During pulse-label and pulse-chase experiments, translation initiation was synchronized by treatment of cells for 30 min with medium containing 200 mM NaCl, as previously described (9). At the end of the radiolabeling or chase period, lysates of whole cells were immunoprecipitated in immunoprecipitation buffer containing 10 mM Tris HCl, 150 mM NaCl, 1% sodium dodecyl sulfate (SDS), 1% Nonidet P-40, and 1% deoxycholate, and the immunoprecipitated proteins were analyzed on SDS–5 to 18% gradient acrylamide gels and prepared for fluorography as previously described (9).
Labeling and detection of newly synthesized cellular and viral RNA.
New cellular and viral RNA was detected by incorporation of bromo-UTP (BrUTP; Sigma) into nascent strands, by a modification of the approach described by Haukenes et al. (19). DBT cells were grown on coverslips and infected or mock infected as described above. At 5 h postinfection (p.i.), the medium was replaced with Optimem medium containing 30 μl of a phosphatidylethanolamine-dimethyldioctadecylammonium bromide lipofection reagent per ml and 5 μg of BrUTP (Sigma) per ml. The transfection medium was preincubated for 15 min at 37°C in the dark before it was added to the cells. Infection was continued for 1.5 h in the presence of BrUTP prior to rinsing, methanol fixation, and processing for immunofluorescence microscopy. Cellular RNA synthesis was abolished by the addition of actinomycin D (5 μg/ml) to the transfection medium.
Immunofluorescence detection of protein and RNA.
For the detection of Hel, DBT cell monolayers were grown on glass coverslips for 36 h, until there were areas of light confluency but the majority of cells remained noncontiguous. Cells were infected with MHV-A59 at a multiplicity of infection of 10 PFU/cell, and the inoculum-containing medium was left on the cells for the duration of infection. Cells were prepared for immunofluorescence at 6 h p.i., when syncytia were detected in confluent areas of the monolayer. Cells were rinsed with 37°C phosphate-buffered saline (PBS), the PBS was aspirated, and −20°C 100% methanol was added to the wells. Cells were kept at −20°C for 1 h prior to processing for immunofluorescence or stored at −20°C under methanol for later use. For detection of Hel, coverslips of infected or mock-infected cells were transferred to wells containing 5% bovine serum albumin in PBS and rehydrated for 15 min. All subsequent steps were at 4°C in PBS with 1% bovine serum albumin–0.05% Nonidet P-40. The rehydration solution was aspirated, and cells were incubated with the B1 antiserum at a 1:100 dilution, either at room temperature for 45 min or at 4°C for 16 h. Cells were then rinsed twice for 15 min and incubated with 2°C antibody, usually Cy-2-conjugated mouse anti-rabbit monoclonal antibody (MAb) (Amersham), at a 1:1,000 dilution for 45 min at room temperature. Cells were rinsed three times, followed by rinses in PBS and water, and the coverslips were mounted with Aquapolymount (Polysciences). When dual-labeling experiments were performed, cells were first processed for detection of BrUTP-labeled RNA, followed by processing for detection of Hel. The primary antibody for detection of BrUTP-labeled RNA was mouse antibromodeoxyuridine MAb (Boehringer) used at a 1:100 dilution, and the secondary antibody was Cy-2-labeled goat anti-mouse antibody. During dual-labeling experiments, Cy-5-conjugated mouse anti-rabbit MAb was used to detect Hel.
Laser confocal microscopy.
Laser confocal microscopy was performed in the Vanderbilt Cell Imaging Resource of the Vanderbilt Cancer Center. A Zeiss LSM 410 laser scanning confocal microscope equipped with argon, krypton, and helium-neon lasers was used for immunofluorescence detection. For detection of Cy-2-labeled proteins or RNA, a 488-nm laser was used, and for detection of Cy-5-labeled helicase, a 647-nm laser was used. Fixed contrast, brightness, and exposure times were used for all data collection and image analysis. For colocalization studies, narrow pinhole sizes (20 Airy units) were used to minimize the depth of field. Images were saved as green (488 nm) and far-red (647 nm) and merged in the LSM software and in Photoshop 4.0 (Adobe). Coronal and axial reconstructions were performed in NIH Image 1.62b (39).
RESULTS
Expression and processing of Hel in MHV-infected DBT cells.
Two polyclonal antisera directed against the putative Hel, B1 (fusion protein) and 96.8 (peptide), were used to immunoprecipitate proteins from whole-cell lysates of MHV-A59-infected DBT cells (Fig. 1). At 5.5 h p.i., a 67-kDa protein was detected in infected cells by both antisera (Fig. 2). The apparent mass of the immunoprecipitated protein was the same as the calculated mass of the predicted Hel based on the deduced amino acid sequence between nt 16355 and nt 18154 from the 5′ end of the genome RNA (Hel domain). The 67-kDa protein was not detected in mock-infected cells by either antiserum, nor in infected cells by preimmune sera, indicating that it was a viral protein. Both sera precipitated proteins in addition to the 67-kDa viral product. The MHV structural nucleocapsid protein (N) was detected in immunoprecipitates by both anti-Hel antisera. We have previously observed coprecipitation of N with other gene 1 antibodies including p1a-22 and 3CLpro (ORF 1a) from infected cells (29, 30), but N appeared to coprecipitate to a greater extent than we have observed with other gene 1 antibodies (see below and Discussion). With the exception of N, the additional proteins detected by 96.8 also were precipitated by preimmune serum in infected cells or by immune serum in mock-infected cells. The B1 immune serum detected a protein of 180 kDa that appeared to be specific to infected cells and immune serum but which was also slightly visible in the infected-preimmune serum lane and was easily seen on prolonged exposure of the gel (data not shown). In addition, three light bands of >200 kDa were present in infected cells immunoprecipitated with B1 immune serum. However, in contrast to the 67-kDa Hel protein and N, which were easily detectable in multiple independent experiments, the trio of bands above 200 kDa was evanescent and not reproducible. Certainly, it is possible that there were polyprotein precursors of >200 kDa, but since the observed protein bands were not reproducible and were not seen at all during the pulse-chase experiment or with the 96.8 antibody, it is unlikely that they represented authentic viral proteins.
FIG. 2.

Identification of a 67-kDa protein by anti-Hel antibodies in MHV-infected DBT cells. (A) Cells were mock infected (mock) or infected (inf.) as described in Materials and Methods. Cells were incubated with actinomycin D for 1 h beginning at 5 h p.i., followed by addition of [35S]methionine for 2 h. Following cell lysis, proteins were immunoprecipitated with immune serum (B1 or 96.8) or preimmune serum (inf. pre). Molecular mass markers (in kilodaltons) are to the left of the gel, and the locations of the 67-kDa protein and nucleocapsid (N) are shown. (B) Radiolabeling of infected cells was performed in the absence (−) or presence (+) of the proteinase inhibitor E64d (400 μg/ml). Mass markers (in kilodaltons) are to the right of the gel.
When radiolabeling was performed in the presence of the cysteine proteinase inhibitor E64d, the 67-kDa protein was not detected (Fig. 2B). E64d has been shown to inhibit cleavage of the gene 1 polyprotein by the ORF 1a-encoded 3C-like proteinase (3CLpro) (30, 31). This result confirmed that the 67-kDa protein was a proteolytic cleavage product that likely was cleaved by 3CLpro. No additional proteins were detected in the presence of E64d, but N still coprecipitated when cleavage of the 67-kDa protein was blocked, and since N was only minimally detected by preimmune sera, the data suggested that N was associated with the mature 67-kDa protein, with noncleaved precursors, or both. The results demonstrated that the 67-kDa protein was the product of translation and processing of polyprotein 1ab, specifically from the region of the polyprotein predicted to encode the viral Hel. The 67-kDa protein will be referred to as Hel in the remainder of this report.
Pulse-label and pulse-chase translation of Hel.
We determined the pattern of Hel expression and processing during virus infection by performing pulse-label and pulse-chase translation (Fig. 3). To demonstrate that the label and chase conditions were optimal for [35S]Met labeling during the pulse and for abolishing labeling during the chase, we analyzed the incorporation of [35S]Met into trichloroacetic acid (TCA)-precipitable polypeptides or protein (Fig. 3A). During labeling, incorporation rapidly increased for the first 120 min and then plateaued for 1 h followed by a slow decline. In contrast, the chase was quite effective in immediately blocking additional incorporation of radiolabel, resulting in a rapid decline in TCA-precipitable protein over a 2-h period. During the pulse-label (Fig. 3B), Hel was first detected at 30 min of labeling, and since high-salt synchronization resulted in a delay of 10 min before reinitiation of translation (9, 41), between 20 and 30 min appeared to be required for translation and processing of detectable amounts of mature Hel. To define precursor-product relationships and to determine the stability of processed Hel, pulse-chase translation was performed concurrently with the pulse-labeling studies, by chasing replicate plates for up to 5 h in the presence of excess unlabeled l-methionine following the labeling period (Fig. 3C). By 3 h of chase (9.5 h p.i.), approximately 75% of the cells were involved in syncytia. By 5 h of chase (11.5 h p.i.), >90% of the cells in the monolayer were involved in syncytia.
FIG. 3.

Pulse-label and pulse-chase translation of p67 Hel in MHV-infected DBT cells. (A) TCA-precipitable counts per minute of [35S]Met during pulse-label and pulse-chase translation. MHV-infected DBT cells were labeled in suspension beginning at 5.5 h p.i., and samples were taken at the time indicated in duplicate for TCA precipitation and scintillation counting. At 60 min, the suspended cells were split and the chase cells were rinsed twice, followed by resuspension in chase medium and sampling for TCA-precipitable counts at the times indicated. Data points are the means of two samples with standard deviations shown. (B) Pulse-label translation. At 5.5 h p.i., replicate plates of MHV-infected DBT cells were incubated in medium containing 200 mM excess NaCl for 30 min, then the medium was changed to medium containing normal concentrations of NaCl with added [35S]Methionine, and plates were harvested for lysis and immunoprecipitation at the times in minutes shown above the lanes. Proteins were immunoprecipitated with the 96.8 antiserum, followed by electrophoresis on an SDS–5 to 18% acrylamide gradient gel and fluorography. Molecular mass markers (m) (in kilodaltons) are to the left of the gel, and the locations of p67 Hel and N are indicated to the right. (C) Pulse-chase translation. Replicate plates were infected and radiolabeled concurrently with the pulse-label plates. Cells were all pulsed with [35S]methionine for 30 min (see panel A, 30 min) followed by chase in medium containing 10-fold excess unlabeled l-methionine for the times in minutes indicated, prior to lysis, immunoprecipitation, and electrophoresis. Numbers at right are molecular masses in kilodaltons.
During the chase, the amount of Hel detected was increased over the amount seen at the end of the labeling period (Fig. 3C, 30 min) at every time point. The amount of Hel detected increased at each point up to 180 min and even at 300 min of chase was equivalent to that seen at 30 min of chase. In contrast, the amount of nonspecifically precipitated proteins (210 and 150 kDa) was stable or decreased during the first 180 min of chase, consistent with the TCA precipitation results that showed decreased [35S]Met labeling of proteins. No intermediate precursors were detected at any point during the chase, consistent with the results seen in the presence of the proteinase inhibitor E64d. There was an increase in the amount of label detected at the top of the gel during the first 120 min of chase, possibly due to completion of translation of polyprotein precursor molecules that were too large to be resolved on the 5 to 18% acrylamide gradient gel. These results together indicated that new molecules of Hel were cleaved, likely from a large polyprotein precursor, over at least a 3-h period and that once cleaved were stable for several hours. This was in contrast to the overall rather rapid turnover of newly synthesized proteins in the cell during the same period. Though the balance between Hel processing and degradation remains to be determined, the pattern of expression and processing of Hel was consistent with our previous analyses of the mature gene 1 (ORF 1a) proteins 3CLpro and p22 (29, 30).
Nucleocapsid was detected during both experiments. The amount of N detected during the pulse-label was significant even before Hel was visible, and the amount of N seen during both pulse-label and pulse-chase remained fairly constant. Since the labeling experiments were performed at late times of infection, N synthesis from mRNA 7 might be expected to occur quickly, and nonlabeled Hel that would be precipitated and could result in coprecipitation of N but would not itself be detected by fluorography would likely be present. This result supported our conclusion that N may be specifically coprecipitated with Hel.
Hel localization in MHV-infected DBT cells.
To define the localization of Hel, MHV-infected DBT cells were probed at 6 h p.i. with anti-Hel antisera and mouse anti-rabbit MAbs conjugated to Cy-2 fluorescent dye (Amersham), followed by detection on the confocal microscope with a 488-nm He-Ne laser. In MHV-infected cells, brightly fluorescing complexes were identified in virus-induced syncytia and in individual infected cells with the B1 antiserum (Fig. 4), and cytoplasmic complexes also were detected with the 96.8 antiserum (data not shown). In contrast, when mock-infected cells were probed, no specific labeling was observed with either antiserum. In the DBT cells with polygonal morphology, the Hel-containing complexes were predominantly perinuclear in distribution but in many cells also were widely distributed throughout the cell cytoplasm. In infected cells with fusiform morphology, the complexes concentrated at the poles of the nuclei. In virus-induced syncytia, Hel was abundant and tended to cluster around the coalesced nuclei or form clusters of multiple discrete foci between the nuclei. The distribution and appearance of the Hel-containing complexes were consistent with membrane-bound vesicles. It was not possible to say whether the protein identified was mostly mature Hel, nascent polyprotein containing Hel, or a combination of both, since the serum antibodies used were polyclonal, and the Hel must exist within the nascent polyprotein prior to cleavage. However, the biochemical data clearly demonstrated that the antibodies used were able to detect mature Hel and did not detect discrete precursors less than 250 kDa in mass. Although it is possible that the specificity of the antibody in confocal immunofluorescence was different from that during immunoprecipitation, it is most likely that the mature protein was being identified.
FIG. 4.

Confocal immunofluorescence detection of Hel in MHV-infected DBT cells. Monolayers of DBT cells were infected with MHV-A59 for 6 h and then fixed and processed for immunofluorescence as described in Materials and Methods. The B1 antibody was used for detection of Hel. Imaging was performed on a Zeiss LSM 410 laser confocal microscope, with a 488-nm laser to excite the Cy-2 dye. The images were obtained with a 63× objective. Phase-contrast images were obtained with a Nomarski polarizer. Separate fluorescent and transmitted images were obtained and merged with Photoshop 4.0. (A) Fluorescent images of mock-infected cells (mock) and two different fields of the infected-cell monolayer (infected), showing individual cells and virus-induced syncytia. (B) Superimposition of transmitted light and fluorescent images. The fluorescent images from panel A were merged with transmitted light images.
Because the Hel-containing complexes were abundant at late times of infection, we sought to define the extent and organization of the complexes. z sectioning of individual infected cells was performed, and the sections were used to reconstruct the cell in sagittal (xy), axial (yz), and coronal (xz) planes (Fig. 5). The two adjacent infected, nonfused cells in Fig. 5A were imaged as 58 sections at 0.2-μm intervals, with every third section shown beginning closest to the coverslip (section 11 of 58), and extending to the “top” of the cell (section 44 of 58). When the z sections were compared, it was clear that Hel-containing complexes surrounded the nucleus. When Hel was identified in serial sections of the cell, there was apparent continuity of the complexes in both cells over a distance of several micrometers. With the cell on the right of the image as an example, the bright Hel signal noted in section 26 extended at least through section 38, a distance of 2.5 μm. To better visualize the extent of the complexes, coronal and axial reconstructions were derived from the stack of sections presented in Fig. 5A (Fig. 5B and C). Both coronal (xz) and axial (yz) reconstructions confirmed that the Hel-containing complexes varied from small and discrete to large, apparently continuous structures that surrounded the nucleus and extended in some instances through more than half the diameter of the cell. This was particularly apparent in the axial reconstruction, where Hel-containing structures as large as 3 by 7 μm were observed (Fig. 5C, frame 4). Because of the limits of light resolution, it was not possible to determine whether the larger complexes were accumulations of smaller membranous complexes or were confluent or fused structures.
FIG. 5.

Three-dimensional sectioning of MHV-infected cells and characterization of Hel-containing inclusions. Cells were imaged for Hel with the 488-nm laser to detect Cy-2. (A) Sagittal (xy) sections of the cells were obtained by z sectioning at 0.2-μm intervals, with resolution in the x, y, and z planes of 0.12 μm/pixel. The total z distance of 12 μm was imaged with 58 sections. The sections were transformed into a stack with NIH Image 1.62b. Section 1 was at the level of the coverslip, and section 58 was at the “top” of the cell most distant from the coverslip. The numbers in each panel are the numbers of the sections: 11 indicates 11 of 58. (B) Coronal (xz) reconstruction of a cell. The cell on the right of the panels in panel A was isolated, and the z stack was used to reconstruct the cell in the xz (coronal) plane with NIH Image 1.62b. Section 29 was used as the reference because it is in the middle of the cell in z dimension and shows the complexes clearly. The levels of the xz slices are shown by white lines 1 through 6, and the panels to the right show the respective slices. The viewpoint is indicated by the arrow, and the orientation of the left and right sides of the respective images is shown. The white line through the panels shows the level of section 29. (C) Axial (yz) reconstruction of the cell. This was performed as described for panel B, except in the yz plane.
Colocalization of Hel and nucleocapsid in cytoplasmic complexes.
Since N was detected by both anti-Hel antibodies, we sought to determine if N was also present in cytoplasmic complexes that contained Hel and also to demonstrate that the detection of N occurred because of coprecipitation rather than cross-reactivity of the anti-Hel antibodies. Sucrose gradient-purified MHV virions were used in Western blot experiments with antibodies directed against whole virions, matrix, nucleocapsid, the 3C-like proteinase (α3CLpro), and anti-Hel (B1) (Fig. 6). The M, N, and S structural proteins were readily detected with anti-MHV polyclonal antibodies, and the M and N proteins were detected by the respective MAbs. A small amount of anti-M binding to N occurred, as well as a small amount of anti-N binding to M. In contrast, the polyclonal antibodies directed against Hel or 3CLpro did not directly bind to the structural proteins, notably N. Only after extreme overexposure of the gel could a faint band of N be detected in the 3CLpro and Hel lanes. The antibodies were used in the same dilutions in both Western blot and immunofluorescence experiments. This experiment indicated that the gene 1 antibodies were not directly cross-reacting with N or other MHV structural proteins and that the fluorescence observed in these experiments was due to direct detection of the cognate gene 1 protein.
FIG. 6.

Immunoblotting of MHV virion proteins by structural and gene 1 protein antibodies. MHV virions were purified by sucrose gradient centrifugation. Virions were lysed in SDS buffer, separated on an SDS–15% acrylamide gel, transferred to polyvinylidene difluoride, and incubated with antibodies directed against the proteins as indicated above the lanes. Equivalent amounts of virion proteins were present in each lane. The anti-M and anti-N mouse MAbs J.1.3 and J.3.3, obtained from John Fleming, were used as primary antibodies at a 1:1,000 dilution. The rabbit polyclonal antibodies directed against 3CLpro (SP9) and Hel (B1) were used as primary antibodies at a 1:100 dilution.
Dual-labeling experiments were then performed with B1 and anti-N antibodies (Fig. 7). The patterns for Hel and N were almost identical. The Hel and N images were merged with an algorithm in NIH Image 1.62 that was essentially a Boolean “and” function and therefore showed white pixels in the merged image only if white pixels were present in the precise location in both images. While this excluded grey pixels, it was most stringent for colocalization of pixels. Extensive, almost complete colocalization of Hel and N was observed by this approach (Hel and N panel). Thus, it appeared that N was tightly associated with the complexes formed during translation and processing of the gene 1 polyprotein. The colocalization of Hel and N corroborated the biochemical experiments demonstrating coprecipitation of the two proteins.
FIG. 7.
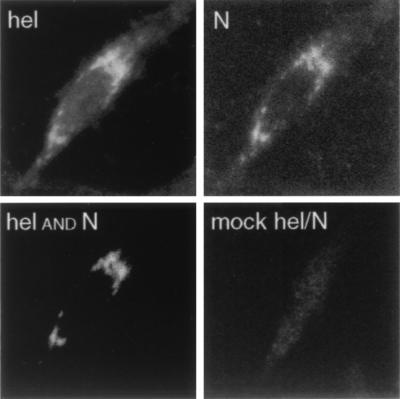
Colocalization of Hel and N in MHV-infected cells. MHV-infected DBT cells were fixed in 100% methanol at 5.5 h p.i. and prepared for confocal immunofluorescence with the anti-N MAb and the anti-Hel rabbit polyclonal serum B1. Hel was imaged at 647 nm (far red), and N was imaged at 488 nm (green). For colocalization (Hel and N), the images were merged with an “and” function requiring the presence of white pixels in both N and Hel images for the white pixels to be seen in the merged image (NIH Image 1.62). Thus, the white pixels in this image were colocalized in the N and Hel images. For the mock Hel-N image, the Hel and N signals were overlapped without filtering, to show the maximum signal from both channels in the merged image.
Colocalization of new viral RNA and Hel in membranous complexes.
The colocalization of Hel and N in discrete cytoplasmic complexes, along with the known involvement of N in encapsidation of genome RNA and its predicted role in RNA transcription, led us to determine whether the Hel- and N-containing complexes were active in viral RNA synthesis. We first assessed whether Hel and new viral RNA were localizing to membranous structures, as the confocal imaging of Hel and N suggested (Fig. 8). Infected cells labeled with [35S]Met or [3H]uridine were used to quantitate Hel and viral RNA, respectively, in membranous pellets and S100 cytosol. The cell monolayers were homogenized in the absence of detergent, and differential centrifugation was performed to separate the cellular membranes from the cytosolic supernatant, including a final spin at 100,000 rpm with a TLA 20.2 rotor in a Beckman TLX Optima centrifuge. The cytosol and membrane pellet were analyzed by immunoprecipitation and densitometry for Hel and by TCA-precipitable [3H]uridine for new viral RNA in the presence of actinomycin D. Both viral RNA and Hel were detected almost exclusively in the membrane pellet, confirming that the observed Hel-containing complexes were associated with cellular membranes.
FIG. 8.

Detection of Hel and viral RNA in membrane fractions of MHV-infected cells. MHV-infected DBT cell monolayers were homogenized in the absence of detergent, and differential centrifugation was performed to separate cellular membranes and cytosol, including a final spin at 100,000 rpm with a TLA 20.2 rotor in a Beckman TLX Optima centrifuge. The combined membrane pellets (memb.) and the post-100,000-rpm cytosol were assessed for Hel by immunoprecipitation with the B1 antibody followed by fluorography and densitometric analysis (NIH Image 1.62) with a calculation of area in pixels × the mean density of the pixels. Quantitation of new viral RNA was performed by determination of TCA-precipitable [3H]uridine in the presence of actinomycin D. Mock-infected cells were used as controls.
To more precisely define sites of MHV RNA synthesis in cells by confocal microscopy, BrUTP was used to metabolically label cellular and viral RNA (Fig. 9B). No cellular RNA was detected in the cytoplasm in the absence of actinomycin D, and the addition of actinomycin D completely abolished labeling and detection of cellular RNA in nuclei as well (Fig. 9D). Thus, we were able to define localization of viral RNA both in the presence and in the absence of actinomycin D, allowing us to clearly identify the nuclei and still distinguish sites of viral RNA synthesis in the cytoplasm.
FIG. 9.

Detection of RNA and Hel in MHV-infected DBT cells. MHV-infected or mock-infected DBT cells were metabolically labeled with BrUTP and processed for confocal immunofluorescence as described in Materials and Methods. (A) Transfection of infected cells in the absence of actinomycin D, to allow for incorporation into both viral and cellular RNA. Cells were imaged for BrUTP incorporation (Cy-2, green, RNA column) and helicase (Cy-5, red, Hel column). The images were merged (“RNA/helicase”), with yellow pixels representing areas of signal colocalization. The white brackets in the “RNA/helicase” panel indicate the area of enlargement in the second row of panels. (B) Mock-infected cells were labeled with BrUTP in the absence of actinomycin D and imaged for RNA and Hel as described for panel A. Only the merged image is shown. (C) Transfection of infected cells in the presence of actinomycin D. Imaging and processing were performed as described for panel A. The small white brackets in the merged image indicate the area of enlargement in the second row. (D) Mock-infected cells were labeled with BrUTP in the presence of actinomycin D and imaged for RNA and Hel as described for panel A. Only the merged image is shown.
To determine the localization of viral RNA synthesis and the relationship with sites of Hel localization, dual-labeling experiments were performed in MHV-infected cells. Metabolic labeling of viral RNA with BrUTP was performed in the absence (Fig. 9A) or presence (Fig. 9C) of actinomycin D. Infected cells were labeled with BrUTP beginning at 5.5 h p.i. When MHV-infected cells were labeled in the absence of actinomycin D, labeling of cellular RNA in nuclei was identical to that in mock-infected DBT cells (Fig. 9A, RNA column). In addition, multiple foci of new RNA were detected in the cytoplasm of isolated cells and syncytia. The cytoplasmic RNA was not detected in all discrete cells where the cell nuclei were labeled with BrUTP, but cytoplasmic RNA was detected in all observed virus-induced syncytia. In individual nonfused cells, the RNA-containing complexes were largely perinuclear in distribution, but in syncytia the complexes were more widely distributed and also were frequently detected as coalesced aggregates in the central portions of the syncytia. When actinomycin D was added to the medium, labeling of new cellular RNA in the nucleus was abolished but the amount and extent of cytoplasmic RNA detection were unaffected (Fig. 9C, RNA column). These results demonstrated that the cytoplasmic complexes were sites of new, actinomycin D-resistant viral RNA synthesis.
The dual-labeling experiments demonstrated that Hel-containing complexes had a distribution almost identical to that of viral RNA (Fig. 9A and C, Hel column). When the Hel and RNA images were merged, the vast majority of the signals for viral RNA and Hel colocalized. The ability to detect viral RNA in the absence of actinomycin D allowed us to confirm that lack of detection of viral RNA in some cells where Hel was abundant (Fig. 9A, Hel column) was due to lack of uptake of BrUTP rather than to lack of RNA synthesis. In all cells where cell nuclei were labeled and Hel was detected, viral RNA was also detected, indicating that Hel detection may be a good surrogate for viral RNA synthesis. To confirm colocalization of the Hel and RNA signals, sequential sections (z sections) were obtained, typically 16 sections at 0.2-μm intervals. In any cell section where both Hel and RNA were detected, the colocalization of Hel and new viral RNA was confirmed. In some cells and primarily in syncytia, foci of viral RNA that lacked detectable Hel were seen. In contrast, although there were limited areas of diffuse staining for Hel that did not have detectable signal for RNA, there were no discrete, Hel-containing complexes that did not contain new viral RNA. Overall, the dual-labeling studies demonstrated that the vast majority of Hel and viral RNA synthetic activity were tightly associated.
DISCUSSION
The replication programs of positive-strand RNA viruses occur in the cytoplasm of infected cells, and the transcription and replication of positive-strand RNA are associated with and require cellular cytoplasmic membranes (2, 11, 12, 18, 33, 40). Positive-strand RNA viruses also may direct the modification of cellular membranes and formation of novel cytoplasmic structures containing viral replicase proteins, cellular membranes, and cellular proteins (26, 34, 36, 43). Since the gene 1 polyprotein of the coronaviruses is presumed to contain all of the viral replicase functions, it is likely that one or more proteins processed from the polyprotein are involved in formation and function of the replication complexes, presumably on cellular membranes. Gene 1 proteins of the nidoviruses equine arteritis virus, HCV-229E, and MHV have all been shown to localize to perinuclear complexes with the appearance of membranous vesicles (3, 20, 47). The hydrophobic proteins that flank 3CLpro in the MHV gene 1 polyprotein have been shown to create a requirement for the presence of membranes for 3CLpro activity when they are expressed along with the proteinase in vitro (38). Recently, it was shown that full-length and subgenomic minus-strand RNAs of the coronavirus transmissible gastroenteritis virus partitioned into a membrane-protected population during cesium chloride gradient fractionation, suggesting that they may be in membrane-associated replication complexes that are active in plus-strand RNA synthesis (44).
Despite the abundance of data indicating that coronaviruses and arteriviruses direct the formation of transcriptionally active complexes containing gene 1 proteins, it had not been experimentally demonstrated for the coronaviruses that the gene 1 protein-containing complexes were sites of viral RNA transcription or replication. We therefore defined the pattern of expression, processing, and localization of the MHV Hel and determined if sites of Hel localization were active in viral RNA synthesis. We have identified the 67-kDa protein translated and processed from the ORF 1ab polyprotein that has been predicted to possess nucleoside triphosphatase and Hel activities (13, 14, 27). Our experiments demonstrated that the 67-kDa Hel was processed from the polyprotein and was easily detectable at late times of infection. The Hel was detected in increasing amounts during prolonged chase, with evidence for continued processing of new molecules of Hel for up to 3 h after radiolabel was removed. Surprisingly, Hel was still easily detectable for up to 5 h of chase (11 h p.i.), a time during a single-cycle infection when the monolayer was almost entirely fused. Although the mechanism for such prolonged detection could not be determined from these experiments, they suggested that the Hel was resistant to degradation following processing. It has previously been shown that continued coronavirus RNA synthesis is dependent on continued polyprotein translation and processing (23, 37, 42). Our results further suggest that the requirement for processing may be independent from that for translation and that some gene 1 proteins, such as Hel, may be stably available once processed.
When localization of Hel and that of new viral RNA synthesis were directly compared, it was clear that Hel localized to complexes that were also sites of synthesis of new viral RNA. There was a change in the appearance and distribution of the Hel-containing complexes over time, from a few punctate perinuclear foci early in infection to widespread cytoplasmic complexes at later times of infection (data not shown). The Hel-containing complexes were detected as multivesicular aggregations in the fused cytoplasm of the virus-induced syncytia, as well as large structures that surrounded the nucleus in discrete cells and occupied a significant portion of the cell cytoplasm. These results suggested that relocalization, aggregation, and possibly fusion of the Hel-containing complexes were occurring and that despite these significant alterations of the cell the complexes remained active in new viral RNA synthesis. Although metabolic labeling with BrUTP cannot differentiate among plus, minus, genomic, and subgenomic RNA species, the fact that a vast majority of incorporation of BrUTP into new RNA was detected within the Hel-containing complexes suggests that an evolving multifunctional complex may be the site of multiple MHV RNA transcription and replication activities.
In contrast, we did detect foci of newly synthesized viral RNA that did not contain detectable Hel. These sites may represent RNA that was synthesized and released from the Hel-containing complexes during the 1.5-h labeling period, or they may have been sites of RNA synthesis that did not require the presence of Hel. Finally, it is possible that Hel was present in these areas of new RNA localization in amounts that were below the limits of detection. These foci of new viral RNA were detected mostly in syncytia, and thus, it seems most plausible that they represented RNA that was not tightly associated with replication complexes or may have been recently released from the complexes. In support of this conclusion, isolated foci of new viral RNA were always directly juxtaposed with complexes containing both Hel and RNA. Further, the finding of viral RNA “outside” membranous complexes is consistent with the recent observations with transmissible gastroenteritis virus demonstrating lack of association of some RNA species with membranous vesicles (44).
Perhaps the most surprising finding was the intimate association of nucleocapsid protein with the Hel-containing membranous complexes. This association was confirmed by confocal microscopic colocalization, by coprecipitation, and by the lack of cross-reactivity of the gene 1 antibodies with N. The coprecipitation of N by gene 1 antibodies was not eliminated by 1% SDS in the lysis and immunoprecipitation solutions, stringent washes, or even boiling of lysate in immunoprecipitation buffer prior to addition of antibodies. These results indicated that N was likely tightly interacting with Hel in membranous complexes, either within the membrane or by protein-protein interactions. We previously have reported that N was immunoprecipitated by antibodies against other gene 1 proteins including p28 and p1a-22 (9, 30), and N has been shown to be involved in MHV RNA synthesis and to bind specifically to MHV RNA sequences (1, 46). However, the almost exclusive localization of N to the Hel- and RNA-containing complexes was remarkable. We expected to see areas of N localization separate from sites of presumed RNA synthesis, notably in presumed areas of virion assembly; we did not observe this in any of our experiments. This suggested that N synthesis, localization, and function may be focused at sites of RNA synthesis, and that once viral nucleocapsids (RNA+N) are formed, they are rapidly incorporated into virions for export and thus do not accumulate to levels detectable in our system. Alternatively, it is possible that the formation and incorporation of nucleocapsid structures into nascent virions render them undetectable by the antibodies. In either case, the abundance of N in all types and stages of replication complex formation and the lack of detection of any diffuse “cytosolic” N suggest that N may be critical for the formation and/or function of the replication complexes.
Our results raise important questions about gene 1 polyprotein expression and processing and the functions of distinct mature gene 1 proteins. It is not yet clear if regulation of polyprotein processing has emerged because precursor forms may perform important alternative functions. The antisera used in this study did not detect intermediate precursors to Hel, but clearly precursors must exist since Hel is cleaved from the gene 1 polyprotein 1ab. In addition, the pulse-chase studies demonstrated the continued processing of new mature Hel molecules. It remains to be determined if the precursors to Hel serve specific functions during viral replication other than as a source of new mature proteins. We have shown a clear association between Hel and new viral RNA, but the function of Hel in the complex during RNA synthesis remains to be determined. The origin and composition of the cellular membranes recruited to MHV replication complexes have not been defined, nor do we know the identity of gene 1 proteins involved in RNA synthesis and replication complex formation. Our results are provocative in that they suggest an amplification process of replication complex formation that is mediated during the ongoing maturation of gene 1 proteins. Our continuing imaging and biochemical experiments are designed to define the precise components of the replication complex and to determine how it can function in the highly complex translation and processing of an 800-kDa polyprotein, while at the same time directing the transcription and replication of a 32-kb RNA genome.
ACKNOWLEDGMENTS
This work was supported by Public Health Service grants AI-26603 and AI01479 (M.R.D.).
We acknowledge the assistance of Jonathan Sheehan in the Molecular Imaging Shared Resource of the Vanderbilt Cancer Center (IP30CA68485). We thank Fred Wassenaar and Sasha Gorbalenya for technical assistance and John Fleming for providing MAbs J.1.3 and J.3.3.
REFERENCES
- 1.Baric R S, Nelson G W, Fleming J O, Deans R J, Keck J G, Casteel N, Stohlman S A. Interactions between coronavirus nucleocapsid protein and viral RNAs: implications for viral transcription. J Virol. 1988;62:4280–4287. doi: 10.1128/jvi.62.11.4280-4287.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barton D J, Sawicki S G, Sawicki D L. Solubilization and immunoprecipitation of alphavirus replication complexes. J Virol. 1991;65:1496–1506. doi: 10.1128/jvi.65.3.1496-1506.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bi W, Bonilla P J, Holmes K V, Weiss S R, Leibowitz J L. Intracellular localization of polypeptides encoded in mouse hepatitis virus open reading frame 1A. Adv Exp Med Biol. 1995;380:251–258. doi: 10.1007/978-1-4615-1899-0_40. [DOI] [PubMed] [Google Scholar]
- 4.Bonilla P J, Gorbalenya A E, Weiss S R. Mouse hepatitis virus strain A59 RNA polymerase gene ORF 1a: heterogeneity among MHV strains. Virology. 1994;198:736–740. doi: 10.1006/viro.1994.1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Breedenbeek P J, Pachuk C J, Noten A F H, Charite J, Luytjes W, Weiss S R, Spaan W J M. The primary structure and expression of the second open reading frame of the polymerase gene of the coronavirus MHV-A59; a highly conserved polymerase is expressed by an efficient ribosomal frameshifting mechanism. Nucleic Acids Res. 1990;18:1825–1832. doi: 10.1093/nar/18.7.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brierley I, Boursnell M E G, Binns M M, Billimoria B, Blok V C, Brown T D K, Inglis S C. An efficient ribosomal frame-shifting signal in the polymerase-encoding region of the coronavirus IBV. EMBO J. 1987;6:3779–3785. doi: 10.1002/j.1460-2075.1987.tb02713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denison M R, Hughes S A, Weiss S R. Identification and characterization of a 65-kDa protein processed from the gene 1 polyprotein of the murine coronavirus MHV-A59. Virology. 1995;207:316–320. doi: 10.1006/viro.1995.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Denison M R, Perlman S. Translation and processing of mouse hepatitis virus virion RNA in a cell-free system. J Virol. 1986;60:12–18. doi: 10.1128/jvi.60.1.12-18.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Denison M R, Zoltick P W, Hughes S A, Giangreco B, Olson A L, Perlman S, Leibowitz J L, Weiss S R. Intracellular processing of the N-terminal ORF 1a proteins of the coronavirus MHV-A59 requires multiple proteolytic events. Virology. 1992;189:274–284. doi: 10.1016/0042-6822(92)90703-R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Vries A A F, Horzinek M C, Rottier P J M, de Groot R J. The genome organization of the nidovirales: similarities and differences between arteri-, toro, and coronaviruses. Semin Virol. 1997;8:33–47. doi: 10.1006/smvy.1997.0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doedens J, Maynell L A, Klymkowsky M W, Kirkegaard K. Secretory pathway function, but not cytoskeletal integrity, is required in poliovirus infection. Arch Virol Suppl. 1994;9:159–172. doi: 10.1007/978-3-7091-9326-6_16. [DOI] [PubMed] [Google Scholar]
- 12.Froshauer S, Kartenbeck J, Helenius A. Alphavirus RNA replicase is located on the cytoplasmic surface of endosomes and lysosomes. J Cell Biol. 1988;107:2075–2086. doi: 10.1083/jcb.107.6.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gorbalenya A, Koonin E. Comparative analysis of amino-acid sequences of key enzymes of replication and expression of positive-strand RNA viruses: validity of approach and functional and evolutionary implications. Sov Sci Rev Sect D. 1993;11:1–81. [Google Scholar]
- 14.Gorbalenya A E, Koonin E V, Donchenko A P, Blinov V M. Coronavirus genome: prediction of putative functional domains in the nonstructural polyprotein by comparative amino acid sequence analysis. Nucleic Acids Res. 1989;17:4847–4861. doi: 10.1093/nar/17.12.4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gorbalenya A E, Koonin E V, Lai M M-C. Putative papain-related thiol proteases of positive-strand RNA viruses. FEBS Lett. 1991;288:201–205. doi: 10.1016/0014-5793(91)81034-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gorbalenya A E, Koonin E V. Viral proteins containing the purine NTP-binding sequence pattern. Nucleic Acids Res. 1989;17:8413–8440. doi: 10.1093/nar/17.21.8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grotzinger C, Heusipp G, Ziebuhr J, Harms U, Suss J, Siddell S G. Characterization of a 105-kDa polypeptide encoded in gene 1 of the human coronavirus HCV 229E. Virology. 1996;222:227–235. doi: 10.1006/viro.1996.0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grun J B, Brinton M A. Separation of functional West Nile virus replication complexes from intracellular membrane fragments. J Gen Virol. 1988;69:3121–3127. doi: 10.1099/0022-1317-69-12-3121. [DOI] [PubMed] [Google Scholar]
- 19.Haukenes G, Szilvay A-M, Brokstad K A, Kanestrom A, Kalland K-H. Labeling of RNA transcripts in eukaryotic cells in culture with BrUTP using a liposome transfection reagent (DOTAP) BioTechniques. 1997;22:308–312. doi: 10.2144/97222st03. [DOI] [PubMed] [Google Scholar]
- 20.Heusipp G, Grotzinger C, Herold J, Siddell S G, Ziebuhr J. Identification and subcellular localization of a 41 kDa, polyprotein 1ab processing product in human coronavirus 229E-infected cells. J Gen Virol. 1997;78:2789–2794. doi: 10.1099/0022-1317-78-11-2789. [DOI] [PubMed] [Google Scholar]
- 21.Heusipp G, Harms U, Siddell S G, Ziebuhr J. Identification of an ATPase activity associated with a 71-kilodalton polypeptide encoded in gene 1 of the human coronavirus 229E. J Virol. 1997;71:5631–5634. doi: 10.1128/jvi.71.7.5631-5634.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirano N, Fujiwara K, Matumoto M. Mouse hepatitis virus (MHV-2); plaque assay and propagation in mouse cell line DBT cells. Jpn J Microbiol. 1976;20:219–225. [PubMed] [Google Scholar]
- 23.Kim J C, Spence R A, Currier P F, Lu X T, Denison M R. Coronavirus protein processing and RNA synthesis is inhibited by the cysteine proteinase inhibitor e64d. Virology. 1995;208:1–8. doi: 10.1006/viro.1995.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koonin E V, Gorbalenya A E, Chumakov K M. Tentative identification of RNA-dependent RNA polymerases of ds RNA viruses and their relationship to positive strand RNA viral polymerases. FEBS Lett. 1989;252:42–46. doi: 10.1016/0014-5793(89)80886-5. [DOI] [PubMed] [Google Scholar]
- 25.Lai M M, Cavanagh D. The molecular biology of coronaviruses. Adv Virus Res. 1997;48:1–100. doi: 10.1016/S0065-3527(08)60286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lavi E, Wang Q, Weiss S R, Gonatas N K. Syncytia formation induced by coronavirus infection is associated with fragmentation and rearrangement of the Golgi apparatus. Virology. 1996;221:325–334. doi: 10.1006/viro.1996.0382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee H-J, Shieh C-K, Gorbalenya A E, Koonin E V, LaMonica N, Tuler J, Bagdzhadhzyan A, Lai M M C. The complete sequence (22 kilobases) of murine coronavirus gene 1 encoding the putative proteases and RNA polymerase. Virology. 1991;180:567–582. doi: 10.1016/0042-6822(91)90071-I. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu D X, Tibbles K W, Brown T D K. A 100-kilodalton polypeptide encoded by open reading frame (ORF) 1b of the coronavirus infectious bronchitis virus is processed by ORF 1a products. J Virol. 1994;68:5772–5780. doi: 10.1128/jvi.68.9.5772-5780.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu X T, Lu Y Q, Denison M R. Intracellular and in vitro translated 27-kDa proteins contain the 3C-like proteinase activity of the coronavirus MHV-A59. Virology. 1996;222:375–382. doi: 10.1006/viro.1996.0434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu X T, Sims A C, Denison M R. Mouse hepatitis virus 3C-like protease cleaves a 22-kilodalton protein from the open reading frame 1a polyprotein in virus-infected cells and in vitro. J Virol. 1998;72:2265–2271. doi: 10.1128/jvi.72.3.2265-2271.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu Y, Lu X, Denison M R. Identification and characterization of a serine-like proteinase of the murine coronavirus MHV-A59. J Virol. 1995;69:3554–3559. doi: 10.1128/jvi.69.6.3554-3559.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu Y Q, Denison M R. Determinants of mouse hepatitis virus 3C-like proteinase activity. Virology. 1997;230:335–342. doi: 10.1006/viro.1997.8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maynell L A, Kirkegaard K, Klymkowsky M W. Inhibition of poliovirus RNA synthesis by brefeldin A. J Virol. 1992;66:1985–1994. doi: 10.1128/jvi.66.4.1985-1994.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McBride A E, Schlegel A, Kirkegaard K. Human protein Sam68 relocalization and interaction with poliovirus RNA polymerase in infected cells. Proc Natl Acad Sci USA. 1996;93:2296–2301. doi: 10.1073/pnas.93.6.2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pachuk C J, Breedenbeek P J, Zoltick P W, Spaan W J M, Weiss S R. Molecular cloning of the gene encoding the putative polymerase of mouse hepatitis coronavirus, strain A59. Virology. 1989;171:141–148. doi: 10.1016/0042-6822(89)90520-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pedersen K W, van der Meer Y, Roos N, Snijder E J. Open reading frame 1a-encoded subunits of the arterivirus replicase induce endoplasmic reticulum-derived double-membrane vesicles which carry the viral replication complex. J Virol. 1999;73:2016–2026. doi: 10.1128/jvi.73.3.2016-2026.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perlman S, Reese D, Bolger E, Chang L J, Stoltzfus C M. MHV nucleocapsid synthesis in the presence of cycloheximide and accumulation of negative strand MHV RNA. Virus Res. 1987;6:261–272. doi: 10.1016/0168-1702(86)90074-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pinon J, Mayreddy R, Turner J, Khan F, Bonilla P, Weiss S. Efficient autoproteolytic processing of the MHV-A59 3C-like proteinase from the flanking hydrophobic domains requires membranes. Virology. 1997;230:309–322. doi: 10.1006/viro.1997.8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rasband, W. 1999. NIH Image 1.62b. [Online.] http://rsb.info.nih.gov/nih-image/. [26 May 1999, last date accessed.]
- 40.Restrepo-Hartwig M A, Ahlquist P. Brome mosaic virus helicase- and polymerase-like proteins colocalize on the endoplasmic reticulum at sites of viral RNA synthesis. J Virol. 1996;70:8908–8916. doi: 10.1128/jvi.70.12.8908-8916.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saborio J L, Pong S-S, Koch G. Selective and reversible inhibition of initiation of protein synthesis in mammalian cells. J Mol Biol. 1974;85:195–211. doi: 10.1016/0022-2836(74)90360-x. [DOI] [PubMed] [Google Scholar]
- 42.Sawicki D L, Sawicki S G. Coronavirus minus-strand RNA synthesis and effect of cycloheximide on coronavirus RNA synthesis. J Virol. 1986;57:328–334. doi: 10.1128/jvi.57.1.328-334.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schlegel A, Giddings T J, Ladinsky M S, Kirkegaard K. Cellular origin and ultrastructure of membranes induced during poliovirus infection. J Virol. 1996;70:6576–6588. doi: 10.1128/jvi.70.10.6576-6588.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sethna P B, Brian D A. Coronavirus genomic and subgenomic minus-strand RNAs copartition in membrane-protected replication complexes. J Virol. 1997;71:7744–7749. doi: 10.1128/jvi.71.10.7744-7749.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Snijder E J, Meulenberg J M. The molecular biology of arteriviruses. J Gen Virol. 1998;79:961–979. doi: 10.1099/0022-1317-79-5-961. [DOI] [PubMed] [Google Scholar]
- 46.Stohlman S A, Baric R S, Nelson G N, Soe L H, Welter L M, Deans R J. Specific interaction between coronavirus leader RNA and nucleocapsid protein. J Virol. 1988;62:4288–4295. doi: 10.1128/jvi.62.11.4288-4295.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van der Meer Y, van Tol H, Krijnse Locker J, Snijder E J. ORF1a-encoded replicase subunits are involved in the membrane association of the arterivirus replication complex. J Virol. 1998;72:6689–6698. doi: 10.1128/jvi.72.8.6689-6698.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Dinten L C, Wassenaar A L, Gorbalenya A E, Spaan W J, Snijder E J. Processing of the equine arteritis virus replicase ORF1b protein: identification of cleavage products containing the putative viral polymerase and helicase domains. J Virol. 1996;70:6625–6633. doi: 10.1128/jvi.70.10.6625-6633.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]